

SUMMARY
Genotoxic stress in mammalian cells, including that caused by anti-cancer chemotherapy, can induce temporary cell cycle arrest, DNA damage-induced senescence (DDIS) or apoptotic cell death. Despite obvious clinical importance, it is unclear how the signals emerging from DNA damage are integrated together with other cellular signaling pathways monitoring the cell’s environment and/or internal state to control different cell fates. Using single cell-based signaling measurements combined with tensor PLSR/PCA analysis, we show that JNK and Erk MAPK signaling regulates the initiation of cell senescence through the transcription factor AP-1 at early times after doxorubicin-induced DNA damage, and the Senescence-Associated Secretory Phenotype at late times after damage. These results identify temporally distinct roles for signaling pathways beyond the classic DNA damage response that control the cell senescence decision and modulate the tumor microenvironment, and reveal fundamental similarities between signaling pathways responsible for oncogene-induced senescence and senescence caused by topoisomerase-II inhibition. A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
eTOC Blurb
Netterfield and colleagues used a two-pronged tensor PLSR and PCA approach to link upstream signaling events to downstream cell fates after topoisomerase-II inhibition in cancer cells, identifying roles for γH2AX in apoptosis, early JNK and Erk activity in senescence induction, and late JNK/Erk activity in cytokine secretion during the SASP.
Graphical Abstract
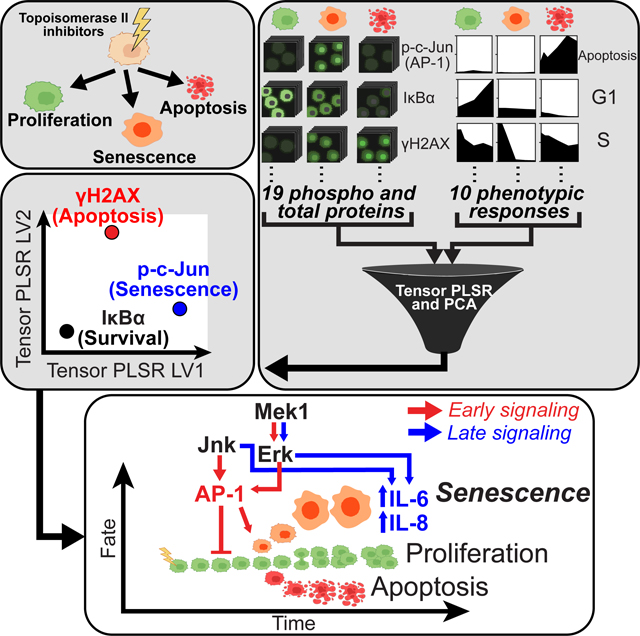
INTRODUCTION
Eukaryotic cells recognize and respond to DNA damage by activating an evolutionarily conserved set of signaling pathways that are essential for maintaining genomic integrity and preventing cancer1,2. These DNA Damage Response (DDR) signaling pathways regulate DNA damage induced-cellular activities and outcomes, including DNA repair, cell cycle arrest, senescence, and apoptosis3,4. Cellular senescence and apoptosis are actively regulated cellular responses that reduce the likelihood of cancer by preventing cells with genomic damage (or cells at risk of genomic damage) from proliferating5–8. Mutations and/or acquired defects that compromise the function of these DDR pathways result in enhanced mutagenesis, and underlie the development and progression of cancer9–11.
In the canonical DDR signaling pathway, double stranded breaks (DSBs) in DNA stimulate the kinase activity of ATM that phosphorylates and recruits a suite of proteins, including the histone variant H2AX, thereby creating detectable foci of DNA damage response proteins in the area adjacent to the DSB1–3,12. ATM effectors include the checkpoint kinases Chk2, Chk1 and MK2, and the multi-functional transcription factor p53, which together communicate DNA damage to the cellular machinery responsible for cell cycle arrest and the induction of programmed cell death13. p53 is a central node in the DDR signaling network that contributes to transient cell cycle arrest and senescence by up-regulating the cyclin-dependent kinase inhibitor p21Waf1 and to apoptosis by transactivation of pro-apoptotic Bcl-2 protein family members14–17. Tumor cells often have mutations in DDR components, including p53, which allows evasion of normal cell cycle control mechanisms and contributes to genomic instability. However, such defects can also sensitize tumor cells to killing and/or cell cycle arrest and senescence by classical DNA-damaging agents, such as ionizing radiation and chemotherapies used to treat cancer18–21.
Cellular senescence is a “catch-all” term that refers to three classes of irreversible cell cycle arrest – replicative senescence (RS), oncogene-induced senescence (OIS) and DNA damage induced senescence (DDIS)22. RS occurs after eukaryotic cells have undergone sufficient rounds of replication to cause exposure of unprotected telomeric DNA, resembling an un-repairable DNA double strand break (DSB). This triggers DDR signaling, resulting in permanent cell cycle arrest23–25. OIS occurs when oncogene expression results in inappropriately high levels of proliferation, leading to DNA replication stress and collapsed replication forks. The resulting DSBs induce a DDR-dependent permanent cell cycle arrest5,9. DDIS occurs after exposure to sub-apoptotic, “intermediate” levels of DNA damage inducing agents, such as ionizing radiation (IR) and doxorubicin, that are too high for repair and cell cycle re-entry but not high enough to induce cell death6,15. Senescent cells of all three classes are viable, metabolically active, enlarged and/or flattened in morphology, and strongly positive for cyclin dependent kinase inhibitors (CDKIs), persistent DNA damage induced foci (PDDF) and senescence-associated heterochromatic foci (SAHF)26. In addition, senescent cells secrete a panel of inflammatory cytokines featuring high levels of IL-6 and IL-827–31. Campisi and co-workers showed that this senescence-associated secretory phenotype (SASP) requires DDR28 and p38/NF-кB32 signaling. Whereas DDR signaling is fundamental to regulation of the senescent cell fate, the additional role of cytokine signaling pathways has not been as clearly defined.
DNA damage-induced cell commitment to either cell cycle arrest, senescence, or apoptosis likely involve integrating DDR signaling with additional signaling pathways governing general stress and survival responses such as the Akt pathway, the NF-kB pathway, and the stress- and mitogen-activated protein kinase (SAPK/MAPK) pathways. Since cell stress and extracellular signals are both transduced through these pathways, they may serve as information processing junctions that integrate signals from the microenvironment with DDR signaling. Previous studies have indicated roles for the ERK, JNK and p38 SAPK/MAPK pathways in cell fate determination after DNA damage33–37. However, the relative importance of these additional signal transduction pathways, and the manner and timing by which their signals are integrated together with those from the canonical DDR pathways to control the outcome of DNA-damaged cells, is not well understood. Therapeutic re-wiring of these pathways could lead to engineering of cellular outcomes and improving the clinical response of tumors to canonical genotoxic therapies38–42.
To investigate cell fate determination after DNA damage in a systematic manner, we undertook a quantitative time-resolved cell signaling and phenotypic response study in the U2OS osteosarcoma cell line exposed to different levels of doxorubicin-induced DNA damage with the intention of using data-driven models to suggest novel relationships between signaling activity and cellular outcomes. We were particularly interested in identifying signaling events that denoted different temporal stages on the paths to senescence and apoptosis. A senescing cell actively re-wires its molecular signaling networks to permanently arrest its cell cycle, become insensitive to serum stimulation, change its morphology, and produce the associated secretory phenotype. We sought to identify the signaling pathways and their activity dynamics that control the transition from a proliferating cancer cell to a senescent cell.
Here we report that the transcription factor AP-1, and its upstream activators, the stress and mitogen activated protein kinases JNK and Erk, play important roles in the cell fate decision to senesce at arrest-inducing (but non-lethal) doses of DNA damage at early times, and contribute to the DNA damage induced SASP at later times. Our findings derive from a structured, multidimensional dataset (tensor) of signaling and DNA damaged-induced responses collected in U2OS cells treated with various doses of doxorubicin at multiple times. The data were modeled by tensor- and matrix-factorization approaches to yield predictions about critical time-dependent signals, which were verified by independent experimental tests. Taken together, this systems analysis points to a fundamental role for SAPK/MAPK signaling in the regulation of DDIS and the SASP along separable distinct timescales, and reveals similarities between the signaling events responsible for both OIS and DDIS.
RESULTS
A Cue-Signal-Response Framework for Interrogating Cell Fate Choice After DNA Damage
DNA damaging agents induce cells to undergo cell cycle arrest, followed by either DNA repair and cell cycle re-entry, DNA damage induced senescence (DDIS) or apoptotic cell death. To quantitatively map the DNA damage cue-signal-response landscape43, we systematically profiled human U2OS osteosarcoma cells treated with a range of doxorubicin doses (Fig. 1A). Doxorubicin—a commonly used chemotherapeutic agent used to treat a variety of human malignancies including osteosarcoma—induces DNA double-strand breaks (DSBs) primarily by inhibiting topoisomerase II, but is also known to generate reactive oxygen species44 and act as an intercalating agent45. U2OS cells were selected for study because they are widely used in studies of DNA damage signaling37,40,46, express wild-type p53, and undergo a full range of DNA damage induced cellular responses, including p53-dependent cell cycle arrest and apoptosis. U2OS cells do not express the cyclin-dependent kinase inhibitor p16, because their INK4 locus has been silenced by methylation; nonetheless, this cell line is fully capable of undergoing senescence5,47.
Figure 1. A cue-signal-response framework for cell fate decisions after DNA damage.

A) Cartoon of cell fates of interest vs. time after DNA damage, created with BioRender.com.
B) A schematic of how the signaling and response data was collected, using icons created in BioRender.com. For details, see text.
C) U2OS cells were treated with a 4-hour pulse of either with DMSO (0 μM) or 0.5, 2, or 10 μM doxorubicin, and fixed 4 days after treatment. Hoechst and Whole Cell Blue (WCB) dye were used to visualize cell morphology, while immunofluorescence for BrdU DNA incorporation was used to label proliferating cells. Images are representative of 20–400 individual cells, depending on the doxorubicin dose.
D) β-galactosidase activity was measured by colorimetric staining in fixed U2OS cells 4 days after doxorubicin treatment. Black arrows denote positively stained cells. Images are representative of 100–500 individual cells, depending on the doxorubicin dose.
E) Cells were either treated with DMSO (0 μM) or with 0.5 μM doxorubicin, and then fixed 6 days later. Cell morphology was visualized with HCS CellMask Blue, and proliferation with EdU DNA incorporation. CellMask Blue channel was processed with a gamma of 0.5 to better visualize the cytoplasmic compartment. Images are representative of 100 (0.5 μM) and 1000 (0 μM) individual cells.
U2OS cells were treated with 0.5, 2 or 10 μM doxorubicin or carrier for 4 hours, followed by media replacement. Individual cells were assayed for signaling events and phenotypic outcomes at 6, 12, 24, 48, 72 and 96 hours after the start of treatment (Fig. 1B). In addition, cell morphology was examined by Whole Cell Blue staining, and cell proliferation assessed by BrdU labeling and immunohistochemical detection at 96 hours after treatment. Mock treated U2OS cells actively proliferated during the 4 days after treatment as shown by increased cell density and BrdU incorporation (Fig. 1C). In contrast, both the 0.5 and 2 μM doxorubicin treatments arrested proliferation, as shown by the absence of nuclear BrdU incorporation. These doses of doxorubicin induced a change in morphology that is consistent with cellular senescence—cell size and nuclear size increased over the 4 day time course, and the cells assumed a ‘fried egg’ appearance48. Interestingly, non-proliferating cells showed perinuclear BrdU staining after 24 hours of exposure to BrdU, likely resulting from BrdU incorporation into RNA49 that suggests metabolically activity is retained even though the cells are not proliferating. Further evidence indicating induction of cell senescence following treatment with these lower doses of doxorubicin was obtained by combined EdU labeling and CellMask Blue staining, and by staining the cells for β-galactosidase (Fig. 1D, E). In contrast, treatment with 10 μM doxorubicin resulted in profound cell death, with nearly all cells eliminated by day 4 of the time course (Fig. 1C).
To further quantify the cellular responses to varying doses of doxorubicin in a manner appropriate for distinguishing the behavior of distinct sub-populations of cells, we used quantitative live cell imaging (Incucyte™) to measure cell proliferation, and flow cytometry to monitor apoptotic and cell cycle responses at the single cell level. Flow cytometry measurements of apoptosis were performed by immunostaining the cells for simultaneous activation of the executioner caspase, caspase-3, and cleavage of PARP, a caspase-3 substrate50 (Fig. 2A). DMSO and 0.5 μM doxorubicin treatments did not induce apoptotic cell death, while 2 μM doxorubicin induced a small fraction of U2OS cells to undergo apoptosis at early times after treatment, but this early burst of apoptosis subsided by 72 hours after treatment (Fig. 2Bi and S1). In contrast, 10 μM doxorubicin treatment induced a significant fraction of cells to apoptose over the entire four-day time course with a biphasic response, resulting in the complete absence of proliferation and the death of nearly all cells by the conclusion of the experiment (Fig. 2Bi–ii). The cell number of DMSO treated cells continued to increase over the four-day time course, consistent with ongoing proliferation, which was eliminated by treatment with as little as 0.5 μM doxorubicin (Fig. 2Bii).
Figure 2. DNA damage induces G2/M arrest with dose-dependent differences between senescent p21hi/cyclin Blo cells and apoptotic p21lo cells.

A) Representative flow cytometry scatter plot measuring apoptotic cells with cleaved caspase-3 (CC3) and cleaved PARP (cPARP) double positivity in U2OS cells 48 hours after doxorubicin treatment.
B) Summary plots of mean phenotypic values (normalized to the maximum value across time and drug treatments) vs. time. Blue indicates a measurement that increases over time, red indicates a measurement that decreases, and gray indicates a measurement that remains the same. Tick marks represent 6, 24, 48, 72, and 96 hours after doxorubicin treatment. For boxplots of raw response data overlaid with replicate values, see supplemental figure S1.
C) Histograms of DNA content in U2OS cells stained with propidium iodide (PI) 48 hours after doxorubicin treatment. 2N and 4N DNA content are annotated on the x-axis of the histogram plots.
D) Representative flow cytometry scatter plots of BrdU antibody staining vs. PI staining in U2OS cells 48 hours after doxorubicin treatment.
E) Representative flow cytometry scatter plot of p21 antibody staining vs PI staining in U2OS cells 96 hours after doxorubicin treatment.
F) Representative flow cytometry scatter plot of cyclin B antibody staining vs PI staining in U2OS cells 96 hours after doxorubicin treatment.
Flow cytometry of propidium iodide-stained cells was used to monitor progression of U2OS cells through the cell cycle following treatment with doxorubicin (Fig. 2C). DNA replication activity was independently measured by analyzing the fraction of cells that incorporate BrdU in a 4-hour pulse (Fig. 2D). Mock-treated cells continue to proliferate, with cells distributed through all phases of the cell cycle. Cells treated with 0.5 μM and 2 μM doxorubicin proceeded through S phase and arrested with ~80% or more of the cells in G2/M and the remaining cells in G1 (Fig. 2C, D), which developed over the course of 24 to 48 hours (Fig. 2Biii–v and S1). In contrast, under these treatment conditions, cells treated with 10 μM doxorubicin did not develop a pronounced G2 arrest, but instead entered and remained arrested in S phase, where they incorporated only low levels of BrdU and did not progress to G2 (Fig. 2Biv–v, 2D). This finding is consistent with our previous work indicating that this type of pulse doxorubicin treatment induced U2OS cells to undergo apoptosis in early S phase35. Cells were then stained for the presence or absence of Cyclin B and the CDK inhibitor p21Waf1 (Fig. 2Bvi–ix, 2E, and 2F). Consistent with the observed G2 arrest, 0.5 and 2 μM dox treatments caused marked up-regulation of the p21Waf1, while the apoptosis-inducing 10 μM dose did not (Fig. 2Bix, E). Cells arrested in G2 initially possessed high levels of cyclin B as would be expected, however, concomitant with the increase in p21, the level of cyclin B in these cells decreased to that of G1 cells (Fig. 2Bvi–vii, ix, 2F, and S1), indicating that low and intermediate doses of doxorubicin caused the cells to gradually lose the ability to transition from G2 to M phase. Taken together, these data indicate that 0.5 μM and 2 μM doxorubicin treatments arrest proliferation by inducing a G2 arrest that prevents cells from progressing into mitosis. Similarly to other cancer cell lines in which p16 has been silenced by methylation51,52, a fraction of U2OS cells endoreduplicated in response to DNA damage as indicated by uptake of BrdU in cells containing >4N DNA (Fig. 2Bx).
To quantify the cell signaling response to different levels of DNA damage, 26 total measurements including the relative protein levels, protein phosphorylation, sub-cellular localization within the nuclear and cytoplasmic compartments, and heterogeneity between cellular sub-populations, for 19 signaling proteins representing key regulatory network nodes for cell cycle control, apoptosis, DNA damage response (DDR), and stress response signaling events (Fig. 3A) were quantified in 96-well plates using singleplex immunofluorescence microscopy at 6 time points: 6, 12, 24, 48, 72 and 96 hours after doxorubicin treatments. Representative signaling data at the 48 hour time point are shown in Figure 3B, with the complete quantified time courses shown in Figures 3C, S2 and S3. As expected, treatment of U2OS cells with doxorubicin strongly activated the DNA damage response, including up-regulation of γH2AX, stabilization of p53, and phosphorylation of the effector kinase Chk2 (Fig. 3B, C and S3). Interestingly, the DDIS-inducing doses of doxorubicin (0.5 and 2 μM) increased the level of p53 more than the apoptosis-inducing dose (10 μM). In addition, these DDIS-inducing doses induced a large increase in the level of the cyclin dependent kinase inhibitor p21Waf1, which likely contributes to the U2OS cell cycle arrest, given the silencing by methylation of the p16 gene in this cell line (Fig. 3B, C)47.
Figure 3. Quantitative single cell measurements define the DNA damage signaling landscape.

A) A wiring diagram of the signaling network that modulates cell-cycle progression and apoptosis after DNA damage, created with BioRender.com.
B) Representative immunofluorescence images of a subset of signaling proteins measured 48 hours after doxorubicin treatment. Each field of view represents a distinct well on a 96-well plate that was immunostained for 1 total or phosphoprotein. Images representative of 250–6000 individual cells per biological replicate, depending on the dose of doxorubicin.
C) Quantification of the mean fluorescence intensity (normalized to the maximum value across time and drug treatments) over time in either the nuclear or cytoplasmic compartment, depending on the given protein measured. Blue indicates a measurement that increases over time, red indicates a decreasing measurement, and gray indicates a measurement that has remained the same. Tick marks on the x-axis represent 6, 24, 48, 72, and 96 hrs. For boxplots of raw signal data overlaid with replicate values, see supplemental figure S2.
Cell cycle arrest is a fundamental component of the DNA damage response. Consequently, we monitored the levels and localization of cyclins A, B, D and E, as well as phosphorylation of the retinoblastoma protein (Rb) (Fig. 3B, C and S3). In DMSO treated cells, the levels of these cell cycle regulating proteins remained largely unchanged for the 4 day duration of the experiment. The G2 arrest of the cells treated with 0.5 μM and 2 μM doxorubicin was reflected in the accumulation of cyclins A and B. Interestingly, U2OS cells undergoing DDIS accumulate cyclin E, indicating that they may be primed for another round of DNA replication. This is consistent with our previous data that HCT116 cells, which like U2OS cells have wild-type p53 and lack p16, also arrest in G2 after DNA damage and accumulate cyclin E, putting them in a 4N pseudo-G1 state that likely contributes to their propensity for endoreduplication52. A small percentage (3–4%) of U2OS cells also endoreduplicated after DNA damage as shown by a population of 8N cells after the 0.5 and 2 μM doxorubicin treatments (Fig. S1).
