

Abstract
Chromosome segregation errors in mammalian oocyte meiosis lead to developmentally compromised aneuploid embryos and become more common with advancing maternal age. Known contributors include age‐related chromosome cohesion loss and spindle assembly checkpoint (SAC) fallibility in meiosis‐I. But how effective the SAC is in meiosis‐II and how this might contribute to age‐related aneuploidy is unknown. Here, we developed genetic and pharmacological approaches to directly address the function of the SAC in meiosis‐II. We show that the SAC is insensitive in meiosis‐II oocytes and that as a result misaligned chromosomes are randomly segregated. Whilst SAC ineffectiveness in meiosis‐II is not age‐related, it becomes most prejudicial in oocytes from older females because chromosomes that prematurely separate by age‐related cohesion loss become misaligned in meiosis‐II. We show that in the absence of a robust SAC in meiosis‐II these age‐related misaligned chromatids are missegregated and lead to aneuploidy. Our data demonstrate that the SAC fails to prevent cell division in the presence of misaligned chromosomes in oocyte meiosis‐II, which explains how age‐related cohesion loss can give rise to aneuploid embryos.
Keywords: ageing, aneuploidy, KIF18A, oocyte meiosis, spindle assembly checkpoint
Subject Categories: Molecular Biology of Disease
Ineffectiveness of the spindle assembly checkpoint in meiosis‐II permits anaphase in oocytes with severely misaligned chromosomes. SAC insensitivity to misaligned chromosomes becomes particularly detrimental with age, leading to age‐related aneuploidy in the embryo.
Introduction
Chromosome segregation is accomplished by the spindle, a barrel‐shaped molecular machine made of microtubules and associated molecules (Compton, 2000; Walczak et al, 2010; Dumont & Desai, 2012; Heald & Khodjakov, 2015; Bennabi et al, 2016). Ensuring correct chromosome segregation during cell division and thus the formation of euploid daughter cells is paramount, and the impact of errors that cause aneuploidy is highly context‐dependent. Whereas in somatic cells aneuploidy is associated with cancer (Potapova & Gorbsky, 2017; Ben‐David & Amon, 2020), in germ cells aneuploidy compromises developmental potential of the embryo and thus is a major cause of reproductive failure (Hassold & Hunt, 2001; MacLennan et al, 2015).
Chromosome segregation in oocytes occurs over two successive meiotic divisions. Meiosis‐I (M‐I) comprises germinal vesicle breakdown (GVBD) and ends with the segregation of homologous chromosomes. Meiosis‐II (M‐II) follows thereafter without an intervening interphase, the oocyte immediately assembles the M‐II spindle, and the cell cycle arrests at metaphase (Met‐II) as a result of the oocyte specific cell cycle regulator Cytostatic Factor (CSF; Madgwick & Jones, 2007). M‐II is ultimately completed upon fertilisation, wherein sister chromatids are separated, and embryonic development is initiated. Chromosome segregation in female meiosis is notoriously error‐prone, and the likelihood of segregation errors in oocytes is increased by advancing maternal age, therein causing developmentally compromised aneuploid embryos (Hassold & Hunt, 2001; Greaney et al, 2018; Mihajlović & FitzHarris, 2018; Charalambous et al, 2022). Landmark work revealed an important clue as to how ageing leads to aneuploidy, that oocytes exhibit a progressive age‐related loss of cohesin, the molecular ties that maintain chromosomal cohesion prior to segregation in anaphase (Angell, 1991, 1997; Angell et al, 1993; Chiang et al, 2010; Lister et al, 2010; Duncan et al, 2012; Merriman et al, 2012; Sakakibara et al, 2015). Progressive cohesion loss during ageing leads to premature separation of homologous chromosomes in M‐I (Fig EV1), resulting in bioriented sister pairs on the spindle that separate prematurely in anaphase‐I (A‐I; Sakakibara et al, 2015). However, how premature chromosome disjunction in oocytes from ageing females ultimately leads to aneuploidy remains unclear.
Figure EV1. Age‐related cohesion loss leads to chromosome misalignment in meiosis‐II.
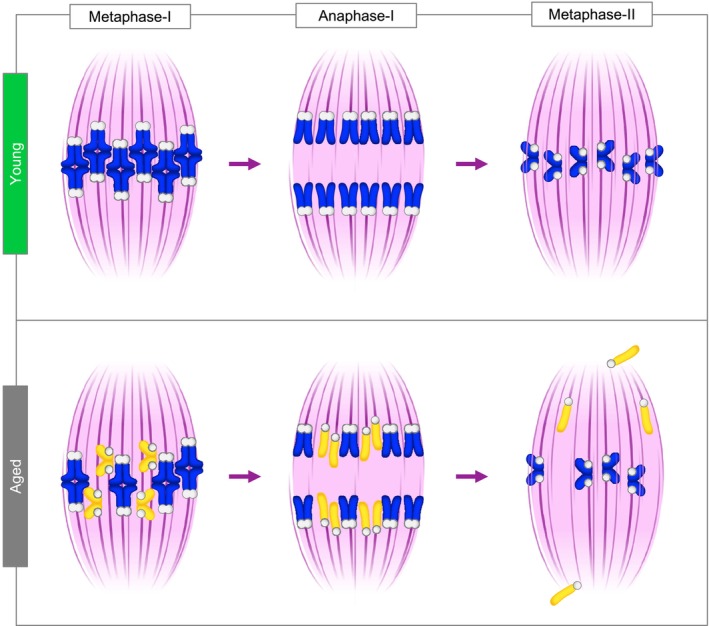
In meiosis‐I, homologous chromosomes (sister chromatid pairs) are held together by the chromosome cohesion. In young oocytes, cohesion is normally intact allowing homologous chromosomes to biorient and segregate towards the opposing spindle poles during anaphase‐I. As a result, sister chromatid pairs remain intact in metaphase of meiosis‐II, biorient and align. In aged oocytes, cohesion loss leads to premature individualisation of homologous chromosomes and consequent biorientation of sister chromatids (yellow) in meiosis‐I. As a result, sister chromatids prematurely separate during anaphase‐I and the resulting individualised chromatids randomly misalign at the metaphase‐II spindle in aged oocytes.
A second well‐established idiosyncrasy of mammalian oocytes that likely contributes to aneuploidy is that the spindle assembly checkpoint (SAC), which prevents anaphase until all chromosomes achieve proper biorientation and alignment in most cells (Musacchio & Salmon, 2007; Lara‐Gonzalez et al, 2012; Musacchio, 2015) is weak in oocytes. This has been extensively established in oocyte M‐I, where treatments and manipulations that cause chromosome misalignment fail to prevent anaphase and completion of M‐I (Gui & Homer, 2012; Kolano et al, 2012; Lane et al, 2012; Sebestova et al, 2012; Kyogoku & Kitajima, 2017; Lane & Jones, 2017). However, whether the SAC can function in M‐II, and how this could contribute to aneuploidy genesis, has been harder to address due to absence of tools and approaches to specifically interrogate M‐II without perturbing other aspects of oogenesis. Here we have developed genetic and small molecule approaches to specifically address the SAC in M‐II. Our results show a stark failure of the SAC to effectively police chromosome segregation fidelity in M‐II. This failure is not intrinsically oocyte age‐dependent but plays a major role in age‐related aneuploidy by permitting errors arising from chromosome cohesion loss to translate into embryonic aneuploidy.
Results and Discussion
Establishment of a genetic model to interrogate the spindle assembly checkpoint in oocyte meiosis‐II
We first sought to develop a mouse model with chromosome alignment defects specifically in oocyte M‐II, thus allowing us to examine SAC function in M‐II without having disrupted chromosome segregation in M‐I. We decided to generate an oocyte‐specific knockout of kinesin‐8 family member, KIF18A. In somatic cells, KIF18A ensures correct biorientation and alignment of sister chromatids at the metaphase plate (Stumpff et al, 2008, 2012; Janssen et al, 2018). Given that chromosome alignment in M‐II comprises biorienting sister chromatids similar to mitosis, whereas M‐I biorients homologues chromosomes on a bivalent chromosome, we reasoned that KIF18A may play a major role in chromosome alignment in M‐II but not M‐I. To test this, we created a transgenic mouse line carrying a Kif18a GFP allele, wherein endogenous KIF18A is N‐terminally tagged with GFP, allowing us to visualise the endogenous protein, but also where the Kif18a GFP allele harbours loxP sites to allow tissue‐specific deletion (Fig EV2A). Interestingly, GFP‐KIF18A was undetectable in GV‐stage oocytes, and first observed only in late M‐I at the spindle midzone, where it remained throughout anaphase‐I/telophase‐I (A‐I/T‐I), and was readily detected in metaphase‐II (Met‐II; Figs 1A and EV2B). To conditionally delete Kif18a, we crossed Kif18a GFP mice with mice that express Cre‐recombinase under the oocyte‐specific Zp3 promoter during oocyte growth (Zp3‐Cre), prior to both meiotic divisions (Fig EV2C and D). Kif18a deletion in oocytes was confirmed by PCR (Fig EV2E) and a lack of GFP‐KIF18A signal (Figs 1B and EV2F; hereafter these oocytes are referred to as KIF18A‐cKO). To determine whether KIF18A knock‐out affected M‐I, we analysed spindles, finding no impact of Kif18a deletion upon spindle length (Fig EV3A). Moreover, we found no difference in chromosome alignment (Fig 1B) nor any change in inter‐kinetochore distance (Fig EV3A). Furthermore, KIF18A‐cKO oocytes underwent GVBD and extruded the first polar body with normal timing (Fig 1C). Importantly, Kif18a deletion did not increase the level of aneuploidy in the resulting Met‐II eggs (Fig 1C; 2.6% in KIF18A‐cKO vs. 3.6% in wild‐type [WT], 5.1% in GFP knock‐in [GFPin], and 3.8% in heterozygotes [HET]). We therefore conclude that KIF18A is dispensable for spindle formation and chromosome segregation fidelity in M‐I.
