

Abstract
Compact CRISPR-Cas systems offer versatile treatment options for genetic disorders, but their application is often limited by modest gene-editing activity. Here we present enAsCas12f, an engineered RNA-guided DNA endonuclease up to 11.3-fold more potent than its parent protein, AsCas12f, and a third of the size of SpCas9. enAsCas12f shows higher DNA cleavage activity than wild-type AsCas12f in vitro and functions broadly in human cells, delivering up to 69.8% insertions and deletions at user-specified genomic loci. Minimal off-target editing is observed with enAsCas12f, suggesting that boosted on-target activity does not impair genome-wide specificity. We determine the Cryo-EM structure of the AsCas12f-sgRNA-DNA complex at 2.9 Å resolution, which reveals dimerization-mediated substrate recognition and cleavage. Structure-guided single guide RNA (sgRNA) engineering leads to sgRNA-v2, which is 33% shorter than the full-length sgRNA, but with on par activity. Together, the engineered hypercompact AsCas12f system enables robust and faithful gene editing in mammalian cells.
Introduction
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems, initially discovered as prokaryotic adaptive immune machinery1–4, have become the new frontier for genome engineering in higher eukaryotes5. In addition to the inherent nuclease activity, CRISPR systems have been functionalized with various effector proteins to enable programmed editing of the genome6–8, epigenome (summarized in ref. 9), transcriptome (summarized in ref. 10), and epitranscriptome11, 12 in a wide range of organisms. Recently, CRISPR technology has moved beyond the laboratory to rewrite pathogenic DNA sequences in patients, offering a powerful solution to various genetic diseases13–15.
CRISPR-Cas systems are broadly distributed in bacteria and archaea with remarkable evolutionary plasticity and functional diversity16. Among the Cas proteins characterized to date, Streptococcus pyogenes Cas9 (SpCas9, type II-A)17–19 and Acidaminococcus sp. BV3L6 Cas12a (AsCas12a, type V-A)20 have repeatedly shown potent gene-editing activity and broad tissue compatibility ex vivo21. Their therapeutic applications in vivo, however, demand safe and efficient delivery strategies. Adeno-associated viruses (AAV) are the leading candidates for in vivo delivery of gene-editing agents22, 23, owing to their long application history in the clinic, lack of pathogenicity and immunogenicity, and programmable tissue tropism. AAV vectors have a maximum packaging capacity of 4.7 kb, insufficient to accommodate SpCas9 (1,368 amino acids) or AsCas12a (1,307 amino acids) and their essential auxiliary components (Fig. 1a). The packaging obstacle can be partially addressed using split Cas proteins24–27, but these designs often lead to lower efficiency as a cell must be infected by at least two different AAV particles to acquire the intact CRISPR complex. Cas proteins of comparable nuclease activity but smaller sizes provide a more straightforward solution to the delivery challenge and may further advance clinical applications of gene-editing agents.
Fig. 1 |. Engineering AsCas12f for increased genome-editing efficiency.

a, Domain organization of AsCas12f compared with SpCas9, AsCas12a, DpbCasX, and UnCas12f. HNH, REC, and RuvC domains are indicated. Protein lengths are drawn to scale. aa: amino acid. b, Sequence alignment of AsCas12f and its homologous proteins. Representative regions are shown, with candidates for mutagenesis highlighted in red boxes. c, Workflow to determine the cellular activity of AsCas12f and its variants. d, e, Indel levels at TP53-1 (d) and HEXA (e) loci generated by AsCas12f variants that bear one, two, three, four, or five single-point mutations. A list of mutations included in each AsCas12f variant is provided in Supplementary Fig. 4. Two independent replicates were carried out in HEK293T cells. f, g, Time-course in vitro DNA cleavage by wild-type AsCas12f and enAsCas12f at 37 °C (f) and 50 °C (g). Data points were fitted to one-phase exponential association curves. Two independent replicates were carried out. Gel images are provided in Supplementary Fig. 5.
Several compact Cas proteins have been reported, including CasX (Cas12e, V-E, 986 amino acids)28, CasΦ (Cas12j, V-J, 700–800 amino acids)29, and Cas12f (also known as Cas14, V-F, 400–700 amino acids)30–32. The IscB and TnpB family proteins (~400 amino acids), putative ancestors of Cas9 and Cas12a, also confer RNA-guided nuclease activity33–38. Of these, Cas12f proteins are of particular interest given their small sizes (Fig. 1a) and unique dimerization-mediated DNA-targeting mechanism39, 40. Initially identified as single-stranded DNA (ssDNA)-specific nucleases30, Cas12f proteins were later demonstrated capable of cleaving double-stranded DNA (dsDNA) with 5′ T- or C-rich protospacer adjacent motifs (PAMs)31, 41. Several family members, including uncultured archaeon Cas12f1 (UnCas12f1, 529 amino acids, hereafter referred to as UnCas12f), Acidibacillus sulfuroxidans Cas12f1 (AsCas12f1, 422 amino acids, hereafter referred to as AsCas12f for simplicity), Oscillibacter sp. Cas12f1 (OsCas12f1, 433 amino acids), and Ruminiclostridium herbifermentans Cas12f1 (RhCas12f1, 415 amino acids), have been experimentally validated for programmed DNA cleavage in mammalian cells41–45. The DNA-targeting activity of UnCas12f was further improved by protein and guide RNA (gRNA) engineering43, 44. AAV-mediated delivery of Cas12f proteins and their cognate gRNAs resulted in successful genome editing in human embryonic kidney (HEK) 293, U2-OS, Huh-7 cells, and laboratory mice41, 42, 44, 45, highlighting the therapeutic potential of the Cas12f family. Albeit promising, Cas12f systems have a significant margin for improvement as gene-editing agents. Compared to the well-characterized Cas9 and Cas12a complexes, Cas12f systems are less potent in cleaving dsDNA and show larger activity variations when targeting different genomic loci41–44.
In this study, we take a rational approach to engineer the AsCas12f system and obtain AsCas12f variants that generate programmed double-stranded breaks (DSBs) 2- to 11-fold more efficiently than the wild-type protein in the human genome. We determine a cryo-EM structure of AsCas12f in complex with the single guide RNA (sgRNA) and the target DNA at 2.9 Å resolution, advancing our mechanistic understanding of type V-F CRISPR systems. Guided by the cryo-EM structure, we truncate 72 nt sequences from the 194 nt sgRNA without compromising the DNA-targeting and cleavage activity of the ribonucleoprotein complex. While highly potent in editing the target genomic loci, engineered AsCas12f gives rise to minimal off-target edits, as assessed by genome-wide, unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq)46. Collectively, we report an engineered hypercompact CRISPR system for robust and faithful gene editing in mammalian cells.
Results
AsCas12f variants with boosted DNA cleavage activity
Nuclease-based genome-editing agents disrupt gene function by introducing DSBs site-specifically in the genome. DSBs are sensed by DNA damage repair machinery and are fixed primarily by nonhomologous end-joining (NHEJ), resulting in the formation of random insertions and deletions (indels)5. Although AsCas12f has demonstrated capable of introducing DSBs in the human genome, its activity is modest and varies substantially among different loci41–43.
We hypothesize that AsCas12f-mediated DNA targeting and cleavage can be improved by increasing the affinity of AsCas12f to the gRNA and target DNA. Protein-nucleic acid engagement is frequently mediated by electrostatic interactions between phosphodiester backbones of nucleic acids and positively charged patches on proteins47. As such, introduction of basic residues such as lysine (K) and arginine (R) into Cas proteins may increase their affinity to nucleic acids, resulting in boosted DNA-targeting and cleavage activity. Similar strategies have been successfully applied to AsCas12a for improved activity and targeting ranges48.
To properly place positively charged residues, we compared AsCas12f with naturally occurring homologous proteins (Fig. 1b, Supplementary Fig. 1 and Fig. 2). We focused on aligned positions that are occupied by neutral or negatively charged residues in AsCas12f but harbor positively charged residues in AsCas12f homologs and generated 32 variants, each bearing a single amino acid substitution (Supplementary Fig. 3). Plasmids encoding AsCas12f variants and its sgRNA targeting two genomic loci, TP53 and HEXA, were delivered into HEK293T cells through lipid-mediated transfection. A nuclear localization signal (NLS) peptide was appended to AsCas12f at both termini (Fig. 1c). Gene-editing activity of AsCas12f variants was assayed by targeted DNA sequencing and quantified by CRISPResso49. Although many AsCas12f variants showed similar or lower activity compared to the wild-type protein, eight single-point mutations, including D196K, N199K, G276R, D281K, T327K, N328G, D364K, D364R, increased the indel frequency at one or both target sites (Fig. 1d, Supplementary Fig. 3).