Since signal transduction pathways other than the canonical DDR signaling network also contribute to cell fate determination, we measured the post-translational modifications and proteins levels reflective of Akt, Erk, JNK, p38 and NF-κB activity (Fig. 3B, C and S2). These pathways exhibit complicated, time- and doxorubicin-dose dependent behaviors that required data-driven modeling to integrate with the canonical DDR signaling network and downstream cell-fate responses 53.
A Tensor PLSR Model Distinguishes Alternative Cell Fates after DNA Damage
To relate these complex time- and dose-dependent changes in cellular signaling to the observed phenotypic responses, we used tensor partial least square regression (t-PLSR) to identify signals, responses, and time points that correlate with specific cell fates. Traditional “unfolded” PLSR is a widely used dimensional reduction modeling method in which the relationship between measured signaling events and phenotypic responses is inferred from maximizing the covariance between the two50,54. Independent signals, dependent cellular responses, and timepoints are weighed separately in the PLSR matrix formulation. In contrast, t-PLSR specifically preserves the natural structure of the data, regressing the stimulus-timepoint-response/cell fate tensor on the stimulus-timepoint-signaling tensor, linking these tensors via regression coefficients55,56. In addition to providing insight into how particular aspects of temporally evolving signaling activities are important for making predictions, tensor PLSR models use fewer parameters than unfolded PLSR, resulting in less overfitting, which is of particular importance when modeling a small number of treatments, as in our case.
In t-PLSR, the signal (X) and response (Y) tensors are simultaneously decomposed into three distinct individual matrices for each tensor: treatment scores (sx or sy), signal (wsx) or response (wsy) weights, and time weights (wtx or wty) (Fig. 4A). To generate these matrices, the signaling tensor X and the cellular response tensor Y are jointly factored using a linear relationship between sx and sy in which the regression coefficients matrix is the slope within this relation, and this factorization occurs iteratively until the covariance between X and Y has been maximized. Upon convergence, these values are considered the score and weight values for latent variable #1, which is conceptually analogous to principal component 1 in traditional unfolded PLSR. The residuals are then subtracted from X and Y to compose the tensors used for the next round of factorization, which will be used to compute latent variable #2, and so on until a majority of the variance in the data has been explained. Treatment scores describe where certain treatments fall in the latent variable space, while signal and response weights describe the contribution of each individual signal or response to a specific latent variable. Time weights offer additional insight into which timepoints are weighted more heavily across all signals and responses in constructing a specific latent variable, information that is difficult to parse in traditional unfolded PLSR. As shown in figure 4A, the appropriate product of these scores and weights, when added over all latent variables, recapitulates the original signaling or response tensor.
Figure 4. A Tensor PLSR model identifies latent variables that define a survival-apoptosis axis, and a cycling-senescence axis.

A) A schematic of the “tensor” PLSR (t-PLSR) algorithm. The transpose of Khatri-Rao product of the computed wsx and wtx multiplied by the computed sx should be able to fairly recapitulate the original tensor, as illustrated.
B) Bar plots of the percent variance explained by each latent variables (LV).
C) The root mean square of the prediction (RMSEP) of when adding each LV to the model.
D) Experimental vs. predicted scatter plots for the values used for calibration (left) and those used for leave-one-out cross-validation (right). Error bars represent standard error of the mean (SEM). Computed R2, Q2, and Pearson correlation are also shown.
E) Treatment scores from the signal tensor visualized on a scatter plot of latent variable #2 vs. latent variable #1.
F) Response weights from the model plotted on a scatter plot of latent variable #2 vs. latent variable #1. Solid gray line indicates the mean (μ) of 500 null models, while dotted gray lines indicate +/− 1 standard deviation (σ) from the average of null models (see text for details).
G) Signal weights from the model plotted on a scatter plot of latent variable #2 vs. latent variable #1. Solid gray lines indicate the mean (μ) of signal weights in the model. Dotted lines indicate one standard deviation (σ) from the mean.
H) Bar plot of variable importance in projection (VIP) scores of signals. Underlining of the signal name indicates a cytoplasmic signal, while the absence of underlining indicates a nuclear signal. Blue bars indicate significant senescent signals, red bars indicate significant apoptotic signals, black bars indicate significant proliferation signals, gray bars indicate non-significant signals as seen in t-PLSR.
A t-PLSR model containing three latent variables captured greater than 80% of the variance in the response data while also minimizing the root-mean square error of prediction (Fig. 4B and 4C), with over 75% of the variance explained by latent variables #1 (LV1) and 2 (LV2). There was good concordance between the predicted and experimentally observed phenotypic responses during both model calibration and cross-validation (Fig. 4D). The largest discrepancies between the experimental and predicted values were observed at the 2 μM doxorubicin dose, likely due in part to the heterogeneity of cell fate responses observed at this particular dose (~25% cumulative apoptosis and ~75% senescence) (Fig. S4A). When examining different phenotypic responses, rather than specific drug doses, the model performed best at predicting the percentage of cells in G1, S and G2/M in both the calibration and cross-validation data with Q2 values greater than 0.64 (Fig. S4B).
To gain insight into how these latent variables correlated with cell fates, the treatment scores and the response weights from the model were explored. Plotting the treatment scores on LV1 vs LV2 revealed that DMSO treatment fell in the LV 1 and 2 negative quadrant, while the treatment scores of both doses of doxorubicin that induced senescence (0.5 and 2 μM) were LV1 positive and LV2 negative. Treatment with the 2 μM dose, which induces a more heterogeneous mix of senescent and apoptotic cells, projected less positively on LV1 than did the 0.5 μM dose. In contrast, the treatment score of the apoptotic dose of doxorubicin (10 μM) projected negatively on the LV1 axis, but was strongly LV2 positive (Fig. 4E). Thus, treatments involving senescent doses of this genotoxic agent are distributed along LV1 while LV2 distinguished the apoptotic dose treatment from the rest.
To further refine the biological meaning of LV1 and 2, the cellular response weights were plotted, and the significance of their projections on these axes evaluated using statistical bootstrapping (Fig. 4F)57. 500 separate null models were constructed from randomly shuffled data, and the observed response weights corresponding to the real data then compared with those obtained from the null models. Responses whose weights were greater than one standard deviation from the mean of the null models were considered significant. Using this cut-off, proliferation emerged as significant within the negative LV1 and 2 quadrant, while the G2 and G2 p21+ state emerged as the only significant responses that were LV1 positive (Fig. 4F). This observation indicates that G2 arrested/senescent cells are separated from cycling proliferative cells by progression along the LV1 axis, in excellent agreement with the distribution of senescence-inducing treatment scores along this axis (2 μM and 0.5 μM doxorubicin; Fig. 4E). In contrast, apoptosis emerged as the only response that was significantly positive along the LV2 axis, in excellent agreement with the observation that the treatment score for the apoptotic dose of doxorubicin projected strongly in the positive LV2 direction. Measurements of the G1- and S-phase cell populations were not significantly distributed along the LV2 axis, they were significant on the negative LV1 axis (Fig. 4F), consistent with the location of actively cycling cells. The apoptosis response was narrowly below the cut-off for significance on the LV1 axes, which is consistent with the observation that cells treated with the apoptotic dose of doxorubicin maintained steady levels of cells in G1 and S that were not dissimilar from cells treated with the DMSO vehicle control (Fig. 2Biii–iv). Taken together, these observations, paired with the treatment scores, indicate that LV1 reflects a cycling versus senescence axis, while LV2 reflects survival versus apoptosis.
Tensor PLSR and PCA Identify Signaling Pathways that Dictate Cell Fate
Interrogation of where specific molecular signals fall on the LV1 and 2 axes of the model can infer potential causal relationships between signaling pathways and cell fates. Signals contributing strongly to the model were identified by weightings on LV1 or LV2 that were one or more standard deviations above the mean signal cut-off (Fig. 4G), and the significance of contributing signals further assessed using Variable Importance in Projection (VIP) scores (Fig. 4H)58. Both nuclear cyclin D and cytoplasmic IκBα levels were strongly negatively weighted on LV1 with high VIP scores, thus significantly correlating with cell proliferation (Fig. 4G). These two elements in the t-PLSR model are in strong agreement with our interpretation of the LV1 axis, since the G1 cyclin, Cyclin D, is well-established as a key regulator of cell proliferation59,60, while activation and nuclear translocation of NF-κB, which is inhibited by IκBα, has been shown to be elevated in senescent cells61–63.
Nuclear γH2AX and phospho-Hsp27, together with levels of activated p38MAPK, JNK, and Erk in the nucleus projected positively along LV2, correlating with apoptosis (Fig. 4G). These findings further support our biological interpretation of the LV2 axes, since γH2AX intensity reflects the extent of DNA damage, and we and others have shown previously that p38MAPK (along with its downstream targets MAPKAP Kinase-2 and Hsp27), Erk, and JNK have complex, context-dependent roles in cellular stress and DNA damage responses34,35,37,64–66, with JNK commonly associated with certain types of stress-associated cell death67–70. Furthermore, Hsp27 is a molecular chaperone that is a direct substrate of the p38MAPK-MAPKAP Kinase-2 (MK2) signaling axis activated downstream of DNA damage, which we and others have previously shown to be phosphorylated after DNA damage37,71,72. Thus, the projection of phospho-Hsp27 along LV2 further confirms the strong correlation between the p38MAPK pathway and apoptosis in our system.
Finally, nuclear levels of phospho-Chk2, phospho-Rb, Cyclin E, Cyclin A, p53, and p21Waf1 projected strongly along LV1 (Fig. 4G), had some of the highest VIP scores amongst the positive LV1 signal weights (Fig. 4H), and closely correlated with cell senescence. Chk2 has been previously associated as a driver of replicative senescence73,74. p53, a critical regulator of senescence75–77, transcriptionally upregulates expression of p21Waf1 78, which is both a canonical marker of senescence and the CDK inhibitor likely contributing to cell cycle arrest in this system79–81. This rationalizes the observed high levels of Cyclins E and A, in these 4N G2-arrested p21+ cells (Fig. 4G, F). Phosphorylation of Rb releases E2F transcription factors to facilitate progression of 2N G1 cells through S-phase82–85, consistent with the observed accumulation of senescent cells in a 4N G2-arrested state. The most unanticipated finding, however, was the very strong contribution of nuclear phospho-c-Jun to LV1, where it emerged as the strongest correlate with cell senescence based on the projection of its weight on LV1 and its VIP score, ranking as a more important contributor than p21Waf1 (Fig. 4G and H). c-Jun is a major component of the AP-1 transcription factor family, and has been well-characterized as a modulator of cell proliferation, cell-cycle progression, and cell death in a context-dependent manner86–89. However, its potential role in modulating DNA damage-induced cell senescence has not been explored.
Phosphorylation and nuclear translocation of c-Jun is known to be mediated by JNK, Erk, and p38MAPK90–93, suggesting that the time-dependent activity of one or more of these kinases might be involved in regulating DDIS. Because time weights in t-PLSR reflect the aggregate measurements contributed by all signals or responses at one particular point in time, and are not amenable for dissecting the specific time-dependent contributions of any individual molecular signal, we instead applied principal component analysis (PCA) to the signaling data to parse how individual signals co-varied and separated by time point. Two principal components (PCs) captured greater than 70% of the variance in the signaling data. When each of the doxorubicin treatments was scored along these signal-generated PCs, the senescence-inducing doses (0.5 and 2 μM) mainly segregated positively along PC1 with respect to the origin in PC space (Fig. 5A, blue vector), while the apoptotic dose (10 μM) separated negatively along PC1 and positively along PC2 (Fig. 5A, red vector).
Figure 5. Kinases upstream of c-Jun, JNK and Erk, regulate senescence after DNA damage.

A) PCA scores of doxorubicin treatments plotted on a scatterplot of principal component 2 (PC 2) vs. principal component 1 (PC 1).
B) Schematic of PCA loading vector (dotted arrow) in relation to apoptosis axis (red arrow) and senescence axis (blue arrow), the angles between the loading vector and senescence axis (blue theta), and the loading vector and apoptosis axis (red theta). The cosine of the blue theta is the projection along the senescence axis, and the cosine of red theta is the projection along the apoptosis axis.
C-F) PCA loadings and projections of significant C) senescent signals, D) survival signals, E) apoptotic signals, and F) time-dependent signals. Larger PCA loadings correspond to later timepoints. Apoptosis projections (red curves) and senescence projections (blue curves) were plotted over time for each signal.
G) Representative immunofluorescence of cells treated with doxorubicin and either DMSO, 10 μM SP600125, 10 μM PD98059, 10 μM SB203580, or 5 mM caffeine. Doxorubicin was left on for 4 hours and then washed off, while inhibitors were left on the entire duration of the six-day experiment. Cell morphology was visualized with Hoechst staining and Whole Cell Blue (WCB) dye, and proliferative cells were visualized with nuclear BrdU antibody staining. Images representative of 150–5,000 individual cells per biological replicate, depending on the condition.
H) Quantification of the percent of cells with that are positive for nuclear BrdU in the DMSO, 10 μM SP600125, and 10 μM PD98059 co-treatment conditions 6 days after doxorubicin treatment. Bars represent mean of three biological replicates, error bars represent SEM. ***: p < 0.001, ns: p > 0.05 with a two-way ANOVA and post-hoc Dunnett’s test vs. DMSO inhibitor control at the same dose of doxorubicin.
I) Representative immunofluorescence for p-c-Jun(Ser63) and γH2AX of cells treated in the same conditions as in subpanel E. Images representative of 150–5,000 individual cells per biological replicate, depending on the condition.
PCA loadings of each of the molecular signals previously determined to be statistically significant contributors to LV1 and 2 by t-PLSR were then examined for their contributions to PC1 and PC2. In order to quantitatively evaluate the extent of correlation of each PCA loading with either senescence or apoptosis, we defined senescence and apoptosis axes in PC space using the 0.5 μM and 10 μM treatment scores, respectively, since these were the most homogenous senescence and apoptosis-inducing doses (Fig. 5A). We then vectorized each PCA loading with respect to the PC1 and 2 origin and calculated the projection of this loading vector with these senescence or apoptosis axes (Fig. 5B).
Senescence and apoptosis projections were calculated for each PCA loading, and plotted over time to gain insight into time-dependent associations. This revealed that several signals identified by t-PLSR as significantly correlated with senescence or apoptotic cell fates mapped onto the corresponding PCA senescence and apoptosis axes across the majority of sampled timepoints. Levels of phospho-c-Jun, and cyclins A and E, for example, projected strongly along the PCA senescence axis at nearly all timepoints (Fig. 5C, lower panels), further confirming their correlation with senescence as revealed in the t-PLSR analysis (Fig. 4G). Cyclin D and IκBα, which were correlated with survival in the t-PLSR analysis, had the most negative projections along the apoptosis axis of any other signal at intermediate and late timepoints, further suggesting that these signals are highly anti-correlated with apoptosis (Fig. 5D, lower panels). In contrast, γH2AX had larger projections along the apoptosis axis than projections along the senescence axis at most timepoints, consistent with its correlation with apoptosis as seen in t-PLSR (Fig. 5E, lower panel).
The remaining signals that were significantly weighted on LV1 and 2 in t-PLSR exhibited complex, time-dependent correlations with senescence and apoptosis. Signals such as phospho-Chk2 and p21 had positive projections along the apoptosis axis at early times, but negative projections along the apoptosis axis and strong positive projections along the senescence axis at middle and late timepoints, while phospho-Hsp27 and phospho-Rb had one and two timepoints, respectively, where the senescence or apoptosis projections dramatically crossed over. p53 projected positively along both the apoptosis and senescence axes in PC space, with a stronger apparent projection towards senescence (Fig. 5F). This fits with the trends seen in the immunofluorescence data, which show that while there is lesser p53 accumulation with the 10 μM dose in comparison to the 0.5 and 2 μM doses, the p53 levels at this dose are still ~2 fold greater than the vehicle condition (Fig. 3C, S2). This correlation with both senescence and apoptosis is consistent with the known role for p53 in both senescence and apoptosis94–96.
Both JNK and Erk MAPKs showed strong projections along the senescence axis at very early times. This was subsequently followed by strong projections for both MAPKs along the apoptosis axis at intermediate times, and increased projection along the senescence axis again at late times (Fig. 5F, lower panels). In contrast, p38MAPK exhibited highly oscillatory apoptosis and senescence axis projections that continued to cross over at middle and late timepoints. Taken together, these results suggest nuanced, time-dependent roles for the majority of signals significantly weighted on LV1 and 2 in t-PLSR, particularly the p38MAPK, JNK, and Erk MAPKs in DDIS.
Early, but not Late JNK and Erk Signaling Controls Senescence After Low-Dose Doxorubicin Treatment
The strong correlation of phospho-c-Jun levels with cell senescence observed after doxorubicin treatment (Figs. 4G and 5C), together with the potential time-dependent roles of JNK, Erk, and p38MAPK in senescence, as revealed by the PCA analysis led us to experimentally investigate whether p38MAPK, Erk, and JNK directly contribute to regulating DNA damage-induced senescence through phosphorylation of c-Jun. U2OS cells were treated with doxorubicin and the kinase activities of p38, Mek1, and JNK blocked with the specific small molecule inhibitors SB203580, PD98059, and SP600125, respectively, with kinase inhibition validated by Western blotting (Fig. S5A–G). In these experiments, U2OS cells were treated with a 4 hour doxorubicin pulse, and given fresh media on days 1 and 3 post-damage. Kinase inhibitors were applied simultaneously with doxorubicin and maintained throughout the 6 day duration of the experiment. Proliferation and senescence were evaluated by measuring the extent of BrdU incorporation over 24 hours starting 5 days after doxorubicin treatment. As observed previously, DMSO-treated U2OS cells showed strong BrdU uptake in spite of reaching high cell densities, while 0.5 μM doxorubicin treatment alone drives DDIS as indicated by morphological changes and the lack of BrdU incorporation into nuclear DNA (Fig. 5G). Inhibition of JNK using SP600125 markedly abrogated this DDIS response. In contrast, following treatment with 2 μM doxorubicin, the DDIS could not be reversed by SP600125 (Fig. 5G and H). Blocking Erk signaling by inhibiting Mek1 similarly abrogated the senescence response to 0.5 μM doxorubicin, but, as was the case for JNK inhibition, PD98059 did not abrogate 2 μM doxorubicin-induced senescence (Fig. 5G and 5H). These pharmacologic perturbations suggested a role for Erk and JNK in DDIS triggered by low-dose doxorubicin.
To simultaneously visualize DNA damage signaling and signaling through c-Jun on a cell-by-cell basis, cells were co-stained for the γH2AX and p-c-Jun(Ser-63) (Fig. 5I). DMSO-treated cells exhibited a uniform, small size and low levels of γH2AX and phospho-c-Jun, while treatment with 0.5 μM doxorubicin induces a population of large, flat senescent cells that stained positively for γH2AX and phospho-c-Jun (Fig. 5I). Although some heterogeneity in the extent of phospho-c-Jun staining was noted in the senescent cells, treatment with either SP600125 or PD98059, which abrogated DDIS, markedly reduced γH2AX staining and nearly completely eliminated nuclear phospho-c-Jun staining.
