

Keywords: bronchopulmonary dysplasia, Gdf15, lung, neonatal, prematurity, sex
Abstract
Growth differentiation factor 15 (GDF15) is a divergent member of the transforming growth factor-β (TGF-β) superfamily, and its expression increases under various stress conditions, including inflammation, hyperoxia, and senescence. GDF15 expression is increased in neonatal murine bronchopulmonary dysplasia (BPD) models, and GDF15 loss exacerbates oxidative stress and decreases cellular viability in vitro. Our overall hypothesis is that the loss of GDF15 will exacerbate hyperoxic lung injury in the neonatal lung in vivo. We exposed neonatal Gdf15−/− mice and wild-type (WT) controls on a similar background to room air or hyperoxia (95% ) for 5 days after birth. The mice were euthanized on postnatal day 21 (PND 21). Gdf15−/− mice had higher mortality and lower body weight than WT mice after exposure to hyperoxia. Hyperoxia exposure adversely impacted alveolarization and lung vascular development, with a greater impact in Gdf15−/− mice. Interestingly, Gdf15−/− mice showed lower macrophage count in the lungs compared with WT mice both under room air and after exposure to hyperoxia. Analysis of the lung transcriptome revealed marked divergence in gene expression and enriched biological pathways in WT and Gdf15−/− mice and differed markedly by biological sex. Notably, pathways related to macrophage activation and myeloid cell homeostasis were negatively enriched in Gdf15−/− mice. Loss of Gdf15 exacerbates mortality, lung injury, and the phenotype of the arrest of alveolarization in the developing lung with loss of female-sex advantage in Gdf15−/− mice.
NEW & NOTEWORTHY We show for the first time that loss of Gdf15 exacerbates mortality, lung injury, and the phenotype of the arrest of alveolarization in the developing lung with loss of female-sex advantage in Gdf15−/− mice. We also highlight the distinct pulmonary transcriptomic response in the Gdf15−/− lung including pathways related to macrophage recruitment and activation.
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is one of the most common causes of morbidity among surviving premature infants. Despite the decreased mortality rate among preterm neonates due to advances in neonatal care, the incidence of BPD among this vulnerable population has stayed the same, leading to an increased disease burden (1). The key pathological feature in “New BPD” is the arrest in lung development, characterized by aberrant vascular development and abnormal alveolar septation (2). BPD also leads to long-term morbidities and may predispose to the early development of chronic lung diseases in adulthood (3).
Growth differentiation factor 15 (GDF15) is a divergent member of transforming growth factor-β (TGF-β) superfamily cytokines. It is also known as macrophage inhibitory cytokine-1 (MIC-1) (4), nonsteroidal anti-inflammatory drug-activated gene-1 (5), and placental transforming growth factor-β (PTGFβ) (6). GDF15 expression increases during pregnancy, and under pathological and stress-related conditions such as hypoxia, inflammation, hyperoxia, infection, senescence, and intense exercise (7, 8). GDF15 plays a central role in the coordination of tolerance to inflammation by initiating metabolic adaptation (9), including triglyceride metabolism and energy homeostasis (10).
In adult literature, GDF15 has been extensively studied and has been associated with multiple cardiopulmonary disorders such as chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), pulmonary viral infections, and pulmonary hypertension (11–14). GDF15 has been identified as a potential biomarker, prognostic factor, and a possible therapeutic target (8, 15). GDF15 was upregulated in human IPF lungs (16) and was identified as mediating the association between age, interstitial lung abnormality, and mortality in two large prospective cohorts: Framingham Heart Study and the COPD gene cohort (17).
Our previous studies in vivo have shown increased pulmonary GDF15 expression in mice when exposed to hyperoxia (18). Hyperoxia also induces GDF15 expression in pulmonary endothelial and epithelial cells, and GDF15 loss increases oxidative stress and decreases cellular viability in vitro (19). Whether increased serum concentrations indicate ongoing cellular injury or represent a protective response to biological stress is still an open question, and the answer might depend on the organ and cellular environment. Gdf15 is a part of the in vivo gene expression signature of oxidative stress (20). GDF15 has been shown to have anti-inflammatory (21–23), proangiogenic (24), and antiapoptotic (25) effects. Whether the loss of GDF15 modulates the response of the neonatal murine lung to postnatal hyperoxia is not known. Using global knockouts for the Gdf15−/− gene, we hypothesized that loss of GDF15 will exacerbate neonatal hyperoxic lung injury with reduced survival and will adversely impact alveolar and pulmonary vascular development.
METHODS
Animals
The study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine. Wild-type mice (C57BL/6) were obtained from Charles River Laboratories (Wilmington, DE). Gdf15−/− mice were obtained from Dr. Se-Jin Lee from Johns Hopkins University (Baltimore). Active colonies of mice were maintained by breeding in our animal facility. Newborn mice were randomly allocated, wild-type mice and Gdf15−/−, to the normoxia or hyperoxia groups within 12 h of birth.
Mouse Model of BPD
Mouse pups were exposed to hyperoxia (95% ) within 12 h of birth up to postnatal day 5 (PND 5) to induce arrest in alveolarization with the goal to replicate the lung phenotype seen in human infants with BPD. Mice are born at the saccular stage of lung development, which corresponds to 26–36 wk of fetal lung development in humans (26). Exposing neonatal mice to hyperoxia in the first 5 days and then transitioning them to room air mimics the majority of preterm human neonates’ clinical course in the neonatal intensive care unit (27, 28), albeit most of the preterm neonates do not need 95% oxygen until 36 wk postmenstrual age, which is a drawback of this model. The mouse pups were randomly assigned to a normoxia group (21% oxygen) and a hyperoxia group (95% oxygen). The litter sizes were limited to six pups in all groups to avoid overcrowding and limit its effects on nutrition and growth. The dams were rotated daily between room air- and hyperoxia-exposed litters to prevent oxygen toxicity and eliminate the maternal effects among different groups. Hyperoxia was achieved using Plexiglass chambers, and the oxygen analyzer was used to measure the in the chambers. The hyperoxia-exposed mice were transitioned to normoxia to recover in room air till PND 21. All mice were euthanized at PND 5 (immediately after hyperoxia exposure) and PND 21 (after recovery from normoxia). A schematic illustration of the experiment design is shown in Fig. 1. Lung weight/body weight ratios were measured at euthanasia on PND 5. Body weights were measured for all the surviving mice at PND 21.
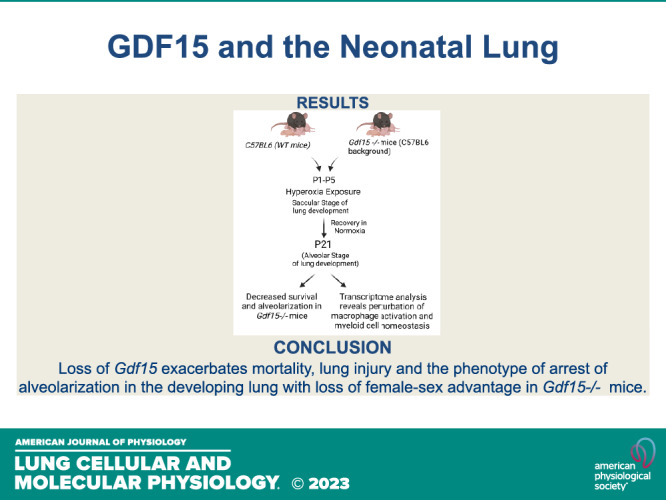
Figure 1.

Does loss of Gdf15 worsen arrest in alveolarization and pulmonary vascular development neonatal hyperoxic lung injury? Schematic showing exposure of WT and Gdf15−/− neonatal mice to hyperoxia (95% , PND 1–5) during the saccular stage of lung development. Euthanasia was performed at PND 21 with evaluation of alveolarization, pulmonary vascular development, and assessment of the pulmonary transcriptome. , fraction of inspired oxygen; Gdf15, growth differentiation factor 15; PND, postnatal day; WT, wild type. [Image created with BioRender.com and published with permission.]
Lung Histology and Morphometry
During euthanasia on PND 21, the trachea was cannulated, and the lungs were inflated with 4% paraformaldehyde administered endotracheally at 25 cmH2O pressure for 15 min. The lungs were collected, and 5 µm sections were obtained and stained with hematoxylin-eosin stain. The slides were examined under a light microscope. Mean linear intercept (MLI) and radial alveolar count (RAC) were used to measure the alveolar development at PND 21 using methods previously described (28). We used 10–15 randomly nonoverlapping fields per biological replicate for analysis. Fields with large airways and vessels were excluded.
Pulmonary vascular development.
The pulmonary vessels were identified using immunofluorescence for von Willebrand factor (vWF), an endothelial cell-specific marker (1:4,000 dilution, Abcam; Cat. No. ab6994). Pulmonary vessel density was measured by counting the stained vessels with external diameters of <100 µm (×40 magnification) per lung field. Ten random nonoverlapping fields per specimen were used for analysis. Fields with large airways and vessels were excluded.
Analysis of inflammation.
To measure the degree of inflammation, we quantified the macrophages per high power field in the distal lung by immunofluorescent staining using F4/80 antibody, a specific antibody for macrophages (1:500 dilution, Bio-Rad Laboratories: Cat. No. MCA497GA). Fifteen random nonoverlapping fields per biological replicate were used for analysis. Fields with large airways and vessels were excluded.
