

Key Points
Question
Do apolipoprotein E ε4 (ApoE4) carriers show accelerated amyloid-related tau spreading?
Findings
In this cohort study of 2 longitudinal tau positron emission tomography samples (N = 367), ApoE4 carriers showed an acceleration of amyloid-mediated tau spreading at a lower amyloid threshold compared to ApoE4 noncarriers, controlling for age and sex.
Meaning
ApoE4 carriage was associated with earlier amyloid-induced tau spreading, indicating that the timing of therapeutic windows in antiamyloid therapies may need special consideration in ApoE4 carriers compared to noncarriers to successfully attenuate tau spreading.
This cohort study evaluates amyloid-related tau spreading in apolipoprotein E ε4 carriers and amyloid positron emission tomography thresholds at which tau spreading may accelerate.
Abstract
Importance
For the Alzheimer disease (AD) therapies to effectively attenuate clinical progression, it may be critical to intervene before the onset of amyloid-associated tau spreading, which drives neurodegeneration and cognitive decline. Time points at which amyloid-associated tau spreading accelerates may depend on individual risk factors, such as apolipoprotein E ε4 (ApoE4) carriership, which is linked to faster disease progression; however, the association of ApoE4 with amyloid-related tau spreading is unclear.
Objective
To assess if ApoE4 carriers show accelerated amyloid-related tau spreading and propose amyloid positron emission tomography (PET) thresholds at which tau spreading accelerates in ApoE4 carriers vs noncarriers.
Design, Setting, and Participants
This cohort study including combined ApoE genotyping, amyloid PET, and longitudinal tau PET from 2 independent samples: the Alzheimer’s Disease Neuroimaging Initiative (ADNI; n = 237; collected from April 2015 to August 2022) and Avid-A05 (n = 130; collected from December 2013 to July 2017) with a mean (SD) tau PET follow-up time of 1.9 (0.96) years in ADNI and 1.4 (0.23) years in Avid-A05. ADNI is an observational multicenter Alzheimer disease neuroimaging initiative and Avid-A05 an observational clinical trial. Participants classified as cognitively normal (152 in ADNI and 77 in Avid-A05) or mildly cognitively impaired (107 in ADNI and 53 in Avid-A05) were selected based on ApoE genotyping, amyloid-PET, and longitudinal tau PET data availability. Participants with ApoE ε2/ε4 genotype or classified as having dementia were excluded. Resting-state functional magnetic resonance imaging connectivity templates were based on 42 healthy participants in ADNI.
Main Outcomes and Measures
Mediation of amyloid PET on the association between ApoE4 status and subsequent tau PET increase through Braak stage regions and interaction between ApoE4 status and amyloid PET with annual tau PET increase through Braak stage regions and connectivity-based spreading stages (tau epicenter connectivity ranked regions).
Results
The mean (SD) age was 73.9 (7.35) years among the 237 ADNI participants and 70.2 (9.7) years among the 130 Avid-A05 participants. A total of 107 individuals in ADNI (45.1%) and 45 in Avid-A05 (34.6%) were ApoE4 carriers. Across both samples, we found that higher amyloid PET–mediated ApoE4-related tau PET increased globally (ADNI b, 0.15; 95% CI, 0.05-0.28; P = .001 and Avid-A05 b, 0.33; 95% CI, 0.14-0.54; P < .001) and in earlier Braak regions. Further, we found a significant association between ApoE4 status by amyloid PET interaction and annual tau PET increases consistently through early Braak- and connectivity-based stages where amyloid-related tau accumulation was accelerated in ApoE4carriers vs noncarriers at lower centiloid thresholds, corrected for age and sex.
Conclusions and Relevance
The findings in this study indicate that amyloid-related tau accumulation was accelerated in ApoE4 carriers at lower amyloid levels, suggesting that ApoE4 may facilitate earlier amyloid-driven tau spreading across connected brain regions. Possible therapeutic implications might be further investigated to determine when best to prevent tau spreading and thus cognitive decline depending on ApoE4 status.
