

Abstract
Aims/hypothesis
Metformin is increasingly used therapeutically during pregnancy worldwide, particularly in the treatment of gestational diabetes, which affects a substantial proportion of pregnant women globally. However, the impact on placental metabolism remains unclear. In view of the association between metformin use in pregnancy and decreased birthweight, it is essential to understand how metformin modulates the bioenergetic and anabolic functions of the placenta.
Methods
A cohort of 55 placentas delivered by elective Caesarean section at term was collected from consenting participants. Trophoblasts were isolated from the placental samples and treated in vitro with clinically relevant doses of metformin (0.01 mmol/l or 0.1 mmol/l) or vehicle. Respiratory function was assayed using high-resolution respirometry to measure oxygen concentration and calculated . Glycolytic rate and glycolytic stress assays were performed using Agilent Seahorse XF assays. Fatty acid uptake and oxidation measurements were conducted using radioisotope-labelled assays. Lipidomic analysis was conducted using LC-MS. Gene expression and protein analysis were performed using RT-PCR and western blotting, respectively.
Results
Complex I-supported oxidative phosphorylation was lower in metformin-treated trophoblasts (0.01 mmol/l metformin, 61.7% of control, p<0.05; 0.1 mmol/l metformin, 43.1% of control, p<0.001). The proton efflux rate arising from glycolysis under physiological conditions was increased following metformin treatment, up to 23±5% above control conditions following treatment with 0.1 mmol/l metformin (p<0.01). There was a significant increase in triglyceride concentrations in trophoblasts treated with 0.1 mmol/l metformin (p<0.05), particularly those of esters of long-chain polyunsaturated fatty acids. Fatty acid oxidation was reduced by ~50% in trophoblasts treated with 0.1 mmol/l metformin compared with controls (p<0.001), with no difference in uptake between treatment groups.
Conclusions/interpretation
In primary trophoblasts derived from term placentas metformin treatment caused a reduction in oxidative phosphorylation through partial inactivation of complex I and potentially by other mechanisms. Metformin-treated trophoblasts accumulate lipids, particularly long- and very-long-chain polyunsaturated fatty acids. Our findings raise clinically important questions about the balance of risk of metformin use during pregnancy, particularly in situations where the benefits are not clear-cut and alternative therapies are available.
Graphical Abstract

Supplementary Information
The online version contains peer-reviewed but unedited supplementary material available at 10.1007/s00125-023-05996-3.
Keywords: Gestational diabetes mellitus, Metabolic, Metformin, Placenta, Pregnancy

Introduction
Metformin exposure during pregnancy is increasing worldwide. Hyperglycaemia, the most common reason for metformin treatment during pregnancy, affects as many as one in six pregnancies globally [1]. Data from the Metformin in the Women with Type 2 Diabetes in Pregnancy (MiTy) trial [2] show that metformin improves glycaemic control and reduces pregnancy weight gain in the context of type 2 diabetes in pregnancy. Other indications for which metformin is increasingly used during pregnancy include polycystic ovarian syndrome [3]. There is also promising evidence that metformin may be beneficial in treating severe early-onset pre-eclampsia [4], a major global cause of maternal and fetal morbidity and mortality, for which no other drug treatment is currently available [5]. In view of the high and increasing global rates of gestational diabetes and other indications for metformin therapy in pregnancy, it is likely that a considerable proportion of the next generation will be exposed to metformin whilst in utero. Previous studies have established that metformin can cross the placenta and is detectable in the fetal circulation [6, 7].
Metformin is a biguanide drug, most commonly prescribed for the treatment of type 2 diabetes [8, 9], and acts via various molecular pathways to produce tissue-specific effects (e.g. activating AMP-activated protein kinase [AMPK] [10], reducing oxidative stress [11], increasing circulating growth differentiation factor-15 (GDF15) [12] and potentially inhibiting mitochondrial electron transport chain activity [13]). However, in the highly metabolically active placenta, the net impact of metformin treatment is under-studied and remains unclear.
Metformin administered to a mother during pregnancy crosses the placenta and results in reduced offspring birthweight independent of maternal glycaemic control [14], possibly leading to catch-up growth, which confers an increased risk for childhood adiposity [3, 15]. A possible explanation for these findings is that metformin decreases placental ATP production [7], leading to reduced support for fetal growth (e.g. via reduced transfer of key nutrients). Hence, it is critically important that the implications of metformin treatment for placental metabolic function are understood, especially in view of the high and rising numbers of pregnancies exposed to metformin treatment.
We address this important knowledge gap by assessing the direct impact of metformin on third-trimester placenta. We use an established model of primary human trophoblasts cultured from term placentas and treated in vitro with metformin at clinically relevant concentrations [7] to profile a range of metabolic consequences.
Methods
Placental sampling
Placentas were donated by women undergoing full-term elective Caesarean section in Cambridge UK who met the study inclusion and exclusion criteria (Table 1). Demographic characteristics of the donors (n=55), which were in keeping with those of the maternity population as a whole, are presented in Table 2. Informed consent was obtained by a research midwife who collected the placentas immediately after birth and transferred them to the laboratory for immediate processing.
Table 1.
Inclusion and exclusion criteria
| Inclusion criteria | Exclusion criteria |
|---|---|
| Caesarean section with no prior labour | Multiple pregnancy |
| Known major fetal anomaly | |
| Severe pre-eclampsia | |
| Any form of diabetes in pregnancy | |
| <37 weeks gestation | |
| Treatment with metformin during pregnancy |
Table 2.
Demographics data
| Variable | Mean (IQR) or n (%) |
|---|---|
| Maternal age, years | 34 (32–38) |
| Maternal BMI, kg/m2 | 28.4 (24–31) |
| Maternal weight category | |
| BMI ≤25 kg/m2 | 20 (36) |
| BMI >25 kg/m2 | 35 (64) |
| Sex of baby | |
| Female | 30 (55) |
| Male | 25 (45) |
| Placental weight, g | 617.5 (540–682) |
| Birthweight, g | 3637.4 (3265–3872) |
| Birthweight centile | 72 (52–92) |
Isolation of primary trophoblasts
For all experiments, primary cytotrophoblasts were isolated from freshly collected placental tissue using a DNase digestion and centrifugation methodology, described in detail elsewhere [7]. Cells were plated in culture medium (electronic supplementary material [ESM] Table 1) at various densities (ESM Table 2) and were allowed to differentiate into syncytiotrophoblasts. Trophoblasts were cultured at 37°C under 5% CO2 and atmospheric oxygen. Cells were cultured for 96 h (4 days) before treatments. Cells were treated with metformin from 96 h (day 4) to 120 h of culture (day 5), then harvested at 120 h. Metformin concentrations were based on previous in vivo measurements during pregnancy [7]. The control group was treated with deionised water (1:1000 vol/vol). The medium was changed every 24 h.