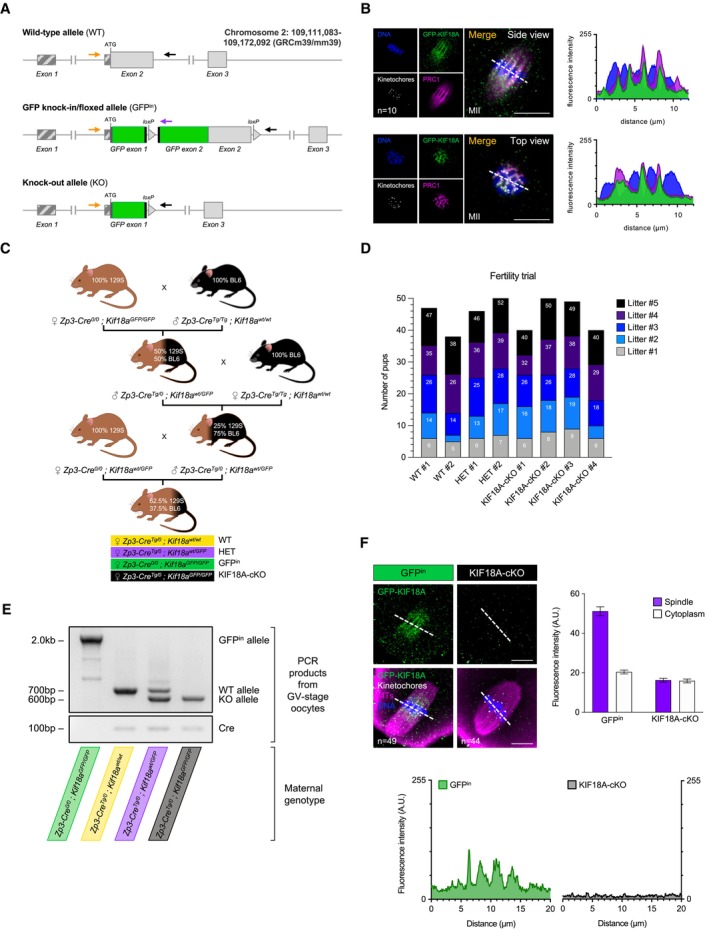
Figure EV2. Generation of Kif18a GFP mouse line and Kif18a conditional deletion in mouse oocytes.
-
ASchematic representation of Kif18a alleles. Top: Wild‐type Kif18a allele (WT). Middle: GFP knock‐in/floxed (GFPin) allele with two GFP exons and an artificial intron inserted immediately after the start codon to produce N‐terminally tagged fusion protein (GFP‐KIF18A). Bottom: Kif18a knock‐out (KO) allele produced following simultaneous Cre recombinase‐mediated excision of GFP‐ and Kif18a‐exon 2. Arrows indicate the position of genotyping primers.
-
BGFP‐KIF18A colocalizes with the midzone protein PRC1 in Kif18a wt/GFP Met‐II eggs (no Cre‐recombinase). Chromosomes are labelled with Hoechst33342 (blue), kinetochores with CREST (grey), midzone with PRC1 (magenta) and GFP‐KIF18A with chicken‐GFP (green) antibodies. ‘n’ is the total number of Met‐II eggs per group. Scale bars, 10 μm.
-
CBreeding strategy to generate female littermates with 4 denoted genotypes.
-
DGraph shows no notable difference in fertility between Zp3‐Cre Tg/0 ; Kif18a GFP/GFP females (KIF18A‐cKO oocytes) and controls (Zp3‐Cre Tg/0 ; Kif18a wt/wt [WT] and Zp3‐Cre Tg/0 ; Kif18a wt/GFP [HET]) after 5 generations of pups (one‐way ANOVA with multiple comparisons), consistent with our observation that a majority of KIF18A‐cKO eggs correctly align their chromosomes.
-
EPCR genotyping confirms successful conditional Kif18a deletion (cKO) in GV‐stage oocytes (10 oocytes per group).
-
FGFPin but not KIF18A‐cKO oocytes show significantly increased GFP‐KIF18A levels at spindle vs. cytoplasm (paired t‐test, P < 0.0001 and P = 0.31, respectively). DNA is labelled with Hoechst33342 (blue) and GFP‐KIF18A, kinetochores and microtubules (MTs) are detected with chicken‐GFP (green), HEC1 (grey) and β‐Tubulin (magenta) antibodies, respectively. Scale bars, 10 μm. Error bars represent SEM. Data from totals of 49 and 44 oocytes across 3 experimental days, for GFPin and KIF18A‐cKO groups, respectively.
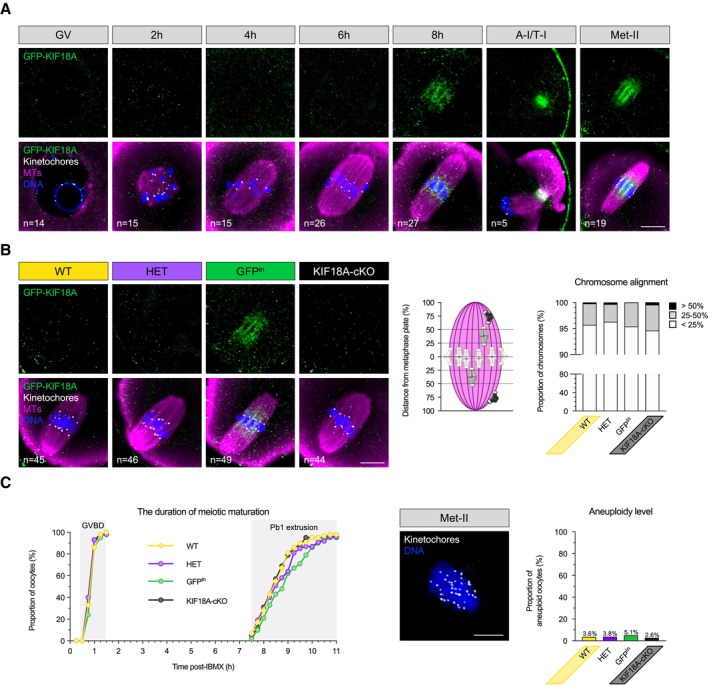
Figure 1. KIF18A removal has no impact on chromosome segregation in meiosis‐I.
-
A, B(A) GFP‐KIF18A protein is first detected in late M‐I (Kif18a wt/GFP oocytes; GV—germinal vesicle; h—hours post‐IBMX release, A‐I/T‐I—anaphase‐I/telophase‐I; Met‐II—metaphase‐II). (B) Late (8 h) M‐I stage spindles in WT (Kif18a wt/wt ), HET (Kif18a wt/Δ ), GFPin (Kif18a GFP/GFP ) and KIF18A‐cKO (Kif18a Δ/Δ ) oocytes. Chart shows no difference in chromosome alignment between KIF18A‐cKO and controls (χ2‐test, P = 0.36). In (A and B), DNA is labelled with Hoechst33342 (blue), kinetochores with CREST (grey) and GFP‐KIF18A and microtubules (MTs) with chicken‐GFP (green) and β‐Tubulin (magenta) antibodies, respectively. ‘n’ is the total number of oocytes per group. Scale bars, 10 μm.
-
CCharts show no difference in meiotic maturation rate (one‐way ANOVA with multiple comparisons) and aneuploidy level (χ2‐test, P = 0.33) between KIF18A‐cKO oocytes (n = 38) and controls (WT [n = 55], HET [n = 52], GFPin [n = 39]; GVBD—germinal vesicle break‐down; Pb1—first polar body). An exemplar WT Met‐II oocyte used in chromosome number assessment. DNA is labelled with Hoechst33342 (blue) and kinetochores with HEC1 antibody (grey). Scale bar, 5 μm.
Source data are available online for this figure.
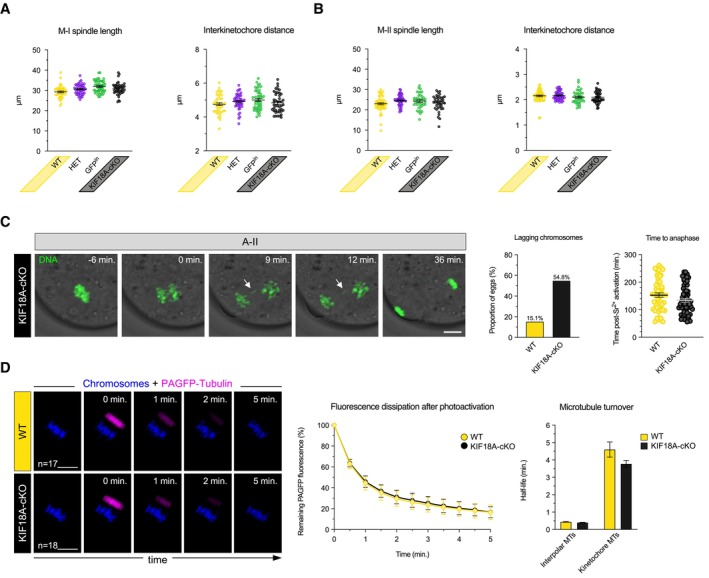
Figure EV3. KIF18A removal has no impact on spindle length and interkinetochore distance in female meiosis, but causes lagging chromosomes in M‐II.