Lung mRNA Extraction and RNA-Seq Analysis
Total RNA from flash-frozen lung samples was isolated using the Zymo micro kit (Zymo Research, Irvine, CA). RNA concentration and quality check were assayed using Nanodrop-8000 (Thermo Scientific, Wilmington, DE). RNA quality parameters were as follows: the 260/280 and 260/230 ratios needed to be >1.9. Furthermore, the RNA integrity number (RIN) was analyzed using the Agilent Bioanalyzer. The samples needed to have RIN values of 7–10 and with a range of 1–1.5. Sequencing was done on the Illumina NovaSeq S4 PE150. Salmon (version 1.9.0) (29) was used to build an index based on the GRCm39 reference transcriptome. It was also used to quantify using—seqBias and—gcBias flags to correct for sequence and fragment-level biases. Tximeta (version 1.16.1) (30) was used to load our quantified data along with our metadata. We perform a prefiltering step to keep only genes that have at least 10 reads present. We then use DESeq2 (version 1.38.3) (31) to perform differential expression analysis. We filtered these results using the results() function from DESeq2 with an α value of 0.05. We only kept genes with less than 0.05 adjusted P value and greater than 0.3219 absolute log2 fold change. We then mapped the resulting ensemble IDs to gene symbols using biomaRt (version 2.54.0) (32) and performed Shrinkage of effect size using DESeq2 lfcShrink() function for better gene ranking and visualization. Fgsea (version 1.24.0) (33) was used for gene set enrichment analysis using the stat value for ranking. We filtered these results to get pathways with a P value less or equal to 0.05 and a gene number of more than 5. Other programs used for sorting, analyzing, and exporting data were Org.Mm.eg.db (version 3.16.0), ggvenn (version 0.1.9), VennDiagram (version 1.7.3), tibble (version 3.1.8), ggplot2 (version 3.4.1), tidyverse (version 2.0.0), and openxlsx (version4.2.5). All the programs, versions, and references are also provided in Supplemental Table S1.
Data Availability
All code used in this manuscript can be found in Supplemental Material; the raw data have been uploaded to National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO); GSE211744. All supplemental data are accessible at https://doi.org/10.6084/m9.figshare.22771034.
Western Blotting and qRT-PCR
Lung tissue from WT and Gdf15−/− mice exposed to room air and hyperoxia as described before was obtained after euthanasia at PND 7. Lung whole protein (20 μg of protein) was prepared as described before and subjected to SDS polyacrylamide gel electrophoresis in 10% acrylamide gels and then transferred to polyvinylidene difluoride membranes, followed by Western blotting. After the membranes were blocked in 5% nonfat dry milk, they were incubated overnight with primary antibodies after which, the membranes were washed and incubated with the appropriate secondary antibodies. Vinculin was used as the loading control. The primary antibodies were GDF15 (Santa Cruz; sc-515675,1:1,200 dilution) and vinculin (Cell Signaling; Cat No. 4650, 1:1,200 dilution). This was followed by electrochemical detection of bands. Band intensities were quantified using Bio-Rad ImageLab Software. Total RNA from lung samples in mice exposed to room air or hyperoxia was isolated on PND 21 using the mRNeasy kit (Qiagen, Valencia, CA). RNA concentration was assayed using a Nanodrop-8000 (Thermo Scientific, Wilmington, DE). NanoDrop spectrophotometer was used to measure RNA concentration and quality and subjected to one-step real-time quantitative TaqMan RT-PCR using 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Gene-specific primers were purchased from life science technologies in the presence of TaqMan reverse transcription reagents and RT reaction mix (Applied Biosystems, Foster City, CA) were used to reverse transcribe RNA, and TaqMan Gene Expression probes and TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA), were used for PCR amplification. Furthermore, 18S was used as the reference gene. The primers used were Ucp1 (Mm01244861_m1), Gdf15 (Mm00442228_m1), Siglec f (Mm00523987_m1), Spp1 (Mm00436767_m1), and 18 s (Hs99999901_s1).
Statistical Analyses
Data analysis was performed using GraphPad Prism 9 software. Survival analysis was performed using the Log-rank (Mantel–Cox) test. Data are displayed as means ± SD. Two-way ANOVA following Tukey’s posttest was performed for statistical evaluation. The main effects of hyperoxia, genotype, and interaction between the independent variables were also assessed. In other analyses, three-way ANOVA was used to assess the main effects of sex, genotype, and treatment, and the interaction among these variables. The significance level was set at P < 0.05. We calculated the sample size based on our preliminary results for hyperoxia-induced alveolar simplification in untreated WT mice. A sample of six mice per group will be needed to detect a 25% increase (two SD) in the MLI in Gdf15−/− mice to provide 80% power with an α of 0.05.
RESULTS
Gdf15−/− Mice Had Higher Mortality and Lower Body Weight than WT When Exposed to Hyperoxia
Gdf15 expression was increased in the hyperoxia-exposed WT lung both at the mRNA and the protein level after hyperoxia exposure (95% till PND 5) (Supplemental Fig. S1). At the time of euthanasia (PND 21), the body weights and lung weights of the surviving mice were measured. Within the Gdf15−/− mice group, hyperoxia-exposed mice had significantly lower body weight than room air-exposed mice. Between the two genotypes, upon exposure to hyperoxia, Gdf15−/− mice had lower body weight than the WT mice. There were no significant differences between the two genotypes at baseline under normoxia. As an independent variable, treatment (hyperoxia or normoxia) was statistically significant, whereas genotype and the interaction term were not (Fig. 2A). Lung weight/body weight ratio (LW/BW) was measured as an indicator of lung injury. LW/BW ratio was increased in both WT and Gdf15−/− mice groups upon exposure to hyperoxia with no differences between the genotypes (Fig. 2B). When analyzed by biological sex, hyperoxia-exposed WT male mice and Gdf15−/− male and female mice showed a significant increase in LW/BW ratio, whereas the WT female mice did not (Fig. 2C). Figure 2D shows the survival curves in WT and Gdf15−/− mice. Under normoxia, WT (n = 32 mice) and Gdf15−/− mice (n = 42 mice) had no significant differences in the survival rates (100% vs. 98%, respectively). Upon exposure to hyperoxia, the survival in Gdf15−/− mice was significantly decreased compared with WT mice, 26% versus 57%, respectively. Survival data by sex was not collected.
Figure 2.

Loss of Gdf15 decreases survival and increases lung injury. A: body weights from room air- and hyperoxia (95% oxygen, PND 1–5)-exposed WT and Gdf15−/− mice at PND 21; n = 11–15/group. B: lung weight/body weight ratios from room air- and hyperoxia-exposed WT and Gdf15−/− mice at PND 21; n = 10–23/group. C: lung weight/body weight ratios from room air- and hyperoxia-exposed WT and Gdf15−/− male and female mice at PND 21; n = 4–12/group. Statistically significant differences between the indicated groups shown by *P < 0.05; #P < 0.05, **P < 0.01 and ***P < 0.001. Statistical analysis performed using two-way or three-way ANOVA. D: survival curves of WT and Gdf15−/− neonatal mice exposed to room air or hyperoxia. WT-room air group n = 32, Gdf15−/− room air group: n = 42, WT hyperoxia group n = 42, Gdf15−/− hyperoxia group n = 54. Values are presented as means ± SD. Survival differed significantly between the four groups, P < 0.0001, using the log-rank test. Gdf15, growth differentiation factor 15; PND, postnatal day; WT, wild type.
Upon Exposure to Hyperoxia, Gdf15−/− Female Mice Had Greater Alveolar Simplification than WT
Lung morphometry was assessed at PND 21, after a period of hyperoxia exposure (95% oxygen) during PND1 5 (saccular stage of lung development). We measured the mean linear intercept (MLI) and radial alveolar count (RAC) to quantify the alveolar simplification as per the techniques described in the methodology. Representative lung sections are shown in Fig. 3, using ×10 and ×20 magnifications. The MLI and RAC were significantly altered upon exposure to hyperoxia in WT and Gdf15−/− mice, however, the effect was greater in Gdf15−/− mice (Fig. 3C). When stratified by biological sex, alveolar simplification was attenuated in WT females compared with WT males and Gdf15−/− female mice. There was no difference between Gdf15−/− male and female mice (Fig. 3, E and F).
Figure 3.
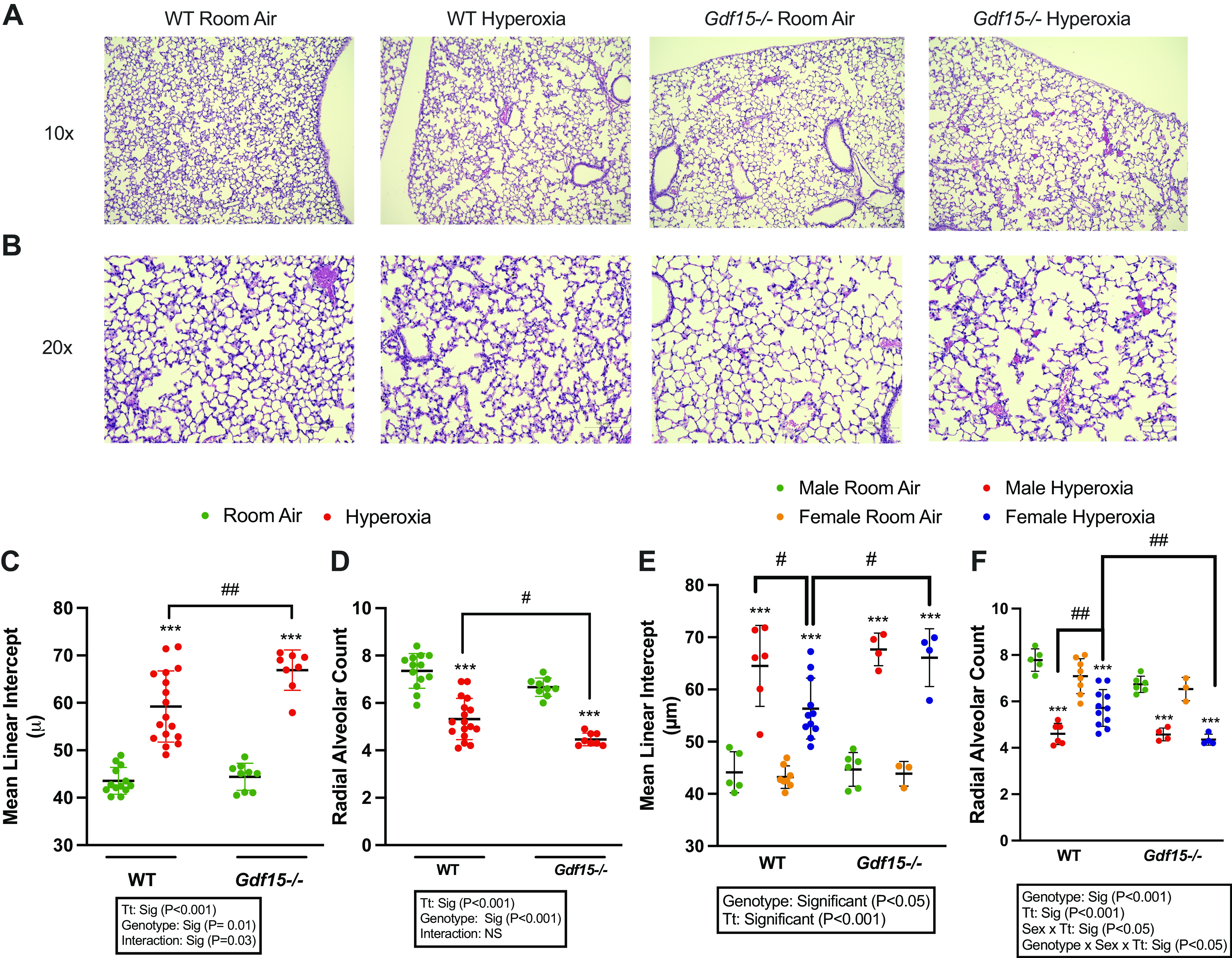
Loss of Gdf15 worsens arrest in alveolarization after hyperoxia exposure during the saccular stage of lung development. Representative hematoxylin and eosin-stained lung sections at ×10 (A) and ×20 (B) magnification from room air and hyperoxia (95% oxygen, PND 1–5)-exposed WT and Gdf15−/− mice euthanized at PND 21. C and D: lung morphometry assessed by mean linear intercept (MLI) and radial alveolar count from room air and hyperoxia (95% oxygen, PND 1–5)-exposed WT and Gdf15−/− mice at PND 21; n = 8–17/group. E and F: lung morphometry in male and female neonatal mice assessed by mean linear intercept (MLI) and radial alveolar count from room air and hyperoxia (95% oxygen, PND 1–5)-exposed WT and Gdf15−/− mice at PND 21; n = 3–11/group. Values are presented as means ± SD. Statistically significant differences between the indicated groups shown by #P < 0.05, ##P < 0.01, and ***P < 0.001. Statistical analysis performed using two-way or three-way ANOVA. Gdf15, growth differentiation factor 15; PND, postnatal day; WT, wild type.
