

Abstract
Fibroblast growth factor receptors (FGFR) are emerging as an important therapeutic target for patients with advanced, refractory cancers. Most selective FGFR inhibitors under investigation show reversible binding, and their activity is limited by acquired drug resistance. This review summarizes the preclinical and clinical development of futibatinib, an irreversible FGFR1-4 inhibitor. Futibatinib stands out among FGFR inhibitors because of its covalent binding mechanism and low susceptibility to acquired resistance. Preclinical data indicated robust activity of futibatinib against acquired resistance mutations in the FGFR kinase domain. In early-phase studies, futibatinib showed activity in cholangiocarcinoma, and gastric, urothelial, breast, central nervous system, and head and neck cancers harboring various FGFR aberrations. Exploratory analyses indicated clinical benefit with futibatinib after prior FGFR inhibitor use. In a pivotal phase II trial, futibatinib demonstrated durable objective responses (42% objective response rate) and tolerability in previously treated patients with advanced intrahepatic cholangiocarcinoma harboring FGFR2 fusions or rearrangements. A manageable safety profile was observed across studies, and patient quality of life was maintained with futibatinib treatment in patients with cholangiocarcinoma. Hyperphosphatemia, the most common adverse event with futibatinib, was well managed and did not lead to treatment discontinuation. These data show clinically meaningful benefit with futibatinib in FGFR2-rearrangement-positive cholangiocarcinoma and provide support for further investigation of futibatinib across other indications. Future directions for this agent include elucidating mechanisms of resistance and exploration of combination therapy approaches.
Keywords: fibroblast growth factor receptor, FGFR inhibitor, futibatinib, cholangiocarcinoma, safety, clinical trials
Fibroblast growth factor receptors (FGFR) are emerging as an important therapeutic target for patients with advanced, refractory cancers. This review summarizes the preclinical and clinical development of futibatinib, an irreversible FGFR1-4 inhibitor.
Implications for Practice.
Patients with FGFR-altered cancers have limited treatment options in advanced stages. Promising responses have been observed with FGFR inhibitors; however, acquired resistance is an emerging concern. This review summarizes data surrounding futibatinib, the only second-generation FGFR1-4 inhibitors in phase II/III clinical development. Futibatinib has shown durable efficacy and tolerability in cholangiocarcinoma with FGFR2 fusions/rearrangements, and recently received approval from the US Food and Drug Administration for this indication. Futibatinib has demonstrated antitumor activity across several FGFR-aberrant tumors, spurring the initiation of several phase II trials of futibatinib or futibatinib-containing combinations in other tumor types.
Introduction
Fibroblast growth factors (FGFs) and their receptors (FGFRs) play an integral role in regulating a wide range of biological processes1 and dysregulation of the FGFR pathway is associated with oncogenesis.2-7 Approximately 7% of all cancers harbor FGFR aberrations, with type and prevalence varying widely.8 Thus, FGFR has emerged as an important therapeutic target. Most FGFR inhibitors in development are ATP-competitive, reversible inhibitors, which are associated with acquired resistance.9,10 Futibatinib, an irreversible FGFR1-4 inhibitor, is the most advanced covalent inhibitor in clinical development for multiple cancer types.11 Here, we briefly describe the role of FGFR and FGFR inhibitors in cancer and discuss recent data supporting futibatinib as a clinically meaningful, second-generation FGFR inhibitor.
FGFR as an Oncologic Target
The FGFR pathway includes a family of 22 FGF ligands, which primarily convey cellular signals through 4 transmembrane tyrosine kinase receptors (FGFR1-4).12,13 Typically, FGFR activation induces cell proliferation and migration,14 but it can also drive cell differentiation or negatively regulate proliferation.15,16 Aberrant FGFR signaling (generally constitutive FGFR activation) can promote tumorigenesis, support tumor survival, and confer resistance to chemotherapy through anti-apoptotic signaling,2-7,17,18 rendering FGFR-altered tumors difficult to treat.19
Analysis of 17 different cancer types showed that 7% had FGFR aberrations, found most commonly in urothelial, breast, endometrial, and squamous cell lung cancer (Fig. 1). Gene amplifications, mutations, and rearrangements accounted for 66%, 26%, and 8% of FGFR aberrations identified, respectively.8 These findings suggest FGFR inhibition as a potential therapeutic strategy in multiple tumor types.
Figure 1.
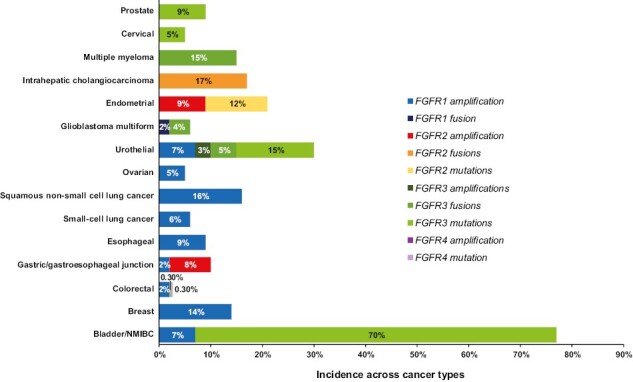
Incidence of FGFR aberrations across cancer types. This bar chart shows the incidence of the indicated FGFR aberrations across various cancer types, data collated from a number of published reports.2,8,13,20-38 In cases where a range of incidences were noted, the upper limit of the range is indicated in this chart. Abbreviation: NMIBC, non-muscle invasive bladder cancer.
Selective, Small-Molecule FGFR Inhibitors
FGFR inhibitors mostly target the FGFR kinase domain, inhibiting FGFR signaling. Although several therapeutic modalities are being investigated for FGFR inhibition (reviewed elsewhere39), small-molecule FGFR inhibitors remain the most widely investigated. These inhibitors vary in their selectivity (specific to FGFR or multikinase) and mode of binding to the FGFR kinase domain (type I, type II, reversible, or irreversible).40 Reversible ATP-competitive FGFR inhibitors currently under investigation, including derazantinib, erdafitinib, pemigatinib, and infigratinib (Table 1; Supplementary Table S1), engage primarily in noncovalent interactions with amino acids in the hinge and surrounding regions of the ATP-binding pocket in the FGFR kinase domain. Irreversible inhibitors, such as PRN1371, futibatinib, and fisogatinib, form a covalent bond, generally with a conserved cysteine in the FGFR kinase domain.40,54
Table 1.
Overview of published studies reporting clinical activity of FGFR inhibitors.
| FGFR inhibitor | Type/isoform selectivity | Tumor/FGFR aberration | Trial phase/ number |
n | Results | Ref | |||
|---|---|---|---|---|---|---|---|---|---|
| ORR (%) | mDOR (mo) | mPFS (mo) |
mOS (mo) | ||||||
| AZD4547 | Reversible FGFR 1-3 inhibitor | Squamous cell non-small cell lung cancer/FGFR1-3 amplification, fusion, or substitution | Phase II NCT02965378 |
27a | 7 | 1.5, 2.9b | 2.7 | 7.5 | 41 |
| Refractory cancers, lymphomas, or myelomas/ Any FGFR1-3 aberration |
Phase II NCT02465060 |
48 | 8 | 7.9 | 3.4 | 7.2 | 42 | ||
| Derazantinib | Reversible FGFR1-3 inhibitor | iCCA/ FGFR2 fusions |
Phase I/II NCT01752920 |
29 | 20.7 | 4.6 | 5.7 | NR | 43 |
| Debio1347 | Reversible FGFR1-3 inhibitor | Advanced solid tumors/ FGFR1-3 aberrations | Phase I NCT01948297 |
58c | 10.5 | NR | NA | NA | 44 |
| Erdafitinib | Reversible FGFR1-4 inhibitor | CCA/ FGFR2/3 alterations |
Phase II NCT02699606 |
12d | 50.0 | 6.8 | 5.6 | NR | 45 |
| Urothelial/FGFR3 mutation or FGFR2/3 fusions | Phase II NCT02365597 |
99 | 34 | 5.6 | 5.5 | 13.8 | 46 | ||
| Fisogatinib | Irreversible FGFR4 | Hepatocellular/ FGF19 positive |
Phase I | 66e | 17 | 5.3 | 3.3 | NA | 47 |
| Futibatinib | Irreversible FGFR1-4 inhibitor | iCCA/ FGFR2 fusions/rearr. |
Phase II NCT02052778 |
103 | 41.7 | 9.7 | 9.0 | 21.7 | 48 |
| Infigratinib | Reversible FGFR1-3 inhibitor | CCA/ FGFR2 fusions/rearr. |
Phase II NCT02150967 |
108 | 23.1 | 5.0 | 7.3 | 12.2 | 49 |
| Urothelial/FGFR3 alterations | Phase I expansion NCT02657486 |
67 | 25.4 | 5.1 | 3.8 | 7.8 | 50 | ||
| Pemigatinib | Reversible FGFR1-3 inhibitor | CCA/FGFR2 fusions/rearr. | Phase II NCT02924376 |
107f | 35.5 | 7.5g | 6.9 | 21.1 | 51 |
| Urothelial/FGF or FGFR alterations | Phase II NCT02872714 |
100 | 25 | NR | NR | NR | 52 | ||
| Rogaratinib | Reversible FGFR1-4 inhibitor | Adv cancers/FGFR mRNA-overexpressing | Phase I NCT01976741 |
126h | 15 | NR | 93 d | NR | 53 |
aAZD4547-treated evaluable patients.
bPartial responses were observed in 2 patients, response durations are listed for each patient.
c57 patients were evaluable for response.
d12 evaluable patients (Asian population).
eResponse evaluable patients who were FGF19-positive.
fSubgroup of population with FGFR2 fusions or rearrangements.
gUpdated median DOR of 9.1 months is reported in the US Prescribing Information.
hTotal enrollment of 126 pts. A total of 121 pts had available progression-free survival. One hundred pts were evaluable for response assessment.
