

Summary
Adherens junctions between tubular epithelial cells are disrupted in renal ischemia/reperfusion (I/R) injury. Syndecan-1 (SDC-1) is involved in maintaining cell morphology. We aimed to study the role of SDC-1 shedding induced by renal I/R in the destruction of intracellular adherens junctions. We found that SDC-1 shedding was increased while the expression of E-cadherin was decreased. This observation was accompanied by the activation of STAT3 in the kidneys. Inhibiting the shedding of SDC-1 induced by I/R could alleviate this effect. Mild renal I/R could induce more severe renal injury, lower E-cadherin expression, damaged cell junctions, and activated STAT3 in knockout mice with the tubule-specific deletion of SDC-1 mice. The results in vitro were consistent with those in vivo. Inhibiting the shedding of SDC-1 could alleviate the decreased expression of E-cadherin and damage of cell adherens junctions through inhibiting the activation of STAT3 during ischemic acute kidney injury.
Subject areas: Nephrology, Medical Microbiology
Graphical abstract
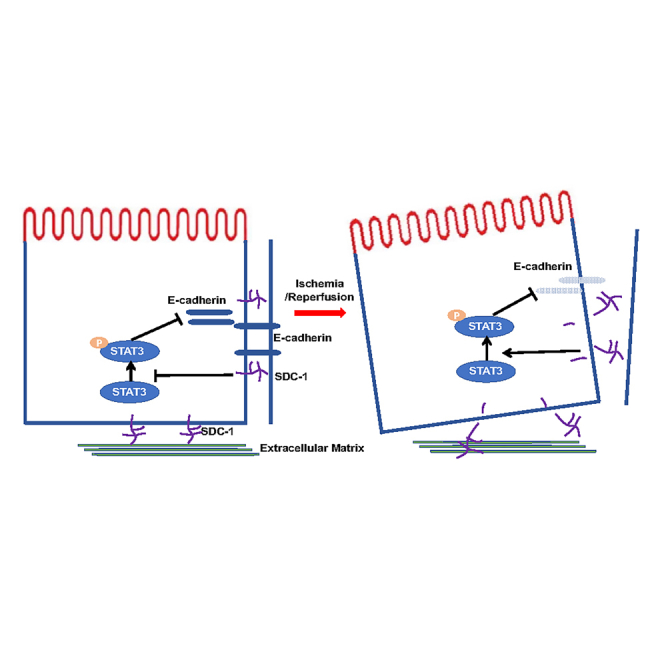
Highlights
-
•
Syndecan-1 (SDC-1) and tubular adherens junction are destroyed in renal I/R injury
-
•
Knockout of SDC-1 aggravates I/R injury and disrupts cell junctions in mice kidney
-
•
SDC-1 shedding decreases level of E-cadherin by activating STAT3 in renal I/R injury
Nephrology; Medical Microbiology
Introduction
Ischemia/reperfusion (I/R) injury is a common cause of acute kidney injury (AKI).1 The destruction of intracellular junctions including adherens junctions among tubular epithelial cells due to ischemia or hypoxia could result in apoptosis or necrosis, which are hallmarks of the early stage of ischemic AKI.1
E-cadherin, a calcium-dependent cell adhesion molecule that ensures intercellular adhesion among neighboring epithelial cells, has been extensively studied in cancer and renal fibrosis.2,3 In the kidney, E-cadherin plays a crucial role in maintaining the integrity and polarity of renal tubular epithelial cells,4 and is also required for epithelial cell polarization and lumen formation during the culture of three-dimensional renal tubules.5
STAT3, a vital component in the STAT family of proteins, plays an important role in cell growth, migration, and apoptosis.6 The activation of STAT3 can promote renal fibrosis and increase renal inflammatory infiltration.7 STAT3 signaling is also involved in regulating the expression of several junction proteins.8 Studies in colorectal cancer have also suggested a negative regulatory effect between STAT3 and E-cadherin9
Syndecan-1 (SDC-1), a transmembrane glycoprotein with heparan sulfate, expressed on the basolateral surface of epithelial cells, plays an important role in maintaining cell morphology and linking the cytoskeleton to the extracellular matrix. In the previous study, we verified that the shedding of SDC-1 could be induced by I/R injury and result in the loss of cell polarity and increases apoptosis in renal tubular epithelial cells.10 It is suggested that SDC-1 was involved in regulating epithelial tight junctions11 and expression of adherens junctions protein E-cadherin.12 However, the role of SDC-1 in the intracellular adherens junctions between renal epithelial cells was unclear. In our current work, we used transgenic mice, 3D tubules combined with clinical data to uncover the relationship between SDC-1 and E-cadherin based on adherens junctions and potential mechanisms in renal I/R injury.
