

Abstract
Metazoan development relies on intricate cell differentiation, communication, and migration pathways, which ensure proper formation of specialized cell types, tissues, and organs. These pathways are crucially controlled by ubiquitylation, a reversible post-translational modification that regulates the stability, activity, localization, or interaction landscape of substrate proteins. Specificity of ubiquitylation is ensured by E3 ligases, which bind substrates and co-operate with E1 and E2 enzymes to mediate ubiquitin transfer. Cullin3-RING ligases (CRL3s) are a large class of multi-subunit E3s that have emerged as important regulators of cell differentiation and development. In particular, recent evidence from human disease genetics, animal models, and mechanistic studies have established their involvement in the control of craniofacial and brain development. Here, we summarize regulatory principles of CRL3 assembly, substrate recruitment, and ubiquitylation that allow this class of E3s to fulfill their manifold functions in development. We further review our current mechanistic understanding of how specific CRL3 complexes orchestrate neuroectodermal differentiation and highlight diseases associated with their dysregulation. Based on evidence from human disease genetics, we propose that other unknown CRL3 complexes must help coordinate craniofacial and brain development and discuss how combining emerging strategies from the field of disease gene discovery with biochemical and human pluripotent stem cell approaches will likely facilitate their identification.
Keywords: ubiquitin, CRL, CRL3, BTB, craniofacial development, brain development, developmental diseases
Introduction: Cullin3-RING ubiquitin ligases regulate many aspects of human development
Metazoan development depends on highly complex cellular differentiation, communication, and migration pathways that enable an embryo to develop from a fertilized egg into a complex multicellular organism. To dynamically control and coordinate these pathways, embryos frequently employ ubiquitylation, a versatile posttranslational modification essential in all eukaryotes. Synthesized by an enzymatic cascade of E1, E2, and E3 enzymes and reversed by deubiquitylases, different ubiquitin modifications dynamically regulate the stability, function, localization, or interaction landscape of protein substrates that play key roles in intracellular signaling networks [1, 2]. In this sense, ubiquitylation is often referred to as a cellular code [3, 4] and research over the last four decades has been geared towards elucidating its underlying signaling principles. While many questions remain outstanding, it is firmly established that substrates can be modified with either one or several ubiquitin molecules (mono- and multimono-ubiquitylation) or with polymeric chains in which successive ubiquitin moieties are linked to specific lysine residues or the N-terminal methionine through isopeptide bonds (polyubiquitylation). These different ubiquitin modifications adopt unique topologies and mediate distinct substrate fates within cells. Well understood examples include mono- or multi-monoubiquitylation, which change interacting proteins of substrates and have important roles in gene expression, vesicular trafficking, and ribosome biogenesis [2, 5–7], K48- or K11-linked-polyubiquitylation, which target substrates for degradation by the 26S proteasome [8, 9], and K63- or M1-linked polyubiquitylation, which allow for formation of signaling complexes during NF-κB activation and DNA repair [10, 11]. In addition, K63-linked polyubiquitylation mediates autophagy-dependent degradation of protein aggregates and dysfunctional organelles [12].
Specificity of ubiquitylation is conferred by ubiquitin E3 ligases, which catalyze transfer of ubiquitin from E2 enzymes typically to lysine residues of substrates [2, 13]. The human genome encodes ~600 E3s that are divided into subclasses according to the domain used for E2 recruitment and mechanism of catalysis [14]. With ~350 members, Cullin-RING ubiquitin ligases (CRLs) are the most abundant family of E3s. CRL complexes have a modular architecture comprised of a cullin protein, which connect an E2-binding RING protein with an interchangeable substrate adaptor module [14–16]. There are seven cullin proteins that can serve as such scaffolds (CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5 and CUL7), which form CRL complexes (CRL1–7) [17–21]. Additionally, two other proteins contain cullin homology domains and form active ligase complexes: APC2 (as subunit of the ubiquitin E3 ligase APC/C [22]) and CUL9/PARC [23, 24].
Many CRLs play critical roles during developmental processes of metazoans [25–33]. In particular, emerging evidence from human genetic, animal, and mechanistic studies suggest a prevalent role of CRL3s in craniofacial and brain development [7, 34–36], the focus of this review. CUL3 is highly conserved in all eukaryotes and while its deletion is compatible with growth in yeast [37, 38], it has been shown to be essential in a variety of different multicellular model organisms. In C. elegans, loss of CUL3 functions leads to defects of cytokinesis in single cell embryos [39] and in mice, CUL3 deletion leads to early embryonic lethality [40]. CUL3 uses substrate adaptor proteins that contain BTB/POZ domains (hereafter referred to as BTB) [37, 41–43] (Fig 1A), which were originally identified as a conserved motif in the Drosphila melongaster Broad-Complex, tramtrack, bric à brack, and transcriptional regulators and in many pox virus zinc finger proteins [44]. Through these CUL3-BTB protein interactions, distinct CRL3s have been shown to ubiquitylate and regulate a wide range of substrates in key cellular processes including transcription [45, 46], translation [7], vesicular trafficking [47], mitosis [48], and migration [49] to orchestrate cellular differentiation and facilitate multicellular growth. It is therefore not surprising that dysregulation of CRL3-mediated ubiquitylation lies at the heart of many human pathologies such as cancer [17], hypertension [50], myopathies [51], retinal degradation [52], or skin fragility [53]. In particular, recent reports show that mutations in CUL3 and BTB adaptors cause and are associated with numerous neurodevelopmental or craniofacial conditions [7, 34–36], suggesting manifold roles of CRL3 ligases during neuroectodermal differentiation. We here propose that the ubiquitin E3 ligase CUL3, through dynamically associating with different BTB substrate adaptors, integrates multiple steps required for human craniofacial and brain development. We summarize regulatory mechanisms underlying CRL3 assembly, substrate recruitment, and ubiquitylation and review our current mechanistic understanding of how particular CRL3 complexes function in formation and maintenance of neuroectodermal tissues. We also discuss how emerging strategies from the field of human disease genetics will provide further insights into CRL3 function in craniofacial and brain development and facilitate identification of novel associated BTB adaptors.
Figure 1: Structure and assembly of CRL3s.
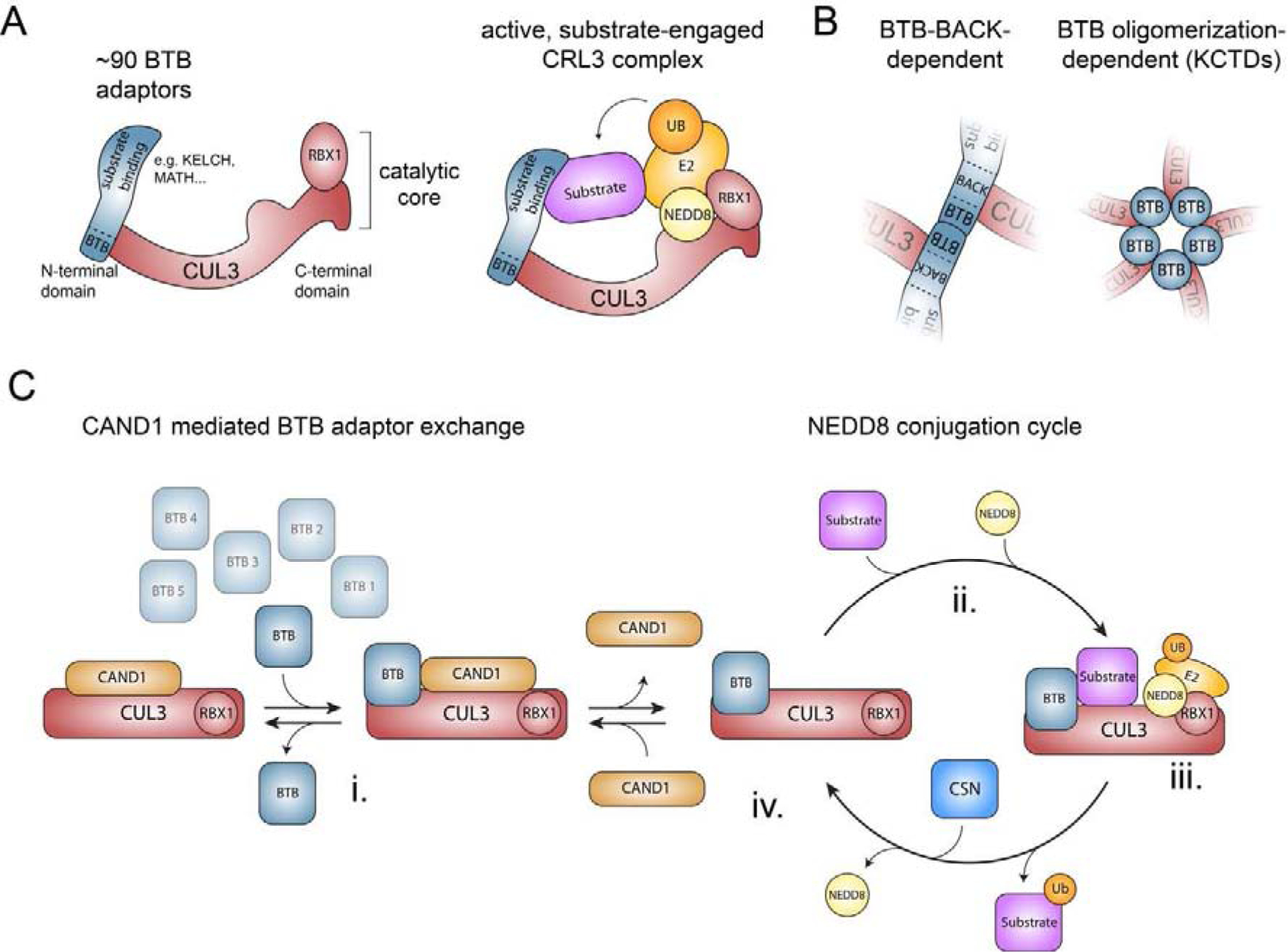
A) Schematic of the structure of a multi-subunit CRL3 complex. Left panel depicts a CRL3 that consists of CUL3 and the RING domain containing protein RBX1 (catalytic core) and one of ~90 interchangeable BTB proteins, which use their BTB domain to bind to the N-terminal domain of CUL3 and variable protein interaction domains to recruit specific sets of substrates. Right panel illustrates a model of an active, substrate-engaged CRL3 complex. Modification of CUL3 with NEDD8 at Lys-712 induces conformational changes and formation of ubiquitylation assemblies, in which RBX1 recruits an ubiquitin-charged E2 enzyme and the BTB protein position substrates for ubiquitin transfer. B) Schematic model of the two different modes how BTB proteins can interact with CUL3. C) Model of how CAND1-mediated BTB adaptor exchange and reversible NEDD8 modification are thought to regulate the cellular CRL3 repertoire (see text for details).
Structure and assembly of CRL3 E3s
Similar to all CRLs, CRL3 complexes are comprised of 3 modules: the scaffold cullin protein CUL3, the interchangeable substrate adaptor module given by BTB-domain containing proteins, and the RING-box protein RBX1 (Figure 1A). A unique aspect of CRL3 is that the BTB proteins directly bind to CUL3, while other cullins rely on a linker proteins to recruit their respective substrate adaptors. The catalytic core of CRL3s is formed through tight association of CUL3’s C-terminal cullin homology domain with RBX1, which is important for recruitment of E2 enzymes [16, 18]. Substrate adaptors are recruited to CUL3 via its N-terminal cullin repeats. This occurs through the BTB domain of the substrate adaptor, which recruits ubiquitylation targets through an adjacent protein-interaction domain.
The human genome encodes ~180 BTB domain-containing proteins, which are further subdivided into families that share common domain architecture. This defines the ZBTB family (BTB-zinc finger proteins), the KLHL/KBTBD family (BTB-BACK KELCH proteins), the T1/Kv family (T1 K+ channel integral membrane proteins), the KCTD family (potassium channel tetramerization domain proteins), the MATH-BTB family (BTB proteins with N-terminal MATH domain), the RhoBTB family (BTB proteins with Ras homology GTPase domain) and the BTB-only family (BTB domain proteins with no recognizable other domain) [44]. While it is unclear how many of these BTB proteins are used as substrate adaptors in cells, elegant biochemical and structural studies have identified several underlying molecular determinants. CUL3 can either recognize conserved motifs in the BTB and the immediately adjacent BACK domain [54–56] or interact with surfaces formed through adjacent BTB subunits in pentameric assemblies of KCTD family proteins [57, 58] (Figure 1B). It is thus thought that at least ~90 human BTB proteins can function as CUL3 adaptors with the largest class being the KLHL/KBTBDs, which use KELCH-repeat based β-propellers as substrate interaction domains [44].
