

Abstract
Background –
Long QT syndrome (LQTS) has been associated with sudden cardiac death likely caused by early afterdepolarizations (EADs) and polymorphic ventricular tachycardias (PVTs). Suppressing the late sodium current (INaL) may counterbalance the reduced repolarization reserve in LQTS and prevent EADs and PVTs.
Methods –
We tested the effects of the selective INaL blocker GS967 on PVT induction in a transgenic rabbit model of LQTS type 2 (LQT2) using intact heart optical mapping, cellular electrophysiology and confocal Ca2+ imaging, and computer modeling.
Results –
GS967 reduced (ventricular fibrillation) VF induction under a rapid pacing protocol (n=7/14 hearts in control vs. 1/14 hearts at 100 nM) without altering APD or restitution and dispersion. GS967 suppressed PVT incidences by reducing Ca2+-mediated EADs and focal activity during isoproterenol perfusion (at 30 nM, n=7/12 and 100 nM n=8/12 hearts without EADs and PVTs). Confocal Ca2+ imaging of LQT2 myocytes revealed that GS967 shortened Ca2+ transient duration via accelerating Na+/Ca2+ exchanger (INCX)-mediated Ca2+ efflux from cytosol, thereby reducing EADs. Computer modeling revealed that INaL potentiates EADs in the LQT2 setting through 1) providing additional depolarizing currents during AP plateau phase, 2) increasing intracellular Na+ (Nai) that decreases the depolarizing INCX thereby suppressing the AP plateau and delaying the activation of slowly-activating delayed rectifier K+ channels (IKs), suggesting important roles of INaL in regulating Nai.
Conclusions –
Selective INaL blockade by GS967 prevents EADs and abolishes PVT in LQT2 rabbits by counterbalancing the reduced repolarization reserve and normalizing Nai.
Journal Subject Term: Arrhythmias, Ion Channels/Membrane Transport, Mechanisms
Keywords: long QT syndrome, ventricular tachycardia, electrophysiology, sodium channels, potassium channels, early afterdepolarization
Graphical Abstract
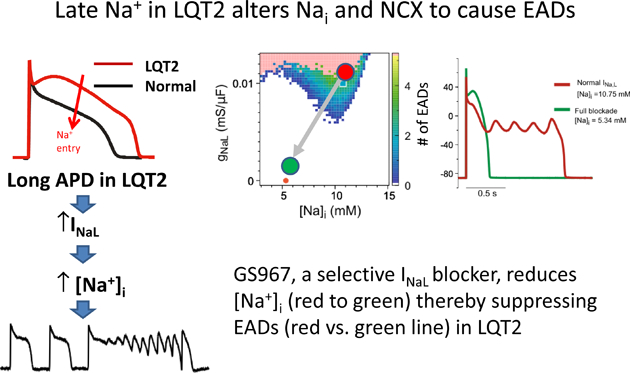
Introduction
Long QT syndrome type 2 (LQT2) is a congenital disease caused by the mutation of KCNH2 or the human ether-a-go-go related gene (hERG) α subunit, which encodes the rapid delayed rectifying potassium current (IKr)1, 2. The loss of function of IKr in LQT2 hearts leads to prolongation of action potential duration (APD), and is associated with polymorphic ventricular tachycardia (PVT) and sudden cardiac death (SCD).1
The hallmark of long QT syndrome is early afterdepolarizations (EADs).2 EADs occur in the setting of delayed repolarization, which promotes regenerative depolarizing currents.3, 4 The main ion current responsible for the depolarization phase of the EADs is the window current of L-type Ca2+ channel (ICaL).4 However, the late Na+ current (INaL) could also, along with the ICaL window current, promote EADs by providing additional depolarizing force during the plateau phase of action potentials (APs).5–8
INaL is carried by persistent openings (or re-openings) of small numbers of Na+ channels during the plateau phase of the AP. Although the amplitude of INaL is small (less than 0.1% of peak INa), its contribution to APD is not negligible5. Tetrodotoxin (TTX), known to block peak INa with a moderate preference for INaL (10 times higher selectivity to INaL)9, suppressed EADs in a drug-induced experimental model of LQTS.10 Ranolazine, an anti-anginal medication that preferentially blocks INaL, effectively reduced EADs and VTs in drug-induced animal models of LQT2 and LQT3.11, 12 GS-458967 (GS967), an INaL blocker with high specificity and selectivity for late vs peak INa, has been reported to abolish EADs and VTs in drug-induced animal models of LQT2 and LQT3 without deleterious effects.13 However, in a recent clinical trial (NCT02104583), the selective INaL blocker, GS6615, did not reduce the number of VTs in patients with implanted ICDs suggesting that INaL blockade should be tested in a genotype- and disease-specific context.
The purpose of the current study was to test the effect of the INaL blocker GS967 in suppressing EADs in a transgenic rabbit model of LQT2. LQT2 rabbits were previously shown to suffer SCD caused by frequent EADs and PVTs.14–16 EADs in LQT2 rabbits are associated with remodeling in Ca2+ handling, particularly in the SR Ca2+ release channel, ryanodine receptor (RyR2), which causes elevation of intracellular Ca2+ (Cai) during the plateau phase of APs, thereby promoting EADs.17 Previous studies suggested that inhibiting INaL can reduce Na+ overload, which can enhance Ca2+ exclusion from cytosol via Na+/Ca2+ exchanger (NCX1), thereby reducing Ca2+ overload and EADs.13, 18 Here, we used GS967 to selectively block INaL in LQT2 rabbits and found that GS967 suppressed EADs and PVTs through modulating NCX1 and IKs. This study suggests that the selective inhibition of INaL could be explored as a novel effective therapy for preventing arrhythmias in LQT2 syndrome.
Methods
The data, analytic methods, and study materials will be/have been made available to other researchers for purposes of reproducing the results or replicating the procedure. The source code of computer modeling is available at https://github.com/MingwangZhong/Late-Sodium.
Heart preparation and optical mapping
Littermate control (LMC, n=7 females) and transgenic rabbits (LQT2, n=9 females and 5 males), averaging 9 months old / 3.33kg body weight / 8.46g heart weight, were euthanized with buprenorphene (0.03 mg/kg IM), acepromazine (0.5 mg kg−1 IM), xylazene (15 mg kg−1 IM), ketamine (60 mg kg−1 IM), pentothal (35 mg kg−1 IV), and heparin (200 U kg−1). This investigation conformed to the current Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, revised 1996). Blebbistatin (5 μmol L−1) was perfused to reduce movement artifact.19 The AV node was ablated to control heart rate, and hearts were paced at 350 ms basic cycle length (CL).
GS967, selective late Na+ channel blocker, was provided from Gilead Science. The optical apparatus and analysis of AP have been previously described16 (see detail in the Data Supplement). All hearts (LMC, LQT2) underwent three stimulation protocols in the following order: 1) S1S2 protocol to reduce CL by 10 ms for APD restitution and tissue refractory period, 2) 10 min rest followed by ramp pacing in which CL was decreased by 10 ms until loss of 1:1 capture or induction of VF, and 3) a bolus injection of isoproterenol (ISO) into the bubble trap (~140 nM for 20 seconds) to induce EADs and PVTs. The stock solution of GS967 (1 mM) was prepared in DMSO13 and added to Tyrode’s solution to have the final concentrations of 30 nM and 100 nM (30 and 100 μL of stock solution to 1 L of Tyrodes’ solution). The stimulation and isoproterenol protocols were repeated and EAD/PVT events were monitored by optical mapping and ECG recording. The vehicle group was treated with 100 μL DMSO only.
Confocal Ca2+ imaging and INaL measurements from single myocytes
Detailed description of single cell Ca2+ imaging and voltage clamp experiments are available in the Supplemental Material. Cytosolic Ca2+ changes from isolated cardiomyocytes were monitored using a Leica SP5 confocal system using the cell-permeable Ca2+ indicator Rhod-2 AM (Life Technologies, Grand Island, NY).17 Myocytes were perfused with 50 nM ISO (10 min.) and paced at 0.5 Hz. The time of the decay to 25% of Ca2+ transient amplitude after the peak (t75) was used as a parameter describing the decay kinetics of Ca2+ transients in LQT2.17 The efflux of Ca2+ through NCX1 was assessed using the time of decay to 50% of peak amplitude of caffeine-induced Ca2+ transient (10 mM).