Introduction
In Alzheimer disease (AD), amyloid-β (Aβ) is thought to initiate the spreading of neurofibrillary tau pathology1 from temporal-lobe epicenters to connected cortical regions,2,3,4,5,6 driving neurodegeneration and cognitive decline.7,8,9,10 Accordingly, Aβ-targeting therapies should ideally be applied at low tau levels to efficiently attenuate the AD cascade and slow tau-related neurodegeneration11 and clinical progression.9,12 It is therefore crucial to determine Aβ thresholds at which tau spreading is triggered to potentially inform treatment decisions.9
However, tau spreading from the medial temporal lobe to the cortex is modulated by patient-specific factors, including sex,13,14 vascular comorbidities,15 brain network architecture,16 and genetic predispositions. Aβ thresholds marking tau spreading may vary individually.17,18,19 Carriage of the apolipoprotein E ε4 (ApoE4) allele,20 the strongest known risk factor for sporadic AD, has been linked to abnormal Aβ-independent tau biomarker levels19,21,22 and cortical tau spreading patterns that closely align with cerebral APOE messenger RNA expression.23 Yet, ApoE4 carriage is neither linked to higher risk of developing primary tauopathies nor to spreading of age-related medial temporal lobe tau to the cortex in the absence of Aβ24,25; therefore, tau spreading in ApoE4 carriers is seemingly associated with Aβ. However, it is unclear whether ApoE4 carriage lowers the Aβ threshold for tau spreading, leading to earlier symptom onset and faster clinical progression.19,26 Given that clinical AD progression is thought to be largely driven by tau rather than Aβ,9,10 anti-Aβ interventions may require earlier intervention within disease progression in ApoE4 carriers to effectively intercept tau spreading and consequent cognitive deterioration.27 Addressing this is critical since 40% to 60% of patients with sporadic AD carry at least 1 ApoE4 allele28 and will likely seek antiamyloid treatment at some point.
The major goal of this study was to investigate whether ApoE4 carriage is associated with earlier and faster Aβ-related tau spreading throughout the cortex. We assessed ApoE4 status, baseline [18F]florbetaben/florbetapir amyloid-positron emission tomography (PET), and longitudinal [18F]flortaucipir tau PET in individuals without dementia across the spectrum of aging and AD, including patients at the earliest stages of amyloidosis—a potentially promising target group for anti-Aβ treatments owing to less progressed pathology than that typically found in patients with dementia in which tau accumulation is more likely driven by local replication rather than Aβ-related transneuronal spread.29,30,31 Samples were taken from the Alzheimer’s Disease Neuroimaging Initiative (ADNI; n = 237) and Avid-A05 (n = 130) to independently validate findings. To capture AD-related tau aggregation and spread via tau PET, we used Braak stage–specific readouts, temporal-lobe tau meta regions of interest (ROIs),32 and individualized connectivity-based tau stages (Q1-Q4), which specifically capture the gradual spread of tau across connected brain regions.3,29 Using these data, we first assessed whether faster tau accumulation in ApoE4 carriers was associated with higher Aβ and second, whether tau accumulation was accelerated at lower Aβ levels in ApoE4 carriers vs noncarriers. Based on this analysis, we determined PET-based Aβ cut points at which tau accumulation rates diverged between ApoE4 carriers vs noncarriers. Third, we ran simulated trials to determine whether therapeutic effects on tau accumulation can be detected at lower Aβ levels in ApoE4 carriers vs noncarriers.
Methods
ADNI Participants
We included 237 ADNI participants without dementia with at least 2 [18F]flortaucipir tau PET scans and baseline [18F]florbetapir (n = 175) or [18F]florbetaben (n = 62) amyloid PET within 6 months of the baseline tau PET. Centiloids were estimated from global [18F]florbetapir and [18F]florbetaben standardized uptake value ratios (SUVRs) according to ADNI guidelines.68 ADNI investigators diagnosed patients as cognitively normal (Mini-Mental State Examination [MMSE] score ≥24, Clinical Dementia Rating [CDR] score of 0, and not depressed), mild cognitive impairment (MMSE score ≥24, CDR score of 0.5, objective memory impairment on education-adjusted Wechsler Memory Scale II, and preserved activities of daily living). Participants with the ApoE ε2/ε4 genotype were excluded to avoid confounding effects of the potentially protective ε2 allele.33 ADNI investigators obtained ethical approval from their respective local institutional ethical review board; all participants provided written informed consent. Supplementary analyses included an additional 34 patients diagnosed with dementia (MMSE score of 20-26, CDR score ≥0.5, National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association criteria for probable AD) fulfilling the same PET data availability criteria (21 with [18F]florbetapir and 13 with [18F]florbetaben).