Abbreviations: Adv, advanced; CCA, cholangiocarcinoma; DOR, duration of response; FGFR, fibroblast growth factor receptors; iCCA, intrahepatic CCA; mo, months; mDOR, median DOR; mOS, median overall survival; mPFS, median progression-free survival; NA, not applicable; NR, not reported; ORR, objective response rate; pts, patients; ref, references.
Selective FGFR inhibitors have shown promising activity in various FGFR-aberrant cancer types. To date, the US Food and Drug Administration (FDA) has approved erdafitinib in patients with metastatic urothelial carcinoma harboring FGFR2/3 aberrations who were previously treated,55 and pemigatinib and infigratinib for second- or later-line treatment of unresectable cholangiocarcinoma (CCA) with FGFR2 fusions or rearrangements.49,55 An emerging concern with these inhibitors is acquired resistance, which leads to disease progression.9,56,57 One mechanism of acquired resistance is the development of secondary “gatekeeper” mutations in the FGFR kinase domain that “block” FGFR inhibitor binding through steric hindrance.9,39,56,58 Reversible inhibitors, such as erdafitinib, infigratinib, and pemigatinib, are largely ineffective against these mutations.56 Second-generation inhibitors that retain activity against these mutations and have a lower susceptibility to resistance are sorely needed.
Futibatinib, a Potent, Irreversible FGFR1-4 Inhibitor
Futibatinib is a structurally novel, highly selective, and potent FGFR inhibitor,11 which binds covalently and irreversibly to a conserved cysteine residue in the FGFR kinase domain within the ATP-binding pocket54 (Fig. 2). As this cysteine residue is conserved across all FGFR receptors, futibatinib inhibits the kinase activity of all 4 FGFR isoforms. The distinct binding site and irreversible binding render futibatinib less susceptible to drug resistance mutations than reversible, ATP-competitive inhibitors.
Figure 2.
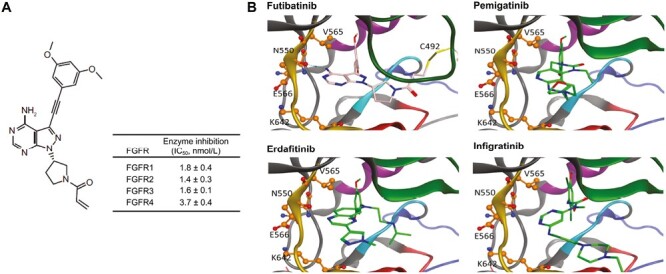
Futibatinib structure and predicted binding of futibatinib and other reversible FGFR inhibitors to the FGFR kinase domain. (A) Chemical structure of futibatinib and in vitro inhibitory activity. Adapted from Cancer Research, 2020, 80(22), 4986-4997, Sootome H, Fujita H, Ito K, et al., Futibatinib Is a Novel Irreversible FGFR 1-4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors, with permission from AACR.11(B) Predicted interactions of futibatinib, erdafitinib, and pemigatinib with the ATP binding pocket of the FGFR2 wild-type kinase domain. Amino acid residues altered in identified resistance mutations are labeled and shown as ball and stick models. Kinase domain regions are depicted as follows: gold, hinge region; red, catalytic loop; blue, activation domain; purple, c-alpha-helix; green, P-loop; cyan, DFG motif. Futibatinib (pink and blue stick figure) binds covalently to C492 in the P-loop (yellow stick), enabling it to persist in the ATP-binding pocket irrespective of the presence of resistance mutations, which block access of reversible FGFR inhibitors such as erdafitinib, pemigatinib, or infigratinib (green and blue stick figures). Reproduced with permission from Goyal et al. 2023.48 Abbreviations: DFG, Asp-Phe-Gly; FGFR, fibroblast growth factor receptors; IC50, half-maximal inhibitory concentration. From The New England Journal of Medicine, Goyal L, Meric-Bernstam F, Hollebecque A, et al., Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma, 388, 228-239. Copyright © (2023) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Preclinical Development
In vitro characterization of futibatinib against a panel of 296 kinases demonstrated high selectivity and potent inhibition of all 4 FGFR isoforms with half-maximal inhibitory concentration values ranging from 1.4 nmol/L to 3.7 nmol/L.11 Futibatinib selectively inhibited cancer cell lines of diverse tissue origins (gastric, lung, multiple myeloma, bladder, endometrial, and breast) harboring a variety of FGFR aberrations. Additionally, futibatinib treatment led to significant dose-dependent tumor reductions and sustained FGFR kinase inhibition in FGFR-aberrant human tumor xenograft mice models.
In vitro futibatinib treatment of gastric cancer cells was associated with a lower risk of developing drug resistance due to FGFR escape mutations than the reversible FGFR inhibitor AZD4547.11 Futibatinib also demonstrated greater inhibition of secondary FGFR2 kinase domain drug-resistant mutations, including the gatekeeper mutation V565I/L, than AZD4547, infigratinib, pemigatinib, or erdafitinib.11
In an unbiased library-based analysis, the activity of futibatinib and other FGFR inhibitors were examined against drug-resistant FGFR2 kinase domain mutations generated by random mutagenesis59 and transfected in a Ba/F3 cell system dependent on FGFR2 signaling for growth. Futibatinib showed the most robust inhibition of drug-resistant FGFR2 kinase domain mutations (also clinically relevant9,56,57,60) as well as the lowest propensity for emergence of resistant clones with prolonged treatment.
Futibatinib Early Clinical Data: Dose Selection and Pharmacology
A first-in-human, phase I dose-escalation study (NCT02052778) evaluated futibatinib safety and pharmacokinetics/pharmacodynamics in 86 patients with advanced solid tumors (83% with FGF/FGFR aberrations) who were heavily pretreated.61 Futibatinib was administered on daily (QD) continuous dosing (4-24 mg QD; n = 44) and 3 times a week (TIW) intermittent dosing (8-200 mg TIW; n = 42) schedules. Dose-limiting toxicities (DLTs), all related to liver enzyme elevations, occurred in 3 patients receiving futibatinib 24 mg QD. No DLTs were observed with TIW dosing. All QD doses tested showed dose-proportional pharmacokinetics, whereas TIW dosing was associated with saturation between 80 mg and 200 mg TIW. As renal handling of phosphorus is mediated by FGF23 signaling,62 serum phosphorus levels were evaluated as an on-target effect and chosen as a pharmacodynamic marker. While serum phosphorus levels correlated positively with futibatinib dose and exposure for both QD and TIW dosing, this correlation was stronger with QD vs. TIW dosing. Similar data were observed in a phase I dose-escalation study in patients with advanced solid tumors from Japan (JapicCTI-142552).63 Based on these data, futibatinib 20 mg QD was selected as the recommended phase II dose.
Futibatinib showed a manageable safety profile.61 The most common treatment-emergent adverse events were hyperphosphatemia, diarrhea, and constipation. In addition, encouraging preliminary antitumor activity was observed in this heavily pretreated population, particularly in those with intrahepatic CCA (iCCA). Across cohorts, 5 patients (6%) experienced partial responses (PRs) and 48% (n = 41) achieved stable disease (SD). Most patients with PRs or SD had tumors harboring FGF/FGFR aberrations; those with PRs included 3 patients with iCCA, all harboring FGFR2 fusions, and 2 patients with FGFR1-mutant brain tumors. Among patients with CCA, 75% (18/24) experienced a PR or SD.
Futibatinib pharmacokinetics were evaluated in healthy adult volunteers in multiple open-label, phase I studies. An absorption, distribution, metabolism, and excretion study with [14C]-futibatinib identified futibatinib as the most abundant component circulating in plasma; other metabolites accounted for 9%-13% of circulating components.64 [14C]-futibatinib was mainly excreted through the fecal route after metabolism, and no unmetabolized futibatinib was detected in the feces or urine.