The assembly of active CRL3 complexes competent to mediate ubiquitin transfer from E2 enzymes to substrates is thought to occur in multiple regulated steps. Research efforts by numerous groups have identified cullin modification by the posttranslational modifier NEDD8, the deneddylase activity of the COP9 signalosome (CSN), and the substrate adaptor exchange factor CAND1 as key factors that allow for controlled assembly and disassembly of CRL complexes in vitro and in cells [59]. Much of this work has focused on CRL1s and CRL4s [60–63]. However, given the shared ability of cullins to bind CAND1 and to be reversibly modified by NEDD8 [64], it is reasonable to presume that the core regulatory principles will be conserved amongst all CRLs. Based on this assumption, we here outline how reversible CRL3 assembly is thought to occur in cells (Figure 1C). Substrate-unbound BTB adaptors are in a dynamic binding equilibrium with CUL3/RBX1, a reaction that is catalyzed by the substrate adaptor exchange factor CAND1 [65, 66] (Figure 1Ci). Though rapid, this exchange reaction can be interrupted by substrate binding to a BTB adaptor (before or after binding to CUL3), which is followed by modification of CUL3 with NEDD8 (Figure 1Cii). Neddylation blocks CAND1 from displacing the BTB adaptor and stimulates substrate ubiquitylation by recruiting an E2 enzyme and coordinating conformational changes in the cullin protein to mediate formation of active ubiquitin ligation assemblies [67–69] (Figure 1Ciii). Once the ubiquitylated substrate is released, the CSN can gain access to the neddylated CRL3 complex and remove NEDD8 [70–74]. This renders the catalytic core CUL3/RBX1 available for CAND1-mediated recycling and interactions with further BTB adaptors (Figure 1Civ). One important feature of this adaptive exchange model is that substrate availability drives the formation of a particular CRL complex, which has been experimentally demonstrated for CRL1s and CRL4s [60–63]. Thus, substrates, reversible neddylation cycles, and CAND1-mediated BTB adaptor exchange are thought to collaborate to maintain the cellular demand for a particular CRL3 repertoire.
Regulatory principles underlying CRL3 substrate recruitment and ubiquitylation
To ensure proper function of the CRL3 family during human development and homeostasis, cells have the challenging task to build at least 90 distinct CRL3 complexes that appropriately recognize and ubiquitylate their cognate substrates. As described in detail in the previous section, two major strategies cells employ to solve this problem are reversible NEDD8 conjugation and CAND1-mediated substrate adaptor exchange, which are thought to allow for rapid and dynamic assembly of specific CRL3 complexes in a substrate-dependent manner. In addition, cells have evolved further layers of regulation of complex components that impinge on substrate recognition and ubiquitylation. Some of these regulatory layers have been shown to be particularly prevalent for CRL3s (e.g. transcriptional regulation [7] and dimerization/oligomerization [26]), while others impinge on all CRLs (e.g. regulated localization and posttranslational modifications [20]).
Transcriptional regulation of BTBs
Experiments in murine and human embryonic stem cells have shown that, while transcript levels of CUL3 and other substrate adaptors of other CRLs are relatively unchanged during differentiation, many CUL3-interacting BTB proteins exhibit differential mRNA expression [7, 75]. For instance, KBTBD8 expression is restricted to stem cells and is silenced once neural crest specification occurs. Conversely, expression of other BTB proteins is upregulated at stages of differentiation or in specialized cell types when they are functionally required. Examples for this include spermatogenesis (KLHL10 [76–78]), myogenic differentiation and muscle tissues (KBTBD5/KLHL40, KBTBD10/KLHL41, KBTBD13 [51, 79–81]), neural differentiation and neuronal function (KLHL16/gigaxonin, KCTD13, KCTD10 [82–85]), or kidney function (KLHL3 [50, 86]). Increasing and decreasing mRNA transcription is thus a common means by which cells alter substrate adaptor concentrations to promote assembly of particular CRL3 complexes in specific developmental stages or tissues.
Regulation of subcellular localization of BTBs
Targeted localization is a common mode of regulation in eukaryotic cells [87]. By directing a particular protein pool to a specific site, cells can accomplish rapid changes in local signaling. Studies in Drosophila melanogaster have shown that local activation of CRL3-KLHL10 is an important feature of spermatogenesis [77, 78, 88]. During terminal sperm differentiation, interconnected spermatids undergo a caspase-dependent process called “individualization”, during which individual spermatids are separated, elongated, and most of their cytoplasmic proteins and organelles are degraded [89]. Caspase activation during spermatogenesis is regulated by CRL3-KLHL10, which mediates polyubiquitylation and proteasomal degradation of the caspase inhibitor dBruce [76]. For this, KLHL10 and CUL3 are recruited to the outer membrane of mitochondria through binding of the β-subunit of the ATP-specific form of the Succinyl-CoA synthetase (A-Sβ). Interaction with A-Sβ at this site also stimulates CRL3-KLHL10-mediated ubiquitylation, thus ensuring local activation of caspase signaling that does not trigger cell death pathways [88]. Such local CRL3 activation has also been shown to be important for ensuring proper actin dynamics during cellular motility [90]. Conversely, regulated localization of BTB adaptors can also inhibit CRL3-dependent ubiquitylation by sequestering CRL3 complexes from substrates. This has been demonstrated during WNT signaling [91] and during IFN-induced apoptotic and autophagic cell death [92]. Thus, targeted localization of CRL3 adaptors can either promote or inhibit substrate recruitment and enables spatial restriction of ubiquitin signaling.
Post-translational modification of substrates and BTBs
Ubiquitin E3 ligases often control intracellular signaling nodes and integrate signals to drive developmental processes. A prevalent regulatory mechanism that allows CRLs to achieve this task is modulation of the adaptor - substrate interaction by post-translational modifications [20]. For CRL3s, there are several well studied examples how substrate recognition is facilitated or inhibited through stimulus-induced modifications (Figure 2A). For example, KBTBD8 only recognizes its targets, the ribosome biogenesis factors NOLC1 and TCOF1, upon their multi-site phosphorylation by CK2 [93]. This allows CRL3-KBTBD8 to convert a rise in embryonic CK2 into monoubiquitylation-dependent remodeling of translational networks that are required for neural crest specification (discussed in more detail below). Similarly, CRL3-KLHL20 relies on substrate phosphorylation to transduce neurotrophin signals into cytoskeletal changes during neuronal morphogenesis [94]. In other examples, phosphorylation can also be employed to inhibit CLR3 substrate recruitment. This has been demonstrated for the substrate adaptors KEAP1/KLHL19 and SPOP, for which phosphorylation in or near the recognition sites of their substrates have been shown to disrupt interaction [56, 95, 96]. However, physiological stimuli that elicit such phosphorylation-modulated recognition in cells remain to be established.
Figure 2: Regulatory principles underlying CRL3 function and substrate recruitment.
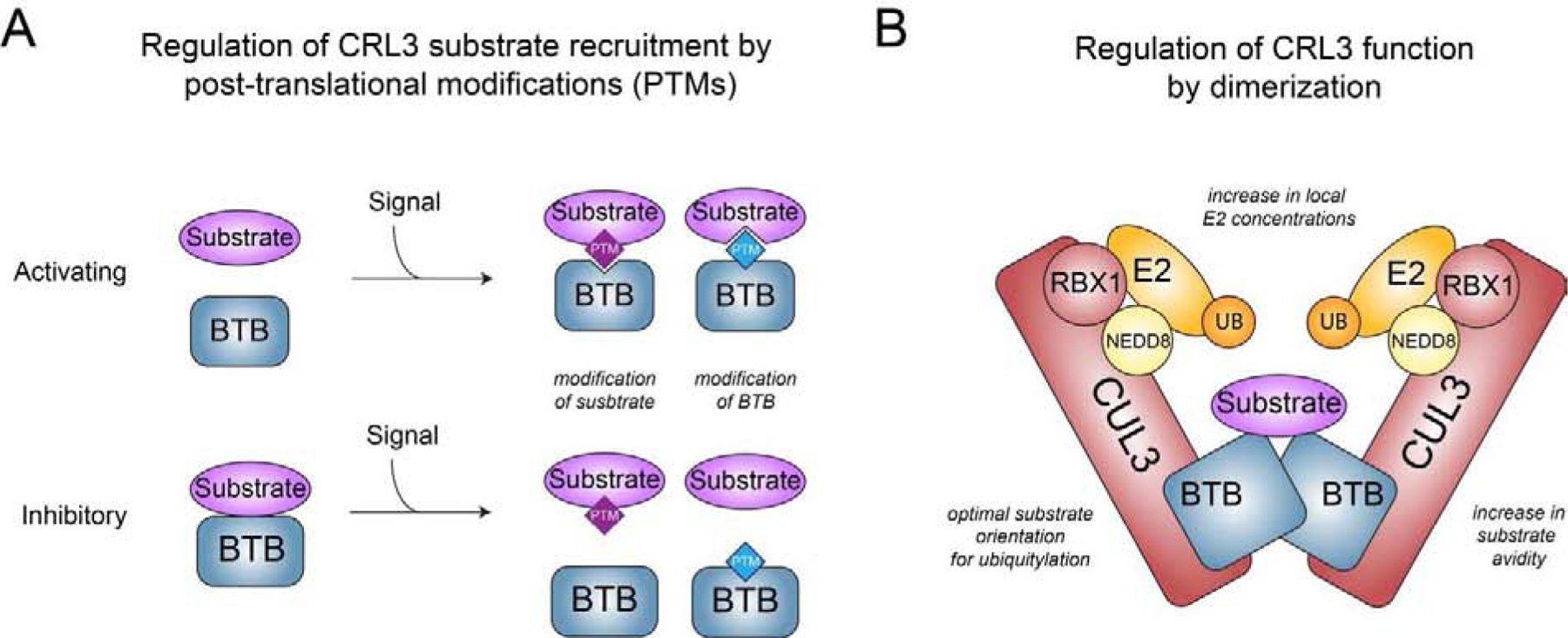
A) Scheme depicting how signal-induced posttranslational modification (PTMs, e.g. phosphorylation, glycosylation, cysteine modifications) of BTB adaptors or substrates can promote or inhibit interaction and thus CRL3 function. B) Model of a homo-dimeric CRL3 complex highlighting the different mechanisms how dimerization is thought to regulate CRL3 function.
In addition to phosphorylation other posttranslational modification are also employed to regulate CRL3 substrate recruitment. A well-studied example is given by CRL3-KEAP1, which constitutively ubiquitylates the transcription factor NRF2 to promote its proteasomal degradation [97, 98]. This reaction is prevented by cysteine modification of KEAP1 during oxidative stress likely through a combination of loss of both, KEAP1-NRF2 and CUL3-KEAP1 interaction [99]. More recently, nutrient-dependent O-GlcNAcylation of serine and threonine residues in BTB proteins has been shown to regulate interactions of BTB proteins with substrates [100] and CUL3 [101], respectively.
Taken together, recruitment of substrates to CRL3 complexes is modulated by a variety of posttranslational modifications of either adaptor or substrate, which allows CRL3s to convert a particular signaling input into downstream cellular responses.
Dimerization and Oligomerization of BTBs
Most, if not all, CRL3 complexes dimerize through the BTB domain of their substrate adaptors, a feature that has emerged as essential for the function of many distinct CRL3s [26]. Initial structural and biochemical studies have shown that homodimers of both KEAP1 and SPOP are able to engage with two binding sites within a single substrate molecule [56, 102–104]. This mode of interaction is thought to promote CRL3-dependent ubiquitylation by several mechanisms, including 1) facilitating optimal positioning of substrates for ubiquitylation [103], 2) increasing the avidity to substrates, thus strengthening binding and allowing recognition of substrates containing multiple suboptimal binding sites [56], and 3) increasing the local concentration of E2 enzymes [54–56] (Figure 2B). Accordingly, dimerization defective CRL3-KEAP1 and CRL3-SPOP are catalytically impaired in vitro and in cells [99, 105]. Recent studies have extended these observations to other CRL3s. Mutations in KLHL10 that impair dimerization are found in patients suffering from male infertility [78], suggesting an important role for dimerization during CRL3-KLHL10-mediated dBruce ubiquitylation (see above). In addition, the function of CRL3-KBTBD8 and CRL3-KLHL12 during neural crest specification requires homodimerization, which enables engagement of multiple binding sites in a single substrate [93] and binding of a substrate with help of a substrate-specific co-adaptor complex [106], respectively (see below). Heterodimeric CRL3 complexes formed through KBTBD6/KBTBD7 or KLHL9/KLHL13 have also been shown to exist and are important for regulating actin-based cell migration [90] and mitosis [107]; however, the underlying molecular details of how these heterodimers engage their substrates remain to be established. The important functions for self-association of CRL3s is highlighted by the existence of a dedicated dimerization quality control (DQC) pathway, which actively monitors BTB complex composition and can discriminate between functional and aberrant BTB dimers. This is achieved through the action of the ubiquitin E3 ligase CRL1-FBXL17, which is able to mediate polyubiquitylation and degradation of improperly assembled dimers through recognition of residues in the interface of BTB subunits that are usually buried in native BTB dimers [108]. As expected, DQC is important for faithful development, as demonstrated by injection of morpholinos targeted against FBXL17 in Xenopus tropicalis embryos. Interestingly, FBXL17-depeletion induced phenotypes manifested mainly during neural crest differentiation and nervous system development, suggesting a particularly important role for BTB dimerization and thus CRL3 function during neuroectodermal differentiation, as further discussed below.