The INaL current was recorded at 34–36°C with Axopatch-200B amplifier, Digidata 1440A and pCalmp 10 (Axon Instruments). The sodium current was activated by 220 ms depolarizing pulses from −120 mV holding potential to +40 mV in 10 mV increments (see the Supplemental Material and Figure S1 for detail). For each cell, INaL was normalized to appropriate cell capacitance and expressed as the current density expressed in pA/pF.
Computational model of INaL
We used the rabbit ventricular myocyte detailed model of Zhong et al20 with dynamically changed Nai. We included the INaL equations described in21. Since the activation and inactivation time scales of the normal Na model are very small at voltage larger than −30 mV, we adopted the rapid kinetics approximation (m∞ = m h∞ = h and, Eq. 1). The expressions of m∞ and h∞ as a function of voltage (Figure S3 in the Supplemental Material) are quantitatively fitted with our patch-clamp measurements for LMC and LQT2 myocytes. The parameters of the late sodium model and the modified parameters of the detailed model are provided in the Supplemental Data.
Statistical Analysis
Continuous variables of optical mapping data are represented as mean ± standard deviation. The parameters of Ca2+ transients recorded from isolated myocytes are represented as mean ± SEM. The values of APD, APD dispersion, conduction velocity, alternans, and Ca2+ handling before and after GS967 were compared using ANOVA.
Results
APD characteristics in LMC and LQT2 with GS967
We first determined whether GS967 changes the basic electrophysiological characteristics of LQT2 rabbit hearts. Hearts were paced at 350 ms CL, and GS967 was added to the perfusate at 30 and 100 nM concentrations. At 350ms CL, GS967 did not have a significant effect on the APD or APD dispersion in either LMC or LQT2 rabbits (p=0.366 and 0.313 for APD and APD dispersion respectively, Figure 1). Figure 2A and 2B shows APD and conduction velocity (CV) restitution curves from LMC and LQT2 rabbits under the S1S2 protocol. The left and right ventricular refractory periods were similar before and after GS967 administration in LMC rabbits (panel A, Δrefractoriness = 1±21 and 6±5 ms at 30 and 100 nM, respectively). APD restitution curves in the range from the basic cycle length of 350 ms to tissue refractoriness were not affected by GS967 in either LMC or LQT2 hearts. However, tissue refractoriness was slightly prolonged in LQT2 hearts despite no change in APD under 350 ms CL pacing (the increase in refractoriness = 13±11 and 29±15 ms at 30 and 100 nM respectively, paired t-test, p < 0.05). CV was not affected by GS967 in the broad range of diastolic interval (DI, 50 – 150 ms) but showed a tendency towards slowing at shorter DIs in both LMC and LQT2 hearts (Figure 2B). Table 1 summarizes the changes in APD and CV before and after GS967. APDs did not show statistically significant differences among 0, 30, and 100 nM at CL=350 and 250 ms. CV was modestly reduced at short CL by 100 nM GS967 (from 0.68±0.04 to 0.59±0.06 m/s, p < 0.05; see Table 1).
Figure 1.
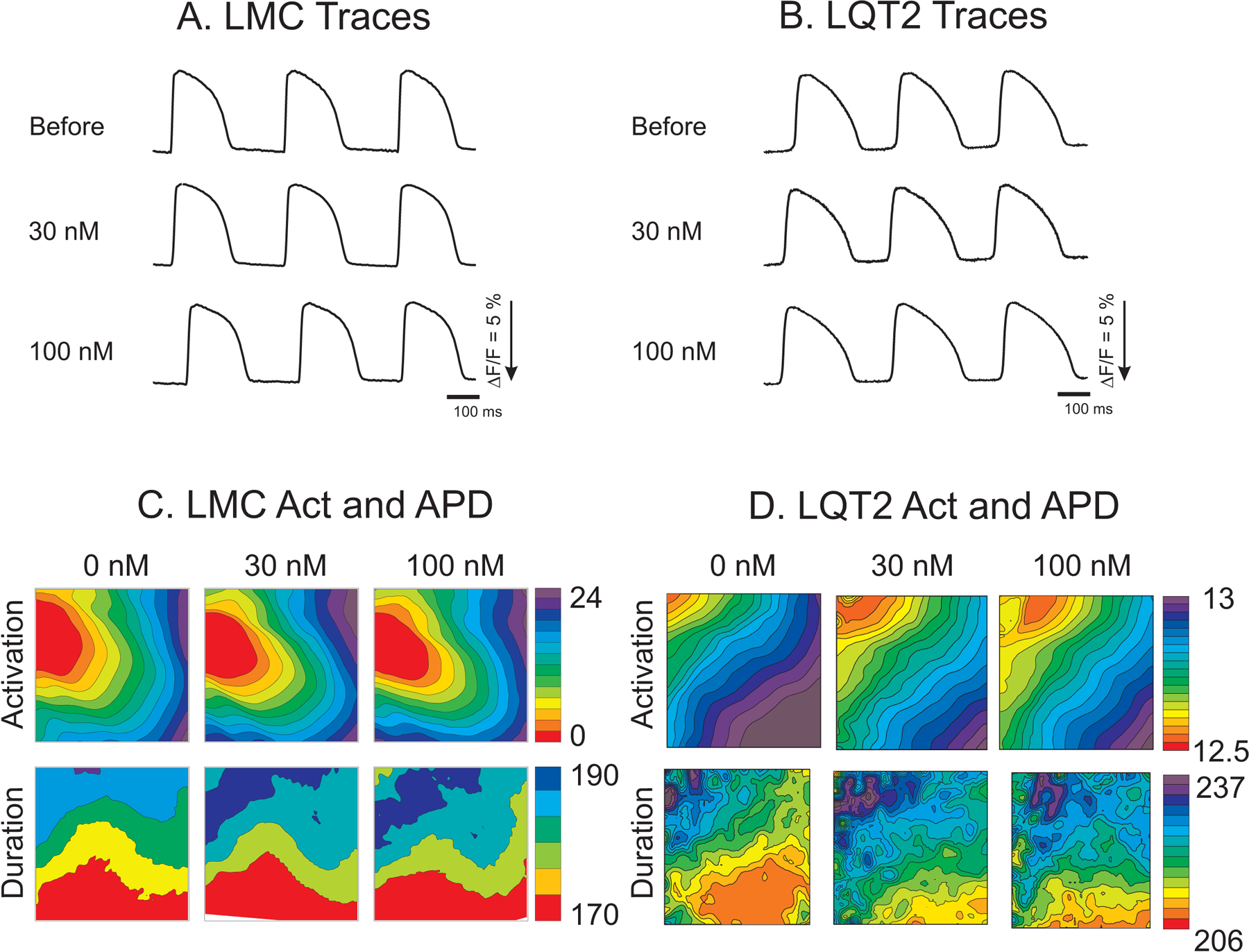
Action potential traces and maps of activation and duration. A & B) Action potential traces before and after GS967 from LMC and LQT2 hearts. C & D) Maps of activation and APD from LMC and LQT2 hearts in the absence and the presence of 30 and 100 nM GS967.
Figure 2.