In a food-effect and drug-drug interaction (DDI) study with a proton pump inhibitor (PPI; lansoprazole), consuming a high-fat, high-calorie meal slightly lowered futibatinib oral bioavailability, and delayed time to futibatinib maximum plasma concentration, but the differences were not clinically meaningful. Coadministration of lansoprazole had no clinically relevant effect on futibatinib pharmacokinetics, indicating that futibatinib can be coadministered with PPIs.64,65
Other phase I studies in healthy adult volunteers evaluated the involvement of futibatinib in the common drug metabolizing CYP3A pathway.66 DDIs were assessed between futibatinib and midazolam (a sensitive CYP3A substrate), itraconazole (a strong dual inhibitor of CYP3A and P-gp), and rifampin (a strong dual inducer of CYP3A and P-gp).65 Multiple doses of futibatinib did not affect the pharmacokinetics of midazolam; therefore, futibatinib is not expected to affect the exposure of concomitant medications metabolized via CYP3A. However, itraconazole coadministration resulted in higher peak plasma concentrations and significant increases in plasma exposure of futibatinib compared with futibatinib alone, and coadministration of rifampin decreased futibatinib exposure. Thus, coadministering futibatinib with strong dual inhibitors or inducers of CYP3A and P-gp should be avoided because of potential significant DDIs.
Activity of Futibatinib in CCA
Based on results from the phase I dose escalation study,61 the phase I dose expansion study evaluated futibatinib in a larger population of patients with advanced solid tumors harboring FGF/FGFR aberrations, including a sizeable CCA population.67 Among 64 patients with FGFR-altered CCA who received futibatinib 20 mg QD, the objective response rate (ORR) was 15.6%, and in the subgroup of patients with iCCA harboring FGFR2 fusions/rearrangements (n = 42), the ORR was 16.7%. Median duration of response (DOR) was 5.3 months and 6.9 months, respectively, and disease control rate (DCR) was 72% and 79%, respectively. In patients with FGFR2 fusion/rearrangement–positive iCCA treated with either futibatinib 20 mg or 16 mg QD, the overall ORR was 25.4% (15/59). These data formed the basis for further study of futibatinib in patients with FGFR2-rearrangement–positive iCCA.
The pivotal phase II FOENIX-CCA2 study investigated futibatinib in 103 patients with advanced unresectable iCCA harboring FGFR2 fusions or rearrangements after one or more lines of systemic chemotherapy.48 FOENIX-CCA2 surpassed its primary endpoint target with an ORR of 41.7% (43/103; 95% CI, 32.1-51.9), as assessed by independent central review (Fig. 3A). Responses were rapid and durable: median time to response was 2.5 months (range, 0.7-7.4), median DOR was 9.7 months (95% CI, 7.6-17.0), and 72% (31/43) of responders had responses lasting at least 6 months (Fig. 3B). Objective responses were consistent across subgroups, including patients with poor prognostic factors, such as patients 65 years and older or who were heavily pre-treated (≥3 prior therapies). Preliminary survival data were promising; after a median follow-up of 17.1 months, median progression-free survival (PFS) was 9.0 months (95% CI, 6.9-13.1) and median overall survival (OS) was 21.7 months (Fig. 3C, 3D). The 1-year OS rate was 72%. Results were similar at extended follow-up (median 25.0 months) with a confirmed ORR of 41.7%, mature median OS of 20.0 months (12-month OS rate, 73%), and median PFS of 8.9 months.68 Based on these data, futibatinib was granted accelerated approval by the FDA for patients with previously treated, unresectable, locally advanced, or metastatic FGFR2-fusion/rearrangement-positive iCCA.69
Figure 3.
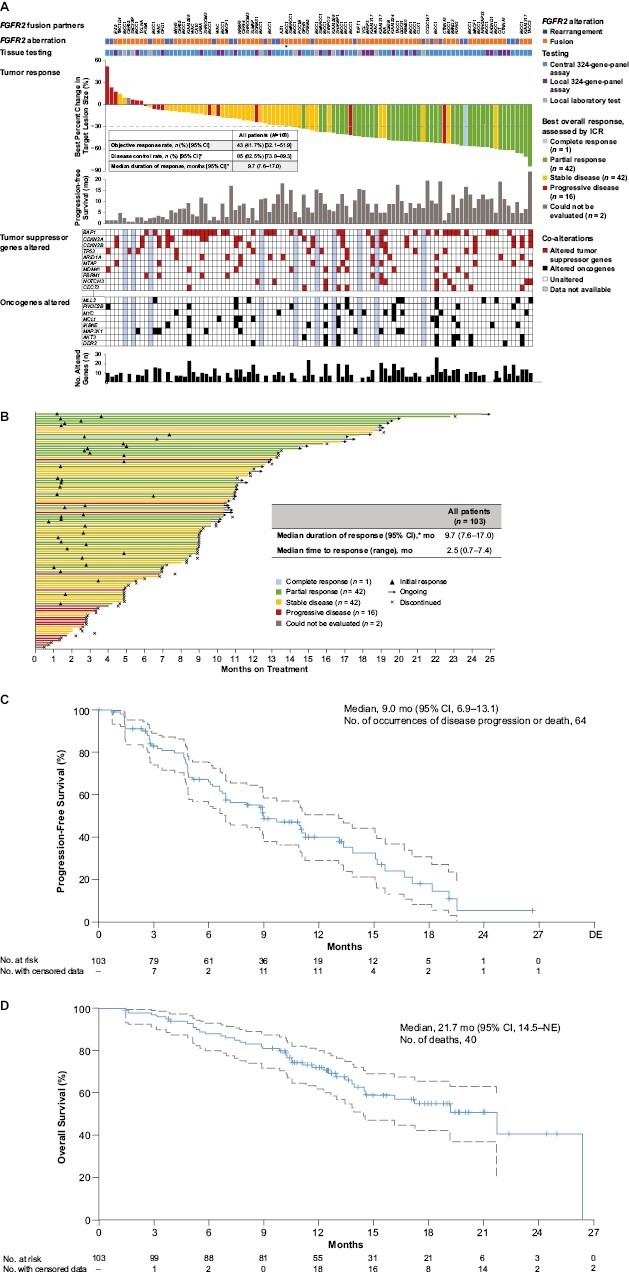
Futibatinib efficacy in the FOENIX-CCA2 study. (A) Best percentage change from baseline in target lesion size in individual patients with bars color coded to indicate best overall response assessed per ICR. Horizontal line represents the threshold for PR (≥30% reduction in lesion size) per RECIST v1. FGFR2 aberrations were assessed by testing of tumor tissue in local labs or using FoundationOne CDx (FoundationOne) assays in central or local labs as shown. The FGFR2 aberration for each patient (rearrangement or fusion) is indicated along with the fusion partner where identified. One patient had an FGFR2 S799fs*22 mutation in addition to an FGFR2 fusion (indicated with an asterisk). The most frequently altered oncogene or tumor suppressor genes are indicated. Three patients were not included in the figure because they were missing tumor assessments: 1 did not have a post-baseline assessment, and 2 had no target lesions available per ICR. (B) Duration and type of response per patient. (C) Kaplan-Meier plot of PFS. Upper and lower 95% CIs indicated as dotted lines. Tick marks represent data censored at the time of the last tumor assessment for patients who did not progress or die. (D) Kaplan-Meier plot of OS. Upper and lower 95% CIs indicated as dotted lines. Tick marks represent data censored at the date of the last follow-up (or data cutoff date, whichever is earlier) for patients who were alive or whose death was not confirmed. *The widths of the CIs have not been adjusted for multiplicity. Abbreviations: CR, complete response; ICR, independent central review; mo, month; NE, not evaluable; no, number; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease. From The New England Journal of Medicine, Goyal L, Meric-Bernstam F, Hollebecque A, et al, Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma, 388, 228-239. Copyright © (2023) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Genomic Profiling of Futibatinib Clinical Activity in CCA
Exploratory molecular profiling analyses from FOENIX-CCA2 examined futibatinib activity by FGFR aberration type or in the context of co-occurring genomic alterations (Fig. 3A).48 Futibatinib response did not appreciably vary with fusion partner type: ORRs were 41.7% and 44.6% in patients with BICC1 and non-BICC1 fusions, respectively.
ORRs were consistent regardless of the presence of co-alterations in TP53 (ORR, 38.5%; 43.8% with unaltered TP53), CDKN2A (40.0%; 43.8% with unaltered CDKN2A), and CDKN2B (43.8%; 42.9% with unaltered CDKN2B) (Fig. 3A).48 Median PFS with futibatinib was 7.0 and 9.0 months in TP53-altered and unaltered populations, respectively, 4.9 and 9.7 months in CDKN2A-altered and unaltered populations, respectively, and 4.8 and 11.0 months in CDKN2B-altered and unaltered populations, respectively.48 While cross-trial comparisons should be made with caution, a similar analysis of pemigatinib treatment in patients with CCA harboring FGFR2 fusions/rearrangements found no response to pemigatinib treatment and a lower median PFS (2.8 months) in patients with TP53 co-alterations, while patients without TP53 co-alterations experienced an ORR of 38.8% and a median PFS of 9.0 months.57 Patients with CDKN2A/B alterations treated with either pemigatinib or futibatinib had a lower median PFS, and those treated with pemigatinib experienced a lower ORR, than patients without alterations.48,57 These data provide interesting information about the activity of these treatments in the context of the CCA genetic landscape; however, the findings are limited by the small number of patients with co-alterations and the post hoc exploratory nature of these analyses.