In addition to homo- and heterodimers, CRL3 complexes have also been shown to engage in higher order oligomers. Several members of the KCTD family of BTB adaptors can form a range of oligomeric assemblies, most frequently involving 5:5 heterodecamers with CUL3 [57, 58]. Furthermore, the BTB and BACK domains of SPOP synergistically allow formation of linear, higher-order CRL3-SPOP oligomers. As with dimerization, formation of such higher order ubiquitin ligase self-assemblies is thought to increase both, the affinity to substrates via simultaneous binding of multiple recognition sites by avidity effects and the rate and processivity of ubiquitylation by increased local E2 concentrations. Indeed, as demonstrated for CRL3-SPOP, oligomerization enhances ubiquitylation of its substrates in vitro and in cells [109]. Interestingly, CLR3-SPOP oligomerization can be modulated through heterodimerization of SPOP with its closely related paralog SPOPL. SPOPL is not able to self-associate through its BACK domain and thus caps SPOP oligomers, which decreases ubiquitylation efficiency in vitro [55]. This offers an elegant mechanism of how CRL3-SPOP oligomerization and function could be regulated in cells. In addition, emerging evidence suggests that SPOP oligomers, through multivalent interactions with substrates containing multiple recognition sites, can undergo liquid-liquid phase separation above a particular saturation concentration in vitro and in cells [105]. Liquid-liquid phase separation is a critical mechanism that allows formation of membrane-less organelles or so called biomolecular condensates in cells [110, 111]. Indeed, above a particular saturation concentration CRL3-SPOP co-localizes with its substrate DAXX in nuclear speckles, where it likely mediates ubiquitylation and subsequent DAXX degradation [105]. As cancer-associated SPOP mutations interfere with phase separation and co-localization in membrane-less organelles, this seems to be a biologically relevant mechanism allowing to limit substrate concentrations to a particular threshold [112].
Taken together, self-association is an essential feature of many CRL3 complexes to ensure proper ubiquitin signaling during development.
Collaboration with different E2 and E3 enzymes
CRL3 complexes have been shown to modify substrates with a variety of different ubiquitin signals, including mono-, multi-mono- and different types of polyubiquitylation (recently summarized in [27]). This different outcome in target modifications is thought to occur through recruitment of specific E2 enzymes, with the UBCH5 and Cdc34 family of E2 enzymes being shown to work with CRL3 in vitro [14, 113, 114]. However, similar to other CRLs, the underlying mechanisms and sets of E2 enzymes that co-operate with specific CRL3s in cells have remained largely elusive. First insights into how different CRL3 complexes could mediate modification of substrates with different ubiquitin modifications derive from recent studies describing that the RBR ligase ARIH1 is a component of several human CRLs, including many CRL3s [115]. Elegant biochemical and cellular studies demonstrated that ARIH1 binds to neddylated CRL complexes and, together with the E2 enzyme UBCH7, facilitates monoubiquitylation of CRL substrates. The ubiquitin moiety on these CRL substrates could then be used for ubiquitin chain elongation; a process mediated by recruitment of the E2 enzyme CDC34 to the RING domain of the CRL. These findings offer the exciting possibilities that CRL3-mediated mono-ubiquitylation of substrates could be a generally achieved through ARIH1 and that ARIH1, through substrate priming, could also be involved in regulating CRL3-dependent polyubiquitin modifications. Future work will be required to test which of these mechanisms is at play for particular CRL3 complexes in cells and how chain elongation would be prevented for substrates whose fate relies on monoubiquitylation.
CRL3s integrate multiple steps to drive neuroectodermal development
During embryogenesis, ectoderm gives rise to neural progenitors, which ultimately develop into neurons and glia of the central nervous system, as well as neural crest cells that generate various cell types and tissues, including the peripheral nervous system and the facial cartilage and bone [116]. Thus, the formation of the nervous system and the craniofacial complex are highly interrelated, as further evidenced by the observation that neurodevelopmental disorders are often associated with craniofacial dysmorphisms [117]. There is mounting evidence that CRL3s are major players in regulating differentiation and maintenance of neuroectodermal tissues: Genetic analyses have identified high risk variants in CUL3 for autism [34, 118] and schizophrenia [36], and loss-of-function mutations in CUL3 can cause intellectual disability [35] (Figure 3A). This is further supported by studies demonstrating that brain region-specific deficiency of CUL3 in mice leads to behavioral problems and impaired neuronal function [119, 120]. A number of recent reports have also described functions and underlying mechanisms of particular CRL3 complexes during neural crest and neuronal differentiation and survival (Table 1). In addition, mutations in several BTB proteins have been shown to cause neurodevelopmental and craniofacial diseases (Table 1). A common theme that emerges from these studies is that mutations in BTB proteins frequently result in loss of protein function. This can occur through nonsense variants that create truncated proteins or through missense variants in functional domains that reduce BTB dimerization, CUL3 binding, or substrate recruitment (exemplified for KLHL16/gigaxonin in Figure 3B). We here summarize our current view of how distinct CRL3 complexes work together to ensure faithful craniofacial and nervous system development.
Figure 3: Mutations in CUL3 and BTB proteins lead to human diseases that affect the differentiation and maintenance of neuroectodermal tissues.
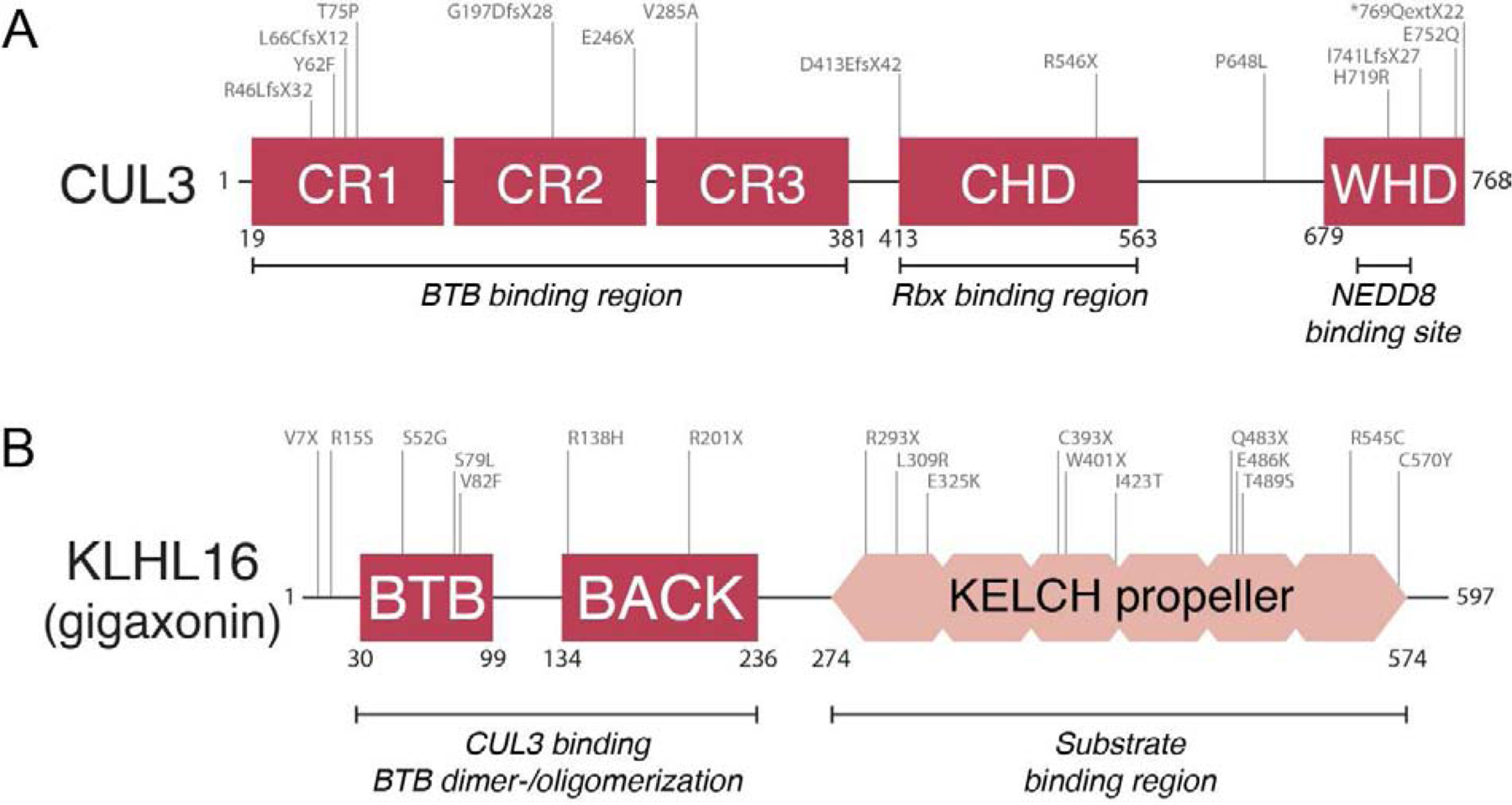
A) Schematic overview of mutations in CUL3 (NM_003590.5) that cause / are associated with autism, schizophrenia, and neurodevelopmental disease. Adopted from [35], and only displaying probable pathogenic mutations and not deletions and non-coding variants. These mutations are generally considered to be loss-of-function, and frequently result in truncated protein versions, but can also inactivate CUL3 function through decreasing BTB binding (e.g. as experimentally demonstrated for V285A [35]) B) Schematic overview of mutations in KLHL16/GAN (NM_022041.4) that have been shown to cause giant axonal neuropathy (adopted from [82] and additional disease-causing variants added as described in [169–171]). These mutations result in loss of protein function through generating truncated protein versions or variants that are deficient in homodimerization, CUL3 binding, and substrate recruitment. Mutations in other BTBs such as LZTR1 and KCTD7 have been shown to cause craniofacial and neurodevelopmental diseases in a similar manner.
Table 1: CRL3s implicated in formation and maintenance of neuroectodermal tissues:
Genes highlighted in green regulate neural crest development, genes highlighted in red regulate formation of maintenance of neural tissues. If not otherwise specified, diseases listed are associated/caused by mutations in the respective BTB protein.
| Name | Other Common Names | Substrates | Molecular Process | (Patho-)Physiological Process | Associated Diseases |
|---|---|---|---|---|---|
| KBTBD8 | TAKRP | TCOF1, NOLC1 [7, 93] | Ribosome biogenesis [7, 93] | Neural crest specification [7, 93] | Treacher Collins (caused by mutations in TCOF1) [7] |
| LZTR1 | BTBD29 | pan-RAS (MRAS, HRAS. NRAS, KRAS), RIT1 [129–132, 135] | RAS signaling [129–131, 135] | Neural crest development [131, 135] | Noonan syndrome [133–136] |
| KLHL12 | - | SEC31 [75, 106] | COPII vesicle size/collagen secretion [75, 106] | bone formation [75, 106] | Cranio-lenticulo-sutural dysplasia (caused by mutations in COPII vesicle protein SEC23A)[137–139] |
| KLHL12 | - | Dsh [91, 140] | WNT signaling [91, 140] | Formation of neural tissues [91, 140] | - |
| BTBD6 | - | PLZF/ZBTB16 [148] | - | Neurogenesis [147, 148] | - |
| KCTD13 | Bacurdl | RND2 [172], RND3 [172], RhoA [49] | Cytoskeletal dynamics [49] | Dendritic spine formation, dendrite branching, synaptic integrity [83] | Microcephaly, macrocephaly, epilepsy, autism (copy number associations) [151], microcephaly in zebrafish [159], reduced synaptic transmission in mouse and zebrafish [83] |
| KLHL16 | Gigaxonin (GAN) | Intermediate Filaments [155, 157, 158], Ptch [146] | Intermediate filament dynamics [156, 173], Sonic Hedgehog signaling [146] | Neural patterning [146], neuronal survival [153] | Giant Axonal Neuropathy [82] |
| TNFAIP1 | Bacurd2 | RND2 [172, 174], RND3 [172], RhoA [49] | - | Neurogenesis [172, 174] | - |
| KLHL17 | Actinfilin | GluR6 [175] | Kianate receptor regulation [175], Neuronal actin dynamics [176] | Neuronal morphology [176] | Possible cause of infantile spasms [177] |
| KLHL20 | KLEIP | PDZ-RhoGEF [94] | RhoA signaling [94] | Neurite outgrowth [94] | - |
| KLHL37 | ENC1 | - | Autophagy [178] | Forebrain development [179] | - |
| KCTD2 | - | c-MYC [180] | Transcription factor regulation [180] | Cell proliferation in glioma stem cells [180] | Alzheimer’s disease association [181, 182] |
| KCTD7 | CLN14 | - | Neuronal autophagy [183] | Neuronal ion channel conductance [184] | Early onset progressive myoclonic epilepsy [183], neuronal ceroid lipofuscinosis, opsoclonus-myoclonus syndrome [186] |
| KCTD11 | REN, KCASH1 | HDAC1 [142] | Sonic Hedgehog signaling [142, 187] | Granule cell proliferation in cerebellar development [187] | - |
| KCTD21 | KCASH2 | HDAC1 [188] | Sonic Hedgehog signaling [188] | Growth/development of Medulloblastoma [188] | Associated with autism [189], and schizophrenia [190] |
| KLHL1 | MRP2 | - | Actin dynamics [191] | Oligodendrogenesis [191] | - |
| KLHL2 | Mayven | - | Actin dynamics [192, 193] | Oligodendrogenesis [192, 193] | - |
CRL3 complexes in controlling neural crest specification and craniofacial development
During early embryogenesis, neural crest cells derived from the neural plate border migrate into the embryonic territory of the future craniofacial skeleton. These cranial neural crest cells differentiate into chondrocytes and osteoblasts, which secrete collagen networks required for bone formation [121]. Recent reports have shown that regulation by CUL3 is required at multiple steps of this developmental process (Figure 4A).
Figure 4: CRL3s controlling neuroectodermal development.