It should be noted that the abrogation of 0.5 μM doxorubicin-induced senescence by either SP600125 or PD98059 was not complete. The sub-population of cells that escaped senescence and proliferated after exposure to 0.5 μM doxorubicin upon treatment with either SP600125 or PD98059 were characterized by “normal” U2OS morphology, incorporation of BrdU, and low levels of both γH2AX and phospho-c-Jun (Fig. 5G and I). In contrast, the residual sub-population of senescent cells that persisted demonstrated an enlarged “fried-egg” cellular morphology, lack of nuclear BrdU incorporation, and increased levels of both γH2AX and phospho-c-Jun, further supporting the correlation of phospho-c-Jun with induction of DDIS. Inhibition of p38MAPK with SB203580 failed to suppress DNA damage-induced senescence, nuclear γH2AX intensity, or phospho-c-Jun staining, while addition of 5 mM caffeine, which served as a positive control, abrogated all three, as would be expected from its ability to simultaneously inhibit all three of the DNA damage response kinases ATM, ATR and DNA-PK (Fig. 5G and I). Taken together, we interpret these data as evidence that signaling though JNK and Erk, but not p38MAPK, plays a causal role in the induction of DDIS, likely through the phosphorylation of c-Jun (see below).
The DNA damage-induced senescent state, which is characterized by morphological changes, elevated cyclins E and A, stable G2 arrest, and high levels of p21 emerges 3–4 days after DNA damage (Fig. 1–3). This multi-day time course between DNA damage and the emergence of the senescence state suggests a series of dynamic temporally-regulated signaling events and regulatory transitions that coordinate progression to senescence. Notably, in our PCA analysis, both JNK and Erk had strong senescence-associated signals 6 hours after doxorubicin treatment, suggesting possible early pro-senescence roles for JNK and Erk activity (Fig. 5F). To experimentally investigate at what point after DNA damage JNK and Erk signaling control the senescence cell fate decision, small molecule inhibitors were added either during the first 12 hours after the onset of doxorubicin-induced DNA damage, or added 12 hours after doxorubicin treatment and removed at 24 hours (Fig. 6A). As shown in figures 6B and C, addition of the JNK inhibitor SP600125, the Mek1 inhibitor PD98059, or caffeine, during the first 12 hours after DNA damage significantly reduced the fraction of cells that underwent DDIS in response to the 0.5 μM doxorubicin treatment, with a smaller but still significant inhibition of DDIS in response to the 2 μM treatment as well. The p38MAPK inhibitor SB203580 did not reverse the DDIS phenotype, consistent with the prior results (Fig. 5G). In contrast, if added 12 hours after genotoxic stress, the JNK and Mek inhibitors and caffeine were unable to abrogate the DDIS in U2OS cells (Fig. 6D and E). Taken together, these observations suggest that JNK, Erk and DNA damage signaling – but not p38 signaling – are required within the first 12 hours after DNA damage to initiate DDIS after low dose doxorubicin treatment.
Figure 6. JNK and Erk signaling in the first 12 hours after DNA damage controls the senescence-proliferation decision, while late JNK and Erk signaling controls the SASP.

A) Schematic of experimental workflow for U2OS and NCI-H1299 cells. (Icons created with BioRender.com)
B) Representative immunofluorescence (IF) of BrdU incorporation into DNA in U2OS cells treated with inhibitors (10 μM for SP600125, PD98059, and SB203580 drugs, and 5 mM of caffeine) for 0–12 hours and co-stained with HCS CellMask Blue. Images representative of 650–30,000 individual cells per biological replicate, depending on the condition.
C) Quantification of the percent of nuclear BrdU+ in U2OS cells after the 0–12 hour inhibitor condition.
D) Representative IF of BrdU incorporation into DNA in U2OS cells treated with inhibitors for 12–24 hours and co-stained with HCS CellMask Blue. Images representative of 650–30,000 individual cells per biological replicate, depending on the condition.
E) Quantification of the percent of nuclear BrdU+ in U2OS cells after the 12–24 hour inhibitor condition.
F) Representative IF of BrdU incorporation into DNA in NCI-H1299 cells treated with inhibitors (10 μM for SP600125, PD98059, and SB203580 drugs, and 1 mM of caffeine) for 0–12 hours and co-stained with HCS CellMask Blue. Images representative of 1,500–15,000 individual cells per biological replicate, depending on the condition.
G) Quantification of the percent of nuclear BrdU+ in NCI-H1299 cells after the 0–12 hour inhibitor condition.
H) Representative IF of BrdU incorporation into DNA in NCI-H1299 cells treated with inhibitors for 12–24 hours and co-stained with HCS CellMask Blue. Images representative of 1,500–15,000 individual cells per biological replicate, depending on the condition.
I) Quantification of the percent of nuclear BrdU+ in NCI-H1299 cells after the 12–24 hour inhibitor condition.
J) IL-6 and K) IL-8 levels in culture media of U2OS cells 2, 4, and 6 days after doxorubicin treatment as measured by ELISA.
L) Schematic of experimental workflow for ELISA-based co-inhibitor experiment
M) IL-6 and N) IL-8 levels as measured by ELISA in U2OS tissue culture media 6 days after doxorubicin addition. DMSO inhibitor control bars in subpanels M and N are the same values as seen in subpanels J and K six days after doxorubicin. For all bar graphs in this figure, bars represent mean ± SEM of three biological replicates. ***: p < 0.001, **: p < 0.01 *: p < 0.05, ns: p > 0.05 with a two-way ANOVA and post-hoc Dunnett’s test for subpanels C, E, G, I, M, and N. All comparisons were vs. the DMSO treatment at the same dose of doxorubicin.
To examine whether the roles of JNK and Erk signaling in driving DDIS were unique to U2OS cells, similar studies were performed in NCI-H1299 non-small cell lung cancer cells, a tumor type that is commonly treated with doxorubicin in combination with other cytotoxic drugs97–99. Treatment with 2 μM of doxorubicin resulted in cells with enlarged nuclei and cytoplasm (i.e. fried egg appearance) that failed to incorporate nuclear BrdU (Fig. 6F and G) and exhibit high levels of the CDK inhibitors p21 and p27 (Fig. S6A). Addition of SP600125, PD98059, or caffeine during the first 12 hours after doxorubicin treatment resulted in a decrease in cells that underwent DDIS, indicating that JNK, Erk, and DDR inhibition early after damage can reverse the DDIS in this cell type (Fig. 6F and G), recapitulating the results obtained in U2OS cells. Similar to what was observed in U2OS cells, addition of JNK or Mek1 inhibitors, or treatment with caffeine, was unable to abrogate DDIS in NCI-H1299 cells if these were added later than 12 hours after doxorubicin treatment (Fig. 6H and I). Similar results were seen in OVCAR-8 high grade serous ovarian cancer cells and HUVEC cells, which are non-cancerous primary endothelial cells (Fig. S6B–J). Together, these data indicate that, in multiple cell lines cells, JNK and Erk signaling contribute to the early information processing that results in DDIS, and this commitment is made within 12 hours after doxorubicin treatment.
Late JNK and Erk Activity Contribute to the Secretory Associated Secretory Phenotype
Phospho-JNK and phospho-Erk levels are elevated at early timepoints, and the activity of these kinases influences the senescence/proliferation fate decision (Fig. 5 and 6A–I). However, as noted previously, the projections for JNK and Erk along the senescence axis also become positive at late timepoints in our PCA analysis (Fig. 5F), raising the question of whether there may be another senescence-associated function for these signaling pathways at late times. Since senescent cells are known to secrete pro-inflammatory cytokines, particularly IL-6 and IL-8, in response to oncogenic stress or DNA damage28,30,31,100, and the JNK and Erk pathways are known to regulate cytokine secretion and mediate cytokine signaling in response to other non-DNA damage stimuli101,102, we hypothesized that the late phase JNK and Erk signaling after genotoxic stress might contribute to regulating the DDIS SASP.
To examine this, U2OS cells were treated with senescence-inducing doses of doxorubicin, and the levels of IL-6 and IL-8 assayed in the media at 2, 4, and 6 days after treatment. As shown in Fig. 6J and 6K, both 0.5 μM and 2 μM doxorubicin treatment induced IL-6 and IL-8 secretion that became significantly elevated by 6 days after treatment. Cells treated with 0.5 μM doxorubicin secreted more IL-6 and IL-8 than cells treated with 2 μM doxorubicin, and the kinetics of cytokine release came after the senescence-associated morphology change. Addition of JNK or Mek1 inhibitors was therefore performed 72 hours after DNA damage to allow for the initiation of senescence, but prior to the detection of SASP-associated cytokines (Fig. 6L). As shown in figure 6M and N, addition of either the JNK inhibitor SP600125, or the Mek1 inhibitor PD98059 reduced the senescence-associated secretion of IL-6 and IL-8. Together with the data in panels A-I, this data indicates that JNK and Erk signaling regulate two different properties of cells undergoing DDIS on two distinct timescales, with early signaling implicated in the senescence decision, and late signaling involved in the senescence associated cytokine secretory response.
JNK and Erk Signal Through AP-1 to Drive Cellular Senescence After Doxorubicin-Induced DNA Damage
Based on the findings in U2OS cells using t-PLSR and PCA analysis, we initially hypothesized that JNK, Erk, and p38MAPK acted through the phosphorylation of c-Jun to drive DDIS, and subsequently demonstrated through inhibition experiments that JNK and Erk activity, but not p38MAPK, were critical for the decision between senescence and proliferation within the first 12 hours after low-dose doxorubicin treatment. To demonstrate that c-Jun phosphorylation was directly regulated by these kinases during this early timeframe, Western blotting and immunofluorescence assays for phospho-c-Jun were performed at 6 and 12 hours following treatment of U2OS cells with 0.5 μM doxorubicin in the presence or absence of JNK or Mek1 inhibitors (Figs. S5A and B, S7). SP600125 caused a marked decrease in the levels of phospho-c-Jun after DNA damage in both assays, while PD98059 caused a more moderate, but also statistically significant reduction in c-Jun phosphorylation.
Next, to examine whether inhibition of JNK or Mek1 activity in the presence of DNA damage controls the downstream transcriptional activity of c-Jun, we used an AP-1 mCherry-based transcriptional reporter containing three canonical AP-1 motifs within a minimal promoter upstream of mCherry. U2OS cells stably transfected with the AP-1 reporter were treated with DMSO or 0.5 μM doxorubicin for four hours, in the presence or absence of SP600125 or PD98059, and mCherry expression quantified by flow cytometry. As shown in figure 7A, doxorubicin treatment induced a ~3 fold-increase in mCherry expression, similar to that observed in the PMA-treated positive control. Both the JNK inhibitor SP600125, and the Mek1 inhibitor PD98059, suppressed AP-1 transcriptional upregulation following DNA damage, mirroring the extent of suppression of c-Jun phosphorylation seen using these inhibitors by both immunoblotting and immunofluorescence (Figs. 7A, S5A–B and S7). To further validate that c-Jun is the relevant AP-1 family member in U2OS cells, the cells were transfected using a dominant negative c-Jun construct lacking the transactivation domain and sites of JNK and Erk phosphorylation, hereafter referred to as JunDN103. Expression of JunDN reduced AP-1-driven transcription in both the absence and presence of doxorubicin treatment, but notably, reduced AP-1 transcription following doxorubicin treatment to the same level as that seen in untreated control cells (Fig. 7A).
Figure 7. c-Jun, together with JNK and Erk directly controls the senescence-proliferation switch after treatment with replication stress-inducing DNA damaging agents.

A) Mean-fluorescence intensity (MFI) in the mCherry channel as measured by flow cytometry in U2OS cells expressing AP-1-mCherry reporter 24 hours after doxorubicin addition or continuous treatment with 100 nM (PMA). Bars represent mean ± SEM of biological replicates n = 4 (DMSO, SP, PD, and PMA) or n = 3 (JunDN). ***: p < 0.001, *: p < 0.05, ns: p > 0.05 with a two-way ANOVA and post-hoc Bonferroni’s multiple comparisons test.
B) Representative immunoblot for c-Jun and β-tubulin 48 hours post-transfection. Images are representative of 1,800–33,000 individual cells per biological replicate, depending on the condition.
C) Representative immunofluorescence images of U2OS stained with anti-BrdU and HCS CellMask Blue six days after either 0.5 μM doxorubicin treatment (+) or DMSO (−). Red arrows indicate BrdU negative cells.
D) Quantified proliferation index, calculated as the percentage of BrdU-positive cells after treatment with 0.5 μM doxorubicin divided by the percentage of BrdU-positive cells after vehicle treatment (DMSO) for each respective siRNA. Bars represent the mean ± SEM of three biological replicates.
E) Schematic of experimental workflow for inducible JunDN over-expression experiment. (Icons created in BioRender.com).
F) Representative immunofluorescence images of U2OS stably transfected with either the TRE-GFP or the TRE-JunDN-T2A-GFP stained with anti-BrdU and HCS CellMask Blue six days after doxorubicin treatment. Images are representative of 3,300–33,000 individual cells per biological replicate, depending on the condition.
G) Quantification of nuclear BrdU incorporation in TRE-GFP and TRE-JunDN-T2A-GFP cells six days after doxorubicin treatment. Bars represent the mean ± SEM of biological replicates (n = 3). ***: p < 0.001, ns: p > 0.05 with a two-way ANOVA and post-hoc Dunnett’s test vs. the TRE-GFP condition with no doxycycline at the same dose of doxorubicin.
H) Workflow of timed JNK and Mek1 inhibitor experiments, similar to Fig 6A–I. U2OS cells were treated with 0.1 μM mitoxantrone or vehicle control, in the presence or absence of SP600125, PD98059 or DMSO for the indicated times. Cells were stained with anti-BrdU and HCS CellMask Blue 6 days after mitoxantrone treatment, and nuclear BrdU positivity quantified. Bars represent the mean ± SEM of three biological replicates. ***: p < 0.001, **: p < 0.01, ‡: ns, p = 0.073, §: ns, p = 0.141, ns: p > 0.05 using two-way ANOVA and post-hoc Dunnett’s test vs. DMSO treatment at the same dose of mitoxantrone. CellMask blue was visualized with gamma=0.6 to better visualize the cytoplasmic compartment. Images are representative of 3,500–25,000 individual cells per biological replicate, depending on the condition.
I) Western blot of phospho-RPA2 (Ser-8) and total RPA2 in U2OS 6 hours after addition of doxorubicin (0.5 μM), mitoxantrone (0.1 μM), neocarzinostatin (NCS, 0.25 μg/mL), or cisplatin (20 μM) and their respective vehicle controls (−).
J) Quantification of western blots for phospho- and total RPA2, JNK, Erk, and phospho-c-Jun(Ser-73) 6 hours after the addition of doxorubicin (0.5 μM), mitoxantrone (0.1 μM), NCS (0.25 μg/mL), or cisplatin (20 μM) and their respective vehicle controls (−). Bars represent the mean ± SEM of biological replicates (n = 3 or 4). ***: p < 0.001, **: p < 0.01, *: p < 0.05 using an unpaired, two-way t-test vs. vehicle with Bonferroni correction. See figure S8B for representative western blots.
K) Proposed mechanism for JNK and Erk signaling in DDIS after doxorubicin or mitoxantrone treatment. (Created with BioRender.com.)
To directly test whether c-Jun controls the early cell fate decision after DNA damage, U2OS cells were transfected with two independent siRNAs targeting c-Jun, resulting in good knockdown of c-Jun at the protein level (Fig. 7D). When proliferation was measured by BrdU incorporation six days after a 4 hour pulse of either vehicle or 0.5 μM doxorubicin, a significant increase in the percentage of BrdU positive cells was observed that correlated with extent of knockdown (Fig. 7B and 7C). However, even in the absence of doxorubicin treatment, c-Jun knockdown resulted in a sub-population of cells that were BrdU negative (Fig. 7B, merge panel), likely as a consequence of prolonged doubling times that exceeded the 24 hour BrdU pulse. To mitigate the impact of continual c-Jun suppression on basal cell proliferation, we next generated U2OS cells stably expressing a tetracycline-inducible (tet-on) construct containing JunDN linked with a T2A cleavable linker to GFP (JunDN-T2A-GFP). Expression of JunDN was induced by doxycycline for 3 days prior to a 4 hour drug pulse of 0.5 μM doxorubicin, and the cells then maintained in doxycycline for 12 hours after doxorubicin treatment. The media was then changed to doxycycline-free media, which was replaced with fresh doxycycline-free media 1 and 3 days after doxorubicin treatment (Fig. 7E). As shown in figure 7F, control cells expressing a tet-on GFP-only construct exhibited no increase in BrdU positive cells six days after DNA damage, regardless of doxycycline treatment. In contrast, cells expressing the JunDN-T2A-GFP construct demonstrated a significant increase in the percentage of BrdU positive cells after DNA damage only in the presence of doxycycline (Fig. 7F and G). These data, combined with the c-Jun knockdown results in panels B-D confirms that AP-1 has a direct role in the senescence cell-fate decision after low-dose doxorubicin treatment, while the AP-1 reporter results, coupled with the phospho-c-Jun measurements in the presence or absence of inhibitors, indicate that JNK and Erk are the upstream kinases activating AP-1 in this context.
Curiously, the levels of phospho-JNK and Erk were also elevated in response to the 2 and 10 μM doxorubicin doses at all measured timepoints, yet the levels of phospho-c-Jun were moderately reduced at the 2 μM dose, and strongly reduced at the 10 μM dose in comparison to the 0.5 μM dose (Fig. 3C). At the suggestion of a reviewer, we therefore measured the levels of total c-Jun in U2OS cells by Western blotting at early time points after treatment with each drug dose (Fig. S5H–I). Relative to the 0.5 μM dose, the levels of total c-Jun progressively decreased at higher doxorubicin doses, becoming statistically significantly reduced by 6 hours, which is well within the timeframe of when c-Jun determines the senescence-fate decision (Figs. 6 and 7F, G). These reduced levels of total c-Jun can at least partially rationalize why high JNK and Erk activity fails to induce the same extent of cell senescence at higher drug doses than that seen with the 0.5 μM dose.
JNK and Erk Signaling Drives Cell Senescence in Response to a Subset of DNA-Damaging Agents
To investigate whether JNK and Erk signaling in response to doxorubicin is a conserved general mechanism for initiating DDIS in response to all types of genotoxic stress, we next examined mitoxantrone, a topoisomerase II inhibitor that is structurally distinct from doxorubicin, neocarzinostatin (NCS), a radiomimetic enediyne agent that directly cleaves DNA to form double-strand breaks, and of cisplatin, a common DNA crosslinking agent. Doses of each drug were selected that induced U2OS cell senescence based on cell morphology and lack of BrdU uptake (Fig. 7H, S8A). Cell senescence induced by mitoxantrone was reversed upon treatment with JNK and Mek1 inhibitors, particularly if added within the first 12 hours, similar to that observed with doxorubicin (Fig. 7H). In contrast, neither the senescence induced by NCS or cisplatin could be reversed by inhibiting JNK or Erk activity (Fig. S8A).
We noted that topoisomerase II inhibitors, such as doxorubicin and mitoxantrone, are particularly strong inducers of replication stress104–107. We therefore examined whether the amount of replication stress that occurred at early times after treatment differed between drugs that responded to JNK and Mek1 inhibitors relative to those that did not. Levels of replication stress were analyzed using phospho-RPA2, a marker of single-stranded DNA exposed at stalled replication forks108–110, by Western blotting after each of these four drug treatments. At 6 hours after the addition of doxorubicin and mitoxantrone, we observed a 50-fold and 100-fold increase in phospho-RPA2 respectively compared to vehicle controls, while NCS and cisplatin exhibited much smaller average fold-changes of 3.5 and 1.4, respectively (Fig. 7I and J). To further delineate features that distinguished drugs whose senescence was reduced by JNK and Mek1 inhibition compared to drugs whose senescence did not, the levels of total and phospho-JNK, Erk, and c-Jun were measured by western blot 6 hours after treatment. As seen in figure 7J, both doxorubicin and mitoxantrone treatment significantly increased phospho-JNK, Erk, and c-Jun levels relative to vehicle controls, while NCS and cisplatin did not (Fig. 7J, S8B). Taken together, these results suggest that the induction of senescence after DNA damage is heavily influenced by signaling through the JNK and Erk pathways particularly in response to treatment with genotoxic drugs that induce early replication stress.