Response to Futibatinib in Patients With iCCA With Prior FGFR Inhibitor Treatment
Preliminary data suggest that futibatinib showed antitumor activity in patients with iCCA with progression after previous FGFR inhibitor treatment. In the dose-escalation study, one responder had a history of disease progression on prior infigratinib before receiving futibatinib treatment for 15.6 months.61 In the dose expansion study, 5 of 28 patients with prior FGFR inhibitor therapy (17.9%) experienced objective responses with futibatinib.67 Duration of response ranged from 3.5 months (with response ongoing at data cutoff) to 20.4 months. Of the 5 responders, 3 had FGFR2 fusions, 1 a FGFR2 mutation, and 1 a FGFR2 amplification/rearrangement. An additional 15 patients previously treated with an FGFR inhibitor had stable disease. Of note, mechanisms of acquired resistance to prior FGFR inhibitor therapy were not captured because immediate pretreatment and post-progression biopsies were not required in the study.
In a separate analysis, clonal dynamics using cell-free circulating tumor DNA (ctDNA) were evaluated in 4 patients from a single site within the phase I patient population.60 These patients received prior infigratinib or Debio 1347, and each experienced clinical benefit with futibatinib (2 PR, 2 SD) with SD or PR lasting 5.1-17.2 months. In 3 patients, analysis of ctDNA at progression on the prior FGFR inhibitor indicated the development of acquired resistance mutations: E566A, H683L, K660M, K715R, M538I, N550H, N550K, N550T, and V565F.60 Analyses of ctDNA at the start of futibatinib treatment and upon subsequent progression in all 4 patients suggested that futibatinib had differential activity against individual FGFR2 secondary mutations compared with the prior FGFR inhibitors. The mutation allele frequency of V565F increased upon futibatinib treatment, whereas levels of E566A and N550K were unchanged. These results suggest that the spectrum of acquired resistance mutations varies and may influence choice of therapy. In follow-up experiments, futibatinib retained activity against FGFR2 kinase domain mutations in preclinical iCCA models. The investigators concluded that these preliminary investigations support the clinical utility of futibatinib in patients with acquired resistance to ATP-competitive reversible FGFR inhibitors.
These analyses were consistent with preclinical experiments showing superior activity of futibatinib against acquired resistance mutations.11,59 However, data on mechanisms of futibatinib resistance remain limited and further research is needed to understand the role of futibatinib after progression on FGFR inhibitors.
Activity of Futibatinib in Tumor Types Other Than CCA
In addition to CCA, futibatinib activity has been observed in at least 7 other tumor types harboring 10 different categories of FGFR1-4 aberrations (Table 2). Among 19 patients with urothelial cancer in the phase I expansion study,67 3 patients had PRs (16% ORR), all with FGFR1/3-mutant tumors. The ORR in this urothelial cohort was numerically lower than that in trials of other selective FGFR inhibitors,46,71 possibly because these patients were heavily pretreated: 58% received ≥3 prior regimens, with 42% previously treated with an FGFR inhibitor. Based on these data, a phase II study (NCT04601857) was initiated to study futibatinib in combination with pembrolizumab in patients with advanced or metastatic urothelial cancer.72
Table 2.
Futibatinib activity in patients receiving futibatinib once daily.
| Tumor type | Study | QD dose (mg) | n | FGF/FGFR aberrationsa | PR (n) | SD (n) |
ORR (%) | DCR (%) |
|---|---|---|---|---|---|---|---|---|
| CCA | Global phase I expansion67 | 20 | 64 |
FGFR1 fusion/rearr. (n = 1) FGFR2 fusion/rearr. (n = 43) FGFR2 mutation (n = 13) FGFR3 fusion/rearr. (n = 1) FGF1 amplification (n = 7) FGF3 amplification (n = 6) FGF4 amplification (n = 5) FGF19 amplification (n = 8) |
10 | 36 | 16 | 72 |
| 16 | 19 |
FGFR2 fusion/rearr. (n = 8)b FGFR2 amplification (n = 1)b |
8 | 8 | 42 | NR | ||
| Global phase II48 | 20 | 103 | FGFR2 fusion/rearr. (n = 103) | 43c | 42 | 42 | 83 | |
| Global phase I escalation61 | 4-24 | 17 | FGFR2 fusion (n = 3)b | 3d | NR | NR | NR | |
| Japanese phase I expansion/ escalation63 | 16-20 | 3 |
FGFR2 mutation (n = 1) FGFR2 fusion/rearr. (n = 2) |
1 | NR | NR | NR | |
| Gastric | Global phase I expansion67 | 20 | 9 |
FGFR2 fusion/rearr. (n = 1) FGFR2 amplification (n = 8) FGFR3 fusion/rearr. (n = 1) FGF1 amplification (n = 2) FGF3 amplification (n = 2) FGF4 amplification (n = 1) FGF19 amplification (n = 2) |
2 | 3 | 22 | 56 |
| Japanese phase I expansion/ escalation63 | 20e | 21 |
FGFR2 amplification CNV ≥10 (n = 11)e FGFR2 amplification CNV <10 (n = 10) |
4e 0 |
2 1 |
36 0 |
54 10 |
|
| Urothelial | Global phase I expansion67 | 20 | 19 |
FGFR1 mutation (n = 1) FGFR1 amplification (n = 1) FGFR3 mutation (n = 13) FGFR3 fusion/rearr. (n = 3) FGF1 amplification (n = 5) FGF3 amplification (n = 5) FGF4 amplification (n = 2) FGF19 amplification (n = 4) |
3 | 6 | 16 | 47 |
| Primary CNS | Global phase I expansion67 | 20 | 36 |
FGFR1 fusion/rearr. (n = 2) FGFR1 mutation (n = 9) FGFR2 mutation (n = 1) FGFR3 fusion/rearr. (n = 23) FGFR3 mutation (n = 1) FGFR3 amplification (n = 1) |
1 | 6 | 3 | 19 |
| Global phase I escalation61 | 4-24 (PR: 24) | 4 | FGFR1 mutation (n = 1)f | 2 | NR | NR | NR | |
| Breastg | Global phase I expansion67 | 20 | 11 |
FGFR1 mutation (n = 1) FGFR1 amplification (n = 1) FGFR2 fusion/rearr. (n = 2) FGFR2 amplification (n = 5) FGFR3 amplification (n = 1) FGFR4 mutation (n = 1) |
0 | 3 | - | 27 |
| 16 | 1 | FGFR2 amplification (n = 1) | 1 | - | - | - | ||
| Head and neck | Global phase I expansion | 20 | 2 | FGFR1 fusion/rearr. (n = 1)b | 1 | - | - | - |
| Myeloid neoplasm | Single patient protocol70 | 20 | 1 | FGFR1 fusion/rearr. (n = 1) | 1 | - | - | - |
Only tumor types with either at least one responder reported or DCR reported are shown.
a In the global phase I expansion study, patients may have more than one FGFR or FGF aberration.
b FGFR aberrations were only reported for responders.
cOne patient experienced a complete response.
dThe 3 responding patients received the following doses of futibatinib: 16 mg, 16 mg, and 24 mg once daily.
eOne patient had received futibatinib 80 mg TIW.
fA second patient with anaplastic oligodendroglioma experienced a PR with futibatinib 160 mg TIW dosing.
gThe Japanese phase I expansion/escalation study included one patient with breast cancer harboring FGFR2 amplifications who had a partial response with futibatinib.
Abbreviations: CCA, cholangiocarcinoma; CNS, central nervous system; CNV, copy number variant; DCR, disease control rate; FGFR, Fibroblast growth factor receptors; NR, not reported; ORR, objective response rate; PR, partial response; QD, once daily; rearr, rearrangements; SD stable disease; TIW, 3 times a week.
Futibatinib showed activity in gastric cancer in 2 phase I studies. In the phase I expansion study, 2 of 9 patients achieved a PR (ORR 22%).67 One responder had an FGFR2 amplification and the other had an FGFR3 fusion. In the Japanese phase I dose-expansion study, patients with gastric cancer harboring an FGFR2 amplification with a copy number ≥10 experienced an ORR of 36.4% and DCR of 54.5% vs. 0% and 10% in those with FGFR2 amplification copy number <10.63
Responses to futibatinib were also observed in primary central nervous system (CNS), breast, and head and neck tumors (Table 2).67 In the phase I dose-escalation study, 2 patients with primary CNS tumors (glioblastoma and anaplastic oligodendroglioma) harboring FGFR1 mutations experienced PRs,61 while in the phase I expansion study, 1 patient with glioblastoma harboring an FGFR1 fusion experienced a PR. A patient with triple-negative breast cancer with FGFR2 amplification from the phase I expansion study (16-mg cohort) and another with FGFR2-amplified breast cancer from the phase I study in Japan experienced durable responses with futibatinib.63,67 In a compassionate use program, a patient with an FGFR1-rearranged myeloid neoplasm treated with futibatinib had complete hematologic and cytogenetic remission.70 Notably, these phase I trials helped to identify previously uncharacterized FGFR aberrations and tumor types as potential FGFR inhibitor targets, including FGF-amplified and FGFR1-mutated urothelial carcinoma and FGFR-fusion positive head and neck cancer.