Results
Renal I/R increased the shedding of SDC-1, accompanied with decreased expression of E-cadherin and activation of STAT3
The renal I/R injury could increase the shedding of SDC-1 from tubular epithelial cells (Figure 1A) and impair renal function including the histopathology damage (Figures 1B and 1C) and increased serum creatinine (SCr) (Figure 1D) at 12 h and 24 h after reperfusion in mice. Moreover, increased apoptosis of tubular cells could be found in kidneys after I/R injury (Figures 1Ji and 1K). Transmission electron microscopy (TEM) indicated that intracellular junctions between tubular epithelial cells were damaged in mice from the I/R groups and the most disruption was shown in the kidney at 24 h after reperfusion (Figure 1L).
Figure 1.

The shedding of syndecan-1 (SDC-1), activation of STAT3 and impaired intracellular junctions induced by renal ischemia/reperfusion (I/R) injury
(A) The shedding of SDC-1 into serum was increased in the I/R mice, n = 6.
(B and C) The renal histopathological injury was aggravated in the I/R mice, Bar = 50μm, n = 6.
(D) The serum creatinine (SCr) was increased in the I/R mice, n = 6.
(E and F) The mRNA and protein expression of SDC-1 in mice kidneys from the I/R group and the Sham group, n = 6.
(G–I) Quantitative analysis of STAT3 activation and E-cadherin expression in I/R and Sham mice kidneys, n = 6–7.
(J) The histochemical staining of E-cadherin expression (n = 4) and TUNEL staining (n = 3) in the I/R and Sham mice kidneys, Bar = 50μm.
(K) The statistical analysis of TUNEL staining.
(L) The representative image of TEM for intracellular junctions between tubular epithelial cells in the I/R and Sham mice, n = 2, Bar = 1μm. ∗p < 0.05, ∗∗p < 0.01 vs. Sham group.
The compensatory mechanism, denoted by increased mRNA expression of SDC-1(Figure 1E), and decreased SDC-1 protein were shown in the I/R kidneys (Figure 1F), which might be due to SDC-1 shedding from the kidney after I/R. Meanwhile, I/R injury resulted in reduced expression of E-cadherin protein significantly (Figures 1Jii and 1I) and increased the expression of phosphorylated STAT3 (pSTAT3) (Figure 1H) in the kidney.
Inhibiting SDC-1 shedding alleviated renal I/R injury with the activation of STAT3 and restored the expression of E-cadherin
We pre-treated the mice with GM6001 before inducing I/R insult. It was shown that the administration of GM6001 could improve renal pathological injury outcomes and maintain renal function (Figures 2B–2D) by inhibiting the shedding of SDC-1 induced by renal I/R in mice (Figure 2A), as we performed previously.13 While GM6001 pre-treatment had no influence on the renal function of the sham mice (Figure S1). We also found that the significantly increased expression of E-cadherin and decreased expression of phosphorylated STAT3 in the I/R mice with GM6001 pre-treatment, compared to that of the control group (Figures 2E and 2F). Meanwhile, the decreased E-cadherin expression in the renal epithelial cells was alleviated by GM6001 pre-treatment, as shown in the immunohistochemical results (Figure 2Gii). The inhibition of SDC-1 shedding in the I/R mice alleviated the apoptotic response and impaired intercellular junctions in the kidneys (Figures 2Gi–2I).
Figure 2.

GM6001 pre-treatment alleviated the damaged intracellular junctions through suppressing the shedding of syndecan-1 (SDC-1) and activation of STAT3
(A) GM6001 pre-treatment inhibiting the shedding of SDC-1 in the ischemia/reperfusion (I/R) mice, n = 6.
(B–D) The serum creatinine (SCr) and renal pathological injury were alleviated after GM6001 pre-treatment in the I/R mice, Bar = 50μm, n = 6.
(E and F) Protein expression of STAT3 and E-cadherin after inhibition of SDC-1 shedding in the I/R mice, n = 5–6.
(G) Histochemical staining of E-cadherin expression (n = 4) and TUNEL staining (n = 3) of apoptotic tubular epithelial cells in mice kidneys from the GM6001+I/R and DMSO+I/R group, Bar = 50μm.
(H) The statistical graph of the TUNEL result.
(I) The intracellular junctions between tubular epithelial cells in the I/R mice shown using TEM, n = 2, Bar = 1μm. ∗p < 0.05, ∗∗p < 0.01 vs. DMSO+I/R group.