DISCUSSION
In this manuscript, we interrogated canonical and non-canonical DNA damage signaling pathways for their influence on cell fate decisions in response to different levels of DNA damage. Since activation of a common set of DDR-activated signaling molecules, including among others ATM, Chk2, H2AX, Nbs1, 53BP1, p53 and p212,111–114 results in diverse phenotypic outcomes, we hypothesized that cross-talk from additional signaling pathways likely influenced the cell fate decision process. We were particularly interested in examining the signaling responses at the single cell level under conditions where subpopulations of cells underwent different cell fates using quantitative microscopy, immunofluorescence, and flow cytometry, combined with computational modeling. The resulting compendium of data was used to construct a t-PLSR model, using fewer parameters to predict the responses than are required for a conventional unfolded PLSR models39,50. Our results demonstrate that a relatively small number of treatments was sufficient to construct a model that was robust and provided biological insight. Using t-PLSR modeling and PCA analysis paired with subsequent experimental validation, we identified unexpected roles for the MAP kinases JNK and Erk in modulating the early fate decision between DDIS and proliferation in the setting of low-dose DNA damage through the common downstream target of c-Jun, as well as a later role for these kinases in controlling the SASP cytokines IL-6 and IL-8 in committed senescent cells.
Our finding of a central role for JNK in enhancing the cell fate choice of senescence after modest DNA damage was unexpected, since JNK is best known to function as a stress-responsive regulator of apoptotic and non-apoptotic cell death. JNK promotes intrinsic apoptotic cell death, both in vitro and in vivo68,115,116 through a variety of mechanisms, including direct phosphorylation and activation of pro-apoptotic BH3 proteins117,118, inactivation of the central scaffolding molecule 14–3-3119,120, and phosphorylation and activation of p53 and/or p73 to increase the transcription of the pro-apoptotic BH3 family member PUMA121,122. In addition, novel roles for JNK in promoting necroptosis, pyroptosis, ferroptosis and autophagic cell death have recently been observed123. JNK activity even prevents p53-mediated cell senescence in unperturbed MEFs in culture124. Our findings of a specific role for JNK in inducing senescence, rather than preventing senescence or promoting cell death, following modest levels of doxorubicin- or mitoxantrone-induced DNA damage, suggest a very specific context-dependence in which JNK signaling controls the fate choice between senescence, proliferation, and death.
Conversely, the Erk MAP kinase pathway has typically be associated with enhancement of cell proliferation and differentiation, rather than senescence induction, through the direct Erk-dependent phosphorylation of a large number of transcription factors, including members of the TCF, ERG, ERF, PEA3, AP-1, EGR families as well as Runx2 and c-Myc themselves125, resulting in their nuclear translocation, enhanced DNA binding, and transcription of immediate early genes and G1 cyclins. In addition, Erk phosphorylation of the stem cell transcription factors Oct4, Klf4, and Klf2 has been shown to decrease their stability and thus lead to loss of pluripotency126–128. In the setting of DNA damage, our lab and others have shown previously that Erk contributes to both G1/S arrest and subsequent apoptotic cell death after DNA damage using high doses of doxorubicin or cisplatin35,64,129. To our knowledge, a clear role for Erk in contributing to cell senescence following extrinsic DNA damage induced by low-dose cytotoxic chemotherapy has not been reported. Erk activity has, however, been shown to contribute to the induction of senescence in p53 wild-type cells in response to expression of oncogenic Ras and/or Raf130–132. Interpretation of our findings in light of the findings from Bartek, Lucas, and colleagues, who showed that oncogene activation results in replication stress and intrinsic DNA damage, suggests that a similar Erk-dependent senescence pathway as that observed following oncogene activation is being activated by genotoxic drugs that induce large amounts of replication stress5.
Unexpectedly, we did not detect a quantitatively significant role for the p38MAPK pathway in controlling the senescence decision at early times following low-dose DNA damage, despite the fact that this pathway is known to be activated by many different types of stress, including DNA damage133. In response to more extensive DNA damage induced by higher doses of doxorubicin (or cisplatin), p38MAPK, acting through MAPKAP Kinase-2, is known to be required for sustained cell cycle arrest and cell survival, but this effect results from signaling at late times greater than 12 hours, and is important only in cells in which p53 activity is at least partially defective40,134–136. This delayed temporal activation of the p38MAPK-MK2 pathway following DNA damage134 rationalizes the lack of effect of p38MAPK inhibition on low dose DNA damage-induced senescence onset, and instead indicates roles for this pathway primarily at later times. Consistent with this, a major role for p38MAPK in regulating the SASP has been reported by Campisi and colleagues32.
The AP-1 transcription factor in general, and c-Jun in particular, emerged as key determinants of senescence in our experiments. The AP-1 transcription factor is a hetero- or homo-dimeric complex comprised of members of the Jun, Fos, ATF, and MAF protein families, that plays an important role in oncogenesis and tumor proliferation, and is known to regulated by JNK, Erk, and p38MAPK in a context-specific manner137. The major AP-1 family member that emerged from our experimental findings and computational t-PLSR and PC analyses of low-dose DNA damage in U2OS cells was c-Jun, whose phosphorylation and nuclear accumulation correlated with the early cell fate decision towards senescence. Suppression of c-Jun using siRNA partially reversed the senescence phenotype after DNA damage, and this partial bypass of senescence was further confirmed using inducible expression of a non-phosphorylatable dominant negative form of c-Jun, which suppressed AP-1 activity in the cells.
Two recent papers have implicated a role for AP-1 in oncogene induced senescence. Martinez-Zamudio et al.138 used a combination of transcriptomic, ATAC-Seq and ChIP-Seq data to nominate AP-1 as a pioneering transcription factor that altered chromatin structure and allowed the establishment of a senescence-inducing enhancer landscape following the inducible expression of a RasG12V mutant oncogene in WI-38 human fibroblasts. siRNA knock-down of c-Jun had a larger effect on the Ras-induced senescence transcriptional program than did knock-down of the non-pioneering transcription factors ETS1 and RelA, primarily suppressing transcription of SASP-related genes, but also partially re-activating proliferation-associated genes. Those authors did not examine upstream regulatory kinases, or demonstrate reversion of a senescence cellular phenotype. Nonetheless, their extensive and comprehensive epigenetic and transcriptional analysis of oncogene-induced senescence is in good potential agreement with our findings of an important role for c-Jun in doxorubicin-induced senescence in U2OS cells.
In a separate study, Han et al. used inducible expression of a mutant RasG12V oncogene in hTERT-immortalized BJ fibroblasts and observed upregulation of enhancer RNAs that were enriched for the binding motif of AP-1139, suggesting an important role for AP-1-driven gene transcription in response to oncogenic stress. They then identified a specific AP-1 driven enhancer region controlling the expression of FOXF1as critical for the onset of oncogene-induced senescence. Taken together, these two studies support our findings that JNK and Erk modulate an AP-1-driven program of senescence, and suggest strong parallels between oncogene-induced senescence and senescence induced by extrinsic DNA damaging drugs that cause extensive replication stress. Interestingly, Davis and colleagues have shown, using a PTEN inactivation-dependent model of prostate cancer, that genetic elimination of JNK, or its upstream activators MKK4 and 7, results in marked enhancement of prostate tumor growth by suppressing the senescence response of prostate cells upon PTEN loss140. Those findings are in excellent agreement with our proposed model for an important role of JNK signaling in promoting senescence (Fig. 7K), caused either by oncogenic replication stress, or by low doses of replication stress-inducing genotoxic drugs. Future work should focus on elucidating the molecular links between replication stress and activation of the JNK and Erk pathways, especially in the context of oncogene-induced senescence and DDIS.
Heterogeneity in the behavior and response of cancer cells is now well established141,142, and recent work indicates a similar heterogeneity is present within senescent cells143. In this regard, it is interesting that we observed heterogeneity in the proliferative response of U2OS, NCI-H1299, OVCAR-8, and HUVEC cells following JNK and Mek inhibition in response to treatment with doxorubicin, with only a fraction of the inhibitor-treated cells escaping from senescence onset. The molecular basis for this heterogeneous response is unclear, and future work will be required to better define the underlying mechanisms, which represent a general challenge for predicting the clinically relevant biology of complex tumors during both their development and their response to treatment144–149.
Nonetheless, our finding of distinct early and late roles for JNK and Erk in senescence progression is of clear clinical utility, given the recent interest in the use of senolytics and SASP inhibitors for the treatment of cancer150–152. For example, administering a JNK or Mek inhibitor days or weeks after chemotherapy could favorably lower IL-6 and IL-8 levels secreted by treatment-induced senescent cells, and thus decrease the IL-6/IL-8-mediated signaling events in the tumor microenvironment that favor cancer progression31,153–155; conversely, administering these inhibitors at the same time as chemotherapy could drive cells towards proliferation rather than senescence due to the early cell fate decision-making role of JNK and Erk, resulting in tumor resistance to cytotoxic agents and hindering the efficacy of senolytic therapies later on. These findings of distinct temporal and context-dependent roles for JNK and Erk MAP kinases in the setting of extrinsic genotoxic stress, however, further illustrate the extraordinary complexity and plethora of cellular responses mediated by these highly conserved signaling cascades in controlling the DNA damage response.
STAR METHODS
Resources Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Yaffe (myaffe@mit.edu).
Materials Availability
Plasmids and cell lines generated in this study are available upon request.
Data and Code Availability
Source data statement: All data reported in this paper will be shared by the lead contact upon reasonable request.
Code statement: Raw quantified data and MATLAB scripts used to construct t-PLSR and PCA results can be found at: github.com/yaffelab/CellFate-TensorPLSR. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-γ-H2AX | Millipore | Cat#05–636, RRID:AB_309864 |
| Rabbit monoclonal anti-phospho-Akt | Cell Signaling Technology | Cat#4058, RRID:AB_331168 |
| Rabbit monoclonal anti-phospho-Erk | Cell Signaling Technology | Cat#4377, RRID:AB_331775 |
| Rabbit monoclonal anti-phospho-Erk | Cell Signaling Technology | Cat#4370, RRID:AB_2315112 |
| Mouse monoclonal anti-Erk | Cell Signaling Technology | Cat#4696, RRID:AB_390780 |
| Rabbit polyclonal anti-phospho-JNK | Cell Signaling Technology | Cat#9251, RRID:AB_331659 |
| Rabbit polyclonal anti-JNK | Cell Signaling Technology | Cat#9252, RRID:AB_2250373 |
| Mouse monoclonal anti-NF-κB | Santa Cruz | Cat#sc-8008, RRID:AB_628017 |
| Rabbit monoclonal anti-phospho-p38 | Cell Signaling Technology | Cat#4631, RRID:AB_331765 |
| Rabbit polyclonal anti-p38 | Cell Signaling Technology | Cat#9212, RRID:AB_330713 |
| Rabbit monoclonal phospho-MK2 | Cell Signaling Technology | Cat#3007, RRID:AB_490936 |
| Rabbit anti-MK2 | Cell Signaling Technology | Cat#3042, RRID:AB_10694238 |
| Rabbit monoclonal anti-phospho-S6 | Cell Signaling Technology | Cat#4856, RRID:AB_2181037 |
| Mouse monoclonal anti-p27 | Santa Cruz | Cat#sc-1641, RRID:AB_628074 |
| Rabbit monoclonal anti-p27 | Cell Signaling Technology | Cat#3686, RRID:AB_2077850 |
| Rabbit polyclonal anti-phospho-Hsp27 | Cell Signaling Technology | Cat#2401, RRID:AB_331644 |
| Mouse monoclonal anti-p53 | Santa Cruz | Cat#sc-263, RRID:AB_628084 |
| Rabbit polyclonal anti-phospho-c-Jun(Ser 73) | Cell Signaling Technology | Cat#9164, RRID:AB_330892 |
| Rabbit monoclonal anti-phospho-c-Jun(Ser 73) | Cell Signaling Technology | Cat#3270, RRID:AB_2129575 |
| Rabbit polyclonal anti-phospho-c-Jun(Ser 63) | Cell Signaling Technology | Cat#9261, RRID:AB_2130162 |
| Rabbit monoclonal anti-c-Jun | Cell Signaling Technology | Cat#9165, RRID:AB_2130165 |
| Rabbit polyclonal anti-phospho-Chk2 | Cell Signaling Technology | Cat#2661, RRID:AB_331479 |
| Mouse monoclonal anti-IκBα | BD Biosciences | Cat#610691, RRID:AB_398014 |
| Mouse monoclonal anti-IκBα | Cell Signaling Technology | Cat#4814, RRID:AB_390781 |
| Mouse monoclonal anti-Cyclin D | Santa Cruz | Cat#sc-20044, RRID:AB_627346 |
| Rabbit polyclonal anti-phospho-Rb | Cell Signaling Technology | Cat#9308, RRID:AB_331472 |
| Mouse monoclonal anti-Cyclin E | Santa Cruz | Cat#sc-247, RRID:AB_627357 |
| Rabbit polyclonal anti-Cyclin A | Santa Cruz | Cat#sc-751, RRID:AB_631329 |
| Mouse monoclonal anti-p21 | Millipore | Cat#05–345, RRID:AB_309684 |
| Rabbit monoclonal anti-p21 | Cell Signaling Technology | Cat#2947, RRID:AB_823586 |
| Rabbit monoclonal anti-p16 | Cell Signaling Technology | Cat#18769, RRID:AB_2935679 |
| Rabbit polyclonal anti-Cyclin B | Santa Cruz | Cat#sc-752, RRID:AB_2072134 |
| Rabbit monoclonal anti-Cyclin B | Cell Signaling Technology | Cat#12231, RRID:AB_2783553 |
| Mouse monoclonal anti-BrdU | BD Biosciences | Cat#555627, RRID:AB_395993 |
| Mouse monoclonal anti-BrdU | Cell Signaling Technology | Cat#5292S, RRID:AB_10548898 |
| Rabbit polyclonal anti-phospho-RPA2(Ser 8) | Cell Signaling Technology | Cat#83745, RRID:AB_2800029 |
| Rabbit monoclonal anti-RPA2 | Cell Signaling Technology | Cat#35869, RRID:AB_2799086 |
| Mouse monoclonal anti-β-tubulin | Sigma-Aldrich | Cat#T7816, RRID:AB_261770 |
| Mouse monoclonal anti-GAPDH | Cell Signaling Technology | Cat#97166, RRID:AB_2756824 |
| Rabbit monoclonal anti-cleaved caspase-3 | BD Biosciences | Cat#559565, RRID:AB_397274 |
| Mouse monoclonal anti-cleaved PARP | BD Biosciences | Cat#558710, RRID:AB_1645431 |
| Rabbit polyclonal anti-Cyclin B | Santa Cruz | Cat#sc-752, RRID:AB_2072134 |
| Mouse monoclonal anti-p21 | Millipore | Cat#05–345, RRID:AB_309684 |
| Rabbit monoclonal anti-cleaved caspase-3 conjugated to Pacific Blue | Cell Signaling Technology | Cat#8788S, RRID:AB_2797665 |
| Rabbit monoclonal anti-cleaved PARP conjugated to Alexa Fluor 647 | Cell Signaling Technology | Cat#6987S, RRID:AB_10858215 |
| Goat polyclonal anti-rabbit conjugated to Alexa Fluor 488 | Invitrogen | Cat#A-11008, RRID:AB_143165 |
| Goat polyclonal anti-rabbit conjugated to Alexa Fluor 647 | Invitrogen | Cat#A-21245, RRID:AB_141775 |
| Goat polyclonal anti-mouse conjugated to Alexa Fluor 647 | Invitrogen | Cat#A-21236, RRID:AB_2535805 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–68070, RRID: RRID:AB_10956588 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–32211, RRID:AB_621843 |
| IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–68071, RRID:AB_10956166 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–32210, RRID:AB_621842 |
| Bacterial and virus strains | ||
| Competent DH5α E. coli | This paper | NA |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Corning | 10–013-CV |
| Fetal bovine serum (FBS) | VWR | 97068–085 |
| Penicillin-streptomycin | Gibco | 15070063 |
| RPMI | Gibco | 11875093 |
| EBM-2 endothelial cell growth medium | Lonza | CC-3162 |
| Doxorubicin hydrochloride | Sigma-Aldrich | PHR1789; CAS: 25316–40-9 |
| SP600125 | Sigma-Aldrich | S5567; CAS: 129–56-6 |
| Neocarzinostatin | Sigma-Aldrich | N9162; CAS: 9014–02-2 |
| Cisplatin | Sigma-Aldrich | P4394; CAS: 15663–27-1 |
| Caffeine | Sigma-Aldrich | C0750; CAS: 58–08-2 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | P1585; CAS: 16561–29-8 |
| Anisomycin | Sigma-Aldrich | A9789; CAS: 2286276–6 |
| PD98059 | Cell Signaling Technology | 9900; CAS: 167869- 21–8 |
| SB203580 | Tocris | 1202; CAS: 152121- 47–6 |
| Mitoxantrone | Tocris | 4250; CAS: 70476- 82–3 |
| Doxycycline | Clontech | 631311; CAS: 564- 25–0 |
| DMSO | Sigma-Aldrich | D8418; CAS: 67–68-5 |
| 5-bromo-2’-deoxyuridine (BrdU) | ThermoFisher | B23151; CAS: 59–143 |
| 16% Paraformaldehyde | Electron Microscopy Science |
15710; CAS: 30525- 89–4 |
| PBS | GenClone | 25–507B |
| Triton-X 100 | Sigma-Aldrich | T9284; CAS: 9036–19-5 |
| HCS CellMask Blue | ThermoFisher | H32720 |
| Hoechst 33342 | Invitrogen | H3570 |
| Whole Cell Blue | ThermoFisher | 8403502 |
| 5-ethynyl-2’-deoxyuridine (EdU) | ThermoFisher | A10044 |
| ProLong Gold Antifade Mount | ThermoFisher | P36980 |
| Odyssey Blocking Buffer | LI-COR Biosciences |
927–50000 |
| Propidium iodide with RNase A | Cell Signaling Technology | 4087 |
| Tween 20 | AmericanBio | AB02038; CAS: 9005–64-5 |
| PhosSTOP™ tablets | Roche | 4906837001 |
| cOmplete™ ULTRA Tablets, Mini, EDTA-free, EASYpack Protease Inhibitor Cocktail | Roche | 05892791001 |
| Ponceau stain | Sigma-Aldrich | P7170 |
| Intercept blocking buffer | Licor | 927–70001 |
| NewBlot nitrocellulose stripping buffer | Licor | 928–40030 |
| Lipofectamine RNAiMAX | ThermoFisher | 13778150 |
| Opti-MEM | ThermoFisher | 11058021 |
| Lipofectamine 2000 | ThermoFisher | 11668019 |
| Puromycin | InvivoGen | ant-pr-1; CAS: 58–58-2 |
| Hydrochloric acid | Sigma-Aldrich | 258148; CAS: 7647- 01–1 |
| Citric acid | Sigma-Aldrich | C-4540; CAS: 77–929 |
| Sodium phosphate monobasic | Sigma-Aldrich | S0751; CAS: 7558–80-7 |
| Phusion® High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S |
| NheI-HF® | NEB | R3131L |
| EcoRI-HF® | NEB | R3101L |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | E2621S |
| Critical commercial assays | ||
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | 9860 |
| Click-iT Plus Alexa Fluor 555 Picolyl Azide Toolkit | ThermoFisher | C10642 |
| IL-6 Human ELISA Kit | ThermoFisher | KHC0061 |
| IL-8 Human ELISA Kit | ThermoFisher | KHC0081 |
| BCA assay | ThermoFisher | 23225 |
| CalPhos™ Mammalian Transfection Kit | Clontech | 631312 |
| Experimental models: Cell lines | ||
| Human: U-2 OS (U2OS) | HTS core at the Koch Institute | RRID:CVCL_T429 |
| Human: OVCAR-8 | Laboratory of Dr. Paula Hammond |
RRID:CVCL_1629 |
| Human: NCI-H1299 | ATCC | RRID:CVCL_0060 |
| Human: HUVEC | Lonza | RRID:CVCL_2959 |
| Human: HEK293T | ATCC | RRID:CVCL_0063 |
| Oligonucleotides | ||
| Non-targeting siRNA | Invitrogen | AM4636 |
| Jun #1 siRNA | Life Technologies |
s7658 |
| Jun #2 siRNA | Life Technologies |
s7659 |
| Recombinant DNA | ||
| pAK-Tol2-TRE-JunDN-HA-T2A-GFP (pTREJunDN-HA-T2A-GFP) | This paper | NA |
| pAK-Tol2-TRE-GFP (pTRE-GFP) | This paper | NA |
| pMIEG3-JunDN | Addgene # 40350 | Ref. 156 |
| pCMV-Tol2 | Addgene # 31823 | Ref. 157 |
| pAK-Tol2-TRE-Puro | Addgene # 130259 | NA |
| pSIRV-AP-1-mCherry | Addgene # 118095 | Ref. 158 |
| pUMVC | Addgene # 8849 | Ref. 159 |
| pCMV-VSV-G | Addgene # 8454 | Ref. 159 |
| Software and algorithms | ||
| Fiji distribution of ImageJ | Ref. 160 | https://fiji.sc/ |
| CellProfiler (version 4.0.6) | Ref. 161 | https://cellprofiler.org/ |
| MATLAB (2019b) | MathWorks | https://www.mathworks.com |
| FlowJo (version 10) | BD Biosciences | https://www.flowjo.com/ |
| Python (version 3.7) | Python Software Foundation | https://www.python.org/ |
| N-way Toolbox (version 1.8.0.0) | Ref. 56 | https://www.mathworks.com/matlabcentral/fileexchange/1088the-n-way-toolbox |
| Multi-way VIP | Ref. 58 | Obtained from corresponding authoruponreques |
| Tensor PLSR and PCA code | This paper | https://doi.org/10.5281/zenodo.7927324 |
| ilastik | Ref. 162 | https://www.ilastik.org/ |
| GraphPad Prism (version 8.4.1) | Dotmatics | https://www.graphpad.com/ |
| NIS Viewer | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/viewer |
| Image Studio Lite (version 5.2) | LI-COR Biosciences |
https://www.licor.com/bio/image-studio-lite/ |
| Other | ||
| Corning™ BioCoat™ 12mm #1 German Glass Coverslips, Round | Fisher Scientific | 08–774-383 |
| 96 Well Black/Clear Bottom Plate, TC Surface | ThermoFisher | 165305 |
Experimental Model and Subject Details
U-2 OS (U2OS) and HEK293T cells were purchased from the American Type Culture Collection (ATCC), and cultured in complete media consisting of Dulbecco’s Modified Eagle’s Medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS, VWR) and 1% penicillin-streptomycin (pen-strep, Gibco). NCI-H1299 cells were purchased from ATCC, and cultured in RPMI (Gibco) supplemented with 10% FBS and 1% pen-strep. HUVEC cells were purchased from Lonza and cultured in complete EBM-2 endothelial cell growth medium (Lonza). OVCAR-8 cells were a gift from Dr. Paula Hammond, identity-confirmed by STR profiling, and cultured in RPMI supplemented with 10% FBS and 1% pen-strep. Cell cultures were maintained in a humidified incubator at 37°C with 5% CO2, and cells with less than 30 passages were used for experiments.
