

Abstract
We previously identified the indole 264 as a potent in vitro antagonist of the human OXE receptor that mediates the actions of the powerful eosinophil chemoattractant 5-oxo-ETE. No antagonists of this receptor are currently commercially available or are being tested in clinical studies. The lack of a rodent ortholog of the OXE receptor has hampered progress in this area because of the unavailability of commonly used mouse or rat animal models. In the present study, we examined the feasibility of using the cynomolgus monkey as an animal model to investigate the efficacy of orally administered 264 in future in vivo studies. We first confirmed that 264 is active in monkeys by showing that it is a potent inhibitor of 5-oxo-ETE-induced actin polymerization and chemotaxis in granulocytes. The major microsomal metabolites of 264 were identified by cochromatography with authentic chemically synthesized standards and LC-MS/MS as its ω2-hydroxy and ω2-oxo derivatives, formed by ω2-oxidation of its hexyl side chain. Small amounts of ω1-oxidation products were also identified. None of these metabolites have substantial antagonist potency. High levels of 264 appeared rapidly in the blood following oral administration to both rats and monkeys, and declined to low levels by 24 h. As with microsomes, its major plasma metabolites in monkeys were ω2-oxidation products. We conclude that the monkey is a suitable animal model to investigate potential therapeutic effects of 264. This, or a related compound with diminished susceptibility to ω2-oxidation, could be a useful therapeutic agent in eosinophilic disorders such as asthma.
Keywords: 5-Oxo-ETE, OXE receptor antagonist, Eicosanoids, 5-Lipoxygenase products, Eosinophils, Drug metabolism
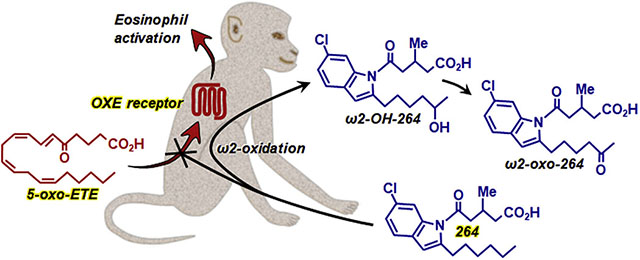
Graphical Abstract
1. Introduction
Arachidonic acid is converted to a variety of pro- and anti- inflammatory mediators that have potent effects on both inflammatory and structural cells. Oxidation of arachidonic acid by 5-lipoxygenase (5-LO) results in the formation of proinflammatory leukotrienes (LTs) and 5-oxo-6,8,11,14-eicosatetraenoic acid (5-oxo-ETE) as well as anti-inflammatory lipoxins. 5-Oxo-ETE is formed by oxidation of 5S-hydroxy-6,8,11,14-eicosatetraenoic acid (5S-HETE) by 5-hydroxyeicosanoid dehydrogenase, which selectively oxidizes the 5S-hydroxyl group in the presence of NADP+ (Powell et al., 1992). The synthesis of 5-oxo-ETE is regulated by the intracellular concentration of NADP+, which normally is very low, but is rapidly increased under conditions of oxidative stress, activation of the respiratory burst in phagocytic cells, and cell death (Powell and Rokach, 2013).
The 5-LO pathway is important in host defence (Bailie et al., 1996) and contributes to the pathophysiology of inflammatory diseases, in particular asthma, in which case the 5-LO inhibitor zileuton and antagonists of the cysLT1 receptor for LTD4 (montelukast, zafirlukast, and pranlukast) are used clinically. Asthma is an inflammatory disease usually characterized by high levels of airway eosinophils, although other phenotypes also exist (Chung, 2016; Robinson et al., 2017). Eosinophils contribute to the pathology of asthma by releasing toxic mediators and proinflammatory cytokines (McBrien and Menzies-Gow, 2017). Interleukin-5 is important for the differentiation and survival of eosinophils, and antibodies against this cytokine or its receptor have been used therapeutically. Chemoattractants also play a critical role in the infiltration of eosinophils into the lungs. Among 5-LO products, 5-oxo-ETE and its metabolite 5-oxo-15-HETE are the only ones that have appreciable chemotactic effects on human eosinophils (Powell et al., 1995; Powell and Rokach, 2013). Furthermore, 5-oxo-ETE promotes the transendothelial migration of eosinophils (Dallaire et al., 2003) due to both its chemoattractant effects and its ability to induced the release of MMP-9 from these cells (Langlois et al., 2009). These effects are mediated by the G protein-coupled OXE receptor, which is highly selective for 5-oxo-ETE (Bäck et al., 2014) and is most highly expressed on eosinophils (Jones et al., 2003) and basophils (Iikura et al., 2005; Sturm et al., 2005) and to a lesser extent on neutrophils (Jones et al., 2003), monocytes (Sturm et al., 2005), and macrophages (Jones et al., 2003), as well as various tumor cell lines (Sarveswaran and Ghosh, 2013).
Because of the potential involvement of 5-oxo-ETE in asthma and other eosinophilic diseases, the OXE receptor is an attractive therapeutic target. As no selective OXE receptor antagonists were available, we initiated a program to synthesize compounds that could selectively block this receptor. We initially identified a series of N-acylindoles that selectively blocked the activation of the OXE receptor in human neutrophils by 5-oxo-ETE. These compounds contained 5-oxo-valerate and hexyl groups in the 1- and 2-positions of the indole, respectively, mimicking the first 5 carbons of 5-oxo-ETE and the hydrophobic ω-end of the molecule (Gore et al., 2013). Addition of a chloro substituent in the 6-position of the indole increased potency from about 1.6 μM to 0.4 μM (Gore et al., 2013), and addition of a methyl group in the 3-position of the 5-oxovalerate side chain (compound 264, Fig. 1) further increased potency to about 30 nM (Gore et al., 2014).
Figure 1.
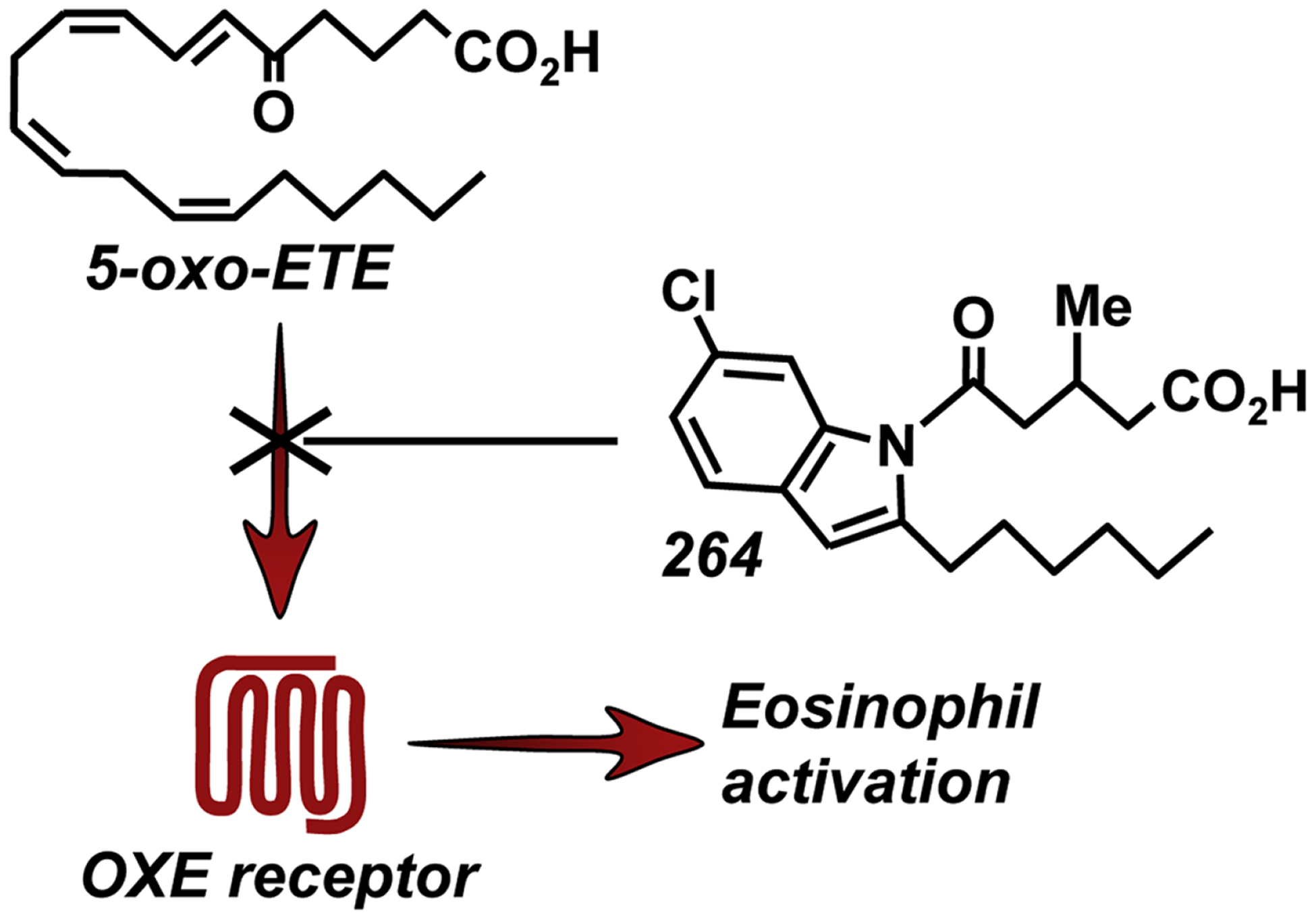
Inhibition of the actions of 5-oxo-ETE by the OXE receptor antagonist 264
Unfortunately, we do not have the option of testing OXE receptor antagonists in mouse or rat models of asthma, because rodents do not possess an ortholog of the OXE receptor. Cats do have an OXE receptor with an amino acid sequence that is about 75% identical to the human OXE receptor. However, although 5-oxo-ETE is a very potent activator of feline eosinophils, 264 is only a weak antagonist of the feline OXE receptor (Cossette et al., 2015). We are therefore investigating the feasibility of using the monkey as an animal model to test the in vivo effects of 264. The objectives of the current study were first to ensure that 264 is active in blocking 5-oxo-ETE-induced OXE receptor activation in monkeys and then to identify its major vivo metabolic pathways and determine whether significant levels appear in the blood following oral administration. We found that 264 is a potent OXE receptor antagonist in monkeys that appears rapidly in the blood following oral administration. Its major route of metabolism is ω2-oxidation of the hexyl side chain.
2. Materials and Methods
2.1. Animals
Male and female cynomolgus monkeys weighing between 2.7 to 3.5 kg, housed at INRS-Institut Armand-Frappier, Laval, Quebec, and male Sprague-Dawley rats (200 to 250 g; 6 to 8 weeks old), housed at the Meakins-Christie Laboratories, were used for these studies. All experiments were performed in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the appropriate local institutional animal care committee.
2.2. Preparation of monkey leukocytes
Whole blood, collected in heparinized tubes from monkeys, was treated with Dextran 500 (Sigma-Aldrich) for 45 min at room temperature. After hypotonic lysis of red blood cells and centrifugation, the leukocytes were suspended in phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 8.1 mM Na2HPO4, pH 7.4).
2.3. Evaluation of actin polymerization in monkey eosinophils and neutrophils
Leukocytes prelabeled with allophycocyanin (APC)-labeled mouse antihuman CD49d (BioLegend) as described previously (Cossette et al., 2015) were suspended in PBS containing Ca2+ (1.8 mM) and Mg2+ (1 mM). Aliquots of the leukocyte suspension (100 μl, 5 × 106 cells/ml) were preincubated for 5 min at 25°C with either vehicle (1 μl DMSO) or 264 followed by the addition of 5-oxo-ETE (10 nM). The incubations were terminated after 20 s by the addition of formaldehyde (final concentration, 8.5%), and the samples were kept on ice for 30 min. Cytosolic F-actin was stained by treatment with a mixture of lysophosphatidylcholine (30 μg in 23.8 μl of PBS) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-phallacidin (40.7 pmol in 6.18 μl methanol; final concentration, 1.36 μM) overnight in the dark at 4°C. F-actin levels were measured by flow cytometry in eosinophils and neutrophils, which were distinguished from one another and from other leukocytes on the basis of side scatter and CD49d expression.
2.4. Measurement of leukocyte migration
Leukocyte migration was assessed as previously described (Monneret et al., 2001) using 48-well microchemotaxis chambers (Neuro Probe, Cabin John, MD) and Sartorius cellulose nitrate filters (8 μm pore size; 140-mm thickness; Neuro Probe). 5-Oxo ETE along with either vehicle (PBS containing 0.5% DMSO) or 264 were added to the bottom well in 30 μl PBS containing 1.8 mM CaCl2, 1 mM MgCl2, 0.05% BSA and 0.5% DMSO. Leukocytes (150,000 cells in PBS containing 0.05% BSA) and either vehicle or 264 were added in a total volume of 55 μl to each of the top wells. After incubation for 2 h at 37°C, the filters were fixed with HgCl2 and stained with hematoxylin and chromotrope 2R.(Kay, 1970) The numbers of cells on the bottoms of the filters were counted in 5 different fields at a magnification of 400x for each incubation, each performed in triplicate.
2.5. Measurement of calcium mobilization in human neutrophils
The effects of 264 metabolites on 5-oxo-ETE-induced Ca++ mobilization in human neutrophils were examined as described previously.(Gore et al., 2014) Neutrophils were prepared by dextran sedimentation and centrifugation over Ficoll-Paque and prelabeled with indo-1. Aliquots of the suspension were placed in a cuvette at 37°C and the baseline fluorescence allowed to stabilize. 264 or one of its synthetic metabolites was then added, followed 2 min later by 5-oxo-ETE (10 nM) and, 1 min later, by digitonin (final concentration 0.1%). Fluorescence (λex, 331 nm; λem, 410 nm) was measured with a Cary Eclipse spectrofluorometer (Agilent Technologies, Santa Clara, CA) equipped with a temperature-controlled cuvette holder and a magnetic stirrer.
2.6. RP-HPLC analysis of microsomal metabolites of 264
Monkey liver microsomes (0.5 mg protein/ml) from a pool of male cynomolgus monkeys (ThermoFisher Scientific) were suspended in PBS and incubated for 4 h at 37 °C with 264 (100 μM) and NADPH (2 mM). Reactions were stopped by the addition of MeOH and cooling to 0 °C. Before analysis the concentration of MeOH was adjusted to 30% by the addition of water. The products were analyzed by precolumn extraction/RP-HPLC using a Waters 2695 Alliance system with a Kinetex C18 column as described in the legend to Fig. 5.