Gdf15−/− and WT Mice Showed Greater Arrest in Angiogenesis upon Exposure to Hyperoxia than Mice Kept on Room Air, but with No Significant Differences between the Two Genotypes
We sought to determine whether the loss of Gdf15 affected vascular development in response to hyperoxia. Pulmonary vascular density was decreased within both genotypes upon exposure to hyperoxia, Gdf15−/− and WT mice. These results are shown in Fig. 4. The degree of hyperoxia-induced arrest in angiogenesis was not significantly different between the two genotypes (P > 0.05). Interestingly, at baseline under normoxia, Gdf15−/− mice had lower vascular density compared with WT mice.
Figure 4.
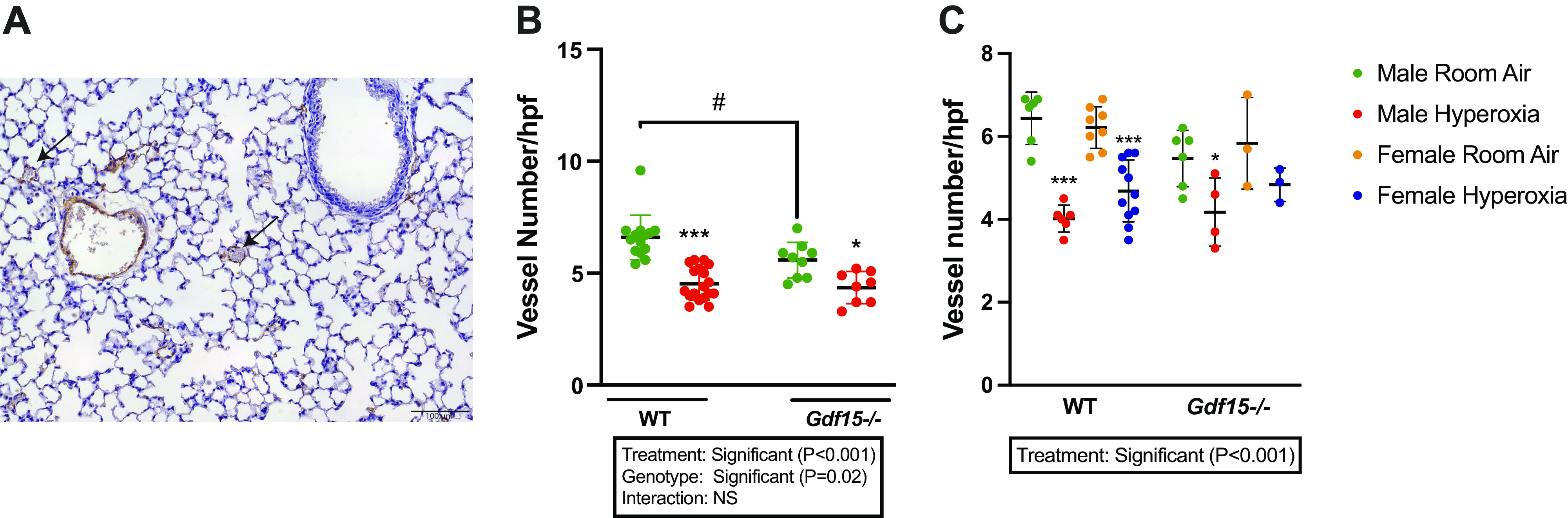
Loss of Gdf15 worsens arrest in pulmonary vascular development after hyperoxia exposure during the saccular stage of lung development. A: representative immunohistochemistry image for pulmonary vascular density using immunohistochemistry for von Willebrand factor (vWF). Arrows point to the pulmonary blood vessels. Scale bar = 100 µm. B: quantitative analysis of the pulmonary vascular density. Green dots: Room Air; Red dots: Hyperoxia. n = 8–17/group (number of vWF-stained vessels per high power field). C: pulmonary vascular density in male and female WT and Gdf15−/− mice exposed to room air or hyperoxia (n = 3/11 per group). Values are presented as means ± SD. Statistically significant differences between the indicated groups shown by *, #P < 0.05, and ***P < 0.001. Statistical analysis performed using two-way or three-way ANOVA. Gdf15, growth differentiation factor 15.
Gdf15−/− Showed Lower Macrophage Recruitment in Response to Hyperoxia Compared with WT Mice
We next explored if the loss of Gdf15 altered the inflammatory response induced by hyperoxia. We assessed lung macrophages by immunohistochemistry (Fig. 5). Interestingly, under normoxic conditions, Gdf15−/− mice had a lower macrophage count in the lung tissue compared with WT mice. Upon exposure to hyperoxia, Gdf15−/− mice showed no increase in macrophage recruitment compared with the room air group. WT mice showed a significant increase in macrophage recruitment in response to hyperoxia compared with the room air group. Treatment, genotype, and interaction term were significant in the statistical analysis.
Figure 5.
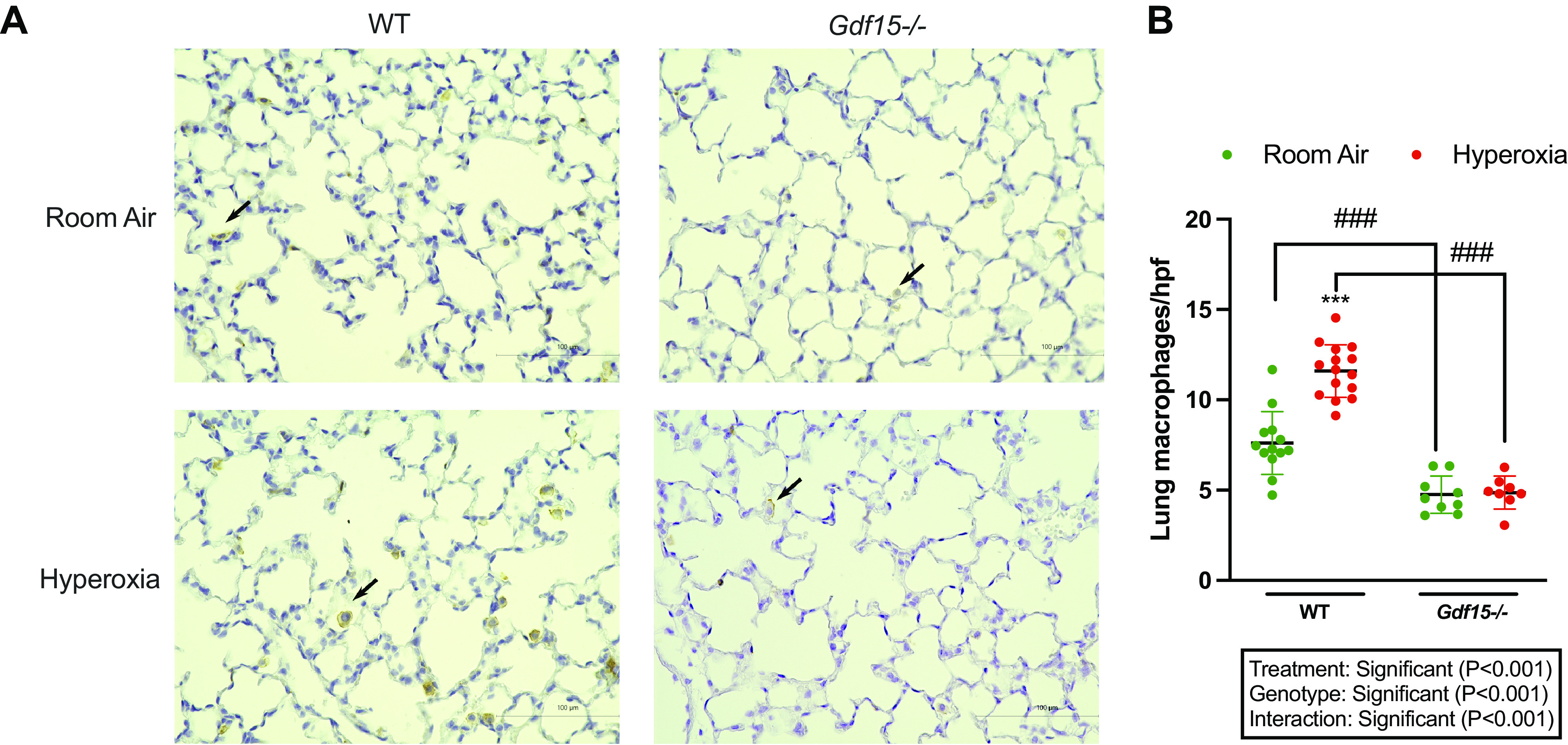
Decreased lung macrophages following loss of Gdf15. A: representative lung sections from wild-type (WT) and Gdf15−/− mice exposed to normoxia or hyperoxia (95% oxygen, postnatal days 1–5). Immunohistochemistry performed for macrophages using F4/80 antibody. Scale bar = 100 µm. B: quantitative analysis of macrophage recruitment (macrophage count per high power field). Values are presented as means ± SD. Statistically significant differences between the indicated groups shown by ###P < 0.001 and ***P < 0.001. Statistical analysis performed using two-way ANOVA. Gdf15, growth differentiation factor 15.