Tubule selective knockout of SDC-1 aggravated the I/R injury and disrupted cell junctions in mice kidneys
We successfully constructed mice with selective SDC-1 gene knockouts in renal tubular epithelial cells (Figure S2). We induced a mild renal I/R insult with ischemia for 20 min and reperfusion for 12 h in the SDC-1fL/fl/Cre+ and SDC-1fL/fL/Cre− genotype mice. The results showed that mild renal I/R could still cause higher SCr (Figure 3C) and worse histopathological injury (Figures 3A and 3B) in the SDC-1fL/fl/Cre+ mice than those in the SDC-1fL/fL/Cre− group. Additionally, there was no difference in the level of SCr between the SDC-1fL/fL/Cre− and the SDC-1fL/fl/Cre+ mice in the sham group, suggesting that renal tubule-specific knockout of SDC-1 did not affect the baseline renal function (Figure 3C). Meanwhile, compared with the SDC-1fL/fL/Cre− group, there was decreased expression of E-cadherin and activation of STAT3 in the SDC-1fL/fl/Cre+ group (Figures 3D–3F). Meanwhile, the apoptotic response in the kidneys in the SDC-1fL/fl/Cre+ group was severe compared to that of the SDC-1fL/fL/Cre− group mice (Figures 3G and 3H), with more damage to the cell junctions shown in the TEM (Figure 3I).
Figure 3.

The effect of tubule selective syndecan-1 (SDC-1) knockout on renal I/R injury, intracellular junctions and activation of STAT3
(A and B) The renal histopathological injury was aggravated in mild ischemia/reperfusion (I/R) injury in the SDC-1fL/fl/Cre+ mice, Bar, 50μm, n = 4.
(C) Mild I/R injury could result in significantly increased serum creatinine (SCr) in the SDC-1fL/fl/Cre+ mice, n = 4.
(D and E) The activation of STAT3 was detected in SDC-1fL/fl/Cre+ mice after mild I/R injury, n = 3–4.
(F) The expression of E-cadherin was decreased in SDC-1fL/fl/Cre+ I/R mice, n = 3–4.
(G and H) the apoptotic cells were increased by mild I/R injury in the SDC-1fL/fl/Cre+ I/R mice, n = 3.
(I) The damaged intracellular junctions of tubular cells by mild I/R injury in SDC-1fL/fL mice kidneys were detected by TEM, n = 2, Bar, 1μm. ∗p < 0.05.
SDC-1 was involved in the E-cadherin expression through STAT3 activation in the renal tubular epithelial cells
We treated PTECs with hypoxia for 24 h and then normoxia for 8 h according our previous study.10 Our results showed that the hypoxia/reoxygenation (H/R) caused apoptosis marker, the ratio of Bax/Bcl2 and the expression of cleaved caspase-3 increased (Figure 4A), and upregulation of pSTAT3 expression and down-regulation of E-cadherin expression in the PTECs (Figure 4B). While GM6001 pretreatment could decrease the ratio of Bax/Bcl2 and the expression of cleaved caspase-3 (Figure 4C), and inhibited the activation of STAT3 and increased the expression of E-cadherin (Figure 4D), which culminated in the inhibition of SDC-1 shedding. Furthermore, S3I-201 and AG490 could inhibit phosphorylation of STAT3 and then prevent the decreased E-cadherin expression induced by H/R insult (Figures 4E and 4F), which implied that activated STAT3 signaling could be involved in the regulation of E-cadherin expression in the tubular epithelial cells under H/R condition.
Figure 4.

Down-regulation of E-cadherin was induced by shedding of syndecan-1 (SDC-1) and activation of STAT3 signaling in hypoxia/reoxygenation (H/R) treated proximal tubular epithelial cells (PTECs)
(A) H/R resulted in the increased apoptosis in PTES, ∗p < 0.05.
(B) H/R could result in the phosphorylation of STAT3 and decreased expression of E-cadherin in the PTECs, ∗p < 0.05.
(C) GM6001 pretreatment could alleviate the apoptosis of PTES, ∗p < 0.05.
(D) Western blot analysis showed that the activation of STAT3 was suppressed. Decreased E-cadherin was alleviated after inhibition of the SDC-1 shedding by GM6001. ∗p < 0.05, ∗∗p < 0.01.
(E) Expression of E-cadherin was increased after S3I-201 (5μM) treatment under H/R condition. ∗p < 0.05 vs. DMSO+H/R group.
(F) AG490 (100μM) was also used to inhibit the activation of STAT3, which could restore the decreased expression of E-cadherin in the PTECs, ∗p < 0.05 vs. DMSO+H/R group, n = 3–4.
We further confirmed the role of SDC-1 in the expression of E-cadherin in the 3D tubules with Madin-Daby canine kidney (MDCK) cells. It was shown that SDC-1 was mainly expressed in the basolateral membrane, while E-cadherin was expressed in the lateral membrane (Figure 5A). The expression of E-cadherin was decreased (Figure 5B) after the knockdown of SDC-1 by siRNA in the 3D tubules (Figure S5). Furthermore, there was lower expression of SDC-1 and E-cadherin in the renal tubular epithelial cell from the patients pathologically diagnosed with acute tubular necrosis (ATN) than those from patients with minor lesion under light microscope (Figure 5C).