Figure 5.
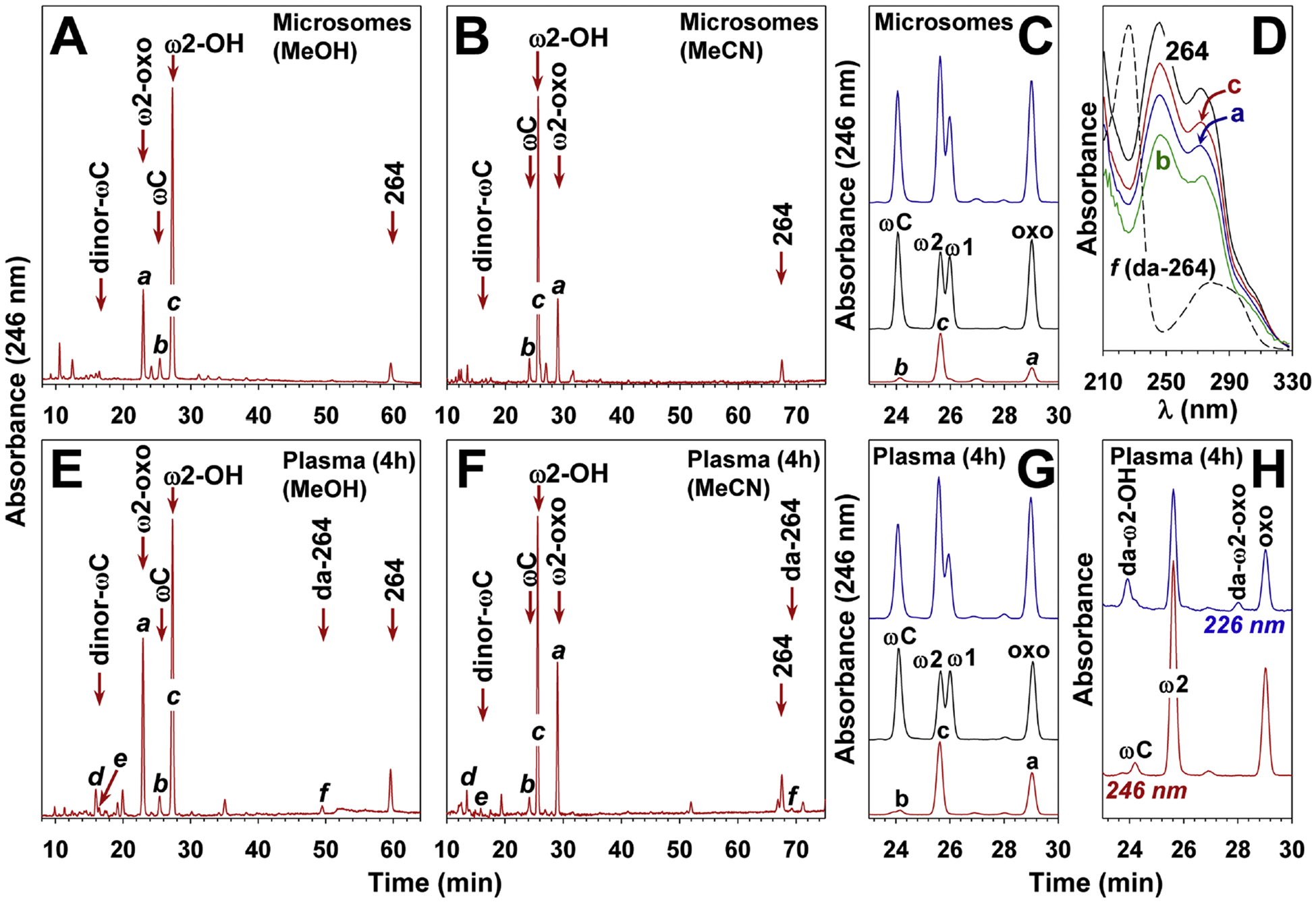
Metabolism of 264 in monkeys. A and B: Monkey liver microsomes were incubated with 264 (100 mM) for 4 h in the presence of NADPH (2 mM) as described in Methods. The products were analyzed by precolumn extraction/RP-HPLC on a Kinetex C18 column (5 μm; 250 × 4.6 mm) using either methanol (A: 61 to 82% MeOH over 63 min + 5 min isocratic) or acetonitrile (B: 38 to 70% MeCN over 78 min), both containing 0.02% HOAc. The flow rate was 1 ml/min and the column temperature 35 °C. ωC, ω-carboxy-264. C: Cochromatography of microsomal metabolites of 264 with standards. The supernatant obtained after incubation of 264 with monkey liver microsomes was chromatographed in the absence (bottom) or presence (top) of authentic standards (conditions as in panel B). Standards alone are shown in the middle chromatogram. ω1, ω1-hydroxy-264; ω2, ω2-hydroxy-264; oxo, ω2-oxo-264. D: UV spectra of metabolites a, b, c, and f compared to 264. The absolute values for each product were adjusted to facilitate comparison. E and F: Plasma obtained 4 h after oral administration of 264 (30 mg/kg) to a monkey was extracted as described in Methods and the products analyzed by precolumn extraction/RP-HPLC using either methanol (E) or acetonitrile (F) gradients as described above. G: Cochromatography of plasma metabolites of 264 with authentic standards. An extract of plasma taken 4 h after administration of 264 in the absence (bottom) or presence (top) of authentic standards was analyzed by RP-HPLC as for panel C. Standards alone are shown in the middle chromatogram. H: Chromatogram of a sample of plasma taken 4 h after administration of 264 showing UV absorbance at 246 nm and 226 nm (da, deacyl). The arrow in panels A, B, E, and F show the elution positions of standards run separately.
2.7. Preparation of 264 suspensions for in vivo administration
Since 264 was not very soluble in aqueous solutions, even as the carboxylate anion, it was administered orally as a suspension in 20 mM NaHCO3, pH 8.0. For the preliminary rat experiments, various amounts of 264 were first completely dissolved in EtOH (100 μl) in amounts to give final doses between 5 and 30 mg/kg. The ethanolic solutions were then added to 20 mM NaHCO3 (1 ml). The resulting suspensions were vortexed and the entire volume was administered by gavage to the rats. In the case of monkeys, a stock solution of 264 (75 mg/kg) was made up in EtOH. The volume of this solution required to achieve doses of either 30 or 75 mg/kg 264 was then added to 10 volumes of 20 mM NaHCO3. After vortexing, the resulting suspension was administered to monkeys by oral gavage. Separate suspensions of 264 were made up for each animal and used immediately.
2.8. Measurement of 264 in rat plasma
Suspensions of 264 (1.1 ml) in 9% EtOH in bicarbonate, prepared as described above, were administered by gavage to Sprague-Dawley rats (200–250 g) at doses between 5 and 30 mg/kgThe rats were fasted for 3 h prior to gavage. The animals were euthanized with CO2 after different times and blood was withdrawn by cardiac puncture. The internal standard 190 (6-chloro-2-hexyl-1H-indol-1-yl)-2,2-dimethyl-5-oxopentanoic acid) was synthesized by N-acylation coupling of 6-chloro-2-hexyl-1H-indole, synthesized as described previously (Gore et al., 2013) with 2,2-dimethylglutaric anhydride using KOH as a base. 190 (1 μg) was immediately added to each blood sample, which was then centrifuged at 1700 × g for 15 min to remove blood cells. The resulting plasma was added to MeOH (2 volumes) and stored overnight at −80 °C. After removal of the sample from the freezer the protein precipitate was removed by centrifugation and the analytes were extracted on a C18 SepPak cartridge (Waters Corp) (Powell, 1980) which was washed with 30% MeOH prior to elution of 264 and its metabolites with 100% MeOH. The eluate was analyzed by precolumn extraction/RP-HPLC (Powell, 1987) using the Waters Alliance system described above with a Novapak column (4 μm particle size; 150 × 3.9 mm). The mobile phase was a gradient between 70 and 100% MeOH containing 0.02% HOAc over 15 min at a flow rate of 1 ml/min and a column temperature of 30 °C.
2.9. Measurement of 264 and its metabolites in monkey plasma
Suspensions of 264 in 9% EtOH in bicarbonate, prepared as described in section 2.7, were administered by oral gavage to cynomolgus monkeys at a dose of either 30 or 75 mg/kg. Blood samples (1 ml) were collected in heparinized tubes and plasma was obtained after centrifugation. After solid-phase extraction using C18 Sep-Paks as described above, samples were analyzed by precolumn extraction/RP-HPLC using a Waters Alliance system and a Kinetex C18 column (see legend to Fig. 5 for further details). 190 was used as an internal standard.
2.10. Chiral HPLC of 264
Following RP-HPLC analysis of plasma samples, the material in the peak corresponding to 264 was analyzed by chiral HPLC using a Cellulose-1 column (250 mm × 4.6 mm; 5 μm particle size; Phenomenex, Torrance, CA). The mobile phase was hexane/methanol/acetic acid (98.5:1.5:0.1) at a flow rate of 1.5ml/min and the column temperature 30 °C.
2.11. Analysis of 264 metabolites by LC-MS/MS
LC-MS/MS analysis was carried out using a model 1100 HPLC system (Agilent Technologies, Santa Clara, CA) connected to an LTQ Velos Orbitrap high resolution mass spectrometer by a heated electrospray ionization source (Thermo Scientific, San Jose, CA). The stationary phase was a Phenomenex Kinetex C18 column (2.6 μm particle size; 50 × 2.1 mm) and the mobile phase was H2O and MeCN (each containing 0.02% HOAc). The gradient used for separation was as follows: 0 min, 30% MeCN; 1–20 min, linear increase from 30% to 65% MeCN; 20–25 min, isocratic at 65% MeCN, re-equilibration for 7 min at 30% MeCN. The flow rate was 0.3 ml/min, the column temperature 25°C and the injection volume 10 μL. Unless otherwise indicated, all analyses were performed in negative electrospray ionization (ESI) mode with the following settings: capillary temperature: 350 °C; source heater temperature: 300 °C; sheath gas flow: 30; auxiliary gas flow: 10; source voltage: − 3.5 kV. The MS settings were: S lens RF level: 60%; automatic gain control (AGC) target: 1 × 106 ions; mass range: m/z 200 to m/z 700; resolution: 100,000. Multiple levels of MSn analysis with data dependent acquisition (DDA) mode were used for the identification and elucidation of 264 metabolites. In DDA mode the selection of the precursor ion for MS2 analysis was based on the chlorine isotope pattern and/or isolation of the top three most intense ions from the full MS scan for fragmentation. MS2 settings also included collision-induced dissociation (CID); signal threshold: 5,000; normalized collision energy: 35; isolation width: 2 Da; activation time: 30 ms. MS3 used parent and product mass lists to trigger MS3 for selected ions and was performed with CID as activation type; minimal signal threshold: 5000; isolation width: 2 Da; activation time: 30 ms; normalized collision energy: 45.
For the identification of ω2-oxo-264, the Orbitrap instrument and LC conditions were as described above. However, positive ion ESI was used for ionization: capillary temperature: 275°C; source heater temperature: 300 °C; sheath gas flow: 20; auxiliary gas flow: 5; source voltage: 4 kV; S-lens RF level 62%. Target detection and identification was based on the full MS scan using FTMS analyser and MSn ion trap experiment. The full MS scan used the mass range from 350 m/z to 475 m/z and resolution: 100,000. MS2 was performed on the protonated parent ion of the oxo metabolite with m/z 378.1467 using CID as activation type, normalized collision energy: 20, activation time: 10 ms, and mass range from 100 m/z to 400 m/z. MS3 was performed using product ion with m/z 250.1848, which was fragmented using CID as activation type, normalized collision energy: 35, activation time: 10 ms, and mass range from 65 m/z to 400 m/z.
2.12. Chemical synthesis of OXE antagonists
2.12.1. Reagents and Methods
All reactions were carried out under an argon atmosphere using dry solvents. DMSO was purchased from Aldrich as anhydrous grade and used without further purification. Potassium tert-butoxide solution (1.0 M in THF), titanium tetrachloride (1.0 M in CH2Cl2), tetraethylammonium iodide, tert-Butyl (chloro)diphenylsilane (TBDPS-Cl), and 3,4-Dihydro-2H-pyran (DHP) were purchased from Aldrich. The catalysts, p-toluenesulfonic acid monohydrate and 10% Pd/C were purchased from Aldrich. All compounds were analyzed by TLC, NMR and HRMS. 1H NMR and 13C NMR spectra were recorded at rt on a BRUKER AMX 400 MHz spectrometer in CDCl3 using TMS as an internal standard. High-resolution mass spectra were recorded using a JEOL DART-AccuTOF mass spectrometer (JEOL USA, Inc, Peabody, MA). Prior to biological assay, the purity of all final compounds was determined to be >95% by a combination of HPLC, NMR and HRMS.
2.12.2. 6-(6-Chloro-1H-indol-2-yl)-hex-5-enoic acid (3)
To a stirred suspension of phosphonium salt 2 (3.1 g, 6.99 mmol) in anhydrous THF (20 ml) was added t-BuOK (1.0 M in THF, 13.91 ml, 13.91 mmol) at −78 °C under argon. The mixture was gradually warmed to rt and stirred for 45 min, cooled to −78 °C, and a solution of aldehyde 1 (500 mg, 2.78 mmol) in dry THF (5 ml) was added. The reaction mixture was warmed to rt, stirred for 5 h, and then quenched with a saturated solution of NH4Cl (13 ml). The aqueous layer was extracted with EtOAc (3 × 20 ml) and the combined organic layers were washed with brine (6 ml) and dried over Na2SO4. The combined organic solvents were evaporated under reduced pressure and the crude residue purified by silica gel column chromatography using 50% EtOAc/n-hexane, affording 3 (609 mg, 83%) as a pale yellow solid containing an inseparable pair of trans/cis (1:1) isomers.
2.12.3. 6-(6-Chloro-1H-indol-2-yl)-hexanoic acid (4)
To a stirred solution of 3 (500 mg, 1.89 mmol) in anhydrous benzene (10 ml) was added 10% Pd/C (50 mg) at rt under H2 atm and stirred for 3 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated under reduced pressure to give the crude residue 4 as a pale yellow solid, which was directly used for the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 7.95 (s, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.28 (s, 1H), 7.03 (dd, J = 8.4, 1.7 Hz, 1H), 6.20 (s, 1H), 2.75 (t, J = 7.5 Hz, 2H), 2.39 (t, J = 7.3 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.49 – 1.40 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 179.6, 140.3, 136.2, 127.3, 126.6, 120.5, 120.2, 110.3, 99.6, 33.7, 28.6, 28.4, 27.7, 24.1.