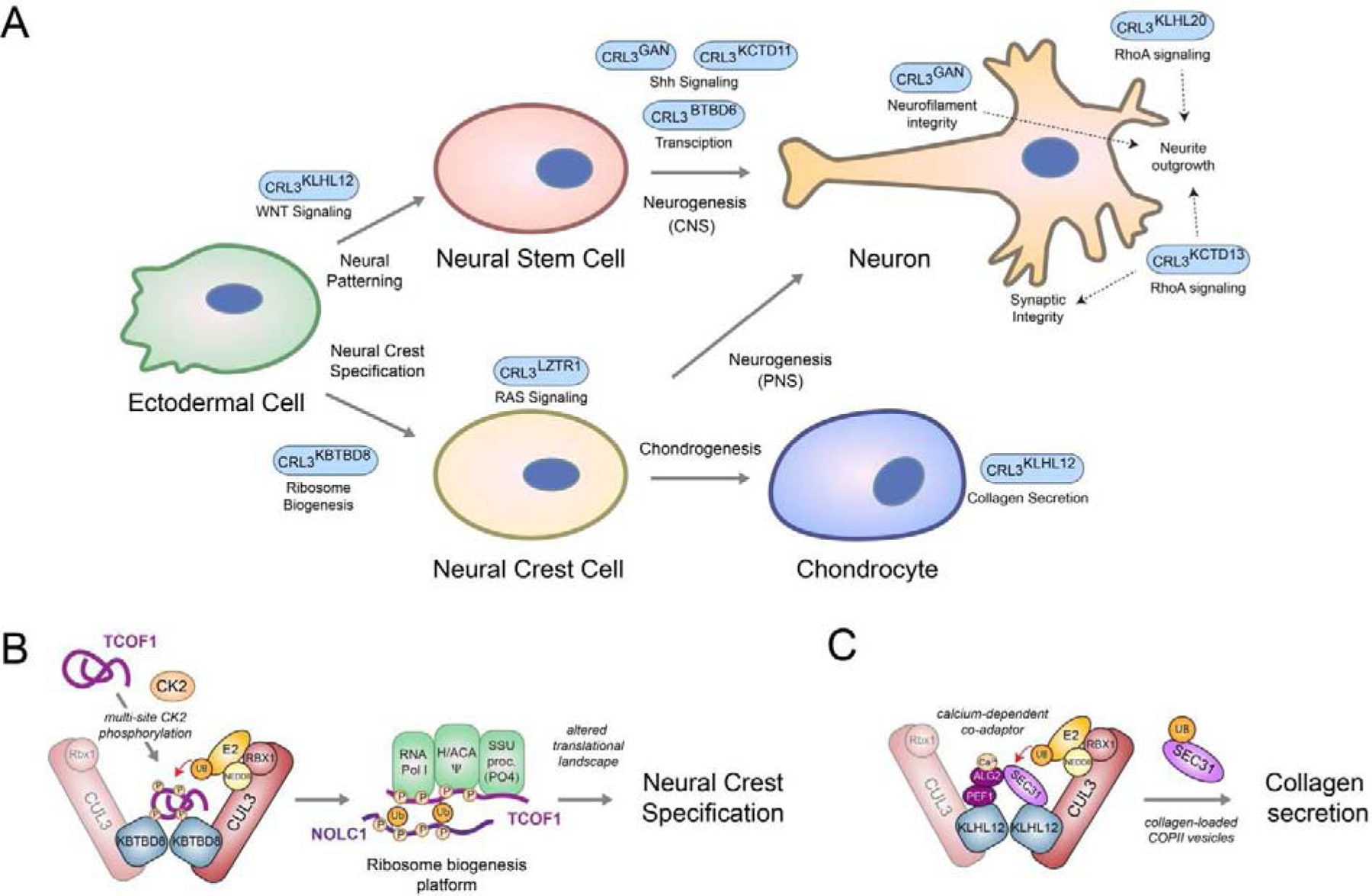
A) Schematic overview of various CRL3 complexes implicated in regulating aspects of neuroectodermal differentiation. B) Model of CRL3-KBTBD8-dependent neural crest specification. During embryonic development, multisite CK2-phopshorylation of the ribosome biogenesis factors TCOF1 and NOLC1 allows their recognition and monoubiquitylation by homo-dimeric CRL3-KBTBD8. This is thought to open the compact conformation of these ampholytic, intrinsically disordered proteins to promote recognition of ribosome biogenesis factors, including the RNA polymerase I, the pseudouridylation machinery (Ψ), and the SSU processome. These ubiquitin-dependent changes in ribosome biogenesis then trigger remodeling of translational networks required for neural crest specification, which likely occurs through newly synthesized, modified ribosomes C) Model of calcium-induced CRL3-KLHL12-dependent collagen secretion. During bone formation, homo-dimeric CRL3-KLHL12 complexes are thought to engage their substrate SEC31 using a co-adaptor complex consisting of PEF1 and ALG2. The ALG2 subunit is only able to engage SEC31 after calcium has been released from the endoplasmic reticulum, thus allowing CRL3-KLHL12 to mono-ubiquitylate and promote formation of large, collagen-loaded COPII vesicles in a calcium-dependent manner.
CRL3-KBTBD8 in neural crest development:
In co-operation with the vertebrate-specific BTB adaptor KBTBD8, CUL3 promotes neural crest specification and chondrocyte differentiation [7]. This occurs, in part, through mono-ubiquitylation of the ribosome biogenesis regulators NOLC1 and TCOF1, which induces the formation of a ribosome biogenesis platform that connects RNA Pol I with ribosome modification and ribosomal processing enzymes (Figure 3B). These ubiquitin-dependent assemblies are important to induce changes in translation of specific mRNAs, which facilitates neural crest specification. This likely occurs through production of newly synthesized, modified ribosomes [7, 122]. The importance of this translation regulation is highlighted by the fact that mutations in both, TCOF1 and RNA polymerase I cause Treacher Collins Syndrome, a neurocristopathy characterized by aberrant craniofacial development [123–125]. Further biochemical and proteomic studies are required to identify how ribosomes produced by CRL3-KBTBD8-dependent biogenesis platforms differ in their protein content and modifications compared to the bulk pool of ribosomes, how they recognize their select mRNAs during differentiation, and how this pathway is dysregulated in Treacher Collins Syndrome. CRL3-KBTBD8-dependent neural crest specification is embedded into the developmental program of the embryo through activation by CK2 [93], a kinase which gradually increases during formation of the nervous system [126]. Biochemical reconstitution experiments demonstrated that TCOF1 and NOLC1 contain 10 or more CK2 motifs, each of which could only be recognized by CRL3-KBTBD8 upon phosphorylation by CK2 (Figure 4B). However, efficient interaction and monoubiquitylation in cells as well as neural crest formation during human embryonic stem cell differentiation required multisite phosphorylation of at least 7 CK2 motifs in CRL3-KBTBD8 substrates. This dependency on multisite phosphorylation allows CRL3-KBTBD8 to react to increases in CK2 activity only upon reaching a certain threshold, thus allowing for switch-like cell-fate transitions [93].
CRL3-LZTR1 in neural crest development:
Upon cell-fate commitment, neural crest cells undergo epithelial-to-mesenchymal transition to become migratory and undergo differentiation. These steps are crucially controlled by RAS-ERK signaling, an important developmental signaling pathway, whose dysregulation leads to neoplasms and congenital syndromes often associated with aberrant neural crest formation [127, 128]. Together with the BTB adaptor LZTR1, CUL3 has been shown to participate in the regulation of RAS-ERK signaling by ubiquitylating several family members of the RAS-GTPase family, including MRAS, HRAS, NRAS, KRAS, and RIT1 [129–132]. CRL3-LZTR1 inactivates RAS proteins by ubiquitylation with different types of polyubiquitin chains. This was proposed to occur through both, proteasomal degradation of RAS proteins [129, 132] and sequestration of ubiquitylated RAS proteins from membranes [130, 131]. The importance of CRL3-LZTR1-dependent inactivation of RAS proteins for neural crest development is highlighted by the fact that mutations in LZTR1 cause Noonan syndrome, which is a RASopathy characterized by multiple congenital anomalies, including distinctive facial features and bone malformations, as well as cardiac disease [133–136]. These mutations decrease CUL3 binding, homodimerization, or substrate binding of LZTR1, resulting in enhanced RAS protein levels and plasma membrane association, increased ERK phosphorylation, and dysregulation of downstream transcriptional responses that are thought to lead to the developmental phenotypes observed in Noonan syndrome. Mutations in several other RAS-ERK pathway components that hyperactivate signaling have been shown to cause Noonan syndrome [128]. This includes mutations in RIT1, which have been shown to prevent its binding to and degradation by CRL3-LZTR1 [132]. Thus, CRL3-LZTR1-dependent regulation of RAS-ERK signaling is essential for proper neural crest and craniofacial development.
CRL3-KLHL12 in craniofacial development:
At later stages of the formation the craniofacial complex, CUL3-KLHL12 mono-ubiquitylates the COPII protein SEC31 to promote generation of large COPII carriers for collagen secretion [75]. SEC31 monoubiquitylation in cells requires homodimerization of CRL3-KLHL12 and two additional interacting proteins, PEF1 and ALG2, which form a complex that bridges interactions between KLHL12 and SEC31 [106] (Figure 4C). The ALG2 and PEF1 co-adaptor interaction with SEC31 is dependent on calcium, which allows CRL3-KLHL12 to convert transient changes in intracellular calcium concentrations into more persistent mono-ubiquitylation signals that promote formation of large COPII coats and collagen secretion. Thus, CRL3-KLHL12 is an important integrator of calcium signals during craniofacial development [19, 106]. Consistent with this notion, mutations in this SEC23A, a COPII component and SEC31 interactor, prevent collagen secretion during chondrogenesis and lead to the craniofacial genetic disease cranio-lenticulo-sutural dysplasia [137–139].
CRL3 complexes in controlling neuronal differentiation, specification, and function
The differentiation of neurons from neuroectodermal cells relies on coordinated changes in transcriptional networks, signaling cascades, and cytoskeletal dynamics. Multiple CRL3 complexes have been implicated in the regulation of these processes to ensure faithful formation and function of different types of neurons (Figure 4A).
CRL3-KLHL12 in neuroectodermal differentiation:
CRL3-KLHL12, in addition to its role during collagen secretion in chondrocytes, has been shown to have independent functions in WNT-induced proteasomal degradation of Disheveled or Dsh, the cytoplasmic adaptor of the WNT receptor Frizzeled [91, 140]. Knockdown of KLHL12 in zebrafish embryos resulted in dysregulated WNT-β-catenin signaling and various developmental defects, the most common being the loss of anterior neural tissues. Together with the key roles of WNT signaling during neural induction [141], this strongly suggests that negative regulation of this pathway by CRL3-KLHL12 is important for establishing neuroectodermal cells (Figure 4A).
CRL3s in differentiation from neural progenitor cells to neurons:
CUL3, in complex with the BTB adaptor KCTD11/REN, was reported to participate in termination of Sonic hedgehog (SHH) signaling in cerebellar granule cell progenitors (GCPs) [142]. During active SHH signaling, HDAC1 deacetylates and thereby promotes gene expression changes by the transcription factors GLI1/2. This is counteracted by CRL3-KCTD11-mediated proteasomal degradation of HDAC1. The negative regulatory impact of CRL3-KCTD11 on the SHH pathway ensures proper cell cycle exit and differentiation of GPCs and, if dysregulated by loss-of-function mutations in KCTD11, leads to medulloblastoma, the most common brain malignancy in childhood [143, 144]. Interestingly, mutations concentrated in the KELCH domain of KBTBD4 also frequently causes medulloblastomas [145], implying ubiquitin-dependent functions of CRL3-KBTBD4 in ensuring faithful neural stem cell-fate transitions. Other reports suggest further roles of CUL3 in controlling the SHH pathway (in cooperation with KLHL16/gigaxonin [146]) as well as in mediating proteasomal degradation of the transcriptional repressor PLZF/ZBTB16 (in cooperation with BTBD6 [147, 148]) to promote neurogenesis. Thus, multiple CRL3s co-operate to regulate transcriptional networks and signaling pathways to orchestrate differentiation of neurons from neural progenitor cells (Figure 4A).