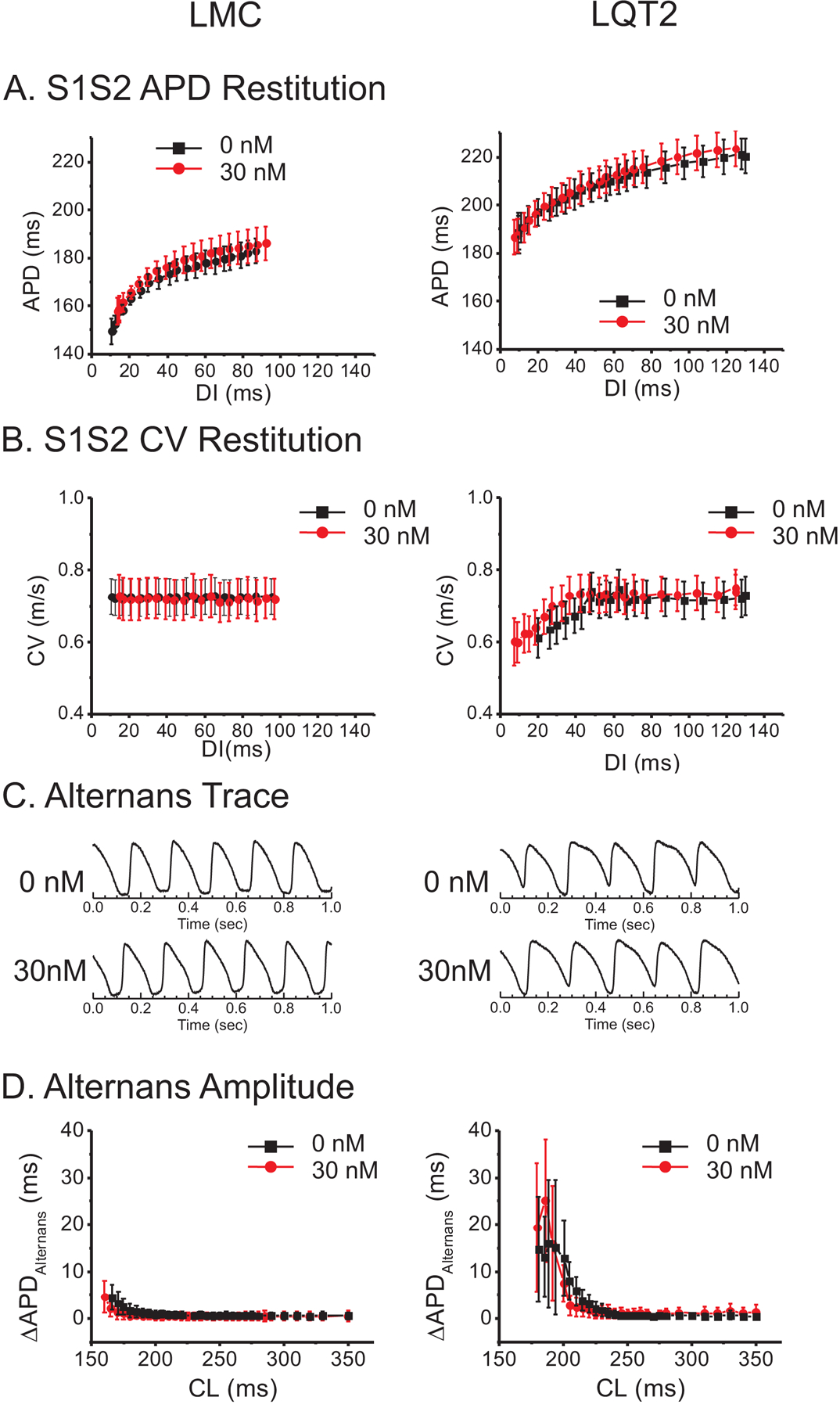
Lack of effect of GS967 on APD, CV restitution, or alternans in LMC (left column) and LQT2 hearts (right column). A) APD restitution in the absence and presence of 30 nM GS967. APD restitution curves did not show statistical differences in either LMC or LQT2 rabbits before or after GS967. Black; 0 nM, Red; 30 nM GS967. B) CV restitution from LMC and LQT2. The CV was not affected at CL=350 ms but showed slight reduction at short DIs. C) Sample traces of action potential alternans from LMC and LQT2 rabbits during ramp pacing. D) APD differences between odd and even beats (ΔAPDalternans) before (black) and after 30 nM GS967 (red).
Table 1.
APD and CV in the absence and presence of GS967 at CL = 350 ms and 250 ms in LQT2 rabbits.
| 0 nM | 30 nM | 100 nM | ||||
|---|---|---|---|---|---|---|
| CL | 350 ms | 250 ms | 350 ms | 250 ms | 350 ms | 250 ms |
| APD (ms) | 221±23 | 197 ± 12 | 237±27 | 203±15 | 227±31 | 201±8 |
| CV (m/s) | 0.75±0.06 | 0.68±0.04 | 0.75±0.03 | 0.64±0.03 | 0.74±0.03 | 0.59±0.06* |
Values are mean ± STDEV obtained from 7 hearts. APDs were not affected by GS967, but CVs were modestly reduced (13%) at fast CL (250 ms) by 100 nM GS967 (n=7, * indicates paired t-test, p<0.05 between 0 nM and 100 nM at CL=250 ms).
APD alternans and VF induction under GS967
We investigated the effect of GS967 on APD and conduction under a ramp pacing protocol in which the stimulation CL was sequentially reduced in 10-msec steps. The pacing protocol often elicited 2:1 block at shorter CL in the presence of 100 nM GS967; hence we focused on the 30 nM concentration for the ramp pacing protocol. Figure 2C shows representative traces of alternans from LMC and LQT2 hearts under the ramp pacing protocol. The amplitude of APD alternans (ΔAPDalternans) defined by |APDodd – APDeven| was greater in LQT2 hearts and sharply increased at short CL below 200 ms. ΔAPDalternans in the presence GS967 (red lines) shows a trend toward reduction under GS967 in both LMC and LQT2 hearts, particularly at short CL (panel D) but was not statistically significant. Of note, during the ramp pacing protocol, the episodes of VF were inducible in 7 of 13 LQT2 hearts (53.8%). At concentrations of 30 nM and 100 nM GS967, the incidence of VF induction during the ramp pacing was reduced to 5 (38.5%) and 1 (7.7%) out of 13 LQT2 hearts (fisher’s exact test between 0 and 100 nM GS967, p < 0.05, see Figure S2 in the Supplemental Material), respectively.
GS967 Prevented EAD and PVT Events in LQT2
We sought to determine whether GS967 could prevent EADs and PVT events at concentrations of 30 nM and 100 nM. A bolus injection of ISO to LQT2 hearts causes numerous EADs and PVTs (Figure 3). As shown in panel E, a total of 62 EAD/PVT events occurred among 12 LQT2 hearts. After adding GS967 to the perfusate, EAD/PVT events were markedly reduced (30 and 7 EAD/PVT events at 30 nM and 100 nM GS967, respectively). Note that the reduction in EAD/PVT events was associated with noticeable APD shortening by GS967 in the presence of ISO (panel D). This is in contrast to no noticeable changes in APD in the absence of ISO (see Figure 1 and Figure 2). GS967 at 30 nM prevented EADs in most LQT2 hearts (n=7 of 12 hearts), and 100 nM of GS967 further reduced EADs and PVTs in 1 out of 5 hearts compared with 30 nM (panel E). In 4 out of 12 LQT2 hearts the EADs led to the development of monomorphic VT in the presence of 100 nM GS967 (panel F). However, 300 nM GS967 suppressed these EADs and the VT in all hearts (averaged EAD/VT events per heart = 8.2±9.6, 4.7±7.2, 1.4±2.1, and 0.3±0.94 for 0, 30, 100, and 300 nM GS967, ANOVA test p<0.05 between 0 and 30, 100 nM, 300 nM). None of the LMC hearts (n=7) showed EAD/PVT events during ISO in the presence of 0, 30, or 100 nM GS967.
Figure 3.