Method Details
Antibodies and chemicals
Doxorubicin hydrochloride, SP600125, neocarzinostatin, cisplatin, caffeine, PMA, and anisomycin were purchased from Sigma-Aldrich. PD98059 was purchased from Cell Signaling Technology. SB203580 and mitoxantrone were purchased from Tocris. Doxycycline was purchased from Clontech. The following antibodies were used for immunofluorescence measurements:
| Antibody | Vendor | Catalog # | Dilution |
|---|---|---|---|
| γ-H2AX | Millipore | 05–636 | 1:200 |
| p-Akt | Cell Signaling Technology | 4058 | 1:200 |
| p-Erk | Cell Signaling Technology | 4377 | 1:200 |
| p-JNK | Cell Signaling Technology | 9251 | 1:100 |
| NF-κB | Santa Cruz | sc-8008 | 1:50 |
| p-p38 | Cell Signaling Technology | 4631 | 1:200 |
| p-S6 | Cell Signaling Technology | 4856 | 1:200 |
| p27 | Santa Cruz | sc-1641 | 1:50 |
| p27 | Cell Signaling Technology | 3686 | 1:400 |
| p-Hsp27 | Cell Signaling Technology | 2401 | 1:200 |
| p53 | Santa Cruz | sc-263 | 1:50 |
| p-c-Jun(Ser 73) | Cell Signaling Technology | 9164 | 1:200 |
| p-c-Jun(Ser 63) | Cell Signaling Technology | 9261 | 1:200 |
| p-Chk2 | Cell Signaling Technology | 2661 | 1:200 |
| IκBα | BD Biosciences | 610691 | 1:200 |
| IκBα | Cell Signaling Technology | 4814 | 1:200 |
| Cyclin D | Santa Cruz | sc-20044 | 1:50 |
| p-Rb | Cell Signaling Technology | 9308 | 1:200 |
| Cyclin E | Santa Cruz | sc-247 | 1:50 |
| Cyclin A | Santa Cruz | sc-751 | 1:50 |
| p21 | Millipore | 05–345 | 1:200 |
| p21 | Cell Signaling Technology | 2947 | 1:400 |
| p16 | Cell Signaling Technology | 18769 | 1:800 |
| Cyclin B | Santa Cruz | sc-752 | 1:50 |
| Cyclin B | Cell Signaling Technology | 12231 | 1:400 |
| BrdU | BD Biosciences | 555627 | 1:400 |
| BrdU | Cell Signaling Technology | 5292S | 1:1000 |
For western blots, the following antibodies were used:
| Antibody | Vendor | Catalog # | Dilution |
|---|---|---|---|
| p-c-Jun(Ser 73) | Cell Signaling Technology | 3270 | 1:1000 |
| c-Jun | Cell Signaling Technology | 9165 | 1:1000 |
| p-Erk | Cell Signaling Technology | 4370 | 1:2000 |
| Erk | Cell Signaling Technology | 4696 | 1:2000 |
| p-JNK | Cell Signaling Technology | 4668 | 1:1000 |
| JNK | Cell Signaling Technology | 9252 | 1:1000 |
| p-MK2 | Cell Signaling Technology | 3007 | 1:1000 |
| MK2 | Cell Signaling Technology | 3042 | 1:1000 |
| p-p38 | Cell Signaling Technology | 4631 | 1:1000 |
| p38 | Cell Signaling Technology | 9212 | 1:1000 |
| p-RPA2(Ser 8) | Cell Signaling Technology | 83745 | 1:1000 |
| RPA2 | Cell Signaling Technology | 35869 | 1:1000 |
| β-tubulin | Sigma-Aldrich | T7816 | 1:2000 |
| GAPDH | Cell Signaling Technology | 97166 | 1:1000 |
For flow cytometry, the following antibodies were used:
| Antibody | Vendor | Catalog # | Dilution |
|---|---|---|---|
| Cleaved Caspase-3 | BD Biosciences | 559565 | 1:200 |
| Cleaved PARP | BD Biosciences | 558710 | 1:200 |
| Cyclin B | Santa Cruz | sc-752 | 1:50 |
| p21 | Millipore | 05–345 | 1:200 |
| BrdU | BD Biosciences | 555627 | 1:200 |
| Pacific Blue cleaved caspase-3 (OVCAR-8 data) |
Cell Signaling Technology | 8788S | 1:50 |
| Alexa fluor 647 cleaved PARP (OVCAR-8 data) | Cell Signaling Technology | 6987S | 1:50 |
The following secondary antibodies were used for both immunofluorescence and flow cytometry in this study:
| Antibody | Vendor | Catalog # | Dilution |
|---|---|---|---|
| Alexa Fluor 488 goat anti-rabbit | Invitrogen | A-11008 | 1:200 (CC3)-2000 (BrdU, CycB) for FC, 1:1000 for IF |
| Alexa Fluor 647 goat anti-rabbit | Invitrogen | A-21245 | 1:1000 for IF |
| Alexa Fluor 647 goat anti-mouse | Invitrogen | A-21236 | 1:2000 for FC, 1:1000 for IF |
Doxorubicin treatment for signaling and response measurements
Cells were plated at least 24 hours before treatment in complete culture media. Doxorubicin or DMSO (Sigma-Aldrich) was directly added to culture media, and media was then removed 4 hours later and replaced with media containing 1% fetal bovine serum for the duration of the experiment for immunofluorescence, cell cycle, apoptosis, and proliferation assays.
β-galactosidase activity assay
U2OS cells were plated in a 24-well plate 24 hours before doxorubicin treatment in complete media. Doxorubicin was directly added to the culture media, and 4 hours later the media was replaced with media containing 1% FBS for the duration of the experiment. The plate was harvested 4 days after doxorubicin, and the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) was used according to the manufacturer’s protocol. Images were taken on an EVOS fluorescence microscope (ThermoFisher) in the brightfield channel.
Immunofluorescence for BrdU
In experiments where 5-bromo-2’-deoxyuridine (BrdU, ThermoFisher) incorporation into DNA was measured with immunofluorescence, cells were seeded in optical bottom 96-well plates (ThermoFisher) at the beginning of the experiment, and then were incubated in 10 μM BrdU for 24 hours before fixation. Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 minutes at room temperature, washed 3x with phosphate-buffered saline (PBS, GenClone), and then incubated with 1 N HCl on ice for 10 minutes. Afterwards, the HCl solution was removed, and cells were incubated with 2 N HCl for 10 minutes at room temperature. The HCl was then neutralized with equal amounts of neutralizing buffer [91% sodium phosphate monobasic, 9% citric acid in water], and incubated on cells for 10 minutes at room temperature. Buffer was then removed from the wells, and cells were washed with 1x PBS three times. Afterwards, cells were then permeabilized with PBS containing 0.1% Triton-X (Sigma-Aldrich) for 20 minutes at room temperature. Cells were then incubated with a primary antibody solution containing BrdU antibody diluted 1:400 or 1:1000 in PBS containing 5% goat serum (Abcam,) and 0.1% Triton-X for 1 hour at room temperature. Afterwards, cells were washed 3x with PBS and incubated with an anti-mouse antibody conjugated to Alexa Fluor 647 diluted 1:1000 in PBS for 1 hour at room temperature. Cells were then washed 3x with PBS, and incubated either in HCS CellMask Blue (ThermoFisher) or Hoechst 33342 (Invitrogen, catalog # H3570) with Whole Cell Blue (ThermoFisher) according to the manufacturers’ protocol. Cells were then washed 3x with PBS, and images were either taken in the DAPI and Cy5 channels using an cellWoRx automated imaging platform (Applied Precision) or in the DAPI and Cy5 channels of an Cellomics ArrayScan VTi (Thermo Fisher).
Detecting EdU incorporation with Click-iT chemistry
U2OS cells were seeded on sterile coverslips coated with poly-L-lysine (Fisher Scientific). The doxorubicin treatment protocol described above was followed, with the exception that media was replaced with 10% FBS media 24 and 72 hours after doxorubicin treatment. 10 μM of 5-ethynyl-2’deoxyuridine (EdU, ThermoFisher) was added to the media 5 days after doxorubicin, and cells were fixed with 4% formaldehyde (Sigma-Aldrich) for 15 minutes at room temperature. Cells were then permeabilized with ice-cold methanol for 10 minutes at −20°C. Cells were then washed with PBS 3x, and coverslips were then transferred to a new 24-well plate. Coverslips were stained with HCS CellMask Blue diluted 1:5000 in PBS for 30 minutes, and then washed 3x in PBS. During CellMask incubation, EdU labeling solution was prepared with Click-iT Plus Alexa Fluor 555 Picolyl Azide Toolkit (ThermoFisher) with the composition according to the manufacturer’s instructions, with 2% CuSO4 and 2.5 μM Alexa Fluor 555 picolyl azide. Coverslips were incubated in EdU labeling solution for 30 minutes at room temperature, and then washed 3x in PBS. Coverslips were then mounted onto glass slides using ProLong Gold Antifade Mount. Slides were then imaged on a Nikon spinning-disk confocal microscope.
Flow cytometry for cell cycle and apoptosis measurements
For all flow cytometry samples, cells were seeded in 15 cm tissue culture dishes 24 hours before doxorubicin treatment. At the indicated time points after doxorubicin treatment, media from the dishes were transferred to 50 mL conical tubes on ice. Cells were then washed with PBS, and afterwards the PBS was added to the respective conical tube (1 tube per plate). Cells were then trypsinized until all cells were detached, and trypsin was subsequently quenched with complete media. Cell suspension was then transferred to the respective conical tube, which was then centrifuged at 1,500 RPM for 5 minutes. The supernatant was then aspirated off, and the cell pellet was resuspended in ice-cold PBS. The cell suspension was then transferred to 1.5 mL Eppendorf tubes, and then centrifuged at 5,000 RPM for 1 minute.
For samples stained for cleaved caspase-3, cleaved PARP, p21, and cyclin B antibody, the pellet supernatant was aspirated, and the cell pellet was resuspended in 4% formaldehyde in PBS and incubated at room temperature for 15 minutes. Formaldehyde solution was removed by centrifuging the cells at 5,000 RPM for 1 minute and removing the supernatant. The cell pellet was then resuspended in ice-cold PBS, and the cell suspension was centrifuged at 5,000 RPM for 1 minute. The supernatant was aspirated off, and cells were resuspended in 90% ice-cold methanol and stored at −20°C until staining. For cleaved caspase-3 and cleaved PARP double staining, cells were washed twice in PBS-T, and then incubated with both antibodies diluted in blocking buffer consisting of 1:1 Odyssey blocking buffer overnight at 4°C, while cells stained for p21 and cyclin B were blocked in blocking buffer for 1 hour at room temperature before proceeding to the overnight primary antibody incubation. For all samples, after the primary antibody step, cells were then washed once with PBS-T, and incubated in respective fluorophore-conjugated secondary antibody diluted in blocking buffer for 1 hour at room temperature for 1 hour. Cells were then washed with PBS-T, and then cleaved caspase-3 and cleaved-PARP stained cells were resuspended in PBS-T for flow cytometry. For the p21 and cyclin B samples, cells were incubated in propidium iodide and RNase A (Cell Signaling Technology) for 1 hour at room temperature before being resuspended in PBS-T for flow cytometry.
For samples stained with BrdU antibody, the pellet supernatant from the ice-cold PBS wash step was aspirated off, and the cell pellet were resuspended in 200 μL ice-cold PBS. 800 μL of ice-cold ethanol was then added drop-by-drop to the Eppendorf tube while vortexing, and samples were then stored at −20°C until staining. Cells were then washed with PBS-T (PBS + 0.5% Tween) and simultaneously permeabilized and denatured with 2 M HCl and 0.5% Triton-X-100 in water for 30 minutes. Cells were washed twice with PBS, and then blocked in blocking buffer consisting of 1:1 Odyssey blocking buffer and PBS-T for 1 hour at room temperature. Blocking buffer was then removed, and cells were then incubated with anti-BrdU antibody diluted in blocking buffer overnight at 4°C. Cells were then washed once in PBS-T, and then were incubated with Alexa Fluor 647 goat anti-mouse antibody diluted in blocking buffer for 1 hour at room temperature. Secondary antibody was removed, and cells were washed once in PBS-T. Cells were then stained with 100 μg/mL propidium iodide with RNase A for 1 hour to visualize DNA content.
All flow cytometry measurements were collected on a BD FacsCaliber flow cytometer (BD Biosciences) or a BD LSR II HTS (BD Biosciences).
Proliferation assay
Cells were seeded at 5,000 cells/well into a tissue culture-treated 96-well plate (ThermoFisher) 24 hours before doxorubicin treatment. Doxorubicin or DMSO was directly added to culture media, and media was changed to media containing 1% FBS media 4 hours after drug addition. The plate was placed back in the incubator, and then 6 hours after doxorubicin treatment was moved to an incubator connected to an Incucyte microscope (Sartorius). Brightfield images of wells were taken 6, 12, 24, 48, 72, and 96 hours after doxorubicin treatment, and cell number was calculated from these images using ilastik software 162. Proliferation values were normalized across all treatments and time points to the 6 hour DMSO cell count values.
Immunofluorescence measurements for signaling proteins
U2OS cells were seeded into optical bottom 96-well plates (ThermoFisher) 24 hours before doxorubicin treatment. DMSO or doxorubicin were added directly to the media, and then 4 hours later the media was replaced with DMEM containing 1% FBS, which was maintained for the rest of the experiment. At each indicated timepoint, the media was removed and cells were fixed with 4% formaldehyde in PBS for 15 minutes. Cells were then washed with PBS-Tween (PBS-T) once, and then permeabilized with ice-cold methanol for 10 minutes. Afterwards, cells were again washed once with PBS-T, and then blocked in blocking buffer, which consisted of a 1:1 mix of Odyssey blocking buffer (LI-COR Biosciences) and PBS-T. Cells were blocked for 1 hour at room temperature.
Afterwards, the blocking buffer was removed and replaced with primary antibody solution consisting of antibody diluted in blocking buffer. Cells were incubated in primary antibody solution overnight at 4°C on a shaker. The primary antibody solution was then removed, and cells were washed once with PBS-T. Cells were then incubated with a secondary antibody solution containing both goat anti-mouse conjugated to Alexa Fluor 488 and goat anti-rabbit conjugated to Alexa Fluor 647 diluted in blocking buffer. Cells were incubated in secondary antibody solution overnight at 4°C on a shaker, and then washed once with PBS-T. To stain both the nuclear and the cytosolic compartments, cells were incubated with either 1:1000 Whole Cell Blue with 1:10,000 Hoechst 33342 in PBS or 1:5000 HCS CellMask Blue in PBS for 1 hour at 4°C. Cells were then washed with PBS, and wells were replaced with PBS for imaging. Cells were imaged in the DAPI, GFP, and Cy5 channels on either a cellWoRx automated imaging platform (Applied Precision) or in the DAPI and Cy5 channels of a Cellomics ArrayScan VTi (Thermo Fisher).
Signal and response data processing
To generate the signaling dataset, raw immunofluorescence images were processed in Fiji 160 and subsequently run through a pipeline in CellProfiler for intensity quantification (version 4.0.6) 161. Mean intensity of the nuclear and cytoplasmic compartments (where applicable) of total and phospho-proteins at all time points were normalized to the 2 μM doxorubicin value at the 6 hour time point across a single experiment. The mean of all biological replicates across experiments for each signal was used for subsequent data visualization and modeling.