2.12.4. 6-[1-(4-Carboxy-3-methyl-butyryl)-6-chloro-1H-indol-2-yl]-hexanoicacid (6)
To a stirred solution of 4 (100 mg, 0.37 mmol) in anhydrous DMSO (5 ml) was added t-BuOK (1.0 M in THF, 1.88 ml, 1.88 mmol) at 0 °C under argon. The reaction mixture was stirred for 30 min followed by addition of 5 (192 mg, 1.5 mmol) at rt and the stirring was continued for 4 h. The reaction mixture was quenched with saturated NH4Cl (8 ml) and the aqueous layer was extracted with EtOAc (3 × 10 ml), and the combined organic layers dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue purified by silica gel column chromatography using 70% EtOAc/n-hexane to afford 6 (118 mg, 80% over two steps) as a white solid. HRMS (ESI) m/z calcd for [C20H24ClNO5 -H]+: 392.1265, found 392.4913. 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.38 (s, 1H), 7.19 (d, J = 8.3 Hz, 1H), 6.36 (s, 1H), 3.13 (dd, J = 16.4, 6.4 Hz, 1H), 2.97 (t, J = 7.0 Hz, 2H), 2.92 (dd, J = 16.2, 6.7 Hz, 1H), 2.83 – 2.70 (m, 1H), 2.50 (dd, J = 15.4, 6.5 Hz, 2H), 2.38 (t, J = 7.1 Hz, 2H), 1.76 – 1.66 (m, 4H), 1.54 – 1.44 (m, 2H), 1.17 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 180.1, 178.7, 172.1, 142.8, 136.5, 129.3, 128.2, 123.4, 120.7, 115.2, 108.0, 44.9, 40.4, 33.9, 30.3, 28.7, 28.5, 27.1, 24.3, 20.1.
2.12.5. 4-(6-Chloro-1H-indol-2-yl)-but-3-enoic acid (8)
To a stirred suspension of phosphonium salt 7 (6.9 g, 16.61 mmol) in anhydrous THF (30 ml) was added t-BuOK (1.0 M in THF, 27.83 ml, 27.83 mmol) at −78 °C under argon. The mixture was stirred for 40 min at rt and then cooled to −78 °C, and a solution of the aldehyde 1 (1 g, 5.56 mmol) in 10 ml of anhydrous THF was added. The reaction mixture was stirred at room temperature for 5 h and then quenched with saturated NH4Cl solution (15 ml), extracted with EtOAc (3 × 20 ml) and the combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 50% EtOAc/n-hexane to yield 8 (787 mg, 60%) as pale yellow solid with trans/cis: 90/10. 1H NMR (400 MHz, CDCl3): δ 8.18 (s, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.29 (s, 1H), 7.04 (dd, J = 8.4, 1.7 Hz, 1H), 6.53 (d, J = 16.2 Hz, 1H), 6.43 (s, 1H), 6.11 (dt, J = 16.0, 7.2 Hz, 1H), 3.34 (dd, J = 7.1, 1.0 Hz, 2H). 13C NMR (100 MHz CDCl3): δ 174.7, 139.6, 136.8, 132.8, 127.6, 126.8, 122.6, 121.7, 115.4, 111.0, 104.0, 37.0.
2.12.6. 4-(6-Chloro-1H-indol-2-yl)-butyric acid (9)
To a solution of compound 8 (600 mg, 2.54 mmol) in anhydrous benzene (12 ml), 10% Pd/C (60 mg) was added and stirred at room temperature under a H2 atm for 3 h. After completion of the reaction, the mixture was filtered through a Celite pad and the solvent was removed under reduced pressure to afford the crude product 9, which was directly used as such for further reaction without purification. 1H NMR (400 MHz, CDCl3): δ 8.02 (s, 1H), 7.42 (d, J = 8.4 Hz,1H), 7.29 (s, 1H), 7.03 (dd, J = 8.4, 1.4 Hz, 1H), 6.24 (s, 1H), 2.82 (t, J = 7.4 Hz, 2H), 2.45 (t, J = 7.1 Hz, 2H), 2.05 (m, 2H). 13C NMR (100 MHz CDCl3): δ 178.2, 139.0, 136.3, 127.2, 126.6, 120.6, 120.3, 110.4, 100.2, 32.8, 27.2, 24.1.
2.12.7. 5-[2-(3-Carboxy-propyl)-6-chloro-indol-1-yl]-3-methyl-5-oxo-pentanoicacid (10)
To a stirred solution of 9 (300 mg, 1.26 mmol) in anhydrous DMSO (8 ml) was added t-BuOK (1.0 M in THF, 6.31 ml, 6.31 mmol) at 0 °C under argon. The reaction mixture was stirred for 30 min followed by addition of 5 (600 mg, 4.68 mmol), and further stirring at rt for 4 h. The reaction mixture was quenched with saturated NH4Cl solution (6 ml), extracted with ethyl acetate (4 × 15 ml) and the combined organic layers dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 70% EtOAc/n-hexane to afford 7 (358 mg, 78% over two steps) as a white solid. HRMS (ESI) m/z calcd for [C18H20ClNO5 - H]+: 364.0952, found 364.2153. 1H NMR (400 MHz, CDCl3): δ 7.86 (s, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.19 (dd, J = 8.3, 1.6 Hz, 1H), 6.39 (s, 1H), 3.17 (dd, J = 16.3, 6.5 Hz, 1H), 3.13 – 2.99 (m, 2H), 2.94 (dd, J = 16.3, 6.8 Hz, 1H), 2.81 – 2.69 (m, 1H), 2.57 – 2.42 (m, 4H), 2.07 – 1.96 (m, 2H), 1.17 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz CDCl3): δ 179.3, 178.5, 172.0, 142.2, 136.3, 129.5, 128.0, 123.5, 120.9, 115.1, 108.8, 44.7, 40.1, 33.2, 29.7, 27.1, 23.8, 20.2.
2.12.8. 6-(6-Chloro-1H-indol-2-yl)-hexan-1-ol (11)
To a stirred solution of 4 (400 mg, 1.50 mmol) in THF (20 ml) at −20 °C was added lithium aluminum hydride (114 mg, 3.01 mmol) under argon. The reaction mixture was gradually warmed to rt over a period of 16 h and then quenched with aqueous NH4Cl solution (12 ml) and extracted with EtOAc (3 × 15 ml). The combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 40% EtOAc/n-hexane to give 11 (352 mg, 93%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.27 (s, 1H), 7.03 (dd, J = 8.4, 1.4 Hz, 1H), 6.20 (s, 1H), 3.65 (t, J = 6.5 Hz, 2H), 2.73 (t, J = 7.6 Hz, 2H), 1.76 – 1.68 (m, 2H), 1.62 – 1.54 (m, 2H), 1.43 – 1.40 (m, 4H). 13C NMR (100 MHz CDCl3): δ 140.6, 136.2, 127.4, 126.6, 120.50, 120.1, 110.3, 99.5, 62.8, 32.5, 28.9, 28.0, 25.4.
2.12.9. 6-Chloro-2-[6-(tetrahydro-pyran-2-yloxy)-hexyl]-1H-indole (12)
To a stirred solution of 11 (200 mg, 0.79 mmol) in CH2Cl2 (10 ml) at 0 °C was added a catalytic amount of PTSA (30 mg, 0.15 mmol) under argon followed by addition of 3,4-dihydro-2H-pyran (1.33 ml, 1.58 mmol). The mixture was stirred for 30 min at rt, quenched with a saturated solution of NaHCO3 (2 ml) and the aqueous layer extracted with EtOAc (2 × 10 ml). The combined organic layers were dried over Na2SO4. The crude compound was concentrated in vacuo and purified by silica gel column chromatography using 15% EtOAc/n-hexane to afford 12 (237 mg, 89%) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.97 (s, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.26 (s, 1H), 7.02 (d, J = 8.4 Hz, 1H), 6.19 (s, 1H), 4.57 (s, 1H), 3.92 – 3.83 (m, 1H), 3.79 – 3.69 (m, 1H), 3.54 – 3.46 (m, 1H), 3.43 – 3.35 (m, 1H), 2.72 (t, J = 7.6 Hz, 2H), 1.93 – 1.65 (m, 4H), 1.62 – 1.47 (m, 6H), 1.46 – 1.37 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 140.7, 136.2, 127.4, 126.59, 120.4, 120.1, 110.2, 98.4, 94.6, 67.5, 62.9, 31.9, 30.7, 28.9 (3C), 25.9, 25.3, 19.7.
2.12.10. 5-{6-Chloro-2-[6-(tetrahydro-pyran-2-yloxy)-hexyl]-indol-1-yl}−3-methyl-5-oxo-pentanoic acid (13)
To a solution of 12 (150 mg, 0.44 mmol) in anhydrous DMSO (5 ml) was added KOH (50 g, 0.89 mmol) at 0 °C under argon. The reaction mixture was stirred for 45 min followed by addition of 5 (114 mg, 0.89 mmol). Stirring was continued for 4 h and then the mixture was quenched with saturated NH4Cl (6 ml), extracted with EtOAc (4 × 10 ml) and the combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 30% EtOAc/n-hexane to give 13 (146 mg, 71%) as colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.94 (s, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.18 (d, J = 8.3, 1.7 Hz, 1H), 6.36 (s, 1H), 4.63 – 4.54 (m, 1H), 4.12 (q, J = 7.1 Hz, 1H), 3.94 – 3.83 (m, 1H), 3.75 (dt, J = 9.5, 6.8 Hz, 1H), 3.55 – 3.46 (m,1H), 3.42 (dt, J = 9.6, 6.4 Hz, 1H), 3.13 (dd, J = 15.8, 5.5 Hz, 1H), 3.01 – 2.93 (m, 2H), 2.93 – 2.85 (m, 1H), 2.81 – 2.68 (m, 1H), 2.55 (ddd, J = 15.6, 6.6, 2.6 Hz, 1H), 2.44 (ddd, J = 15.7, 6.6, 1.8 Hz, 1H), 1.76 – 1.66 (m, 3H), 1.66 – 1.57 (m, 3H), 1.57 – 1.48 (m, 3H), 1.48 – 1.39 (m, 4H), 1.16 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 176.9, 172.2, 142.8, 136.6, 129.3, 128.21, 123.4, 120.6, 115.3, 107.9, 99.0, 62.5, 44.8, 40.3, 30.7, 30.4, 29.5, 29.1, 28.91, 27.2, 26.0, 25.4, 20.1, 19.7, 14.1.
2.12.11. 5-[6-Chloro-2-(6-hydroxy-hexyl)-indol-1-yl]-3-methyl-5-oxo-pentanoic acid (14)
A catalytic amount of PTSA (8.2 mg, 0.04 mmol) was added successively to a solution of 13 (100 mg, 0.21 mmol) in a mixture of THF:H2O (4:1) (10 ml). The reaction mixture was stirred at rt for 30 min, quenched with water, and the aqueous layer was extracted with EtOAc (4 × 20 ml). The crude residue was concentrated in vacuo and purified by silica gel column chromatography using 50% EtOAc/n-hexane to give 14 (67 mg, 95%) as a white solid. HRMS (ESI) m/z calcd for [C20H26ClNO4 + H]+: 380.1629, found 380.3787. 1H NMR (400 MHz, CDCl3): δ 7.97 (s, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.18 (dd, J = 8.3, 1.4 Hz, 1H), 6.36 (s, 1H), 3.68 (t, J = 6.2 Hz, 2H), 3.16 (dd, J = 16.3, 5.7 Hz, 1H), 3.03 – 2.92 (m, 2H), 2.89 (dd, J = 16.3, 7.6 Hz, 1H), 2.79 – 2.67 (m, 1H), 2.55 – 2.39 (m, 2H), 1.75 – 1.66 (m, 2H), 1.64 – 1.55 (m, 2H), 1.49 – 1.40 (m, 4H), 1.15 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 176.5, 172.3, 142.63, 136.8, 129.4, 128.1, 123.4, 120.6, 115.4, 107.9, 62.6, 44.7, 40.4, 32.2, 30.3, 28.9, 27.2, 25.2, 20.1, 14.2.
2.12.12. 5-Iodo-pentan-2-ol (16). To a solution of 2-methyltetrahydrofuran
15 (3 g, 34.83 mmol) in CH2Cl2 (30 ml) was added tetraethyl ammonium iodide (10.7 g, 41.66 mmol) followed by addition of boron trifluoride diethyl etherate (4.95 g, 34.87 mmol) at rt. The reaction mixture was stirred at rt for 16 h and then quenched with saturated solution of NaHCO3 (12 ml). The aqueous layer was extracted with EtOAc (4 × 20 ml) and the combined organic layers dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 20% EtOAc/n-hexane to give 16 (3.72 g, 50%) as colorless liquid. 1H NMR (400 MHz, CDCl3): δ 3.95 – 3.84 (m, 1H), 3.59 (br s, 1H), 3.23 (t, J = 6.9 Hz, 2H), 2.05 – 1.81 (m, 2H), 1.64 – 1.52 (m, 2H), 1.24 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 67.3, 39.8, 29.8, 23.6, 6.9.
2.12.13. Tert-Butyl-(4-iodo-1-methyl-butoxy)-diphenyl-silane (17)
Imidazole (1.90 g, 28.03 mmol), TBDPSCl (4.23 ml, 15.41 mmol) and a catalytic amount of DMAP (342 mg, 2.80 mmol) were added to a stirred solution of compound 16 (3 g, 14.01 mmol) in CH2Cl2 (30 ml) at 0 °C. Stirring was continued at rt for 12 h and then the mixture was diluted with CH2Cl2 (20 ml). Evaporation of the solvent under reduced pressure followed by silica gel column chromatography using 10% EtOAc/n-hexane afforded 17 (5.0 g, 80%) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.70 – 7.652 (m, 5H), 7.44 – 7.33 (m, 5H), 3.87 (dd, J = 11.7, 5.9 Hz, 1H), 3.06 (td, J = 7.0, 1.6 Hz, 2H), 1.87 – 1.77 (m, 2H), 1.58 – 1.47 (m, 2H), 1.05 (s, 12H). 13C NMR (100 MHz, CDCl3): δ 136.0 (4C), 135.0 (2C), 129.8 (2C), 127.8 (4C), 68.7, 40.3, 29.5, 27.3, 23.5, 19.2, 7.4.