Avid-A05 Participants
A total of 130 patients without dementia were selected from the Avid-1451-A05 phase 2/3 trial,69,70 with at least 2 [18F]flortaucipir tau PET scans and 1 [18F]florbetapir amyloid PET scan within 30 days of the initial tau PET scan. All tau PET follow-up scans were taken at fixed intervals (9 and 18 months). Centiloid values were estimated from global [18F]florbetapir SUVRs according to Avid guidelines.34 Participants were classified as cognitively normal (MMSE score ≥29, no history of cognitive impairment) or mildly cognitively impaired (MMSE score 24 to less than 29, showing mild cognitive impairment according to the National Institute on Aging and the Alzheimer's Association working group’s diagnostic guidelines35). Individuals with the ApoE ε2/ε4 genotype were excluded. The study was approved by each clinical investigator’s local institutional review board; all participants provided written informed consent. Supplementary analyses included an additional 35 patients diagnosed with dementia (MMSE score 10-24, showing possible or probable AD based on the National Institute on Aging and the Alzheimer's Association working group’s diagnostic guidelines) fulfilling the same PET data availability criteria. For transparency and generalizability, we have reported self-reported racial and ethnic distributions using predefined categories to depict the demographic composition of both groups.
Connectivity Assessment
Functional connectivity of 42 cognitively normal participants who were Aβ negative from the same ADNI sample was assessed using the 200 ROI Schaefer atlas36 as Fisher z–transformed Pearson moment correlations between ROI pairs. Participant-specific connectivity matrices were averaged to determine a connectivity template, and negative values and autocorrelations were set to 0, following our preestablished approach.37 All neuroimaging acquisition and processing is described in the eAppendix in Supplement 1. Participant-specific tau-spreading ROIs were generated by grouping 95% of regions into quartiles according to their template-based connectivity to 5% of brain regions with highest baseline tau PET (ie, the participant-specific tau epicenter, where Q1 indicates the strongest connectivity to the epicenter and Q4 indicates the weakest connectivity to the epicenter).3,16,29
Statistical Analysis
Group demographic characteristics were compared using analyses of variance for continuous and χ2 tests for categorical variables. ROI-specific annual tau PET change rates were estimated using linear mixed models with longitudinal tau PET SUVR values as the dependent variable and time from baseline as the independent variable, including random slope and intercept.3,16,29
To confirm previous evidence that ApoE4 carriers have elevated Aβ and faster tau accumulation, we assessed differences in baseline centiloids and global tau PET change rates between ApoE4 carriers and noncarriers using analyses of covariance controlling for age and sex.19
To test our main hypothesis that ApoE4 carriers show accelerated Aβ-related tau accumulation and spread, we first investigated whether faster ApoE4-related tau accumulation was associated with higher Aβ. Bootstrapped mediation models with 1000 iterations conducted in R version 4.2.2 (R Foundation) were fitted for both samples separately with ApoE4 status as the independent variable, centiloids as the mediator, and the annual rate of global tau PET change as the dependent variable (ie, the average tau PET change across all 200 cortical Schaefer ROIs). These models were additionally stratified by Braak stage to determine whether ApoE4 associations with tau accumulation were most present in early tau-susceptible regions. All mediation models were controlled for age and sex, and a significance threshold was set at P ≤ .05.
To further assess whether ApoE4 was associated with an acceleration of tau spreading at lower Aβ levels, we tested the centiloids by ApoE4 interaction on annual tau PET SUVR change rates through Braak and connectivity stage ROIs (Q1-Q4). In line with evidence for nonlinear progression of tau according to amyloid,38 quadratic interaction models fit the data better than linear interactions (according to Akaike information criterion and analyses of variance); therefore, we report quadratic regression models controlled for age and sex (eTable 2 in Supplement 1). Centiloid thresholds in which ApoE4 carrier and noncarrier tau accumulation trajectories diverged were defined in all regression models according to a nonparametric resampling technique, which involved identifying where 95% CIs of the regression lines diverged or reconverged on the y-axis averaged across 1000 bootstrapped regressions to ensure thresholds were robust and not influenced by specific observations. To explore whether ApoE4 was associated with an acceleration of tau spreading at lower Aβ levels across an extended Aβ spectrum, linear models with CI thresholds were repeated with additional participants diagnosed with dementia. All analyses were carried out in both ADNI and Avid-A05 participants.
Lastly, we tested in the larger ADNI sample with good coverage of patients with early-stage and preclinical AD whether the sensitivity for detecting potential treatment effects on tau accumulation was higher among ApoE4 carriers at lower Aβ levels. To this end, simulated interventions with attenuated tau PET change rates of 30% (ie, simulated effect of interest) were carried out for global tau PET increase or tau PET change rates in the temporal meta32 or Q1 ROI.3,29 Simulated interventions were performed using 2 approaches, by defining a 70-centiloid–wide window with an upper and lower boundary that was shifted from −20 centiloids to 140 centiloids using a sliding window approach vs defining a lower centiloid boundary for defining the sample of interest, which was systematically increased from −20 centiloids to 70 centiloids in increments of 10. The required sample sizes were estimated using the R package pwr (settings: 2-sample t test, comparing actual vs attenuated cognitive changes; 2-tailed α = .05, power = 0.8). Statistical analyses were computed using R statistical software version 4.0.2 (R Foundation). Our primary hypothesis-driven analyses were not controlled for multiple comparisons due to our independent validation approach across 2 samples, yet false discovery rate–corrected P values are also reported in Tables 1 and 2.39 The study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Table 1. Mediation Resultsa.