-
A, BCharts show no impact of KIF18a removal on spindle length and interkinetochore distance in late M‐I (A) and M‐II (B) (one‐way ANOVA with multiple comparisons). Error bars represent SEM. For late M‐I, data from totals of 45, 46, 49 and 44 oocytes across 3 experimental days, for WT, HET, GFPin and KIF18A‐cKO groups, respectively. For M‐II, data from totals of 55, 53, 39 and 39 oocytes across 3 experimental days, for WT, HET, GFPin and KIF18A‐cKO groups, respectively.
-
CTime‐lapse confocal images show anaphase with lagging chromosome (white arrow) in KIF18A‐cKO Met‐II egg. Charts show significant increase in lagging chromosomes formation (χ2‐test, P < 0.00001) and no difference in anaphase onset timing (unpaired t‐test, P = 0.07) in KIF18A‐cKO (n = 62) vs. WT (n = 53) Met‐II eggs. DNA is visualised with SiR‐DNA (green) and indicated time (min) is relative to the anaphase onset. Error bars represent SEM.
-
DTime‐lapse confocal images show dissipation of PAGFP‐Tubulin fluorescent signal (magenta) in WT and KIF18A‐cKO Met‐II eggs. Chromosomes are visualised with H2B‐RFP (blue). The ‘0 min’ marks the first frame after photoactivation. The fluorescence dissipation after photoactivation curve shows the average decay rate of PAGFP‐Tubulin in WT and KIF18A‐cKO Met‐II eggs (as mean ± SEM for each time‐point). Chart shows no significant difference in the stable/non‐stable MT turnover rates between the groups (unpaired t‐test, P = 0.09 for stable and P = 0.08 for non‐stable MTs). ‘n’ is the total number of Met‐II eggs per group.
Data information: In (C and D), scale bars, 10 μm.
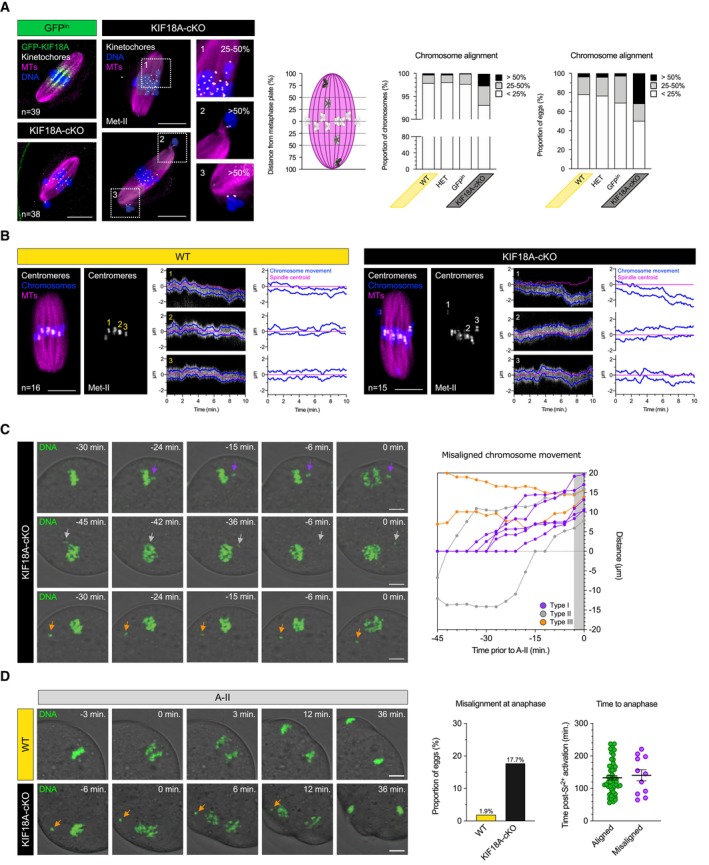
Next, we assessed the requirement for KIF18A in Met‐II eggs. Whilst there was no difference in Met‐II spindle length and inter‐kinetochore distance between KIF18A‐cKO and control groups (Fig EV3B) or microtubule (MT) turnover as measured using photoactivatable‐GFP assay (Fig EV3D), the frequency of misaligned chromosomes was significantly increased in KIF18A‐cKO Met‐II eggs (Fig 2A). In somatic cells, KIF18A depletion causes misalignment by increasing the amplitude of rapid stereotyped oscillations of bioriented sister chromatid pairs about the metaphase plate (typical frequency 1 min−1; Mayr et al, 2007; Stumpff et al, 2008). In Met‐II oocytes however, by performing high temporal resolution centromere live‐imaging (8 s intervals), we found that chromosomes do not undergo stereotyped rapid oscillatory movements irrespective of KIF18A presence or absence (Fig 2B). However, using longer term imaging we observed three distinct chromosome behaviours that gave rise to misalignment (Fig 2C). First, the majority of misaligned chromosomes (56%) were initially aligned but became displaced from the metaphase plate ~ 18–33 min prior to anaphase onset. Second, in 22% of cases, misaligned chromosome transiently aligned, only to misalign at the opposite spindle pole. Third, the remaining 22% of chromosomes were misaligned near the same spindle pole throughout the whole movie. Notably, the chromosome misalignment persists until anaphase onset and is thus different from the misalignment that occurs transiently during Met‐II spindle formation (Kouznetsova et al, 2019). Thus, KIF18A is required for maintaining the equatorial positioning of aligned chromosomes, exclusively in M‐II, and so its conditional depletion in oocytes provides a tool for examining the impact of chromosome misalignment in Met‐II eggs.
Figure 2. Misaligned chromosomes fail to delay anaphase onset in KIF18A‐cKO eggs.
-
AMet‐II‐spindles in GFPin and KIF18A‐cKO oocytes. Insets show mildly (25–50%) and severely (> 50%) misaligned chromosomes in KIF18A‐cKO Met‐II eggs. Note that misaligned chromosomes are pairs of sister chromatids. DNA is labelled with Hoechst33342 (blue) and GFP‐KIF18A, kinetochores and microtubules (MTs) are detected with chicken‐GFP (green), HEC1 (grey) and β‐Tubulin (magenta) antibodies, respectively. Charts show increased chromosome misalignment in KIF18A‐cKO (n = 38) vs. control Met‐II eggs (WT [n = 55], HET [n = 51], GFPin [n = 39]; χ2‐test, P < 0.02). Scale bars, 10 μm.
-
BHigh temporal resolution live imaging of chromosome movements in WT and KIF18A‐cKO Met‐II eggs. Chromosomes are visualised with H2B‐RFP (blue), centromeres with Maj.Sat.‐mClover (grey) and spindle with SiR‐Tubulin (magenta). Kymographs show individual chromosome movement along the vertical spindle axis. Charts show centromere movements (blue) relative to spindle centroid (magenta). ‘n’ is the total number of Met‐II eggs per group. Scale bars, 10 μm.
-
CTime‐lapse confocal images depicting three types of misaligned chromosome behaviour in KIF18A‐cKO Met‐II eggs (left) with graphical representation of their movement (right). Top: aligned chromosome wanders of the metaphase plate shortly prior anaphase (purple; n = 5/9). Middle: misaligned chromosome travels to the opposite spindle pole (grey; n = 2/9). Bottom: chromosome remains misaligned near the same spindle pole throughout the movie (orange; n = 2/9). Purple, grey and orange arrows respectively indicate exemplary misaligned chromosomes for each type of movement.
-
DAnaphase‐II (A‐II) with ostensibly normal chromosome segregation in WT (top) and severely misaligned chromosome (orange arrow) in KIF18A‐cKO (bottom) eggs. Charts show significantly increased proportion of KIF18A‐cKO (n = 62) vs. WT (n = 53) eggs with misaligned chromosomes (χ2‐test, P < 0.01) and no anaphase delay in KIF18A‐cKO eggs (unpaired t‐test, P = 0.66). Error bars represent SEM.
Data information: In (C and D), DNA is visualised with SiR‐DNA (green) and indicated time (min) is relative to the A‐II onset. Scale bars, 10 μm.
Source data are available online for this figure.
KIF18A‐cKO eggs demonstrate SAC fallibility in meiosis‐II
In somatic cells, a single misaligned chromosome can activate the spindle assembly checkpoint, the failsafe pathway that prevents M‐phase exit, thus preventing anaphase until all chromosomes are aligned. Briefly, unattached kinetochores recruit SAC components including MAD2, which leads to the production of a Mitotic Checkpoint Complex (MCC) that prevents anaphase promoting complex (APC)‐mediated Cyclin B and Securin destruction, and thus postpones anaphase until all chromosomes are correctly aligned and attached (Musacchio & Salmon, 2007). To determine the impact of chromosome misalignment on the completion of M‐II we parthenogenetically activated KIF18A‐cKO eggs and monitored misaligned chromosome behaviour (Fig 2D). Surprisingly, misaligned chromosomes had no impact on the timing of anaphase onset, consistent with a lack of SAC activation (Figs 2D and EV3C). Strikingly, in all cases where the chromosome was sufficiently distanced from the metaphase plate at anaphase onset to allow reliable assessment (n = 8), the misaligned chromosome travelled in its entirety to the nearest spindle pole (Fig 2D). Interestingly, kinetochore labelling revealed that misaligned chromosomes in KIF18A‐cKO Met‐II eggs were always non‐bioriented sister‐chromatid pairs (Fig 2A), and thus their delivery to only one daughter cell inevitably leads to aneuploidy. Notably, the frequency of anaphase lagging chromosome also increased in KIF18A‐cKO Met‐II eggs (Fig EV3C; 54.8% in KIF18A‐cKO vs. 15.1% in WT), suggesting KIF18A has dual role in preventing chromosome segregation errors in M‐II. Thus, KIF18A‐cKO eggs undergo anaphase despite misaligned chromosomes following parthenogenetic activation, suggesting an absence of a robust SAC in oocyte M‐II.