Fig. 2 |. Genome editing facilitated by engineered AsCas12f systems.
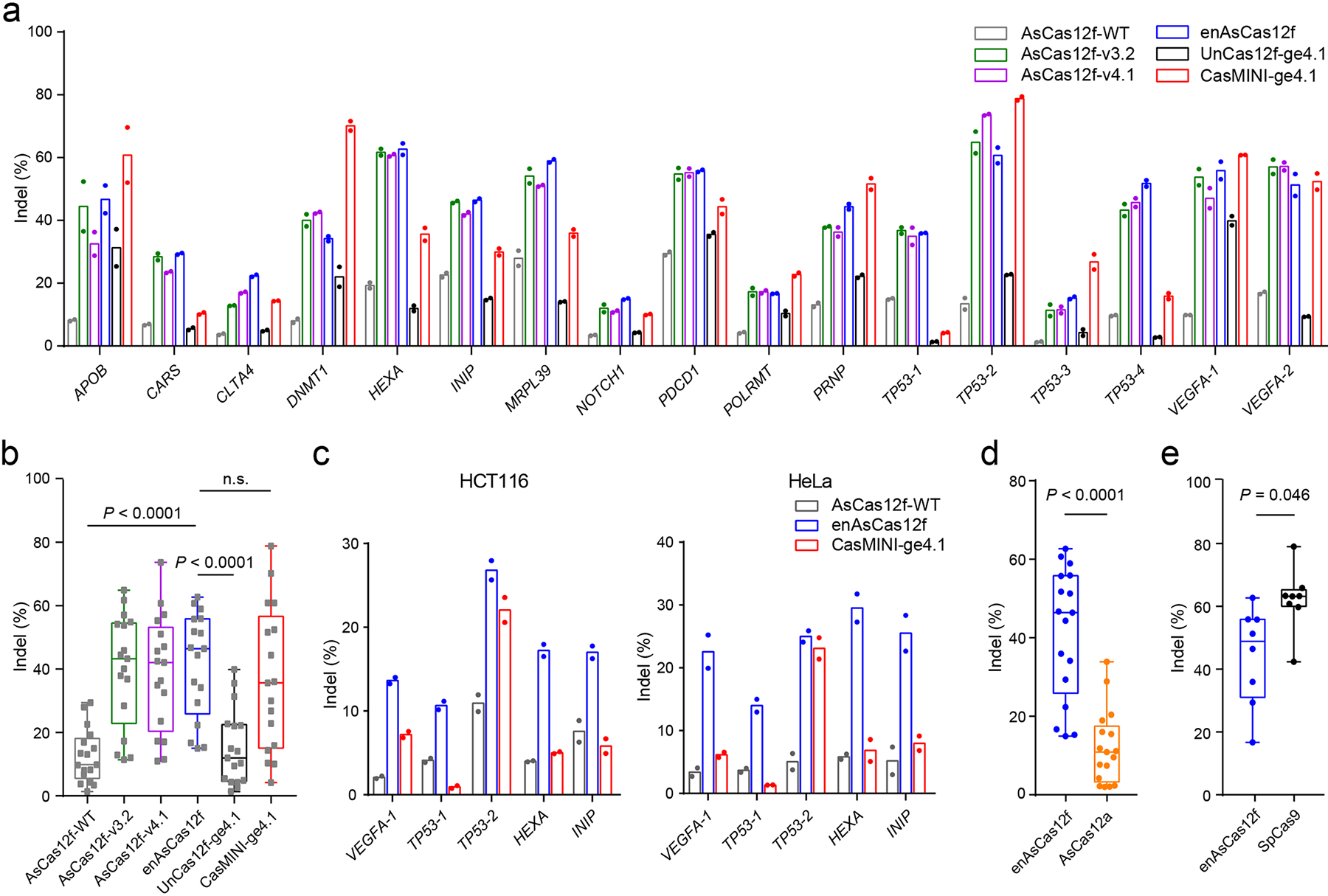
a, Indel frequencies mediated by wild-type and engineered AsCas12f, in comparison to wild-type UnCas12f and CasMINI with an engineered UnCas12f sgRNA (ge4.1), in HEK293T cells. b, Box-and-whisker plot of indel frequencies delivered by AsCas12f and UnCas12f systems shown in a. c, Indel frequencies mediated by wild-type AsCas12f, enAsCas12f, and CasMINI-ge4.1 in HCT116 (left) and HeLa (right) cells. d, Box-and-whisker plot of indel frequencies delivered by enAsCas12f and AsCas12a. e, Box-and-whisker plot of indel frequencies delivered by enAsCas12f and SpCas9. Two independent replicates were carried out in a and c. For b, d, and e, all data points (n = 17 target sites in b and d, n = 8 target sites in e) were plotted, with the centerline representing the median and the whiskers showing the minimum to the maximum. The boundaries of the box indicate the first and third quantiles. P values were determined by two-tailed paired Student’s t-test.
We next queried if we could further increase AsCas12f activity by combining beneficial mutations and tested a collection of AsCas12f variants harboring double, triple, quadruple, and quintuple mutations. Many assayed combinations gave rise to greater levels of indels (up to 73.6%), with the best variant (AsCas12f-v5.2: D196K/N199K/G276R/N328G/D364R) exhibiting 2.5- to 3.5-fold higher gene-editing activity at all three tested target sites (Fig. 1d–e, Supplementary Fig. 4). The observed improvement in activity cannot be attributed to differences in protein expression or stability, as Flag-tagged wild-type AsCas12f, AsCas12f-v4.1, and AsCas12f-v5.2 exhibited similar protein levels in HEK293T cells (Supplementary Fig. 5a). We name our best variant (AsCas12f-v5.2) enhanced AsCas12f (enAsCas12f).
We heterologously expressed and purified wild-type AsCas12f and enAsCas12f in Escherichia coli. enAsCas12f was more active than wild-type AsCas12f in cleaving dsDNA at both 37 °C and 50 °C (Fig. 1f, g, Supplementary Fig. 5b–d). In HEK293T cells, enAsCas12f forms a similar indel pattern as wild-type AsCas12f, indicating that the preferred sites of cleavage in the target DNA remain unchanged (Supplementary Fig. 6a–b). Deletion signals center at 19–24 bp downstream of the PAM and extend beyond the 3’ end of the protospacer (Supplementary Fig. 6a–c). Consistent with a previous study42, minimal insertions were detected with all AsCas12f variants (Supplementary Fig. 6a, c). Therefore, enAsCas12f boosts gene-editing efficiency while maintaining key features of AsCas12f.
Genome editing facilitated by the enAsCas12f system
We next evaluated whether enAsCas12f functions broadly in the human genome and chose 17 genomic loci, covering TTTG, TTTA, ATTG, and CTTG PAM sequences, for characterization. In addition to enAsCas12f, we included two engineered AsCas12f variants bearing three (AsCas12f-v3.2: D196K/N199K/N328G) and four (AsCas12f-v4.1 D196K/N199K/N328G/D364R) mutations that generated high indel rates in our screening.
In line with a previous report42, wild-type AsCas12f generated <20% indels at most target sites (Fig. 2a, 12.6±8.4%; mean±s.d.). All three engineered AsCas12f proteins showed notably higher indel levels than the wild-type protein at all tested loci across different PAMs (AsCas12f-v3.2: 39.8±17.8%, AsCas12f-v4.1: 38.8±18.3%, enAsCas12f: 41.4±16.6%, Fig. 2a, Supplementary Fig. 7a–c). Among them, enAsCas12f led to highest editing efficiency at 12 of 17 target sites (Fig. 2a), delivering up to 11.3-fold more indels than the wild-type protein with a 4.5-fold improvement on average (Supplementary Fig. 7d, e).