2.12.14. 4-(tert-Butyl-diphenyl-silanyloxy)-pentyl]-triphenyl-phosphonium iodide salt (18)
To a solution of 17 (4 g, 8.84 mmol) in acetonitrile (40 ml) was added PPh3 (2.78 g, 10.60 mmol) and the reaction mixture was refluxed for 48 h at 80 °C. Evaporation of the solvent under reduced pressure followed by silica gel column chromatography using 5% MeOH/CH2Cl2 to afford 18 (3.79 g, 60%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.84 – 7.60 (m, 15H), 7.57 – 7.45 (m, 4H), 7.42 – 7.32 (m, 2H), 7.27 (td, J = 7.9, 3.2 Hz, 4H), 3.89 (dd, J = 9.8, 6.4 Hz, 1H), 3.58 (dt, J = 12.5, 8.0 Hz, 2H), 1.95 – 1.70 (m, 3H), 1.57 – 1.44 (m, 1H), 1.06 (d, J = 6.2 Hz, 3H), 0.90 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 135.7 (4C), 135.0 (6C), 134.2 (2C), 133.7 (6C), 130.5 (3C), 130.4 (3C), 129.5 (2C) 127.6, 127.4, 118.5, 117.7, 69.1, 39.9, 26.9 (3C), 23.5, 23.4, 22.9, 19.2.
2.12.15. 2-[5-(tert-Butyl-diphenyl-silanyloxy)-hex-1-enyl]-6-chloro-1H-indole (19)
To a stirred suspension of phosphonium iodide salt 18 (3 g, 4.19 mmol) in anhydrous THF (30 ml) was added t-BuOK (1.0 M in THF, 10 ml, 10 mmol) at −78 °C under argon. The mixture was allowed to warm at rt for 40 min, cooled to −78 °C, and a solution of aldehyde 1 (600 mg, 5.58 mmol) in anhydrous THF (15 ml) was added. The reaction mixture was warmed to rt and stirred for 4 h. The reaction mixture was quenched with saturated NH4Cl solution (15 ml), extracted with EtOAc (3 × 20 ml) and the combined organic layers were dried over Na2SO4. The solvents were evaporated under reduced pressure and the crude residue was purified by silica gel chromatography using 25% EtOAc/n-hexane to afford 19 (1.1 g, 68%) with cis/trans: 80/20. 1H NMR (400 MHz, CDCl3): δ 7.96 (s,1H), 7.72–7.66 (m, 5H), 7.45 – 7.32 (m, 6H), 7.22 (s, 1H), 7.04 (dd, J = 8.4, 1.7 Hz, 1H), 6.42 (s, 1H), 6.25 – 6.19 (m, 1H), 5.60 (dt, J = 11.6, 7.2 Hz, 1H), 4.00 – 3.87 (m, 1H), 2.63 – 2.33 (m, 2H), 1.77 – 1.60 (m, 2H), 1.06 (s, 9H), 1.04 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 136.1, 135.9 (2C), 134.8 (4C), 133.5, 129.6 (2C), 129.5, 127.7 (7C), 121.2, 119.0, 110.5, 102.9, 69.3, 39.1, 27.0 (3C), 25.6, 23.3, 19.3.
2.12.16. 2-[5-(tert-Butyl-diphenyl-silanyloxy)-hexyl]-6-chloro-1H-indole (20)
To a stirred solution of 19 (1.0 mg, 2.04 mmol) in anhydrous benzene (20 ml) was added 10% Pd/C (100 mg) at rt under a H2 atm and stirred at rt for 3 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated under reduced pressure and the crude product as a white solid 20 was directly used for the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 7.71 (s, 1H), 7.68 – 7.64 (m, 6H), 7.43 – 7.32 (m, 6H), 7.03 (dd, J = 8.36, 1.84 Hz, 1H), 6.13 (s, 1H), 3.85 – 3.81 (m, 1H), 2.62 (t, J = 7.6 Hz, 2H), 1.61 – 1.48 (m, 2H), 1.46 – 1.29 (m, 4H), 1.09 – 1.02 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 140.5, 136.1, 135.9 (4C), 134.7 (2C), 129.4 (2C), 127.6, 127.4 (4C), 126.6, 120.4, 120.1, 110.2, 99.5, 69.3, 39.0, 29.0, 28.0, 27.0 (3C), 25.2, 23.3, 19.2.
2.12.17. 6-(6-Chloro-1H-indol-2-yl)-hexan-2-ol (21)
To a solution of 20 (200 mg, 0.4 mmol) in anhydrous CH2Cl2 (10 ml) was added TiCl4 (1.0 M solution in CH2Cl2, 0.8 ml, 0.8 mmol) at 0 °C. The reaction mixture was stirred for 30 min and then quenched with water, extracted with EtOAc (4 × 10 ml), and the combined organic layers dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue purified by silica gel chromatography using 25% EtOAc/n-hexane to afford 22 (86 mg, 84% over two steps) as a colorless liquid. HRMS (ESI) m/z calcd for [C14H18ClNO+H]+: 252.1155, found 252.4972. 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.25 (s, 1H), 7.03 (dd, J = 8.4, 1.8 Hz, 1H), 6.20 (s, 1H), 3.84–3.77 (m, 1H), 2.76 (t, J = 7.5 Hz, 2H), 1.78 –1.68 (m, 2H), 1.54 – 1.47 (m, 4H), 1.20 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 140.5, 136.2, 127.3, 127.70, 120.5, 120.2, 110.2, 96.6, 68.0, 38.8, 29.0, 28.1, 25.3, 23.6.
2.12.18. 5-{2-[5-(tert-Butyl-diphenyl-silanyloxy)-hexyl]-6-chloro-indol-1-yl}-3-methyl-5-oxo-pentanoic acid (22)
To a stirred solution of 20 (600 mg, 1.22 mmol) in anhydrous THF (20 ml) was added t-BuOK (1.0 M in THF, 2.44 ml, 2.44 mmol) at 0 °C under argon. The reaction mixture was stirred for 30 min followed by addition of 5 (313 mg, 2.44 mmol) and further stirring for 4 h. The reaction mixture was quenched with saturated NH4Cl (10 ml), extracted with EtOAc (3 × 15 ml), and the combined organic layers dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 40% EtOAc/n-hexane to afford 22 (643 mg, 85% over two steps) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.90 (s, 1H), 7.70 – 7.63 (m, 4H), 7.43 – 7.31 (m, 7H), 7.19 (dd, J = 8.3, 1.5 Hz, 1H), 6.29 (s, 1H), 3.85–3.82 (m,1H), 3.08 (dd, J = 16.2, 6.3 Hz, 1H), 2.96 – 2.84 (m, 3H), 2.75 (m, 1H), 2.58 (dd, J = 15.7, 6.2 Hz, 1H), 2.42 (dd, J = 15.6, 7.1 Hz, 1H), 1.63 – 1.33 (m, 6H), 1.15 (d, J = 6.7 Hz, 3H), 1.07 (d, J = 6.1 Hz, 3H), 1.03 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 176.7, 172.1, 142.9, 136.5, 135.8 (4C), 135.8, 135.3, 134.8 (2C), 130.1, 129.4 (2C), 127.7 (4C), 120.6, 115.2, 107.9, 69.4, 40.1, 39.1, 30.5, 28.7, 27.2, 27.0 (3C), 26.9, 24.9, 24.0, 23.2, 19.2.
2.12.19. 5-[6-Chloro-2-(5-hydroxy-hexyl)-indol-1-yl]-3-methyl-5-oxo-pentanoicacid (23)
To a solution of 22 (150 mg, 0.242 mmol) in anhydrous THF (10 ml) was added TiCl4 (1.0 M solution in CH2Cl2, 0.48 ml, 0.48 mmol) at 0 °C. The reaction mixture was stirred for 2 h and then quenched with water and extracted with ethyl acetate (4 × 10 ml). The combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel column chromatography using 50% EtOAc/n-hexane to afford 23 (27.6 mg, 30%) as a white solid. HRMS (ESI) m/z calcd for [C20H26ClNO4 + H]+: 380.1629, found 380.1979. 1H NMR (400 MHz, CDCl3): δ 7.91 (s, 1H), 7.29 (s, 1H), 7.12 (d, J = 8.2 Hz, 1H), 6.29 (s, 1H), 3.86 – 3.73 (m, 1H), 3.16–3.05 (m, 1H), 2.97–2.75 (m, 3H), 2.74–2.61 (m, 1H), 2.40 (d, J = 6.4 Hz, 2H), 1.74–1.35 (m, 6H), 1.14 (d, J = 6.1 Hz, 3H), 1.09 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 177.2, 172.2, 142.4, 136.8, 129.5, 128.1, 123.5, 120.1, 115.5, 108.4, 68.1, 40.0, 31.9, 29.7, 28.3, 26.5, 25.7, 23.6, 22.7, 19.8.
2.12.20. 5-[6-Chloro-2-(5-oxo-hexyl)-indol-1-yl]-3-methyl-5-oxo-pentanoic acid (24)
To the above obtained alcohol 23 (20 mg, 0.052 mmol) in 3 ml of dry CH2Cl2, Dess-Martin periodinate (26 mg, 0.063 mmol) was added at 0 °C and stirred for 4 h. After completion, the reaction was quenched with aqueous NaHCO3 (1 ml). The reaction mixture was extracted with CH2Cl2 (3 × 3 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography using 40% EtOAc/hexane to afford 24 (9.9 mg, 50%) as a white solid. HRMS (ESI) m/z calcd for [C20H24ClNO4 + H]+: 378.1472, found 378.2272. 1H NMR (400 MHz, CDCl3): δ 7.86 (s, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.20 (ddd, J = 11.0, 9.4, 1.6 Hz, 1H), 6.38 (s, 1H), 3.13 (dd, J = 16.0, 6.2 Hz, 1H), 3.05–2.90 (m, 2H), 2.82–2.71 (m, 1H), 2.60–2.40 (m, 4H), 2.15 (s, 3H), 1.75–1.58 (m, 5H), 1.17 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 209.2, 175.8, 172.3, 142.6, 136.4, 129.4, 128.2, 123.4, 120.8, 115.2, 108.4, 45.2, 43.3, 40.2, 30.0, 29.53, 28.4, 27.2, 23.2, 14.1.
2.12.21. 5-(6-Chloro-2-hexyl-indol-1-yl)-2,2-dimethyl-5-oxo-pentanoic acid (190)
To a stirred solution of 27 (400 mg, 1.70 mmol) in DMSO (10 ml) was added KOH (114 mg, 2.04 mmol) at 0 °C. After 30 min, 28 (314 mg, 2.21 mmol) in DMSO (5 ml) was added and the reaction mixture stirred at rt for 4 h. The reaction was quenched by adding saturated NH4Cl (8 ml). The aqueous layer was extracted with EtOAc (4 × 20 ml), and the combined organic layers were washed with brine (6 ml) and dried over Na2SO4. The solvents were evaporated under reduced pressure to obtain the crude product that was purified by silica gel column chromatography using 50% EtOAc/n-Hex to afford 190 (416 mg, 65%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.81 (s, 1H), 7.25 (d, J = 8.2 Hz, 1H), 7.07 (dd, J = 8.2, 1.6 Hz, 1H), 6.26 (s, 1H), 2.88 (dt, J = 29.8, 7.9 Hz, 4H), 2.05- (t, J = 6.0 Hz, 2H), 1.63–1.56 (m, 2H), 1.34–1.29 (m, 2H), 1.27–1.24 (m, 4H), 1.24 (s, 6H), 0.81 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 183.3, 173.0, 143.0, 136.6, 129.3, 128.2, 123.3, 120.6, 115.2, 107.8, 41.4, 35.0, 34.8, 31.6, 30.5, 29.1, 28.9, 25.1 (2C), 22.5, 14.0.
3. Results
3.1. 264 blocks 5-oxo-ETE-induced granulocyte activation
Because we hope to use the monkey as an experimental model to explore the in vivo effects of 264 we first wanted to ensure that monkey granulocytes respond to this antagonist in the same way as human granulocytes. Eosinophils were gated out from a population of mixed leukocytes based on their forward and side scatter and their ability to bind fluorescently labeled CD49d and polymerized F-actin was measured using NBD-phallicidin. 5-Oxo-ETE (10 nM) strongly stimulated actin polymerization in these cells and this was almost completely blocked by 264 (300 nM) (Fig. 2A). The IC50 for this effect (13 ± 4 nM) (Fig. 2B) is similar to what we previously observed (Gore et al., 2014) for human eosinophils (Fig. 2B). 264 similarly inhibited 5-oxo-ETE-induced actin polymerization in neutrophils (33 ± 9 nM) and also the migration of leukocytes (40% neutrophils and 1% eosinophils) induced by 5-oxo-ETE (100 nM) with an IC50 of 260 ± 50 nM (Fig. 2C).
Figure 2.