CRL3s in regulating neuronal functions:
The formation of the nervous system requires coordinated proliferation, migration, and differentiation of neurons, which undergo major developmental changes as they migrate, form axons and dendrites, and establish synaptic connections [149]. These functional and morphological changes are coordinated by dynamic changes in the structural organization of the neuronal cytoskeleton, consisting of microtubules, actin filaments, and intermediate filaments. Initiated by discoveries from human disease genetics [82, 150, 151], research over the last decades has firmly established CUL3, in complex with KLHL16/gigaxonin and KCTD13/BACURD1, as a key regulator of the neuronal cytoskeleton. Mutations in KLHL16/gigaxonin cause giant axonal neuropathy [82], a severe neurodegenerative disorder primarily characterized by deterioration of the nervous system and extensive aggregation of intermediate filaments throughout the body [152]. Initially, this disease was attributed to loss of CRL3-Gigaxonin-mediated proteasomal degradation of microtubule components leading to microtubule instability [153, 154]. However, these findings could not be verified in patient cells or gigaxonin knock out mice [155, 156] and studies in these models rather suggested a direct role of CRL3-gigaxonin in the regulation of intermediate filament networks. Indeed, several groups showed that gigaxonin co-immunoprecipitates with intermediate filaments and mediates their proteasomal degradation in neuronal and non-neuronal cells [155, 157, 158]. Thus, while biochemical reconstitution studies are required to confirm that CRL3-gigaxonin directly ubiquitylates intermediate filaments, these reports clearly establish this ubiquitin E3 ligase as an essential factor for the degradation of neuronal intermediate filaments (Figure 4A). Similar to gigaxonin, human genetic studies instigated mechanistic investigation into CRL3-KCTD13 and its role during neurodevelopment. KCTD13 is located in the 16p11.2 locus, which was shown to contribute to risk of epilepsy, autism and autism spectrum disorder when deleted [151] or to autism and schizophrenia when duplicated [150]. Importantly, zebrafish and mouse studies showed that out of the 29 genes comprised within the 16p11.2 locus that is subject to copy number variants in patients, KCTD13 is a major driver for the associated neurodevelopmental phenotypes [83, 159]. CRL3-KCTD13 has been reported to ubiquitylate and mediate proteasomal degradation of RhoA [49], a small GTPase protein that regulates actin- or tubulin-based cytoskeletal dynamics important for various aspects of neuronal differentiation, including neurite outgrowth and synapse formation [160]. Consistent with these observations in tissue culture cells, deletion of KCTD13 in mice resulted in increased RhoA levels, loss of dendritic spines, and reduced synaptic activity in the CA1 region of the hippocampus [83]. Importantly, reduced synaptic transmission could be restored through RhoA inhibition. Together with results from spatiotemporal 16p11.2 protein network analyses of brain subregions [85], these findings identify an essential function of CRL3-KCTD13 in regulating RhoA signaling required for neuronal network formation during brain development (Figure 4A). Interestingly, CRL3-KLHL20 also controls RhoA signaling in neurons, but by degrading its activator PDZ-RhoGEF [94], suggesting different CRL3s can regulate the same signaling pathway at distinct steps to promote a developmental process.
Conclusions and perspective
Since the initial discovery of BTB proteins as substrate adaptors of CUL3-based ubiquitin E3 ligases almost 20 years ago [37, 41–43], numerous studies have revealed intricate regulatory principles underlying the assembly and substrate recruitment of CRL3s that allow these enzymes to orchestrate various aspects of human development. In particular, we here highlight how distinct CRL3s dynamically regulate diverse cellular processes, including transcription, translation, secretion, signaling, and cytoskeletal dynamics, to integrate multiple steps required for faithful differentiation and maintenance of neuroectodermal tissues. Despite these advances and this emerging concept, many open questions remain. First, since most experiments for the adaptive exchange model for CRL assembly originates from CRL1 and CRL4s, further investigations are required to determine how the CRL3 network is remodeled in cells and changed during differentiation. Insights from these studies will be particularly important to understand how autism-associated mutations that reduce overall CUL3 concentrations [118, 161] affect the cellular CRL3 repertoire. Second, what are the endogenous E2s that cooperate with CRL3s to synthesize the known mono- and polyubiquitin modifications on their cognate substrates? Third, there are likely more BTB adaptors that contribute to neuroectodermal differentiation, e.g. as suggested by recent studies that either reported functions of CUL3 on neuronal translation without identification of the associated BTB adaptor(s) [119] or mutations in CUL3 that affect BTB binding and lead to developmental delays, intellectual disability, and infantile spasms [35]. Identification of these substrate adaptors and the spectrum and dynamics of CRL3 complexes will provide additional insights into this multi-layer regulatory process.
As described in this review for CRL3-gigaxonin and CRL-KCTD13, initial findings from human disease genetics have often driven investigations into targets and mechanisms of action of E3 ligases [21]. Recent bioinformatics efforts have leveraged the availability of large datasets from sequencing studies to determine the mutational constraint spectrum of human genes [162–164].By sequencing and comparing a large number of healthy individuals, genomic constraint quantifies the depletion of variation in every gene within control populations. These metrics allow for identification of genes intolerant to variation that, when mutated, have a high likelihood of causing embryonic lethality or disease. We propose that targeted searches for missense mutations of highly constrained BTB proteins in whole exome sequencing data of patients with undiagnosed neurodevelopmental and craniofacial diseases will be a powerful means to identify novel CUL3 adaptors controlling neuroectodermal differentiation. Combining such genotype first approaches with state-of-the art techniques of ubiquitin research [165] will allow identification of targets and underlying mechanisms of these CRL3s. Given the rising interest in ubiquitin E3 ligases as drug targets and the growing ability to modulate CRL activity with small molecules [166–168], such insights might be useful to develop novel therapeutic approaches to ameliorate the symptoms of diseases associated with dysregulation of ubiquitylation.[185]
Highlights:
CRL3s are a family of multi-subunit E3 ligases that utilize ~90 interchangeable BTB proteins as substrate adaptors
Distinct CRL3-BTBs ubiquitylate and control the fate of specific sets of substrates to control diverse aspects of development
Intricate regulatory mechanisms impinge on CRL3 assembly, substrate recruitment, and ubiquitylation
CRL3s integrate multiple steps required for the differentiation and formation of neuroectodermal tissues and their dysregulation underlies various congenital craniofacial diseases and neurodevelopmental disorders
Emerging tools from the fields of human disease genetics will be a powerful means to identify novel BTB adaptors required for neuroectodermal differentiation and enable dissection of their underlying mechanisms
Acknowledgements:
We apologize to all scientists whose work could not be discussed within the space confines of this review. We thank members of our lab for continuous help and discussions, and we are grateful to Regina Baur and Laura Kerosuo for comments on this manuscript. A.J.A, D.B.B., and A.W. are funded by the intramural program of the NIDCR and NHGRI, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
The authors declare no conflict of interest.
Citations
- [1].Clague MJ, Urbe S, Komander D, Breaking the chains: deubiquitylating enzyme specificity begets function, Nat Rev Mol Cell Biol 20 (2019) 338–352. [DOI] [PubMed] [Google Scholar]
- [2].Oh E, Akopian D, Rape M, Principles of Ubiquitin-Dependent Signaling, Annu Rev Cell Dev Biol 34 (2018) 137–162. [DOI] [PubMed] [Google Scholar]
- [3].Komander D, Rape M, The ubiquitin code, Annu Rev Biochem 81 (2012) 203–229. [DOI] [PubMed] [Google Scholar]
- [4].Yau R, Rape M, The increasing complexity of the ubiquitin code, Nat Cell Biol 18 (2016) 579–586. [DOI] [PubMed] [Google Scholar]
- [5].Di Fiore PP, Polo S, Hofmann K, When ubiquitin meets ubiquitin receptors: a signalling connection, Nat Rev Mol Cell Biol 4 (2003) 491–497. [DOI] [PubMed] [Google Scholar]
- [6].Haglund K, Dikic I, Ubiquitylation and cell signaling, EMBO J 24 (2005) 3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Werner A, Iwasaki S, McGourty CA, Medina-Ruiz S, Teerikorpi N, Fedrigo I, Ingolia NT, Rape M, Cell-fate determination by ubiquitin-dependent regulation of translation, Nature 525 (2015) 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A, A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein, Science 243 (1989) 1576–1583. [DOI] [PubMed] [Google Scholar]
- [9].Jin L, Williamson A, Banerjee S, Philipp I, Rape M, Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex, Cell 133 (2008) 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K, Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation, Nat Cell Biol 11 (2009) 123–132. [DOI] [PubMed] [Google Scholar]
- [11].Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ, TAK1 is a ubiquitin-dependent kinase of MKK and IKK, Nature 412 (2001) 346–351. [DOI] [PubMed] [Google Scholar]
- [12].Grumati P, Dikic I, Ubiquitin signaling and autophagy, J Biol Chem 293 (2018) 5404–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pickart CM, Mechanisms underlying ubiquitination, Annu Rev Biochem 70 (2001) 503–533. [DOI] [PubMed] [Google Scholar]
- [14].Deshaies RJ, Joazeiro CA, RING domain E3 ubiquitin ligases, Annu Rev Biochem 78 (2009) 399–434. [DOI] [PubMed] [Google Scholar]
- [15].Lydeard JR, Schulman BA, Harper JW, Building and remodelling Cullin-RING E3 ubiquitin ligases, EMBO Rep 14 (2013) 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zimmerman ES, Schulman BA, Zheng N, Structural assembly of cullin-RING ubiquitin ligase complexes, Curr Opin Struct Biol 20 (2010) 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng J, Guo J, Wang Z, North BJ, Tao K, Dai X, Wei W, Functional analysis of Cullin 3 E3 ligases in tumorigenesis, Biochim Biophys Acta Rev Cancer 1869 (2018) 11–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Petroski MD, Deshaies RJ, Function and regulation of cullin-RING ubiquitin ligases, Nat Rev Mol Cell Biol 6 (2005) 9–20. [DOI] [PubMed] [Google Scholar]
- [19].Werner A, Manford AG, Rape M, Ubiquitin-Dependent Regulation of Stem Cell Biology, Trends Cell Biol 27 (2017) 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Skaar JR, Pagan JK, Pagano M, Mechanisms and function of substrate recruitment by F-box proteins, Nat Rev Mol Cell Biol 14 (2013) 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rape M, Ubiquitylation at the crossroads of development and disease, Nat Rev Mol Cell Biol 19 (2018) 59–70. [DOI] [PubMed] [Google Scholar]
- [22].Watson ER, Brown NG, Peters JM, Stark H, Schulman BA, Posing the APC/C E3 Ubiquitin Ligase to Orchestrate Cell Division, Trends Cell Biol 29 (2019) 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Z, Pei XH, Yan J, Yan F, Cappell KM, Whitehurst AW, Xiong Y, CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity, Mol Cell 54 (2014) 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Skaar JR, Florens L, Tsutsumi T, Arai T, Tron A, Swanson SK, Washburn MP, DeCaprio JA, PARC and CUL7 form atypical cullin RING ligase complexes, Cancer Res 67 (2007) 2006–2014. [DOI] [PubMed] [Google Scholar]
- [25].Dubiel D, Bintig W, Kahne T, Dubiel W, Naumann M, Cul3 neddylation is crucial for gradual lipid droplet formation during adipogenesis, Biochim Biophys Acta Mol Cell Res 1864 (2017) 1405–1412. [DOI] [PubMed] [Google Scholar]
- [26].Genschik P, Sumara I, Lechner E, The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications, EMBO J 32 (2013) 2307–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jerabkova K, Sumara I, Cullin 3, a cellular scripter of the non-proteolytic ubiquitin code, Semin Cell Dev Biol 93 (2019) 100–110. [DOI] [PubMed] [Google Scholar]
- [28].Holt RJ, Young RM, Crespo B, Ceroni F, Curry CJ, Bellacchio E, Bax DA, Ciolfi A, Simon M, Fagerberg CR, van Binsbergen E, De Luca A, Memo L, Dobyns WB, Mohammed AA, Clokie SJH, Zazo Seco C, Jiang YH, Sorensen KP, Andersen H, Sullivan J, Powis Z, Chassevent A, Smith-Hicks C, Petrovski S, Antoniadi T, Shashi V, Gelb BD, Wilson SW, Gerrelli D, Tartaglia M, Chassaing N, Calvas P, Ragge NK, De Novo Missense Variants in FBXW11 Cause Diverse Developmental Phenotypes Including Brain, Eye, and Digit Anomalies, Am J Hum Genet 105 (2019) 640–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, Dagoneau N, Chemaitilly W, Superti-Furga A, Dos Santos H, Megarbane A, Morin G, Gillessen-Kaesbach G, Hennekam R, Van der Burgt I, Black GC, Clayton PE, Read A, Le Merrer M, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V, Identification of mutations in CUL7 in 3-M syndrome, Nat Genet 37 (2005) 1119–1124. [DOI] [PubMed] [Google Scholar]