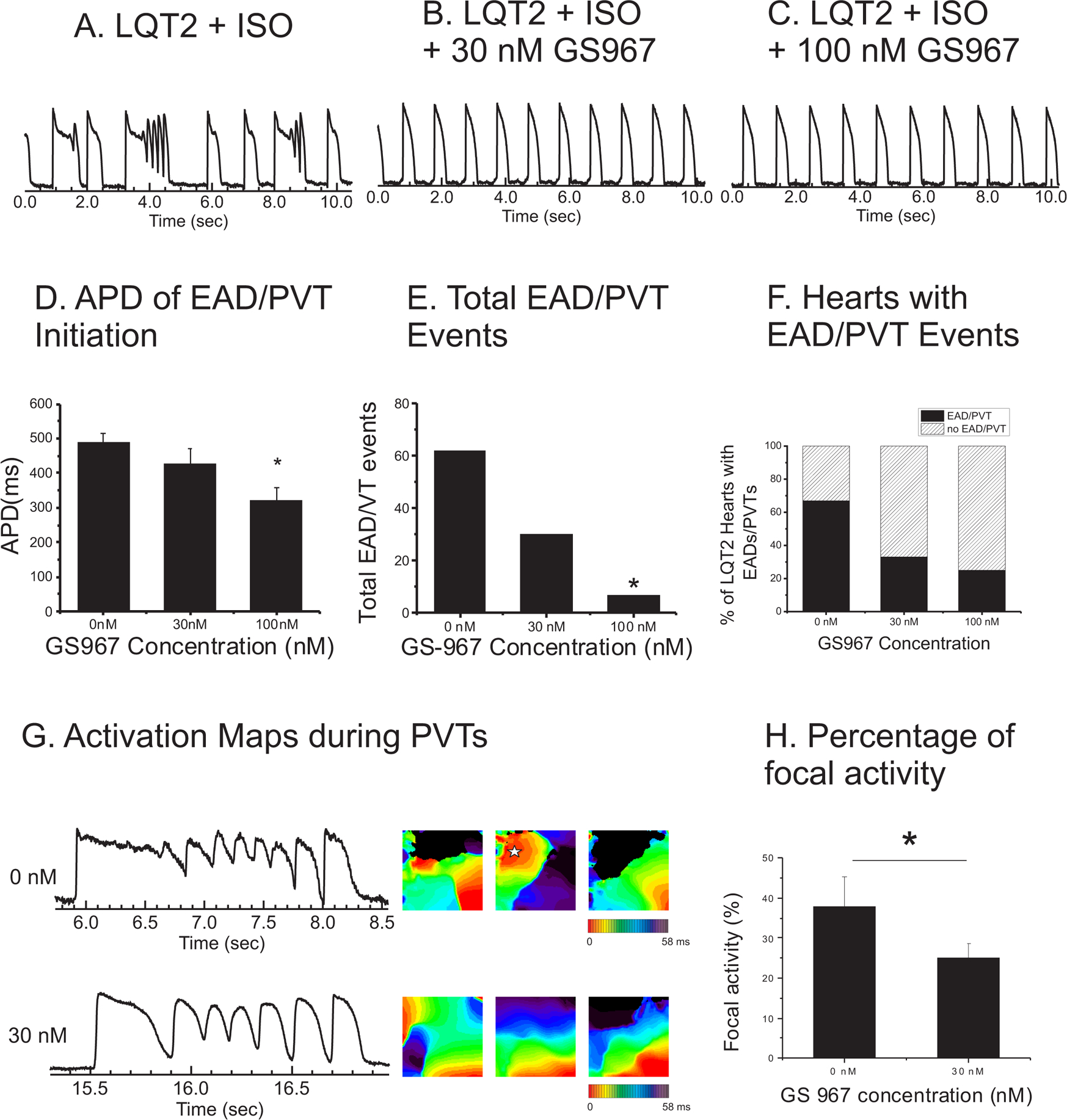
Prevention of EADs and pVTs in LQT2 rabbits by GS967. A-C) Representative traces of LQT2 AV-node-ablated heart with continuous pacing at 0.5 Hz in the absence and presence of 0 nM, 30 nM, and 100 nM GS967. D) APDs at 0.5 Hz after isoproterenol (n=12, 0 nM; 490.0±28.4 ms, 30 nM; 427.0±43.1 ms, 100 nM; 321.2±37.2 ms, p<0.05). E) Total number of EAD/PVT events in LQT2 rabbits (n=12) administered 0 nM, 30 nM, and 100 nM of GS967. F) Percentage of LQT2 rabbit hearts that showed EAD/PVT events at 0 nM, 30 nM, and 100 nM GS967 (8/12 at 0 nM, 5/12 at 30 nM, and 4/12 at 100 nM). G) Representative traces and activation maps in the absence and presence of 30 nM GS967 in LQT2 hearts. PVT in the absence of GS967 demonstrated complex activation patterns of mixed focal activity (star in the middle panel) and reentry. After 30 nM GS967 administration, PVT showed relatively repetitive activation patterns (panel G bottom activation maps and also see online supplementary movies S1 and S2). H) Reduced focal activity in 30 nM GS967 (t-test, p<0.05, n=10 hearts).
GS967 Suppresses EADs and Multi-focal Activity
We investigated the activation pattern and dynamics of PVTs before and after administration of GS967 to better understand its protective mechanisms. Due to the limited number of PVT events in the presence of 100 nM GS967, only PVT events that occurred in the presence of 30 nM GS967 were analyzed. Under the control condition (0 nM), PVTs showed complex wave dynamics with multiple focal activity and beat-to-beat changes in activation pattern (Figure 3G upper panels). The traces of PVTs show that Vm oscillates at a relatively high plateau phase under control conditions. However, GS967 at 30 nM (Figure 3B) markedly changed action potential shapes and activation patterns of PVTs, shortened APD of the first beat (from 596 ms to 296 ms, see Figure 3D and G), reduced small-amplitude Vm oscillations (panel G), and reduced the complexity in activation patterns of PVTs (panel G, activation maps). The activation maps during PVTs frequently showed focal activity (star in the 3rd activation map under 0 nM). However, focal activity was markedly reduced (panel H) under 30 nM GS967. This result indicates that the blockade of INaL by GS967 is beneficial by suppressing EADs and PVTs via reducing focal activity.
GS967 Normalizes Defective Ca2+ Homeostasis
A previous study by our group showed that abnormal Ca2+ handling in LQT2 myocytes potentiates EADs.17 Therefore, using confocal Ca2+ imaging of single myocytes, we investigated whether blocking INaL has a major impact on Ca2+ handling in LQT2 rabbits, thereby suppressing EADs in LQT2 hearts. Figure 4A shows representative traces of Ca2+ transients from LQT2 myocytes treated with ISO (50 nM, 10 min) in the presence (red) and absence (black) of GS967 (100 nM). GS967 accelerated Ca2+ transient decay (panel B), resulting in a significant shortening of Ca2+ transients without affecting Ca2+ transient amplitude.
Figure 4.
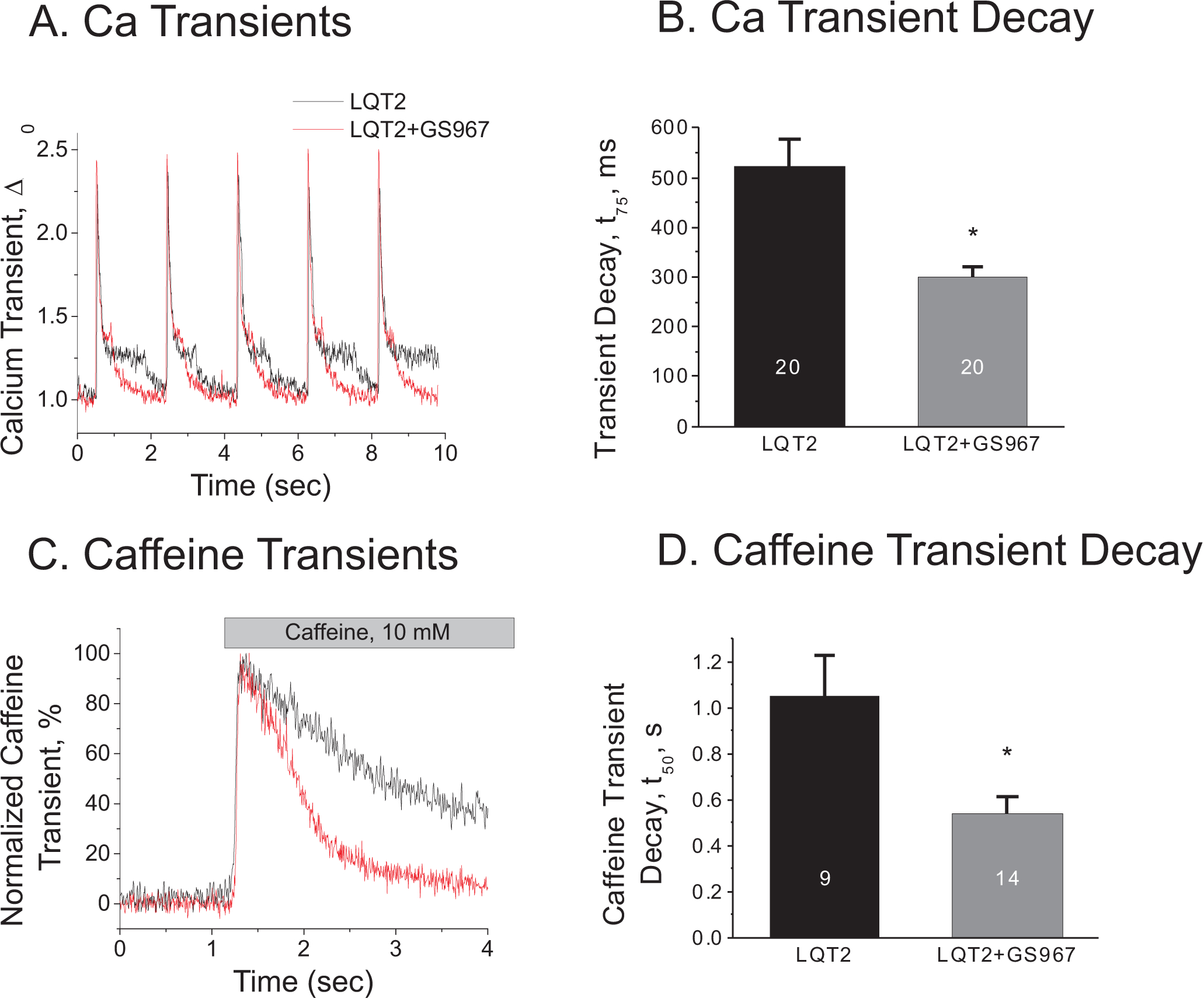
GS967 accelerates Ca2+ transient decay in LQT2 in the presence of 50 nM ISO. A) Representative Ca2+ transients recorded from isolated cardiac myocytes before (black) and after (red) 100 nM GS967 at CL = 2000 ms. B) Ca2+ transient decay at 0.5 Hz. (τ75=523.8±35.0 vs. 300.7±19.3 for 0 and 100 nM GS967 respectively. N=20 cells from 3 LQT2 hearts in each group, p < 0.05). C-D) Caffeine-induced Ca2+ release and its decay. GS967 (red) accelerated Ca2+ decay (τ50 =1.051±0.179 vs. 0.540±0.070 s for 0 and 100 nM GS967 respectively. n=9 and 14 cells from 3 LQT2 hearts. p<0.05).