The Pulmonary Transcriptome Shows Marked Differences in the Gdf15−/− Developing Lung upon Exposure to Hyperoxia Compared with WT Mice
We subjected lungs from room air- and hyperoxia-exposed WT and Gdf15−/− mice at PND 21 to RNA-Seq analysis. The number of differentially expressed genes (DEGs) in each genotype (hyperoxia vs. room air) was analyzed. We then analyzed the overlap of the hyperoxia response between the WT and Gdf15−/− mice. Among the upregulated genes, there were 42 genes (out of 475) and 23 downregulated genes (among 424) that were common between the two genotypes highlighting the marked differences in the transcriptomic response to hyperoxia (Fig. 6, A and B). The enriched biological pathways also had minor overlap with seven pathways common between WT and Gdf15−/− mice (Fig. 6C). Volcano plots showing the DEGs in WT and Gdf15−/− mice are shown in Fig. 6, D and E. The WT hyperoxia response has been described in previous publications (34). One of the upregulated genes in the Gdf15−/− mice is Suv39h2, a histone lysine methyltransferase that plays a role in chromatin remodeling. Data from LungMAP (35) localize expression in the myeloid/macrophage population at PND 7, 14, and 28 (Supplemental Fig. S2). Increased Suv39h2 expression decreased trained immunity in monocytes (36). Endoglin (Eng) was one of the top downregulated genes in the Gdf15−/− mice, which is expressed on macrophages (37), and mice lacking Eng in macrophages show an impaired immune response, including macrophage recruitment (38, 39). Representative biological pathways are unique to WT and Gdf15−/− mice and pathways modulated in opposite directions between WT and Gdf15−/− mice are highlighted in Fig. 6F. Circulatory system development and organ morphogenesis-related pathways were negatively enriched in the WT hyperoxia-exposed mice. Interestingly, macrophage activation and regulation of cellular response to stress were negatively enriched in Gdf15−/− mice. Differentially expressed genes and biological pathways in all genotypes, biological sex, and treatment groups are provided in Supplemental Table S2.
Figure 6.

Distinct pulmonary transcriptomic differences in the Gdf15−/− mice exposed to hyperoxia (95% oxygen, PND 1–5): Differentially expressed genes (DEGs) upregulated (A) and downregulated (B) in WT and Gdf15−/− mice. Venn diagrams showing genes that are common to and distinct in WT and Gdf15−/− mice. C: Venn diagrams showing biological pathways that are common to and distinct in WT and Gdf15−/− mice. Volcano plots showing upregulated and downregulated genes in WT (D) and Gdf15−/− (E) mice exposed to hyperoxia compared with room air controls. F: biological pathways enriched in WT and Gdf15−/− mice exposed to hyperoxia compared with room air controls. Normalized enrichment score (NES) plotted on the x axis. Gdf15, growth differentiation factor 15; PND, postnatal day; WT, wild type.
Sex as a Biological Variable Modulates the Changes in the Pulmonary Transcriptome in Response to Hyperoxia and Is Distinct in WT and Gdf15−/− Mice
We next wanted to analyze how biological sex modulates the transcriptomic response to hyperoxia in WT and Gdf15−/− mice. The number of up- and downregulated DEGs in WT and Gdf15−/− male and female mice are shown in Fig. 7A. In the WT mice, there was a 5% (20/404 DEGs) and 11.5% (50/433 DEGs) overlap between male and female mice for up- and downregulated genes, respectively. Gdf15−/− mice showed 7% (22/321 DEGs) and 8% (18/234 DEGs) for up- and downregulated genes between male and female mice. Similarly, when examined for overlap in biological pathways this was very different among male and female mice with WT mice showing only 18% overlap and Gdf15−/− mice with 3% overlap (Fig. 7B). This is further highlighted in Fig. 7C, where the gene expression and the biological pathways common to and distinct in each genotype and biological sex are represented. These results underscore the crucial role of sex as a biological variable in regulating the gene expression in the lung upon exposure to hyperoxia. Differences between the transcriptomic responses in WT male and female neonatal lungs have been highlighted in our prior publications (34). Volcano plots showing the DEGs in male and female WT and Gdf15−/− mice are shown in Fig. 7D. One of the top downregulated genes in Gdf15−/− male mice is Adarb1 (Adar2), which is involved in myeloid cell differentiation (40). SiglecF was decreased in Gdf15−/− female mice, a known marker for lung alveolar macrophages (41). Representative enriched biological pathways in WT and Gdf15−/− male and female mice are shown in Fig. 7E. Noticeably, immune system development and myeloid cell differentiation are negatively enriched in Gdf15−/− male mice, as in response to oxidative stress. Lipid metabolic pathways were negatively enriched in WT males, whereas carbohydrate-derivative metabolic process was positively enriched in WT females. We validated our results from the RNA-Seq experiment with qRT-PCR of some relevant genes based on the curation of our enriched biological pathways. With the marked decrease in the number of macrophages in the Gdf15−/− lung both under room air and upon exposure to hyperoxia, we measured the gene expression for Spp1 and SiglecF. Expression of both genes was decreased in Gdf15−/− hyperoxia-exposed mice compared with WT. On the other hand, the expression of Ucp1 was significantly increased in the hyperoxia-exposed Gdf15−/− mice compared with WT (Fig. 8). Uncoupling protein 1 (Ucp1), a well-known thermogenic and mitochondrial gene was increased in the Gdf15−/− mice. This gene is also highly expressed in the myeloid/macrophage population in the developing lung (Supplemental Fig. S2). Uncoupling proteins are members of the family of mitochondrial anion carrier proteins, that separate oxidative phosphorylation from ATP synthesis with energy dissipated as heat. Deficiency of Ucp1 promotes macrophage recruitment (42). The role of GDF15 as a mitochondrial stress-induced cytokine is well established (43), but the role in the setting of neonatal hyperoxic lung injury in a cell-specific manner needs to be elucidated (44, 45).
Figure 7.
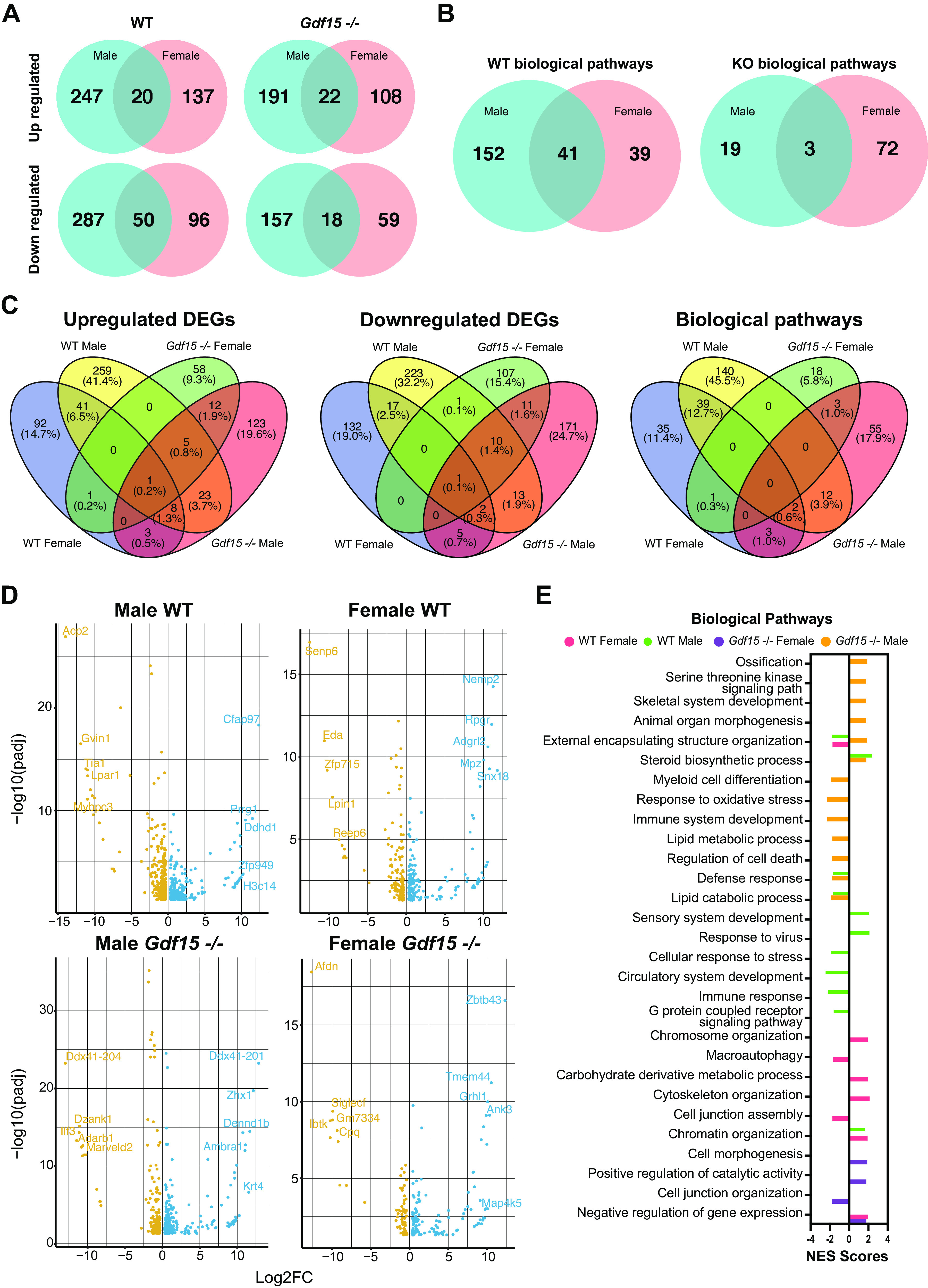
Remarkable sex-specific differences in the pulmonary transcriptome in WT and Gdf15−/− mice exposed to hyperoxia (95% oxygen, PND 1–5). A: differentially expressed genes (DEGs) upregulated and downregulated in male and female WT and Gdf15−/− mice exposed to hyperoxia compared with room air controls. Venn diagrams showing genes that are common to and distinct in WT and Gdf15−/− mice. B: Venn diagrams showing biological pathways that are common to and distinct in hyperoxia-exposed WT and Gdf15−/− male and female mice compared with room air controls. C: four-way Venn plots showing overlap and distinct DEGs and pathways between male and female WT and Gdf15−/− mice exposed to hyperoxia compared with room air controls. D: volcano plots showing upregulated and downregulated genes in WT and Gdf15−/− male and female mice exposed to hyperoxia compared with room air controls. E: biological pathways enriched in WT and Gdf15−/− male and female mice exposed to hyperoxia compared with room air controls. Normalized enrichment score (NES) plotted on the x axis. Gdf15, growth differentiation factor 15; PND, postnatal day; WT, wild type.