The response dataset consisted of normalized fold-change data for the proliferation measurements, and percent positive as gated in flow cytometry for the rest of the response measurements collected. The mean was calculated for each individual response and time point across all biological replicates and experiments. Then, percent positive measurements were divided by 100 to scale values between 0 and 1, and a logit transformation was applied to all data points. Values of 0 were converted to 0.00001 for the logit transformation.
For principal component analysis (PCA), the normalized signals data was shaped into a 4×156 matrix, with rows representing the 4 distinct treatments and columns representing individual signals (26) and timepoints (6). The z-score was taken of this 4×156 matrix, which was used for principal component analysis.
For tensor PLSR, the raw signal data (X) was shaped into a 4×6×26 tensor, with mode 1 representing treatments (n = 4), mode 2 representing time points (n = 6), and mode 3 representing each individual signal (n = 26). The response data (Y) was shaped into a 4×6×10 tensor, with the same modes 1 and 2, and mode 3 representing individual responses (n = 10).
Tensor PLSR and VIP score calculation
The X and Y tensors were mean-centered across mode 1 and variance scaled across modes 2 and 3. The tensor PLSR model was constructed in MATLAB 2019b using the N-way Toolbox (version 1.8.0.0) as described in 55. All scripts and packages are available for download from the Github repository listed in the “Code Availability” statement.
The VIP scores were calculated as described in 58,163 using the Multi-Way VIP package in MATLAB 58.
Principal component analysis
The z-scored signaling matrix of 4 × 156 was run through the pca() function in MATLAB using three principal components and with no additional centering. Scores and loadings were plotted on principal components one and two, as these explained the majority of the variance in the signals (> 70%). All of the scores and loadings of PC 2 were multiplied by −1 to improve the interpretability in comparison to the t-PLSR results. To calculate the senescence and apoptosis projections of PCA loadings, the angle between the respective axis and the PCA loading vector was calculated, and then the cosine of this value was calculated. See the code on the Github repository for more details.
Inhibitor co-treatment experiments
Cells were seeded at 5,000 cells/well into an optical bottom 96-well plate (ThermoFisher, catalog # 165305) 24 hours before doxorubicin treatment. For the 0–12 hour inhibitor pulse, cells were treated with either vehicle (DMSO) or doxorubicin in addition to 10 μM SP600125, 10 μM PD98059, 10 μM SB203580, caffeine (5 mM for U2OS cells and 1 mM for OVCAR-8 cells), or vehicle (DMSO). Cells were maintained in this media for 4 hours, and then media was replaced with 1% FBS media containing either the respective inhibitor or vehicle, media was replaced with 10% FBS media 1 and 3 days after doxorubicin treatment, and inhibitor was maintained for the entire experiment for the results seen in figure 5G–I. For figures 6 and S6, 12 hours after the addition of doxorubicin, the media was replaced with 1% FBS media containing no inhibitors or vehicle. For the 12–24 hour inhibitor pulse, cells were treated with either vehicle (DMSO) or doxorubicin for 4 hours in the absence of inhibitors, and then the media was replaced with 1% FBS media. 12 hours after the addition of doxorubicin, the media was replaced with media containing the above inhibitors at the listed concentrations. Media was replaced with 10% FBS media 1 day and 3 days after the addition of doxorubicin for both the 0–12 and the 12–24 hour inhibitor condition. 5 days after the addition of doxorubicin, 10 μM BrdU was added to all wells, and 24 hours later plates were harvested for BrdU immunofluorescence, which was performed as described above.
ELISA for IL-6 and IL-8
U2OS cells were seeded at 5,000 cells/well into a 96-well plate 24 hours before doxorubicin treatment. Cells were treated with either vehicle or doxorubicin for 4 hours, and then media was replaced with drug-free, 1% FBS media. 24 hours after the addition of doxorubicin, media was replaced with 10% FBS media. Three days after doxorubicin addition, 10% FBS media containing either vehicle (DMSO), 10 μM SP600125, or 10 μM PD98059 was used to replace the growth media. Five days after doxorubicin treatment, the media was changed in wells to media without serum, and six days after doxorubicin treatment, media was transferred to a v-bottom 96-well plate, and stored at −80°C. ELISA assays for IL-6 (Invitrogen) and IL-8 (Invitrogen) levels in the media samples were conducted according to the manufacturer’s protocol.
Western blotting
Cells were treated as indicated in 6-well plates. At the given time points, the culture media was aspirated and cells were immediately lysed on ice. For the siRNA knockdown blots, RIPA buffer [50 mM Tris-HCl (pH 8), 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% Triton-X] was used, while for the rest of the shown blots high SDS (0.7%) lysis buffer [10 mM Tris (pH 7.5), 50 mM NaCl, 0.5% NP-40, 0.5% sodium deoxycholate, 0.7% SDS, 10 mM iodoacetamide] was used. Both types of lysis buffer were supplemented with phosSTOP (Roche, catalog # 4906837001) and cOmplete (Roche, catalog # 5892791001) tabs. 100 μL of lysis buffer was added to each well after aspiration, and afterwards was evenly distributed within the well. The plate was then incubated on ice for 5 minutes, and a cell scraper was used to scrape remaining cells while the plate was still on ice. The lysates were transferred to a cold Eppendorf tube and subsequently sonicated and centrifuged at 18,000 x g for 10 minutes at 4°C for the RIPA buffer lysates. For the high SDS lysates, lysates were vortexed for 15 seconds and then put back on ice for 10 minutes after the sonication step. This vortexing step was repeated twice for a total of 30 minutes, and then lysates were centrifuged at 18,000 x g for 10 minutes at 4°C. After centrifugation, the supernatants were transferred over to a new, pre-chilled Eppendorf tube and stored at −20°C.
The protein concentration of lysates was measured using a BCA assay (ThermoFisher), and the protein concentration was normalized across all samples. Samples were boiled for 5 minutes in 1x sample buffer [34 mM Tris, pH 6.8, 7% glycerol, 500 mM 2-mercaptoethanol, 1.6% SDS], and then loaded on a 10% SDS PAGE gel. After the samples ran through the gel at 100 V, a wet transfer step was run at 100 V for 1 hour at 4°C to transfer protein to a 0.22 μM nitrocellulose membrane. The quality of the transfer was checked with a Ponceau staining (Sigma-Aldrich), and after destaining the membranes were blocked in a blocking buffer consisting of a 1:1 mix of Intercept Blocking Buffer (LI-COR Biosciences) and PBS-T (PBS + 0.05% Tween) at room temperature for 1 hour on a shaker. Primary antibody was diluted in blocking buffer, and incubated with the membrane overnight at 4°C on a shaker. The membrane was then washed five times quickly with PBS-T, and then washed three times for 5 minutes each in PBS-T on a shaker at room temperature. Membranes were then incubated with secondary antibody (LI-COR Biosciences) diluted in blocking buffer for 1 hour at room temperature on a shaker, and afterwards the same wash steps as described above were performed, with the exception that the last wash step was conducted with PBS instead of PBS-T. Membranes were then kept in PBS, and then imaged in the 680 and 800 channel of an Odyssey CLx machine (LICOR-Biosciences). The mean, background subtracted signal intensity of respective bands were quantified in Image Studio Lite (LI-COR Biosciences, version 5.2), and intensity values were used for described ratio calculations.
For blots of phospho and total c-Jun, JNK, and RPA2 the blot was probed first using the phospho antibody, and then the membrane was stripped with 1x NewBlot nitrocellulose stripping buffer (LI-COR Biosciences), washed 5x quickly in PBS, and then washed three times for 10 minutes each in PBS on a shaker at room temperature. The membrane was then blocked again in 1:1 Odyssey blocking buffer to PBS-T, and reprobed with the total antibody on a shaker overnight at 4°C. The total MK2 blots were stripped and reprobed for p38 as described above on the same membrane, which was also done for the p-MK2 blots with p-p38.
siRNA transfection
Non-targeting (catalog #AM4636) was purchased from Invitrogen, and Jun #1 (catalog # s7658),and Jun #2 (catalog # s7659) siRNAs were purchased from Life Technologies. U2OS cells were plated in 6-well plates at a density of 150,000 cells/well 24 hours prior to transfection. Lipofectamine RNAiMAX was purchased from ThermoFisher Scientific, and used according to the manufacturer’s protocol to dose cells with 10 nM of siRNA-Lipofectamine duplex. Cells were incubated in siRNA/Lipofectamine mixture for 24 hours, and then each well was seeded into a 96-well plate at a density of 5,000 cells/well in triplicate. A concurrent 6-well plate transfected with siRNAs had media changed 24 hours post-transfection, and cells were harvested for immunoblotting 48 hours after transfection.
Cells in the 96-well plate were treated with doxorubicin 24 hours after seeding, and media was changed to media containing 1% FBS 4 hours after doxorubicin treatment. 24 hours after doxorubicin treatment, media was changed to complete media containing 10% FBS. Immunofluorescence for BrdU was conducted six days after doxorubicin treatment, as described above.
Cloning and Tol2-mediated stable transfection
pAK-Tol2-TRE-JunDN-HA-T2A-GFP (pTRE-JunDN-HA-T2A-GFP) construct was assembled using Gibson assembly with NEBuilder HiFi DNA Assembly Master Mix (NEB,) and inserted into a leid="P91">ntiviral pLVX-CMV empty backbone. pMIEG3-JunDN was a gift from Alexander Dent (Addgene plasmid # 40350; http://n2t.net/addgene:40350; RRID:Addgene_40350) 156, and was used as a template to amplify JunDN-HA and GFP fragments used in the assembly. JunDN-HA-T2A-GFP was amplified from the pLVX-CMV backbone using Phusion Mastermix (NEB), and inserted into the empty backbone of pAK_Tol2_TRE_Puro (Addgene, plasmid #130259) with digestion and ligation cloning into the NheI (NEB) and EcoRI (NEB) sites. GFP amplified from pLVX-CMV-JunDN-HA-T2A-GFP was inserted as described above into the same backbone for the TRE-GFP control construct. Positive colonies for both plasmids were confirmed with Sanger sequencing.
U2OS cells were seeded at a density of 200,000 cells/well into a 6-well plate 24 hours before transfection. Cells were co-transfected with 1.25 μg pCMV-Tol2 and 1.25 μg of either pAK-Tol2-TRE-JunDN-HA-T2A-GFP or pAK-Tol2-TRE-GFP (pTRE-GFP) with Lipofectamine 2000 (ThermoFisher Scientific) according to the manufacturer’s instructions at a 1:2 DNA (μg) to Lipofectamine 2000 reagent (μL) ratio. Media was replaced 24 hours later, and cells were selected with 2 μg/mL puromycin (Invivogen) 96 hours post-transfection for 3 days. Cells were then induced with 1 μg/mL doxycycline for 3 days, and GFP positive cells were harvested by FACS using a BD FACSAria III machine (BD Biosciences) for further experiments.
c-Jun dominant negative experiments
U2OS cells stably expressing either the pTRE-GFP or the pTRE-JunDN-HA-T2A-GFP construct and were FACS-sorted for GFP positivity as described above were seeded in a 96-well plate at 1,000 cells/well 96 hours before doxorubicin treatment. 72 hours before doxorubicin treatment, media was changed to complete media with or without 1 μg/mL doxycycline. 24 hours before doxorubicin treatment, media was changed again to complete media with or without 1 μg/mL doxycycline. Doxorubicin or DMSO was added directly to the wells, and media was changed to media containing 1% FBS with or without 1 μg/mL doxycycline. 12 hours after doxorubicin treatment, media was changed to media containing 1% FBS without doxycycline, and 24 and 72 hours after doxorubicin treatment, the media was changed to complete media containing 10% FBS. Immunofluorescence for BrdU was conducted as described above.
Retrovirus production and transduction
pSIRV-AP-1-mCherry was a gift from Peter Steinberger (Addgene plasmid # 118095; http://n2t.net/addgene:118095; RRID:Addgene_118095) 158, and pUMVC was a gift from Bob Weinberg (Addgene plasmid # 8449; http://n2t.net/addgene:8449; RRID:Addgene_8449) 159. HEK293T cells were co-transfected with 15 μg pSIRV-AP-1-mCherry, 10 μg pUMVC, and 2.5 μg pCMV-VSVG using the calcium phosphate transfection system (Clonetech), and media was changed 24 hours later. Media was harvested 24 hours after the media changed, and filtered through a 0.45 μm filter with a syringe. Filtered media was given to U2OS cells for 24 hours, and two weeks post-transduction, mCherry positive cells were harvested by FACS using a BD FACSAria III machine (BD Biosciences) for further experiments.
To generate U2OS cells containing both AP-1-mCherry and JunDN-IRES-eGFP, FACS-sorted AP-1-mCherry cells from above were infected with pMIEG3-JunDN construct packaged in retrovirus. pMIEG3-JunDN was packaged with HEK293Ts as described above, and filtered media containing virus was diluted 1:1 into complete media. U2OS cells were incubated with virus for 24 hours, and two weeks later, a GFP high population was harvested by FACS for further experiments using a BD FACSAria III machine (BD Biosciences).
AP-1-mCherry experiments
U2OS cells stably transfected with the AP-1 mCherry reporter were seeded into 6-well plates at a density of 150,000 cells/mL 24 hours before treatment. Cells were treated with either DMSO or doxorubicin for 4 hours, and then media was replaced with DMEM containing 1% FBS. For the entire experiment, cells were co-treated with either DMSO, 10 μM SP600125, or 10 μM PD98059. AP-1-mCherry cells co-expressing JunDN-IRES-GFP were also treated with either DMSO or doxorubicin. As a positive control, AP-1-mCherry cells were treated with 100 nM phorbol 12-myristate 13-actetate (PMA) for 24 hours and was administered at the same time as the doxorubicin. Cells were harvested 24 hours after treatment, and flow cytometry was performed using a BD LSR II HTS flow cytometer (BD Biosciences).
Software
For image processing, either Fiji or NIS Viewer were used. CellProfiler was used for downstream image quantification, and FlowJo v10 was used to process flow cytometry-based response data. For Incucyte-based proliferation data, ilastik was used to count the number of cells in brightfield images. MATLAB 2019b was used for modeling and PCA analysis, and Python 3.7 was used to analyze and compile CellProfiler outputs as well as generate boxplots. All cartoon illustrations were made directly in Adobe Illustrator, or by using BioRender under an academic license. Publication licenses are available upon request.
Quantification and Statistical Analysis
All statistical testing was performed in GraphPad Prism (version 8.4.1), which specific tests performed listed in the relevant figure caption. Data was quantified as described in in the respective method subsections and/or captions.
Supplementary Material
Highlights.
DNA damage from topoisomerase-II inhibitors induces senescence through phospho-c-Jun
JNK and Erk MAPK activity within 12 hours of damage controls senescence through c-Jun
Late JNK and Erk MAPK activity after 12 hours controls cytokine release during the SASP
Tensor PLSR and PCA can be used to map cell phenotypes to specific signaling dynamics
ACKNOWLEDGEMENTS
We gratefully acknowledge Bjorn Millard for assistance with microscopy, Leslie Gaffney for assistance with figure design, and all members of the Yaffe lab for support and suggestions. We also specifically would like to acknowledge the reviewers for their invaluable input, which greatly improved this work. We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Flow Cytometry Facility, High Throughput Sciences Facility, and Microscopy Facility, which are supported in part by the Koch Institute Support Grant P30-CA14051 from the National Cancer Institute. This work was supported by NIH grants R35-ES028374 and R01-GM104047 to M.B.Y., U54-CA112967 to M.B.Y and D.A.L., P50-GM68762 to P.K.S. and D.A.L., an NSF Graduate Research Fellowship 1122374 to T.S.N, a Ludwig Center Graduate Fellowship to T.S.N., the Ovarian Cancer Research Fund, the Charles and Marjorie Holloway Foundation, and the MIT Center for Precision Cancer Medicine.