Previous studies by Kornyeyev et al.18 showed that blocking INaL with GS967 reduces intracellular [Na+] (Nai), which increases the driving force of Na+ ions for NCX1, in turn accelerating Ca2+ extrusion. To verify that GS967 increases Ca2+ efflux through NCX1, Ca2+ release was elicited by 10 mM caffeine, and the decay rate of the caffeine-induced Ca2+ transient was measured as described in the Methods section. Panel C in Figure 4 shows representative traces of caffeine-elicited Ca2+ release. The amplitudes of caffeine-induced Ca2+ transients, i.e., the measure of SR Ca2+ content, were not statistically significantly (normalized caffeine transients = 97.9±7.3 vs. 121.0±11% for control and 100 nM GS967, p=0.07). However, the decay rate of caffeine transients was about twice as fast in the presence of GS967 (see Figure 4D), supporting the contention that accelerating NCX1 by GS967 can play an important role in the suppression of EADs in LQT2 rabbits.
INaL modulates APDs through altering Nai
Our experimental data shows that INaL blockade slightly affects APDs at physiological heart rate, but largely affects APDs at slow heart rate during which EADs occur, associated with suppressing PVTs robustly through altering Ca2+ handling in LQT2 rabbits. We next examined INaL in LQT2 rabbits using patch clamp technique (see supplemental material).64 Figure 5A and B shows representative traces of the GS967-sensitive current from isolated LQT2 myocytes during a depolarizing −20 mV pulse (panel A) under 50 nM isoproterenol and IV-curve of INaL.
Figure 5.
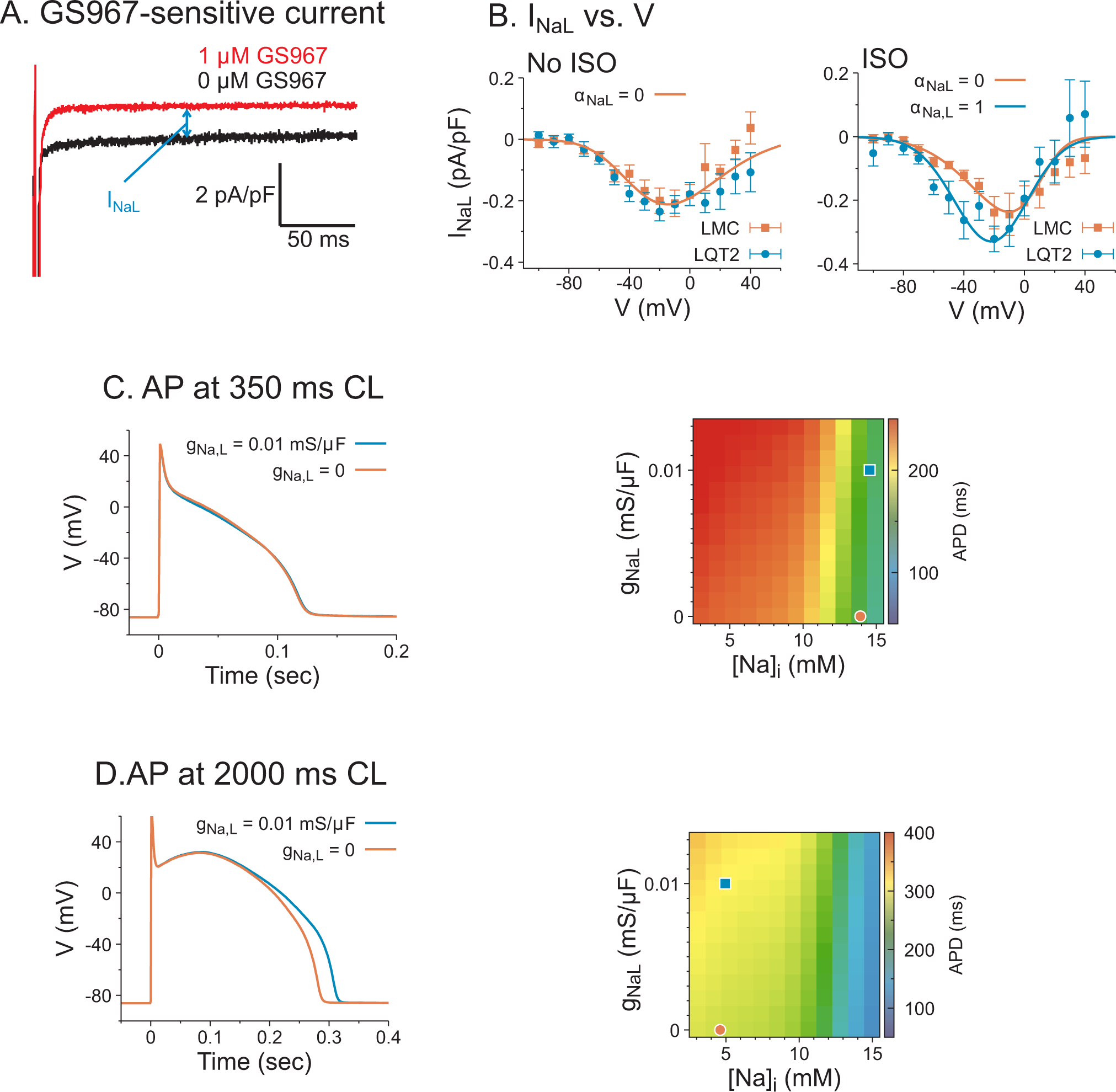
The effect of INaL on APDs in computer modeling. A) Representative traces of INaL by measuring GS967-sensitive current during a depolarizing 200 ms pulse to −20 mV from the holding potential −120 mV measured from LQT2 myocytes. B) The IV curve of INaL, which was fitted to equation (1). INaL has no significant difference between LMC and LQT2 when ISO was absent, and INaL was 32% greater in LQT2 than LMC when ISO was present (−0.32±0.11 vs −0.24±0.05 pA/pF, two way ANOVA, factor genotype, p=0.003). C) The effect of INaL on APDs at CL=350 ms in LQT2. Left panel: AP traces with gNaL=0 (red), 0.01 mS/μF (blue). Right panel: APDs in the parameter space of gNaL vs Nai at CL=350 ms. The red circle and blue square correspond to the results on the left panel. D) AP traces and APDs of computer simulation at CL=2 sec in LQT2 myocytes without ISO.