Mitochondrial respiratory function
Trophoblasts were trypsinised (TrypLE; Thermo Fisher Scientific, Waltham, USA), centrifuged and then re-suspended in warmed mitochondrial respiration medium (miR-05 solution; ESM Table 1) at 2×106 cells/ml. Trophoblast suspensions were added to pre-calibrated chambers of an Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria) at 37°C and stirred (750 rev/min), as described previously [16]. Cells were permeabilised with 10 µg/ml digitonin before commencing a substrate-uncoupler-inhibition titration assay (detailed in ESM Table 3). Initially, malate (2 mol/l) and pyruvate (25 mol/l) were added to support LEAK state respiration, followed by ADP (10 mol/l) to stimulate pyruvate-supported oxidative phosphorylation (OXPHOS). Next, glutamate (10 mol/l) was added to saturate complex I-supported respiration, followed by cytochrome c (0.01 mol/l) to assess outer mitochondrial membrane integrity and, finally, rotenone (0.5 µmol/l) to inhibit complex I. Between additions, respiratory rates were allowed to stabilise. Respiration rates were corrected for cell number. Data were processed using DatLab (version 6.1; Oroboros Instruments).
Cell media glucose and lactate levels
Trophoblast cell culture medium was collected and centrifuged to remove debris. Glucose levels were measured using the QPulse glucose assay (Siemens Healthcare, Sudbury, UK) and lactate levels were measured using the Siemens Lactate Assay (Siemens Healthcare). Assays were conducted at the MRC MDU Mouse Biochemistry Laboratory, Cambridge, UK.
Glycolytic rate and glycolytic stress assays
Glycolytic rate, glycolytic stress and non-mitochondrial respiration in trophoblasts were assayed using the Agilent Seahorse XF-96 Extracellular Flux Analyser (Agilent Technologies, Oxford, UK) according to the manufacturer’s suggested protocols (Wave Software v2.6, Agilent Technologies). Cell culture media was replaced with freshly prepared XF media (ESM Table 1). For the glycolytic rate assay, rotenone (mitochondrial complex I inhibitor 0.5 mol/l) or antimycin A (mitochondrial complex III inhibitor 0.5 mol/l) and 2-deoxy-d-glucose (2-DG 50 mol/l) were added. For the glycolytic stress test, glucose (100 µmol/l), oligomycin (100 µmol/l) and 2-DG (500 µmol/l) were added. The mito stress assay was conducted as described previously [7]. Representative examples of each assay are shown in ESM Fig. 1.
Fatty acid uptake and oxidation assays
Cultured trophoblast cells were washed with PBS, then cultured in fresh, low-glucose DMEM containing 0.1 μCi/ml [1-14C]oleic acid (PerkinElmer, Waltham, MA, USA) with 0.0025% fatty acid-free BSA(wt/vol) for 1 h. The medium was transferred to scintillation tubes containing scintillation fluid. Cells were washed three times in ice-cold 0.1% BSA (wt/vol) solution in PBS and lysed in RIPA buffer. Cell lysates were centrifuged (10 min/16,000 g) and the supernatant fractions were added to scintillation fluid. Disintegrations per minute from [1-14C]oleic acid were measured in both the cell media and supernatant fractions using a TRI-CARB 5110TR Liquid Scintillation Counter system (PerkinElmer).
Fatty acid oxidation was measured as described previously [17]. Trophoblast cells were incubated in low-glucose DMEM containing 12.5 mol/l HEPES, 0.3% fatty acid-free BSA (wt/vol), 1 mol/l l-carnitine, 100 µmol/l oleic acid and 0.4 μCi/ml [1-14C]oleic acid. Filter paper discs saturated with 1 M NaOH were fixed to the inside of sealed wells and cells were incubated at 37°C for 3h. NaOH saturated disks were then carefully removed and added to scintillation fluid. Disintegrations per minute from 1-14C-labelled CO2 converted to sodium salts in the filter disks were measured using a TRI-CARB 5110TR Liquid Scintillation Counter system (PerkinElmer). All results were normalised to protein content and the mean of four technical replicates was reported for each sample.
Trophoblast lipid content
Trophoblast cells were fixed with 4% buffered paraformaldehyde. Fixed cells were stained with diluted Nile Red solution (Sigma; 1 µg/ml), then incubated at room temperature, protected from light for 5 min. After washing, fluorescence was measured using a plate reader (emission λ 585 nm, excitation λ 515 nm; Tecan, Reading, UK) (ESM Fig. 2).
Lipidomic analysis
Lipids were isolated from trophoblasts using an adapted liquid–liquid extraction protocol [18]. Briefly, chloroform–methanol (2:1) was added and cells were homogenised. Internal standards of lipid (1–10 µmol/l in methanol) and acylcarnitine (5 µmol/l in methanol) were added, and the mixture was homogenised. Following centrifugation (21,000 g/5 min), the organic lower layer (lipid extract) was removed and dried down using a ConcentratorPlus system (Eppendorf, Stevenage, UK) at 60°C for 180 min. Samples were reconstituted in propan-2-ol, acetonitrile and water (2:1:1 ratio) for LC-MS analysis. Full chromatographic separation of intact lipids used Waters Acquity H-Class HPLC System with the injection of 10 µl onto a Waters Acquity UPLC (CSH C18 column; 1.7 µm i.d., 2.1 mm×50 mm) maintained at 55°C. ESM Table 4 details the different phases used. The mass spectrometer (Exactive Orbitrap; Thermo Fisher Scientific) was calibrated immediately before sample analysis using a positive and a negative ionisation calibration solution (Thermo Fisher Scientific). The mass spectrometer scan rate was set at 4 Hz, giving a resolution of 25,000 (at 200 m/z) with a full-scan range of m/z 100–1800.