Altogether, these data support further investigation of futibatinib in multiple FGFR-aberrant tumor types and as a disease-agnostic treatment for patients with FGFR-altered advanced solid tumors.
Futibatinib Safety and Tolerability
Safety data in the 2 largest populations of patients who received futibatinib 20 mg QD, the phase I expansion 20-mg cohort (n = 170)67 and the phase II iCCA study (n = 103),48 indicated a manageable safety profile for futibatinib consistent with that of other FGFR inhibitors.43,44,46,51,73 Adverse events (AEs) were common in both studies (reported in >98% of patients), including hyperphosphatemia, diarrhea, constipation, fatigue, dry mouth, and alopecia (Table 3). In the phase I expansion and phase II iCCA studies, any-cause grade ≥3 AEs were reported in 72% and 77% of patients, respectively; grade ≥3 treatment-related AEs (TRAEs) occurred in 43% and 57% of patients, respectively, with grade 3 hyperphosphatemia most commonly reported in ≥10% of patients (phase I expansion, 22%; phase II, 30%). One grade 4 TRAE was reported in each study (increased gamma glutamyl-transferase and increased alanine aminotransferase). Serious TRAEs were reported in 6% and 10% of patients in the phase I expansion and phase II iCCA studies, respectively; no treatment-related deaths occurred in either study.
Table 3.
Treatment-emergent AEs occurring in ≥10% of all patients in futibatinib studies.
| TEAE (%) | Phase I dose escalation combined QD cohort61 (n = 44) |
Phase I dose expansion 20 mg QD cohort67 (n = 170) |
Japanese phase I expansion/escalation QD cohort (n=43) | Phase II iCCA 20 mg QD48 (n = 103) |
||||
|---|---|---|---|---|---|---|---|---|
| Any-grade | Grade 3a | Any-grade | Grade ≥3b | Any-grade | Grade ≥3 | Any-grade | Grade ≥3c | |
| Hyperphosphatemia | 68 | 16 | 81 | 22.4 | 100 | 5 | 85 | 30 |
| Constipation | 41 | 2 | 32 | 1.2 | 40 | 0 | 39 | 0 |
| Diarrhea | 34 | 2 | 33 | <1 | 30 | 0 | 36 | 1 |
| Nausea | 30 | 5 | 28 | 0 | 30 | 0 | 24 | 2 |
| ALT increased | 25 | 7 | 24 | 10 | 21 | 0 | 17 | 6 |
| AST increased | 25 | 5 | 24 | 5 | 21 | 2 | 25 | 10 |
| Dry mouth | 25 | 0 | 18 | 0 | - | - | 35 | 0 |
| Vomiting | 25 | 5 | 25 | 1 | 23 | 2 | 19 | 1 |
| Anemia | 20 | 5 | 14 | 5 | 19 | 12 | 16 | 5 |
| Asthenia | 20 | 0 | 16 | 4 | - | - | - | - |
| Stomatitis | 20 | 2 | 15 | 3 | 16 | 0 | 24 | 6 |
| Decreased appetite | 16 | 0 | 19 | 2 | 58 | 5 | 23 | 3 |
| Fatigue | 16 | 2 | 25 | 5 | 9 | 0 | 34 | 8 |
| Abdominal pain | 14 | 2 | 19 | 3 | - | - | 21 | 3 |
| Alopecia | 14 | 0 | 19 | 0 | - | - | 34 | 0 |
| Dry skin | 14 | 0 | 13 | 0 | 14 | 0 | 29 | 0 |
| Back pain | 11 | 0 | - | - | - | - | 17 | 2 |
| Cough | 11 | 0 | - | - | - | - | - | - |
| Dry eye | 11 | 0 | - | - | - | - | 21 | 1 |
| PPE syndrome | - | - | 13 | 4 | - | - | 21 | 5 |
| Increased blood creatinine | - | - | 12 | 0 | 16 | 2 | 15 | 0 |
| Arthralgia | - | - | 11 | 0 | 5 | 0 | 22 | 0 |
| Hypercalcemia | - | - | 11 | 1 | - | - | 16 | 2 |
| Dysgeusia | - | - | 11 | 0 | 14 | 9 | 20 | 0 |
| Decreased weight | - | - | 10 | <1 | 12 | 9 | 18 | 4 |
| Urinary-tract infection | - | - | - | - | - | - | 19 | 3 |
| Hyponatremia | - | - | - | - | 14 | 0 | 16 | 11 |
| Nail disorder | - | - | - | - | - | - | 16 | 0 |
| Onycholysis | - | - | - | - | - | - | 16 | 0 |
| Muscle spasms | - | - | - | - | - | - | 15 | 1 |
| Myalgia | - | - | - | - | - | - | 15 | 0 |
| Onychomadesis | - | - | - | - | - | - | 15 | 1 |
| Nail discoloration | - | - | - | - | - | - | 14 | 0 |
| Peripheral edema | - | - | - | - | 14 | 2 | 14 | 0 |
| Pyrexia | - | - | - | - | 7 | 0 | 14 | 0 |
| Blood alkaline phosphatase increase | - | - | - | - | - | - | 13 | 3 |
| Hypophosphatasemia | - | - | - | - | 7 | 5 | 13 | 5 |
| Dizziness | - | - | - | - | - | - | 11 | 1 |
| Thrombocytopenia | - | - | - | - | - | - | 11 | 2 |
| Blood creatine phosphokinase increase | - | - | - | - | 16 | 2 | 10 | 3 |
| Oropharyngeal pain | - | - | - | - | - | - | 10 | 0 |
| Peripheral sensory neuropathy | - | - | - | - | - | - | 10 | 1 |
| Hypoalbuminemia | - | - | - | 14 | 9 | - | - | - |
| Insomnia | - | - | - | 12 | 0 | - | - | - |
| Tumor pain | - | - | - | 14 | 0 | - | - | - |
Dashed lines (-) indicate the TEAE was reported in fewer than 10% of patients.
aNone of the TEAEs reported in this table were grade 4 or 5. Among patients treated with the once daily dose in this study, 3 patients had grade 4 TEAEs (one of which was considered treatment-related [increased creating phosphokinase]) and 4 patients had grade 5 events (none of which were considered treatment related).
bAmong any-grade TEAEs reported in >10% of patients, there was one grade 4 event (increased ALT) and there were no grade 5 events. Overall, 9 patients had grade TEAEs, one of which was considered treatment-related (increased gamma glutamyltransferase). None of the grade 5 TEAEs (n = 16) were considered treatment related.
cAmong any-grade TEAEs reported in ≥10% of patients, 4 grade 4 events were reported (1 patient each of increased ALT, hypercalcemia, hyponatremia, and hypophosphatasemia), and 1 grade 5 event was reported (decreased appetite). Overall, 6 patients had grade 4 TEAEs, 1 of which was considered treatment-related (increased ALT). None of the grade 5 TEAEs (n = 5) were treatment related.
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartine aminotransferase; iCCA, intrahepatic cholangiocarcinoma; PPE, Palmar–plantar erythrodysesthesia syndrome; QD, once daily; TEAEs, treatment-emergent adverse events.
The most common AE across studies was hyperphosphatemia (Table 3)48,59,63,67 similar to findings with pemigatinib and infigratinib.49,51,74 Hyperphosphatemia is an on-target effect of FGFR inhibition because decreased FGF23–FGFR1 signaling leads to increased phosphate reabsorption and hyperphosphatemia in proximal tubules.62 The numerically higher rates of hyperphosphatemia reported with futibatinib vs. pemigatinib and infigratinib49,51,74 may be related to between-study differences in dosing schedules, safety assessments, and grade definitions. Hyperphosphatemia was not defined in the National Cancer Institute Common Criteria for Adverse Events version 4.03, the version used for safety assessments in these trials. In the futibatinib studies, hyperphosphatemia was graded by serum phosphate levels regardless of symptoms,48,67 which was not the case for the pemigatinib trial.49,51,74 In the 2 futibatinib studies, hyperphosphatemia was managed with phosphate binders (75%-78%), dose interruptions (17%-20%), or dose reductions (8%-20%). All grade 3-4 events of hyperphosphatemia resolved, except in 2 patients in the phase I expansion study in whom resolution could not be assessed as they discontinued the study because of disease progression and withdrawal of consent.
Eye and nail toxicities are also considered AEs of special interest for FGFR inhibitors.48,49 Similar to agents targeting the MAPK pathway, FGFR inhibitors can cause central serous retinopathy (CSR)/retinal pigment epithelial dystrophy (RPED). Patients with CSR/RPED can be asymptomatic; however, more severe cases manifest with acute central vision decrease/loss and metamorphopsia.75 In the phase I expansion study, 26% of patients had eye-related AEs, most commonly dry eye (9%), and blurred vision (6%). All but 2 cases (grade 3 cataract [treatment-related]; grade 3 macular fibrosis/grade 4 ocular ischemic syndrome [unrelated]) were grade 1-2. Seven patients experienced grade 1-2 central serous retinopathy.67 In the phase II iCCA study, retinal disorders were reported in 8% of patients (grade 1-2 events).48 In the phase I expansion study, 20% of patients had nail toxicities, all but one (grade 3 onychalgia) grade 1 or 2. In the phase II study, 47% of patients developed nail toxicities (including nail disorder, onycholysis, nail discoloration, and paronychia), with grade 3 cases in 2%.48 Similar data on eye and nail-related toxicities have been reported with other FGFR inhibitors.76,77
Palmar-plantar erythrodysesthesia syndrome, clinically notable in patients treated with FGFR inhibitors, was reported in 13% (grade ≥3, 4%) and 21% (grade ≥3, 5%) of patients in the phase I expansion and phase II studies, respectively. No grade 5 AEs of special interest were reported in either study.