We then used computer modeling to investigate the influence of INaL on action potential dynamics. We developed an INaL equation25 similar to the Na channel model, with parameters chosen to fit the patch clamp data in panel B. The simulation results show that the effect of INaL on APDs is greatly dependent on Nai modulation. When the pacing cycle length is at 350 ms, the steady state Nai is around 14.5 mM and APDs are similar for gNaL = 0.01 mS/μF vs. 0, masking the effect of additional depolarizing currents by INaL (panel C) as seen experimentally in Table 1 and Figure 1. When the pacing cycle length is set to 2 sec, blockade of INaL (by reducing gNaL to 0) shortens APDs (panel D) as seen experimentally Figure 3D. These computational modeling results demonstrate the important roles of INaL regulating APD through modulating Nai. Similar behaviors were also observed in LMC myocytes (Figure S4 in the Supplemental Material).
INaL blockade prevents EADs through modulating Nai and INCX
To dissect the two potential contributions of INaL to EAD formation through Nai and additional depolarizing current, we implemented computer simulations with fixed Nai values, which are chosen from steady-states of simulations where Nai is unclamped (Figure S5 in the Supplemental Material). Figure 6A shows EAD formation in the parameter space of gNaL vs. Nai. Multiple EADs were observed when gNaL is greater than 0.007 mS/μF in the range of 7 ~ 12 mM Nai. The voltage traces corresponding to the squares and circles in Figure 6A are shown in Figure 6B, where the squares and thick lines represent normal INaL in LQT2 at different Nai levels, and the circles and thin lines represent fully blocked INaL (100 nM GS967). When Nai was fixed at 10.75 mM, the blockade of INaL eliminates EADs (blue thin line). The complete abolishment of EADs can also be obtained by reducing Nai to 5.34 mM (red thick line) or the combination of blocking INaL and reducing Nai. Note that when Nai is reduced to 5.34 mM, the plateau Vm is elevated (blue vs. red lines in panel B) due to changes in INCX. Panel D shows the outward NCX current (reverse mode, blue lines) at 10.75 mM Nai. INCX becomes inward current (forward mode, red lines) throughout plateau when Nai is reduced to 5.34 mM, which elevates Vm during the initial AP plateau phase, causing rapid activation of IKs (panel E, red lines) to repolarize APs quickly and prevent EADs. This computer modeling study emphasizes the important roles of INaL in Nai homeostasis: the blockade of INaL alleviates Nai overload in addition to depolarizing currents during AP plateau phase. The same mechanism was observed when INaL was reduced to the LMC level by reducing αNaL from 1 to 0 (Fig. S6 in the Supplemental Material).
Figure 6.
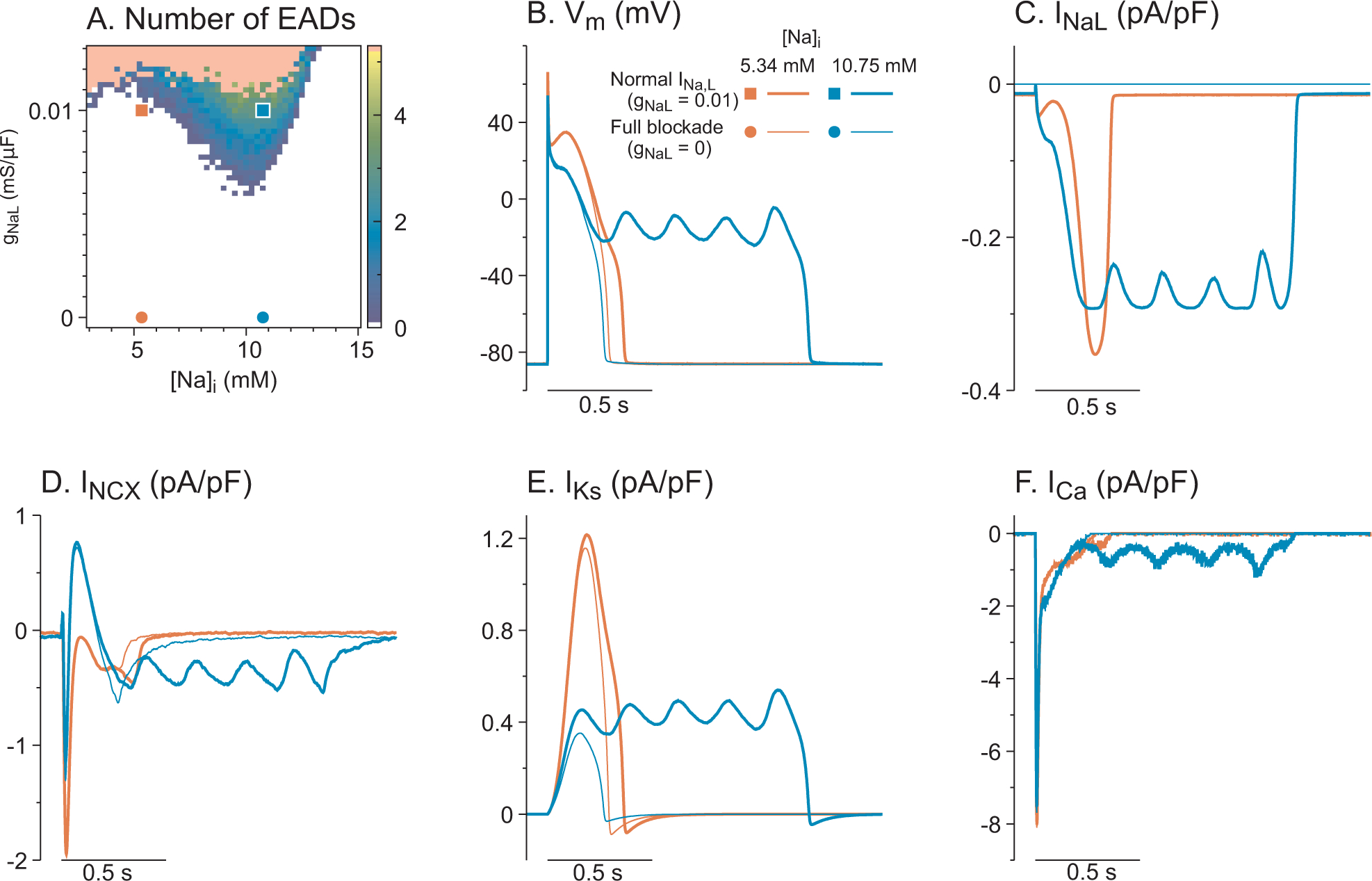
Computer modeling study of EADs reproduces INaL-dependent EADs in LQT2 myocytes with 50 nM ISO. A) EAD formation in the parameter space of gNaL vs. Nai. Representative traces corresponding to the squares and circles are shown in panels B-F. The Nai values are chosen from steady states for different αNaL values. The pink region represents infinite plateaus. B) Vm traces for different gNaL and Nai. Both lowering gNaL from 0.01 mS/μF (thick blue line) and reducing Nai from 10.75 mM to 5.34 mM eliminate EADs. C-F) Similar to panel B, INa, INCX, IKs, and ICa during pacing. Lowering Nai to 5.34 mM (blue lines) increases forward mode INCX, which elevates Vm during plateau and largely activates IKs, resulting in rapid repolarization without EADs.
Discussion
Targeting INaL to accelerate repolarization and suppress EADs has been tested in many different animal models of cardiac diseases.5–8, 13, 22–28 In the current study, we tested GS967, a selective INaL blocker, to explore its anti-arrhythmic potential in the transgenic rabbit model of LQT2. GS967 markedly reduced the incidence of EADs and PVTs in LQT2 rabbit hearts by increasing Ca2+ efflux. These anti-arrhythmic effects were accompanied by minimal changes in APD, CV, or their restitution kinetics, revealing the important role of INaL in LQT-related arrhythmias and inhibition of INaL as a potential therapeutic target for LQT2.
Proarrhythmic role of INaL in LQT2 rabbits
The proarrhythmic role of INaL has been implicated in many pathological conditions in which INaL is enhanced such as in the ischemic and/or failing heart and LQT3.7, 8, 11, 28 INaL is also thought to influence Vm and EAD genesis when repolarization reserve such as IKr is reduced.24, 29 Ranolazine, an antianginal drug, is known to block INaL preferentially relative to the peak of INa and effectively reduce arrhythmias.5 However, the efficacy of ranolazine has been interpreted with caution due to a) non-specific blockade such as IKr24, 30, b) use-dependent blockade of peak INa30 and c) anti-adrenergic effect.31. Studies with the more-selective INaL blocker GS967 showed promising results in preventing arrhythmia with negligible effects on peak INa and IKr.13, 26 In the present study testing GS967 on a transgenic rabbit model of LQT2, the results clearly demonstrated that INaL is an important contributor to triggering EADs and PVTs which can be effectively abolished by selective inhibition of INaL with GS967.