Gene expression analysis
RNA was extracted from trophoblasts, quantified using a Nano-drop spectrophotometer (Thermo Fisher Scientific) and checked for integrity by running an agarose gel to check for the presence of the 18S and 28S ribosomal RNA bands (ESM Fig. 3), then synthesised to cDNA as detailed previously [19]. Gene expression was determined by RT-PCR using custom-designed primers (Merck, Darmstadt, Germany; ESM Table 5), which were designed to fall within one exon and run against human gDNA standard curves to determine copy numbers. SYBR Green PCR master mix (Applied Biosystems, Waltham, MA, USA) was used as previously described [19]. Equal efficiency of the reverse transcription of RNA from all groups was confirmed through quantification of expression of the housekeeping gene B2M, the expression of which did not differ between groups (vehicle, 5,143,615±943,441 copies; 0.01 mmol/l metformin, 4,998,756±760,223 copies; 0.1 mmol/l metformin, 5,153,399±556,920 copies).
Protein expression analysis
Protein was extracted from trophoblasts as described previously [20]. Protein (20 µg) was loaded onto 10% polyacrylamide gels, electrophoresed, transferred to polyvinylidene difluoride membranes and detected using a variety of primary antibodies (ESM Table 6). Anti-rabbit IgG secondary antibodies were used (1:5000; Jackson Laboratories, Bar Harbor, ME, USA). Original blot images are shown in ESM Fig. 4. All antibodies were validated with positive and negative controls. All antibodies were diluted with 2% BSA (wt:vol) in Tris-buffered saline with Tween 20. Band densitometry was determined using chemiluminescence detection and Alpha Ease Imaging Software v4.1.0 (Alpha Innotech, Watford, UK).
Statistical analyses
Data were analysed using hierarchal mixed linear regression models, with fixed effect for treatment group and random effect for placenta of origin. Each biological sample was treated with each experimental condition. No randomisation was required due to the triad structure of the data. Researchers were blinded to experimental groups during all experiments. Demographic factors (Table 1) were tested as covariates but these were not retained in our models as they made no difference to the results. Modelling results are presented as β±SEM. Numeric data are represented as mean±SEM. In graphical representations, box plots show median and IQR, with whiskers representing 1.5×IQR. Where p values are reported, α<0.05 was pre-specified as statistically significant. Data analysis was conducted using R v4.2.2 [21].
Ethics approval
Study-specific ethics approval was granted under REC21/SC/0025 ‘The impact of metformin exposure on placenta ageing, metabolism and mitochondrial function’. Fresh placental collections were obtained via the biobanks of the Centre for Trophoblast Research (REC17/EE/0151) and from the Cambridge Blood and Stem Cell Biobank (REC18/EE/0199).
Results
Respiratory function
We initially assayed respiratory function in trophoblasts exposed to either 0.01 mmol/l or 0.1 mmol/l metformin for 24 h vs vehicle. Using high-resolution respirometry, we directly measured oxygen concentration and calculated following exposure to a bespoke substrate-inhibitor-uncoupler titration. Pyruvate-supported OXPHOS was lower in trophoblasts treated with 0.1 mmol/l metformin than in trophoblasts treated with vehicle (control) (p<0.001); the OXPHOS was also lower in trophoblasts treated with 0.01 mmol/l metformin although this did not reach statistical significance (p=0.23; Fig. 1a). OXPHOS specifically supported by complex I was assessed by the rate of following addition of glutamate to maximally stimulate complex I minus the rate of when complex I was inhibited by the addition of rotenone. Complex I-supported OXPHOS was lower in both groups of metformin-treated trophoblasts compared with vehicle-treated controls (0.01 mmol/l p<0.05; 0.1 mmol/l p<0.001, Fig. 1b). Complex I-supported OXPHOS in the experimental groups was reduced to 61.7% (0.01 mmol/l metformin) and 43.1% (0.1 mmol/l metformin) of the control value. There was no significant difference in the LEAK rate between treatment and control groups (Fig. 1c), indicating that the differences observed were not due to metformin-induced alterations in proton leak or other components of basal state respiration.
Fig. 1.
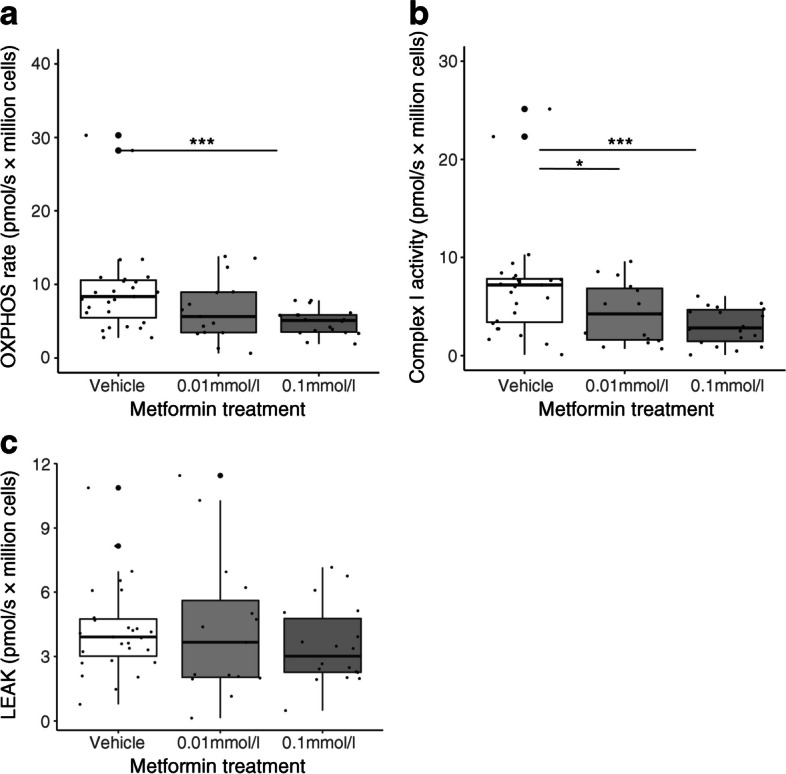
OXPHOS in human trophoblasts cultured with metformin. (a) Rate of OXPHOS (respiration rate when mitochondria are saturated with pyruvate, malate, ADP and oxygen). (b) Complex I activity (rate of respiration when complex I is maximally stimulated minus the rate of respiration when complex I is inhibited). (c) LEAK rate (basal oxygen flux that compensates for proton leak, proton slip, cation cycling and electron leak). Control group n=14; 0.01 mmol/l metformin n=12; 0.1 mmol/l metformin n=15. Technical replicates are plotted where applicable to display the total data distribution, but n refers to the number of placental samples. *p<0.05, ***p<0.001. Box plots represent median with IQR, and whiskers the max/min values excluding outliers, i.e. Q1/3±1.5×1QR
Glycolytic rate and glycolytic capacity
In view of reduced OXPHOS in trophoblasts following metformin treatment, we next explored whether there might be a compensatory increase in glycolysis to maintain cellular energy production. Glucose concentrations in the trophoblast culture medium were reduced following treatment with both 0.01 mmol/l (p<0.001) and 0.1 mmol/l metformin (p<0.001) (Fig. 2a). Lactate accumulation increased following treatment with both 0.01 mmol/l (p<0.001) and 0.1 mmol/l metformin (p<0.001) (Fig. 2b), strongly suggesting dose-dependent upregulation of glycolysis in response to metformin treatment.