Another Cas12f family member, UnCas12f, shows minimal gene-editing activity when assayed in mammalian cells44. Systematic gRNA engineering has led to ge4.1, which, when complexed with wild-type UnCas12f, delivers significantly enhanced gene-editing activity44. In a separate work, researchers carried out structure-guided protein engineering and obtained a variant, namely CasMINI, that increases DNA-binding and cleavage activity of the UnCas12f system43. We therefore tested UnCas12f and CasMINI in combination with the engineered gRNA scaffold. We observed 1.3- to 6.2-fold more indels with CasMINI than wild-type UnCas12f when paired with ge4.1 (Fig. 2a, Supplementary Fig. 7f), confirming that the engineered gRNA scaffold is broadly compatible with UnCas12f and its derivatives.
We compared our engineered AsCas12fs with UnCas12f-ge4.1 and CasMINI-ge4.1 across 17 genomic loci. The side-by-side comparison is possible because both AsCas12f and UnCas12f recognize T-rich PAMs. enAsCas12f generated 41.4±16.6% indels at 17 targets, higher than indel rates delivered by UnCas12f-ge4.1 (15.2±12.0%) and CasMINI-ge4.1 (36.8±22.8%, Fig. 2a–b). In particular, enAsCas12f beat UnCas12f-ge4.1 at all times and outperformed CasMINI-ge4.1 at 10 of 17 target sites (Fig. 2a, b). Notably, CasMINI exhibited more scattered indel levels than enAsCas12f (Fig. 2a, b, Supplementary Fig. 7a–c), likely indicating a stronger context dependence.
We also compared enAsCas12f with two commonly used Cas proteins, AsCas12a and SpCas9. AsCas12a recognizes T-rich PAMs located upstream of the protospacer. We therefore assayed AsCas12a-mediated indel formation directly at target sites designed for AsCas12f. AsCas12a generated 12.2±9.4% indels across 17 target sites, defeated by enAsCas12f in all cases (Fig. 2d, Supplementary Fig. 8a). SpCas9 recognizes 5’-NGG PAMs at the 3’ end of target sites. We selected five loci that carry 5’-NGG PAMs among the 17 loci and designed three additional spacers for SpCas9 that recognize sites adjacent to those targeted by enAsCas12f. SpCas9 showed activity comparable to or higher than that of enAsCas12f at all eight target sites (62.2±10.0% indels, Fig. 2e, Supplementary Fig. 8b).
Cas12f systems have recently been repurposed for base editing, transcription repression and activation43, 45, 50, 51, offering a versatile toolbox for genome engineering. However, transcription activation of endogenous genes using AsCas12f has not been reported. To this end, we constructed CRISPR activation (CRISPRa) systems by fusing a transcription activator complex VP64-p65-Rta (VPR)52 to dead AsCas12f variants (D225A) (Supplementary Fig. 8c) and compared them with previously established dCas9- and dCas12a-based CRISPRa systems at three genomic loci. Wild-type AsCas12f modestly activated transcription of HBB in HEK293T cells, but was unable to do so to HBG and IL1RN. In comparison, enAsCas12f increased transcription of all three genes by 2- to 23-fold. Meanwhile, AsCas12a gave rise to >100-fold activation of IL1RN and HBB, and SpCas9 led to >1,000-fold activation of all three genes (Supplementary Fig. 8d). The moderate gene activation capability observed for AsCas12f in our CRISPRa assays may be due to suboptimal protein fusion strategies, which could impede dimerization and/or DNA binding of AsCas12f. Note that we directly used spacer sequences previously reported for CasMINI43 in our assays for AsCas12a and enAsCas12f. Spacer screening, which is often necessary to achieve optimal transcription activation, may improve the performance of AsCas12a and enAsCas12f in CRISPRa experiments.
We next evaluated enAsCas12f in HCT116 and HeLa cells. enAsCas12f showed markedly higher gene-editing efficiency than wild-type AsCas12f in both cell lines and delivers indel frequencies higher than CasMINI-ge4.1 at 5 of 6 loci (Fig. 2e). Collectively, enAsCas12f is a potent gene-editing agent that functions broadly in human cells.
Cryo-EM structure of the AsCas12f complex
To gain mechanistic insight into the AsCas12f system and the functional impact of enAsCas12f mutations, we purified ribonucleoprotein consisting of nuclease-deficient AsCas12f (AsCas12f-D225A), sgRNA (194 nt), and a target dsDNA (42 bp) with a TTTG PAM sequence. Cryo-electron microscopy (cryo-EM) and single-particle analysis were performed (Supplementary Fig. 9a, Supplementary Table 5), which resulted in a complex structure determined at 2.9 Å resolution (Supplementary Fig. 9b).
AsCas12f contains an N-lobe that spans the wedge (WED) domain and the recognition (REC) domain, and a C-lobe constituting the RuvC endonuclease domain and a zinc finger motif (ZF) (Fig. 3a). Two copies of AsCas12f, hereby referred to as AsCas12f.1 and AsCas12f.2, are identified in the cryo-EM map, which form an asymmetric dimer wrapping around the sgRNA and the target DNA (Fig. 3b). Similar to UnCas12f39, 40, the N-lobe of AsCas12f.1 is involved in template recognition by interacting with the PAM, whereas the C-lobe of AsCas12f.1 and the N-lobe of AsCas12f.2 make extensive contacts with both the sgRNA and the target DNA (Fig. 3b, c). The C-lobe of AsCas12f.2 is situated close to the cleavage site of the target DNA, but is poorly resolved due to structural flexibility (Supplementary Fig. 10a, b). The folding of monomeric AsCas12f and UnCas12f is similar (Supplementary Fig. 11a–c), while AsCas12f is smaller than UnCas12f because the REC domain of UnCas12f hosts an additional zinc finger motif (78 aa) close to its N-terminus (Supplementary Fig. 11c).
Fig. 3 |. Cryo-EM structure of the AsCas12f-sgRNA-DNA complex.
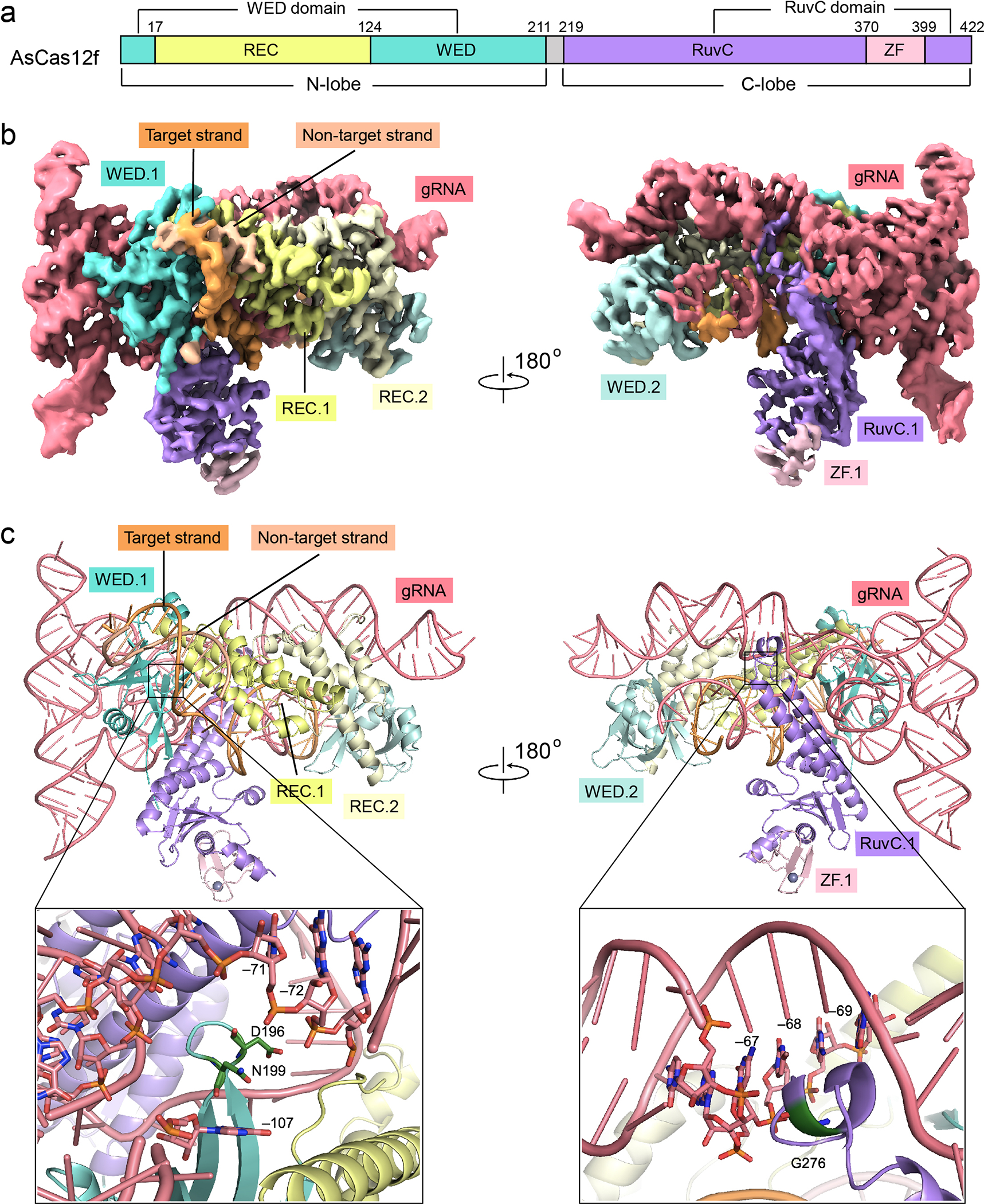
a, Domain structure of AsCas12f. The N-lobe contains a wedge (WED) domain and a recognition (REC) domain. The C-lobe includes a RuvC nuclease domain and a zinc finger (ZF) motif. b, Unsharpened cryo-EM map for the AsCas12f-gRNA-DNA complex (contoured at a level of 0.020). c, Top: atomic model of the AsCas12f-gRNA-DNA complex. Bottom: close-up views of residues mutated in enAsCas12f (D196K, N199K, G276R). Note that these residues are close to the backbone of the sgRNA. In b and c, AsCas12f domains are colored as shown in a.