Figure 5.

The relationship between syndecan-1 (SDC-1) and E-cadherin in the three dimensional (3D) tubule and ATN patients
(A) The location of SDC-1 and E-cadherin in the 3D tubule were detected by confocal microscopy, Bar = 20μm.
(B) Knockdown of SDC-1 expression by siRNA resulted in the decreased expression of E-cadherin.
(C) The lower expression of SDC-1 and E-cadherin were detected in the kidneys from the ATN patient compared with that from the minor lesion patient, Bar = 50μm.
Discussion
AKI is a common critical disease involving all clinical departments, and I/R injury is the most common cause of hospitalization AKI, which can increase the incidence and death rate of AKI. In various diseases such as heart and liver surgery, shock, and kidney transplantation, renal I/R injury is induced by renal blood flow interruption and then reperfusion,14 which results in the incidence of perioperative AKI.
Cell junctions among tubular epithelial cells are involved in maintaining the function, polarity, and morphology of the kidney, which contributes to the overall renal physiology.15 The loss of intracellular junctions among the tubular epithelial cells, due to the loss of energy in the process of ischemic AKI, could result in the apoptosis, shedding of epithelial cells, and the subsequent degradation of tubular integrity.1 Therefore, the restoration of intracellular junctions in the tubule could be considered as an important treatment for AKI.16 Limited studies have focused on the overall structural integrity of renal tubules in this pathological process. In this study, we determined the effects of SDC-1 shedding on the structural integrity of renal tubules in ischemic AKI, as well as the underlying mechanism.
The transmembrane glycoprotein SDC-1 is involved in regulating a variety of diseases, by binding to mediators through its extracellular chain of heparin sulfate. There have been several studies regarding the role of SDC-1 in AKI. In rat renal I/R injury, it was shown that increased shedding of SDC-1 was positively correlated with the injury index of renal function.17 In our previous study, we found that shedding of SDC-1 was potentially involved in apoptosis and inflammation in ischemic renal injury while the inhibition of SDC-1 shedding could protect against I/R injury.13 Furthermore, elevated SDC-1 in plasma was related with progressive AKI in the patients after cardiac surgery.18 Considering the vital role of SDC-1 in cell morphology, we have confirmed that SDC-1 played significant role in maintaining cell polarity of tubular epithelial cells in renal I/R injury.10 We further investigated the function of SDC-1 in maintaining cell junctions in ischemic AKI in this study.
Several studies have been described the role of E-cadherin in AKI. In cisplatin-induced AKI, the maintenance and preservation of E-cadherin was considered to have a protective role against cisplatin-induced apoptosis of epithelial cells.19 Furthermore, cisplatin-induced AKIs resulted in the decreased expression of E-cadherin also triggered an inflammation response.16 In ischemic AKI, it was found that the long non-coding RNA TUG1 inhibited apoptosis and played a protective role in kidneys by positively regulating the expression of E-cadherin.20 These studies suggested that reduced E-cadherin expression could play a protective role in AKI through inhibiting the inflammation response and apoptosis. As a hallmark of adhesive junctions, the decreased expression of E-cadherin has been reported to be involved in initiating the process of epithelial mesenchymal transition.21
SDC-1 might participate in the regulation of E-cadherin expression in breast cancer malignant tumor cells22 and NMuMg cells,12 however, without clear mechanism. In our study, we found that the increased shedding of SDC-1 would result in the decreased expression of E-cadherin, which might be regulated through the STAT3. STAT is a family of protein with multiple highly conserved domains.7 As an important member of the STAT family, STAT3 could be activated by several upstream factors, including cytokines, chemokines, and growth factors.23 The role of STAT3 in AKI has thus far been inconclusive. Some studies suggested that the inhibition of STAT3 activation in ischemic AKI could play a protective role in renal I/R injury, which was mainly due to the role of STAT3 in mediating the immune inflammatory response by promoting T cells differentiating toward Th17 subsets.24 In rats with I/R injury, blocking the JAK/STAT pathway could reduce the recruitment of macrophages and play a protective role in the kidney.25 Activated STAT3 could lead to aggravated kidney damage, and nicotine was found to play a protective role in the kidney by inhibiting STAT3 activation.26 However, some researchers have suggested a protective role of STAT3 activation in ischemic AKI. For example, activated STAT3 has been reported to protect PTECs by inhibiting apoptosis in ischemic AKIs27 and nephrotoxic injury.28 In IL-22 overexpression mice, the activation of STAT3 could reduce the apoptotic damage of the kidney during I/R, and thus play a protective role in the kidney.29
In conclusion, SDC-1 shedding induced by I/R injury could lead to the inhibition of E-cadherin via STAT3 activation. Inhibiting the shedding of SDC-1 could reduce the activation of STAT3, thereby alleviating the downregulation of E-cadherin and subsequent renal dysfunction.