Fig. 2.
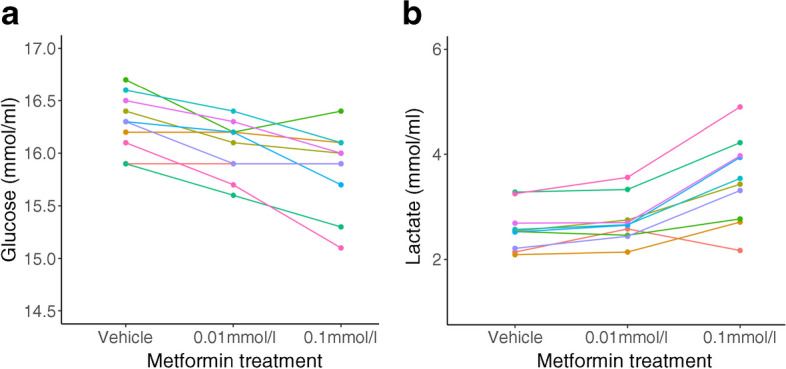
(a) Glucose concentration in trophoblast cell culture medium following 24 h treatment with vehicle, 0.01 mmol/l metformin or 0.1 mmol/l metformin. (b) Lactate concentration in cell culture medium following 24 h treatment with vehicle, 0.01 mmol/l metformin or 0.1 mmol/l metformin. Difference between groups p<0.001 for all comparisons. n=10 placental samples (each shown in a different colour) per group with a single technical replicate for each
We explored glycolysis in metformin-treated trophoblasts further by measuring glycolytic rates under both physiological and stress conditions using Seahorse XF assays. There was a dose-dependent increase in the basal rate of glycolysis following metformin treatment (Fig. 3a). The proton efflux rate arising from glycolysis under physiological conditions was increased following metformin treatment, up to 23±5% above control conditions following treatment with 0.1 mmol/l metformin (p<0.01; Fig. 3b). Glycolytic reserve was reduced in metformin-treated trophoblasts compared with controls (0.01 mmol/l, non-significant; 0.1 mmol/l, p<0.001; Fig. 3c) glycolytic capacity was not affected by metformin (Fig. 3d). This suggests that metformin treatment causes trophoblasts to operate at a higher level within their glycolytic range rather than altering their overall potential to perform glycolysis.
Fig. 3.
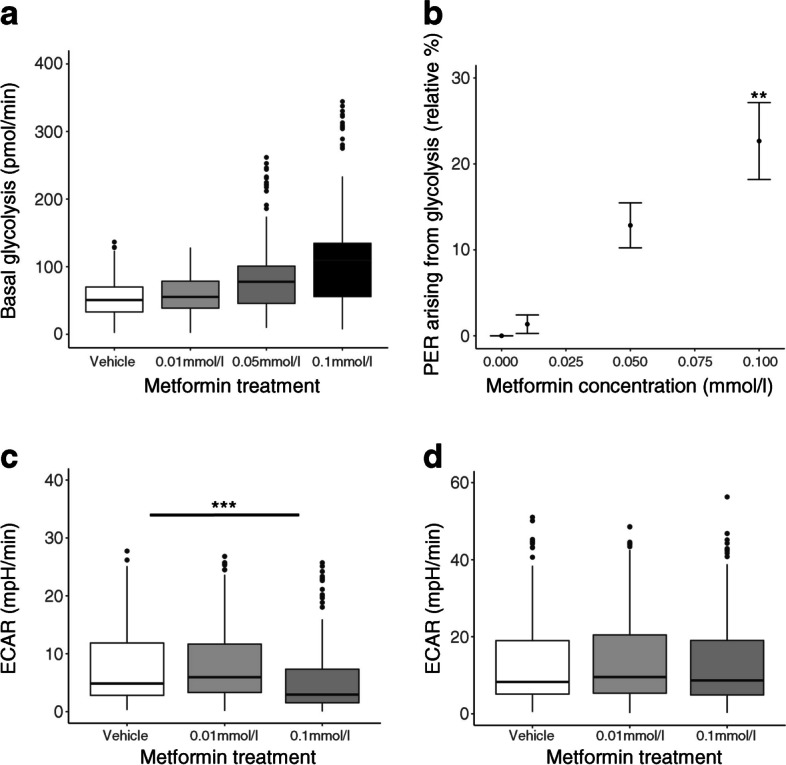
Glycolysis in human trophoblasts cultured with metformin. (a) Basal rate of glycolysis derived from rate and extracellular acidification rate under basal conditions minus acidification due to mitochondrial activity. (b) Proton efflux rate arising from glycolysis expressed as a percentage of the control values. (c) Glycolytic reserve (difference between maximal glycolytic capacity and basal glycolytic rate). (d) Glycolytic capacity expressed as the maximal extracellular acidification rate following inhibition of mitochondrial ATP production minus the non-glycolytic acidification rate. **p<0.01, ***p<0.001. n=7 placental samples for each group, each of which was run in 16 technical replicate wells. Box plots represent median with IQR, and whiskers the max/min values excluding outliers, i.e. Q1/3±1.5×1QR. ECAR, extracellular acidification rate; PER, proton efflux rate
Alongside our finding of increased rates of glycolysis, the expression of genes encoding placental glucose transporters (GLUT1, GLUT3, GLUT4, also known as SLC2A1, SLC2A3, SLC2A4, respectively) and hexokinases (HK2, HK3 and HK4, also known as GCK) were unchanged following metformin treatment (Table 3). However, HK1 expression was significantly reduced following treatment of the trophoblasts with 0.1 mmol/l metformin (p<0.001), consistent with reduced shuttling of substrate into the mitochondrial matrix [22]. Levels of phosphorylated pyruvate dehydrogenase (PDH) protein increased following treatment with 0.1 mmol/l metformin compared with vehicle (p<0.05), indicating PDH inhibition, but the increase did not reach statistical significance following treatment with 0.01 mmol/l metformin (p=0.30; ESM Table 7). There was no difference between groups in total PDH. Greater phosphorylation (inactivation) of PDH is in keeping with channelling of pyruvate away from mitochondrial oxidation towards lactate production [23].