AsCas12f dimerizes through an extensive interface in the REC domain (Supplementary Fig. 12a). Alanine substitutions that disrupt the dimer interface, including E44A, D51A, and Y52A, led to decreased indel frequencies when assayed in HEK293T cells (Supplementary Fig. 12b), suggesting that proper dimerization is essential for AsCas12f to engage and cleave DNA.
AsCas12f recognizes T-rich PAMs by interacting with both the non-target strand (K80, S92 in REC.1, Supplementary Fig. 13a) and the target strand (K96, S92 in REC.1, Supplementary Fig. 13b). Apart from the PAM, AsCas12f also pervasively interacts with the phosphodiester groups of the target DNA (Supplementary Fig. 13a, b). Mutating residues that interact with the target DNA again led to reduced indel frequencies (Supplementary Fig. 13c, blue bars).
The sgRNA of AsCas12f, initially created by fusing a 49-nt CRISPR RNA (crRNA) and a 138-nt trans-activating CRISPR RNA (tracrRNA), is comprised of five stem loops (Supplementary Fig. 14a, b). Among them, stem 2 engages both AsCas12f monomers, making major contributions to protein-sgRNA assembly (Fig. 3c). Notably, AsCas12f sgRNA is much longer and adopts a tertiary structure distinct from UnCas12f sgRNA (Supplementary Fig. 14b–d). Superimposition of AsCas12f and UnCas12f sgRNAs suggests that the two RNA sequences differ mainly by their 3’ ends (Supplementary Fig. 14d). A single turn in stem 5 of UnCas12f sgRNA directs the spacer towards the target DNA (Supplementary Fig. 14c, d), while a similar task is fulfilled by a long stretch of stems 3–5 in AsCas12f sgRNA (Supplementary Fig. 14b, d). Alanine substitutions of AsCas12f residues interacting with the sgRNA, such as W17, H72, R121, and Y351 (Supplementary Fig. 15a–e), decreased indel frequencies (Supplementary Fig. 13c, orange bars). Notably, three mutated residues in enAsCas12f, D196, N199, and G276, are located in close proximity to the phosphodiester backbone of the sgRNA (Fig. 3c), providing mechanistic support to our hypothesis that supplementing electrostatic interactions at these positions facilitates complex formation and target DNA engagement.
Structure-guided sgRNA engineering
In addition to protein engineering, modifications to the gRNA have improved the gene-editing performance of several CRISPR systems32, 43, 44, 53, 54. Based on our cryo-EM structure, we hypothesized that truncation of the sgRNA, especially in regions that do not directly interact with AsCas12f, might reduce the flexibility of the complex and consolidate key interactions with enAsCas12f. The poorly resolved cryo-EM density of U(–47)-U(–15) in stem 5 (Supplementary Fig. 10a, b, grey box in Supplementary Fig. 14a) suggests this segment is flexible and does not intimately interact with AsCas12f. Indeed, truncation of U(–47)-U(–15) did not impact indel formation efficiency (stem 5–1 and stem 5–2, Fig. 4a, Supplementary Fig. 16a). In contrast, truncating ≥ 3 bp from the spacer-proximal region of stem 5 (yellow box in Supplementary Fig. 14a) abolished DNA cleavage (stem 5–4, stem 5–5, and stem 5–6, Fig. 4a, Supplementary Fig. 16b). Intriguingly, removal of the entire stem 5 (stem 5–3, Supplementary Fig. 16c) resulted in a slightly higher indel level than trimming 5 bp from the spacer-proximal region (stem 5–6, Fig. 4a), suggesting that the complex may adopt a different conformation to compensate large truncations. Single base-pair modifications of stem 2 led to decreased indel levels (stem 2–1, stem 2–2, and stem 2–3, Fig. 4a, Supplementary Fig. 16d), reinforcing stem 2 as a major contributor to sgRNA-protein interactions.
Fig. 4 |. Structure-guided engineering of the AsCas12f gRNA.