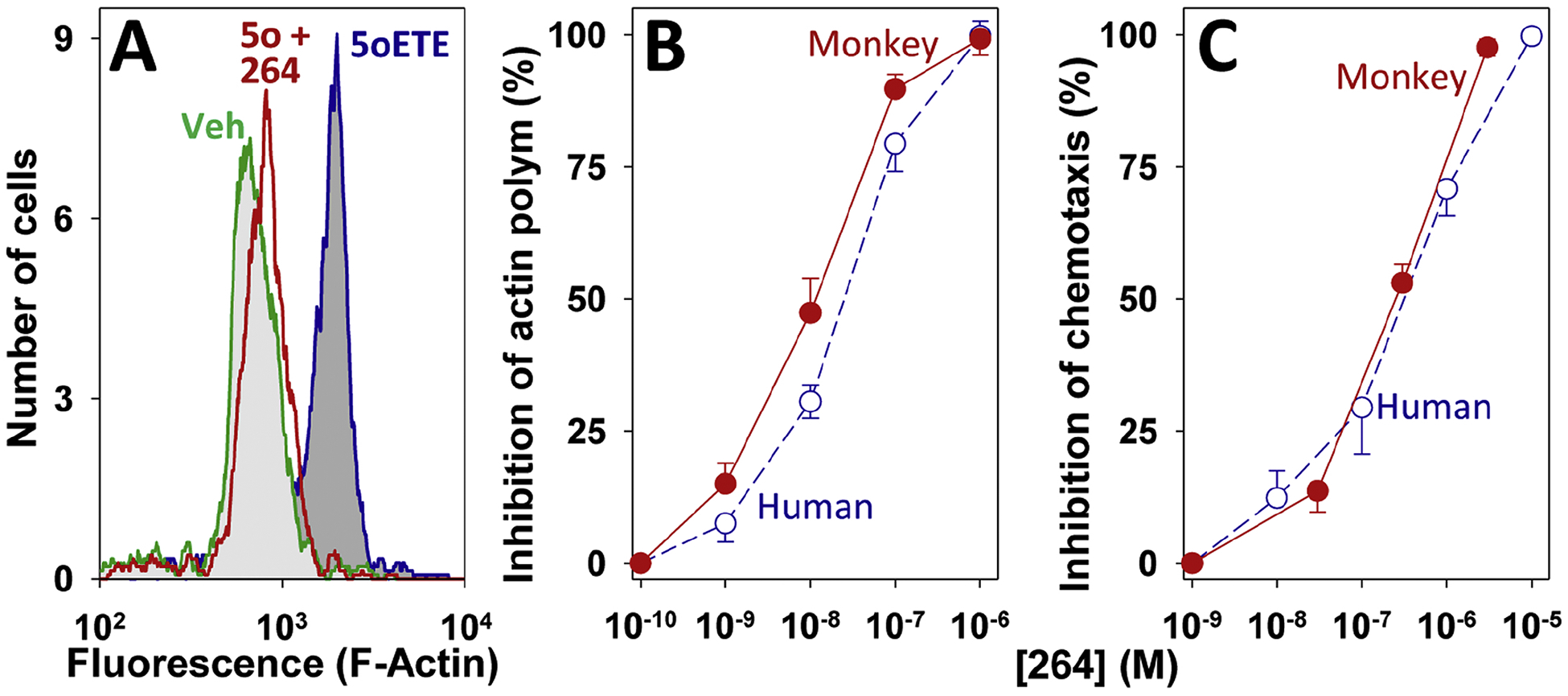
Inhibition of 5-oxo-ETE-induced granulocyte activation by 264. A: Histograms showing F-actin levels in eosinophils, measured by flow cytometry, following incubation of leukocytes with either vehicle (green; light grey fill), 5-oxo-ETE (10 nM; blue; dark grey fill) or 5-oxo-ETE (10 nM) in the presence of 300 nM 264 (red; no fill). B: Concentration-response curves for the inhibition of actin polymerization in response to 5-oxo-ETE (10 nM) in monkey (●) and human (○) eosinophils. C: Concentration-response curves for the inhibition of the chemotactic response to 5-oxo-ETE (100 nM) in monkey (●) and human (○) leukocytes. The data for human cells were taken from ref. 12 for comparison.
3.2. 264 rapidly appears in the blood after oral administration to rats
A pilot experiment in rats revealed that appreciable blood levels of 264 were achieved following oral administration. Fig. 3A shows a chromatogram of a plasma sample obtained 4 h after administration of 264 (10 mg/kg) by oral gavage. The largest peak had a tR (12.02 min) and UV spectrum (Fig. 3B) identical to those of authentic 264. Four incompletely resolved polar metabolites (1 – 4) were also detected (tR, 6.41, 6.52, 6.92, and 7.02 min), all of which had UV spectra identical to 264 (Fig. 3B), suggesting that the indole chromophore was intact. As shown in Fig. 3C, 264 appeared rapidly in the blood after oral administration, reaching a plasma concentration of just over 2 μM by 30 min, and a maximum of 7.5 μM by 4 h, after which time it diminished. The plasma concentration of 264 after 1 h was proportional to the dose administered (Fig. 3C, inset).
Figure 3.
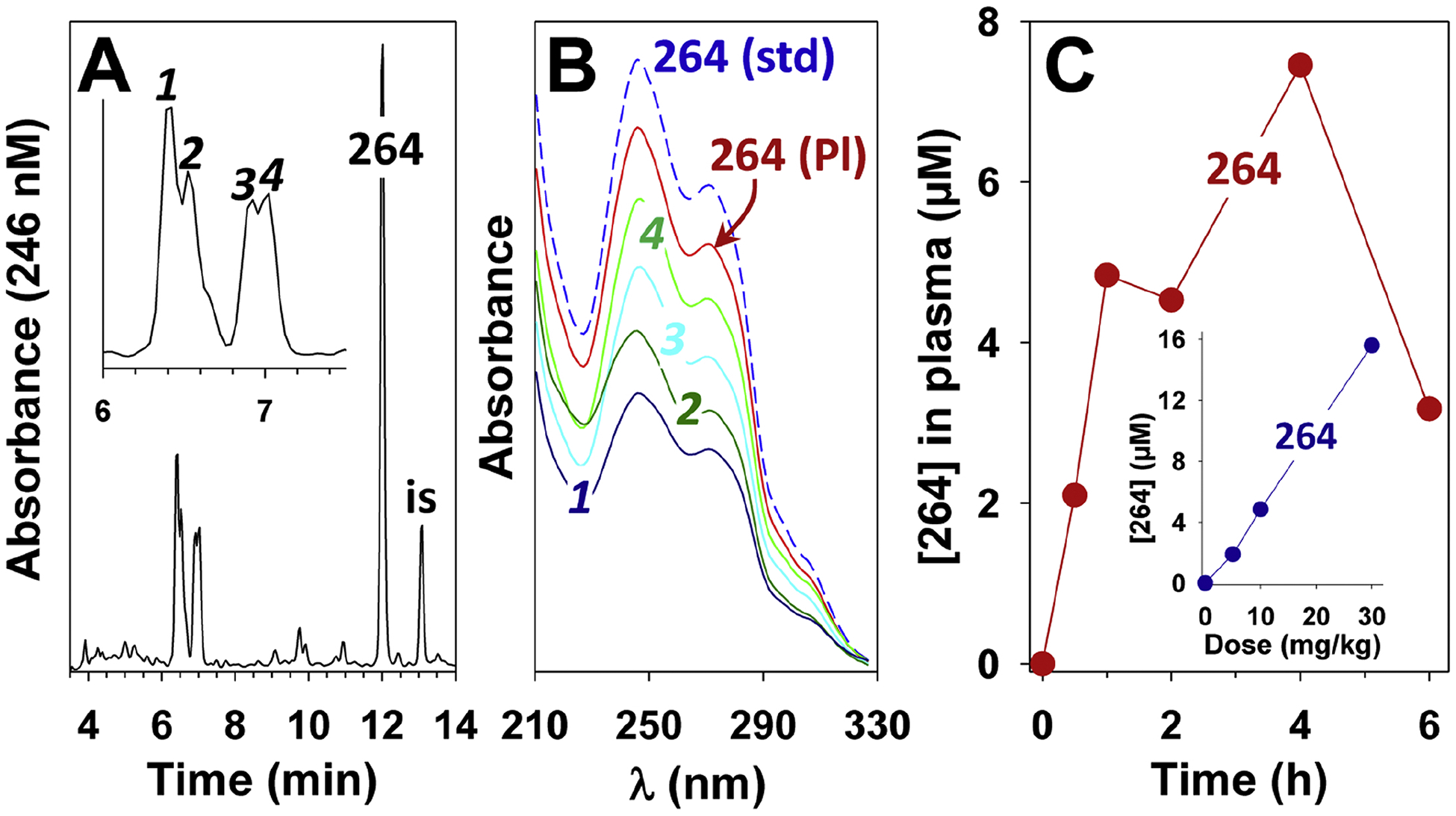
Metabolism and pharmacokinetics of 264 in rats. 264 was administered by oral gavage to rats as described in the Methods section. The rats were sacrificed after different times and plasma samples were analyzed by RP-HPLC on a Novapak C18 column following solid-phase extraction using 190 as an internal standard (is) as described in Methods. A: Chromatogram of an extract of plasma obtained 4 h after administration of 264 (10 mg/kg). The region containing metabolites 1–4 is expanded in the inset. B: UV spectra of metabolites 1–4 from panel A along with the peak labeled “264” in panel A (264 (Pl)) and authentic 264. C: Plasma levels of 264 after oral administration of a dose of 10 mg/kg (●). The inset shows the plasma levels of 264 (●) 1 h after administration of doses of 5, 10, and 30 mg/kg. Each point represents a single rat (blood was taken only once at the time the animal was sacrificed).
3.3. Preparation of synthetic ω1 oxidation products of 264
Since β-oxidation of the acyl side chain of 264 should be blocked by the 3-methyl group, we anticipated that the hexyl side chain would be the most likely target for metabolism. From the rat study we suspected that 264 might be converted to polar metabolites by hepatic cytochrome P450 enzymes. To facilitate the identification of such products we prepared a series of potential metabolites by total chemical synthesis, including the ω1-oxidation products ω1-hydroxy-264, ω-carboxy-264, and dinor-ω-carboxy-264 (Fig. 4). ω-Carboxy-264 was synthesized by reaction of the Wittig salt 2 with the 6-chloroindole aldehyde 1, followed by introduction of the second side chain by reaction with 3-methyl glutaric anhydride 5. Dinor-ω-carboxy-264 was synthesized in an analogous manner using the Wittig salt 7, whereas ω1-hydroxy-264 was prepared by reduction of the carboxyl group of 4, followed by protection of the resultant hydroxyl group and coupling with 3-methyl glutaric anhydride 5. Note that in this paper we have designated the terminal methyl group of the alkyl side chain as “ω1” and the adjacent methylene group as “ω2”.
Figure 4.
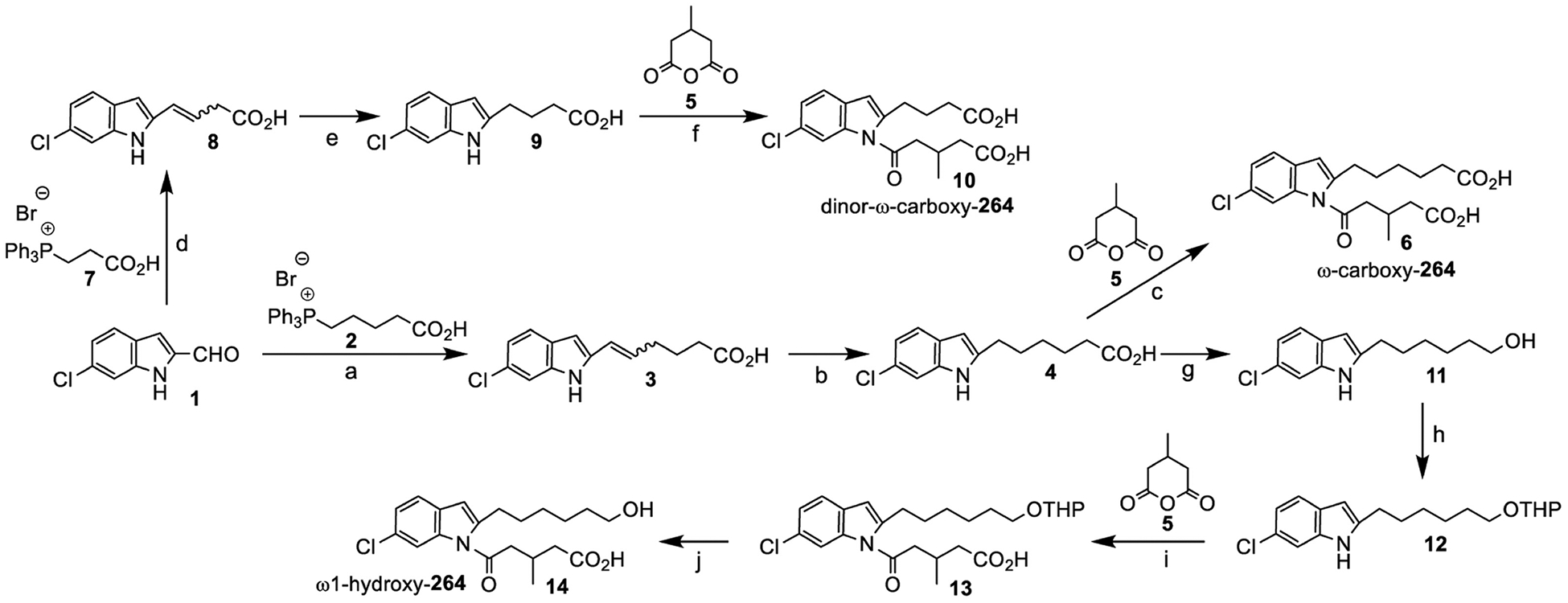
Synthesis of ω1-oxidation products of 264. Reagents and conditions: (a) t-BuOK, THF, 5 h, −78 °C-rt, 83%; (b) Pd/C, benzene, 3 h, rt; (c) t-BuOK, DMSO, 4 h, 0 °C-rt, 80% over two steps; (d) t-BuOK, THF, 5 h, −78 °C-rt, 60%; (e) Pd/C, benzene, 3 h, rt; (f) t-BuOK, DMSO, 4 h, 0 °C-rt, 78% over two steps; (g) LiAlH4, THF, −20 °C-rt, 16 h, 93%; (h) DHP, Cat. PTSA, CH2Cl2, 0 °C, 0.5 h, 89%; (i) KOH, DMSO, 0 °C-rt, 4 h, 71%; (j) Cat. PTSA, THF, rt, 16 h, 95%.
3.4. Metabolism of 264 by monkey liver microsomes
264 was incubated with monkey liver microsomes in the presence of NADPH for 4 h at 37 °C and the products were analyzed by RP-HPLC using a water/methanol gradient (Fig. 5A). 264 was almost completely converted to more polar metabolites, including 2 major products (a and c), and a minor product (b). The tR of the most abundant product (c) was identical to that of ω1-hydroxy-264, whereas that of the minor product (b) was identical to that of ω-carboxy-264. The UV spectra of all 3 products were virtually identical to that of 264, with absorbance maxima at 247 and 271 nm (3D), suggesting that the indole chromophore was unchanged. Several very small peaks were observed with tRs close to that of authentic dinor-ω-carboxy-264, but it was difficult to identify this compound conclusively by HPLC/UV alone. When acetonitrile was substituted for methanol in the mobile phase, the tR of a shifted dramatically compared to b and c, and was now the longest (Fig. 5B). This is typical of oxoeicosanoids, which have shorter tRs than the corresponding hydroxyeicosanoids with methanol gradients, but longer tRs with acetonitrile gradients (Powell et al., 1997). With acetonitrile, the tR of b was still identical to that of ω-carboxy-264, but that of c was slightly shorter than that of ω1-hydroxy-264, which was confirmed in a cochromatography experiment (Fig. 5C).