| b (95% CI)b | P value | |
|---|---|---|
| ADNI | ||
| Global | 0.15 (0.05 to 0.28) | .01c |
| Braak I | 0.03 (−0.09 to 0.14) | .68 |
| Braak III | 0.19 (0.07 to 0.32) | <.001c |
| Braak IV | 0.17 (0.06 to 0.30) | .01c |
| Braak V | 0.12 (0.01 to 0.24) | .03c |
| Braak VI | 0.09 (−0.02 to 0.21) | .11c |
| Avid-A05 | ||
| Global | 0.33 (0.14 to 0.54) | <.001c |
| Braak I | −0.02 (−0.17 to 0.13) | .78 |
| Braak III | 0.33 (0.14 to 0.54) | <.001c |
| Braak IV | 0.28 (0.11 to 0.48) | <.001c |
| Braak V | 0.34 (0.15 to 0.55) | <.001c |
| Braak VI | 0.27 (0.10 to 0.46) | <.001c |
Abbreviation: ADNI, Alzheimer’s Disease Neuroimaging Initiative.
The mediation models are controlled for age and sex. Additional mediation models correcting for diagnosis are shown in eTable 3 in Supplement 1.
Values are average causal mediation effect values derived from mediation analyses with apolipoprotein E ε4 risk as predictor, centiloid as mediator, and the annual tau standardized uptake value ratio rate of change in the respective Braak stage as the dependent variable.
Significant at false discovery rate–corrected P < .05.
Table 2. Interaction Effects Estimated by Linear Regressiona.
| b | t value | P valueb | Partial R2 | Mean (95% CI) | ||
|---|---|---|---|---|---|---|
| Lower cut point | Upper cut point | |||||
| Braak stage | ||||||
| ADNI | ||||||
| Braak III | −0.34 | −3.35 | <.001a | 0.05 | 16.10 (15.43 to 16.76) | 75.90 (75.05 to 76.75) |
| Braak IV | −0.36 | −3.51 | <.001a | 0.05 | 12.44 (11.74 to 13.15) | 74.20 (73.33 to 75.06) |
| Braak V | −0.28 | −2.64 | .009a | 0.03 | 12.43 (11.43 to 13.42) | 79.80 (78.76 to 80.84) |
| Braak VI | −0.18 | −1.63 | .104 | 0.01 | NA | NA |
| A05 | ||||||
| Braak III | −0.27 | −2.19 | .030 | 0.04 | 15.42 (14.14 to 16.70) | 60.47 (58.32 to 62.61) |
| Braak IV | −0.16 | −1.28 | .204 | 0.02 | NA | NA |
| Braak V | −0.26 | −2.23 | .028 | 0.05 | 11.03 (9.89 to 12.17) | 63.96 (61.82 to 66.10) |
| Braak VI | −0.09 | −0.71 | .480 | 0.03 | NA | NA |
| Connectivity stage | ||||||
| ADNI | ||||||
| Q1 | −0.39 | −3.78 | <.001a | 0.06 | 11.20 (10.61 to 11.80) | 83.60 (82.74 to 84.45) |
| Q2 | −0.27 | −2.66 | .008a | 0.03 | 13.29 (12.39 to 14.19) | 80.77 (79.72 to 81.83) |
| Q3 | −0.20 | −1.94 | .053 | 0.02 | NA | NA |
| Q4 | −0.14 | −1.26 | .208 | 0.01 | NA | NA |
| A05 | ||||||
| Q1 | −0.23 | −2.02 | .046 | 0.03 | 13.31 (12.25 to 14.37) | 69.70 (67.67 to 71.74) |
| Q2 | −0.31 | −2.67 | .008a | 0.07 | 11.93 (10.86 to 12.99) | 56.48 (54.38 to 58.57) |
| Q3 | −0.20 | −1.67 | .098 | 0.04 | NA | NA |
| Q4 | −0.02 | −0.15 | .884 | 0.02 | NA | NA |
Abbreviations: ADNI, Alzheimer’s Disease Neuroimaging Initiative; NA, not applicable.