Limitations of the study
The sample size of renal biopsy specimens included was limited due to ethic limitation. Moreover, inhibiting the activation of STAT3 might be better to be evaluated in the SDC-1fL/fl/Cre+ mice and worthy of further study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Syndecan-1 antibody [EPR6454] | Abcam | Cat# ab128936; RRID:AB_11150990 |
| Beta Actin antibody | Genetxt | Cat#109639; RRID:AB_1949572 |
| BAX Polyclonal antibody | Proteintech | Cat#50599-2-Ig; RRID:AB_2061561 |
| Anti-Bcl-2 antibody | Abcam | Cat#ab182858; RRID: AB_2715467 |
| Caspase 3/p17/p19 Polyclonal antibody | Proteintech | Cat# 19677-1-AP; RRID: AB_10733244 |
| Anti-STAT3(phospho Y705) antibody [EP2147Y] | Abcam | Cat# ab76315; RRID:AB_1658549 |
| Anti-STAT3 [EPR787Y] antibody | Abcam | Cat# ab68153; RRID:AB_2889877 |
| Anti-Aquaporin 1 antibody [ERP11588(B)] | Abcam | Cat# ab168387; RRID:AB_2810992 |
| Purified Mouse Anti-E-Cadherin | BD bioscience | Cat#: 610181; RRID: AB_397580 |
| Biological samples | ||
| Human kidney samples | Zhongshan hospital, Fudan university | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel matrix | Corning | Cat#:354277 |
| GM6001 | Abcam | Cat#: ab120845 |
| S3I-201 | Selleck | Cat#: S1155 |
| AG490 | Selleck | Cat#: S1143 |
| Trizol | Invitrogen | Cat#: 15596018 |
| Critical commercial assays | ||
| PrimeScriptä RT Master Mix | Takara | Cat#: RR036A |
| TB Greenä Premix Ex Taqä II (Tli RNaseH Plus) | Takara | Cat#: RR820A |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | Cat#: L3000001 |
| Mouse SDC-1 ELISA kit | Diaclone | Cat#: 860.090.096 |
| Pierce ECL Western blotting substrate | Thermo fisher Scientific | Cat#:32209 |
| Quantichrom™ Creatinine Assay Kit | Bioassay System | Cat#: DICT-500 |
| Experimental models: Organisms/strains | ||
| SDC-1flox/flox | Shanghai Southern Model Biological | N/A |
| Cdh16cre | Shanghai Southern Model Biological | N/A |
| Oligonucleotides | ||
| Primers see method details | This paper | N/A |
| Software and algorithms | ||
| ImageJ | ImageJ software | Version 1.51 |
| Graphpad Prism | Graphpad software | Version |
| Photoshop | Adobe | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xialian Xu (xu.xialian@zs-hospital.sh.cn).
Materials availability
This study did not generate new unique materials.
Experimental model and study participant details
Animals
Male C57BL/6J mice, 6–8 weeks of age and weighing 19 g to 22 g, were approved by the Institutional Animal Care and Use Committee of Fudan University in a clean environment. Animal experiment procedures were strictly complied with the standards of the Shanghai Animal Experiment Management Committee. The mouse I/R model was constructed as described below.30 Briefly, bilateral renal pedicles were clamped with vascular clips for 35 minutes in wild-type (WT) mice, and the body temperature was maintained at 36°C to 37°C throughout the entire operation. In sham operation group, the abdomen was sutured immediately after renal pedicle separation without clamping. In the I/R+GM6001 group, GM6001 was intraperitoneally injected into the mice 18h before the surgery for a dosage of 100 mg/kg14.
SDC-1fl/fl mice and CDH16-Cre mice were purchased from Shanghai Southern Model Biological. SDC-1fl/fl mice with C57BL/6 background were constructed using CRISPR-Cas9 technology. SDC-1fl/fl mice and CDH16-Cre mice were hybridized to produce a tubule-specific knockout of SDC-1 mice (SDC-1fl/fl/Cre+). Floxed SDC-1 homozygous mice not recognized by CDH16-Cre recombinase (SDC-1fl/fl/Cre-) were used as controls. DNA was extracted from mice by clipping their tails and detected by PCR to verify the genotype. SDC-1fl/fl and WT mice were distinguished with primer sequences 5'-TATCCAGCTGCAAGCCACTC-3' and 5'-GCTCTCCCCATTCTTCTGGG-3'. The primers 5'-GCAGATCTGGCTCTCCAAAG-3' and 5'-AGGCAAATTTTGGTGTACGG-3' were used to detect CDH16-Cre.