3.5. Identification of ω2-oxidation as the major pathway for metabolism of 264 by liver microsomes
Since ω1-oxidation was not a major metabolic pathway for 264, we synthesized potential ω2-oxidation products. ω2-Hydroxy-264 was synthesized by the regioselective ring opening of 2-methyltetrahydrofuran 15, followed by addition to 6-chloroindole using a Wittig reaction (Fig. 6). The 5-oxo-3-methyl valeric acid side chain was then added as shown for ω1-oxidation products in Fig. 4. ω2-Oxo-264 (24) was synthesized by the DMP oxidation of ω2-hydroxy-264 (23). The tRs of the major microsomal 264 metabolites c and a were identical to those of authentic ω2-hydroxy-264 and ω2-oxo-264, respectively, with both methanol and acetonitrile gradients (Figs. 4A and 4B). Furthermore, the biological and synthetic compounds cochromatographed with one another with both acetonitrile (Fig. 5C) and methanol gradients.
Figure 6.
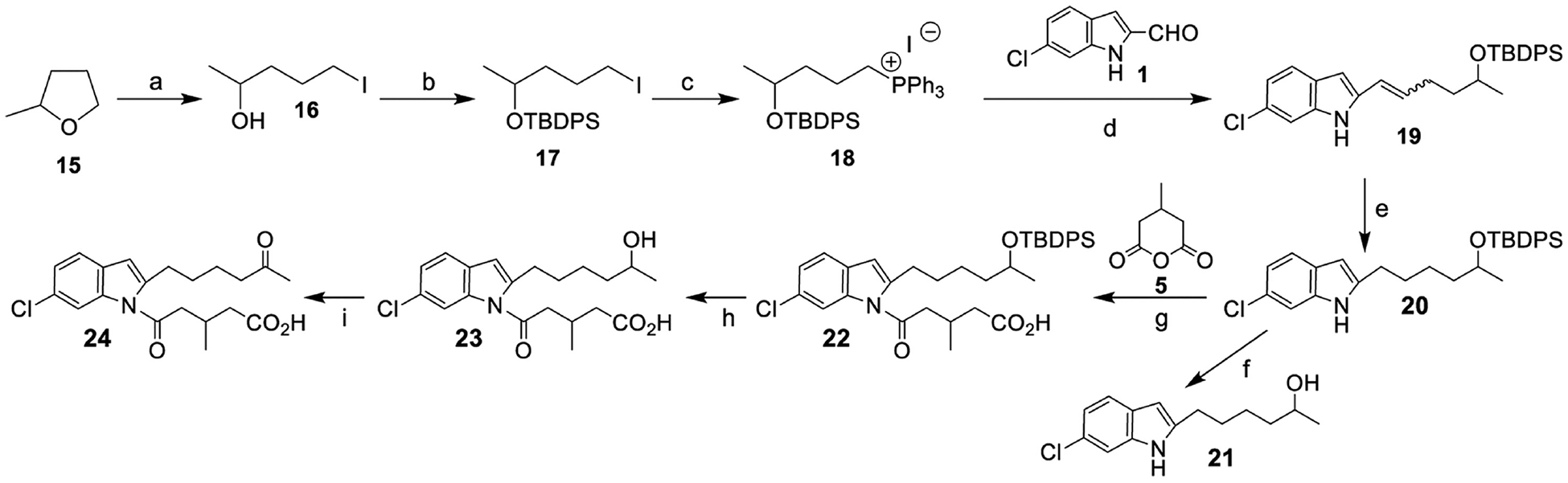
Synthesis of ω2-oxidation products of 264. Reagents and conditions: (a) Et4N+ I−, BF3.Et2O, CH2Cl2, 16 h, rt, 50%; (b) TBDPS-Cl, imidazole, DMAP, CH2Cl2, rt, 12 h, 80%; (c) PPh3, CH3CN, 48 h, reflux, 60%; (d) t-BuOK, THF, −78 °C, 4 h, 68%; (e) Pd/C, benzene, rt, 3 h; (f) TiCl4, CH2Cl2, 0.5 h, 0 °C-rt, 84% over two steps; (g) DMSO, t-BuOK, 4 h, 0 °C, 85% over two steps; (h) TiCl4, CH2Cl2, 2 h, 0 °C-rt, 30%; (i) DMP, CH2Cl2, rt, 4 h, 50%.
3.6. Analysis of the major microsomal 264 metabolites by LC-MS/MS
The identity of c as ω2-hydroxy-264 was confirmed by LC-MS/MS. It had an [M-H]− ion at m/z 378.1460, compared to the theoretical value m/z 378.1478 for a monohydroxy metabolite of 264 (mass accuracy, 4.8 ppm). The MS2 fragmentation pattern of this ion was identical to that of authentic ω2-hydroxy-264 and consisted of only one significant ion at m/z 250, consistent with loss of the acyl side chain (Fig. 7A). MS3 `fragmentation of this ion gave a pattern (Fig. 7B) identical to authentic ω2-hydroxy-264, with major ions at m/z 232 (loss of H2O), 206 (loss of CHOH=CH2), 190 and 164.
Figure 7.

Mass spectra of 264 metabolites. Metabolites were first purified by RP-HPLC as shown in Fig. 5 and then analyzed by LC/MS/MS using negative ion ESI except for metabolite a (panels C and D), in which case positive ion ESI was used. A: MS2 fragmentation of the [M-H]− ion at m/z 378 of c isolated from monkey liver microsomes. B: MS3 fragmentation of m/z 250 from panel A. C: MS2 of the [M+H]+ ion at m/z 378 for metabolite a, isolated from liver microsomes. D: MS3 fragmentation of the ion at m/z 250 in panel C. E: MS2 of the [M-H]− ion at m/z 392 for metabolite b, isolated from monkey plasma. F: MS2 fragmentation of the ion at m/z 264 in panel E. G: MS2 of the [M-H]− ion at m/z 364 for metabolite e from plasma. H: MS2 of the [M-H]− ion at m/z 394 for metabolite d isolated from plasma. I: MS2 of the [M-H]− ion at m/z 234 of metabolite f from plasma.
Using negative ion ESI, we were unable to obtain satisfactory mass spectra of authentic ω2-oxo-264 or metabolite a, with which it cochromatographed. However, positive ion ESI analysis of a revealed an [M+H]+ ion at m/z 378.1464, compared to the theoretical value m/z 378.1472 for an oxo derivative of 264 (mass accuracy, 2.1 ppm). MS2 fragmentation of this ion (Fig. 7C) gave an intense ion at m/z 250 due to loss of the acyl group, along with a much less intense ion at m/z 360 due to loss of H2O. MS3 fragmentation of the ion at m/z 250 resulted in an intense ion at 232 due to loss of H2O together with a less intense ion at m/z 204 due to the subsequent loss of ethylene (Fig. 7D)
3.7. Identification of plasma metabolites of 264
To identify the major in vivo metabolites of 264 in plasma, a dose of 30 mg/kg was administered by oral gavage and a blood sample taken after 4 h. Analysis of a plasma extract by RP-HPLC revealed a pattern very similar to microsomes with both methanol (Fig. 5E) and acetonitrile (Fig. 5F) gradients. The major metabolites (a and c) and the minor metabolites b cochromatographed with ω2-oxo-264, ω2-hydroxy-264, and ω-carboxy-264, respectively (Fig. 5G) and had UV spectra identical to the authentic standards (data not shown). The mass spectra of a and c were identical to those shown in Fig. 7, panels A-D. The identity of metabolite b from plasma was also confirmed by LC-MS/MS. It had an [M-H]− ion at m/z 392.1251 compared to the theoretical value of 392.1265 (mass accuracy 3.5 ppm). The MS2 fragmentation pattern of this ion (Fig. 7E) was identical to that of authentic ω-carboxy-264 and displayed a single significant ion at m/z 264 due to loss of the N-acyl side chain, as well as an ion of weak intensity at m/z 374 (loss of H2O). MS3 fragmentation of the ion at m/z 264 (Fig. 7F) resulted in daughter ions at m/z 246 (loss of H2O) and 220 (loss of CO2), identical to the MS3 spectrum of the authentic standard.
The minor plasma metabolite e had a tR identical to dinor-ω-carboxy-264, but several other peaks with similar tRs were also present, which complicated its identification using UV detection. However, the identity of this metabolite was confirmed by LC-MS/MS. It displayed an [M-H]− ion at m/z 364.0944, very close to the theoretical value of 364.0952 expected for dinor-ω-carboxy-264 (mass accuracy 2.2 ppm). MS2 fragmentation of this ion resulted in a mass spectrum (Fig. 7G) identical to that of the authentic standard, with ions at m/z 346 (loss of H2O), 322 (loss of H2O + CH2=C=O), 304 (loss of CH2C(OH)2 due to a McLafferty rearrangement), 283, and 236 (base peak; loss of the 5-oxo-3-methylvalerate side chain.
A small amount of another polar metabolite (d) was also detected in plasma samples, and had tRs of 16.0 and 13.5 min with methanol (Fig. 5E) and acetonitrile (Fig. 5F) gradients, respectively. Metabolite d had an [M-H]− ion at m/z 394.1423, compared to the theoretical value of 394.1421 expected for a dihydroxy metabolite of 264 (mass accuracy, 0.5 ppm). MS2 fragmentation of this ion resulted in a single intense ion at m/z 266 due to loss of the acyl side chain. These data indicate that d has two hydroxyl groups on its hexyl side chain, one of which we would assume to be in the ω2 position.
Relatively small amounts of deacylation products of 264 and some of its metabolites, formed by loss of the N-acyl group, were also detected in plasma by comparison of their chromatographic properties and UV spectra with those of authentic standards. However, it is unclear whether these products were formed enzymatically, as their amounts increased upon sample storage, especially in the presence of column solvent, which contained low concentrations of acetic acid. Therefore, the column solvent was normally removed as soon as possible and the samples stored in acetonitrile. The deacylated products could readily be detected by their UV spectra (λmax 226 and 278 nm), which were quite distinct, and markedly different from their precursors containing the 3-methyl-5-oxo-valerate group. Metabolite f (Figs. 4E and 4F) cochromatographed with authentic deacyl-264 and had a UV spectrum identical to the authentic standard (Fig. 5D). It had an [M-H]− ion at m/z 234.1043 compared to the theoretical value of 234.1050 (mass accuracy 3.0 ppm). MS2 fragmentation of this ion gave a daughter ion at m/z 163 due to loss of a pentyl group (Fig. 7I), identical to that for the authentic standard.
A chromatogram showing the deacylated derivatives of ω2-hydroxy-264 and ω2-oxo-264 in a 4 h plasma sample is shown in Fig. 5H. These products had retention times identical to those of the corresponding authentic deacyl standards (data not shown). We did not observe significant amounts of deacylated products among microsomal metabolites of 264, possibly because the work-up of these samples was much simpler than that of the plasma samples.
3.7. Pharmacokinetics of 264 in monkeys
To investigate its pharmacokinetics in monkeys, 264 (30 mg/kg) was administered by oral gavage to 4 animals. RP-HPLC of plasma extracts revealed high levels of 264 after 30 min, with maximal levels (26 ± 11 μM) after 1 h (Fig. 8A). The concentrations of 264 declined fairly rapidly thereafter, to about 3 μM by 4 h, less than 1 μM by 8 h, and smaller, but detectable, amounts after 24 h. The major metabolite of 264, ω2-OH-264, also appeared rapidly in the blood, reaching a plateau of ~9 μM between 1 and 4 h, and then declining more slowly than 264. Between 8 and 12 h the plasma concentration ω2-OH-264 was about 5 times that of 264. Plasma concentrations of ω2-oxo-264 initially increased more slowly, reaching maximal levels (~4 μM) by 4 h and then declining, but remaining higher than those of 264 between 4 and 24 h.
Figure 8.
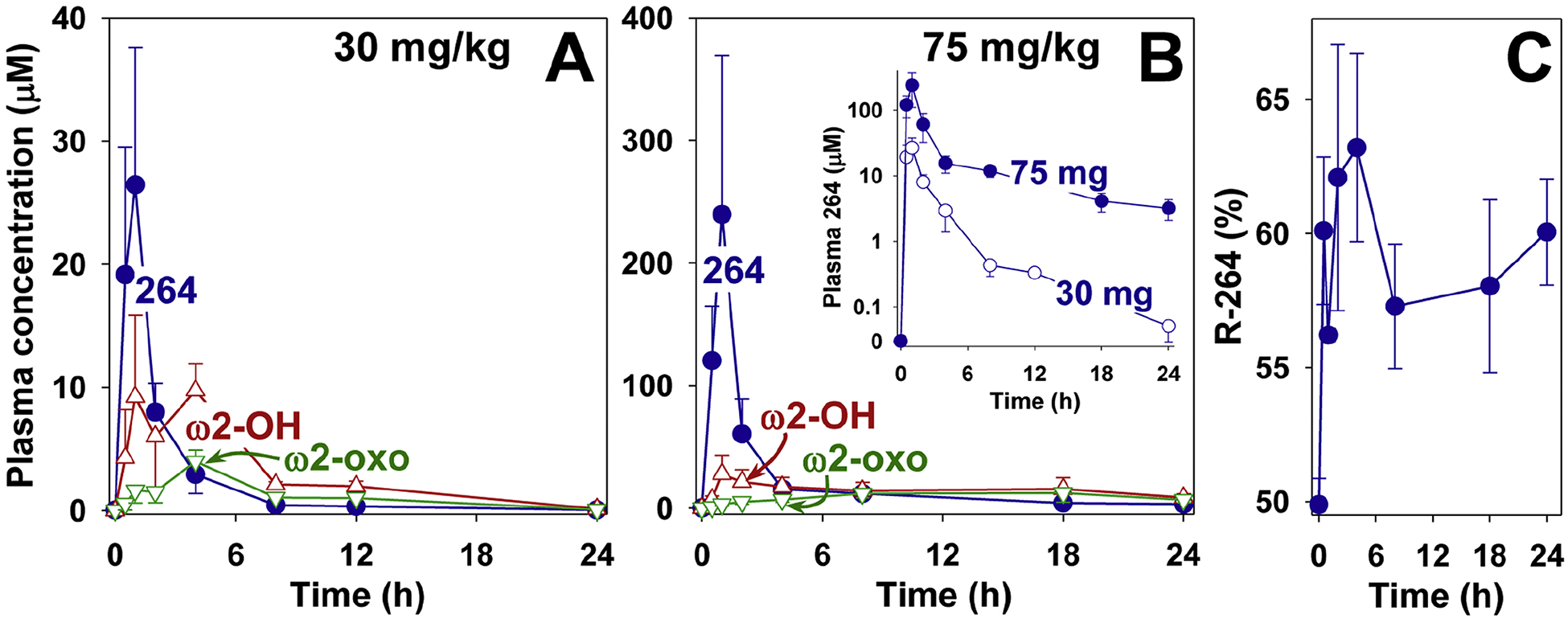
Pharmacokinetics of 264 in monkeys. 264 was administered by oral gavage to cynomolgus monkeys as described in Materials and Methods at doses of either 30 mg/kg (A) or 75 mg/kg (B) and the products were extracted and analyzed by RP-HPLC as shown in the legend to Fig. 5, using 190 as an internal standard. The concentrations of 264 (●), ω2-hydroxy-264 (△), and ω2-oxo-264 (▽) are shown. In the inset to panel B the plasma concentrations of 264 (30 (○) and 75 (●) mg/kg) at different time points are compared using a logarithmic scale. C: The percentage of 264 in the form of the R-enantiomer was determined for each of the time points shown in panel B (264, 75 mg/kg). The S and R enantiomers were separated by chiral HPLC as described in Materials and Methods.