Values derived from regressions fitted with the interaction effect of apolipoprotein E ε4 risk and centiloid on the rate of annual tau accumulation in respective Braak stages and connectivity stages in ADNI and Avid-A05. Lower and upper cut point means and CIs were estimated through selecting the point of no overlap and reoverlap of 95% confidence intervals derived from bootstrapped regressions. The regression models are controlled for age and sex. Additional regression models correcting for diagnosis are shown in eTable 4 in Supplement 1.
Significance at false discovery rate–corrected P < .05.
Results
Sample Characteristics
A total of 237 ADNI participants (mean [SD] age, 73.9 [7.35] years; 1 American Indian [0.4%], 1 Asian [0.4%], 13 Black [5.4%], 5 of more than 1 race or ethnicity [2.1%], and 217 White [91.6%]), including 107 ApoE4 carriers, were included. Analysis of variance and χ2 tests revealed no significant differences between sex, clinical status (cognitively normal vs mildly cognitively impaired), or years of education, but ApoE4 carriers had significantly lower MMSE scores, were significantly younger with a higher proportion meeting amyloid PET positivity thresholds,40,41 and had shorter tau PET follow-up. Of the 130 Avid-A05 participants (mean [SD] age, 70.2 [9.7] years; 2 Asian [1.5%], 12 Black [9.2%], 114 White [87.7%], and 2 other [unspecified] [1.5%]), 45 ApoE4 carriers were included. Sample characteristics were congruent with ADNI except for clinical status; there were significantly fewer cognitively normal ApoE4 carriers than cognitively normal noncarriers or moderately cognitively impaired ApoE4 carriers. Results are summarized in eTable 1 in Supplement 1. As expected, ApoE4 carriers had significantly higher centiloids (ADNI F1,232 = 45.12; P < .001; Avid-A05 F1,125 = 20.73; P < .001) and a faster annual rate of global tau PET SUVR accumulation (ADNI F1,232 = 15.47; P < .001; Avid-A05 F1,125 = 11.38; P < .001), controlling for age and sex. The demographic and clinical characteristics of additional participants with dementia added to supplementary analyses are displayed in eTable 5 in Supplement 1. Tau PET uptake and accumulation rates stratified by diagnostic group and ApoE4 status are shown in Figure 1 (for further stratification by Aβ-status, see eFigure 1 in Supplement 1).
Figure 1. Group Average Tau Positron Emission Tomography (PET) Standardized Uptake Value Ratios (SUVRs) at Baseline Stratified by Apolipoprotein E ε4 (ApoE4) Status and Diagnostic Group.

Tau PET SUVRs are shown as continuous values, white outlines define areas that surpass a preestablished pathological tau SUVR threshold of 1.344 in participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (A) and in the Avid-A05 study (C). Number of patients displayed under each group rendering; group average tau SUVR annual change rates defined by linear mixed models, stratified by ApoE4 carriership and diagnostic group in patients in ADNI (B) and those in the Avid-A05 study (D). Tukey honestly significant difference (95% CI) post hoc tests on region of interest–wise analysis of covariance reveal mean differences between tau PET SUVR values of ApoE4 carriers minus those of noncarriers (E) and their annual tau PET SUVR change (F) stratified by sample. FDR indicates false discovery rate.
Faster Tau Accumulation in ApoE4 Carriers Mediated by Higher Aβ
We first assessed whether higher ApoE4-conferred Aβ burden was associated with faster tau accumulation. Supporting this, bootstrapped mediation analyses revealed that the association between ApoE4 status and faster annual rate of tau PET SUVR accumulation was mediated by higher centiloids (ADNI average causal mediation effect: b, 0.15; 95% CI, 0.05-0.28; P = .001 and Avid-A05 ACME average causal mediation effect: b, 0.33; 95% CI, 0.14-0.54; P < .001) (Figure 2).
Figure 2. Global Mediation Analyses.