To investigate the effect of tubular specific knockout of SDC-1 on renal ischemia/ hypoxia injury, SDC-1fl/fl/Cre+ and SDC-1fl/fl/Cre- mice were randomly divided into sham group and I/R group.A mild I/R renal injury model was established in the SDC-1 KO mice. Briefly, bilateral renal pedicles were clipped for 20 min, and the mice were sacrificed 12h after reperfusion.
Cell culture and treatment
The primary mouse proximal tubular cells (PTECs) were cultured as described below.31 Briefly, the kidneys were harvested form male mice (6-8 weeks), the renal cortex were isolated and digested with an enzyme solution containing collagenase and a trypsin inhibitor. The isolated single cells were transferred to 50% percoll (Coolber) for gradient centrifugation, the PTECs were located in the uppermost fraction.
For hypoxia/reoxygenation (H/R) treatment, the PTECs were treated in hypoxic conditions with 1% O2, 5% CO2 or 94% N2 for 24 h and then transferred to normoxic atmosphere containing 21% O2 for 8 h. S3I-201(inhibiting the DNA-binding activity of STAT3) and AG490 (blocking JAK2 protein to inhibit the activation of the downstream molecule STAT3) were used to inhibit the activation of STAT3 in H/R treated PTECs, respectively.
We used MDCK cells for 3D culture. The cells were mixed with matrigel matrix (Corning) and then were cultured on a matrigel matrix-coated culture plate, when it formed a sphere, this was indicative that a 3D tubules were formed.
Patients
We enrolled one Chinese patient (male, 53 years old) pathologically diagnosed with acute tubular necrosis (ATN) shown as granular or vacuole-like degeneration of renal tubular epithelial cells, cell flattening, lumen dilation, brush edge shedding, severe cases of epithelial cells partially or completely shedding from the basement membrane, transparent, granular or cell tubular in the lumen, epithelial cell apoptosis under light microscope. The other Chinese patient (female, 52 years old) pathologically diagnosed with minor lesion whose serum creatinine were normal and proteinuria was 0.12g/24h. there were no obvious lesions under light microscope in the renal section, while diffuse fusion of foot processes of podocytes could be found under electron microscopy. This study was approved by the Ethics Committee of Zhongshan Hospital, Fudan University (B2021-067R), and informed consents were obtained from all patients.
Method details
Kidney histology and immunohistochemical staining
Harvested kidneys were fixed in 4% formalin, embedded in paraffin and cut into 4-um thick sections. Then, the Haematoxylin-Eosin (HE) staining was performed for injury scoring. The damaged tubules were scored as follows: no injury (0), less than 25% (1), less than 50% (2), less than 75% (3), and more than 75% (4).
Briefly, immunohistochemical staining of kidney slides were performed after dewaxing, antigen extraction and blocking, the sections were incubated with the antibody against E-cadherin (1:200), then incubated with the horseradish peroxidase (HRP)-conjugated secondary antibodies and were detected by a light microscopy.
Immunofluorescent staining
The cultured PTECs were fixed with 4% formalin, and then incubated with primary antibodies of aquaporin 1 (AQP1) (1:200), and then with the secondary antibody, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). the slides were observed with a fluorescence microscope.
Real-time PCR
Trizol was used to extract total RNA samples, and were transcribed into cDNA by using the PrimeScript reverse transcription Kit. The qRT-PCR process was conducted using SYBR TM Premix Ex Taq as per the manufacturer’s instructions on a 7500 real-time PCR System (Thermo Fisher Scientific, Pittsburgh, PA). The primer sequences of SDC-1 showed blow: forward 5'-AACGGGCCTCAACAGTCAG-3', reverse 5'-TGCGGATGAGATGTGA-3'.
Western blotting
The samples were collected with a mixture of radioimmunoprecipitation assay (RIPA) buffer containing phosphatase inhibitors and protease inhibitors, then the collected protein samples were boiled with loading buffer. The samples were loaded and subsequently separated on a sodium dodecyl sulfate-polyacrylamide gel, and transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was incubated at 4°C overnight with the primary antibody against Bcl-2 (1:1000), BAX (1:5000), Caspase3/C-caspase3 (1:1000), STAT3 (1:1000), phospho-STAT3 (1:1000), E-cadherin (1:1000), and SDC-1(1:1000). The membrane was then washed by Tris Buffered Saline with Tween (TBST), and incubated with HRP-labeled secondary antibody at room temperature for 1 h and then exposed on Image Lab software, version 3.0 (Bio-Rad, USA),and the relevant electrophoresis apparatus were purchased from Bio-Rad company as well The density of each protein blots was analyzed by Image Lab software, version 3.0 (Bio-Rad, USA). The results were normalized to β-actin.
Detection of serum creatinine
The blood of mice was collected, the serum was was then separated, and the serum creatinine (SCr) was determinate according to the protocol of Quantichrom™ Creatinine Assay Kit (BioAssay).