Table 3.
Effect of metformin on gene expression of placental glucose transporters and hexokinase isoforms in human trophoblasts
| Gene | 0.01 mmol/l metformin | p value | n | 0.1 mmol/l metformin | p value | n |
|---|---|---|---|---|---|---|
| GLUT1 | 170,670±33,412 | 0.77 | 29 | 191,679±47,195 | 0.65 | 28 |
| GLUT3 | 3331±541 | 0.91 | 30 | 3499±547 | 0.65 | 30 |
| GLUT4 | 2238±729 | 0.32 | 30 | 1249±360 | 0.07 | 29 |
| HK1 | 21,382±1738 | 0.99 | 30 | 15,025±1364 | <0.001 | 30 |
| HK2 | 5052±975 | 0.48 | 31 | 4085±637 | 0.83 | 31 |
| HK3 | 13,694±2487 | 0.97 | 31 | 11,582±1887 | 0.47 | 31 |
| HK4 | 2380±998 | 0.77 | 30 | 1272±529 | 0.68 | 31 |
Copy number is displayed as n±SE. n represents number of biological replicates
p values are derived from mixed-effects linear regression analysis with treatment group included as a fixed effect and placenta of origin as a random effect; the vehicle-treated group acts as the referent
Lipid uptake and oxidation
Having established that metformin causes the placenta to undergo a substantial shift away from OXPHOS and towards glycolysis for energy production, we next investigated how metformin impacts lipid uptake and oxidation. Initially using Nile Red staining on fixed primary trophoblasts, we detected a dose-dependent increase in lipid droplets in trophoblasts treated with metformin compared with vehicle control (0.01 mmol/l, p<0.05; 0.1 mmol/l, p<0.001; ESM Fig. 2). We investigated this result further using lipidomic analysis to determine which lipid species were accumulating in response to metformin treatment. There was a significant increase in abundance of seven out of 23 classes of lipids, including triglycerides, analysed following treatment with 0.1 mmol/l metformin (all p<0.05; Fig. 4a). In all the seven classes there was also an increase following treatment with 0.01 mmol/l metformin but the increase did not reach statistical significance. In no case was there a significant reduction in a lipid class following metformin treatment. Several individual lipid species increased in response to treatment with 0.1 mmol/l metformin, in both adjusted (Fig. 4b) and unadjusted analysis (ESM Fig. 5). These included individual species from nine different lipid classes, including several triglyceride and sphingomyelin species known to be important for fetal development. Triglyceride species were most commonly affected by metformin treatment, in particular those likely to be composed of long- and very-long-chain polyunsaturated fatty acids (PUFAs) (Fig. 4c). There was a clear positive dose–response relationship between metformin and the concentrations of these triglyceride species in primary trophoblasts (Fig. 4c, p<0.01 in 22/23 species with 0.1 mmol/l metformin; ESM Fig. 6 shows results with 0.01 mmol/l metformin).
Fig. 4.

Lipid metabolism in human trophoblasts cultured with metformin. (a) Cellular lipid content by lipid class (normalised to vehicle-treated cells [control]). Values are shown as mean±SD for each group; n=10 in all groups. Light grey bars, control; dark grey bars, 0.01 mmol/l metformin; black bars, 0.1 mmol/l metformin; *p<0.05. (b) Forest plot showing relative abundance (OR±95% CI) of lipid species in 0.1 mmol/l metformin-treated trophoblasts vs control (vehicle-treated trophoblasts). Lipid species where p<0.05 following adjustment for false discovery rate are shown in red, n=10 per group. Analysis was adjusted for fetal sex, maternal BMI and gestational age at delivery. Unadjusted analysis is presented for comparison in ESM Fig. 5. (c) Metformin concentration vs triglyceride concentration for all triglyceride species likely to include long- or very-long-chain PUFAs. R2 for correlation is shown in each panel. Samples included where metformin level <2 SD from mean for either metformin concentration, p>0.01 for all species, except TG_48.5. n=8 placental samples per group with a single technical replicate for each. Cer, ceramide; CL, cardiolipin; CPE, ceramide phosphoethanolamine; DG, di-acylglyceride; GB, globotriaosylceramide; GD, disialodihexosylganglioside; GM, monosialodihexosylganglioside; Hex.Cer, hexosyl-ceramide; Lacto.cer, lactosyl-ceramide; LPC, lyso-phosphatidylcholine; LPE, lyso-phosphatidylethanolamine; Mono.lyso, mono-lyso-cardiolipin; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; S, sulfatides; SM, sphingomyelin; Sph, sphingosines; TG, tri-acylglyceride
Intracellular lipid accumulation could be the result of either increased uptake or reduced utilisation. We therefore performed dynamic assays on our primary trophoblast cultures using radiolabelled lipids to directly assess uptake and oxidation following metformin treatment. Fatty acid uptake from the cell culture medium was unaffected by metformin treatment at either concentration (Fig. 5a). Fatty acid oxidation was reduced by ~50% in trophoblasts treated with 0.1 mmol/l metformin compared with vehicle treatment (p<0.001) but the reduction did not reach statistical significance with 0.01 mmol/l metformin (p=0.16) (Fig. 5b; n=5 per group). Expression levels of genes encoding fatty acid transporters (FATP1–5 [also known as SLC27A1–5]) were unchanged following treatment with 0.01 mmol/l metformin but FATP1, FATP2 and FATP4 were all significantly upregulated following treatment with 0.1 mmol/l metformin (Table 4). Increased fatty acid transporters may be required for increased formation and expansion of intracellular lipid droplets within the metformin-treated trophoblast [24].
Fig. 5.
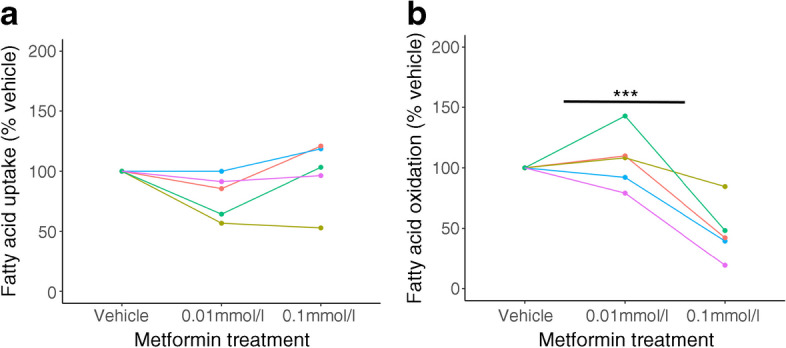
Lipid metabolism in human trophoblasts cultured with metformin. (a) Fatty acid uptake, expressed as percentage of vehicle-treated control; n=5 per group. (b) Fatty acid oxidation, expressed as percentage of vehicle-treated control. n=5 placental samples (each represented by a different colour), each of which was run in four technical replicates. ***p<0.001
Table 4.