- [30].Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, Dicks E, Stevens C, Tofts C, Avis T, Barthorpe S, Buck G, Cole J, Gray K, Halliday K, Harrison R, Hills K, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Varian J, West S, Widaa S, Mallya U, Moon J, Luo Y, Holder S, Smithson SF, Hurst JA, Clayton-Smith J, Kerr B, Boyle J, Shaw M, Vandeleur L, Rodriguez J, Slaugh R, Easton DF, Wooster R, Bobrow M, Srivastava AK, Stevenson RE, Schwartz CE, Turner G, Gecz J, Futreal PA, Stratton MR, Partington M, Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor, Am J Hum Genet 80 (2007) 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H, Li J, Gao G, Guo Y, Yan C, Wei J, Shao C, Gong Y, Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation, Am J Hum Genet 80 (2007) 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Silverman JS, Skaar JR, Pagano M, SCF ubiquitin ligases in the maintenance of genome stability, Trends Biochem Sci 37 (2012) 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Voigt J, Papalopulu N, A dominant-negative form of the E3 ubiquitin ligase Cullin-1 disrupts the correct allocation of cell fate in the neural crest lineage, Development 133 (2006) 559–568. [DOI] [PubMed] [Google Scholar]
- [34].De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Study DDD, Homozygosity A Mapping Collaborative for, Consortium UK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD, Synaptic, transcriptional and chromatin genes disrupted in autism, Nature 515 (2014) 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nakashima M, Kato M, Matsukura M, Kira R, Ngu LH, Lichtenbelt KD, van Gassen KLI, Mitsuhashi S, Saitsu H, Matsumoto N, De novo variants in CUL3 are associated with global developmental delays with or without infantile spasms, J Hum Genet (2020). [DOI] [PubMed] [Google Scholar]
- [36].Schizophrenia C Working Group of the Psychiatric Genomics, Biological insights from 108 schizophrenia-associated genetic loci, Nature 511 (2014) 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Geyer R, Wee S, Anderson S, Yates J, Wolf DA, BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases, Mol Cell 12 (2003) 783–790. [DOI] [PubMed] [Google Scholar]
- [38].Michel JJ, McCarville JF, Xiong Y, A role for Saccharomyces cerevisiae Cul8 ubiquitin ligase in proper anaphase progression, J Biol Chem 278 (2003) 22828–22837. [DOI] [PubMed] [Google Scholar]
- [39].Kurz T, Pintard L, Willis JH, Hamill DR, Gonczy P, Peter M, Bowerman B, Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway, Science 295 (2002) 1294–1298. [DOI] [PubMed] [Google Scholar]
- [40].Singer JD, Gurian-West M, Clurman B, Roberts JM, Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells, Genes Dev 13 (1999) 2375–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Furukawa M, He YJ, Borchers C, Xiong Y, Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases, Nat Cell Biol 5 (2003) 1001–1007. [DOI] [PubMed] [Google Scholar]
- [42].Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M, The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase, Nature 425 (2003) 311–316. [DOI] [PubMed] [Google Scholar]
- [43].Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW, BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3, Nature 425 (2003) 316–321. [DOI] [PubMed] [Google Scholar]
- [44].Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG, Sequence and structural analysis of BTB domain proteins, Genome Biol 6 (2005) R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M, Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2, Mol Cell Biol 24 (2004) 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M, Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex, Mol Cell Biol 24 (2004) 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gschweitl M, Ulbricht A, Barnes CA, Enchev RI, Stoffel-Studer I, Meyer-Schaller N, Huotari J, Yamauchi Y, Greber UF, Helenius A, Peter M, A SPOPL/Cullin-3 ubiquitin ligase complex regulates endocytic trafficking by targeting EPS15 at endosomes, Elife 5 (2016) e13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Maerki S, Olma MH, Staubli T, Steigemann P, Gerlich DW, Quadroni M, Sumara I, Peter M, The Cul3-KLHL21 E3 ubiquitin ligase targets aurora B to midzone microtubules in anaphase and is required for cytokinesis, J Cell Biol 187 (2009) 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen Y, Yang Z, Meng M, Zhao Y, Dong N, Yan H, Liu L, Ding M, Peng HB, Shao F, Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement, Mol Cell 35 (2009) 841–855. [DOI] [PubMed] [Google Scholar]
- [50].Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Valimaki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP, Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities, Nature 482 (2012) 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gupta VA, Beggs AH, Kelch proteins: emerging roles in skeletal muscle development and diseases, Skelet Muscle 4 (2014) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A, Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa, Am J Hum Genet 84 (2009) 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin Z, Li S, Feng C, Yang S, Wang H, Ma D, Zhang J, Gou M, Bu D, Zhang T, Kong X, Wang X, Sarig O, Ren Y, Dai L, Liu H, Zhang J, Li F, Hu Y, Padalon-Brauch G, Vodo D, Zhou F, Chen T, Deng H, Sprecher E, Yang Y, Tan X, Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility, Nat Genet 48 (2016) 1508–1516. [DOI] [PubMed] [Google Scholar]
- [54].Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, Marsden BD, Gileadi O, Knapp S, von Delft F, Bullock AN, Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases, J Biol Chem 288 (2013) 7803–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Errington WJ, Khan MQ, Bueler SA, Rubinstein JL, Chakrabartty A, Prive GG, Adaptor protein self-assembly drives the control of a cullin-RING ubiquitin ligase, Structure 20 (2012) 1141–1153. [DOI] [PubMed] [Google Scholar]
- [56].Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP, Schulman BA, Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases, Mol Cell 36 (2009) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ji AX, Chu A, Nielsen TK, Benlekbir S, Rubinstein JL, Prive GG, Structural Insights into KCTD Protein Assembly and Cullin3 Recognition, J Mol Biol 428 (2016) 92–107. [DOI] [PubMed] [Google Scholar]
- [58].Pinkas DM, Sanvitale CE, Bufton JC, Sorrell FJ, Solcan N, Chalk R, Doutch J, Bullock AN, Structural complexity in the KCTD family of Cullin3-dependent E3 ubiquitin ligases, Biochem J 474 (2017) 3747–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang K, Deshaies RJ, Liu X, Assembly and Regulation of CRL Ubiquitin Ligases, Adv Exp Med Biol 1217 (2020) 33–46. [DOI] [PubMed] [Google Scholar]
- [60].Liu X, Reitsma JM, Mamrosh JL, Zhang Y, Straube R, Deshaies RJ, Cand1-Mediated Adaptive Exchange Mechanism Enables Variation in F-Box Protein Expression, Mol Cell 69 (2018) 773–786 e776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Reichermeier KM, Straube R, Reitsma JM, Sweredoski MJ, Rose CM, Moradian A, den Besten W, Hinkle T, Verschueren E, Petzold G, Thoma NH, Wertz IE, Deshaies RJ, Kirkpatrick DS, PIKES Analysis Reveals Response to Degraders and Key Regulatory Mechanisms of the CRL4 Network, Mol Cell 77 (2020) 1092–1106 e1099. [DOI] [PubMed] [Google Scholar]
- [62].Reitsma JM, Liu X, Reichermeier KM, Moradian A, Sweredoski MJ, Hess S, Deshaies RJ, Composition and Regulation of the Cellular Repertoire of SCF Ubiquitin Ligases, Cell 171 (2017) 1326–1339 e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Straube R, Shah M, Flockerzi D, Wolf DA, Trade-off and flexibility in the dynamic regulation of the cullin-RING ubiquitin ligase repertoire, PLoS Comput Biol 13 (2017) e1005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bennett EJ, Rush J, Gygi SP, Harper JW, Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics, Cell 143 (2010) 951–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dubiel D, Ordemann J, Pratschke J, Dubiel W, Naumann M, CAND1 exchange factor promotes Keap1 integration into cullin 3-RING ubiquitin ligase during adipogenesis, Int J Biochem Cell Biol 66 (2015) 95–100. [DOI] [PubMed] [Google Scholar]
- [66].Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RL, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S, Shan SO, Deshaies RJ, Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins, Cell 153 (2013) 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Baek K, Krist DT, Prabu JR, Hill S, Klugel M, Neumaier LM, von Gronau S, Kleiger G, Schulman BA, NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly, Nature 578 (2020) 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA, Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation, Cell 134 (2008) 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saha A, Deshaies RJ, Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation, Mol Cell 32 (2008) 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith RE, Tichkule RB, Hassiepen U, Abdulrahman W, Pantelic RS, Matsumoto S, Sugasawa K, Stahlberg H, Thoma NH, Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome, Nature 531 (2016) 598–603. [DOI] [PubMed] [Google Scholar]
- [71].Emberley ED, Mosadeghi R, Deshaies RJ, Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism, J Biol Chem 287 (2012) 29679–29689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Enchev RI, Scott DC, da Fonseca PC, Schreiber A, Monda JK, Schulman BA, Peter M, Morris EP, Structural basis for a reciprocal regulation between SCF and CSN, Cell Rep 2 (2012) 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, Iwai S, Gut H, Sugasawa K, Thoma NH, The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation, Cell 147 (2011) 1024–1039. [DOI] [PubMed] [Google Scholar]
- [74].Mosadeghi R, Reichermeier KM, Winkler M, Schreiber A, Reitsma JM, Zhang Y, Stengel F, Cao J, Kim M, Sweredoski MJ, Hess S, Leitner A, Aebersold R, Peter M, Deshaies RJ, Enchev RI, Structural and kinetic analysis of the COP9-Signalosome activation and the cullin-RING ubiquitin ligase deneddylation cycle, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M, Ubiquitin-dependent regulation of COPII coat size and function, Nature 482 (2012) 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Arama E, Bader M, Rieckhof GE, Steller H, A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila, PLoS Biol 5 (2007) e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yan W, Ma L, Burns KH, Matzuk MM, Haploinsufficiency of kelch-like protein homolog 10 causes infertility in male mice, Proc Natl Acad Sci U S A 101 (2004) 7793–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yatsenko AN, Roy A, Chen R, Ma L, Murthy LJ, Yan W, Lamb DJ, Matzuk MM, Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization, Hum Mol Genet 15 (2006) 3411–3419. [DOI] [PubMed] [Google Scholar]
- [79].Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, Ogata K, Hsu C, Clarke NF, Darras BT, Farrar MA, Hashem A, Manton ND, Muntoni F, North KN, Sandaradura SA, Nishino I, Hayashi YK, Sewry CA, Thompson EM, Yau KS, Brownstein CA, Yu TW, Allcock RJ, Davis MR, Wallgren-Pettersson C, Matsumoto N, Alkuraya FS, Laing NG, Beggs AH, Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline Myopathy, Am J Hum Genet 93 (2013) 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ravenscroft G, Miyatake S, Lehtokari VL, Todd EJ, Vornanen P, Yau KS, Hayashi YK, Miyake N, Tsurusaki Y, Doi H, Saitsu H, Osaka H, Yamashita S, Ohya T, Sakamoto Y, Koshimizu E, Imamura S, Yamashita M, Ogata K, Shiina M, Bryson-Richardson RJ, Vaz R, Ceyhan O, Brownstein CA, Swanson LC, Monnot S, Romero NB, Amthor H, Kresoje N, Sivadorai P, Kiraly-Borri C, Haliloglu G, Talim B, Orhan D, Kale G, Charles AK, Fabian VA, Davis MR, Lammens M, Sewry CA, Manzur A, Muntoni F, Clarke NF, North KN, Bertini E, Nevo Y, Willichowski E, Silberg IE, Topaloglu H, Beggs AH, Allcock RJ, Nishino I, Wallgren-Pettersson C, Matsumoto N, Laing NG, Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy, Am J Hum Genet 93 (2013) 6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sambuughin N, Yau KS, Olive M, Duff RM, Bayarsaikhan M, Lu S, Gonzalez-Mera L, Sivadorai P, Nowak KJ, Ravenscroft G, Mastaglia FL, North KN, Ilkovski B, Kremer H, Lammens M, van Engelen BG, Fabian V, Lamont P, Davis MR, Laing NG, Goldfarb LG, Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores, Am J Hum Genet 87 (2010) 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M, The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy, Nat Genet 26 (2000) 370–374. [DOI] [PubMed] [Google Scholar]
- [83].Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F, Liu S, Thyme SB, Lopez-Garcia IA, Mendoza DB, Usui N, Ellegood J, Eisch AJ, Konopka G, Lerch JP, Schier AF, Speed HE, Powell CM, Kctd13 deletion reduces synaptic transmission via increased RhoA, Nature 551 (2017) 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ganay T, Boizot A, Burrer R, Chauvin JP, Bomont P, Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice, Mol Neurodegener 6 (2011) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lin GN, Corominas R, Lemmens I, Yang X, Tavernier J, Hill DE, Vidal M, Sebat J, Iakoucheva LM, Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases, Neuron 85 (2015) 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, Beaurain G, Bonnefond A, Sand O, Simian C, Vidal-Petiot E, Soukaseum C, Mandet C, Broux F, Chabre O, Delahousse M, Esnault V, Fiquet B, Houillier P, Bagnis CI, Koenig J, Konrad M, Landais P, Mourani C, Niaudet P, Probst V, Thauvin C, Unwin RJ, Soroka SD, Ehret G, Ossowski S, Caulfield M,International Consortium for Blood Bruneval PP, Estivill X, Froguel P, Hadchouel J, Schott JJ, Jeunemaitre X, KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron, Nat Genet 44 (2012) 456–460, S451–453. [DOI] [PubMed] [Google Scholar]