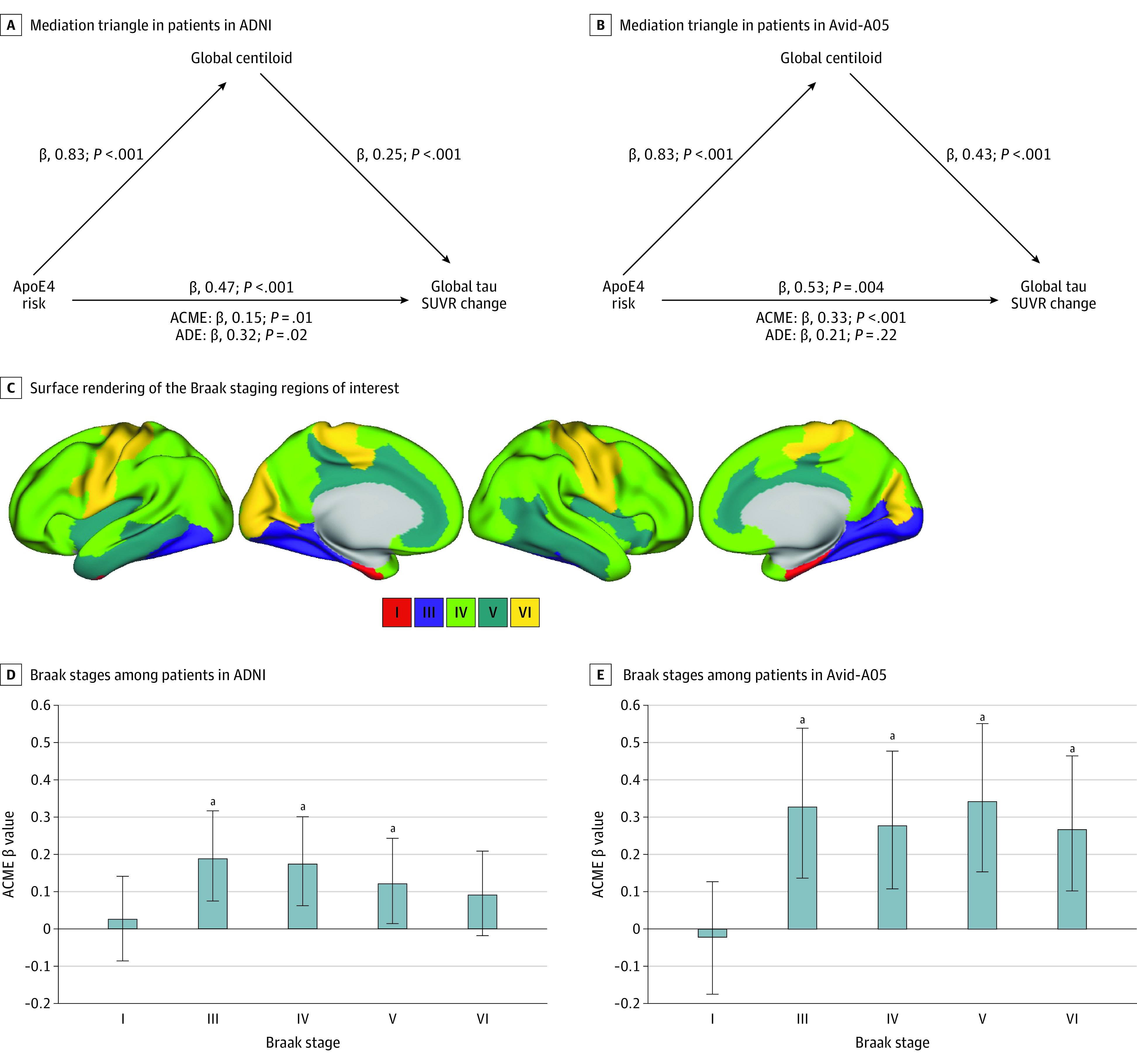
The average causal mediation effect (ACME) and the average direct effect (ADE) are displayed under each mediation triangle as estimated from bootstrapped mediation models. The models are controlled for age and sex and tested in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (A) and Avid-A05 (B). Surface rendering of the Braak staging regions of interest by which longitudinal tau positron emission tomography changes were determined (C). Bar charts present ACME b values of mediation analyses (apolipoprotein E ε4 [ApoE4] risk as predictor, amyloid standardized uptake value ratio (SUVR) as mediator, and the annual tau SUVR rate of change across Braak stages as the dependent variable). Error bars represent 95% CIs of mediation bootstrapping in ADNI (D) and Avid-A05 (E).
When repeating the mediation analysis stratified by Braak ROIs, we found no mediation of amyloid on ApoE4 risk on tau accumulation in Braak I, but found a mediation in Braak III followed by a weakening in subsequent Braak ROIs in both samples (Figure 2; Table 1). This suggests that ApoE4 carriage was not associated with an acceleration in the initial emergence of tau pathology in the earliest tau-vulnerable region, Braak I, but may propel amyloid-related tau accumulation particularly in cortical regions.1,9 For exploratory purposes, we also found no main effect of ApoE4 on tau accumulation in Braak I (ADNI: F = 2.95; P = .09; Avid-A05: F = 2.31; P = .13), suggesting the initial dynamics of tau are not driven by Aβ or ApoE. Sensitivity analyses also controlling for clinical diagnosis are shown in eTable 3 in Supplement 1.