Because of the relatively rapid decline in plasma 264 at longer time points we also investigated the PK profile of a 2.5-fold higher dose (75 mg/kg) (Fig. 8B). This resulted in much higher plasma levels (maximum, 240 ± 130 μM after 1 h) and concentrations in excess of 3 μM over 24 h. The difference between the two doses of 264 is more easily visualized when its plasma concentrations are plotted on a log scale (inset to Fig. 8B). Maximal levels of ω2-hydroxy-264 (~28 μM) were reached by 1 h, and declined more slowly compared to the lower dose. The maximal levels of ω2-oxo-264 (~12 μM) were reached later (between 8 and 12 h), and were nearly equal to those of ω2-hydroxy-264 at 24 h (~7 μM vs ~9 μM). The chirality of carbon-3 of the 3-methyl-5-oxo-valerate side chain of 264 (75 mg/kg dose) was determined using chiral-HPLC. A higher proportion of the R-enantiomer appeared to be present at all time points (~6:4 ratio of R:S; Fig. 8C).
3.8. Antagonist activity of 264 metabolites
We examined the abilities of the major 264 metabolites to block 5-oxo-ETE-induced calcium mobilization in human neutrophils (Fig. 9). All of the ω-oxidized metabolites were much less potent than 264. The major metabolites ω2-hydroxy-264 (IC50, 3.3 μM) and ω2-oxo-264 (IC50, 1.4 μM were nearly 400 and 170 times less potent than 264 (IC50, 8.4 nM). The minor metabolite ω1-hydroxy-264 (IC50, 0.53 μM) was 64 times less potent, whereas its oxidation products ω-carboxy-264 (IC50, 13 μM) and dinor-ω-carboxy-264 (IC50, 5.7 μM) were much less potent.
Figure 9.
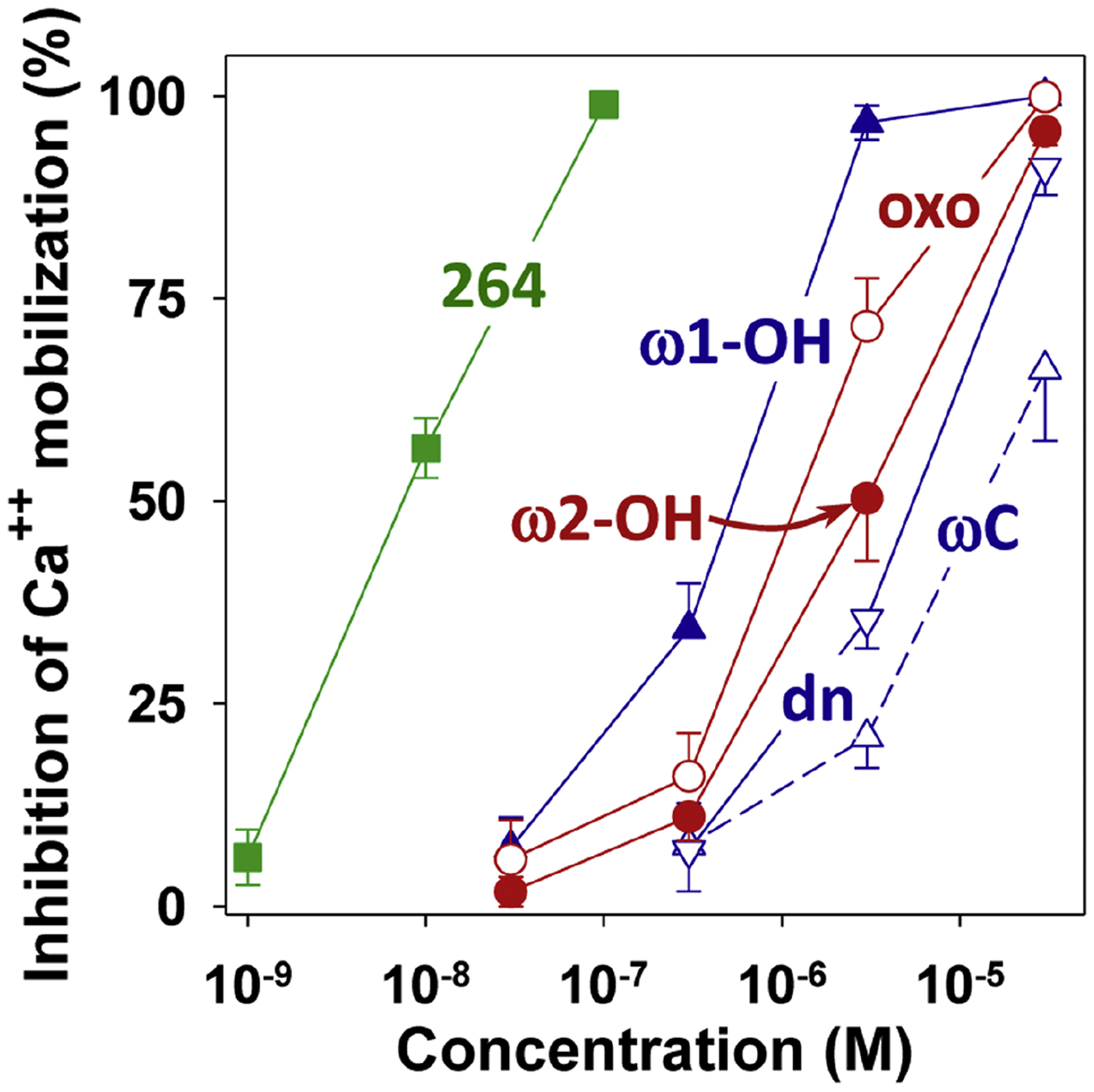
Effects of 264 metabolites on 5-oxo-ETE-induced calcium mobilization in human neutrophils. Intracellular calcium levels were measured in indo-1-loaded neutrophils treated with 264 or one of its synthetic metabolites followed 2 min later by addition of 5-oxo-ETE (10 nM). All compounds are racemic mixtures: 264 (■), ω1-hydroxy-264 (▲, ω-OH); ω-carboxy-264 (△, ωC), dinor-ω-carboxy-264 (▽, dn), ω2-hydroxy-264 (●, ω2-OH), ω2-oxo-264 (○, oxo). Values are means ± SE of data from 4 to 5 independent experiments.
4. Discussion
We recently identified the synthetic indole 264 as a potent and selective antagonist of the OXE receptor for 5-oxo-ETE, suggesting that it could be a potential therapeutic agent in eosinophilic diseases such as asthma (Gore et al., 2014). The next step in the development of this compound is to identify an animal model to test its efficacy in vivo. We had hoped to be able to use cats for this purpose, as they are particularly susceptible to asthma, but, although feline eosinophils are highly responsive to 5-oxo-ETE, 264 is only a weak OXE receptor antagonist in this species, presumably because of significant differences between the feline and human receptors (Cossette et al., 2015). We are therefore now exploring the possibility of the monkey as an experimental model to investigate 264.
We recently showed that monkey leukocytes synthesize 5-oxo-ETE, which is a potent monkey granulocyte chemoattractant (Cossette et al., 2016). To ensure that 264 can block the OXE receptor in monkeys we tested its effects on 5-oxo-ETE-induced activation of monkey leukocytes. We found that 264 inhibits the effects of 5-oxo-ETE on actin polymerization in monkey eosinophils and neutrophils as well as leukocyte migration with potencies similar to what we previously observed with human cells (Gore et al., 2014).
The next question was whether 264 would appear in the blood following oral administration. A preliminary experiment in rats confirmed that this is indeed the case, as 264 could be detected in plasma shortly after oral administration of doses as low as 5 mg/kg. We then investigated the PK of 264 in monkeys, initially using a dose of 30 mg/kg. As with rats, 264 appeared rapidly in the blood but its concentration quickly dropped after 1 h, in contrast to rats, where the plasma concentration was relatively stable over the following 5 h at a dose of 10 mg/kg. However, because of the limited experiments with rats, it is difficult to make a detailed comparison between the two species. At a dose of 30 mg/kg the plasma level of 264 was about 16 μM in rats and 28 μM in monkeys after 1 h. However, after 4 h, the plasma concentration of 264 was about 7.5 μM in rats receiving a dose of 10 mg/kg compared to 2.9 μM in monkeys receiving a dose 3 times higher, indicating that 264 is cleared from the circulation more rapidly in monkeys. By 8 h, the plasma concentration of 264 was below 1 μM. Increasing the dose of 264 in monkeys from 30 to 75 mg/kg resulted in disproportionately higher plasma levels of the unmetabolized antagonist, suggesting saturation of the enzymes responsible for its metabolism. The peak level of 264, reached after 1 h, was 240 μM, 9 times higher than that achieved with the lower dose. After 8 h, the plasma concentration was 12 μM and after 24 h it was 3 μM. Although we did not look specifically for adverse effects due to administration of 264, no untoward effects of the two doses employed were noted, and we did not observe any long-lasting effects in these monkeys.
Although administration of 264 as a suspension by gavage may not be the optimal approach, it served our purposes well in the current study. The suspensions were made up specifically for each animal and were used immediately after preparation. As the entire amount was administered in each case, the doses should be reasonably correct, and this is confirmed by the good relationship between the dose administered and the plasma concentrations of 264 in both rats (inset to Fig. 3C) and monkeys (inset to Fig. 8B). We did some exploratory experiments in monkeys in which 264 was administered in the form of capsules, but the plasma levels achieved were lower than those using suspensions, and this approach was not pursued further. However, the issue of formulation of 264 or related OXE receptor antagonists will need to be addressed at some point during the development of these compounds as therapeutic agents in the future in order to optimize their efficacy.
264 is a racemic mixture of two enantiomers because of the 3-methyl group on the 5-oxovalerate side chain. Because only the S-enantiomer possesses potent antagonist activity we assessed the chirality of plasma 264 and found that the inactive R-enantiomer predominated over the S-enantiomer in a ratio of about 6:4. Therefore, in future studies it would be preferable to use the pure S-enantiomer, which was not possible in the current study because of the relatively large amounts of 264 that were required for in vivo experiments. However, this should be feasible in the future, using a novel procedure that we have recently developed to synthesize the S-enantiomer of the 3-methyl-5-oxovalerate synthon (Reddy et al., 2015).
Chromatographic analysis of plasma samples obtained after administration of 264 revealed that it is converted to two major polar metabolites, which seemed likely to be formed by oxidation of the hexyl side chain (Fig. 10). Although ω2-oxidation is thermodynamically favored over ω1-oxidation because of a weaker C-H bond, many cytochrome P450 enzymes preferentially hydroxylate the terminal methyl group of fatty acids such as lauric acid due to steric constraints imposed by the enzyme (Johnston et al., 2011). For example, many eicosanoids, including 5-oxo-ETE (Powell et al., 1996), leukotriene B4 (Lindgren et al., 1982) and prostaglandins (Powell and Solomon, 1978) are converted to ω1-hydroxy metabolites by cytochrome P450 enzymes. We therefore initially suspected that the polar metabolites of 264 might be formed by ω1-oxidation and we synthesized ω1-hydroxy-264, ω-carboxy-264, and its β-oxidation product dinor-ω-carboxy-264 to serve as authentic standards. However, only small amounts of these metabolites were formed, and the major pathway was instead ω2-oxidation to ω2-hydroxy-264 and ω2-oxo-264, both of which were identified by cochromatography with authentic standards and LC-MS/MS. We also detected very small amounts of other plasma metabolites of 264 with [M-H]− ions at m/z 378 and MS2 spectra consistent with the presence of a hydroxyl group in other positions of the hexyl side (data not shown), but we were unable to determine the positions of the hydroxyl groups of these products. We also detected a dihydroxy metabolite of 264, presumably with one of the hydroxyl groups in the ω2-position. The pattern of metabolites detected after administration of a higher dose of 264 (75 instead of 30 mg/kg) was similar, except that proportionately smaller amounts of metabolites were detected, suggesting that the metabolic enzymes were saturated at the higher dose.
Figure 10.