- [87].Bauer NC, Doetsch PW, Corbett AH, Mechanisms Regulating Protein Localization, Traffic 16 (2015) 1039–1061. [DOI] [PubMed] [Google Scholar]
- [88].Aram L, Braun T, Braverman C, Kaplan Y, Ravid L, Levin-Zaidman S, Arama E, A Krebs Cycle Component Limits Caspase Activation Rate through Mitochondrial Surface Restriction of CRL Activation, Dev Cell 37 (2016) 15–33. [DOI] [PubMed] [Google Scholar]
- [89].Arama E, Agapite J, Steller H, Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila, Dev Cell 4 (2003) 687–697. [DOI] [PubMed] [Google Scholar]
- [90].Genau HM, Huber J, Baschieri F, Akutsu M, Dotsch V, Farhan H, Rogov V, Behrends C, CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling, Mol Cell 57 (2015) 995–1010. [DOI] [PubMed] [Google Scholar]
- [91].Shami Shah A, Batrouni AG, Kim D, Punyala A, Cao W, Han C, Goldberg ML, Smolka MB, Baskin JM, PLEKHA4/kramer Attenuates Dishevelled Ubiquitination to Modulate Wnt and Planar Cell Polarity Signaling, Cell Rep 27 (2019) 2157–2170 e2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM, Chen RH, The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses, EMBO J 29 (2010) 1748–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Werner A, Baur R, Teerikorpi N, Kaya DU, Rape M, Multisite dependency of an E3 ligase controls monoubiquitylation-dependent cell fate decisions, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lin MY, Lin YM, Kao TC, Chuang HH, Chen RH, PDZ-RhoGEF ubiquitination by Cullin3-KLHL20 controls neurotrophin-induced neurite outgrowth, J Cell Biol 193 (2011) 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M, Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling, EMBO J 25 (2006) 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang J, Chen M, Zhu Y, Dai X, Dang F, Ren J, Ren S, Shulga YV, Beca F, Gan W, Wu F, Lin YM, Zhou X, DeCaprio JA, Beck AH, Lu KP, Huang J, Zhao C, Sun Y, Gao X, Pandolfi PP, Wei W, SPOP Promotes Nanog Destruction to Suppress Stem Cell Traits and Prostate Cancer Progression, Dev Cell 48 (2019) 329–344 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Furukawa M, Xiong Y, BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase, Mol Cell Biol 25 (2005) 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tong KI, Kobayashi A, Katsuoka F, Yamamoto M, Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism, Biol Chem 387 (2006) 1311–1320. [DOI] [PubMed] [Google Scholar]
- [99].Taguchi K, Motohashi H, Yamamoto M, Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution, Genes Cells 16 (2011) 123–140. [DOI] [PubMed] [Google Scholar]
- [100].Chen PH, Hu J, Wu J, Huynh DT, Smith TJ, Pan S, Bisnett BJ, Smith AB, Lu A, Condon BM, Chi JT, Boyce M, Gigaxonin glycosylation regulates intermediate filament turnover and may impact giant axonal neuropathy etiology or treatment, JCI Insight 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, Hogue M, Soderblom E, Tang F, Marks JR, Major MB, Swarts BM, Boyce M, Chi JT, Glycosylation of KEAP1 links nutrient sensing to redox stress signaling, EMBO J 36 (2017) 2233–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD, Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex, J Biol Chem 281 (2006) 24756–24768. [DOI] [PubMed] [Google Scholar]
- [103].Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M, Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response, Mol Cell Biol 27 (2007) 7511–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, Jiang J, Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase, Proc Natl Acad Sci U S A 106 (2009) 21191–21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, Schulman BA, Mittag T, Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments, Mol Cell 72 (2018) 19–36 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].McGourty CA, Akopian D, Walsh C, Gorur A, Werner A, Schekman R, Bautista D, Rape M, Regulation of the CUL3 Ubiquitin Ligase by a Calcium-Dependent Co-adaptor, Cell 167 (2016) 525–538 e514. [DOI] [PubMed] [Google Scholar]
- [107].Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, Peter M, A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells, Dev Cell 12 (2007) 887–900. [DOI] [PubMed] [Google Scholar]
- [108].Mena EL, Kjolby RAS, Saxton RA, Werner A, Lew BG, Boyle JM, Harland R, Rape M, Dimerization quality control ensures neuronal development and survival, Science 362 (2018). [DOI] [PubMed] [Google Scholar]
- [109].Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H, Ben-Nissan G, Kolaitis RM, Peters JL, Pounds S, Errington WJ, Prive GG, Taylor JP, Sharon M, Schuck P, Ogden SK, Mittag T, Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles, EMBO J 35 (2016) 1254–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Banani SF, Lee HO, Hyman AA, Rosen MK, Biomolecular condensates: organizers of cellular biochemistry, Nat Rev Mol Cell Biol 18 (2017) 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Shin Y, Brangwynne CP, Liquid phase condensation in cell physiology and disease, Science 357 (2017). [DOI] [PubMed] [Google Scholar]
- [112].Cuneo MJ, Mittag T, The ubiquitin ligase adaptor SPOP in cancer, FEBS J 286 (2019) 3946–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Deol KK, Lorenz S, Strieter ER, Enzymatic Logic of Ubiquitin Chain Assembly, Front Physiol 10 (2019) 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ye Y, Rape M, Building ubiquitin chains: E2 enzymes at work, Nat Rev Mol Cell Biol 10 (2009) 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA, de Jong A, Ovaa H, Alpi AF, Harper JW, Schulman BA, Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation, Cell 166 (2016) 1198–1214 e1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Simoes-Costa M, Bronner ME, Establishing neural crest identity: a gene regulatory recipe, Development 142 (2015) 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wilkie AO, Morriss-Kay GM, Genetics of craniofacial development and malformation, Nat Rev Genet 2 (2001) 458–468. [DOI] [PubMed] [Google Scholar]
- [118].O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE, Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations, Nature 485 (2012) 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Dong Z, Chen W, Chen C, Wang H, Cui W, Tan Z, Robinson H, Gao N, Luo B, Zhang L, Zhao K, Xiong WC, Mei L, CUL3 Deficiency Causes Social Deficits and Anxiety-like Behaviors by Impairing Excitation-Inhibition Balance through the Promotion of Cap-Dependent Translation, Neuron 105 (2020) 475–490 e476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Rapanelli M, Tan T, Wang W, Wang X, Wang ZJ, Zhong P, Frick L, Qin L, Ma K, Qu J, Yan Z, Behavioral, circuitry, and molecular aberrations by region-specific deficiency of the high-risk autism gene Cul3, Mol Psychiatry (2019). [DOI] [PubMed] [Google Scholar]
- [121].Dash S, Trainor PA, The development, patterning and evolution of neural crest cell differentiation into cartilage and bone, Bone 137 (2020) 115409. [DOI] [PubMed] [Google Scholar]
- [122].Genuth NR, Barna M, Heterogeneity and specialized functions of translation machinery: from genes to organisms, Nat Rev Genet 19 (2018) 431–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group, Nat Genet 12 (1996) 130–136. [DOI] [PubMed] [Google Scholar]
- [124].Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer-Haas C, Maiwald R, Zweier C, Kerr B, Cobo AM, Toral JF, Hoogeboom AJ, Lohmann DR, Hehr U, Dixon MJ, Breuning MH, Wieczorek D, Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome, Nat Genet 43 (2011) 20–22. [DOI] [PubMed] [Google Scholar]
- [125].Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA, Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities, Proc Natl Acad Sci U S A 103 (2006) 13403–13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Mestres P, Boldyreff B, Ebensperger C, Hameister H, Issinger OG, Expression of casein kinase 2 during mouse embryogenesis, Acta Anat (Basel) 149 (1994) 13–20. [DOI] [PubMed] [Google Scholar]
- [127].Dinsmore CJ, Soriano P, MAPK and PI3K signaling: At the crossroads of neural crest development, Dev Biol 444 Suppl 1 (2018) S79–S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Tidyman WE, Rauen KA, The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation, Curr Opin Genet Dev 19 (2009) 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Abe T, Umeki I, Kanno SI, Inoue SI, Niihori T, Aoki Y, LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases, Cell Death Differ 27 (2020) 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Bigenzahn JW, Collu GM, Kartnig F, Pieraks M, Vladimer GI, Heinz LX, Sedlyarov V, Schischlik F, Fauster A, Rebsamen M, Parapatics K, Blomen VA, Muller AC, Winter GE, Kralovics R, Brummelkamp TR, Mlodzik M, Superti-Furga G, LZTR1 is a regulator of RAS ubiquitination and signaling, Science 362 (2018) 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, Abbasi Asbagh L, Pastor T, De Troyer M, Simicek M, Radaelli E, Brems H, Legius E, Tavernier J, Gevaert K, Impens F, Messiaen L, Nussinov R, Heymans S, Eyckerman S, Sablina AA, Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination, Science 362 (2018) 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Castel P, Cheng A, Cuevas-Navarro A, Everman DB, Papageorge AG, Simanshu DK, Tankka A, Galeas J, Urisman A, McCormick F, RIT1 oncoproteins escape LZTR1-mediated proteolysis, Science 363 (2019) 1226–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, Trinh QM, Peltekova VD, Reid JG, Tworog-Dube E, Morgan MB, Muzny DM, Stein L, McPherson JD, Roberts AE, Gibbs RA, Neel BG, Kucherlapati R, Next-generation sequencing identifies rare variants associated with Noonan syndrome, Proc Natl Acad Sci U S A 111 (2014) 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, Blair E, Borck G, Brinkmann J, Craigen W, Dung VC, Emrick L, Everman DB, van Gassen KL, Gulsuner S, Harr MH, Jain M, Kuechler A, Leppig KA, McDonaldMcGinn DM, Can NTB, Peleg A, Roeder ER, Rogers RC, Sagi-Dain L, Sapp JC, Schaffer AA, Schanze D, Stewart H, Taylor JC, Verbeek NE, Walkiewicz MA, Zackai EH, Zweier C, Members N of the Undiagnosed Diseases, M. Zenker, B. Lee, L.G. Biesecker, Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants, Genet Med 20 (2018) 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, Borck G, Salviati L, Zenker M, Cirstea IC, Tartaglia M, Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling, Hum Mol Genet 28 (2019) 1007–1022. [DOI] [PubMed] [Google Scholar]
- [136].Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, Abramowicz A, Cristian I, Buscarilli M, Naslavsky MS, Malaquias AC, Zatz M, Bodamer O, Majewski J, Jorge AA, Pereira AC, Kim CA, Passos-Bueno MR, Bertola DR, Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome, J Med Genet 52 (2015) 413–421. [DOI] [PubMed] [Google Scholar]
- [137].Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, Orci L, Eyaid W, Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking, Nat Genet 38 (2006) 1192–1197. [DOI] [PubMed] [Google Scholar]
- [138].Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L, The genetic basis of a craniofacial disease provides insight into COPII coat assembly, Dev Cell 13 (2007) 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW, Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation, Nat Genet 38 (2006) 1198–1203. [DOI] [PubMed] [Google Scholar]
- [140].Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ, Moon RT, The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation, Nat Cell Biol 8 (2006) 348–357. [DOI] [PubMed] [Google Scholar]
- [141].Patapoutian A, Reichardt LF, Roles of Wnt proteins in neural development and maintenance, Curr Opin Neurobiol 10 (2000) 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone EM, Gallinari P, Giorgi A, Steinkuhler C, Vitagliano L, Pedone C, Schinin ME, Screpanti I, Gulino A, Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation, Nat Cell Biol 12 (2010) 132–142. [DOI] [PubMed] [Google Scholar]
- [143].Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, Gallo R, Masuelli L, Napolitano M, Maroder M, Modesti A, Giangaspero F, Screpanti I, Alesse E, Gulino A, REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma, Proc Natl Acad Sci U S A 101 (2004) 10833–10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Gilbertson RJ, Ellison DW, The origins of medulloblastoma subtypes, Annu Rev Pathol 3 (2008) 341–365. [DOI] [PubMed] [Google Scholar]
- [145].Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Grobner S, Segura-Wang M, Zichner T, Rudneva VA, Warnatz HJ, Sidiropoulos N, Phillips AH, Schumacher S, Kleinheinz K, Waszak SM, Erkek S, Jones DTW, Worst BC, Kool M, Zapatka M, Jager N, Chavez L, Hutter B, Bieg M, Paramasivam N, Heinold M, Gu Z, Ishaque N, Jager-Schmidt C, Imbusch CD, Jugold A, Hubschmann D, Risch T, Amstislavskiy V, Gonzalez FGR, Weber UD, Wolf S, Robinson GW, Zhou X, Wu G, Finkelstein D, Liu Y, Cavalli FMG, Luu B, Ramaswamy V, Wu X, Koster J, Ryzhova M, Cho YJ, Pomeroy SL, Herold-Mende C, Schuhmann M, Ebinger M, Liau LM, Mora J, McLendon RE, Jabado N, Kumabe T, Chuah E, Ma Y, Moore RA, Mungall AJ, Mungall KL, Thiessen N, Tse K, Wong T, Jones SJM, Witt O, Milde T, Von Deimling A, Capper D, Korshunov A, Yaspo ML, Kriwacki R, Gajjar A, Zhang J, Beroukhim R, Fraenkel E, Korbel JO, Brors B, Schlesner M, Eils R, Marra MA, Pfister SM, Taylor MD, Lichter P, The whole-genome landscape of medulloblastoma subtypes, Nature 547 (2017) 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Arribat Y, Mysiak KS, Lescouzeres L, Boizot A, Ruiz M, Rossel M, Bomont P, Sonic Hedgehog repression underlies gigaxonin mutation-induced motor deficits in giant axonal neuropathy, J Clin Invest 129 (2019) 5312–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bury FJ, Moers V, Yan J, Souopgui J, Quan XJ, De Geest N, Kricha S, Hassan BA, Bellefroid EJ, Xenopus BTBD6 and its Drosophila homologue lute are required for neuronal development, Dev Dyn 237 (2008) 3352–3360. [DOI] [PubMed] [Google Scholar]