Effect of metformin on gene expression of fatty acid transporters and endothelial lipase in human trophoblasts
| Gene | 0.01 mmol/l metformin | p value | n | 0.1 mmol/l metformin | p value | n |
|---|---|---|---|---|---|---|
| FATP1 | 3520±583 | 0.77 | 30 | 3841±572 | 0.04 | 30 |
| FATP2 | 2195±303 | 0.91 | 30 | 3611±451 | 0.02 | 30 |
| FATP3 | 2894±368 | 0.82 | 30 | 2889±364 | 0.65 | 30 |
| FATP4 | 8377±876 | 0.92 | 32 | 11033±949 | 0.03 | 31 |
| FATP5 | 821±195 | 0.77 | 32 | 679±137 | 0.91 | 32 |
| FATP6 | 29,013±3748 | 0.99 | 32 | 34,663±4871 | 0.53 | 32 |
| LIPG | 8393±727 | 0.21 | 31 | 8001±1034 | 0.83 | 31 |
Copy number is displayed as n±SE. n represents number of placentas per group, run in duplicate
p values are derived from mixed-effects linear regression analysis with treatment group included as a fixed effect and placenta of origin as a random effect, with the vehicle-treated group as the referent
Discussion
We found that in primary trophoblasts derived from term placentas metformin treatment caused a reduction in OXPHOS. Specifically, we demonstrated a reduction in complex I-supported respiration with an associated increase in glycolysis. Metformin-treated trophoblasts operate at higher basal glycolytic rates, closer to the ceiling of their glycolytic capacity. Fatty acid oxidation was also reduced in metformin-treated trophoblasts, likely as a consequence of metformin-induced partial blockade of the electron transport chain. Trophoblasts continue to take up fatty acids at the same rate in the presence of metformin, leading to intracellular fatty acid accumulation in lipid droplets following metformin treatment. Our findings are summarised in ESM Fig. 7.
Previous studies have suggested that metformin is an inhibitor of complex I in other tissues [25–27]; however, this has not previously been investigated in trophoblasts. Some studies suggest that metformin may only be a direct inhibitor of complex I at supratherapeutic concentrations, using concentrations over tenfold higher than those used in our experiments [28]. However, we have shown previously that the concentrations of metformin used in this study overlap with the concentration range measured in serum samples from mothers who were taking metformin therapy during pregnancy [7], suggesting that our finding of complex I inhibition is clinically relevant.
The shift from OXPHOS towards glycolysis for energy production in trophoblasts may have implications for placental function, particularly in meeting both the energetic demands of the third-trimester placenta itself and the energy required to support fetal growth. By the end of pregnancy, under physiological conditions the placenta produces 2.5 kg of ATP daily [29], which is necessary to fulfil its extensive biosynthetic roles in supporting both maternal adaptation to pregnancy and fetal development. We have shown previously that ATP production is reduced in metformin-treated trophoblasts [7], in keeping with a shift from OXPHOS towards glycolysis. Meta-analysis of clinical trials suggests that randomisation to metformin vs other treatments reduces birthweight independent of maternal glycaemic control in the context of gestational diabetes [15]. One potential explanation for this finding is an indirect effect of placental metformin exposure on fetal growth. It is possible that placental ATP production via glycolysis cannot keep pace with the energetic demands of supporting fetal growth thus leading to fetal growth restriction. This would be a clinically important finding, given the considerable short- and long-term risks associated with poor fetal growth in utero [30, 31]. Other potential interpretations include a direct effect of metformin on fetal growth, as it is known to cross into the fetal circulation at concentrations comparable with those in the maternal circulation [7].
The electron transport chain is a convergence point of various pathways of cellular metabolism [32]. Complex I inhibition therefore has multiple impacts, including reducing β-oxidation of fatty acids [33]. We show that metformin treatment reduces β-oxidation in trophoblasts by ~50%. As fatty acid uptake remains unaltered, there is significant intracellular accumulation of triglyceride species. We show that the specific triglycerides accumulating within metformin-treated trophoblasts are esters of long-chain PUFAs. In particular, the triglycerides identified in metformin-treated trophoblast are likely to be esters of arachidonic acid and docosahexaenoic acid (DHA), in addition to several sphingomyelin species, all of which are also essential for fetal central nervous system development [34]. Towards the end of pregnancy, fetal DHA requirements are very high (approaching 70 mg daily) as they are essential components of neural growth and development [35]. Under physiological conditions, DHA levels in the fetal circulation reflect those in the mother [36], although we show that PUFAs accumulate within metformin-treated trophoblasts. Therefore, investigating whether the fetal availability of PUFAs is reduced in metformin-exposed pregnancies is an important area of future research.
Strengths of our study design include the ability to compare the impact of clinically relevant doses of metformin in trophoblasts derived from the same donors. This circumvents questions about how unobserved variation (e.g. genetic factors [37]), maternal smoking or variation in trophoblast growth from different placentas may influence metformin response. The known demographic characteristics of our donors were tested and found not to contribute to inter-sample variation in metformin response. Furthermore, the availability of a large cohort of placentas delivered exclusively by elective Caesarean section increases confidence in our findings. We were also able to take account of the sex of the placentas as a co-variate in our models but did not find significant differences between male and female placentas in their response to metformin.
Our study design also has limitations. In particular we studied trophoblasts that were mainly differentiated in culture to syncytiotrophoblasts [38], whereas previous work suggests that cytotrophoblasts have different baseline glycolytic capacity and energy production [39]. We did not formally assess for any impact of metformin on syncytialisation or apoptosis. We modelled the effect of short-term metformin exposure using trophoblasts from full-term placentas that had already differentiated in culture. However, it is possible that there may be different impacts where trophoblasts are exposed to metformin during most or all of a pregnancy. We were also unable to directly model the fetal impacts of the placental changes we observe in our human trophoblast culture system. Animal models of metformin treatment in pregnancy may contribute to filling this important knowledge gap [40, 41], as will human studies that investigate potential longer-term impacts of metformin treatment on the offspring (e.g. growth [14] and neurodevelopmental outcomes [42]).