Figure 8.
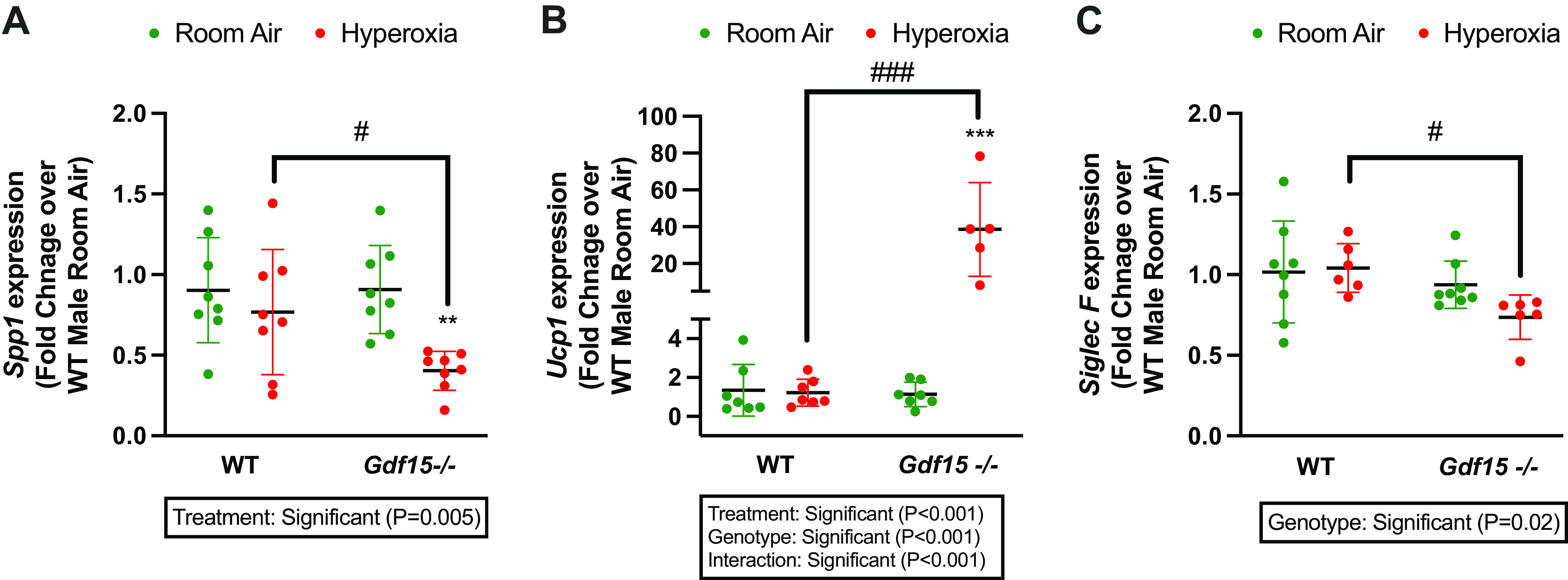
Validation of RNA-Seq data by qRT-PCR. Differentially expressed genes in RNA-Seq were validated by qRT-PCR. A: Spp1 mRNA expression B: Ucp1 mRNA expression, C: Siglec F mRNA expression. Values are represented as means ± SD. Independent biological replicates in each group are shown (n = 6–8/group). Statistical analysis was performed using two-way ANOVA. Significant differences between groups are shown by #P < 0.05, ###P < 0.001, **P < 0.01, and ***P < 0.001.
DISCUSSION
GDF15 is a stress-responsive protein that has recently received significant scientific attention as a key modulator of metabolic adaptation to stress and regulation of appetite (10). The role of GDF15 in many pulmonary diseases has been studied, and they range from disease modulation to a marker of severity and mortality (12). The significance of GDF15 in bronchopulmonary dysplasia (BPD) is yet to be elucidated. To our knowledge, this study is the first that focuses on the novel hypothesis that the loss of Gdf15 exacerbates neonatal hyperoxic lung injury in the mouse model.
Our study showed that Gdf15 deficiency decreased the tolerance to hyperoxia and decreased survival upon exposure to hyperoxia in Gdf15−/− compared with WT mice. This significant result points to the protective effect of GDF15 levels in hyperoxic conditions. The sources of increased GDF15 production could be both pulmonary and extrapulmonary, leading to increased circulating GDF15 levels (46). The survival results of this study comply with our published in vitro study, which showed that GDF15 loss in the pulmonary epithelial and endothelial cells increases cellular oxidative stress and decreases cell viability (19).
Clinically, infants with BPD show poor growth and nutritional status, which are detrimental to postnatal lung growth (47, 48). Experimentally, mouse models of BPD show lower body weight in hyperoxia-exposed neonatal mouse pups (28). GDF15 regulates appetite, food intake, and body weight via its interaction with its receptor, glial cell line-derived neurotrophic factor (GDNF) family receptor α-like (GFRAL), a distant relative of receptors for a distinct class of the TGF-β superfamily ligands, in the hindbrain (49, 50). Tsai et al. (51) and other studies have shown that Gdf15−/− mice weigh more than WT mice, and that this difference manifests at 4 wk of age. In contrast to these findings, our study showed that Gdf15−/− mice had lower body weight than WT mice upon exposure to hyperoxia. That may be because GDF15 deficiency augments the intolerance to hyperoxic lung injury, hence exaggerating the weight loss in that group. Evaluation at PND 21 following neonatal hyperoxia exposure might provide insufficient recovery time, which may halt the expected regain in body weight in the Gdf15−/− mice.
BPD is characterized by the arrest in lung development with aberrant vascular development and abnormal alveolar septation (2). Our results suggest that in the postnatal hyperoxia model, loss of Gdf15 exacerbates neonatal hyperoxic lung injury with respect to alveolarization with a worse phenotype in Gdf15−/− mice compared with WT mice at PND 21. Significantly, the sex-specific differences noted in WT mice with the female mice being more resilient to hyperoxic injury compared with male mice, were not seen in Gdf15−/− mice. Lung weight/body weight ratios immediately after hyperoxia injury at PND 5 also showed similar results with a significant increase in WT male mice, but not in female mice. However, this sex-specific difference was lost in female mice. Prior studies have shown sex-specific differences in hyperoxic lung injury regarding alveolarization, angiogenesis, and inflammation via different molecular mechanisms (28, 34, 52–57).
Hyperoxia causes a decrease in pulmonary vascular development. We did not observe robust differences between Gdf15−/− mice and WT regarding vascular development upon exposure to hyperoxia, but there was a difference at baseline under normoxia. Prior studies showed that GDF15 plays an important role in angiogenesis via the induction of other angiogenic factors, including Hif-1 α and Vegf (24, 58).
Inflammation is one of the major contributors to lung injury and the development of BPD (59). Inhibiting inflammatory cell recruitment in the lung leads to improved alveolarization and angiogenesis (60). Many studies on Gdf15−/− mice showed higher macrophage and inflammatory cell infiltration in different pathological states (21, 61). Interestingly, we observed that Gdf15−/− mice had significantly lower macrophage recruitment compared with WT mice under hyperoxia and normoxia. In addition, Gdf15 deficiency blunted the expected increase in macrophage recruitment in the Gdf15−/− mice in response to hyperoxia. The disparity between study results can be secondary to the significant complexity of the regulatory mechanisms and differences in the time courses of inflammatory cell recruitment and activation. In a model of ricin-induced acute lung injury, Gdf15−/− mice showed decreased alveolar macrophages (24 h postinjury) and decreased number of Ly6clo (circulating/patrolling) monocytes (72 h postinjury). However, the same study showed increased interstitial macrophages in the Gdf15−/− lung following ricin-induced injury compared with WT mice (62). Regarding macrophage function, Gdf15 decreases macrophage production of tumor necrosis factor and nitric oxide by inhibiting TGF-β-activated kinase TAK1 (63). Gdf15 is required for oxidative metabolism in macrophages and leads to M2-like polarization (64). A recent study highlighted the critical role of myeloid-derived GDF15 in the regulation of both the infiltration and transition to a reparative phenotype following injury (65). Gdf15 was identified in the Ly6clo monocyte-derived macrophage population in this study and single-cell sequencing identified a unique Gdf15 expressing repair-type macrophage subpopulation (65). More studies need to explore the interaction and mechanistic role of GDF15 in macrophage modulation and activation in hyperoxic lung injury and BPD with close attention to the time course of macrophage composition and cell state after hyperoxic injury.