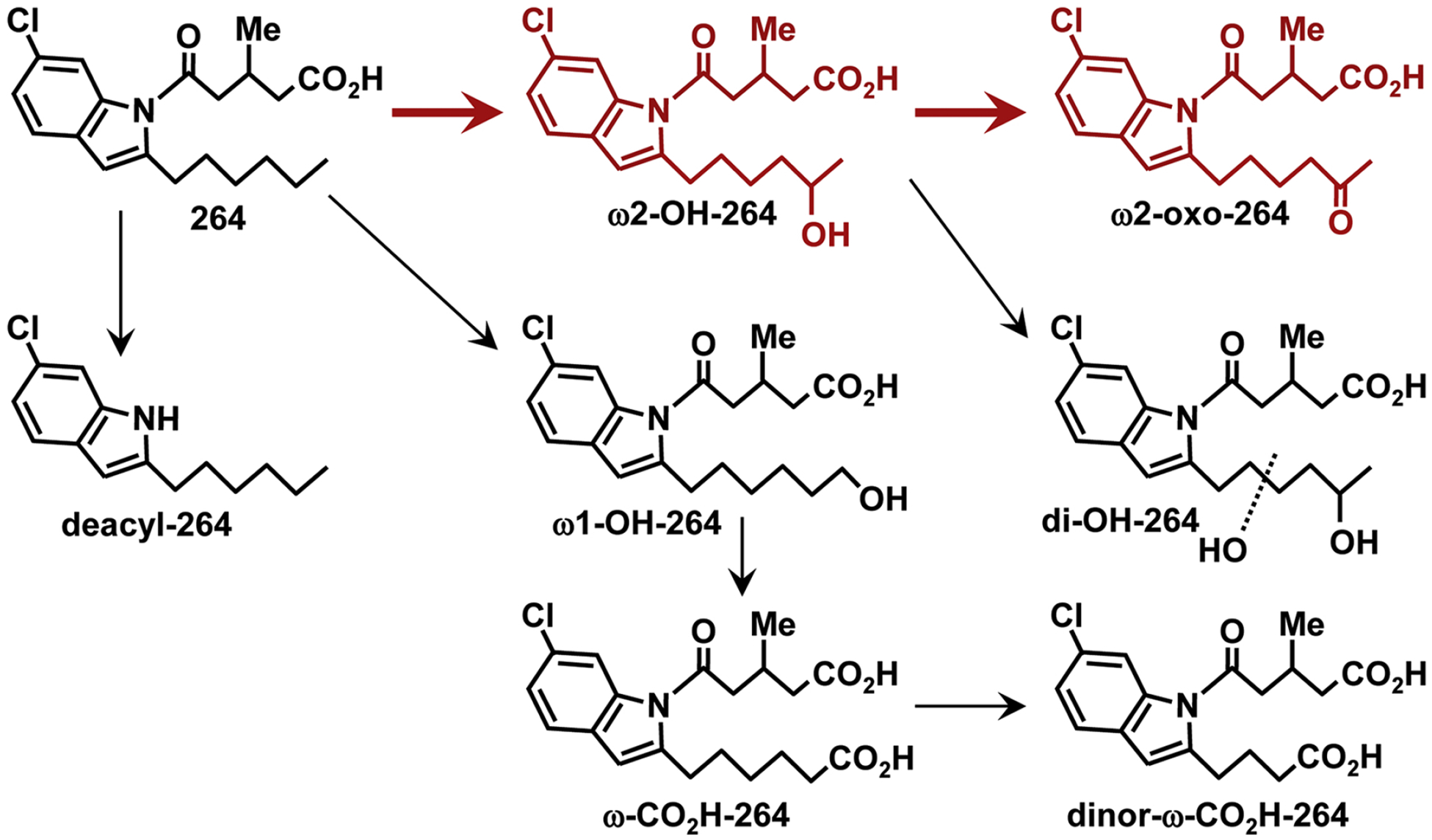
Metabolism of 264 in monkeys. Major pathways are shown in red. Small amounts of deacylated products were also observed.
The increased clearance of 264 from the blood in monkeys compared to rats is likely to be due, at least in part, to an increased rate of metabolism in monkeys. Comparison of the chromatographic profile for rat plasma 4 h after administration of 264 (10 mg/kg; Fig. 3A) with that for monkey plasma at the same time point, but with a 3-fold higher dose (Fig. 5E) reveals that the ratio of 264 metabolites to unmetabolized 264 was much higher in monkeys than in rats. These data suggest that although its absorption rates in rats and monkeys may be similar, 264 persists longer in rats, probably due to a reduced rate of metabolism in this species. Despite the lower rate of clearance of 264 from rat blood, the pattern of metabolism was more complex, as 4 major polar metabolites were formed. Unfortunately, the chromatographic conditions used for these experiments were quite different from the improved conditions used to analyze monkey plasma metabolites, so a direct comparison is not possible. However, we would anticipate that the rat metabolites were formed by cytochrome P450-catalyzed ω-oxidation of 264 at 2 or more positions in the hexyl side chain. Rat leukocytes, for example, convert leukotriene B4 to a complex array of ω2- and ω3- hydroxy and oxo metabolites (Powell and Gravelle, 1990), in contrast to human neutrophils, which form only ω1-oxidation products (Powell, 1984).
We recently examined the metabolism and pharmacokinetics of another OXE receptor antagonist (5-(5-chloro-2-hexyl-1-methyl-1H-indol-3-yl)-3-methyl-5-oxo-pentanoic acid) in monkeys, which also contains a hexyl group in the 2-position of an indole (Cossette et al., 2016). Like 264, this compound was metabolized only to a small extent by ω1-oxidation and to a much greater extent by ω2-oxidation. However, its metabolism was more complex than 264, with substantial hydroxylation also occurring in the ω6 position of the hexyl side chain (i.e. α to the indole) (Chourey et al., 2017).
None of the 264 metabolites that we identified had appreciable antagonist activity. Among the synthetic compounds that we tested, the minor metabolite ω1-hydroxy-264 was the most potent, with an IC50 over 60 times higher than that of 264, with the ω2-oxidation products being 200–400 times less potent. It is therefore clear that the presence of hydroxyl, oxo, or carboxyl groups at the end of the hexyl side chain prevents substantial antagonist activity. Modification of hexyl group to prevent ω1/ω2 oxidation might result in antagonists that are more resistant to metabolism, resulting in more sustained concentrations in the blood.
5. Conclusions
In conclusion, 264 is a potent OXE receptor antagonist in monkeys. Oral administration results in high plasma concentrations within a short time, but its levels subsequently decline due to ω2-oxidation to inactive metabolites. The lifetime of 264 in the blood could likely be substantially enhanced if this metabolic pathway could be blocked by modification of the hexyl group to diminish its oxidation. Because of its potential to inhibit eosinophil accumulation by blocking the effects of the potent eosinophil chemoattractant 5-oxo-ETE, 264 or a related compound could be a useful therapeutic agent in eosinophilic diseases such as asthma, either alone or in combination with other anti-inflammatory drugs targeting IL-5 or other mediators acting by different mechanisms. The monkey, given the lack of the OXE receptor in rodents, would be an excellent animal model for preclinical studies with such compounds, which are currently underway.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research [Grants MOP-6254 and PP2-133388 (WSP)], the American Asthma Foundation [Grant 12-0049 (JR)], the National Heart, Lung, and Blood Institute [Grant R01HL081873 (JR)] and AmorChem (Montreal, QC). The Meakins-Christie Laboratories-MUHC-RI are supported in part by a Centre grant from Le Fond de la Recherche en Santeé du Queébec as well as by the J. T. Costello Memorial Research Fund. JR also wishes to acknowledge the National Science Foundation for the AMX-360 [Grant CHE-90-13145] and Bruker 400 MHz [Grant CHE-03-42251] NMR instruments. DV and IS were supported by the Natural Sciences and Engineering Research Council of Canada [Grant RGPIN/435814-2103]. IS also wishes to acknowledge the Centre for Biological Applications of Mass Spectrometry at Concordia University for PhD scholarship funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
ABBREVIATIONS
- 5-HETE
5S-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 5-oxo-ETE
5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 5-LO
5-lipoxygenase
- DMP
Dess-Martin Periodane
- PTSA
p-toluenesulfonic acid
- DHP
3,4-Dihydro-2H-pyran
- LT
leukotriene
- TBDPS-Cl
tert-Butyldiphenylsilyl chloride
- TMS
tetramethylsilane
- HRMS
High Resolution Mass Spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
JR and WSP hold European and US patents related to 264.
References
- Bäck M, Powell WS, Dahlén SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE, 2014. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Brit J Pharmacol 171, 3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M, 1996. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J. Immunol 157, 5221–5224. [PubMed] [Google Scholar]
- Chourey S, Ye Q, Reddy CN, Cossette C, Gravel S, Zeller M, Slobodchikova I, Vuckovic D, Rokach J, Powell WS, 2017. In vivo alpha-hydroxylation of a 2-alkylindole antagonist of the OXE receptor for the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid in monkeys. Biochem. Pharmacol 138, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KF, 2016. Asthma phenotyping: a necessity for improved therapeutic precision and new targeted therapies. J Intern Med 279, 192–204. [DOI] [PubMed] [Google Scholar]
- Cossette C, Chourey S, Ye Q, Nagendra Reddy C, Gore V, Gravel S, Slobodchikova I, Vuckovic D, Rokach J, Powell WS, 2016. Pharmacokinetics and Metabolism of Selective Oxoeicosanoid (OXE) Receptor Antagonists and Their Effects on 5-Oxo-6,8,11,14-eicosatetraenoic Acid (5-Oxo-ETE)-Induced Granulocyte Activation in Monkeys. J. Med. Chem 59, 10127–10146. [DOI] [PubMed] [Google Scholar]
- Cossette C, Gravel S, Reddy CN, Gore V, Chourey S, Ye Q, Snyder NW, Mesaros CA, Blair IA, Lavoie JP, Reinero CR, Rokach J, Powell WS, 2015. Biosynthesis and actions of 5-oxoeicosatetraenoic acid (5-oxo-ETE) on feline granulocytes. Biochem. Pharmacol 96, 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaire MJ, Ferland C, Page N, Lavigne S, Davoine F, Laviolette M, 2003. Endothelial cells modulate eosinophil surface markers and mediator release. Eur.Respir.J 21, 918–924. [DOI] [PubMed] [Google Scholar]
- Gore V, Gravel S, Cossette C, Patel P, Chourey S, Ye Q, Rokach J, Powell WS, 2014. Inhibition of 5-oxo-6,8,11,14-eicosatetraenoic acid-induced activation of neutrophils and eosinophils by novel indole OXE receptor antagonists. J. Med. Chem 57, 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore V, Patel P, Chang CT, Sivendran S, Kang N, Ouedraogo YP, Gravel S, Powell WS, Rokach J, 2013. 5-Oxo-ETE Receptor Antagonists. J. Med. Chem 56, 3725–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iikura M, Suzukawa M, Yamaguchi M, Sekiya T, Komiya A, Yoshimura-Uchiyama C, Nagase H, Matsushima K, Yamamoto K, Hirai K, 2005. 5-Lipoxygenase products regulate basophil functions: 5-Oxo-ETE elicits migration, and leukotriene B(4) induces degranulation. J. Allergy Clin. Immunol 116, 578–585. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, Podust LM, de Montellano PRO, 2011. Structural control of cytochrome P450-catalyzed omega-hydroxylation. Arch Biochem Biophys 507, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CE, Holden S, Tenaillon L, Bhatia U, Seuwen K, Tranter P, Turner J, Kettle R, Bouhelal R, Charlton S, Nirmala NR, Jarai G, Finan P, 2003. Expression and characterization of a 5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid receptor highly expressed on human eosinophils and neutrophils. Mol. Pharmacol 63, 471–477. [DOI] [PubMed] [Google Scholar]
- Kay AB, 1970. Studies on eosinophil leucocyte migration. II. Factors specifically chemotactic for eosinophils and neutrophils generated from guinea-pig serum by antigen-antibody complexes. Clin. Exp. Immunol 7, 723–737. [PMC free article] [PubMed] [Google Scholar]
- Langlois A, Chouinard F, Flamand N, Ferland C, Rola-Pleszczynski M, Laviolette M, 2009. Crucial implication of protein kinase C (PKC)-delta, PKC-zeta, ERK-1/2, and p38 MAPK in migration of human asthmatic eosinophils. J. Leukoc. Biol 85, 656–663. [DOI] [PubMed] [Google Scholar]
- Lindgren JÅ, Hansson G, Claesson HE, Samuelsson B, 1982. Formation of novel biologically active leukotrienes by omega- oxidation in human leukocyte preparations. Adv.Prostaglandin Thromboxane Leukotriene Res 9, 53–60. [PubMed] [Google Scholar]
- McBrien CN, Menzies-Gow A, 2017. The Biology of Eosinophils and Their Role in Asthma. Front Med (Lausanne) 4, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monneret G, Gravel S, Diamond M, Rokach J, Powell WS, 2001. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 98, 1942–1948. [DOI] [PubMed] [Google Scholar]
- Powell WS, 1980. Rapid extraction of oxygenated metabolites of arachidonic acid from biological samples using octadecylsilyl silica. Prostaglandins 20, 947–957. [DOI] [PubMed] [Google Scholar]
- Powell WS, 1984. Properties of leukotriene B4 20-hydroxylase from polymorphonuclear leukocytes. J. Biol. Chem 259, 3082–3089. [PubMed] [Google Scholar]
- Powell WS, 1987. Precolumn extraction and reversed-phase high-pressure liquid chromatography of prostaglandins and leukotrienes. Anal. Biochem 164, 117–131. [DOI] [PubMed] [Google Scholar]
- Powell WS, Chung D, Gravel S, 1995. 5-Oxo-6,8,11,14-eicosatetraenoic acid is a potent stimulator of human eosinophil migration. J. Immunol 154, 4123–4132. [PubMed] [Google Scholar]
- Powell WS, Gravelle F, 1990. Metabolism of arachidonic acid by peripheral and elicited rat polymorphonuclear leukocytes. Formation of 18- and 19-oxygenated dihydro metabolites of leukotriene B4. J. Biol. Chem 265, 9131–9139. [PubMed] [Google Scholar]
- Powell WS, Gravelle F, Gravel S, 1992. Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J. Biol. Chem 267, 19233–19241. [PubMed] [Google Scholar]
- Powell WS, MacLeod RJ, Gravel S, Gravelle F, Bhakar A, 1996. Metabolism and biologic effects of 5-oxoeicosanoids on human neutrophils. J. Immunol 156, 336–342. [PubMed] [Google Scholar]
- Powell WS, Rokach J, 2013. The eosinophil chemoattractant 5-oxo-ETE and the OXE receptor. Prog. Lipid Res 52, 651–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WS, Solomon S, 1978. Formation of 20-hydroxyprostaglandins by lungs of pregnant rabbits. J. Biol. Chem 253, 4609–4616. [PubMed] [Google Scholar]
- Powell WS, Wang L, Khanapure SP, Manna S, Rokach J, 1997. High-pressure liquid chromatography of oxo-eicosanoids derived from arachidonic acid. Anal. Biochem 247, 17–24. [DOI] [PubMed] [Google Scholar]
- Reddy CN, Ye QJ, Chourey S, Gravel S, Powell WS, Rokach J, 2015. Stereoselective synthesis of two highly potent 5-oxo-ETE receptor antagonists. Tetrahedron Lett. 56, 6896–6899. [Google Scholar]
- Robinson D, Humbert M, Buhl R, Cruz AA, Inoue H, Korom S, Hanania NA, Nair P, 2017. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy 47, 161–175. [DOI] [PubMed] [Google Scholar]
- Sarveswaran S, Ghosh J, 2013. OXER1, a G protein-coupled oxoeicosatetraenoid receptor, mediates the survival-promoting effects of arachidonate 5-lipoxygenase in prostate cancer cells. Cancer Lett. 336, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm GJ, Schuligoi R, Sturm EM, Royer JF, Lang-Loidolt D, Stammberger H, Amann R, Peskar BA, Heinemann A, 2005. 5-Oxo-6,8,11,14-eicosatetraenoic acid is a potent chemoattractant for human basophils. J.Allergy Clin.Immunol 116, 1014–1019. [DOI] [PubMed] [Google Scholar]