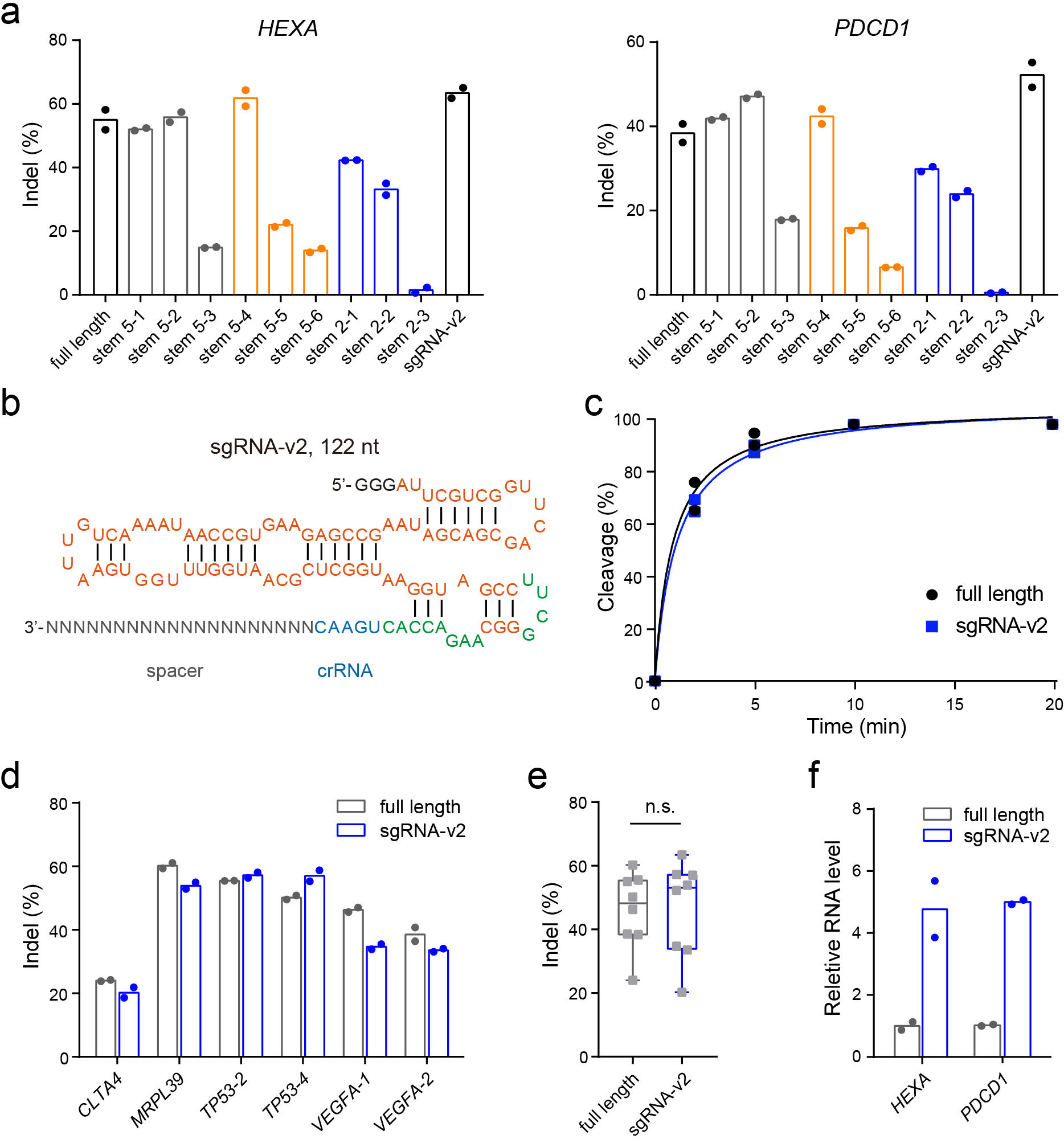
a, Indel frequencies mediated by enAsCas12f with engineered AsCas12f gRNAs at HEXA and PDCD1 loci. The structures of engineered gRNAs are shown in Supplementary Fig. 14. b, Structure of sgRNA-v2. c, Time-course in vitro DNA cleavage using full-length sgRNA and sgRNA-v2. The assay was conducted using enAsCas12f at 37 °C. Data points were fitted to one-phase exponential association curves. Gel images are provided in Supplementary Fig. 16. d, Indel frequencies mediated by the full-length sgRNA and sgRNA-v2 in complex with enAsCas12f at denoted genomic loci in HEK293T cells. e, Box-and-whisker plot of indel frequencies mediated by the full-length sgRNA or sgRNA-v2 in complex with enAsCas12f. All data points (n = 8 target sites) were plotted, with the centerline showing the median and the whiskers showing the minimum to the maximum. The boundaries of the box indicate the first and third quantiles. P values were determined by two-tailed paired Student’s t-test. f, Relative abundance of full-length sgRNA and sgRNA-v2 targeting HEXA and PDCD1 loci in HEK293T cells. Two independent replicates were carried out in a, c, and d.
Considering that stems 3 and 4 do not form strong interactions with AsCas12f, and stem 5 truncation does not impair the complex (Fig. 4a), we removed the entire stems 3 and 4 and modified stem 5 to yield a compact AsCas12f sgRNA. This new sgRNA, which we named sgRNA-v2, is 72 nt shorter than the original sequence (>33% decrease in molecular weight, Fig. 4b). When complexed with enAsCas12f, sgRNA-v2 showed DNA cleavage activity on par with the full-length sgRNA in vitro (Fig. 4c, Supplementary Fig. 16e). The robust activity of sgRNA-v2 extends to indel formation across eight assayed target sites in HEK293T cells (Fig. 4a, d, e).
We next tested whether truncation led to more efficient expression of sgRNA-v2 since the U6 promoter may favor shorter, less structured transcripts. The abundances of full-length sgRNA and sgRNA-v2 in transfected HEK293T cells were examined using quantitative reverse transcription PCR (RT-qPCR). sgRNA-v2 showed ~4-fold higher expression than the full-length sgRNA (Fig. 4f). The improved expression, however, did not translate into higher indel formation frequencies, indicating that the cellular activity of the enAsCas12f system is not limited by sgRNA expression. Altogether, the cryo-EM structure of the AsCas12f-sgRNA-DNA complex enabled rational gRNA engineering, yielding a more compact and potent AsCas12f system.
Off-target effects of engineered Cas12f systems
We next interrogated genome-wide specificity of engineered AsCas12f variants by GUIDE-seq, wherein DNA breakage sites are mapped by integration of a double-stranded oligonucleotide (dsODN). GUIDE-seq has been applied to profile the off-target effects of various Type V CRISPR systems, including AsCas12a and LbCas12a that introduce DSBs of similar patterns as AsCas12f55. We analyzed the 17 target sites assayed in this study using Cas-OFFinder56 and selected five sites with the largest numbers of potential off-target sites for GUIDE-seq (Supplementary Table 6). Consistent with results obtained from lipid-mediated transfection (Fig. 2a, c, d), AsCas12f-v4.1 and enAsCas12f showed high potency at the on-target sites in GUIDE-seq, generating up to 20.7% and 34.6% indels, respectively (Fig. 5a). In contrast, much lower indel frequencies (up to 2.6%) were observed with wild-type AsCas12f. We note that the overall indel rates are lower in GUIDE-seq because delivery is compromised by co-electroporation of large amounts of dsODN. dsODN-bearing reads constitute 0.8–7.5% of indel-containing reads among all GUIDE-seq samples (Supplementary Fig. 17a). These numbers are comparable to dsODN integration efficiency observed for AsCas12a and LbCas12a, and are lower than the levels delivered by SpCas9 in GUIDE-seq46, 55.
Fig. 5 |. Genome-wide specificity of wild-type and engineered AsCas12f.
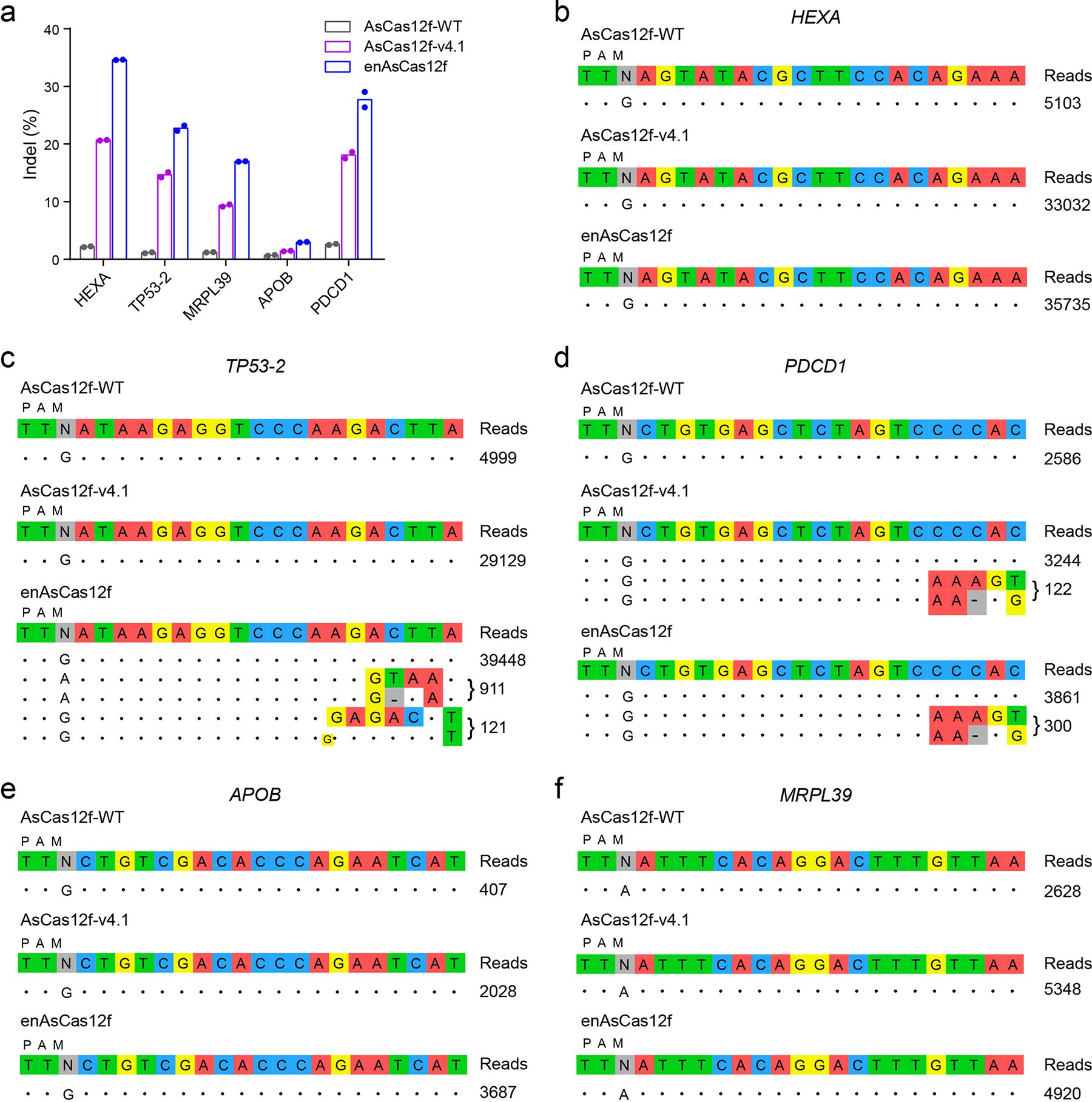
a, On-target indel frequencies in GUIDE-seq samples. b-f, Off-target editing sites for wild-type AsCas12f, AsCas12f-v4.1, and enAsCas12f with sgRNAs targeting HEXA (b), TP53-2 (c), PDCD1 (d), APOB (e), and MRPL39 (f) loci reported by GUIDE-seq in HEK293T cells. Mismatch positions are highlighted in colors. GUIDE-seq experiments were performed in duplicates, with the read counts of one replicate shown to the right of the corresponding sequences. Results from the other replicate are shown in Supplementary Fig. 17. Full-length sgRNAs were used in all GUIDE-seq experiments.