Footnotes
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
DECLARATION OF INTERESTS
Kevin A. Janes, Peter K. Sorger, and Douglas A. Lauffenburger are on the Advisory Board of Cell Systems.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hoeijmakers JHJ (2001). The consequences of DNA injury Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SP, and Bartek J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harper JW, and Elledge SJ (2007). The DNA Damage Response: Ten Years After. Mol. Cell 28, 739–745. 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Matt S, and Hofmann TG (2016). The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell. Mol. Life Sci. 73, 2829–2850. 10.1007/s00018-016-2130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LVF, Kolettas E, Niforou K, Zoumpourlis VC, et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 6.D’Adda Di Fagagna F. (2008). Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522. 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 7.Gorgoulis VG, Vassiliou LVF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA, Kastrinakis NG, Levy B, et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 8.Van Nguyen T, Puebla-Osorio N, Pang H, Dujka ME, and Zhu C. (2007). DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: A mouse model. J. Exp. Med. 204, 1453–1461. 10.1084/jem.20062453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halazonetis TD, Gorgoulis VG, and Bartek J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 10.Kastan MB, and Bartek J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 11.Ciccia A, and Elledge SJ (2010). The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 40, 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, et al. (2003). Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 278, 20303–20312. 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 13.Reinhardt HC, and Yaffe MB (2009). Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255. 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine AJ, and Oren M. (2009). The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 9, 749–758. 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Childs BG, Baker DJ, Kirkland JL, Campisi J, and van Deursen JM (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153. 10.15252/embr.201439245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, Khokhar AR,and Kuang J. (2005). Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene 24, 2929–2943. 10.1038/sj.onc.1208474. [DOI] [PubMed] [Google Scholar]
- 17.Toshiyuki M, and Reed JC (1995). Tumor suppressor p53 is a direct transcriptionalactivator of the human bax gene. Cell 80, 293–299. 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 18.Helleday T. (2008). Amplifying tumour-specific replication lesions by DNA repairinhibitors - A new era in targeted cancer therapy. Eur. J. Cancer 44, 921–927. 10.1016/j.ejca.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 19.Helleday T, Petermann E, Lundin C, Hodgson B, and Sharma RA (2008). DNArepair pathways as targets for cancer therapy. Nat. Rev. Cancer 8, 193–204. 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy RD, and D’Andrea AD (2006). DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J. Clin. Oncol. 24, 3799–3808. 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 21.Powell SN, and Bindra RS (2009). Targeting the DNA damage response for cancer therapy. DNA Repair 8, 1153–1165. 10.1016/j.dnarep.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Campisi J, and D’Adda Di Fagagna F. (2007). Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 23.D’Adda Di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, and Jackson SP (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 24.Herbig U, Jobling WA, Chen BPC, Chen DJ, and Sedivy JM (2004). Telomereshortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 14, 501–513. 10.1016/S1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 25.Shay JW, and Wright WE (2004). Telomeres are double-strand DNA breaks hiddenfrom DNA damage responses. Mol. Cell 14, 420–421. 10.1016/S1097-2765(04)00269-2. [DOI] [PubMed] [Google Scholar]
- 26.Rodier F, and Campisi J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basisty N, Kale A, Jeon O, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Ferrucci L, Campisi J, et al. (2020). A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biology 18, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; Basisty, N., Kale, A., Jeon, O., Kuehnemann, C., Payne, T., Rao, C., Holtz, A., Shah, S., Ferrucci, L., Campisi, J., & Schilling, B. (2020). A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biology, 1–26. 10.2139/ssrn.3380253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR,Freund A, Campeau E, Davalos AR, and Campisi J. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davalos AR, Coppe JP, Campisi J, and Desprez PY (2010). Senescent cells as asource of inflammatory factors for tumor progression. Cancer Metastasis Rev. 29, 273–283. 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coppé JP, Desprez PY, Krtolica A, and Campisi J. (2010). The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol.: Mech. Dis. 5, 99–118. 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS,Desprez PY, and Campisi J. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6. 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freund A, Patil CK, and Campisi J. (2011). P38MAPK is a novel DNA damageresponse-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548. 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim MJ, Choi SY, Park IC, Hwang SG, Kim C, Choi YH, Kim H, Lee KH,and Lee SJ (2008). Opposing roles of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in the cellular response to ionizing radiation in human cervical cancer cells. Mol. Cancer Res. 6, 1718–1731. 10.1158/1541-7786.MCR-08-0032. [DOI] [PubMed] [Google Scholar]
- 34.Spallarossa P, Altieri P, Barisione C, Passalacqua M, Aloi C, Fugazza G,Frassoni F, Podestá M, Canepa M, Ghigliotti G, et al. (2010). P38 MAPK and JNK antagonistically control senescence and cytoplasmic p16ink4a expression in doxorubicin-treated endothelial progenitor cells. PLoS One 5, 1–12. 10.1371/journal.pone.0015583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tentner AR, Lee MJ, Ostheimer GJ, Samson LD, Lauffenburger DA, and Yaffe MB (2012). Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1 / S arrest after genotoxic stress. Mol. Syst. Biol, 1–18. 10.1038/msb.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, and Sorger PK (2006). The Response of Human Epithelial Cells to TNF Involves an Inducible Autocrine Cascade. Cell 124, 1225–1239. 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 37.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AEH, and Yaffe MB (2005). MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G 2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48. 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 38.Mokim Ahmed K, and Jian Li J. (2007). ATM-NF-κB Connection as a Target for Tumor Radiosensitization. Current Cancer Drug Targets 7, 335–342. 10.2174/156800907780809769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, andYaffe MB (2012). Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 149, 780–794. 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinhardt HC, Aslanian AS, Lees JA, and Yaffe MB (2007). p53-Deficient CellsRely on ATM- and ATR-Mediated Checkpoint Signaling through the p38MAPK/MK2 Pathway for Survival after DNA Damage. Cancer Cell 11, 175–189. 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts PJ, and Der CJ (2007). Targeting the Raf-MEK-ERK mitogen-activatedprotein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310. 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 42.Kong YW, Dreaden EC, Morandell S, Zhou W, Dhara SS, Sriram G, Lam FC,Patterson JC, Quadir M, Dinh A, et al. (2020). Enhancing chemotherapy response through augmented synthetic lethality by co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat. Commun. 11, 1–12. 10.1038/s41467-020-17958-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janes KA, Kelly JR, Gaudet S, Albeck JG, Sorger PK, and Lauffenburger DA (2004). Cue-Signal-Response Analysis of TNF-Induced Apoptosis by Partial Least Squares Regression of Dynamic Multivariate Data. J. Comput. Biol. 11, 544–561. [DOI] [PubMed] [Google Scholar]
- 44.Gewirtz DA (1999). A Critical Evaluation of the Mechanisms of Action Proposed forthe Antitumor Effects of the Anthracycline Antibiotics Adriamycin and Daunorubicin. Biochemical Pharmacology 57, 727–741. [DOI] [PubMed] [Google Scholar]
- 45.Bodley A, Liu LF, Israel M, Seshadri R, Koseki Y, Giuliani FC, Kirschenbaum S, Silber R, and Potmesil M. (1989). DNA Topoisomerase II-mediated Interaction of Doxorubicin and Daunorubicin Congeners with DNA. Cancer Res. 49, 5969–5978. [PubMed] [Google Scholar]
- 46.Maki CG, and Howley PM (1997). Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol. Cell. Biol. 17, 355–363. 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park YB, Park MJ, Kimura K, Shimizu K, Lee SH, and Yokota J. (2002). Alterations in the INK4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet. Cytogenet. 133, 105–111. 10.1016/S0165-4608(01)00575-1. [DOI] [PubMed] [Google Scholar]
- 48.Amtmann E, Eddé E, Sauer G, and Westphal O. (1990). Restoration of theresponsiveness to growth factors in senescent cells by an embryonic cell extract. Exp. Cell Res. 189, 202–207. 10.1016/0014-4827(90)90236-4. [DOI] [PubMed] [Google Scholar]
- 49.Wansink DG, Schul W, Van der Kraan I, Van Steensel B, Van Driel R, and DeJong L. (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol. 122, 283–293. 10.1083/jcb.122.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, and Yaffe MB(2005). A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 310, 1646–1653. 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 51.Stewart ZA, Leach SD, and Pietenpol JA (1999). p21 Waf1/Cip1 Inhibition of Cyclin E/Cdk2 Activity Prevents Endoreduplication after Mitotic Spindle Disruption. Mol. Cell. Biol. 19, 205–215. 10.1128/mcb.19.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toettcher JE, Loewer A, Ostheimer GJ, Yaffe MB, Tidor B, and Lahav G. (2009). Distinct mechanisms act in concert to mediate cell cycle arrest. Proc. Natl. Acad. Sci. U. S. A. 106, 785–790. 10.1073/pnas.0806196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janes KA, and Yaffe MB (2006). Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell Biol. 7, 820–828. 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 54.Geladi P, and Kowalski BR (1986). Partial Least-Squares Regression: A Tutorial. Anal. Chim. Acta 185, 1–17. [Google Scholar]
- 55.Chitforoushzadeh Z, Ye Z, Sheng Z, La Rue S, Fry RC, Lauffenburger DA, andJanes KA (2016). TNF-insulin crosstalk at the transcription factor GATA6 is revealed by a model that links signaling and transcriptomic data tensors. Sci. Signal. 9, 1–18. 10.1126/scisignal.aad3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bro R. (1996). Multiway calibration. Multilinear PLS. J. Chemom. 10, 47–61. . [DOI] [Google Scholar]
- 57.Caulk AW, and Janes KA (2019). Robust latent-variable interpretation of in vivo regression models by nested resampling. Sci. Rep. 9, 1–15. 10.1038/s41598-019-557962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Favilla S, Durante C, Vigni ML, and Cocchi M. (2013). Assessing feature relevance in NPLS models by VIP. Chemometrics Intellig. Lab. Syst. 129, 76–86. 10.1016/j.chemolab.2004.07.001. [DOI] [Google Scholar]
- 59.Baldin V, Lukas J, Marcote MJ, Pagano M, and Draetta G. (1993). Cyclin D1 is anuclear protein required for cell cycle progression in G1. Genes and Development 7, 812–821. 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 60.Yang K, Hitomi M, and Stacey DW (2006). Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 1, 1–8. 10.1186/17471028-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL,Medema RH, Hummerich H, and Jat PS (2011). Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 30, 2356–2366. 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. (2012). NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 2601–2612. 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P,Luo W, Chicas A, Lee CS, et al. (2011). Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes and Development 25, 2125–2136. 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, Martindale JL, and Holbrook NJ (2000). Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 275, 39435–39443. 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 65.Wada T, Stepniak E, Hui L, Leibbrandt A, Katada T, Nishina H, Wagner EF,and Penninger JM (2008). Antagonistic control of cell fates by JNK and p38-MAPK signaling. Cell Death Differ. 15, 89–93. 10.1038/sj.cdd.4402222. [DOI] [PubMed] [Google Scholar]
- 66.Xia Z, Dickens M, Raingeaud J, Davis RJ, and Greenberg ME (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331. 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 67.Chen YR, Meyer CF, and Tan TH (1996). Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in γ radiation-induced apoptosis. J. Biol. Chem. 271, 631–634. 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 68.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, and Davis RJ (2000). Requirement of JNK for stress-induced activation of the cytochrome c- mediated death pathway. Science 288, 870–874. 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 69.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E,Zon LI, Kyriakis JM, et al. (1996). Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 380, 75–79. 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 70.Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J,Liu FF, and Woodgett JR (1996). The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr. Biol. 6, 606–613. 10.1016/S0960-9822(02)00547-X. [DOI] [PubMed] [Google Scholar]
- 71.Venkatakrishnan CD, Tewari AK, Moldovan L, Cardounel AJ, Zweier JL,Kuppusamy P, and Ilangovan G. (2006). Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am. J. Physiol. Heart Circ. Physiol. 291, H2680–91. 10.1152/ajpheart.00395.2006. [DOI] [PubMed] [Google Scholar]
- 72.Stokoe D, Engel K, Campbell DG, Cohen P, and Gaestel M. (1992). Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 313, 307–313. 10.1016/00145793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- 73.Gire V, Roux P, Wynford-Thomas D, Brondello JM, and Dulic V. (2004). DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 23, 2554–2563. 10.1038/sj.emboj.7600259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nayak D, Kumar A, Chakraborty S, Ur Rasool R, Amin H, Katoch A, Gopinath V, Mahajan V, Zilla MK, Rah B, et al. (2017). Inhibition of Twist1-mediated invasion by Chk2 promotes premature senescence in p53-defective cancer cells. Cell Death Differ. 24, 1275–1287. 10.1038/cdd.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lane DP (1992). p53, guardian of the genome. Nature Publishing Group UK. 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 76.Kumari R, and Jat P. (2021). Mechanisms of Cellular Senescence: Cell Cycle Arrestand Senescence Associated Secretory Phenotype. Front Cell Dev Biol 9, 645593. 10.3389/fcell.2021.645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 88, 593–602. 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 78.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 79.Brown JP, Wei W, and Sedivy JM (1997). Bypass of senescenoe after disruption ofp21(CIP1)/(WAF1) gene in normal diploid human fibroblasts. Science 277, 831–834. 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 80.Noda A, Ning Y, Venable SF, Pereira-Smith OM, and Smith JR (1994). Cloningof Senescent Cell-Derived Inhibitors of DNA Synthesis Using an Expression Screen. Exp. Cell Res. 211, 90–98. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y, Blandino G, and Givol D. (1999). Induced p21(waf) expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene 18, 2643–2649. 10.1038/sj.onc.1202632. [DOI] [PubMed] [Google Scholar]
- 82.Chen PL, Scully P, Shew JY, Wang JY, and Lee WH (1989). Phosphorylation ofthe retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 58, 1193–1198. 10.1016/0092-8674(89)90517-5. [DOI] [PubMed] [Google Scholar]
- 83.Sellers WR, Rodgers JW, and Kaelin WG Jr (1995). A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc. Natl. Acad. Sci. U. S. A. 92, 11544–11548. 10.1073/pnas.92.25.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu CL, Classon M, Dyson N, and Harlow E. (1996). Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol. Cell. Biol. 16, 3698–3706. 10.1128/MCB.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, and Kaelin WG Jr(1995). The transcription factor E2F-1 is a downstream target of RB action. Mol. Cell. Biol. 15, 742–755. 10.1128/MCB.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kovary K, and Bravo R. (1991). The jun and fos protein families are both required forcell cycle progression in fibroblasts. Mol. Cell. Biol. 11, 4466–4472. 10.1128/mcb.11.9.4466-4472.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson RS, Van Lingen B, Papaioannou VE, and Spiegelman BM (1993). A nullmutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes and Development 7, 1309–1317. 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- 88.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret F-X, Green DR, and Karin M. (1999). Withdrawal of Survival Factors Results in Activation of the JNK Pathway in Neuronal Cells Leading to Fas Ligand Induction and Cell Death. Mol. Cell. Biol. 19, 751–763. 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, and Karin M. (2000). Themammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 103, 897–908. 10.1016/s0092-8674(00)00193-8. [DOI] [PubMed] [Google Scholar]
- 90.Humar M, Loop T, Schmidt R, Hoetzel A, Roesslein M, Andriopoulos N, Pahl HL, Geiger KK, and Pannen BHJ (2007). The mitogen-activated protein kinase p38 regulates activator protein 1 by direct phosphorylation of c-Jun. Int. J. Biochem. Cell Biol. 39, 2278–2288. 10.1016/j.biocel.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 91.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, and Davis RJ(1994). JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025–1037. 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 92.Leppä S, Saffrich R, Ansorge W, and Bohmann D. (1998). Differential regulation ofc-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 17, 4404–4413. 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, and Woodgett JR (1991). Phosphorylation of c-jun mediated by MAP kinases. Nature 353, 670–674. [DOI] [PubMed] [Google Scholar]
- 94.Chen QM, Liu J, and Merrett JB (2000). Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J 347, 543–551. 10.1042/0264-6021:3470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fridman JS, and Lowe SW (2003). Control of apoptosis by p53. Oncogene 22, 9030–9040. 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 96.Vousden KH, and Prives C. (2009). Blinded by the Light: The Growing Complexity ofp53. Cell 137, 413–431. 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 97.Feld R, Rubinstein L, and Thomas PA (1993). Adjuvant chemotherapy with cyclophosphamide, doxorubicin, and cisplatin in patients with completely resected stage I non-small-cell lung cancer. The Lung Cancer Study Group. J. Natl. Cancer Inst. 85, 299–306. 10.1093/jnci/85.4.299. [DOI] [PubMed] [Google Scholar]
- 98.Otterson GA, Villalona-Calero MA, Hicks W, Pan X, Ellerton JA, Gettinger SN, and Murren JR (2010). Phase I/II study of inhaled doxorubicin combined with platinum-based therapy for advanced non-small cell lung cancer. Clin. Cancer Res. 16, 2466–2473. 10.1158/1078-0432.CCR-09-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joss RA, Alberto P, Obrecht JP, Barrelet L, Holdener EE, Siegenthaler P,Goldhirsch A, Mermillod B, and Cavalli F. (1984). Combination chemotherapy for non-small cell lung cancer with doxorubicin and mitomycin or cisplatin and etoposide. Cancer Treat. Rep. 68, 1079–1084. [PubMed] [Google Scholar]
- 100.Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, and Peeper DS (2008). Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 133, 1019–1031. 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 101.Hoffmann E, Thiefes A, Buhrow D, Dittrich-Breiholz O, Schneider H, Resch K, and Kracht M. (2005). MEK1-dependent delayed expression of fos-related antigen-1 counteracts c-Fos and p65 NF-κB-mediated interleukin-8 transcription in response to cytokines or growth factors. J. Biol. Chem. 280, 9706–9718. 10.1074/jbc.M407071200. [DOI] [PubMed] [Google Scholar]
- 102.Krause A, Holtmann H, Eickemeier S, Winzen R, Szamel M, Resch K, Saklatvala J, and Kracht M. (1998). Stress-activated protein kinase/Jun N-terminal kinase is required for interleukin (IL)-1-induced IL-6 and IL-8 gene expression in the human epidermal carcinoma cell line KB. J. Biol. Chem. 273, 23681–23689. 10.1074/jbc.273.37.23681. [DOI] [PubMed] [Google Scholar]
- 103.Thompson EJ, Gupta A, Stratton MS, and Bowden GT (2002). Mechanism of action of a dominant negative c-jun mutant in inhibiting activator protein-1 activation. Mol. Carcinog. 35, 157–162. 10.1002/mc.10090. [DOI] [PubMed] [Google Scholar]
- 104.Tewey KM, Rowe TC, Yang L, Halligan BD, and Liu LF (1984). Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226, 466–468. 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 105.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579. 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Durr FE, Wallace RE, and Citarella RV (1983). Molecular and biochemical pharmacology of mitoxantrone. Cancer Treat. Rev. 10 Suppl B, 3–11. 10.1016/03057372(83)90016-6. [DOI] [PubMed] [Google Scholar]