We show that metformin causes a substantial shift in trophoblast energy production from OXPHOS towards glycolysis, accompanied by the intracellular accumulation of triglycerides. A key feature of the trophoblast response to metformin treatment is the increased amount of glucose required to maintain glycolytic energy production. This may contribute to the efficacy of metformin as a treatment for gestational diabetes [43] via the absorption of additional glucose from the maternal circulation. Given the harms that result from untreated gestational diabetes [44], in situations where alternative treatments such as insulin are not feasible, metformin may be extremely useful to achieve maternal normoglycaemia and thus mitigate these risks [43]. Furthermore, metformin-induced suppression of complex I may result in decreased generation of reactive oxygen species in trophoblasts [7]. The resulting reduction in oxidative stress may underlie the observed efficacy of metformin in delaying delivery in the context of severe early-onset pre-eclampsia [4]. Severe early-onset pre-eclampsia remains a life-threatening condition, in which even small increases in gestation at birth can result in large gains in infant survival rate [45]. Therefore, although our results raise important questions about the long-term impacts of metformin exposure during key developmental phases, these are set against the benefits of metformin treatment in specific high-risk circumstances. However, our findings that metformin reduces energy production and causes trophoblasts to accumulate fatty acids bring into question whether it is an ideal drug in low-risk pregnancy or where alternative treatments are available. Further investigation of the complex impacts of metformin on the bioenergetic and anabolic functions of the placenta will allow more specific targeting of metformin treatment in pregnancy to individuals for whom the benefits outweigh the potential risks.
Supplementary Information
Below is the link to the electronic supplementary material.
Abbreviations
- 2-DG
2-Deoxy-d-glucose
- DHA
Docosahexaenoic acid
- OXPHOS
Oxidative phosphorylation
- PDH
Pyruvate dehydrogenase
- PUFAs
Polyunsaturated fatty acids
Data availability
Data are available upon request to the corresponding author.
Funding
This research was funded by a Medical Research Council New Investigator Grant (MR/T016701/1) to CEA. CEA, BJ and AK are supported by the NIHR Cambridge Biomedical Research Centre (146281). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. SEO is supported by the Medical Research Council (MC_UU_00014/4) and the British Heart Foundation (RG/17/12/33167 and PG/20/11/34957). AJM is supported by the Evelyn Trust (16/33), Gates Foundation, British Heart Foundation (FS/4yPhD/F/20/34124C and FS/19/54/34889C) and Wellcome Trust (220033/Z/19/Z).
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
CEA, SEO, AJM, IGR and AK were responsible for conception and design of the experiments. JLT-A, IGR, LCP, JLA, BDT, LMWH, AEK, SV and BJ performed experiments. CEA, SEO, AJM, IGR, AK and BJ analysed and interpreted the data. CEA and JLT-A drafted the manuscript. All authors critically evaluated and discussed the manuscript. All authors approved the final version. CEA designed experiments, contributed to discussion, analysed and interpreted the data, and wrote and critically evaluated the manuscript. CEA is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis and controlled the decision to publish.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.International Diabetes Federation (2021) IDF Diabetes Atlas. IDF, Belgium: 2021. Available at: https://www.diabetesatlas.org. Last accessed 16/02/2023
- 2.Feig DS, Donovan LE, Zinman B, et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): a multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020;8(10):834–844. doi: 10.1016/S2213-8587(20)30310-7. [DOI] [PubMed] [Google Scholar]
- 3.Fornes R, Simin J, Nguyen MH, et al. Pregnancy, perinatal and childhood outcomes in women with and without polycystic ovary syndrome and metformin during pregnancy: a nationwide population-based study. Reprod Biol Endocrinol. 2022;20(1):30. doi: 10.1186/s12958-022-00905-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cluver CA, Hiscock R, Decloedt EH, et al. Use of metformin to prolong gestation in preterm pre-eclampsia: randomised, double blind, placebo controlled trial. BMJ. 2021;374:n2103. doi: 10.1136/bmj.n2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poon LC, Shennan A, Hyett JA, et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: a pragmatic guide for first-trimester screening and prevention. Int J Gynaecol Obstet. 2019;145(Suppl 1):1–33. doi: 10.1002/ijgo.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charles B, Norris R, Xiao X, Hague W. Population pharmacokinetics of metformin in late pregnancy. Ther Drug Monit. 2006;28(1):67–72. doi: 10.1097/01.ftd.0000184161.52573.0e. [DOI] [PubMed] [Google Scholar]
- 7.Tarry-Adkins JL, Robinson IG, Reynolds RM, et al. Impact of metformin treatment on human placental energy production and oxidative stress. Front Cell Dev Biol. 2022;10:935403. doi: 10.3389/fcell.2022.935403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma M, Nazareth I, Petersen I. Trends in incidence, prevalence and prescribing in type 2 diabetes mellitus between 2000 and 2013 in primary care: a retrospective cohort study. BMJ Open. 2016;6(1):e010210. doi: 10.1136/bmjopen-2015-010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkinson S, Douglas I, Stirnadel-Farrant H, et al. Changing use of antidiabetic drugs in the UK: trends in prescribing 2000–2017. BMJ Open. 2018;8(7):e022768. doi: 10.1136/bmjopen-2018-022768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/jci13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esteghamati A, Eskandari D, Mirmiranpour H, et al. Effects of metformin on markers of oxidative stress and antioxidant reserve in patients with newly diagnosed type 2 diabetes: a randomized clinical trial. Clin Nutr. 2013;32(2):179–185. doi: 10.1016/j.clnu.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Coll AP, Chen M, Taskar P, et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature. 2020;578(7795):444–448. doi: 10.1038/s41586-019-1911-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goel S, Singh R, Singh V, et al. Metformin: activation of 5' AMP-activated protein kinase and its emerging potential beyond anti-hyperglycemic action. Front Genet. 