We allocated a similar number of reads to each sample in deep sequencing. However, more deduplicated reads (1.3- to 7.9-fold) were mapped to the on-target sites for AsCas12f-v4.1 and enAsCas12f than wild-type AsCas12f (Fig. 5b–f), in line with the higher indel rates observed for engineered AsCas12f variants. No off-target integration was detected for wild-type AsCas12f for all assayed sgRNAs (Fig. 5b–f, Supplementary Fig. 17b–f). enAsCas12f hit two potential off-target sites when complexed with the TP53-2-targeting sgRNA – site 1 (911/39,448, reads mapped to off-target/on-target sites) and site 2 (121/39,448, Fig. 5c). Both AsCas12f-v4.1 and enAsCas12f led to potential integration at one off-target site when complexed with sgRNA targeting the PDCD1 locus (site 3, 122/3,244 for AsCas12f-v4.1, 300/3,861 for enAsCas12f, Fig. 5d). Amplicon deep sequencing of site 1 revealed 0.6% indels for enAsCas12f, with wild-type AsCas12f and AsCas12f-v4.1 delivering signals hardly distinguishable from the background (0.1%). No indels were observed at site 2 for all three proteins. Collectively, the engineered AsCas12f proteins are both potent and faithful in editing the human genome.
Discussion
Compact CRISPR-Cas systems are highly desired for gene-editing applications, especially for in vivo delivery using vectors with a cargo-size limit. In this work, we rationally engineer AsCas12f, one of the most compact RNA-guided endonucleases identified so far, for improved gene-editing activity. We choose mutation sites based on sequence alignment with naturally occurring homologous proteins, a strategy that avoids many deleterious mutations. Consequently, we identify beneficial mutations at a rate (8/32) notably higher than random mutagenesis and some structure-guided approaches. These beneficial mutations, when combined, lead to enAsCas12f, an AsCas12f variant up to 11.3-fold more potent in editing the human genome than the wild-type protein.
enAsCas12f generates high indel frequencies at PAM-distal regions, similar to wild-type AsCas12f and UnCas12f. We note that the gene-editing efficiency for both wild-type and engineered AsCas12f can vary across target sites. This observation cannot be fully explained by differences in chromatin states, because UnCas12f appears to favor a different set of target sites. The mechanism through which Cas12f searches and identifies its target DNA requires further investigation. Additionally, both wild-type and engineered AsCas12f proteins show limited compatibility with CRISPRa in our tested condition. Further optimization of protospacer selection and fusion design is necessary to fully capitalize on AsCas12f-mediated gene regulation.
In the cryo-EM structure, two AsCas12f monomers wrap asymmetrically around one copy of gRNA and DNA, serving distinct roles in nucleic acid recognition and possibly DNA cleavage. Interestingly, AsCas12f purifies as a monomer in the absence of nucleic acids, suggesting a dimerization process driven by the gRNA. Indeed, although AsCas12f and UnCas12f share similar folds, the two proteins employ different residues at the dimerization interfaces and recognize structurally distinct gRNAs. These observations may indicate a gRNA-first evolutionary principle for type V-F CRISPR systems.
Our enAsCas12f system delivers activity on par with, or higher than, the combination of CasMINI – an engineered UnCas12f protein – and ge4.1 – an optimized UnCas12f gRNA. While proficient in cleaving on-target sites, enAsCas12f shows minimal off-target editing in the human genome. All three putative off-target sites identified in this study host 5’-TTN PAMs and are capable of forming at least 13 perfect base pairs with the corresponding sgRNA immediately downstream of the PAM, suggesting that the AsCas12f system is sensitive to mismatches in the PAM-proximal region. Future investigations are needed to determine whether the observed PAM-proximal mismatch sensitivity extends to other spacer sequences. AsCas12f is ~30% of the size of SpCas9 and AsCas12a, and is 20% smaller than its ortholog UnCas12f (Fig. 1a). As one of the most efficient and compact CRISPR systems reported to date, enAsCas12f unlocks new territory in CRISPR-based gene editing.
Online Methods
Plasmid construction
AsCas12f gene fragments codon-optimized for Escherichia coli and human expression were synthesized by Genewiz. Oligonucleotides were ordered from Integrated DNA Technologies. For recombinant AsCas12f expression and purification, Escherichia coli-codon-optimized AsCas12f was cloned into a pET47b vector following an N-terminal His6-tag. For genome editing in human cells, CMV-driven AsCas12f and U6-driven sgRNA were cloned into two separate plasmids of pBR322 origins. For CRISPRa, catalytically inactive Cas proteins were fused to VPR with an SV40 NLS linker and cloned into the same vector. DNA fragments for plasmid construction were PCR amplified using Phusion U DNA Polymerase (Thermo Fisher, F555S) and assembled by USER enzyme mix (New England Biolabs, M5505L). AsCas12f mutants and sgRNA plasmids were generated by site-directed mutagenesis.
Cell culture
HEK293T cells were purchased from ATCC (CRL11268) and were cultured in DMEM (Gibco 11995) supplemented with 10% (v/v) fetal bovine serum (Gibco), 1% penicillin and streptomycin (Gibco). HeLa cells were purchased from ATCC (CCL2) and were cultured in DMEM (Gibco 11965) supplemented with 10% (v/v) fetal bovine serum (Gibco), 1% penicillin and streptomycin (Gibco). HCT116 cells were purchased from ATCC (CCL-247) and were cultured in McCoy’s 5A (Gibco 16600) supplemented with 10% (v/v) fetal bovine serum (Gibco), 1% penicillin and streptomycin (Gibco). Cells were grown at 37 °C with 5% CO2.
Evaluation of indel frequencies
Transfection was carried out in 96-well plates. 120 ng plasmid encoding AsCas12f and 120 ng plasmid encoding the sgRNA were transfected using 0.5 μL Lipofectamine 2000 reagent (Thermo Fisher, 11668019) in 50 μL optiMEM (Gibco). Cells were harvested 3 days after transfection and were lysed with 50 μL lysis buffer (10 mM Tris-HCl pH 8.0, 0.05% SDS, 20 μg/mL proteinase K (Thermo Fisher, EO0491)). The lysate was incubated for 60 min at 37 °C, followed by 40 min at 55 °C, 30 min at 85 °C, and 10 min at 95 °C. Target-specific primers were used to amplify 200–400 bp regions surrounding the target site using Taq DNA polymerase (New England Biolabs, M0273L), with 1 μL cell lysate supplied as templates. Spacer sequences for all genomic target sites are listed in Supplementary Tables 2–4. Sequences of target-specific primers are included in Supplementary Data 1. Amplicons were further tagged with Illumina TruSeq indexes through PCR. The final PCR products were gel-purified and subjected to 150-bp pair-ended sequencing on an Illumina Miseq platform. Indel frequencies were calculated by CRISPEResso248 using the Cpf1 mode (for AsCas12f and AsCas12a) or the Cas9 mode (for SpCas9) with 2 bp quantification windows.