- 107.Zhao H, Rybak P, Dobrucki J, Traganos F, and Darzynkiewicz Z. (2012). Relationship of DNA damage signaling to DNA replication following treatment with DNA topoisomerase inhibitors camptothecin/topotecan, mitoxantrone, or etoposide. Cytometry A 81, 45–51. 10.1002/cyto.a.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olson E, Nievera CJ, Klimovich V, Fanning E, and Wu X. (2006). RPA2 Is a Direct Downstream Target for ATR to Regulate the S-phase Checkpoint*. J. Biol. Chem. 281, 39517–39533. 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 109.Vassin VM, Anantha RW, Sokolova E, Kanner S, and Borowiec JA (2009). Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 122, 4070–4080. 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zuazua-Villar P, Ganesh A, Phear G, Gagou ME, and Meuth M. (2015). Extensive RPA2 hyperphosphorylation promotes apoptosis in response to DNA replication stress in CHK1 inhibited cells. Nucleic Acids Res. 43, 9776–9787. 10.1093/nar/gkv835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cuella-Martin R, Oliveira C, Lockstone HE, Snellenberg S, Grolmusova N, and Chapman JR (2016). 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol. Cell 64, 51–64. 10.1016/j.molcel.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fernando RN, Eleuteri B, Abdelhady S, Nussenzweig A, Andäng M, and Ernfors P. (2011). Cell cycle restriction by histone H2AX limits proliferation of adult neural stem cells. Proc. Natl. Acad. Sci. U. S. A. 108, 5837–5842. 10.1073/pnas.1014993108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reyes J, Chen JY, Stewart-Ornstein J, Karhohs KW, Mock CS, and Lahav G. (2018). Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 71, 581–591.e5. 10.1016/j.molcel.2018.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsu CH, Altschuler SJ, and Wu LF (2019). Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 178, 361–373.e12. 10.1016/j.cell.2019.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hübner A, Barrett T, Flavell RA, and Davis RJ (2008). Multisite Phosphorylation Regulates Bim Stability and Apoptotic Activity. Mol. Cell 30, 415–425. 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kennedy NJ, Sluss HK, Jones SN, Bar-Sagi D, Flavell RA, and Davis RJ (2003). Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes and Development 17, 629–637. 10.1101/gad.1062903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lei K, and Davis RJ (2003). JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100, 2432–2437. 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Donovan N, Becker EBE, Konishi Y, and Bonni A. (2002). JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 277, 40944–40949. 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- 119.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, and Gotoh Y. (2004). JNK promotes Bax translocation to mitochondria through phosphorylation of 14–3-3 proteins. EMBO J. 23, 1889–1899. 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sunayama J, Tsuruta F, Masuyama N, and Gotoh Y. (2005). JNK antagonizes Akt-mediated survival signals by phosphorylating 14–3-3. J. Cell Biol. 170, 295–304. 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oleinik NV, Krupenko NI, and Krupenko SA (2007). Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 26, 7222–7230. 10.1038/sj.onc.1210526. [DOI] [PubMed] [Google Scholar]
- 122.Jones EV, Dickman MJ, and Whitmarsh AJ (2007). Regulation of p73mediated apoptosis by c-Jun N-terminal kinase. Biochem. J 405, 617–623. 10.1042/BJ20061778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dhanasekaran DN, and Premkumar Reddy E. (2017). JNK-signaling: A multiplexing hub in programmed cell death. Genes and Cancer 8, 682–694. 10.18632/genesandcancer.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Das M, Jiang F, Sluss HK, Zhang C, Shokat KM, Flavell RA, and Davis RJ (2007). Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 104, 15759–15764. 10.1073/pnas.0707782104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lavoie H, Gagnon J, and Therrien M. (2020). ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632. 10.1038/s41580-020-0255-7. [DOI] [PubMed] [Google Scholar]
- 126.Spelat R, Ferro F, and Curcio F. (2012). Serine 111 phosphorylation regulates OCT4A protein subcellular distribution and degradation. J. Biol. Chem. 287, 38279–38288. 10.1074/jbc.M112.386755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim MO, Kim SH, Cho YY, Nadas J, Jeong CH, Yao K, Kim DJ, Yu DH, Keum YS, Lee KY, et al. (2012). ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat. Struct. Mol. Biol. 19, 283–290. 10.1038/nsmb.2217. [DOI] [PubMed] [Google Scholar]
- 128.Yeo JC, Jiang J, Tan ZY, Yim GR, Ng JH, Göke J, Kraus P, Liang H, Gonzales KAU, Chong HC, et al. (2014). Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell 14, 864–872. 10.1016/j.stem.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 129.Yeh PY, Chuang SE, Yeh KH, Song YC, Chang LLY, and Cheng AL (2004). Phosphorylation of p53 on Thr55 by ERK2 is necessary for doxorubicin-induced p53 activation and cell death. Oncogene 23, 3580–3588. 10.1038/sj.onc.1207426. [DOI] [PubMed] [Google Scholar]
- 130.Lin AW, Barradas M, Stone JC, Van Aelst L, Serrano M, and Lowe SW (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes and Development 12, 3008–3019. 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, and Sun P. (2002). Sequential Activation of the MEK-Extracellular Signal-Regulated Kinase and MKK3/6-p38 Mitogen-Activated Protein Kinase Pathways Mediates Oncogenic ras Induced Premature Senescence. Mol. Cell. Biol. 22, 3389–3403. 10.1128/mcb.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhu J, Woods D, McMahon M, and Bishop JM (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes and Development 12, 2997–3007. 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roux PP, and Blenis J. (2004). ERK and p38 MAPK-Activated Protein Kinases: a Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 68, 320–344. 10.1128/mmbr.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MATM, Wang XZ, Linding R, Ong SE, Weaver D, Carr SA, et al. (2010). DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 40, 34–49. 10.1016/j.molcel.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cannell IG, Merrick KA, Morandell S, Zhu CQ, Braun CJ, Grant RA, Cameron ER, Tsao MS, Hemann MT, and Yaffe MB (2015). A Pleiotropic RNABinding Protein Controls Distinct Cell Cycle Checkpoints to Drive Resistance of p53-Defective Tumors to Chemotherapy. Cancer Cell 28, 623–637. 10.1016/j.ccell.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, and Yaffe MB (2013). A Reversible Gene-Targeting Strategy Identifies Synthetic Lethal Interactions between MK2 and p53 in the DNA Damage Response InVivo. Cell Reports 5, 868–877. 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eferl R, and Wagner EF (2003). AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 138.Martínez-Zamudio RI, Roux PF, de Freitas JANLF, Robinson L, Doré G, Sun B, Belenki D, Milanovic M, Herbig U, Schmitt CA, et al. (2020). AP-1 imprints a reversible transcriptional programme of senescent cells. Nat. Cell Biol. 22, 842–855. 10.1038/s41556-020-0529-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Han R, Li L, Ugalde AP, Tal A, Manber Z, Barbera EP, Chiara VD, Elkon R, and Agami R. (2018). Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Biol. 19, 1–13. 10.1186/s13059-018-1494-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hübner A, Mulholland DJ, Standen CL, Karasarides M, Cavanagh-Kyros J, Barrett T, Chi H, Greiner DL, Tournier C, Sawyers CL, et al. (2012). JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc. Natl. Acad. Sci. U. S. A. 109, 12046–12051. 10.1073/pnas.1209660109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dagogo-Jack I, and Shaw AT (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94. 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 142.Melo FDSE, Vermeulen L, Fessler E, and Medema JP (2013). Cancer heterogeneity - A multifaceted view. EMBO Rep. 14, 686–695. 10.1038/embor.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cohn RL, Gasek NS, Kuchel GA, and Xu M. (2022). The heterogeneity of cellular senescence: insights at the single-cell level. Trends in Cell Biology, 1–9. 10.1016/j.tcb.2022.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, and Morrison SJ (2010). Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523. 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kemper K, Krijgsman O, Cornelissen P, Shahrabi A, Weeber F, Song J, Kuilman T, Vis DJ, Wessels LF, Voest EE, et al. (2015). Intra and inter tumor heterogeneity in a vemurafenib resistant melanoma patient and derived xenografts. EMBO Mol. Med. 7, 1104–1118. 10.15252/emmm.201404914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Morrissy AS, Garzia L, Shih DJH, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FMG, Ramaswamy V, Lindsay PE, et al. (2016). Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529, 351–357. 10.1038/nature16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhao B, Hemann MT, and Lauffenburger DA (2014). Intratumor heterogeneity alters most effective drugs in designed combinations. Proc. Natl. Acad. Sci. U. S. A. 111, 10773–10778. 10.1073/pnas.1323934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, and Polyak K. (2014). Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58. 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chapman A, del Ama LF, Ferguson J, Kamarashev J, Wellbrock C, and Hurlstone A. (2014). Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 8, 688–695. 10.1016/j.celrep.2014.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang L, Lankhorst L, and Bernards R. (2022). Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 22, 340–355. 10.1038/s41568-022-00450-9. [DOI] [PubMed] [Google Scholar]
- 151.Fielder E, Wan T, Alimohammadiha G, Ishaq A, Low E, Weigand BM, Kelly G, Parker C, Griffin B, Jurk D, et al. (2022). Short senolytic or senostatic interventions rescue progression of radiation-induced frailty and premature ageing in mice. Elife 11, 1–29. 10.7554/elife.75492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Short S, Fielder E, Miwa S, and von Zglinicki T. (2019). Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 41, 683–692. 10.1016/j.ebiom.2019.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, Belt BA, Alspach E, Leahy K, Luo J, et al. (2016). Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7. 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wu J, Gao FX, Wang C, Qin M, Han F, Xu T, Hu Z, Long Y, He XM, Deng X, et al. (2019). IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 38, 1–15. 10.1186/s13046-019-1310-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Goulet CR, Champagne A, Bernard G, Vandal D, Chabaud S, Pouliot F, and Bolduc S. (2019). Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 19, 1–13. 10.1186/s12885-019-5353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang Z-Y, Sato H, Kusam S, Sehra S, Toney LM, and Dent AL (2005). Regulation of IL-10 Gene Expression in Th2 Cells by Jun Proteins. The Journal of Immunology 174, 2098–2105. 10.4049/jimmunol.174.4.2098. [DOI] [PubMed] [Google Scholar]
- 157.Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, Wang X, Hackett PB, Largaespada DA, McIvor RS, et al. (2006). Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2, e169. 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jutz S, Leitner J, Schmetterer K, Doel-Perez I, Majdic O, Grabmeier-Pfistershammer K, Paster W, Huppa JB, and Steinberger P. (2016). Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-κB, NFAT and AP-1. J. Immunol. Methods 430, 10–20. 10.1016/j.jim.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 159.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, and Goodman A. (2021). CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 1–11. 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). Ilastik: Interactive Machine Learning for (Bio)Image Analysis. Nat. Methods 16, 1226–1232. 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- 163.Favilla S, Huber A, Pagnoni G, Lui F, Facchin P, Cocchi M, Baraldi P, and Porro CA (2014). Ranking brain areas encoding the perceived level of pain from fMRI data. Neuroimage 90, 153–162. 10.1016/j.neuroimage.2014.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data statement: All data reported in this paper will be shared by the lead contact upon reasonable request.
Code statement: Raw quantified data and MATLAB scripts used to construct t-PLSR and PCA results can be found at: github.com/yaffelab/CellFate-TensorPLSR. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-γ-H2AX | Millipore | Cat#05–636, RRID:AB_309864 |
| Rabbit monoclonal anti-phospho-Akt | Cell Signaling Technology | Cat#4058, RRID:AB_331168 |
| Rabbit monoclonal anti-phospho-Erk | Cell Signaling Technology | Cat#4377, RRID:AB_331775 |
| Rabbit monoclonal anti-phospho-Erk | Cell Signaling Technology | Cat#4370, RRID:AB_2315112 |
| Mouse monoclonal anti-Erk | Cell Signaling Technology | Cat#4696, RRID:AB_390780 |
| Rabbit polyclonal anti-phospho-JNK | Cell Signaling Technology | Cat#9251, RRID:AB_331659 |
| Rabbit polyclonal anti-JNK | Cell Signaling Technology | Cat#9252, RRID:AB_2250373 |
| Mouse monoclonal anti-NF-κB | Santa Cruz | Cat#sc-8008, RRID:AB_628017 |
| Rabbit monoclonal anti-phospho-p38 | Cell Signaling Technology | Cat#4631, RRID:AB_331765 |
| Rabbit polyclonal anti-p38 | Cell Signaling Technology | Cat#9212, RRID:AB_330713 |
| Rabbit monoclonal phospho-MK2 | Cell Signaling Technology | Cat#3007, RRID:AB_490936 |
| Rabbit anti-MK2 | Cell Signaling Technology | Cat#3042, RRID:AB_10694238 |
| Rabbit monoclonal anti-phospho-S6 | Cell Signaling Technology | Cat#4856, RRID:AB_2181037 |
| Mouse monoclonal anti-p27 | Santa Cruz | Cat#sc-1641, RRID:AB_628074 |
| Rabbit monoclonal anti-p27 | Cell Signaling Technology | Cat#3686, RRID:AB_2077850 |
| Rabbit polyclonal anti-phospho-Hsp27 | Cell Signaling Technology | Cat#2401, RRID:AB_331644 |
| Mouse monoclonal anti-p53 | Santa Cruz | Cat#sc-263, RRID:AB_628084 |
| Rabbit polyclonal anti-phospho-c-Jun(Ser 73) | Cell Signaling Technology | Cat#9164, RRID:AB_330892 |
| Rabbit monoclonal anti-phospho-c-Jun(Ser 73) | Cell Signaling Technology | Cat#3270, RRID:AB_2129575 |
| Rabbit polyclonal anti-phospho-c-Jun(Ser 63) | Cell Signaling Technology | Cat#9261, RRID:AB_2130162 |
| Rabbit monoclonal anti-c-Jun | Cell Signaling Technology | Cat#9165, RRID:AB_2130165 |
| Rabbit polyclonal anti-phospho-Chk2 | Cell Signaling Technology | Cat#2661, RRID:AB_331479 |
| Mouse monoclonal anti-IκBα | BD Biosciences | Cat#610691, RRID:AB_398014 |
| Mouse monoclonal anti-IκBα | Cell Signaling Technology | Cat#4814, RRID:AB_390781 |
| Mouse monoclonal anti-Cyclin D | Santa Cruz | Cat#sc-20044, RRID:AB_627346 |
| Rabbit polyclonal anti-phospho-Rb | Cell Signaling Technology | Cat#9308, RRID:AB_331472 |
| Mouse monoclonal anti-Cyclin E | Santa Cruz | Cat#sc-247, RRID:AB_627357 |
| Rabbit polyclonal anti-Cyclin A | Santa Cruz | Cat#sc-751, RRID:AB_631329 |
| Mouse monoclonal anti-p21 | Millipore | Cat#05–345, RRID:AB_309684 |
| Rabbit monoclonal anti-p21 | Cell Signaling Technology | Cat#2947, RRID:AB_823586 |
| Rabbit monoclonal anti-p16 | Cell Signaling Technology | Cat#18769, RRID:AB_2935679 |
| Rabbit polyclonal anti-Cyclin B | Santa Cruz | Cat#sc-752, RRID:AB_2072134 |
| Rabbit monoclonal anti-Cyclin B | Cell Signaling Technology | Cat#12231, RRID:AB_2783553 |
| Mouse monoclonal anti-BrdU | BD Biosciences | Cat#555627, RRID:AB_395993 |
| Mouse monoclonal anti-BrdU | Cell Signaling Technology | Cat#5292S, RRID:AB_10548898 |
| Rabbit polyclonal anti-phospho-RPA2(Ser 8) | Cell Signaling Technology | Cat#83745, RRID:AB_2800029 |
| Rabbit monoclonal anti-RPA2 | Cell Signaling Technology | Cat#35869, RRID:AB_2799086 |
| Mouse monoclonal anti-β-tubulin | Sigma-Aldrich | Cat#T7816, RRID:AB_261770 |
| Mouse monoclonal anti-GAPDH | Cell Signaling Technology | Cat#97166, RRID:AB_2756824 |
| Rabbit monoclonal anti-cleaved caspase-3 | BD Biosciences | Cat#559565, RRID:AB_397274 |
| Mouse monoclonal anti-cleaved PARP | BD Biosciences | Cat#558710, RRID:AB_1645431 |
| Rabbit polyclonal anti-Cyclin B | Santa Cruz | Cat#sc-752, RRID:AB_2072134 |
| Mouse monoclonal anti-p21 | Millipore | Cat#05–345, RRID:AB_309684 |
| Rabbit monoclonal anti-cleaved caspase-3 conjugated to Pacific Blue | Cell Signaling Technology | Cat#8788S, RRID:AB_2797665 |
| Rabbit monoclonal anti-cleaved PARP conjugated to Alexa Fluor 647 | Cell Signaling Technology | Cat#6987S, RRID:AB_10858215 |
| Goat polyclonal anti-rabbit conjugated to Alexa Fluor 488 | Invitrogen | Cat#A-11008, RRID:AB_143165 |
| Goat polyclonal anti-rabbit conjugated to Alexa Fluor 647 | Invitrogen | Cat#A-21245, RRID:AB_141775 |
| Goat polyclonal anti-mouse conjugated to Alexa Fluor 647 | Invitrogen | Cat#A-21236, RRID:AB_2535805 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–68070, RRID: RRID:AB_10956588 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–32211, RRID:AB_621843 |
| IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–68071, RRID:AB_10956166 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences |
Cat#926–32210, RRID:AB_621842 |
| Bacterial and virus strains | ||
| Competent DH5α E. coli | This paper | NA |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Corning | 10–013-CV |
| Fetal bovine serum (FBS) | VWR | 97068–085 |
| Penicillin-streptomycin | Gibco | 15070063 |
| RPMI | Gibco | 11875093 |
| EBM-2 endothelial cell growth medium | Lonza | CC-3162 |
| Doxorubicin hydrochloride | Sigma-Aldrich | PHR1789; CAS: 25316–40-9 |
| SP600125 | Sigma-Aldrich | S5567; CAS: 129–56-6 |
| Neocarzinostatin | Sigma-Aldrich | N9162; CAS: 9014–02-2 |
| Cisplatin | Sigma-Aldrich | P4394; CAS: 15663–27-1 |
| Caffeine | Sigma-Aldrich | C0750; CAS: 58–08-2 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | P1585; CAS: 16561–29-8 |
| Anisomycin | Sigma-Aldrich | A9789; CAS: 2286276–6 |
| PD98059 | Cell Signaling Technology | 9900; CAS: 167869- 21–8 |
| SB203580 | Tocris | 1202; CAS: 152121- 47–6 |
| Mitoxantrone | Tocris | 4250; CAS: 70476- 82–3 |
| Doxycycline | Clontech | 631311; CAS: 564- 25–0 |
| DMSO | Sigma-Aldrich | D8418; CAS: 67–68-5 |
| 5-bromo-2’-deoxyuridine (BrdU) | ThermoFisher | B23151; CAS: 59–143 |
| 16% Paraformaldehyde | Electron Microscopy Science |
15710; CAS: 30525- 89–4 |
| PBS | GenClone | 25–507B |
| Triton-X 100 | Sigma-Aldrich | T9284; CAS: 9036–19-5 |
| HCS CellMask Blue | ThermoFisher | H32720 |
| Hoechst 33342 | Invitrogen | H3570 |
| Whole Cell Blue | ThermoFisher | 8403502 |
| 5-ethynyl-2’-deoxyuridine (EdU) | ThermoFisher | A10044 |
| ProLong Gold Antifade Mount | ThermoFisher | P36980 |
| Odyssey Blocking Buffer | LI-COR Biosciences |
927–50000 |
| Propidium iodide with RNase A | Cell Signaling Technology | 4087 |
| Tween 20 | AmericanBio | AB02038; CAS: 9005–64-5 |
| PhosSTOP™ tablets | Roche | 4906837001 |
| cOmplete™ ULTRA Tablets, Mini, EDTA-free, EASYpack Protease Inhibitor Cocktail | Roche | 05892791001 |
| Ponceau stain | Sigma-Aldrich | P7170 |
| Intercept blocking buffer | Licor | 927–70001 |
| NewBlot nitrocellulose stripping buffer | Licor | 928–40030 |
| Lipofectamine RNAiMAX | ThermoFisher | 13778150 |
| Opti-MEM | ThermoFisher | 11058021 |
| Lipofectamine 2000 | ThermoFisher | 11668019 |
| Puromycin | InvivoGen | ant-pr-1; CAS: 58–58-2 |
| Hydrochloric acid | Sigma-Aldrich | 258148; CAS: 7647- 01–1 |
| Citric acid | Sigma-Aldrich | C-4540; CAS: 77–929 |
| Sodium phosphate monobasic | Sigma-Aldrich | S0751; CAS: 7558–80-7 |
| Phusion® High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S |
| NheI-HF® | NEB | R3131L |
| EcoRI-HF® | NEB | R3101L |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | E2621S |
| Critical commercial assays | ||
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | 9860 |
| Click-iT Plus Alexa Fluor 555 Picolyl Azide Toolkit | ThermoFisher | C10642 |
| IL-6 Human ELISA Kit | ThermoFisher | KHC0061 |
| IL-8 Human ELISA Kit | ThermoFisher | KHC0081 |
| BCA assay | ThermoFisher | 23225 |
| CalPhos™ Mammalian Transfection Kit | Clontech | 631312 |
| Experimental models: Cell lines | ||
| Human: U-2 OS (U2OS) | HTS core at the Koch Institute | RRID:CVCL_T429 |
| Human: OVCAR-8 | Laboratory of Dr. Paula Hammond |
RRID:CVCL_1629 |
| Human: NCI-H1299 | ATCC | RRID:CVCL_0060 |
| Human: HUVEC | Lonza | RRID:CVCL_2959 |
| Human: HEK293T | ATCC | RRID:CVCL_0063 |
| Oligonucleotides | ||
| Non-targeting siRNA | Invitrogen | AM4636 |
| Jun #1 siRNA | Life Technologies |
s7658 |
| Jun #2 siRNA | Life Technologies |
s7659 |
| Recombinant DNA | ||
| pAK-Tol2-TRE-JunDN-HA-T2A-GFP (pTREJunDN-HA-T2A-GFP) | This paper | NA |
| pAK-Tol2-TRE-GFP (pTRE-GFP) | This paper | NA |
| pMIEG3-JunDN | Addgene # 40350 | Ref. 156 |
| pCMV-Tol2 | Addgene # 31823 | Ref. 157 |
| pAK-Tol2-TRE-Puro | Addgene # 130259 | NA |
| pSIRV-AP-1-mCherry | Addgene # 118095 | Ref. 158 |
| pUMVC | Addgene # 8849 | Ref. 159 |
| pCMV-VSV-G | Addgene # 8454 | Ref. 159 |
| Software and algorithms | ||
| Fiji distribution of ImageJ | Ref. 160 | https://fiji.sc/ |
| CellProfiler (version 4.0.6) | Ref. 161 | https://cellprofiler.org/ |
| MATLAB (2019b) | MathWorks | https://www.mathworks.com |
| FlowJo (version 10) | BD Biosciences | https://www.flowjo.com/ |
| Python (version 3.7) | Python Software Foundation | https://www.python.org/ |
| N-way Toolbox (version 1.8.0.0) | Ref. 56 | https://www.mathworks.com/matlabcentral/fileexchange/1088the-n-way-toolbox |
| Multi-way VIP | Ref. 58 | Obtained from corresponding authoruponreques |
| Tensor PLSR and PCA code | This paper | https://doi.org/10.5281/zenodo.7927324 |
| ilastik | Ref. 162 | https://www.ilastik.org/ |
| GraphPad Prism (version 8.4.1) | Dotmatics | https://www.graphpad.com/ |
| NIS Viewer | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/viewer |
| Image Studio Lite (version 5.2) | LI-COR Biosciences |
https://www.licor.com/bio/image-studio-lite/ |
| Other | ||
| Corning™ BioCoat™ 12mm #1 German Glass Coverslips, Round | Fisher Scientific | 08–774-383 |
| 96 Well Black/Clear Bottom Plate, TC Surface | ThermoFisher | 165305 |