AEs were mostly managed with dosing interruption and reductions. In the phase I expansion study, TRAEs led to dosing modifications in 44% of patients and treatment discontinuation in 4% of patients. In the phase II study, TRAEs led to dose interruption, dose reduction, or treatment discontinuation in 50%, 54%, and 2% of patients, respectively.
An integrated analysis of futibatinib safety in 318 patients across the global phase I/II and Japanese phase I studies showed a consistent safety profile: grade ≥3 hyperphosphatemia occurred in 23% of patients, but nearly all grade ≥3 events resolved and only 3% discontinued because of TRAEs.78
Collectively, these results indicate a monitorable and manageable safety profile for futibatinib, rarely requiring treatment discontinuation due to AEs.
Quality of Life with Futibatinib Treatment
Across therapies for iCCA, little data are available on how treatment affects patient quality of life (QoL).79 The phase II study of futibatinib in patients with iCCA harboring FGFR2 fusions/rearrangements was among the first to report patient-reported outcomes (PROs) with an FGFR inhibitor. PROs were measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30 (EORTC QLQ-C30) and Euro QoL Visual analog scale (EQ-VAS).48 Ninety-two of 103 patients (89%) had PRO data with at least one follow-up assessment, with 48 patients (47%) having PRO data at cycle 13 (final cycle assessed). Through 9 months of futibatinib treatment, patient global health status was maintained, with no clinically meaningful changes in individual functional measures (physical, role, cognitive, emotional, and social). Individual symptom measures on the EORTC QLQ-C30 were also stable except for constipation, which met criteria for a clinically meaningful change at cycle 4 only. Mean EQ-VAS scores were sustained and the status across all EQ-5D-3L dimensions remained the same or improved over this period. Most patients (82%-95%) maintained the same or better Eastern Cooperative Oncology Group performance status score relative to baseline. Overall, these data suggest patient QoL was not negatively impacted by AEs while on futibatinib treatment.
Ongoing Studies and Future Directions for Development of Futibatinib
Based on phase I data, phase II studies are examining the safety and activity of futibatinib in various FGFR-aberrant cancer types including metastatic breast cancer and urothelial cancer (Table 4). A tumor-agnostic phase II study will investigate futibatinib as a disease-agnostic treatment option for patients with FGFR-rearranged advanced solid tumors. Building on the phase II iCCA study results, an ongoing open-label, randomized phase III study will assess futibatinib as a first-line treatment vs. gemcitabine–cisplatin for patients with FGFR2 fusion/rearrangement-positive iCCA.
Table 4.
Ongoing clinical studies with futibatinib.
| Study | Cancer type | Study design | Intervention(s)a | Primary outcome(s) | Estimated enrollment | Start date; estimated end date | NCT identifier |
|---|---|---|---|---|---|---|---|
| FOENIX-CCA3 (phase III) | Advanced CCA with FGFR2 rearrangements | Randomized (1:1) | Futibatinib or gemcitabine-cisplatin (control) | PFS | 216 | March 1, 2020; April 2028 | NCT04093362 |
| Phase II study in metastatic breast cancer (MBC) | MBC harboring FGFR2 amplifications (cohorts 1-3) | Open label | Futibatinib | ORR or CBR | 168 across cohorts | August 30, 2019; June 30, 2023 | NCT04024436 |
| MBC harboring FGFR1 amplifications (cohort 4) | Futibatinib + fulvestrant | PFS at 6 months | |||||
| Phase II study in patients with specific FGFR aberrations | Solid tumors with FGFR1-4 rearrangements (cohort A) | Open label | Futibatinib | ORR | 115 across cohorts | August 24, 2020; December, 2022 |
NCT04189445 |
| Gastric/gastroesophageal junction tumors with FGFR2-amplifiication (cohort B) | Futibatinib | ORR | |||||
| Myeloid/lymphoid neoplasms with FGFR1-rearrangement (cohort C) | Futibatinib | CR rate | |||||
| Phase II study of advanced/ metastatic UC (mUC) | mUC with FGFR3 mutation or FGFR1-4 fusions/rearrangements (cohort A) | Open label | Futibatinib + pembrolizumab | ORR | 46 across cohorts | January 21, 2021; December 30, 2023 | NCT04601857 |
| All other mUC not included in cohort Ab (cohort B) | |||||||
| Phase II study in advanced or metastatic hepatocellular carcinoma | Advanced or metastatic FGF19-positive BCLC stage A, B, or C hepatocellular carcinoma | Open-label | Futibatinib + pembrolizumab | PFS at 6 months | 25 | May 7, 2021; May 6, 2024 |
NCT04828486 |
| Phase I/II study of combination therapy | Advanced solid tumors | Open label, dose escalation (TAS-117) | Futibatinib + TAS-117 | Safety and efficacy | 137 | May 1, 2019; June 30, 2023 |
Japic CTI-194864 |
| Phase Ib/II study of advanced KRAS mutant non-small cell lung cancer | Advanced or metastatic solid tumors (part 1) | Open-label | Futibatinib + binimetinib | RP2D (part 1) | 36 across cohorts | September 20, 2021; December 2024 |
NCT04965818 |
| Advanced non-small cell lung cancer with KRAS mutation (part 2) | ORR (part 2) |
aFutibatinib to be administered as 20 mg once daily in all studies.
bIncludes patients with other FGFR/non-FGFR genetic aberrations and patients with wild-type tumors.
Abbreviations: BCLC, Barcelona clinic liver cancer; CBR, clinical benefit rate; CCA, cholangiocarcinoma; CR, complete response; NCT, National Clinical Trials; ORR, objective response rate; PFS, progression-free survival; RP2D, recommended phase II dose; UC, urothelial carcinoma.
Futibatinib combination studies are another important future prospect. The combination of FGFR inhibitors with immunotherapy is supported by preclinical evidence,80 and phase II trials are evaluating futibatinib combined with pembrolizumab in metastatic urothelial carcinoma (NCT04601857) and metastatic hepatocellular carcinoma (NCT04828486). In preclinical models, futibatinib combined with cytotoxic chemotherapy, MEK inhibitors, or PI3K pathway inhibitors induced synergistic tumor regression81-83; trials evaluating the combination of futibatinib with AKT and MEK inhibitors are ongoing (JapicCTI-194864; NCT04965818). There is also rationale for the combination of FGFR inhibitors with VEGF inhibitors.84 Future exploration of futibatinib combined with other treatments could yield additional clinical benefits, particularly to combat tyrosine kinase inhibitor resistance.
Summary
FGFR dysregulation drives oncogenesis across a broad range of tumor types. Although many FGFR inhibitors are currently in clinical development, futibatinib has a unique mechanism of action as an irreversible FGFR1-4 inhibitor with potential activity against acquired secondary FGFR kinase domain mutations. In early studies, futibatinib demonstrated activity in diverse tumor types harboring various FGFR aberrations. Based on durable responses and manageable safety in the phase II FOENIX-CCA2 study futibatinib was approved for patients with iCCA harboring FGFR2 fusions/rearrangements. These data, combined with the unique irreversible mechanism of action, set futibatinib apart as a leading second-generation FGFR inhibitor, while both preclinical evidence and exploratory clinical results suggest a role for futibatinib after failure of prior FGFR inhibitor treatment. Further studies are required to assess mechanisms of futibatinib resistance and combination therapy approaches using this agent.
Supplementary Material
Acknowledgments
Editorial and medical writing assistances were provided under the direction of authors by Meredith Kalish, MD, Vasupradha Vethantham, PhD, and Kathleen Blake, PhD, Ashfield MedComms, an Inizio company, funded by Taiho Oncology, Inc.
Contributor Information
Milind Javle, Department of Gastrointestinal Oncology, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Gentry King, Division of Medical Oncology, University of Washington, Seattle, WA, USA.
Kristen Spencer, Perlmutter Cancer Center of NYU Langone Health, New York, NY, USA; NYU Grossman School of Medicine, New York University, New York, NY, USA.
Mitesh J Borad, Department of Oncology, Mayo Clinic Cancer Center, Phoenix, AZ, USA.