Despite effective suppression of EADs and PVTs by GS967, it is worth noting that in a few cases, the PVTs morphed into transient monomorphic VTs caused by transient single rotors in the field of view (Figure 3G and see supplemental movie S2). This phenomenon is most likely caused by GS967 suppression of EADs (and thereby multi-focal activity) allowing single reentry formation. In the structurally normal heart such as in LQT2 rabbits, this single rotor moved out of boundary to terminate the monomorphic VTs. Further studies are required to evaluate the advantages and risks for inhibiting INaL in other cardiac diseases.
Modulation of APDs by INaL through Nai
The minimal effect on APD under basal conditions and CV restitution is a major advantage of GS967. Earlier studies in rabbits reported that GS967 did not alter QRS duration or activation time in the wide range of physiological heart rates.26, 27 Our further examinations using S1S2 and the ramp pacing protocol revealed that APD and APD dispersion were not affected by GS967. Previous experiments in drug-induced LQT2 models using E4031 demonstrated that inhibition of INaL by ranolazine or GS967 shortens APDs, and this phenomenon was associated with suppression of EADs.29 GS967 also shortened APDs in transgenic LQT2 rabbit hearts but only during slow heart rate and ISO perfusion (Figure 3D) while APDs were not affected at normal heart rate. This heart-rate dependent APD shortening by GS967 in LQT2 rabbit hearts is most likely due to complex APD modulation by INaL through modulating Nai. Previous single cell and computer modeling studies32, 33 reported that the elevated Nai drives INCX and INaK in the outward direction during AP plateau phase to lower the AP plateau. Therefore, the overall effect of INaL blockade on APD will be the result of two competing effects, direct reduction of INaL and also indirect increase of INCX and INaK during the AP plateau. Since Nai increases with heart rate, the effect of Nai on APDs becomes greater at fast heart rate, compensating INaL’s direct impact on APDs. Therefore, INaL blockade by GS967 may have resulted in a negligible effect on APDs in LQT2 rabbit hearts unless the heart rate is slow as shown experimentally in Figure 1 and by computer simulation in Figure 5.
GS967 suppresses EADs via normalizing defective Na+ and Ca2+ homeostasis
Abnormal Ca2+ cycling has been suspected as a major contributing factor for triggering EADs in LQTS.34, 35 The proposed mechanism is that the prolonged APD in LQTS increases Ca2+ influx through L-type Ca2+ channels, resulting in increased intracellular Ca2+. Animal models of LQT2 with IKr blockers showed that Ca2+ elevation often preceded the upstrokes of EADs35 and depletion of SR Ca2+ suppressed EADs, supporting the role of Ca2+ in triggering EADs. Importantly, our previous study showed that unlike in drug-induced LQT2 animal models, cardiomyocytes from transgenic LQT2 rabbits exhibit no Ca2+ overload in SR.17 In this transgenic LQT2 model, Ca2+ transient amplitude is decreased in parallel with a significant decrease in SR Ca2+ content due to the increased SR Ca2+ loss mediated by hyperactive RyR2s. The increased propensity to generate Ca2+-dependent EADs in hereditary LQT2 has been linked to accelerated reactivation of RyR2 clusters after Ca2+ transient peak that via NCX1 contribute to prolonged maintenance of AP plateau at the voltage range suitable for reactivation of L-type Ca2+ channels.17
The importance of NCX1 in EAD genesis has been confirmed by numerous studies.36, 37 We previously reported that blocking NCX1 with SEA0400 abolished EADs in LQT2 myocytes,17 supporting its important role in EAD genesis. The activity of the NCX1 depends on parameters such as the transmembrane concentration gradients of Na+ and Ca2+, as well as the membrane potential. NCX1-mediated removal of [Ca2+] from the dyadic space has been shown to effectively suppress localized spontaneous Ca2+ releases from the RyR2 clusters.38 The present study indicates that the effective suppression of EADs by GS967 is associated with the accelerated NCX1-mediated extrusion of intracellular Ca2+ from dyadic space during the peak and recovery phase of Ca2+ transients.
The potential mechanism underlying the protective effect of GS967 may be related to the role of INaL in modulating Nai homeostasis,13, 18 thereby accelerating NCX1. The enhanced INaL in cardiomyopathy has been linked to Nai overload, leading cytosolic Ca2+overload and afterdepolarizations that can be abolished with ranolazine.39 A previous study by Kornyeyev et al.18 showed that prolongation of APD by E4031 slightly increases Nai (~ 1 mM change from 4 to 5 mM in rabbit myocytes). Since small changes in Nai can greatly alter NCX1 kinetics, it is possible that INaL blockade by GS967 is sufficient to lower Nai, which accelerates forward mode of NCX1 to increase AP plateau as well as activate IKs, thereby suppressing EADs. Further studies are needed to verify the role of INaL in modulating Nai homeostasis and NCX1 kinetics. Importantly, our transgenic LQT2 rabbit model shows major remodeling of cardiac excitation: 1) prolongation of APD in LQT2 rabbits is much smaller compared to drug-induced LQT2 model (18% vs. 40%), which is associated with downregulation of ICa and IKs,14 2) E4031 perfusion in isolated rabbit hearts triggers EADs and PVTs without sympathetic stimulation while transgenic LQT2 rabbits (similar to humans) did not show EADs and PVTs at baseline and required ISO or low K+ to trigger EADs or PVTs,15, 40 3) LQT2 rabbits have remodeling of Ca2+ handling proteins.17 The reduction in protein phosphatase 2 tethering to RyR complex in LQT2 rabbit myocytes result in hyperactive RyRs (leaky RyRs), leading to unique EAD genesis through sustained Ca2+ release during AP plateau phase and may potentially underlie diastolic dysfunction. It is possible that similar loss of PP2a17 due to posttranslational modifications which is known to increase INaL41 may have altered INaL to have higher efficacy of suppressing PVTs by GS967 through Nai regulation. Since multiple pathways can impact INaL such as glycosylation, phosphorylation on Tyr residues, Arg methylation, and mechanosensitivity (see the review by42), it is difficult to identify a single pathway that underlies INaL remodeling in LQT2 rabbits. Overall, these unique features of transgenic LQT2 rabbits may have caused EAD formation and PVT induction more sensitive to Nai and Ca2+ homeostasis shown in our computational modeling.
Conclusion
We found that the selective INaL blocker GS967 abolished EAD/PVT events in LQT2 rabbits. The inhibition of INaL by GS967 normalized Ca2+ homeostasis by accelerating cytosolic Ca2+ extrusion via NCX1 at the single-cell level. Our result suggests that INaL is an important current modulating Ca2+ cycling and bi-excitability, and a potential therapeutic target to prevent triggered activity and PVT events in LQTS. However, transient reentry formation during suppression of focal activity by GS967 could be proarrhythmic under certain circumstances
Supplementary Material
What is known?
Long-QT syndrome type 2 (LQT2), an inherited disease caused by loss-of-function mutations in KCNH2 (hERG) underlying rapidly activating delayed rectifier K+ current (IKr), is associated with sudden cardiac arrest triggered by early afterdepolarizations (EADs).
The late Na+ current (INaL) could also, along with the L-type Ca2+ current (ICaL), promote EADs by providing an additional depolarizing force during the plateau phase of action potentials (APs).
What the study adds?
It tests whether the selective INaL blocker GS967 can prevent EADs and polymorphic ventricular tachycardia (PVT) in a transgenic rabbit model of LQT2.
It shows that the inhibition of INaL by GS967 lessens [Na+]i overload. Decreased [Na+]i accelerates the forward mode of the Na+/Ca2+ exchanger, which elevates action potential plateau Vm and then activates the slow activating delayed rectifier K+ current (IKs), thereby suppressing EADs and PVT in LQT2 rabbits.
It identifies important proarrhythmic roles of INaL via regulating [Na+]i and [Ca2+]i homeostasis in the LQT2 setting.
Sources of Funding:
This study was supported by funding from Gilead Science. The computational studies in this work was supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health R01HL110791 to G.K.