Genes related to macrophage activation, recruitment, and homeostasis were differentially expressed in the Gdf15−/− lung. Spp1 or Osteopontin (Opn) is an arginine-glycine-aspartate-containing adhesive glycoprotein that is secreted as soluble cytokine (66) and is known to be produced by alveolar macrophages as well as other cells in the lung (67, 68). Spp1 also plays an important role in macrophage recruitment, function, and accumulation at sites of injury (69–71). Notably, Spp1 was downregulated in Gdf15−/− mice in our study. Other genes such as Csf1r (macrophage colony-stimulating factor I receptor) were downregulated in the Gdf15−/− male lung upon exposure to hyperoxia. Csf1r plays a crucial role in the development of tissue macrophages and modulates macrophage survival and chemotaxis (72). Other macrophage function-related genes include Eng (38, 39) and Siglec F, both of which are downregulated in the Gdf15−/− mice.
The exact source of GDF15 production in the injured neonatal lung needs to be elucidated. Apart from myeloid cells being a potential source, as detailed earlier, evidence from in vivo and in vitro experiments points toward lung epithelial cells (73–75). Data from single-cell RNA-sequencing data from human IPF lungs identified alveolar epithelial cells as the source for GDF15, and levels were increased both in the murine model and the human disease (76). High Gdf15 expression was reported in aberrant basaloid cells in IPF lungs at the edge of myofibroblastic foci (77). Some evidence also points to the pulmonary endothelial cells as sources of the protein (19, 78). GDF15 levels were recently associated with postcapillary pulmonary hypertension (79). However, the levels and the possible modulatory role of GDF15 in BPD-PH still need to be determined. In an ovalbumin-sensitization-based asthma murine model, GDF15 expression was induced by Notch in Treg cells and mediated a Type 2 innate lymphoid cell-dependent inflammation (80). Gdf15 deficiency attenuated the cigarette smoke-induced pulmonary inflammation (81), but cigarette smoke-induced GDF15 expression increased airway epithelial cell senescence (82) and mucin overexpression (83). In summary, there could be multiple cellular sources of GDF15 in different injury models and they could differ based on the kind of injury/disease process, developmental stage, acute/chronic phase of the disease/injury, and the cellular microenvironment.
The only known receptor for GDF15 is in the hindbrain (GFRAL) (49, 50). Upon binding to this receptor, it mediates anorexic effects and is responsible for weight loss and cachexia associated with GDF15 expression. Whether GDF15 exerts any receptor-mediated effects in the lung are not known. Zhang et al. (76) reported that there was no expression of GFRAL in the lungs. In vitro experiments in A549 cells suggest that the biological effects of GDF15 may be dependent on TGFBR2 expression (84). We recently reported that GDF15 levels are elevated in preterm babies, are inversely related to the gestational age at birth, and that longitudinal GDF15 levels were associated with increased respiratory support and need for oxygen at 36 wk postmenstrual age (85). This suggests that as a biomarker, sustained elevation in GDF15 levels is associated with worse clinical outcomes. The critical windows of intervention postinjury where modulation of GDF15 with either increasing physiological levels acutely or decreasing sustained elevated GDF15 levels need to be elucidated.
Among the limitations of this study is the use of the global knockout model for the loss of Gdf15. Timed loss of Gdf15 after the onset of injury or during the recovery would pinpoint the role of the gene during the acute lung injury and the chronic repair phase. In this study, we are not able to report the cellular source of Gdf15 production within the lung. Studies from adult lungs have mainly pointed to the epithelial origin of this protein. In the developing lung, the expression of Gdf15 is mainly in the myeloid/macrophage cell population (LungMAP database) and perhaps switches to epithelial cells later during development (Supplemental Fig. S3). Among the myeloid cells, Gdf15 expression is higher in the alveolar and interstitial macrophages (Supplemental Fig. S3) (86). Timed scRNA-Seq experiments and lineage tracing experiments using Gdf15 reporter mice will enable the cellular sources and fate of these cells in the lung. We did not assess the acute effects of loss of GDF15 at the acute stage of neonatal hyperoxic lung injury at PND 5. Also, given the role of GDF15 as a biomarker in adult pulmonary arterial hypertension, whether the loss of GDF15 is associated with a worse pulmonary hypertension phenotype following neonatal hyperoxia exposure was not studied. Finally, to answer the question whether the phenotype seen in Gdf15−/− mice is due to the local pulmonary effects or centrally GFRAL-mediated effects of GDF15 will need to be elucidated using GFRAL loss of function murine models.
In conclusion, we show for the first time that loss of Gdf15 exacerbates mortality, lung injury, and the phenotype of the arrest of alveolarization in the developing lung with loss of female-sex advantage in Gdf15−/− mice. We also highlight the distinct pulmonary transcriptomic response in the Gdf15−/− lung including pathways related to macrophage recruitment and activation.
DATA AVAILABILITY
The raw data have been uploaded to the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under Accession No. GSE211744.
SUPPLEMENTAL DATA
Supplemental Tables S1 and S2 and Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.22771034.
GRANTS
This work was supported in part by National Institutes of Health Grants R01-HL144775, R01-HL146395, and R21-HD100862 (to K.L.) and P42 ES0327725 and R01HL129794 (to B.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.L. conceived and designed research; F.A.-M., W.J., L.W., X.D., and K.L. performed experiments; F.A.-M., M.C.G., A.C., W.J., L.W., X.D., B.M., and K.L. analyzed data; F.A.-M., M.C.G., and K.L. interpreted results of experiments; F.A.-M., M.C.G., A.C., L.W., and K.L. prepared figures; F.A.-M., M.C.G., A.C., and K.L. drafted manuscript; M.C.G., B.M., E.S., and K.L. edited and revised manuscript; M.C.G., W.J., E.S., and K.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Graphical abstract image created with BioRender.com and published with permission.
REFERENCES
- 1. Rysavy MA, Horbar JD, Bell EF, Li L, Greenberg LT, Tyson JE, Patel RM, Carlo WA, Younge NE, Green CE, Edwards EM, Hintz SR, Walsh MC, Buzas JS, Das A, Higgins RD; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network and Vermont Oxford Network. Assessment of an updated neonatal research network extremely preterm birth outcome model in the Vermont Oxford Network. JAMA Pediatr 174: e196294, 2020. doi: 10.1001/jamapediatrics.2019.6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr 23: 167–172, 2011. doi: 10.1097/MOP.0b013e3283423e6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Collaco JM, McGrath-Morrow SA. Bronchopulmonary dysplasia as a determinant of respiratory outcomes in adult life. Pediatr Pulmonol 56: 3464–3471, 2021. doi: 10.1002/ppul.25301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc Natl Acad Sci USA 94: 11514–11519, 1997. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baek SJ, Kim JS, Nixon JB, DiAugustine RP, Eling TE. Expression of NAG-1, a transforming growth factor-β superfamily member, by troglitazone requires the early growth response gene EGR-1. J Biol Chem 279: 6883–6892, 2004. doi: 10.1074/jbc.M305295200. [DOI] [PubMed] [Google Scholar]
- 6. Lawton LN, Bonaldo MDF, Jelenc PC, Qiu L, Baumes SA, Marcelino RA, de Jesus GM, Wellington S, Knowles JA, Warburton D, Brown S, Soares MB. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene 203: 17–26, 1997. doi: 10.1016/s0378-1119(97)00485-x. [DOI] [PubMed] [Google Scholar]
- 7. Marjono AB, Brown DA, Horton KE, Wallace EM, Breit SN, Manuelpillai U. Macrophage inhibitory cytokine-1 in gestational tissues and maternal serum in normal and pre-eclamptic pregnancy. Placenta 24: 100–106, 2003. doi: 10.1053/plac.2002.0881. [DOI] [PubMed] [Google Scholar]
- 8. Al-Mudares F, Reddick S, Ren J, Venkatesh A, Zhao C, Lingappan K. Role of growth differentiation factor 15 in lung disease and senescence: potential role across the lifespan. Front Med 7: 594137, 2020. doi: 10.3389/fmed.2020.594137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, Zhang C, Ring AM, Young LH, Medzhitov R. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell 178: 1231–1244. e11, 2019. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsai VWW, Husaini Y, Sainsbury A, Brown DA, Breit SN. The MIC-1/GDF15-GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab 28: 353–368, 2018. doi: 10.1016/j.cmet.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 11. Wu Q, Jiang D, Schaefer NR, Harmacek L, O'Connor BP, Eling TE, Eickelberg O, Chu HW. Overproduction of growth differentiation factor 15 promotes human rhinovirus infection and virus-induced inflammation in the lung. Am J Physiol Lung Cell Mol Physiol 314: L514–L527, 2018. doi: 10.1152/ajplung.00324.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Husebø GR, Grønseth R, Lerner L, Gyuris J, Hardie JA, Bakke PS, Eagan TM. Growth differentiation factor-15 is a predictor of important disease outcomes in patients with COPD. Eur Respir J 49: 1601298, 2017. doi: 10.1183/13993003.01298-2016. [DOI] [PubMed] [Google Scholar]
- 13. Martinez CH, Freeman CM, Nelson JD, Murray S, Wang X, Budoff MJ, Dransfield MT, Hokanson JE, Kazerooni EA, Kinney GL, Regan EA, Wells JM, Martinez FJ, Han MLK, Curtis JL. GDF-15 plasma levels in chronic obstructive pulmonary disease are associated with subclinical coronary artery disease. Respir Res 18: 42, 2017. doi: 10.1186/s12931-017-0521-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nickel N, Kempf T, Tapken H, Tongers J, Laenger F, Lehmann U, Golpon H, Olsson K, Wilkins MR, Gibbs JSR, Hoeper MM, Wollert KC. Growth differentiation factor-15 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 534–541, 2008. doi: 10.1164/rccm.200802-235OC. [DOI] [PubMed] [Google Scholar]