- [148].Sobieszczuk DF, Poliakov A, Xu Q, Wilkinson DG, A feedback loop mediated by degradation of an inhibitor is required to initiate neuronal differentiation, Genes Dev 24 (2010) 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kapitein LC, Hoogenraad CC, Building the Neuronal Microtubule Cytoskeleton, Neuron 87 (2015) 492–506. [DOI] [PubMed] [Google Scholar]
- [150].McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar RA, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, Roccanova P, Bhandari A, Pavon K, Lakshmi B, Leotta A, Kendall J, Lee YH, Vacic V, Gary S, Iakoucheva LM, Crow TJ, Christian SL, Lieberman JA, Stroup TS, Lehtimaki T, Puura K, Haldeman-Englert C, Pearl J, Goodell M, Willour VL, Derosse P, Steele J, Kassem L, Wolff J, Chitkara N, McMahon FJ, Malhotra AK, Potash JB, Schulze TG, Nothen MM, Cichon S, Rietschel M, Leibenluft E, Kustanovich V, Lajonchere CM, Sutcliffe JS, Skuse D, Gill M, Gallagher L, Mendell NR, Wellcome C Trust Case Control, Craddock N, Owen MJ, O’Donovan MC, Shaikh TH, Susser E, Delisi LE, Sullivan PF, Deutsch CK, Rapoport J, Levy DL, King MC, Sebat J, Microduplications of 16p11.2 are associated with schizophrenia, Nat Genet 41 (2009) 1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ, Autism C, Association between microdeletion and microduplication at 16p11.2 and autism, N Engl J Med 358 (2008) 667–675. [DOI] [PubMed] [Google Scholar]
- [152].Kuhlenbaumer G, Timmerman V, Bomont P, Giant Axonal Neuropathy, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews((R)), Seattle; (WA: ), 1993. [Google Scholar]
- [153].Allen E, Ding J, Wang W, Pramanik S, Chou J, Yau V, Yang Y, Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival, Nature 438 (2005) 224–228. [DOI] [PubMed] [Google Scholar]
- [154].Wang W, Ding J, Allen E, Zhu P, Zhang L, Vogel H, Yang Y, Gigaxonin interacts with tubulin folding cofactor B and controls its degradation through the ubiquitin-proteasome pathway, Curr Biol 15 (2005) 2050–2055. [DOI] [PubMed] [Google Scholar]
- [155].Bomont P, Degradation of the Intermediate Filament Family by Gigaxonin, Methods Enzymol 569 (2016) 215–231. [DOI] [PubMed] [Google Scholar]
- [156].Cleveland DW, Yamanaka K, Bomont P, Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway, Hum Mol Genet 18 (2009) 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Israeli E, Dryanovski DI, Schumacker PT, Chandel NS, Singer JD, Julien JP, Goldman RD, Opal P, Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy, Hum Mol Genet 25 (2016) 2143–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, Bomont P, Julien JP, Kuczmarski E, Opal P, Goldman RD, Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation, J Clin Invest 123 (2013) 1964–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, Kamiya A, Beckmann JS, Katsanis N, KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant, Nature 485 (2012) 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Govek EE, Newey SE, Van Aelst L, The role of the Rho GTPases in neuronal development, Genes Dev 19 (2005) 1–49. [DOI] [PubMed] [Google Scholar]
- [161].Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K, Rate of de novo mutations and the importance of father’s age to disease risk, Nature 488 (2012) 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].S. Deciphering Developmental Disorders, Large-scale discovery of novel genetic causes of developmental disorders, Nature 519 (2015) 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database C, Neale BM, Daly MJ, MacArthur DG, The mutational constraint spectrum quantified from variation in 141,456 humans, Nature 581 (2020) 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C, Analysis of protein-coding genetic variation in 60,706 humans, Nature 536 (2016) 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Williamson A, Werner A, Rape M, The Colossus of ubiquitylation: decrypting a cellular code, Mol Cell 49 (2013) 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Burslem GM, Crews CM, Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery, Cell 181 (2020) 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Simonetta KR, Taygerly J, Boyle K, Basham SE, Padovani C, Lou Y, Cummins TJ, Yung SL, von Soly SK, Kayser F, Kuriyan J, Rape M, Cardozo M, Gallop MA, Bence NF, Barsanti PA, Saha A, Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction, Nat Commun 10 (2019) 1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Verma R, Mohl D, Deshaies RJ, Harnessing the Power of Proteolysis for Targeted Protein Inactivation, Mol Cell 77 (2020) 446–460. [DOI] [PubMed] [Google Scholar]
- [169].Abu-Rashid M, Mahajnah M, Jaber L, Kornreich L, Bar-On E, Basel-Vanagaite L, Soffer D, Koenig M, Straussberg R, A novel mutation in the GAN gene causes an intermediate form of giant axonal neuropathy in an Arab-Israeli family, Eur J Paediatr Neurol 17 (2013) 259–264. [DOI] [PubMed] [Google Scholar]
- [170].Kuhlenbaumer G, Young P, Oberwittler C, Hunermund G, Schirmacher A, Domschke K, Ringelstein B, Stogbauer F, Giant axonal neuropathy (GAN): case report and two novel mutations in the gigaxonin gene, Neurology 58 (2002) 1273–1276. [DOI] [PubMed] [Google Scholar]
- [171].Normendez-Martinez MI, Monterde-Cruz L, Martinez R, Marquez-Harper M, Esquitin-Garduno N, Valdes-Flores M, Casas-Avila L, de Leon-Suarez VP, Romero-Diaz VJ, Hidalgo-Bravo A, Two novel mutations in the GAN gene causing giant axonal neuropathy, World J Pediatr 14 (2018) 298–304. [DOI] [PubMed] [Google Scholar]
- [172].Gladwyn-Ng I, Huang L, Ngo L, Li SS, Qu Z, Vanyai HK, Cullen HD, Davis JM, Heng JI, Bacurd1/Kctd13 and Bacurd2/Tnfaip1 are interacting partners to Rnd proteins which influence the long-term positioning and dendritic maturation of cerebral cortical neurons, Neural development 11 (2016) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Bomont P, Koenig M, Intermediate filament aggregation in fibroblasts of giant axonal neuropathy patients is aggravated in non dividing cells and by microtubule destabilization, Human molecular genetics 12 (2003) 813–822. [DOI] [PubMed] [Google Scholar]
- [174].Gladwyn-Ng IE, Li SS, Qu Z, Davis JM, Ngo L, Haas M, Singer J, Heng JI, Bacurd2 is a novel interacting partner to Rnd2 which controls radial migration within the developing mammalian cerebral cortex, Neural development 10 (2015) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Salinas GD, Blair LA, Needleman LA, Gonzales JD, Chen Y, Li M, Singer JD, Marshall J, Actinfilin is a Cul3 substrate adaptor, linking GluR6 kainate receptor subunits to the ubiquitin-proteasome pathway, The Journal of biological chemistry 281 (2006) 40164–40173. [DOI] [PubMed] [Google Scholar]
- [176].Chen Y, Li M, Interactions between CAP70 and actinfilin are important for integrity of actin cytoskeleton structures in neurons, Neuropharmacology 49 (2005) 1026–1041. [DOI] [PubMed] [Google Scholar]
- [177].Paciorkowski AR, Thio LL, Rosenfeld JA, Gajecka M, Gurnett CA, Kulkarni S, Chung WK, Marsh ED, Gentile M, Reggin JD, Wheless JW, Balasubramanian S, Kumar R, Christian SL, Marini C, Guerrini R, Maltsev N, Shaffer LG, Dobyns WB, Copy number variants and infantile spasms: evidence for abnormalities in ventral forebrain development and pathways of synaptic function, European journal of human genetics : EJHG 19 (2011) 1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Lee H, Ahn HH, Lee W, Oh Y, Choi H, Shim SM, Shin J, Jung YK, ENC1 Modulates the Aggregation and Neurotoxicity of Mutant Huntingtin Through p62 Under ER Stress, Molecular neurobiology 53 (2016) 6620–6634. [DOI] [PubMed] [Google Scholar]
- [179].Garcia-Calero E, Puelles L, Enc1 expression in the chick telencephalon at intermediate and late stages of development, The Journal of comparative neurology 517 (2009) 564–580. [DOI] [PubMed] [Google Scholar]
- [180].Kim EJ, Kim SH, Jin X, Jin X, Kim H, KCTD2, an adaptor of Cullin3 E3 ubiquitin ligase, suppresses gliomagenesis by destabilizing c-Myc, Cell death and differentiation 24 (2017) 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Boada M, Antunez C, Ramirez-Lorca R, DeStefano AL, Gonzalez-Perez A, Gayan J, Lopez-Arrieta J, Ikram MA, Hernandez I, Marin J, Galan JJ, Bis JC, Mauleon A, Rosende-Roca M, Moreno-Rey C, Gudnasson V, Moron FJ, Velasco J, Carrasco JM, Alegret M, Espinosa A, Vinyes G, Lafuente A, Vargas L, Fitzpatrick AL, Alzheimer’s Disease Neuroimaging I, Launer LJ, Saez ME, Vazquez E, Becker JT, Lopez OL, Serrano-Rios M, Tarraga L, van Duijn CM, Real LM, Seshadri S, Ruiz A, ATP5H/KCTD2 locus is associated with Alzheimer’s disease risk, Molecular psychiatry 19 (2014) 682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Traylor M, Adib-Samii P, Harold D, I. Alzheimer’s Disease Neuroimaging, U.K.Y.L.S.D.N.A.r. International Stroke Genetics Consortium, Dichgans M, Williams J, Lewis CM, Markus HS, Metastroke, i. International Genomics of Alzheimer’s Project, Shared genetic contribution to Ischaemic Stroke and Alzheimer’s Disease, Annals of neurology 79 (2016) 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Metz KA, Teng X, Coppens I, Lamb HM, Wagner BE, Rosenfeld JA, Chen X, Zhang Y, Kim HJ, Meadow ME, Wang TS, Haberlandt ED, Anderson GW, Leshinsky-Silver E, Bi W, Markello TC, Pratt M, Makhseed N, Garnica A, Danylchuk NR, Burrow TA, Jayakar P, McKnight D, Agadi S, Gbedawo H, Stanley C, Alber M, Prehl I, Peariso K, Ong MT, Mordekar SR, Parker MJ, Crooks D, Agrawal PB, Berry GT, Loddenkemper T, Yang Y, Maegawa GHB, Aouacheria A, Markle JG, Wohlschlegel JA, Hartman AL, Hardwick JM, KCTD7 deficiency defines a distinct neurodegenerative disorder with a conserved autophagy-lysosome defect, Annals of neurology 84 (2018) 766–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Moen MN, Fjaer R, Hamdani EH, Laerdahl JK, Menchini RJ, Vigeland MD, Sheng Y, Undlien DE, Hassel B, Salih MA, El Khashab HY, Selmer KK, Chaudhry FA, Pathogenic variants in KCTD7 perturb neuronal K+ fluxes and glutamine transport, Brain : a journal of neurology 139 (2016) 3109–3120. [DOI] [PubMed] [Google Scholar]
- [185].Staropoli JF, Karaa A, Lim ET, Kirby A, Elbalalesy N, Romansky SG, Leydiker KB, Coppel SH, Barone R, Xin W, MacDonald ME, Abdenur JE, Daly MJ, Sims KB, Cotman SL, A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system, American journal of human genetics 91 (2012) 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Blumkin L, Kivity S, Lev D, Cohen S, Shomrat R, Lerman-Sagie T, Leshinsky-Silver E, A compound heterozygous missense mutation and a large deletion in the KCTD7 gene presenting as an opsoclonus-myoclonus ataxia-like syndrome, Journal of neurology 259 (2012) 2590–2598. [DOI] [PubMed] [Google Scholar]
- [187].Argenti B, Gallo R, Di Marcotullio L, Ferretti E, Napolitano M, Canterini S, De Smaele E, Greco A, Fiorenza MT, Maroder M, Screpanti I, Alesse E, Gulino A, Hedgehog antagonist REN(KCTD11) regulates proliferation and apoptosis of developing granule cell progenitors, The Journal of neuroscience : the official journal of the Society for Neuroscience 25 (2005) 8338–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].De Smaele E, Di Marcotullio L, Moretti M, Pelloni M, Occhione MA, Infante P, Cucchi D, Greco A, Pietrosanti L, Todorovic J, Coni S, Canettieri G, Ferretti E, Bei R, Maroder M, Screpanti I, Gulino A, Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma, Neoplasia 13 (2011) 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Al-Mubarak B, Abouelhoda M, Omar A, AlDhalaan H, Aldosari M, Nester M, Alshamrani HA, El-Kalioby M, Goljan E, Albar R, Subhani S, Tahir A, Asfahani S, Eskandrani A, Almusaiab A, Magrashi A, Shinwari J, Monies D, Al Tassan N, Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families, Scientific reports 7 (2017) 5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Need AC, Ge D, Weale ME, Maia J, Feng S, Heinzen EL, Shianna KV, Yoon W, Kasperaviciute D, Gennarelli M, Strittmatter WJ, Bonvicini C, Rossi G, Jayathilake K, Cola PA, McEvoy JP, Keefe RS, Fisher EM, St Jean PL, Giegling I, Hartmann AM, Moller HJ, Ruppert A, Fraser G, Crombie C, Middleton LT, St Clair D, Roses AD, Muglia P, Francks C, Rujescu D, Meltzer HY, Goldstein DB, A genome-wide investigation of SNPs and CNVs in schizophrenia, PLoS genetics 5 (2009) e1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Jiang S, Seng S, Avraham HK, Fu Y, Avraham S, Process elongation of oligodendrocytes is promoted by the Kelch-related protein MRP2/KLHL1, The Journal of biological chemistry 282 (2007) 12319–12329. [DOI] [PubMed] [Google Scholar]
- [192].Jiang S, Avraham HK, Park SY, Kim TA, Bu X, Seng S, Avraham S, Process elongation of oligodendrocytes is promoted by the Kelch-related actin-binding protein Mayven, Journal of neurochemistry 92 (2005) 1191–1203. [DOI] [PubMed] [Google Scholar]
- [193].Soltysik-Espanola M, Rogers RA, Jiang S, Kim TA, Gaedigk R, White RA, Avraham H, Avraham S, Characterization of Mayven, a novel actin-binding protein predominantly expressed in brain, Mol Biol Cell 10 (1999) 2361–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]