ApoE4 Carriage Associated With a Lower Threshold for Amyloid-Related Tau Spreading
Second, we tested whether ApoE4 not only accelerates tau accumulation via increased amyloid (ie, mediation effect), but whether ApoE4 and amyloid have synergistic effects on accelerating tau accumulation as suggested by previous cross-sectional studies.42,43 Supporting this, we found significant centiloid–by–ApoE4 status interactions on annual tau accumulation rates for Braak ROIs III-V in ADNI and for Braak III and V in Avid-A05 (Figure 3). On average, tau trajectories diverged at approximately 15 centiloids for ADNI and at approximately 12 centiloids for Avid-A05, as determined by nonparametric resampling. This result pattern was consistent for connectivity-based tau stages, which better capture individualized tau spread. Here, significant ApoE4-by-centiloids interactions were found for Q1 AND Q2 in ADNI and avid-A05 (Figure 3), where ApoE4-related tau accumulation accelerated at approximately 13 centiloids for ADNI and approximately 12 centiloids for Avid-A05. The strength of the interaction effect became weaker across Q1 throughQ4, suggesting that ApoE4 carriage was specifically associated with accelerated tau spreading from patient-specific epicenters to closely connected regions (Table 2). Sensitivity analyses also controlling for clinical diagnosis are shown in eTable 4 in Supplement 1. Exploratory analyses also including participants with dementia (271 in ADNI and 165 in Avid-A05) yielded consistent results (eFigure 3 and eTable 6 in Supplement 1). Together, these results suggest that ApoE4 carriage facilitates tau spreading at lower amyloid thresholds. Linear regression models revealed further that higher annual rate of tau SUVR accumulation was associated with faster MMSE decline in ADNI (b, −0.32; P < .001) and Avid-A05 (b, −0.28; P = .002) (eTable 7 in Supplement 1). This suggests that earlier ApoE4-related tau accumulation in the presence of Aβ may translate into faster cognitive decline.
Figure 3. Interaction Effect Between Apolipoprotein E ε4 (ApoE4) Status and Centiloid on the Annual Rate of Tau Standardized Uptake Value Ratio (SUVR) Change Through Braak Stages III to VI .
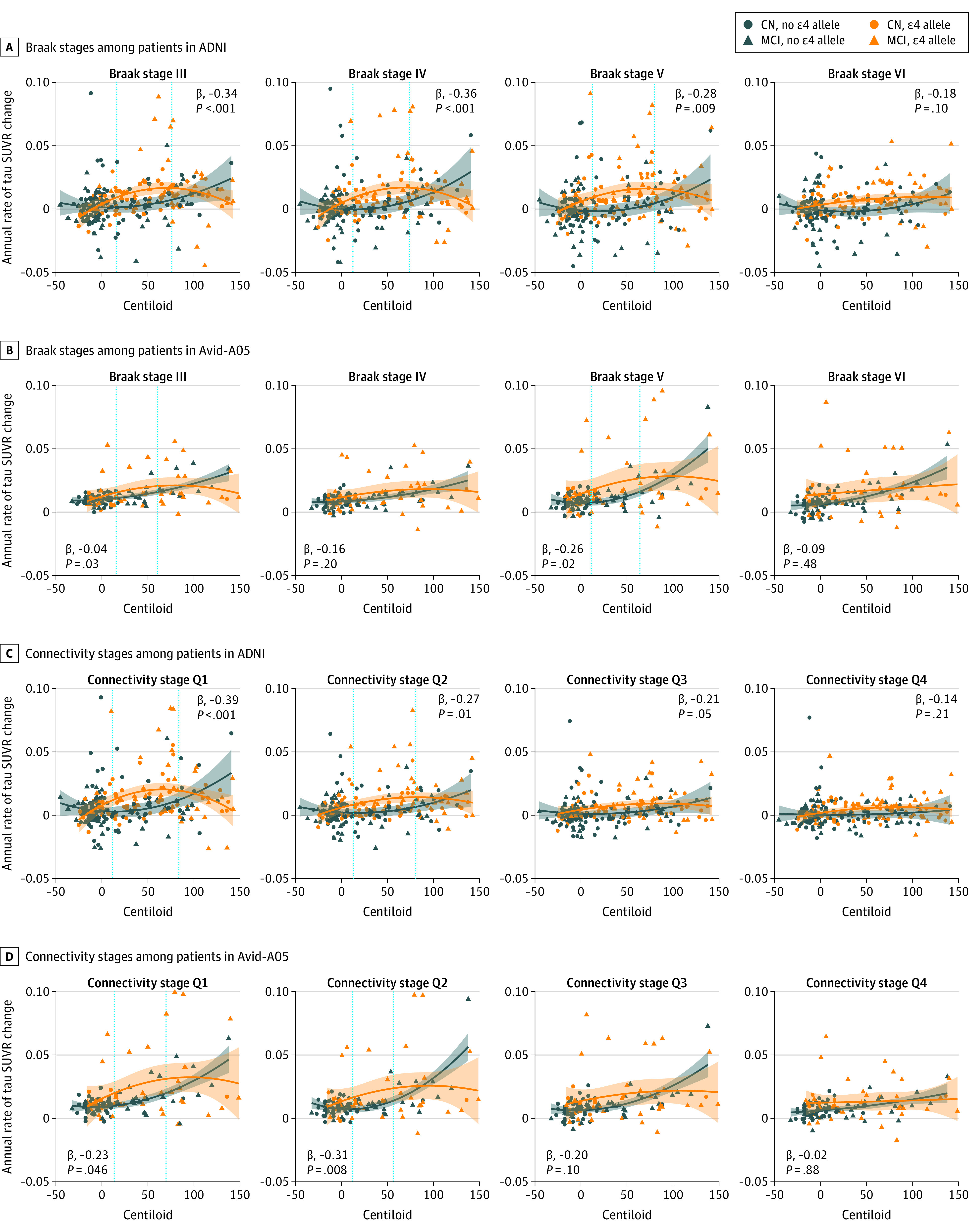
ApoE4 carriers show an amyloid-related increase in tau accumulation in early disease stages. Vertical dashed lines represent the centiloid threshold at which groups diverge and converge, estimated according to a nonparametric bootstrapping technique with 1000 iterations identifying the point at which confidence intervals around regression lines diverge and converge, presented with shaded 95% CI thresholds.
ApoE4 Associated With Higher Sensitivity to Detect Treatment-Related Tau Attenuation at Lower Amyloid Levels