Nonstandard Abbreviations and Acronyms:
- AP
Action potential
- APD
action potential duration
- Cai
intracellular Ca2+ concentration
- CL
Cycle length
- CV
Conduction Velocity
- EAD
Early afterdepolarizations
- hERG
human ether-a-go-go gene
- ICaL
L-type Ca2+ current
- ICD
Implantable Cardioverter Defibrillator
- IKr
the rapidly activating delayed rectifying K+ current
- INa
Na+ current
- INaK
Na+/K+ ATPase current
- INaL
Late Na+ current
- INCX
Na+/Ca2+ exchanger
- ISO
Isoproterenol
- LMC
Littermate control
- LQT2
Long QT Syndrome Type 2
- LQTS
Long QT Syndrome
- Nai
Intracellular Na+ concentration
- PVT
Polymorphic Ventricular Tachycardia
- RyR
Ryanodine Receptor
- SCD
Sudden cardiac death
- VF
Ventricular Fibrillation
Footnotes
Disclosures: None
References:
- 1.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, et al. Spectrum of mutations in long-qt syndrome genes. Kvlqt1, herg, scn5a, kcne1, and kcne2. Circulation. 2000;102:1178–1185. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Bloise R, Crotti L. The long qt syndrome. Europace. 2001;3:16–27. [DOI] [PubMed] [Google Scholar]
- 3.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys J. 1995;68:949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.January CT, Riddle JM. Early afterdepolarizations: Mechanism of induction and block. A role for l-type ca2+ current. Circ Res. 1989;64:977–990. [DOI] [PubMed] [Google Scholar]
- 5.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart. 2006;92 Suppl 4:iv6–iv14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: Studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. [DOI] [PubMed] [Google Scholar]
- 7.Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovascular & hematological agents in medicinal chemistry. 2008;6:348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S169–S177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. [DOI] [PubMed] [Google Scholar]
- 10.Kaseda S, Gilmour RF Jr., Zipes DP. Depressant effect of magnesium on early afterdepolarizations and triggered activity induced by cesium, quinidine, and 4-aminopyridine in canine cardiac purkinje fibers. Am Heart J. 1989;118:458–466. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman ES. Use of ranolazine in long-qt syndrome type 3. J Cardiovasc Electrophysiol. 2008;19:1294–1295. [DOI] [PubMed] [Google Scholar]
- 12.Frommeyer G, Kaiser D, Uphaus T, Kaese S, Osada N, Rajamani S, Belardinelli L, Breithardt G, Eckardt L, Milberg P. Effect of ranolazine on ventricular repolarization in class iii antiarrhythmic drug-treated rabbits. Heart Rhythm. 2012;9:2051–2058. [DOI] [PubMed] [Google Scholar]
- 13.Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, Karpinski S, Li CH, Hu L, Li XJ, et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32. [DOI] [PubMed] [Google Scholar]
- 14.Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, et al. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long qt syndrome. J Clin Invest. 2008;118:2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu GX, Choi BR, Ziv O, Li W, de Lange E, Qu Z, Koren G. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic lqt1 and lqt2 rabbits. J Physiol. 2012;590:1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, Schofield L, Centracchio J, Zehender M, Peng X, et al. Estradiol promotes sudden cardiac death in transgenic long qt type 2 rabbits while progesterone is protective. Heart Rhythm. 2012;9:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley J, et al. Hyperphosphorylation of ryrs underlies triggered activity in transgenic rabbit model of lqt2 syndrome. Circ Res. 2014;115:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornyeyev D, El-Bizri N, Hirakawa R, Nguyen S, Viatchenko-Karpinski S, Yao L, Rajamani S, Belardinelli L. Contribution of the late sodium current to intracellular sodium and calcium overload in rabbit ventricular myocytes treated by anemone toxin. Am J Physiol Heart Circ Physiol. 2016;310:H426–435. [DOI] [PubMed] [Google Scholar]
- 19.Fedorov V, Lozinsky I, Sosunov E, Anyukhovsky E, Rosen M, Balke C, Efimov I. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. [DOI] [PubMed] [Google Scholar]
- 20.Zhong M, Rees CM, Terentyev D, Choi BR, Koren G, Karma A. Ncx-mediated subcellular ca(2+) dynamics underlying early afterdepolarizations in lqt2 cardiomyocytes. Biophys J. 2018;115:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trenor B, Cardona K, Gomez JF, Rajamani S, Ferrero JM, Jr., Belardinelli L, Saiz J. Simulation and mechanistic investigation of the arrhythmogenic role of the late sodium current in human heart failure. PLoS One. 2012;7:e32659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk qt-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. [DOI] [PubMed] [Google Scholar]
- 23.Wu L, Rajamani S, Shryock JC, Li H, Ruskin J, Antzelevitch C, Belardinelli L. Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res. 2008;77:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC, Belardinelli L. Late sodium current contributes to the reverse rate-dependent effect of ikr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. [DOI] [PubMed] [Google Scholar]
- 26.Sicouri S, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of the highly selective late sodium channel current blocker gs-458967. Heart Rhythm. 2013;10:1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang J, Belardinelli L, Rajamani S, Pfeiffer Z, Yang B, Kim T, Centrachio J, Schofield L, Peng X, Kodssidas K, et al. Novel late sodium current blocker, gs-458967, suppresses action potential duration alternans and early afterdepolarization in transgenic rabbit model of long qt type 2. Circulation. 2012;126:A15425. [Google Scholar]
- 28.Fraser H, Belardinelli L, Wang L, Light PE, McVeigh JJ, Clanachan AS. Ranolazine decreases diastolic calcium accumulation caused by atx-ii or ischemia in rat hearts. J Mol Cell Cardiol. 2006;41:1031–1038. [DOI] [PubMed] [Google Scholar]
- 29.Wu L, Rajamani S, Li H, January CT, Shryock JC, Belardinelli L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late na(+) current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–1057. [DOI] [PubMed] [Google Scholar]
- 30.Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao G, Walsh E, Shryock JC, Messina E, Wu Y, Zeng D, Xu X, Ochoa M, Baker SP, Hintze TH, et al. Antiadrenergic and hemodynamic effects of ranolazine in conscious dogs. J Cardiovasc Pharmacol. 2011;57:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faber GM, Rudy Y. Action potential and contractility changes in [na(+)](i) overloaded cardiac myocytes: A simulation study. Biophys J. 2000;78:2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc Res. 2000;46:376–392. [DOI] [PubMed] [Google Scholar]
- 35.Choi BR, Burton F, Salama G. Cytosolic ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long qt syndrome. J Physiol. 2002;543:615–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bogeholz N, Pauls P, Bauer BK, Schulte JS, Dechering DG, Frommeyer G, Kirchhefer U, Goldhaber JI, Muller FU, Eckardt L, et al. Suppression of early and late afterdepolarizations by heterozygous knockout of the na+/ca2+ exchanger in a murine model. Circ Arrhythm Electrophysiol. 2015;8:1210–1218. [DOI] [PubMed] [Google Scholar]
- 37.Jost N, Nagy N, Corici C, Kohajda Z, Horvath A, Acsai K, Biliczki P, Levijoki J, Pollesello P, Koskelainen T, et al. Orm-10103, a novel specific inhibitor of the na+/ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br J Pharmacol. 2013;170:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bovo E, de Tombe PP, Zima AV. The role of dyadic organization in regulation of sarcoplasmic reticulum ca(2+) handling during rest in rabbit ventricular myocytes. Biophys J. 2014;106:1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–584. [DOI] [PubMed] [Google Scholar]
- 40.Odening KE, Kirk M, Brunner M, Ziv O, Lorvidhaya P, Liu GX, Schofield L, Chaves L, Peng X, Zehender M, et al. Electrophysiological studies of transgenic long qt type 1 and type 2 rabbits reveal genotype-specific differences in ventricular refractoriness and his conduction. Am J Physiol Heart Circ Physiol. 2010;299:H643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Refaey M, Musa H, Murphy NP, Lubbers ER, Skaf M, Han M, Cavus O, Koenig SN, Wallace MJ, Gratz D, et al. Protein phosphatase 2a regulates cardiac na(+) channels. Circ Res. 2019;124:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horvath B, Bers DM. The late sodium current in heart failure: Pathophysiology and clinical relevance. ESC Heart Fail. 2014;1:26–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.