2022;13:1022739. doi: 10.3389/fgene.2022.1022739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarry-Adkins JL, Aiken CE, Ozanne SE. Comparative impact of pharmacological treatments for gestational diabetes on neonatal anthropometry independent of maternal glycaemic control: A systematic review and meta-analysis. PLoS Med. 2020;17(5):e1003126. doi: 10.1371/journal.pmed.1003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarry-Adkins JL, Aiken CE, Ozanne SE. Neonatal, infant, and childhood growth following metformin versus insulin treatment for gestational diabetes: a systematic review and meta-analysis. PLoS Med. 2019;16(8):e1002848. doi: 10.1371/journal.pmed.1002848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitehead A, Krause FN, Moran A, et al. Brown and beige adipose tissue regulate systemic metabolism through a metabolite interorgan signaling axis. Nat Commun. 2021;12(1):1905. doi: 10.1038/s41467-021-22272-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantaleão LC, Inzani I, Furse S, et al. Maternal diet-induced obesity during pregnancy alters lipid supply to mouse E18.5 fetuses and changes the cardiac tissue lipidome in a sex-dependent manner. Elife. 2022;11:e69078. doi: 10.7554/eLife.69078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins B, Ronis M, Koulman A. LC-MS lipidomics: exploiting a simple high-throughput method for the comprehensive extraction of lipids in a ruminant fat dose-response study. Metabolites. 2020;10(7):296. doi: 10.3390/metabo10070296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarry-Adkins JL, Chen JH, Smith NS, Jones RH, Cherif H, Ozanne SE. Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB J. 2009;23(5):1521–1528. doi: 10.1096/fj.08-122796. [DOI] [PubMed] [Google Scholar]
- 20.Tarry-Adkins JL, Aiken CE, Ashmore TJ, Ozanne SE. Insulin-signalling dysregulation and inflammation is programmed trans-generationally in a female rat model of poor maternal nutrition. Sci Rep. 2018;8(1):4014. doi: 10.1038/s41598-018-22383-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
- 22.Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu N, Zhang SO, Cole RA, et al. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol. 2012;198(5):895–911. doi: 10.1083/jcb.201201139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol (Lausanne) 2019;10:294. doi: 10.3389/fendo.2019.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. doi: 10.1042/bj3480607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang M, Darwish T, Larraufie P, et al. Inhibition of mitochondrial function by metformin increases glucose uptake, glycolysis and GDF-15 release from intestinal cells. Sci Rep. 2021;11(1):2529. doi: 10.1038/s41598-021-81349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavlovic K, Krako Jakovljevic N, Isakovic AM, Ivanovic T, Markovic I, Lalic NM. Therapeutic vs. suprapharmacological metformin concentrations: different effects on energy metabolism and mitochondrial function in skeletal muscle cells. Front Pharmacol. 2022;13:930308. doi: 10.3389/fphar.2022.930308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aye ILMH, Aiken CE, Charnock-Jones DS, Smith GCS. Placental energy metabolism in health and disease-significance of development and implications for preeclampsia. Am J Obstet Gynecol. 2022;226(2S):S928–S944. doi: 10.1016/j.ajog.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Longo S, Bollani L, Decembrino L, Di Comite A, Angelini M, Stronati M. Short-term and long-term sequelae in intrauterine growth retardation (IUGR) J Matern Fetal Neonatal Med. 2013;26(3):222–225. doi: 10.3109/14767058.2012.715006. [DOI] [PubMed] [Google Scholar]
- 31.Armengaud JB, Yzydorczyk C, Siddeek B, Peyter AC, Simeoni U. Intrauterine growth restriction: clinical consequences on health and disease at adulthood. Reprod Toxicol. 2021;99:168–176. doi: 10.1016/j.reprotox.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7(3):254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 33.Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11(1):102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birch EE, Garfield S, Castañeda Y, Hughbanks-Wheaton D, Uauy R, Hoffman D. Visual acuity and cognitive outcomes at 4 years of age in a double-blind, randomized trial of long-chain polyunsaturated fatty acid-supplemented infant formula. Early Hum Dev. 2007;83(5):279–284. doi: 10.1016/j.earlhumdev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Clandinin MT, Chappell JE, Heim T, Swyer PR, Chance GW. Fatty acid utilization in perinatal de novo synthesis of tissues. Early Hum Dev. 1981;5(4):355–366. doi: 10.1016/0378-3782(81)90016-5. [DOI] [PubMed] [Google Scholar]
- 36.Innis SM, Friesen RW. Essential n-3 fatty acids in pregnant women and early visual acuity maturation in term infants. Am J Clin Nutr. 2008;87(3):548–557. doi: 10.1093/ajcn/87.3.548. [DOI] [PubMed] [Google Scholar]
- 37.Chen S, Zhou J, Xi M, et al. Pharmacogenetic variation and metformin response. Curr Drug Metab. 2013;14(10):1070–1082. doi: 10.2174/1389200214666131211153933. [DOI] [PubMed] [Google Scholar]
- 38.Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF., 3rd Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118(4):1567–1582. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- 39.Bucher M, Kadam L, Ahuna K, Myatt L. Differences in glycolysis and mitochondrial respiration between cytotrophoblast and syncytiotrophoblast in-vitro: evidence for sexual dimorphism. Int J Mol Sci. 2021;22(19):10875. doi: 10.3390/ijms221910875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoonejans JM, Blackmore HL, Ashmore TJ, Aiken CE, Fernandez-Twinn DS, Ozanne SE. Maternal metformin intervention during obese glucose-intolerant pregnancy affects adiposity in young adult mouse offspring in a sex-specific manner. Int J Mol Sci. 2021;22(15):8105. doi: 10.3390/ijms22158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hufnagel A, Fernandez-Twinn DS, Blackmore HL, et al. Maternal but not fetoplacental health can be improved by metformin in a murine diet-induced model of maternal obesity and glucose intolerance. J Physiol. 2022;600(4):903–919. doi: 10.1113/JP281902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feig DS, Sanchez JJ, Murphy KE, et al. Outcomes in children of women with type 2 diabetes exposed to metformin versus placebo during pregnancy (MiTy Kids): a 24-month follow-up of the MiTy randomised controlled trial. Lancet Diabetes Endocrinol. 2023;11(3):191–202. doi: 10.1016/s2213-8587(23)00004-9. [DOI] [PubMed] [Google Scholar]
- 43.Tarry-Adkins JL, Ozanne SE, Aiken CE. Impact of metformin treatment during pregnancy on maternal outcomes: a systematic review/meta-analysis. Sci Rep. 2021;11(1):9240. doi: 10.1038/s41598-021-88650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nijs H, Benhalima K. Gestational diabetes mellitus and the long-term risk for glucose intolerance and overweight in the offspring: a narrative review. J Clin Med. 2020;9(2):599. doi: 10.3390/jcm9020599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, and management. Vasc Health Risk Manag. 2011;7:467–474. doi: 10.2147/vhrm.S20181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request to the corresponding author.