Enzyme-Linked Immunosorbent Assy
We used a commercial Elisa kit (Diaclone, Franch) and followed the manufacturer’s protocol to detect the serum levels of SDC-1, briefly, we harvested serum, and binding the samples and known standards with SDC-1 antibody coated microtiter strip plate, after incubating with secondary antibody, HRP conjugate solution and chromogen substrate successively, adding stop solution. The absorbance of the colour complex is measured, and according the generated OD values of each standard, we formed a standard curve. The concentration of SDC-1 in samples were tested by using standard curve.
TUNEL assay
The cell apoptosis in kidney were detected by using an In Situ Cell Death Detection Kit according to the manufacturer's protocol. We counted the TUNEL-positive cells through 6 random fields of each slide.
Quantification and statistical analysis
GraphPad Prism 9.0 (GraphPad Software, San Diego, CA) was used to perform statistical analysis on our data. Data were expressed as mean ± standard deviation. The normal distribution of data was verified by the Shapiro-Wilk normality test. Comparisons between two groups were performed with unpaired t test or the Mann-Whitney test according to the normal distribution of data. One-way ANOVA followed by a post-hoc test was used for multiple comparisons. All comparisons were two-tailed, P<0.05 was regarded as statistically significant.
Acknowledgments
This work supported by the Natural Science Foundation of Shanghai, China (23ZR1411200), the National Natural Science Foundation of China grants (82102289) to Wuhua Jiang and (81800596) to Shuan Zhao, Shanghai Kidney Disease Clinical Medical Center Construction Project(2017ZZ01015),Shanghai Key Laboratory of Kidney Disease and Blod Purification (14DZ2260200) and Shanghai Municipal Key Clinical Specialty (shslczdzk02501).
Author contributions
X.Q.D. and X.L.X. conceived and designed the study. M.G., D.Q.S., S.Y.Q., S.Z., J.R.X., and W.D.Z. performed the experiment. W.H.J., Y.Q.W., J.L.W., and X.M.G. helped to analysis the date. M.G. wrote the article. All authors have reviewed and approved the final version of article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108211.
Contributor Information
Xiaoqiang Ding, Email: ding.xiaoqiang@zs-hospital.sh.cn.
Xialian Xu, Email: xu.xialian@zs-hospital.sh.cn.
Supplemental information
Data and code availability
-
•
Data: All data reported in this paper will be shared by the lead contact upon request.
-
•
Code: This study did not report original code.
-
•
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Bonventre J.V., Yang L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 3.Onder T.T., Gupta P.B., Mani S.A., Yang J., Lander E.S., Weinberg R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.Can-07-2938. [DOI] [PubMed] [Google Scholar]
- 4.Cho E.A., Patterson L.T., Brookhiser W.T., Mah S., Kintner C., Dressler G.R. Differential expression and function of cadherin-6 during renal epithelium development. Development. 1998;125:803–812. doi: 10.1242/dev.125.5.803. [DOI] [PubMed] [Google Scholar]
- 5.Jia L., Liu F., Hansen S.H., Ter Beest M.B.A., Zegers M.M.P. Distinct roles of cadherin-6 and E-cadherin in tubulogenesis and lumen formation. Mol. Biol. Cell. 2011;22:2031–2041. doi: 10.1091/mbc.E11-01-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H., Pardoll D., Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pace J., Paladugu P., Das B., He J.C., Mallipattu S.K. Targeting STAT3 signaling in kidney disease. Am. J. Physiol. Ren. Physiol. 2019;316:F1151–F1161. doi: 10.1152/ajprenal.00034.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong H., Hong J., Du W., Lin Y.W., Ren L.L., Wang Y.C., Su W.Y., Wang J.L., Cui Y., Wang Z.H., Fang J.Y. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012;287:5819–5832. doi: 10.1074/jbc.M111.295964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong H., Zhang Z.G., Tian X.Q., Sun D.F., Liang Q.C., Zhang Y.J., Lu R., Chen Y.X., Fang J.Y. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia. 2008;10:287–297. doi: 10.1593/neo.07971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo M., Xu J., Zhao S., Shen D., Jiang W., Zhang L., Ding X., Xu X. Suppressing Syndecan-1 Shedding to Protect Against Renal Ischemia/Reperfusion Injury by Maintaining Polarity of Tubular Epithelial Cells. Shock. 2022;57:256–263. doi: 10.1097/shk.0000000000001838. [DOI] [PubMed] [Google Scholar]