Conflict of Interest
Milind Javle reported consulting fees from QED, Incyte, Taiho, Merck, EMD Serono, Basilea, Oncosil, BMS, and AstraZeneca; fees for promotional services from Incyte; institutional research funding from Bayer, Koo Foundation, Sanofi, Cyclacel; and advisory/consulting relationships with Tempus Inc, Pfizer, QED, and Novartis. Gentry King reported institutional funding from Bayer, Koo Foundation, Sanofi, and Cyclacel, and advisory/consulting relationships with Tempus Inc, Pfizer, QED, and Novartis.. Kristen Spencer reported advisory board member with QED Therapeutics and Caris Life Sciences. Mitesh Borad reported stock and other ownership interests with AVEO, Gilead Sciences, Intercept Pharmaceuticals, and Spectrum Pharmaceuticals; consulting or advisory role with Agios (Inst), ArQule (Inst), Celgene (Inst), De Novo Pharmaceuticals, Exelixis, Fujifilm (Inst), G1 Therapeutics, Genentech, Halozyme (Inst), Immunovative Therapies, Imvax, Inspyr Therapeutics, Insys Therapeutics (Inst), Klus Pharma, Lynx Group, Merck, Novartis (Inst), Pieris Pharmaceuticals (Inst), Taiho Pharmaceutical (Inst), and Western Oncolytics; research funding from Adaptimmune (Inst), Agios (Inst), ARIAD (Inst), AstraZeneca (Inst), Basilea (Inst), BiolineRx (Inst), Boston Biomedical (Inst), Celgene (Inst), Dicerna (Inst), Eisai (Inst), EMD Serono (Inst), Halozyme (Inst), ImClone Systems (Inst), Incyte (Inst), Isis Pharmaceuticals (Inst), MedImmune (Inst), Merck Serono (Inst), miRNA Therapeutics (Inst), Puma Biotechnology (Inst), QED Therapeutics (Inst), RedHill Biopharma (Inst), Senhwa Biosciences (Inst), Sillajen (Inst), Sun Biopharma (Inst), Taiho Pharmaceutical (Inst), Threshold Pharmaceuticals (Inst), and Toray Industries (Inst).
Author Contributions
All authors contributed to the conception, design, data analysis, interpretation, manuscript writing, and final approval of manuscript.
Data Availability
No new data were generated or analyzed for this manuscript.
References
- 1. Touat M, Ileana E, Postel-Vinay S, André F, Soria J-C.. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21(12):2684-2694. 10.1158/1078-0432.CCR-14-2329. [DOI] [PubMed] [Google Scholar]
- 2. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427-1434. 10.1002/hep.26890. [DOI] [PubMed] [Google Scholar]
- 3. Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precis Oncol. 2018;2:1-12. 10.1200/PO.17.00080. [DOI] [PubMed] [Google Scholar]
- 4. Ardizzone A, Scuderi SA, Giuffrida D, et al. Role of fibroblast growth factors receptors (FGFRS) in brain tumors, focus on astrocytoma and glioblastoma. Cancers (Basel). 2020;12(12):3825. 10.3390/cancers12123825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. André F, Bachelot T, Campone M, et al. Targeting FGFR with dovitinib (tki258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19(13):3693-3702. 10.1158/1078-0432.ccr-13-0190. [DOI] [PubMed] [Google Scholar]
- 6. Jang JH, Shin KH, Park JG.. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61(9):3541-3543. [PubMed] [Google Scholar]
- 7. Xie L, Su X, Zhang L, et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin Cancer Res. 2013;19(9):2572-2583. 10.1158/1078-0432.CCR-12-3898. [DOI] [PubMed] [Google Scholar]
- 8. Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259-267. 10.1158/1078-0432.CCR-14-3212. [DOI] [PubMed] [Google Scholar]
- 9. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252-263. 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krook MA, Reeser JW, Ernst G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer. 2021;124(5):880-892. 10.1038/s41416-020-01157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sootome H, Fujita H, Ito K, et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020;80(22):4986-4997. 10.1158/0008-5472.CAN-19-2568. [DOI] [PubMed] [Google Scholar]
- 12. Zhang X, Ibrahimi OA, Olsen SK, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281(23):15694-15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turner N, Grose R.. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116-129. 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 14. De Moerlooze L, Spencer-Dene B, Revest JM, et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483-492. 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- 15. Yu K, Xu J, Liu Z, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130(13):3063-3074. 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- 16. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM.. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12(4):390-397. 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 17. Pardo OE, Lesay A, Arcaro A, et al. Fibroblast growth factor 2-mediated translational control of IAPs blocks mitochondrial release of Smac/DIABLO and apoptosis in small cell lung cancer cells. Mol Cell Biol. 2003;23(21):7600-7610. 10.1128/MCB.23.21.7600-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pardo OE, Wellbrock C, Khanzada UK, et al. Fgf-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006;25(13):3078-3088. 10.1038/sj.emboj.7601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Y, Wu C, Lu G, et al. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer. 2020;11(8):2000-2007. 10.7150/jca.40531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simon R, Richter J, Wagner U, et al. High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res. 2001;61(11):4514-4519. [PubMed] [Google Scholar]
- 21. Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9(12):e115383. 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Farshidfar F, Zheng S, Gingras MC, et al. ; Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;19(13):2878-2880. 10.1016/j.celrep.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Courjal F, Cuny M, Simony-Lafontaine J, et al. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res. 1997;57(19):4360-4367. [PubMed] [Google Scholar]
- 24. Knowles MA, Hurst CD.. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15(1):25-41. 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 25. Heist RS, Mino-Kenudson M, Sequist LV, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7(12):1775-1780. 10.1097/JTO.0b013e31826aed28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schultheis AM, Bos M, Schmitz K, et al. Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod Pathol. 2014;27(2):214-221. 10.1038/modpathol.2013.141. [DOI] [PubMed] [Google Scholar]
- 27. Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231-1235. 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su X, Zhan P, Gavine PR, et al. FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer. 2014;110(4):967-975. 10.1038/bjc.2013.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu YJ, Shen D, Yin X, et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer. 2014;110(5):1169-1178. 10.1038/bjc.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hur JY, Chao J, Kim K, et al. High-level FGFR2 amplification is associated with poor prognosis and lower response to chemotherapy in gastric cancers. Pathol Res Pract. 2020;216(4):152878. 10.1016/j.prp.2020.152878. [DOI] [PubMed] [Google Scholar]
- 31. Dutt A, Salvesen HB, Chen TH, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA. 2008;105(25):8713-8717. 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chesi M, Brents LA, Ely SA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97(3):729-736. 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 33. Rosty C, Aubriot MH, Cappellen D, et al. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol Cancer. 2005;4(1):15. 10.1186/1476-4598-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hernandez S, de Muga S, Agell L, et al. FGFR3 mutations in prostate cancer: association with low-grade tumors. Mod Pathol. 2009;22(6):848-856. 10.1038/modpathol.2009.46. [DOI] [PubMed] [Google Scholar]
- 35. von Loga K, Kohlhaussen J, Burkhardt L, et al. FGFR1 amplification is often homogeneous and strongly linked to the squamous cell carcinoma subtype in esophageal carcinoma. PLoS One. 2015;10(11):e0141867. 10.1371/journal.pone.0141867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gött H, Uhl E.. FGFR3-TACCs3 fusions and their clinical relevance in human glioblastoma. Int J Mol Sci. 2022;23(15):8675. 10.3390/ijms23158675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jogo T, Nakamura Y, Shitara K, et al. Circulating tumor DNA analysis detects FGFR2 amplification and concurrent genomic alterations associated with FGFR inhibitor efficacy in advanced gastric cancer. Clin Cancer Res. 2021;27(20):5619-5627. 10.1158/1078-0432.CCR-21-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33(1):125-136.e3. 10.1016/j.ccell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105-122. 10.1038/s41571-018-0115-y. [DOI] [PubMed] [Google Scholar]
- 40. Dai S, Zhou Z, Chen Z, Xu G, Chen Y.. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. 2019;8(6):614. 10.3390/cells8060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aggarwal C, Redman MW, Lara PN Jr, et al. SWOG S1400D (NCT02965378), a phase II study of the fibroblast growth factor receptor inhibitor AZD4547 in previously treated patients with fibroblast growth factor pathway-activated stage iv squamous cell lung cancer (lung-map substudy). J Thorac Oncol. 2019;14(10):1847-1852. 10.1016/j.jtho.2019.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chae YK, Hong F, Vaklavas C, et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH trial (EAY131) subprotocol W. J Clin Oncol. 2020;38(21):2407-2417. 10.1200/JCO.19.02630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165-171. 10.1038/s41416-018-0334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Voss MH, Hierro C, Heist RS, et al. A phase I, open-label, multicenter, dose-escalation study of the oral selective FGFR inhibitor debio 1347 in patients with advanced solid tumors harboring FGFR gene alterations. Clin Cancer Res. 2019;25(9):2699-2707. 10.1158/1078-0432.CCR-18-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park JO, Feng Y-H, Chen Y-Y, et al. Updated results of a phase IIa study to evaluate the clinical efficacy and safety of erdafitinib in Asian advanced cholangiocarcinoma (CCA) patients with FGFR alterations. J Clin Oncol. 2019;37(15_suppl):4117-4117. 10.1200/jco.2019.37.15_suppl.4117. [DOI] [Google Scholar]