Small molecule‐induced spindle errors fail to activate the SAC in oocyte meiosis‐II
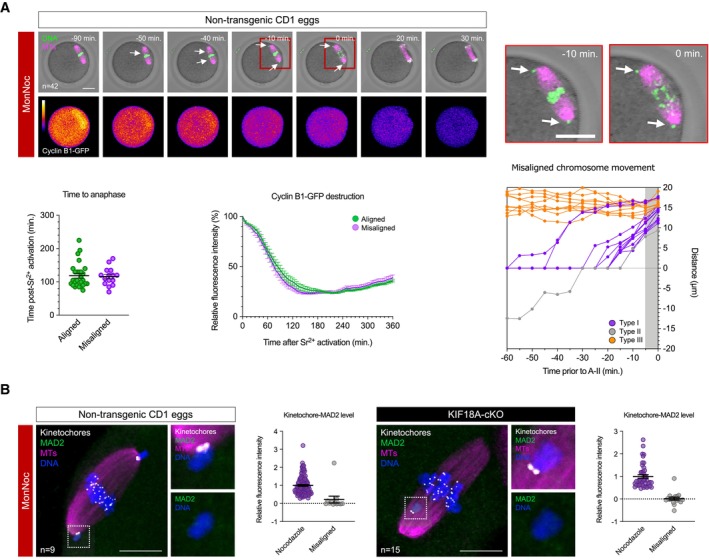
To exclude the possibility that the absence of a SAC response to misaligned chromosomes might be unique to KIF18A‐cKO Met‐II eggs, we also developed pharmacological approach to induce chromosome misalignment in normal (non‐transgenic) Met‐II eggs using consecutive monastrol and nocodazole treatments (MonNoc eggs). Whilst neither of the drugs alone produces individually misaligned chromosomes, 200 μM monastrol‐induced spindle collapse followed by monastrol wash‐out in a low concentration of nocodazole (10 nM) resulted in bipolar spindles that have misaligned pairs of sister chromatids in 35.7% of cases (Fig 3A; n = 15/42 eggs; 1.9 ± 0.3 misaligned chromosomes per egg). To assess the impact of misaligned chromosomes on meiotic exit, we parthenogenetically activated MonNoc eggs, imaging Cyclin B1‐GFP destruction as a means of determining APC activation, and monitored chromosome behaviour in M‐II. In MonNoc eggs, chromosomes were segregated even in eggs with clearly misaligned chromosomes at the time of anaphase onset, and misalignments had no impact on anaphase onset timing or Cyclin B1‐GFP destruction dynamics (Fig 3A), thus further supporting the observation that misaligned chromosomes do not activate the SAC in eggs. Thus, misaligned chromosomes fail to prevent meiotic exit not only in KIF18A‐cKO, but also in normal (non‐transgenic) Met‐II eggs.
Figure 3. Misaligned chromosomes fail to activate SAC in meiosis‐II.
-
ATime‐lapse confocal images show meiotic exit despite misaligned chromosomes in MonNoc Met‐II eggs. White arrows denote misaligned chromosomes and insets depict their behaviour during anaphase. Chromosomes are visualised with SiR‐DNA (green) and spindle with SPY555‐Tubulin (magenta). Bottom panels show changes in Cyclin B1‐GFP levels (colour gradient) over time. Indicated time (min) is relative to anaphase onset. Scale bars, 20 μm. Charts show no change in anaphase onset timing (unpaired t‐test, P = 0.79) or Cyclin B1‐GFP destruction dynamics (unpaired t‐test, P = 0.99) by misaligned chromosomes and three types of misaligned chromosome behaviour in MonNoc eggs. Error bars represent SEM. Data from total of 42 oocytes across 2 experimental days.
-
BMAD2 immunofluorescence (green) in MonNoc and KIF18A‐cKO Met‐II eggs with graphs showing quantification of kinetochore‐MAD2 signal. Note significantly lower levels of kinetochore‐MAD2 at misaligned chromosomes in MonNoc and KIF18A‐cKO eggs compared to eggs treated with nocodazole alone (Mann Whitney test, P < 0.0001 for both MonNoc and KIF18A‐cKO eggs). Chromosomes are labelled with Hoechst33342 (blue), whilst kinetochores (grey) and microtubules (magenta) are detected with CREST and β‐Tubulin antibodies, respectively. Scale bars, 10 μm. Error bars represent SEM. Data from totals of 9 and 15 oocytes across 3 experimental days, for MonNoc and KIF18A‐cKO groups, respectively.
Source data are available online for this figure.
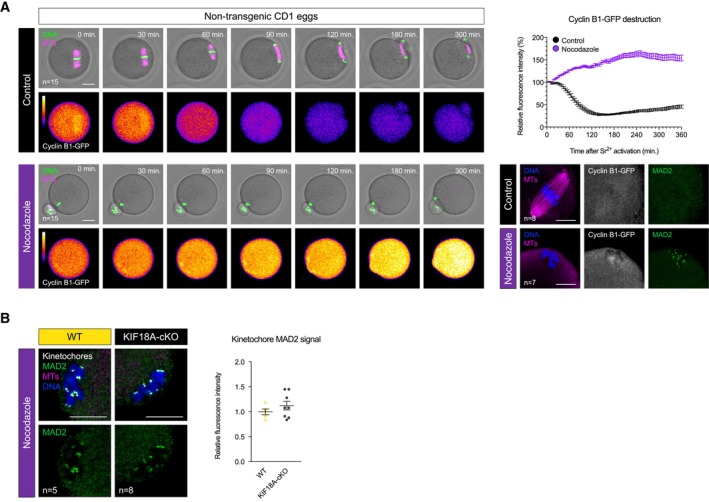
We next wondered why the SAC is insensitive to misaligned chromosomes in oocyte M‐II. We examined MAD2 localisation in both KIF18A‐cKO and MonNoc Met‐II eggs, and observed no MAD2 recruitment at the misaligned kinetochore pairs (Fig 3B). A high concentration of nocodazole (10 μM) to completely collapse the spindle caused kinetochore‐MAD2 recruitment as expected (Fig EV4A and B), and prevented Cyclin B1‐GFP destruction at egg activation (Fig EV4A). Thus, a severe spindle insult can activate the SAC to prevent M‐II completion suggesting the failure of misaligned chromosomes to recruit MAD2 in KIF18A‐cKO or MonNoc eggs is unlikely a simple absence of checkpoint machinery. Taken together, this suggests that although the SAC can be activated in extreme circumstances, small numbers of misaligned chromatid pairs generated by two entirely distinct approaches fail to recruit MAD2, permitting chromosome segregation errors and aneuploidy.
Figure EV4. Complete spindle disruption activates SAC in Met‐II eggs.
-
ATime‐lapse confocal images (left) show nocodazole‐treated and control non‐transgenic Met‐II eggs following Sr2+ activation. Note the lack of Cyclin B1‐GFP destruction and chromosome segregation in nocodazole group. Indicated time (min) is relative to Sr2+ addition. DNA is visualised with SiR‐DNA (green) and spindle with SPY555‐Tubulin (magenta). Scale bars, 20 μm. Graph (top right) shows changes in Cyclin B1‐GFP levels over time. Confocal images (bottom right) show kinetochore‐MAD2 recruitment following nocodazole addition. Chromosomes are labelled with Hoechst33342 (blue), and Cyclin B1‐GFP (grey) and microtubules (magenta) with chicken‐GFP and β‐Tubulin antibodies, respectively. Scale bars, 10 μm.
-
BMAD2 immunofluorescence (green) in nocodazole‐treated WT and KIF18A‐cKO Met‐II eggs. Chart shows no difference in kinetochore‐MAD2 levels between the groups (unpaired t‐test, P = 0.33). DNA is labelled with Hoechst33342 (blue), and kinetochores (grey) and microtubules (magenta) with CREST and β‐Tubulin antibodies, respectively. Scale bars, 10 μm.
Data information: In (A and B), ‘n’ is the total number of Met‐II eggs per group. Error bars represent SEM.
SAC ineffectiveness renders aged eggs error‐prone in M‐II
Misaligned chromosomes are rare in healthy oocytes from young females (Figs 2A and D and 4A; Pan et al, 2008; Shomper et al, 2014). However, several groups have shown that the incidence of misalignment is far more substantial in Met‐II eggs from aged mice (Liu & Keefe, 2008; Pan et al, 2008; Lister et al, 2010; Shomper et al, 2014), and indeed this mirrors the age‐related increase in misalignments in human Met‐II eggs (Battaglia et al, 1996; Volarcik et al, 1998). In mouse, it has been demonstrated that the misaligned chromosomes at Met‐II are prematurely individualised sister chromatids (Shomper et al, 2014; Yun et al, 2014), that result from cohesion loss that causes sisters to erroneously separate in M‐I (Sakakibara et al, 2015). This led us to hypothesise that whereas a weak SAC might be relatively inconsequential in young Met‐II eggs, SAC ineffectiveness in M‐II might render eggs unable to prevent random segregation of the individual sister chromatids that emerge in M‐II specifically in aged eggs.
Figure 4. SAC ineffectiveness permits chromosome segregation error at egg activation.