- 15. Geenen LW, Baggen VJM, Kauling RM, Koudstaal T, Boomars KA, Boersma E, Roos-Hesselink JW, van den Bosch AE. Growth differentiation factor-15 as candidate predictor for mortality in adults with pulmonary hypertension. Heart 106: 467–473, 2020. doi: 10.1136/heartjnl-2019-315111. [DOI] [PubMed] [Google Scholar]
- 16. He J, Li X, Yu M. Bioinformatics analysis identifies potential ferroptosis key genes in the pathogenesis of pulmonary fibrosis. Front Genet 12: 788417, 2021. doi: 10.3389/fgene.2021.788417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanders JL, Putman RK, Dupuis J, Xu H, Murabito JM, Araki T, Nishino M, Benjamin EJ, Levy DL, Ramachandran VS, Washko GR, Curtis JL, Freeman CM, Bowler RP, Hatabu H, O’Connor GT, Hunninghake GM. The association of aging biomarkers, interstitial lung abnormalities, and mortality. Am J Respir Crit Care Med 203: 1149–1157, 2021. doi: 10.1164/rccm.202007-2993OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Jiang W, Wang L, Lingappan K. Sex-specific differences in the modulation of Growth Differentiation Factor 15 (GDF15) by hyperoxia in vivo and in vitro: role of Hif-1 α. Toxicol Appl Pharmacol 332: 8–14, 2017. doi: 10.1016/j.taap.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tiwari KK, Moorthy B, Lingappan K. Role of GDF15 (growth and differentiation factor 15) in pulmonary oxygen toxicity. Toxicol In Vitro 29: 1369–1376, 2015. doi: 10.1016/j.tiv.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han ES, Muller FL, Pérez VI, Qi W, Liang H, Xi L, Fu C, Doyle E, Hickey M, Cornell J, Epstein CJ, Roberts LJ, van Remmen H, Richardson A. The in vivo gene expression signature of oxidative stress. Physiol Genomics 34: 112–126, 2008. doi: 10.1152/physiolgenomics.00239.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, Bolomini-Vittori M, Korf-Klingebiel M, Napp LC, Hansen B, Kanwischer A, Bavendiek U, Beutel G, Hapke M, Sauer MG, Laudanna C, Hogg N, Vestweber D, Wollert KC. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med 17: 581–588, 2011. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- 22. Kim JM, Kosak JP, Kim JK, Kissling G, Germolec DR, Zeldin DC, Bradbury JA, Baek SJ, Eling TE. NAG-1/GDF15 transgenic mouse has less white adipose tissue and a reduced inflammatory response. Mediators Inflamm 2013: 641851, 2013. doi: 10.1155/2013/641851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Preusch MR, Baeuerle M, Albrecht C, Blessing E, Bischof M, Katus HA, Bea F. GDF-15 protects from macrophage accumulation in a mouse model of advanced atherosclerosis. Eur J Med Res 18: 19, 2013. doi: 10.1186/2047-783X-18-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song H, Yin D, Liu Z. GDF-15 promotes angiogenesis through modulating p53/HIF-1α signaling pathway in hypoxic human umbilical vein endothelial cells. Mol Biol Rep 39: 4017–4022, 2012. doi: 10.1007/s11033-011-1182-7. [DOI] [PubMed] [Google Scholar]
- 25. Jin YJ, Lee JH, Kim YM, Oh GT, Lee H. Macrophage inhibitory cytokine-1 stimulates proliferation of human umbilical vein endothelial cells by up-regulating cyclins D1 and E through the PI3K/Akt-, ERK-, and JNK-dependent AP-1 and E2F activation signaling pathways. Cell Signal 24: 1485–1495, 2012. doi: 10.1016/j.cellsig.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 26. Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, Langhe S. D, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E. Lung organogenesis. Curr Top Dev Biol 90: 73–158, 2010. doi: 10.1016/S0070-2153(10)90003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol 307: L936–L947, 2014. doi: 10.1152/ajplung.00159.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lingappan K, Jiang W, Wang L, Moorthy B. Sex-specific differences in neonatal hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 311: L481–L493, 2016. doi: 10.1152/ajplung.00047.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419, 2017. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Soneson C, Hickey PF, Johnson LK, Tessa Pierce N, Shepherd L, Morgan M, Patro R. Tximeta: reference sequence checksums for provenance identification in RNA-seq. PLoS Comput Biol 16: e1007664, 2020. doi: 10.1371/journal.pcbi.1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4: 1184–1191, 2009. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis (Preprint). bioRxiv, 2021. doi: 10.1101/060012. [DOI]
- 34. Coarfa C, Zhang Y, Maity S, Perera DN, Jiang W, Wang L, Couroucli X, Moorthy B, Lingappan K. Sexual dimorphism of the pulmonary transcriptome in neonatal hyperoxic lung injury: identification of angiogenesis as a key pathway. Am J Physiol Lung Cell Mol Physiol 313, 2017. doi: 10.1152/ajplung.00230.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ardini-Poleske ME, Clark RF, Ansong C, Carson JP, Corley RA, Deutsch GH, Hagood JS, Kaminski N, Mariani TJ, Potter SS, Pryhuber GS, Warburton D, Whitsett JA, Palmer SM, Ambalavanan N; LungMAP Consortium. LungMAP: the molecular atlas of lung development program. Am J Physiol Lung Cell Mol Physiol 313: L733–L740, 2017. doi: 10.1152/ajplung.00139.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arts RJW, Blok BA, van Crevel R, Joosten LAB, Aaby P, Benn CS, Netea MG. Vitamin A induces inhibitory histone methylation modifications and down-regulates trained immunity in human monocytes. J Leukoc Biol 98: 129–136, 2015. doi: 10.1189/jlb.6AB0914-416R. [DOI] [PubMed] [Google Scholar]
- 37. Lastres P, Bellon T, Cabañas C, Sanchez‐Madrid F, Acevedo A, Gougos A, Letarte M, Bernabeu C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur J Immunol 22: 393–397, 1992. doi: 10.1002/eji.1830220216. [DOI] [PubMed] [Google Scholar]
- 38. Ojeda-Fernández L, Recio-Poveda L, Aristorena M, Lastres P, Blanco FJ, Sanz-Rodríguez F, Gallardo-Vara E, de las Casas-Engel M, Corbí Á, Arthur HM, Bernabeu C, Botella LM. Mice lacking endoglin in macrophages show an impaired immune response. PLoS Genet 12: e1005935, 2016. doi: 10.1371/journal.pgen.1005935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scharpfenecker M, Floot B, Russell NS, Stewart FA. The TGF-β co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. Radiother Oncol 105: 313–320, 2012. doi: 10.1016/j.radonc.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 40. Rossetti C, Picardi E, Ye M, Camilli G, D'Erchia AM, Cucina L, Locatelli F, Fianchi L, Teofili L, Pesole G, Gallo A, Sorrentino R. RNA editing signature during myeloid leukemia cell differentiation. Leukemia 31: 2824–2832, 2017. doi: 10.1038/leu.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49: 503–510, 2013. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bond LM, Burhans MS, Ntambi JM. Uncoupling protein-1 deficiency promotes brown adipose tissue inflammation and ER stress. PLoS One 13: e0205726, 2018. doi: 10.1371/journal.pone.0205726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahmed DS, Isnard S, Berini C, Lin J, Routy JP, Royston L. Coping with stress: the mitokine GDF-15 as a biomarker of COVID-19 severity. Front Immunol 13: 820350, 2022. doi: 10.3389/fimmu.2022.820350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chrysovergis K, Wang X, Kosak J, Lee SH, Kim JS, Foley JF, Travlos G, Singh S, Baek SJ, Eling TE. NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int J Obes (Lond) 38: 1555–1564, 2014. doi: 10.1038/ijo.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ost M, Igual Gil C, Coleman V, Keipert S, Efstathiou S, Vidic V, Weyers M, Klaus S. Muscle-derived GDF15 drives diurnal anorexia and systemic metabolic remodeling during mitochondrial stress. EMBO Rep 21: e48804, 2020. doi: 10.15252/embr.201948804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Yang L, Qin W, Zhang G, Yuan J, Wang F. Adaptive induction of growth differentiation factor 15 attenuates endothelial cell apoptosis in response to high glucose stimulus. PLoS One 8: e48804, 2013. doi: 10.1371/journal.pone.0065549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moya F, Salas AA. Preterm nutrition and pulmonary disease. World Rev Nutr Diet 122: 400–416, 2021. doi: 10.1159/000514766. [DOI] [PubMed] [Google Scholar]
- 48. Ramel SE, Brown LD, Georgieff MK. The impact of neonatal illness on nutritional requirements-one size does not fit all. Curr Pediatr Rep 2: 248–254, 2014. doi: 10.1007/s40124-014-0059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, Coskun T, Hamang MJ, Sindelar DK, Ballman KK, Foltz LA, Muppidi A, Alsina-Fernandez J, Barnard GC, Tang JX, Liu X, Mao X, Siegel R, Sloan JH, Mitchell PJ, Zhang BB, Gimeno RE, Shan B, Wu X. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med 23: 1215–1219, 2017. doi: 10.1038/nm.4393. [DOI] [PubMed] [Google Scholar]
- 50. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjær SB, Wu X, Huang T, Hultman K, Paulsen SJ, Wang J, Bugge A, Frantzen JB, Nørgaard P, Jeppesen JF, Yang Z, Secher A, Chen H, Li X, John LM, Shan B, He Z, Gao X, Su J, Hansen KT, Yang W, Jørgensen SB. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med 23: 1158–1166, 2017. doi: 10.1038/nm.4394. [DOI] [PubMed] [Google Scholar]