- 46. Loriot Y, Necchi A, Park SH, et al. ; BLC2001 Study Group. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338-348. 10.1056/NEJMoa1817323. [DOI] [PubMed] [Google Scholar]
- 47. Kim RD, Sarker D, Meyer T, et al. First-in-human phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019;9(12):1696-1707. 10.1158/2159-8290.CD-19-0555. [DOI] [PubMed] [Google Scholar]
- 48. Goyal L, Meric-Bernstam F, Hollebecque A, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. 2023;388(3)228-239. 10.1056/NEJMoa2206834. [DOI] [PubMed] [Google Scholar]
- 49. Javle M, Roychowdhury S, Kelley RK, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6(10):803-815. 10.1016/S2468-1253(21)00196-5. [DOI] [PubMed] [Google Scholar]
- 50. Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1-3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018;8(7):812-821. 10.1158/2159-8290.CD-18-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671-684. 10.1016/S1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Necchi A, Pouessel D, Leibowitz-Amit R, et al. Interim results of fight-201, a phase II, open-label, multicenter study of INCB054828 in patients (pts) with metastatic or surgically unresectable urothelial carcinoma (UC) harboring fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations (GA). Ann Oncol. 2018;29(Supp. 8):viii319VIII319-viii319viii320. 10.1093/annonc/mdy283.109. [DOI] [Google Scholar]
- 53. Schuler M, Cho BC, Sayehli CM, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019;20(10):1454-1466. 10.1016/S1470-2045(19)30412-7. [DOI] [PubMed] [Google Scholar]
- 54. Kalyukina M, Yosaatmadja Y, Middleditch MJ, et al. TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem. 2019;14(4):494-500. 10.1002/cmdc.201800719. [DOI] [PubMed] [Google Scholar]
- 55. Cleary JM, Raghavan S, Wu Q, et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov. 2021;11(10):2488-2505. 10.1158/2159-8290.CD-20-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krook MA, Lenyo A, Wilberding M, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther. 2020;19(3):847-857. 10.1158/1535-7163.MCT-19-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Silverman IM, Hollebecque A, Friboulet L, et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov. 2021;11(2):326-339. 10.1158/2159-8290.CD-20-0766. [DOI] [PubMed] [Google Scholar]
- 58. Chen H, Ma J, Li W, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27(5):717-730. 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sootome H, Kato S, Aoyagi Y, et al. Acquired resistance to ATP-competitive and irreversible FGFR inhibitors (FGFRi's): a library-based approach. Cancer Res. 2021;81(Supp 13):1117. 10.3389/fnhum.2015.00181. [DOI] [Google Scholar]
- 60. Goyal L, Shi L, Liu LY, et al. Tas-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064-1079. 10.1158/2159-8290.CD-19-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bahleda R, Meric-Bernstam F, Goyal L, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann Oncol. 2020;31(10):1405-1412. 10.1016/j.annonc.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wohrle S, Bonny O, Beluch N, et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res. 2011;26(10):2486-2497. 10.1002/jbmr.478. [DOI] [PubMed] [Google Scholar]
- 63. Doi T, Shitara K, Kojima T, et al. Phase I study of the irreversible fibroblast growth factor receptor 1-4 inhibitor futibatinib in Japanese patients with advanced solid tumors. Cancer Sci. 2023;114:574-585. 10.1111/cas.15486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamamiya I, Hunt A, Yamashita F, et al. Phase 1, open-label studies of futibatinib excretion, metabolic profiling, and food effects in healthy adult volunteers. Poster Presented at: American Society for Clinical Pharmacology and Therapeutics conference, March 8–12 and 15-17, 2021; Virtual. https://metaballcreative.com/TAGS/wp-content/uploads/2021/02/Yamamiya_ASCPT2021_ADME_foodeffect_poster_2.11.21.pdf. Accessed January 12, 2023.
- 65. Yamamiya I, Laabs J, Hunt A, et al. Evaluation of potential drug–drug interactions (DDIs) between futibatinib and cyp3a inhibitors/inducers, CYP3A substrates, or proton pump inhibitors (PPIs). Cancer Res. 2021;81(13_Supplement):CT125-CT125. 10.1158/1538-7445.am2021-ct125. [DOI] [Google Scholar]
- 66. McDonnell AM, Dang CH.. Basic review of the cytochrome p450 system. J Adv Pract Oncol. 2013;4(4):263-268. 10.6004/jadpro.2013.4.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meric-Bernstam F, Bahleda R, Hierro C, et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 2022;12(2):402-415. 10.1158/2159-8290.CD-21-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goyal L, Meric-Bernstam F, Hollebecque A, et al. Updated results of the FOENIX-CCA2 trial: Efficacy and safety of futibatinib in intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 fusions/rearrangements. J Clin Oncol. 2022;40(16_suppl):4009-4009. 10.1200/jco.2022.40.16_suppl.4009. [DOI] [Google Scholar]
- 69. Lytgobi (futibatinib) tablets, for oral use [prescribing information]. Princeton, NJ: Taiho Oncology, Inc.; 2022. [Google Scholar]
- 70. Kasbekar M, Nardi V, Dal Cin P, et al. Targeted FGFR inhibition results in a durable remission in an FGFR1-driven myeloid neoplasm with eosinophilia. Blood Adv. 2020;4(13):3136-3140. 10.1182/bloodadvances.2020002308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase i, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35(2):157-165. 10.1200/JCO.2016.67.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Futibatinib and pembrolizumab combination in the treatment of advanced or metastatic urothelial carcinoma. ClinicalTrial.gov identifier NCT04601857. Bethesda, MD: U.S. National Library of Medicine, October 26 2020. https://clinicaltrials.gov/ct2/show/NCT04601857. Accessed October 28, 2021. [Google Scholar]
- 73. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276-282. 10.1200/JCO.2017.75.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Javle MM, Roychowdhury S, Kelley RK, et al. Final results from a phase ii study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. J Clin Oncol. 2021;39(3_suppl):265-265. 10.1200/jco.2021.39.3_suppl.265.33503392 [DOI] [Google Scholar]
- 75. King G, Javle M.. FGFR inhibitors: clinical activity and development in the treatment of cholangiocarcinoma. Curr Oncol Rep. 2021;23(9):108. 10.1007/s11912-021-01100-3. [DOI] [PubMed] [Google Scholar]
- 76. Kommalapati A, Tella SH, Borad M, Javle M, Mahipal A.. FGFR inhibitors in oncology: insight on the management of toxicities in clinical practice. Cancers. 2021;13(12):2968. 10.3390/cancers13122968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lacouture ME, Sibaud V, Anadkat MJ, et al. Dermatologic adverse events associated with selective fibroblast growth factor receptor inhibitors: overview, prevention, and management guidelines. Oncologist. 2021;26(2):e316-e326. 10.1002/onco.13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Meric-Bernstam F, Furuse J, Oh D-Y, et al. Pooled analysis safety profile of futibatinib in patients with advanced solid tumors, including intrahepatic cholangiocarcinoma. Ann Oncol. 2021;32(Supp 5):S378S376-S378S381. 10.1016/j.annonc.2021.08.330. [DOI] [Google Scholar]
- 79. Cho Y, Chadwick C, Barrett S, et al. Health-related quality-of-life in patients with cholangiocarcinoma: results of a systematic literature review. J Clin Oncol. 2019;37(15_suppl):e15623-e15623. 10.1200/jco.2019.37.15_suppl.e15623. [DOI] [Google Scholar]
- 80. Palakurthi S, Kuraguchi M, Zacharek SJ, et al. The combined effect of FGFR inhibition and PD-1 blockade promotes tumor-intrinsic induction of antitumor immunity. Cancer Immunol Res. 2019;7(9):1457-1471. 10.1158/2326-6066.CIR-18-0595. [DOI] [PubMed] [Google Scholar]
- 81. Iwasaki J, Kuramoto T, Komori T, Saito H, Hirai H.. Abstract 661: synergistic antitumor activity of futibatinib, an FGFR1-4 inhibitor, and TAS-117, a selective AKT inhibitor, in FGFR-deregulated cancer models. Cancer Res. 2020;80(16_Supplement):661-661. 10.1158/1538-7445.am2020-661. [DOI] [Google Scholar]
- 82. Sootome H, Miura A, Komori T, Hirai H.. Abstract 564: Futibatinib (TAS-120) plus chemotherapy demonstrates a synergistic effect across various FGFR-deregulated cancer cell lines and xenograft models. Cancer Res. 2020;80(16_Supplement):564-564. 10.1158/1538-7445.am2020-564. [DOI] [Google Scholar]
- 83. Miura A, Sootome H, Komori T, Ochiiwa H, Hirai H.. Abstract 659: synergistic antitumor activity of futibatinib (TAS-120), a FGFR1-4 inhibitor, and PI3K pathway inhibitors. Cancer Res. 2020;80(16_Supplement):659-659. 10.1158/1538-7445.am2020-659.31831463 [DOI] [Google Scholar]
- 84. Lieu C, Heymach J, Overman M, Tran H, Kopetz S.. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res. 2011;17(19):6130-6139. 10.1158/1078-0432.CCR-11-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data were generated or analyzed for this manuscript.