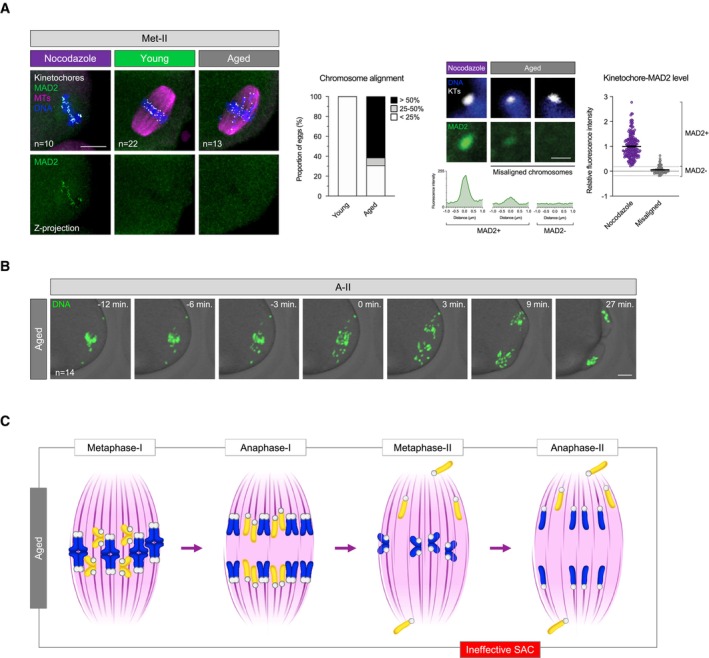
-
AMAD2 immunofluorescence (green) in young and aged Met‐II eggs (nocodazole‐treated young Met‐II eggs served as a positive control). Graph shows age‐related increase in proportion of Met‐II eggs with misaligned chromosomes (Fisher's Exact test, P < 0.0001). Exemplar weak MAD2+ and MAD2− misaligned chromatids are shown. Graph shows significantly lower levels of kinetochore‐MAD2 at misaligned chromatids in aged eggs compared to eggs treated with nocodazole alone (Mann Whitney, P < 0.0001). Chromosomes are labelled with Hoechst33342 (blue), kinetochores (grey) with CREST and microtubules (magenta) with β‐Tubulin antibody. Scale bars, 10 μm (left) and 1 μm (right). Error bars represent SEM. Data from totals of 10, 22 and 13 oocytes across 2 experimental days, for nocodazole, young and aged groups, respectively.
-
BTime‐lapse confocal images show aged Met‐II eggs successfully completing M‐II following Sr2+ activation despite chromosome misalignment. DNA is visualised with SiR‐DNA (green) and indicated time (min) is relative to the anaphase onset. Scale bars, 10 μm. ‘n’ is the total number of aged Met‐II eggs.
-
CSchematic shows a series of events during female meiosis that leads to age‐related aneuploidy in embryo. Age‐related cohesion loss leads to premature separation of homologue chromosomes and consequent biorientation of sister chromatids in meiosis‐I. This causes premature sister chromatid disjunction during anaphase‐I and the presence of individualised misaligned chromatids in Met‐II eggs. SAC ineffectiveness to detect misaligned chromosomes allows anaphase‐II entry and random segregation of individualised chromatids that results in embryo aneuploidy.
Source data are available online for this figure.
Consistent with previous reports, we observed significant increase in the rate of chromosome misalignment in aged Met‐II eggs (from mice aged 14–16 months) compared to young (2–3 months) (Fig 4A; 69.2% of aged vs. 0% of young eggs, Fisher's Exact test; P < 0.0001) and confirmed that the misalignments are indeed prematurely individualised sisters in all cases (Fig 4A; 7.8 ± 1.3 misaligned chromatids per egg). Strikingly, the majority of these misaligned individual chromatids had no detectable MAD2 signal at their kinetochores (57/70), whilst others had only a very weak signal (13/70) (Fig 4A). To directly determine whether aged Met‐II eggs possessing misaligned individual chromatids can complete M‐II, we monitored chromosome behaviour following egg activation (Fig 4B). We found that 78.6% of aged Met‐II eggs (n = 11/14) segregated chromosomes and divided in the presence of misaligned chromatids, all of which travelled to the nearest spindle pole during anaphase. Given that individualisation of sister chromatid pairs precedes M‐II spindle assembly (Yun et al, 2014; Sakakibara et al, 2015), their attachment to spindle microtubules is established independently, and so their segregation is random. Therefore, SAC insensitivity to misaligned chromosomes in M‐II renders aged eggs prone to chromosome segregation errors and puts them at great risk of aneuploidy.
An integrated view of oocyte aneuploidy across M‐I and M‐II
Previous studies have sought to determine the proportional contribution of chromosome segregation errors in M‐I and M‐II to the aneuploidy that leads to infertility (Angell, 1997; Hassold & Hunt, 2001; Fragouli et al, 2011; Handyside, 2012; Greaney et al, 2018). However, our data demonstrate that errors in the two meiotic divisions are inextricably linked. Elegant imaging experiments previously showed that ageing induces premature chromosome separation in oocyte M‐I, which not only predisposes oocytes to segregation errors in M‐I itself (Sakakibara et al, 2015), but also leads to prematurely individualised sister chromatids that have a tendency to misalign on the M‐II spindle (Shomper et al, 2014; Yun et al, 2014), but the fate of these individualised chromatids was unclear. Our data reveal an ineffective SAC in Met‐II eggs that permits M‐II completion despite chromosome misalignment (Fig 4C). In this way, individual sister chromatids that first emerged through premature sister separation in M‐I ultimately lead to aneuploidy at the completion of M‐II. Whilst the lack of a SAC response to chromosome misalignment is less pertinent in oocytes from young and healthy females, where chromosome cohesion is robust and the prolonged metaphase caused by CSF is ample for appropriate chromosome alignment and avoidance of aneuploidy, this becomes detrimental with advancing maternal age when individualised sisters are more common and the incidence of misalignment consequently increases (Fig 4A) (Liu & Keefe, 2008; Lister et al, 2010; Shomper et al, 2014). Our study therefore adds SAC weakness in M‐II as an important piece of the puzzle as to how age‐related defects contribute to oocyte and embryo aneuploidy and demonstrates that segregation errors in M‐I and M‐II are not independent but rather an integrated series of events that should be viewed as a whole in order to understand the aetiology of oocyte aneuploidy.
SAC fallibility is the norm in eggs and embryos
Whereas the SAC is nearly ubiquitous in somatic cells, and efficient to limit chromosome segregation errors, it has long been known that its function is idiosyncratic in early development. In lower vertebrates such as X. laevis and zebrafish, functional SAC is completely absent in oocytes and embryos and becomes acquired during development (Hara et al, 1980; Gerhart et al, 1984; Shao et al, 2013; Zhang et al, 2015). In C. elegans embryos, the SAC is present but its strength varies with cell size and fate (Galli & Morgan, 2016; Gerhold et al, 2018). In mammalian embryos, the SAC is ineffective since anaphase proceeds despite chromosome misalignment even in normal embryos (Vázquez‐Diez et al, 2019). Similarly, in mammalian oocyte M‐I, it is well established that chromosome segregation errors are not robustly prevented by the SAC (Gui & Homer, 2012; Kolano et al, 2012; Lane et al, 2012; Sebestova et al, 2012). This phenomenon remains poorly understood but could relate to a possible re‐purposing of the checkpoint to survey DNA damage in M‐I (Collins et al, 2015; Marangos et al, 2015; Lane et al, 2017). Our data in M‐II bridge the gap, revealing that the SAC is ineffective to prevent aneuploidies throughout all of oogenesis and early embryogenesis in mammals. It is worth underlining that the causes of SAC failure may be different at different stages of development. For example, whereas anaphase takes place despite MAD2 accumulation on the kinetochores of misaligned chromosomes in M‐I (Gui & Homer, 2012; Lane et al, 2012) and embryos (Vázquez‐Diez et al, 2019), MAD2 is apparently not recruited in Met‐II eggs. In somatic cells, MAD2 recruitment at the kinetochore occurs in response to Aurora B kinase‐mediated error correction and their consequent detachment from spindle microtubules (Santaguida et al, 2010). Surprisingly, we found that misaligned chromosomes tend to lack stable kinetochore‐MT attachments, and phosphorylated Aurora‐B targets are detected at misaligned kinetochores (data not shown), suggesting that failure to recruit and activate Aurora kinase is unlikely responsible for SAC activation failure. It is possible that unstable kinetochore attachment to spindle microtubules could satisfy the SAC in eggs and provide a potential explanation for SAC inability to respond to misaligned chromosomes. Nonetheless, we find here that SAC weakness is the norm in early mammalian development. Whether this provides some as‐yet unappreciated selective benefit (Ayala & Coluzzi, 2005; Sirard, 2011), or whether manipulations that might in the future reduce the toll of aneuploidy for some patients might increase reproductive success, remains to be seen.
Material and Methods
Animal models
All the animal work was performed in the animal facility of Centre Hospitalier de l'Université de Montréal (CRCHUM) and approved by the Comité Institutionnel de Protection des Animaux du CHUM (CIPA) under the protocol number IP18034GFs. All non‐transgenic CD1 mice were bred at either Charles Rivers Laboratories (Crl:CD1 [ICR], Raleigh, NC, USA) or Envigo (Hsd:ICR [CD‐1], Indianapolis, IN, USA), delivered to CRCHUM animal facility at 2 or 9 months of age, and used at 2–3 months (young) or 14–16 months (aged), respectively. The conditional Kif18a GFP knock‐in mouse reporter line was generated in Dr. Laura Reinholdt's laboratory (JR#27013, 129S‐Kif18a tm1.1Lgr /Lgr, see below). Zp3‐CreTg transgenic line (C57BL/6‐Tg(Zp3‐cre)93Knw/J, strain#: 003651) was obtained from The Jackson Laboratory (de Vries et al, 2000). All transgenic animals were crossed and bred at CRCHUM animal facility. Animals were maintained in individually ventilated cages (up to 5 animals/cage) at 22 ± 2°C and 40–60% humidity in 12 h light/dark cycle with lights on from 6:30 am to 6:30 pm and ad libitum access to food and water.