- 11.Teng Y.H.F., Aquino R.S., Park P.W. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31:3–16. doi: 10.1016/j.matbio.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couchman J.R. Syndecan-1 (CD138), Carcinomas and EMT. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22084227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Z., Song N., Shen B., Xu X., Fang Y., Shi Y., Ning Y., Hu J., Dai Y., Ding X., et al. Syndecan-1 Shedding Inhibition to Protect Against Ischemic Acute Kidney Injury Through HGF Target Signaling Pathway. Transplantation. 2018;102:e331–e344. doi: 10.1097/tp.0000000000002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonventre J.V., Weinberg J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003;14:2199–2210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 15.Dong Y., Simske J.S. Vertebrate Claudin/PMP22/EMP22/MP20 family protein TMEM47 regulates epithelial cell junction maturation and morphogenesis. Dev. Dynam. 2016;245:653–666. doi: 10.1002/dvdy.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao L., Liu M.M., Zang H.M., Ma Q.Y., Yang Q., Jiang L., Ren G.L., Li H.D., Wu W.F., Wang J.N., et al. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab. Invest. 2018;98:911–923. doi: 10.1038/s41374-018-0052-5. [DOI] [PubMed] [Google Scholar]
- 17.Qin L.L., Xue F., Yin F., Zhao J., Zhang K.Y. Expression of syndecan-1, PKC and VEGF in rats with acute kidney injury and correlation between syndecan-1 and renal function. Eur. Rev. Med. Pharmacol. Sci. 2020;24:12794–12801. doi: 10.26355/eurrev_202012_24180. [DOI] [PubMed] [Google Scholar]
- 18.Xu J., Jiang W., Li Y., Li H., Geng X., Chen X., Hu J., Shen B., Wang Y., Fang Y., et al. Association Between Syndecan-1, Fluid Overload, and Progressive Acute Kidney Injury After Adult Cardiac Surgery. Front. Med. 2021;8 doi: 10.3389/fmed.2021.648397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni J., Hou X., Wang X., Shi Y., Xu L., Zheng X., Liu N., Qiu A., Zhuang S. 3-deazaneplanocin A protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression. Cell Death Dis. 2019;10:355. doi: 10.1038/s41419-019-1589-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L., Xu J.Y., Tan H.B. LncRNA TUG1 regulates the development of ischemia-reperfusion mediated acute kidney injury through miR-494-3p/E-cadherin axis. J. Inflamm. 2021;18:12. doi: 10.1186/s12950-021-00278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong S.H.M., Fang C.M., Chuah L.H., Leong C.O., Ngai S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018;121:11–22. doi: 10.1016/j.critrevonc.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Leppä S., Vleminckx K., Van Roy F., Jalkanen M. Syndecan-1 expression in mammary epithelial tumor cells is E-cadherin-dependent. J. Cell Sci. 1996;109:1393–1403. doi: 10.1242/jcs.109.6.1393. [DOI] [PubMed] [Google Scholar]
- 23.Heinrich P.C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J.W., Bae E., Kwon S.H., Yu M.Y., Cha R.H., Lee H., Kim D.K., Lee J.P., Ye S.K., Yoo J.Y., et al. Transcriptional modulation of the T helper 17/interleukin 17 axis ameliorates renal ischemia-reperfusion injury. Nephrol. Dial. Transplant. 2019;34:1481–1498. doi: 10.1093/ndt/gfy370. [DOI] [PubMed] [Google Scholar]
- 25.Yang N., Luo M., Li R., Huang Y., Zhang R., Wu Q., Wang F., Li Y., Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol. Dial. Transplant. 2008;23:91–100. doi: 10.1093/ndt/gfm509. [DOI] [PubMed] [Google Scholar]
- 26.Yeboah M.M., Xue X., Javdan M., Susin M., Metz C.N. Nicotinic acetylcholine receptor expression and regulation in the rat kidney after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2008;295:F654–F661. doi: 10.1152/ajprenal.90255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J., Ouyang C., Chen X., Fu B., Lu Y., Hong Q. STAT3 inhibits apoptosis of human renal tubular epithelial cells induced by ATP depletion/recovery. Nephron Exp. Nephrol. 2008;108:e11–e18. doi: 10.1159/000112557. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L., Fu P., Huang X.R., Liu F., Lai K.N., Lan H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010;21:31–41. doi: 10.1681/asn.2008111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu M.J., Feng D., Wang H., Guan Y., Yan X., Gao B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. J. Am. Soc. Nephrol. 2014;25:967–977. doi: 10.1681/asn.2013060611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X., Kriegel A.J., Liu Y., Usa K., Mladinov D., Liu H., Fang Y., Ding X., Liang M. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012;82:1167–1175. doi: 10.1038/ki.2012.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S., Jia P., Fang Y., Jin J., Sun Z., Zhou W., Li J., Zhang Y., Wang X., Ren T., et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022;101:987–1002. doi: 10.1016/j.kint.2022.01.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: All data reported in this paper will be shared by the lead contact upon request.
-
•
Code: This study did not report original code.
-
•
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.