CRISPRa
Transfection was carried out in 24-well plates seeded with HEK293T cells. 240 ng plasmid encoding a CRISPRa cassette and 240 ng plasmid encoding the sgRNA were transfected using 1 μL Lipofectamine 2000 reagent (Thermo Fisher, 11668019) in 100 μL optiMEM (Gibco). Total RNA was isolated from cells using TRIzol Reagent (Thermo Fisher, 15596026) 2 days after transfection. 500 ng purified total RNA was subjected to reverse transcription using Maxima H Minus cDNA Synthesis Master Mix (Thermo Fisher, M1661). Relative mRNA levels were determined by qPCR, normalizing to the level of GAPDH. qPCR primer sequences are included in Supplementary Table 1.
Evaluation of sgRNA expression levels
HEK293T cells were seeded in 6-well plates. 300 ng plasmid encoding AsCas12f and 300 ng plasmid encoding the sgRNA were transfected using 1.2 μL Lipofectamine 2000 reagent (Thermo Fisher, 11668019) in 100 μL optiMEM (Gibco). Total RNA was isolated from cells using TRIzol Reagent (Thermo Fisher, 15596026) 3 days after transfection. To a 20 μL reaction, 5 μg total RNA was subjected to poly(A) tailing using 5 units (1 μL) of E. coli Poly(A) Polymerase (New England Biolabs, M0276S) following manufacturer’s protocol. RNA was then purified using the RNA Clean & Concentrator kit (Zymo Research, R1014). 500 ng purified RNA was subjected to reverse transcription using the T6-RT primer and SuperScript III Reverse Transcriptase (Thermo Fisher, 18080044). Relative sgRNA levels were determined by qPCR, normalizing to the level of GAPDH. Sequences of the T6-RT primer and qPCR primers are provided in Supplementary Table 1.
Western blot
Cell lysates were denatured and analyzed by SDS-PAGE. Proteins were transferred to nitrocellulose membranes, which were blocked by 5% milk in PBS containing 0.1% Tween-20 (PBST). Membranes were then incubated with HRP-conjugated monoclonal anti-Flag (Sigma-Aldrich, A8592-.2MG, 1:1000) or HRP-conjugated GAPDH monoclonal (Proteintech, HRP-60004, 1:1000) antibodies. Membranes were washed 5 times with PBST before being applied with ECL and developed.
sgRNA preparation
DNA templates for sgRNA production were generated by PCR. sgRNA sequences are available in Supplementary Table 1. sgRNAs were prepared by in vitro transcription using T7 RNA polymerase (New England Biolabs, M0251L) following the manufacturer’s protocol. In general, 50 μL reactions were set up with 2 μg DNA template, 2 mM NTP mix, 5 mM DTT, and 5 μL T7 RNA polymerase. Reactions were incubated at 37 °C overnight before treated with 0.2 U/μL Turbo DNase (Thermo Fisher, AM2238) at 37 °C for 15 min. sgRNAs were then purified using the RNA Clean & Concentrator kit (Zymo Research, R1014).
In vitro DNA cleavage assay
The dsDNA substrate was prepared by PCR amplification of a 954 bp region spanning the TP53-1 site. In vitro DNA cleavage reactions were set up by mixing gel-purified dsDNA substrate (48 nM), sgRNA (900 μM), and the wild-type or engineered AsCas12f protein (900 μM) in 20 μL 1× reaction buffer (10 mM Tris-HCl pH 7.5, 10 mM MgCl2 and 50 mM NaCl). Reactions were incubated at 37 °C or 50 °C and quenched by adding 1 μL 500 mM EDTA at different time points. The cleavage products were analyzed by 2% agarose gel electrophoresis. Band intensities were quantified by ImageJ. Time-course DNA cleavage efficiency was fitted to a one-phase exponential association curve using Prism 7.
Protein expression
N-terminal His-tagged AsCas12f variants were overexpressed in E. coli BL21(DE3). E. coli harboring the expression plasmid were cultured in Terrific Broth at 37 °C until OD600 reached 1.0. Protein expression was induced by isopropylthio-β-galactoside (IPTG) at 0.25 mM. Bacteria were further cultured at 16 °C for 24 h before harvest. Around 50 g cell pellets were resuspended in 300 mL lysis buffer (20 mM Tris-HCl pH 7.5, 1 M NaCl, 15 mM imidazole, 1 mM DTT) and lysed by sonication. Lysates were cleared by centrifugation and incubated with 3 mL Ni-NTA beads (QIAGEN) that were pre-equilibrated in lysis buffer. After 2–3 h of gentle agitation, the beads were packed into a gravity column and washed with 30 mL wash buffer (20 mM Tris-HCl pH 7.5, 1 M NaCl, 50 mM imidazole, 1 mM DTT). Proteins were eluted with 15 mL elution buffer (20 mM Tris-HCl, pH 7.5, 1 M NaCl, 250 mM imidazole, 1 mM DTT), immediately diluted by adding a 2-fold volume of dilution buffer (20 mM Tris-HCl pH 7.5, 1 M NaCl and 1 mM DTT), and concentrated using a 30 kDa Amicon Ultra-15 Centrifugal Filter (Millipore Sigma). Proteins were further purified by size exclusion chromatography on a Superdex 200 Increase 10/300 GL column (GE Healthcare) using a buffer containing 20 mM Tris-HCl pH 7.5, 1 M NaCl, and 1 mM DTT. Purified AsCas12f proteins were flash-frozen and stored at −80 °C.
Electron microscopy sample preparation
The AsCas12f-sgRNA-DNA complex was assembled by mixing purified AsCas12f (D225A), the 194 nt sgRNA, the 42 nt target DNA, and the 42 nt non-target DNA, at a molar ratio of 1:0.5:1.2:1.2. Sequences of the sgRNA and the target DNA are provided in Supplementary Table 1. The mixture was incubated on ice for 30 min before being loaded onto a Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated with buffer D (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2, and 0.5 mM TCEP). Fractions that contain the pure AsCas12f-sgRNA-DNA complex were pooled and concentrated to roughly 2.5 mg/mL.
Sample vitrification was performed using a Vitrobot Mark IV (Thermo Fisher) operating at 8 °C and 100% humidity. 3.5 μL sample was applied to holey carbon grids (Quantifoil 200 mesh Cu 1.2/1.3) that had been glow-discharged for 30 seconds. The grids were blotted for 4 seconds at a “blotting force” 0 by standard Vitrobot filter paper (Ted Pella, 47000–100) and were then plunge-frozen in liquid ethane.
Cryo-EM data collection
Frozen grids were sent to the Advanced Electron Microscopy Facility at the University of Chicago for data collection. The dataset was acquired as movie stacks using EPU (Thermo Fisher) installed on a Titan Krios transmission electron microscope operating at 300 kV and equipped with a K3 direct detector camera (Gatan). Images were recorded at a nominal magnification of 81,000× and super-resolution counting mode by image shift. The total exposure time was set to 4 s with 40 frames in a single stack and a total exposure of around 50 electrons/Å2. The defocus range was set at −1.0 to −2.5 μm. Detailed parameters for Cryo-EM data collection are summarized in Supplementary Table 5.
Cryo-EM image processing
Stack images were subjected to motion correction by MotionCor257. Motion-corrected micrographs were then imported to a cryoSPARC live session58 for CTF determination and particle picking. Particles were automatically picked using 2D class averages as templates, which were generated from blob picking. The extracted particles were imported to cryoSPARC for further processing. After 2D classification, contamination and poorly aligned classes were disposed. The resulting 3,370,441 particles were used to generate three initial models by ab initio reconstruction. 3D classification was then performed in cryoSPARC using the three initial models as the starting points. The coordinates of the particles from the best class (1,576,757 particles) were imported into RELION59 for particle re-extraction. CTFFIND60 was used to determine the CTF parameters in RELION. Another round of 3D classification was performed using the map generated from cryoSPARC as the initial model. The best class was subjected to 3D refinement, CTF refinement, Bayesian polishing, and postprocessing. The final map of the AsCas12f-sgRNA-DNA complex was resolved at 2.9 Å based on the criteria of FSC = 0.143.