Generation of Kif18a GFP mouse reporter line
To create the conditional GFP N‐terminal tag allele, a BAC clone was obtained from TransgeneOmics (PMID: 18391959, PMID: 23417121, mouse Kif18a‐NFLAP (ENSMUSG00000027115, RP24‐307I16); MCB_4891). This clone contained a Kif18a‐NFLAP allele with an N‐terminal GFP fusion and a neomycin‐kanamycin resistance gene (neo) within an artificial intron flanked by loxP sites. Recombineering was used to retrieve the genomic interval from the BAC clone into a gene‐targeting vector for mESCs. The original neo cassette was replaced with a neo cassette flanked by frt sites to create a new single 5′ loxP site in the artificial intron and 3′ loxP was added to intron 2. The resulting targeting vector was electroporated into CJ7 mESCs (RRID:CVCL_C316, 129S1/SvImJ background), targeted clones were identified by loss of allele assays and confirmed by Southern blotting. Two correctly targeted clones were microinjected into C57BL/6J (RRID:IMSR_JAX:000664) embryos. Because the C57BL/6J genetic background was previously found to reduce penetrance of the Kif18a mutant infertility phenotype (PMID: 25824710), the breeding scheme for the chimeras was designed to minimise C57BL/6J. Chimeras were first bred to C57BL/6J females to identify males with germline transmission (GLT) of the targeted mESC genome by coat colour. One confirmed GLT male was then bred to 129S1/SvImJ (RRID:IMSR_JAX:002448) to establish the breeding colony. After two backcrosses to 129S1/SvImJ, N2 heterozygous males were bred to a FLP recombinase‐expressing strain from a 129 genetic background (RRID:IMSR_JAX:003946) to remove the frt‐neo cassette from the targeted allele. The resulting “GFPin” allele is shown in Fig EV2A and this allele was subsequently maintained through intercrossing and mice from this colony were shipped to CRCHUM for these studies. Sperm from a homozygous male were cryopreserved at The Jackson Laboratory as JR#27013, 129S‐Kif18a tm1.1Lgr /Lgr. The functionality of the conditional allele was tested by crossing 129S‐Kif18a tm1.1Lgr males to Sox2‐cre (RRID:IMSR_JAX:008454) females to excise the translational start site, as well as the coding portion of exon 2, and resulting heterozygous Kif18a KO mice were intercrossed. Heterozygous Kif18a KO mice were also crossed to heterozygous Kif18a gcd2 mice. Homozygous KO mice and compound heterozygous Kif18a KO /Kif18a gcd2 mice exhibited hypogonadism and infertility as described previously, confirming loss of function (PMID: 25824710, PMID: 16822657).
Genotyping
PCR genotyping was performed using MyTaq™ Extract‐PCR Kit (Meridian Bioscience, Cat# BIO‐21126) and MyTaq™ HS Red Mix (Meridian Bioscience, Cat# BIO‐25047) according to manufacturer's instructions. For animal genotyping, genomic DNA was extracted from the ear clips. For gamete genotyping, 10 GV‐stage oocytes retrieved from a single female were pooled together to extract DNA. To detect Kif18a alleles, two PCR assays were used with following primers: (i) KIF18A‐F: 5′‐CCAGTAATACATTTGCTAGGTGGAT‐3′ and KIF18A‐R: 5′‐GTAATGGCAATGCCCTGGGA ‐3′; (ii) KIF18A‐F: 5′‐CCAGTAATACATTTGCTAGGTGGAT‐3′ and GFP‐R: 5′‐TCCTTGAAGAAGATGGTGCG‐3′. The initial DNA denaturation was performed at 95°C for 3 min, followed by 35 cycles of amplification at 95°C for 15 s, 59°C for 15 s, 72°C for 1 min (for assay 1) or 20 s (for assay 2). In assay 1, the WT allele yields a 715 bp product, the GFPin allele a 1,990 bp product and KIF18A‐cKO allele a 627 bp product. In assay 2, GFPin allele yields 682 bp product. Myogenin‐specific primers were used as an internal positive control (MYOG‐F: 5′‐CCTGTAACCCAGGCAGAGAG‐3′ and MYOG‐R: 5′‐AGCCTAGGTAGGGACTAAGGG‐3′; yields 217 bp product). The following pair of primers was used to detect Cre transgene: Cre‐F: 5′‐GCGGTCTGGCAGTAAAAACTATC‐3′ and Cre‐R: 5′‐GTGAAACAGCATTGCTGTCACTT‐3′; 100 bp product). The initial DNA denaturation was done at 95°C for 3 min, followed by 35 cycles of amplification at 95°C for 15 s, 57°C for 15 s, 72°C for 20 s. Interleukin‐2 precursor‐specific primers were used as an internal positive control (IL‐2‐F: 5′‐ CTAGGCCACAGAATTGAAAGATCT‐3′ and IL‐2‐R: 5′‐ GTAGGTGGAAATTCTAGCATCATCC‐3′; 324 bp product).
Fertility trial
Sexually mature females (2 months of age) of three different genotypes: Zp3‐Cre Tg/0 ; Kif18a wt/wt (WT oocytes), Zp3‐Cre Tg/0 ; Kif18a wt/GFP (HET oocytes) and Zp3‐Cre Tg/0 ; Kif18a GFP/GFP (KIF18A‐cKO oocytes) were continuously mated with wild‐type B6D2F1/J (strain#: 100006) male mice of confirmed fertility (2 months of age; Jackson Laboratories) until 5 litters were born. Cages were checked daily for pups.
Oocytes and egg collection and handling
Fully grown, germinal vesicle (GV)‐stage oocytes were collected from 2 to 3 months (young) and 14–16 months (aged) old non‐transgenic CD1 or transgenic mice, 44–46 h following pregnant mare's serum gonadotrophin (PMSG; Genway Biotech, Cat. # GWB‐2AE30A) hormonal stimulation (5 and 10 IU of PMSG/mouse, respectively). Oocyte handling outside the incubator was done in M2 media (Sigma; Cat# M7167) and in the presence of 200 μM 3‐isobutyl‐1‐methylxanthine (IBMX; Sigma, Cat# I5879) to maintain them at GV stage. The surrounding cumulus cells have been removed and the oocytes washed through M16 media (Cat# M7292, Sigma) with 200 μM IBMX and left in the incubator at 37°C and 5% CO2 for approximately 1 h to recover prior any further manipulation. To obtain M‐II oocytes, females were stimulated with 5 IU of human chorionic gonadotropin (hCG; Sigma, Cat# CG10‐1VL) 48 h following PMSG and sacrificed 14 h post‐hCG. Cumulus‐oocyte complexes were released from the oviduct into M2 media containing 0.3 mg/ml hyaluronidase (Sigma; Cat# H4272) to dissociate the oocytes from cumulus cells. MII‐oocytes were then washed through M16 media and left in the incubator at 37°C and 5% CO2 prior any further manipulation.
Drug treatments
To disassemble the spindle in Figs 4A and EV4, eggs were exposed to 10 μM nocodazole (Cat# 487928, Sigma) either for 20 min prior fixation or during the entire live imaging. To induce chromosome misalignment in Fig 3, ovulated Met‐II oocytes (14–16 h post‐hCG stimulation) were exposed to consecutive 200 μM monastrol (Cat# 475879, Sigma) and 10 nM nocodazole treatments (each lasting 2 h), and live imaged in the presence of 10 nM nocodazole.
Microinjection
GV‐stage oocytes and ovulated eggs were microinjected in M2 media with or without 200 μM IBMX, using a picopump (World Precision Instruments), intracellular electrometer (Warner instruments), and micromanipulators (Narishige), mounted on a Leica DMI4000 inverted microscope. The mRNAs used for injections were in vitro synthesised using mMessage mMachine kits (T3 and T7, Cat# AM1348 and AM1344, Ambion), followed by poly‐adenylation using Poly(A) Tailing kit (Cat# AM1350, Invitrogen) according to the manufacturer's instructions. The following linearised plasmids were used as templates: pRN3: H2B‐RFP (a gift from Alex McDougall, Observatoire Océanologique de Villefranche sur Mer, Villefranche sur Mer, France; Prodon et al, 2010), pTALYM3B15 (Maj. Sat.‐mClover; Addgene #47878, Miyanari et al, 2013) and pCMX: Cyclin B1‐GFP (Addgene #26061, Hagting et al, 1999). Injected oocytes were left in the incubator for at least 2 h to allow the time for fluorescent protein expression.