- 51. Tsai VWW, Macia L, Johnen H, Kuffner T, Manadhar R, Jørgensen SB, Lee-Ng KKM, Zhang HP, Wu L, Marquis CP, Jiang L, Husaini Y, Lin S, Herzog H, Brown DA, Sainsbury A, Breit SN. TGF-b superfamily cytokine MIC-1/GDF15 is a physiological appetite and body weight regulator. PLoS One 8: e55174, 2013. doi: 10.1371/journal.pone.0055174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leary S, Das P, Ponnalagu D, Singh H, Bhandari V. Genetic strain and sex differences in a hyperoxia-induced mouse model of varying severity of bronchopulmonary dysplasia. Am J Pathol 189: 999–1014, 2019. doi: 10.1016/j.ajpath.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, Coarfa C, Dong X, Jiang W, Hayward-Piatkovskyi B, Gleghorn JP, Lingappan K, Jp G, Microrna-A LK. MicroRNA-30a as a candidate underlying sex-specific differences in neonatal hyperoxic lung injury: implications for BPD. Am J Physiol Lung Cell Mol Physiol 316: L144–L156, 2019. doi: 10.1152/ajplung.00372.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y, Dong X, Shirazi J, Gleghorn JP, Lingappan K. Pulmonary endothelial cells exhibit sexual dimorphism in their response to hyperoxia. Am J Physiol Heart Circ Physiol 315: H1287–H1292, 2018. doi: 10.1152/ajpheart.00416.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lingappan K, Srinivasan C, Jiang W, Wang L, Couroucli XI, Moorthy B. Analysis of the transcriptome in hyperoxic lung injury and sex-specific alterations in gene expression. PLoS One 9: e101581, 2014. doi: 10.1371/journal.pone.0101581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Balaji S, Dong X, Li H, Zhang Y, Steen E, Lingappan K. Sex-specific differences in primary neonatal murine lung fibroblasts exposed to hyperoxia in vitro: Implications for bronchopulmonary dysplasia. Physiol Genomics 50: 940–946, 2018. doi: 10.1152/physiolgenomics.00075.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheng H, Wang H, Wu C, Zhang Y, Bao T, Tian Z. Proteomic analysis of sex differences in hyperoxic lung injury in neonatal mice. Int J Med Sci 17: 2440–2448, 2020. doi: 10.7150/ijms.42073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang S, Li M, Zhang W, Hua H, Wang N, Zhao J, Ge J, Jiang X, Zhang Z, Ye D, Yang C. Growth differentiation factor 15 promotes blood vessel growth by stimulating cell cycle progression in repair of critical-sized calvarial defect. Sci Rep 7: 9027, 2017. doi: 10.1038/s41598-017-09210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kallapur SG, Jobe AH. Contribution of inflammation to lung injury and development. Arch Dis Child Fetal Neonatal Ed 91: F132–F135, 2006. doi: 10.1136/adc.2004.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jagarapu J, Kelchtermans J, Rong M, Chen S, Hehre D, Hummler S, Faridi MH, Gupta V, Wu S. Efficacy of leukadherin-1 in the prevention of hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol 53: 793–801, 2015. doi: 10.1165/rcmb.2014-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abulizi P, Loganathan N, Zhao D, Mele T, Zhang Y, Zwiep T, Liu K, Zheng X. Growth differentiation factor-15 deficiency augments inflammatory response and exacerbates septic heart and renal injury induced by lipopolysaccharide. Sci Rep 7: 1037, 2017. doi: 10.1038/s41598-017-00902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Deng M, Su D, Xiao N, Zhang Z, Wang Y, Zong F, Li S, Wang J, Zhou D, Zhao Y, Yang H. Gdf15 deletion exacerbates acute lung injuries induced by intratracheal inoculation of aerosolized ricin in mice. Toxicology 469: 153135, 2022. doi: 10.1016/j.tox.2022.153135. [DOI] [PubMed] [Google Scholar]
- 63. Ratnam NM, Peterson JM, Talbert EE, Ladner KJ, Rajasekera P. V, Schmidt CR, Dillhoff ME, Swanson BJ, Haverick E, Kladney RD, Williams TM, Leone GW, Wang DJ, Guttridge DC. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J Clin Invest 127: 3796–3809, 2017. doi: 10.1172/JCI91561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jung SB, Choi MJ, Ryu D, Yi HS, Lee SE, Chang JY, Chung HK, Kim YK, Kang SG, Lee JH, Kim KS, Kim HJ, Kim CS, Lee CH, Williams RW, Kim H, Lee HK, Auwerx J, Shong M. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun 9: 1551, 2018. doi: 10.1038/s41467-018-03998-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Patsalos A, Halasz L, Medina-Serpas MA, Berger WK, Daniel B, Tzerpos P, Kiss M, Nagy G, Fischer C, Simandi Z, Varga T, Nagy L. A growth factor-expressing macrophage subpopulation orchestrates regenerative inflammation via GDF-15. J Exp Med 219: e20210420, 2022. doi: 10.1084/jem.20210420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Regan AW, Hayden JM, Body S, Liaw L, Mulligan N, Goetschkes M, Berman JS. Abnormal pulmonary granuloma formation in osteopontin-deficient mice. Am J Respir Crit Care Med 164: 2243–2247, 2001. doi: 10.1164/ajrccm.164.12.2104139. [DOI] [PubMed] [Google Scholar]
- 67. Takahashi F, Takahashi K, Shimizu K, Cui R, Tada N, Takahashi H, Soma S, Yoshioka M, Fukuchi Y. Osteopontin is strongly expressed by alveolar macrophages in the lungs of acute respiratory distress syndrome. Lung 182: 173–185, 2004. doi: 10.1007/s00408-004-0309-1. [DOI] [PubMed] [Google Scholar]
- 68. Berman JS, Serlin D, Li X, Whitley G, Hayes J, Rishikof DC, Ricupero DA, Liaw L, Goetschkes M, O’Regan AW. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am J Physiol Lung Cell Mol Physiol 286: L1311–L1318, 2004. doi: 10.1152/ajplung.00394.2003. [DOI] [PubMed] [Google Scholar]
- 69. Ophascharoensuk V, Giachelli CM, Gordon K, Hughes J, Pichler R, Brown P, Liaw L, Schmidt R, Shankland SJ, Alpers CE, Couser WG, Johnson RJ. Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int 56: 571–580, 1999. doi: 10.1046/j.1523-1755.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- 70. Nau GJ, Liaw L, Chupp GL, Berman JS, Hogan BLM, Young RA. Attenuated host resistance against Mycobacterium bovis BCG infection in mice lacking osteopontin. Infect Immun 67: 4223–4230, 1999. doi: 10.1128/IAI.67.8.4223-4230.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287: 860–864, 2000. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 72. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 6: a021857, 2014. doi: 10.1101/cshperspect.a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Takenouchi Y, Kitakaze K, Tsuboi K, Okamoto Y. Growth differentiation factor 15 facilitates lung fibrosis by activating macrophages and fibroblasts. Exp Cell Res 391: 112010, 2020. doi: 10.1016/j.yexcr.2020.112010. [DOI] [PubMed] [Google Scholar]
- 74. Song H, Chen Q, Xie S, Huang J, Kang G. GDF-15 prevents lipopolysaccharide-mediated acute lung injury via upregulating SIRT1. Biochem Biophys Res Commun 526: 439–446, 2020. doi: 10.1016/j.bbrc.2020.03.103. [DOI] [PubMed] [Google Scholar]
- 75. Šimečková P, Marvanová S, Kulich P, Králiková L, Neča J, Procházková J, Machala M. Screening of cellular stress responses induced by ambient aerosol ultrafine particle fraction PM0.5 in A549 cells. Int J Mol Sci 20: 6310, 2019. doi: 10.3390/ijms20246310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, Chen X, Sembrat J, Chu Y, Cardenes N, Tuder RM, Herzog EL, Ryu C, Rojas M, Lafyatis R, Gibson KF, McDyer JF, Kass DJ, Alder JK. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 317: L510–L521, 2019. doi: 10.1152/ajplung.00062.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nickel N, Jonigk D, Kempf T, Bockmeyer CL, Maegel L, Rische J, Laenger F, Lehmann U, Sauer C, Greer M, Welte T, Hoeper MM, Golpon HA. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res 12: 62, 2011. doi: 10.1186/1465-9921-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mirna M, Rohm I, Jirak P, Wernly B, Bäz L, Paar V, Kretzschmar D, Hoppe UC, Schulze PC, Lichtenauer M, Jung C, Franz M. Analysis of novel cardiovascular biomarkers in patients with pulmonary hypertension (PH). Heart Lung Circ 29: 337–344, 2020. doi: 10.1016/j.hlc.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 80. Harb H, Stephen-Victor E, Crestani E, Benamar M, Massoud A, Cui Y, Charbonnier L-M, Arbag S, Baris S, Cunnigham A, Manuel Leyva-Castillo J, Geha RS, Mousavi AJ, Guennewig B, Schmitz-Abe K, Sioutas C, Phipatanakul W, Chatila TA. A regulatory T cell Notch4–GDF15 axis licenses tissue inflammation in asthma. Nat Immunol 21: 1359–1370, 2020. [Erratum in Nat Immunol 22: 794–795, 2021]. doi: 10.1038/s41590-020-0777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Verhamme FM, Seys LJM, de Smet EG, Provoost S, Janssens W, Elewaut D, Joos GF, Brusselle GG, Bracke KR. Elevated GDF-15 contributes to pulmonary inflammation upon cigarette smoke exposure. Mucosal Immunol 10: 1400–1411, 2017. doi: 10.1038/mi.2017.3. [DOI] [PubMed] [Google Scholar]
- 82. Wu Q, Jiang D, Matsuda JL, Ternyak K, Zhang B, Chu HW. Cigarette smoke induces human airway epithelial senescence via growth differentiation factor 15 production. Am J Respir Cell Mol Biol 55: 429–438, 2016. doi: 10.1165/rcmb.2015-0143OC. [DOI] [PubMed] [Google Scholar]
- 83. Wu Q, Jiang D, Chu HW. Cigarette smoke induces growth differentiation factor 15 production in human lung epithelial cells: implication in mucin over-expression. Innate Immun 18: 617–626, 2012. doi: 10.1177/1753425911429837. [DOI] [PubMed] [Google Scholar]
- 84. Tarfiei GA, Shadboorestan A, Montazeri H, Rahmanian N, Tavosi G, Ghahremani MH. GDF15 induced apoptosis and cytotoxicity in A549 cells depends on TGFBR2 expression. Cell Biochem Funct 37: 320–330, 2019. doi: 10.1002/cbf.3391. [DOI] [PubMed] [Google Scholar]
- 85. Almudares F, Hagan J, Chen X, Devaraj S, Moorthy B, Lingappan K. Growth and differentiation factor 15 (GDF15) levels predict adverse respiratory outcomes in premature neonates. Pediatr Pulmonol 58: 271–278, 2023. doi: 10.1002/ppul.26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sajti E, Link VM, Ouyang Z, Spann NJ, Westin E, Romanoski CE, Fonseca GJ, Prince LS, Glass CK. Transcriptomic and epigenetic mechanisms underlying myeloid diversity in the lung. Nat Immunol 21: 221–231, 2020. doi: 10.1038/s41590-019-0582-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1 and S2 and Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.22771034.
Data Availability Statement
All code used in this manuscript can be found in Supplemental Material; the raw data have been uploaded to National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO); GSE211744. All supplemental data are accessible at https://doi.org/10.6084/m9.figshare.22771034.
The raw data have been uploaded to the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under Accession No. GSE211744.