Cryo-EM model building, refinement, and validation
Model building was performed in COOT61 using a starting model of AsCas12f predicted by AlphaFold262. One full copy of AsCas12f and a second copy of the N-lobe were identified in the cryo-EM map and modeled. DNA and sgRNA were built into the map based on the knowledge of sequence complementarity, secondary structure prediction by IPknot63, and fragment RNA model generated by RNAComposer64. The final model was refined in real space and validated using PHENIX65. Molecular graphics was prepared using PyMOL (Schrödinger, LLC) and UCSF ChimeraX66. The statistics of model refinement and geometry are available in Supplementary Table 5.
GUIDE-seq
GUIDE-seq experiments were performed following a reported protocol46, 67. Briefly, 1.8 μg plasmid encoding AsCas12f, 1.8 μg plasmid encoding the sgRNA, and 5 μL end-protected double-stranded oligodeoxynucleotide (dsODN, 100 μM) were added to one million HEK293T cells in 100 μL nucleofection buffer. Nucleofection was performed on a 4D-Nucleofector (Lonza) according to the manufacturer’s instructions. Full-length sgRNAs were applied in all GUIDE-seq experiments.
Cells were harvested 3 days post-nucleofection and were subjected to genomic DNA (gDNA) isolation. Targeted deep sequencing was performed as described above to analyze indel and dsODN incorporation frequencies. 1 μg gDNA was applied to fragmentation, end-repair, A-tailing, adapter ligation, and dsODN-specific amplification. The libraries were sequenced for 150 cycles on an Illumina Nextseq platform. Data were analyzed and visualized using open-source guideseq software68. DNA oligos used for GUIDE-seq are provided in Supplementary Table 1.
Supplementary Material
Acknowledgements
We thank all Tang lab members for discussion. We thank the staff at the University of Chicago Advanced Electron Microscopy (RRID: SCR_019198) for helping with cryo-EM data collection. We thank the Research Computing Center at the University of Chicago for providing the computing resources of the Beagle3 HPC cluster funded by NIH (S10OD028655). This work was supported by the National Institutes of Health (NIH) under grant number R35GM143052 to M.Z...W.T. is supported by the Searle Scholars Program (SSP-2021-113), the Cancer Research Foundation Young Investigator Program, the American Cancer Society (RSG-22-043-01-ET), and the David & Lucile Packard Foundation.
Footnotes
Competing interests
T.W., S.Z., and W.T. are inventors on a U.S. provisional patent application on enAsCas12f. T.W. is a shareholder of AccuraDX Inc. The other authors declare no competing interests.
Data availability
Sequencing data are available at NCBI Gene Expression Omnibus (GEO) with accession number GSE211600 and Sequence Read Archive (SRA) with accession number PRJNA962057. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB, www.ebi.ac.uk/pdbe/emdb/) under accession code EMD-27801. The atomic model has been deposited to the Protein Data Bank (PDB, www.rcsb.org) under accession code 8DZJ.
References
- 1.Mojica FJM, Díez-Villaseñor C.s., García-Martínez J & Soria E Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Barrangou R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 109, E2579–E2586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura M, Gao Y, Dominguez AA & Qi LS CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23, 11–22 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Terns MP CRISPR-based technologies: impact of RNA-targeting systems. Mol. Cell 72, 404–412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X-M, Zhou J, Mao Y, Ji Q & Qian S-B Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol. 15, 865–871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson C, Chen PJ, Miao Z & Liu DR Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 38, 1431–1440 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampton T With first CRISPR trials, gene editing moves toward the clinic. JAMA 323, 1537–1539 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Gillmore JD et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Frangoul H et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Koonin EV & Makarova KS Evolutionary plasticity and functional versatility of CRISPR systems. PLOS Biol. 20, e3001481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinek M et al. RNA-programmed genome editing in human cells. eLife 2, e00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickar-Oliver A & Gersbach CA The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin H, Kauffman KJ & Anderson DG Delivery technologies for genome editing. Nat. Rev. Drug Discov. 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Lino CA, Harper JC, Carney JP & Timlin JA Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 25, 1234–1257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright AV et al. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 112, 2984–2989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zetsche B, Volz SE & Zhang F A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 33, 139–142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nihongaki Y, Kawano F, Nakajima T & Sato M Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Chew WL et al. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 13, 868–874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J-J et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 566, 218–223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pausch P et al. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369, 333–337 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrington Lucas B et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karvelis T et al. PAM recognition by miniature CRISPR–Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 48, 5016–5023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HJ, Kim HJ & Lee SJ Miniature CRISPR-Cas12f1-mediated single-nucleotide microbial genome editing using 3′-truncated sgRNA. CRISPR J. 6, 52–61 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapitonov Vladimir V, Makarova Kira S, Koonin Eugene V & Zhulin IB ISC, a novel group of bacterial and archaeal DNA transposons that encode Cas9 homologs. J. Bacteriol. 198, 797–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altae-Tran H et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato K et al. Structure of the IscB–ωRNA ribonucleoprotein complex, the likely ancestor of CRISPR-Cas9. Nat. Commun. 13, 6719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirano S et al. Structure of the OMEGA nickase IsrB in complex with ωRNA and target DNA. Nature 610, 575–581 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karvelis T et al. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 599, 692–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuler G, Hu C & Ke A Structural basis for RNA-guided DNA cleavage by IscB-ωRNA and mechanistic comparison with Cas9. Science 376, 1476–1481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda SN et al. Structure of the miniature type V-F CRISPR-Cas effector enzyme. Mol. Cell 81, 558–570.e553 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Xiao R, Li Z, Wang S, Han R & Chang L Structural basis for substrate recognition and cleavage by the dimerization-dependent CRISPR–Cas12f nuclease. Nucleic Acids Res. 49, 4120–4128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong X et al. Engineered CRISPR-OsCas12f1 and RhCas12f1 with robust activities and expanded target range for genome editing. Nat. Commun. 14, 2046 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Z et al. Programmed genome editing by a miniature CRISPR-Cas12f nuclease. Nat. Chem. Biol. 17, 1132–1138 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Xu X et al. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 81, 4333–4345 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Kim DY et al. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 40, 94–102 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim DY et al. Hypercompact adenine base editors based on a Cas12f variant guided by engineered RNA. Nat. Chem. Biol. 18, 1005–1013 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcovitz A & Levy Y Frustration in protein–DNA binding influences conformational switching and target search kinetics. Proc. Natl. Acad. Sci. USA 108, 17957–17962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleinstiver BP et al. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 37, 276–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xin C et al. Comprehensive assessment of miniature CRISPR-Cas12f nucleases for gene disruption. Nat. Commun. 13, 5623 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang S et al. TadA reprogramming to generate potent miniature base editors with high precision. Nature Commun. 14, 413 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chavez A et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang Y et al. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 16, 280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moon SB, Kim DY, Ko J-H, Kim J-S & Kim Y-S Improving CRISPR genome editing by engineering guide RNAs. Trends Biotechnol. 37, 870–881 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Kleinstiver BP et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 34, 869–874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bae S, Park J & Kim J-S Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 57.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Scheres SHW RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mindell JA and Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sato K, Kato Y, Hamada M, Akutsu T & Asai K IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 27, i85–i93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Popenda M et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 40, e112–e112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malinin NL et al. Defining genome-wide CRISPR–Cas genome-editing nuclease activity with GUIDE-seq. Nat. Protoc. 16, 5592–5615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsai SQ, Topkar VV, Joung JK & Aryee MJ Open-source guideseq software for analysis of GUIDE-seq data. Nat. Biotechnol. 34, 483–483 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available at NCBI Gene Expression Omnibus (GEO) with accession number GSE211600 and Sequence Read Archive (SRA) with accession number PRJNA962057. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB, www.ebi.ac.uk/pdbe/emdb/) under accession code EMD-27801. The atomic model has been deposited to the Protein Data Bank (PDB, www.rcsb.org) under accession code 8DZJ.