Egg activation and live‐cell confocal imaging
For live‐imaging experiments in Figs 2C and D, and 4B, and EV3C, eggs were incubated in M16 media with 1 μM SiR‐DNA alone (Cat# CY‐SC007, Cytoskeleton Inc.) for 2 h to label the chromosomes or, in Figs 3A and EV4A, in combination with 1xSPY555‐Tubulin (Cat# CY‐SC203, Cytoskeleton Inc.) to label the spindle. For live imaging experiment in Fig 2B, eggs were incubated in 300 nM SiR‐Tubulin (Cat# CY‐SC002, Cytoskeleton Inc.) for 1 h to label the spindle. Eggs were parthenogenetically activated using 10 mM SrCl2 (Cat# 439665, Sigma) in Ca2+‐free M2 media and placed in an on‐stage incubating chamber (at 37°C) mounted on a Leica SP8 confocal microscope. Z‐stacks time‐lapse images were acquired with 20× air objective (0.75 NA) using 2 μm optical section and step size at 3 min time‐intervals for experiments in Figs 2C and D, and 4B, and EV3C and 5 min intervals in Figs 3A and EV4A. For Fig 2B, single optical sections were taken at 8 s time‐intervals. For the experiments in Figs 2B and 3A, and EV4A, the oocytes were illuminated with 488, 552, 638 nm light (using respectively, 0.5, 0.15 and 0.5% power in Fig 2B, and 1, 1 and 0.5% in Figs 3A and EV4A) and in Figs 2C and D, and 4B, and EV3C with 638 nm laser (0.5% power) and fluorescence was detected with a HyD detector.
Fixation and immunofluorescence
Oocytes were fixed using 2% paraformaldehyde (PFA; Cat# P6148, Sigma) in phosphate‐buffered saline (PBS; Cat# OXBR0014G, ThermoFisher) for 20 min, followed by permeabilisation step in 0.25% Triton‐X 100 (Cat# T9284, Sigma) in PBS for 10 min at ambient temperature. Blocking was done in 3% BSA (Cat# A7906, Sigma) in PBS for 1 h or overnight at 4°C and was followed by incubation step in antibodies for 1 h at 37°C. Primary antibodies were used at following dilutions: chicken anti‐GFP (1:500; Cat# GFP‐1020, Aves Labs), mouse anti‐β‐Tubulin (1:1,000; Cat# T4026, Sigma), rabbit anti‐PRC1 (1:200; Cat# 15617‐1‐AP, Proteintech), rabbit anti‐MAD2 (1:300; Cat# 924601, BioLegend), rabbit anti‐HEC1 (1:500; a gift from Robert Benezra, Memorial Sloan Kettering Cancer Center, New York, USA) and CREST (1:100; a gift from Marvin J. Fritzler, University of Calgary, Calgary, Canada). Secondary antibodies were used at following dilutions: goat anti‐mouse Alexa 488 (1:1,000; Cat# A‐11029, ThermoFisher), goat anti‐mouse Alexa 568 (1:1,000; Cat# A‐11031, ThermoFisher), goat anti‐mouse Alexa 633 (1:500; Cat# A‐21050, ThermoFisher), goat anti‐rabbit Alexa 568 (1:1,000; Cat# A‐11011, ThermoFisher), donkey anti‐rabbit Alexa 647 (1:1,000; Cat# A‐31573, ThermoFisher), donkey anti‐chicken Alexa 488 (1:500; Cat# 703‐545‐155, Jackson ImmunoResearch), donkey anti‐chicken Alexa 647 (1:500; Cat# 703‐605‐155, Jackson ImmunoResearch) and goat anti‐human Alexa 546 (1:1,000; Cat# A‐21089, ThermoFisher). Chromosomes were labelled with Hoechst 33342 (20 μg/ml; H1399, Invitrogen) for 20 min at ambient temperature. Imaging of fixed samples was carried out on a Leica SP8 confocal microscope using 63xoil‐immersion objective (1.4 NA) using 0.9 μm optical section and step size.
Microtubule turnover measurement
To measure the microtubule turnover in Fig EV3D, PAGFP‐Tubulin was photoactivated with 405 nm laser within a defined rectangular shaped region of interest positioned in between the metaphase plate and one of the spindle poles and live‐imaged at 30 s. time intervals for 5 min to assess the dissipation of the fluorescent signal. The fluorescence decay curves and MT half‐live (T 1/2) were obtained by plotting the fluorescence intensity decay obtained for each spindle against time and fitted into a double exponential curve ƒ(t) = A × exp(−k 1 t) + B × exp(−k 2 t) using the cftool in MATLAB, wherein t represents the time; A is the non‐stable interpolar MT population, B is the stable KT‐MT population and k 1 and k 2 are the decay rates of the two MT populations, respectively. The half‐life (T 1/2) of KT‐MT population was calculated as ln2/k 2. Each measurement was corrected for photobleaching, that was measured by imaging oocytes in the presence of 10 μM MT‐stabilising agent, Taxol (Cat# T7402, Sigma), where the MT‐turnover rate was minimal.
Image analysis and statistics
All image analysis was performed in ImageJ/Fiji (Schindelin et al, 2012) and Imaris (version 9.6.0, Bitplane). To assess spindle length, interkinetochore distance and chromosome alignment in Figs 1B and 2A, and 4A, and EV3A and B, spatial coordinates of the spindle poles and individual kinetochores were manually determined based on β‐Tubulin and CREST/HEC1 immunofluorescence signal, and distances calculated using Pythagoras' theorem. The extent of chromosome alignment was determined by calculating chromosome distance from each spindle pole (expressed as a relative distance from the spindle midpoint) and subdivided into three categories: aligned (< 25%), mildly misaligned (25–50%) and severely misaligned (> 50%). For Fig 1C, the germinal vesicle breakdown and polar body extrusion timing was determined by inspecting oocytes each 15 min within first 1.5 and 7–11 h following IBMX washout and the aneuploidy level was assessed by counting the number of kinetochores in fixed Met‐II eggs. For Fig 2B, chromosomes oscillatory movements were measured in the metaphase of meiosis‐II (in the period 12–18 h post‐IBMX release) based on centromeric Maj. Sat.‐mClover signal and normalised relative to spindle centroid (calculated in ImageJ/Fiji using SiR‐Tubulin fluorescence). For Figs 2C and 3A, values of chromosome metaphase displacement were obtained by measuring chromosome distance from the metaphase plate. For Fig 2D, misalignment at anaphase was determined based on the presence of chromosome optically separate from the metaphase plate at the time frame prior to anaphase onset. For Figs 2D and 3A, and EV3C, time to anaphase was calculated from the moment of Sr2+ addition to the first time‐frame when two main body masses of segregating chromosomes appeared optically separate from each other. For Figs 3B and 4A, and EV4B, kinetochore‐MAD2 immunofluorescent signal intensity was quantified by calculating the ratio between the average fluorescent signal (mean grey value) of individual kinetochores and cytoplasmic background (20 kinetochores per egg). For Fig EV2B, PRC1 signal and GFP‐KIF18A colocalisation assessment was done by drawing a line across the metaphase plate and plotting the fluorescent signal intensity profile in Fiji. For Fig EV3C, anaphase‐II chromosome was considered lagging if it was optically separate from the main body of segregating chromosomes when the spindle was oriented to be planar to the field of view. For Figs 3A and EV4A, CyclinB1‐GFP destruction was assessed by measuring the average cytoplasmic fluorescent signal intensity at the Z‐plane with the largest oocyte membrane perimeter over time. Statistical analysis was performed in GraphPad Prism8 using χ2‐test, unpaired or paired t‐test, Fisher's Exact test, Mann–Whitney test and one‐way ANOVA where appropriate. Error bars represent standard error of the mean (SEM).
Author contributions
Aleksandar I Mihajlović: Conceptualization; data curation; formal analysis; validation; investigation; visualization; writing – original draft; writing – review and editing. Candice Byers: Writing – original draft. Laura Reinholdt: Writing – original draft. Greg FitzHarris: Conceptualization; supervision; funding acquisition; validation; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Gaudeline Remillard‐Labrosse for excellent laboratory support and CRCHUM's transgenesis and animal modelling core facility for genotyping. We thank Anthony Hyman of the Max‐Planck Institute for Cell Biology and Genetics (CBG) for providing the BAC that was used to generate the targeted Kif18a allele, as well as the Genetic Engineering Technologies core at The Jackson Laboratory, and Anne Kneeland for colony management. We also thank Hugh Clarke, Gilles Hickson and John Carroll for critical reading of the manuscript. This research was supported by the grants to GF from Canadian Institute for Health Research (CIHR) and Foundation Jean‐Louis Levesque. AIM was supported by the Fonds de Recherche du Québec‐ Santé (FRQS) and McGill's Centre for Research in Reproduction and Development (CRRD) Postdoctoral Fellowships.
EMBO reports (2023) 24: e57227
Data availability
Our study includes no data deposited in public repositories.
References
- Angell RR (1991) Predivision in human oocytes at meiosis I: a mechanism for trisomy formation in man. Hum Genet 86: 383–387 [DOI] [PubMed] [Google Scholar]
- Angell R (1997) First‐meiotic‐division nondisjunction in human oocytes. Am J Hum Genet 61: 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell RR, Xian J, Keith J (1993) Chromosome anomalies in human oocytes in relation to age. Hum Reprod 8: 1047–1054 [DOI] [PubMed] [Google Scholar]
- Ayala FJ, Coluzzi M (2005) Chromosome speciation: humans, Drosophila, and mosquitoes. Proc Natl Acad Sci USA 102: 6535–6542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia DE, Goodwin P, Klein NA, Soules MR (1996) Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum Reprod 11: 2217–2222 [DOI] [PubMed] [Google Scholar]
- Ben‐David U, Amon A (2020) Context is everything: aneuploidy in cancer. Nat Rev Genet 21: 44–62 [DOI] [PubMed] [Google Scholar]
- Bennabi I, Terret M‐E, Verlhac M‐H (2016) Meiotic spindle assembly and chromosome segregation in oocytes. J Cell Biol 215: 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charalambous C, Webster A, Schuh M (2022) Aneuploidy in mammalian oocytes and the impact of maternal ageing. Nat Rev Mol Cell Biol 24: 27–44 [DOI] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA (2010) Evidence that weakened centromere cohesion is a leading cause of age‐related aneuploidy in oocytes. Curr Biol 20: 1522–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JK, Lane SIR, Merriman JA, Jones KT (2015) DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat Commun 6: 8553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton DA (2000) Spindle assembly in animal cells. Annu Rev Biochem 69: 95–114 [DOI] [PubMed] [Google Scholar]
- Dumont J, Desai A (2012) Acentrosomal spindle assembly and chromosome segregation during oocyte meiosis. Trends Cell Biol 22: 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan FE, Hornick JE, Lampson MA, Schultz RM, Shea LD, Woodruff TK (2012) Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 11: 1121–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragouli E, Alfarawati S, Goodall N‐N, Sánchez‐García JF, Colls P, Wells D (2011) The cytogenetics of polar bodies: insights into female meiosis and the diagnosis of aneuploidy. Mol Hum Reprod 17: 286–295 [DOI] [PubMed] [Google Scholar]
- Galli M, Morgan DO (2016) Cell size determines the strength of the spindle assembly checkpoint during embryonic development. Dev Cell 36: 344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J, Wu M, Kirschner M (1984) Cell cycle dynamics of an M‐phase‐specific cytoplasmic factor in Xenopus laevis oocytes and eggs. J Cell Biol 98: 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhold AR, Poupart V, Labbé J‐C, Maddox PS (2018) Spindle assembly checkpoint strength is linked to cell fate in the Caenorhabditis elegans embryo. Mol Biol Cell 29: 1435–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney J, Wei Z, Homer H (2018) Regulation of chromosome segregation in oocytes and the cellular basis for female meiotic errors. Hum Reprod Update 24: 135–161 [DOI] [PubMed] [Google Scholar]
- Gui L, Homer H (2012) Spindle assembly checkpoint signalling is uncoupled from chromosomal position in mouse oocytes. Development 139: 1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Jackman M, Simpson K, Pines J (1999) Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation‐dependent nuclear import signal. Curr Biol 9: 680–689 [DOI] [PubMed] [Google Scholar]
- Handyside AH (2012) Molecular origin of female meiotic aneuploidies. Biochim Biophys Acta 1822: 1913–1920 [DOI] [PubMed] [Google Scholar]
- Hara K, Tydeman P, Kirschner M (1980) A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc Natl Acad Sci USA 77: 462–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Hunt P (2001) To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2: 280–291 [DOI] [PubMed] [Google Scholar]
- Heald R, Khodjakov A (2015) Thirty years of search and capture: the complex simplicity of mitotic spindle assembly. J Cell Biol 211: 1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LME, Averink TV, Blomen VA, Brummelkamp TR, Medema RH, Raaijmakers JA (2018) Loss of Kif18A results in spindle assembly checkpoint activation at microtubule‐attached kinetochores. Curr Biol 28: 2685–2696.e4 [DOI] [PubMed] [Google Scholar]
- Kolano A, Brunet S, Silk AD, Cleveland DW, Verlhac M‐H (2012) Error‐prone mammalian female meiosis from silencing the spindle assembly checkpoint without normal interkinetochore tension. Proc Natl Acad Sci USA 109: E1858–E1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova A, Kitajima TS, Brismar H, Höög C (2019) Post‐metaphase correction of aberrant kinetochore‐microtubule attachments in mammalian eggs. EMBO Rep 20: e47905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyogoku H, Kitajima TS (2017) Large cytoplasm is linked to the error‐prone nature of oocytes. Dev Cell 41: 287–298.e4 [DOI] [PubMed] [Google Scholar]
- Lane SIR, Jones KT (2017) Chromosome biorientation and APC activity remain uncoupled in oocytes with reduced volume. J Cell Biol 216: 3949–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SIR, Yun Y, Jones KT (2012) Timing of anaphase‐promoting complex activation in mouse oocytes is predicted by microtubule‐kinetochore attachment but not by bivalent alignment or tension. Development 139: 1947–1955 [DOI] [PubMed] [Google Scholar]
- Lane SIR, Morgan SL, Wu T, Collins JK, Merriman JA, ElInati E, Turner JM, Jones KT (2017) DNA damage induces a kinetochore‐based ATM/ATR‐independent SAC arrest unique to the first meiotic division in mouse oocytes. Development 144: 3475–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara‐Gonzalez P, Westhorpe FG, Taylor SS (2012) The spindle assembly checkpoint. Curr Biol 22: R966–R980 [DOI] [PubMed] [Google Scholar]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y et al (2010) Age‐related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol 20: 1511–1521 [DOI] [PubMed] [Google Scholar]
- Liu L, Keefe DL (2008) Defective cohesin is associated with age‐dependent misaligned chromosomes in oocytes. Reprod Biomed Online 16: 103–112 [DOI] [PubMed] [Google Scholar]
- MacLennan M, Crichton JH, Playfoot CJ, Adams IR (2015) Oocyte development, meiosis and aneuploidy. Semin Cell Dev Biol 45: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madgwick S, Jones KT (2007) How eggs arrest at metaphase II: MPF stabilisation plus APC/C inhibition equals cytostatic factor. Cell Div 2: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangos P, Stevense M, Niaka K, Lagoudaki M, Nabti I, Jessberger R, Carroll J (2015) DNA damage‐induced metaphase I arrest is mediated by the spindle assembly checkpoint and maternal age. Nat Commun 6: 8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr MI, Hümmer S, Bormann J, Grüner T, Adio S, Woehlke G, Mayer TU (2007) The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr Biol 17: 488–498 [DOI] [PubMed] [Google Scholar]
- Merriman JA, Jennings PC, McLaughlin EA, Jones KT (2012) Effect of aging on superovulation efficiency, aneuploidy rates, and sister chromatid cohesion in mice aged up to 15 months. Biol Reprod 86: 49 [DOI] [PubMed] [Google Scholar]
- Mihajlović AI, FitzHarris G (2018) Segregating chromosomes in the mammalian oocyte. Curr Biol 28: R895–R907 [DOI] [PubMed] [Google Scholar]
- Miyanari Y, Ziegler‐Birling C, Torres‐Padilla M‐E (2013) Live visualization of chromatin dynamics with fluorescent TALEs. Nat Struct Mol Biol 20: 1321–1324 [DOI] [PubMed] [Google Scholar]
- Musacchio A (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 25: R1002–R1018 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED (2007) The spindle‐assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8: 379–393 [DOI] [PubMed] [Google Scholar]
- Pan H, Ma P, Zhu W, Schultz RM (2008) Age‐associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev Biol 316: 397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova T, Gorbsky GJ (2017) The consequences of chromosome segregation errors in mitosis and meiosis. Biology (Basel) 6: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodon F, Chenevert J, Hébras C, Dumollard R, Faure E, Gonzalez‐Garcia J, Nishida H, Sardet C, McDougall A (2010) Dual mechanism controls asymmetric spindle position in ascidian germ cell precursors. Development 137: 2011–2021 [DOI] [PubMed] [Google Scholar]
- Sakakibara Y, Hashimoto S, Nakaoka Y, Kouznetsova A, Höög C, Kitajima TS (2015) Bivalent separation into univalents precedes age‐related meiosis I errors in oocytes. Nat Commun 6: 7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190: 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebestova J, Danylevska A, Novakova L, Kubelka M, Anger M (2012) Lack of response to unaligned chromosomes in mammalian female gametes. Cell Cycle 11: 3011–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Li R, Ma C, Chen E, Liu XJ (2013) Xenopus oocyte meiosis lacks spindle assembly checkpoint control. J Cell Biol 201: 191–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomper M, Lappa C, FitzHarris G (2014) Kinetochore microtubule establishment is defective in oocytes from aged mice. Cell Cycle 13: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirard M‐A (2011) Is aneuploidy a defense mechanism to prevent maternity later in a woman's life. J Assist Reprod Genet 28: 209–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpff J, von Dassow G, Wagenbach M, Asbury C, Wordeman L (2008) The kinesin‐8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell 14: 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpff J, Wagenbach M, Franck A, Asbury CL, Wordeman L (2012) Kif18A and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension. Dev Cell 22: 1017–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez‐Diez C, Paim LMG, FitzHarris G (2019) Cell‐size‐independent spindle checkpoint failure underlies chromosome segregation error in mouse embryos. Curr Biol 29: 865–873.e3 [DOI] [PubMed] [Google Scholar]
- Volarcik K, Sheean L, Goldfarb J, Woods L, Abdul‐Karim FW, Hunt P (1998) The meiotic competence of in‐vitro matured human oocytes is influenced by donor age: evidence that folliculogenesis is compromised in the reproductively aged ovary. Hum Reprod 13: 154–160 [DOI] [PubMed] [Google Scholar]
- de Vries WN, Binns LT, Fancher KS, Dean J, Moore R, Kemler R, Knowles BB (2000) Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 26: 110–112 [PubMed] [Google Scholar]
- Walczak CE, Cai S, Khodjakov A (2010) Mechanisms of chromosome behaviour during mitosis. Nat Rev Mol Cell Biol 11: 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Y, Lane SIR, Jones KT (2014) Premature dyad separation in meiosis II is the major segregation error with maternal age in mouse oocytes. Development 141: 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Kothari P, Lampson MA (2015) Spindle assembly checkpoint acquisition at the mid‐blastula transition. PLoS One 10: e0119285 [DOI] [PMC free article] [PubMed] [Google Scholar]