Lastly, we assessed whether the sensitivity to detect therapeutic effects on tau accumulation at lower Aβ levels was higher in ApoE4 carriers. We computed required sample sizes to detect simulated intervention effects with 30% attenuation of tau PET change as an end point through global, temporal-meta, and Q1 ROIs in ADNI. When using a sliding window approach spanning 70 centiloids, we found that detecting tau attenuation as a treatment effect would require overall lower sample sizes in ApoE4 carriers compared to ApoE4 noncarriers (eFigure 2 in Supplement 1). For global and Q1 tau PET readouts, the sensitivity to detect treatment effects diverged particularly at approximately 10 centiloids, consistent with the previous analysis in which we showed ApoE4-related tau accumulation acceleration at this centiloid threshold (Figure 3). Congruent results were obtained when using a lower centiloid boundary approach (eFigure 2in Supplement 1), indicating that ApoE4 carrier inclusion in trials using tau PET as a surrogate end point can reduce sample sizes required to detect treatment effects, especially at lower Aβ levels. This exploratory analysis could not be reliably repeated in Avid-A05 owing to skewed sample sizes across the centiloid spectrum leading to biased power estimations.
Discussion
In this cohort study, we demonstrate an association between ApoE4 carriage and enhanced Aβ pathology that mediated faster tau accumulation across early Braak stage regions and an acceleration of cortical tau spreading in ApoE4 carriers at lower Aβ levels. This suggests a potential indirect and direct effect of ApoE4 on tau, first by driving Aβ accumulation (which triggers tau accumulation) and second by lowering the Aβ threshold at which tau spreading accelerates from local epicenters across connected regions. We found that Aβ-related tau trajectories diverged around 12 to 15 centiloids between ApoE4 carriers and noncarriers, at which neuritic plaque pathology is already observed postmortem,44 but below the typical 26-centiloid cutoff for amyloid PET positivity.45 This indicates that tau spreading may be triggered earlier in ApoE4 carriers and therefore may be beneficial to explore disease-modifying anti-Aβ treatments at lower Aβ levels. Supporting this, our simulated trials show that tau accumulation attenuation can be detected at lower Aβ levels in ApoE4 carriers, therefore encouraging earlier disease-modifying intervention in carriers of the strongest risk factor for developing sporadic AD.
Our first major finding that Aβ mediated faster tau-PET increase in ApoE4 carriers vs noncarriers provides evidence that accelerated tau progression in ApoE4 carriers may be partly driven by stronger Aβ deposition. Mediating effects of Aβ on the association between ApoE4 and accelerated tau accumulation were specifically found for cortical Braak stage regions but not for the entorhinal cortex (ie, Braak I), where age-related tau PET increase is also found in the absence of Aβ.46 This is in line with past research demonstrating that tau accumulation in the entorhinal cortex is not mediated by Aβ in ApoE4 carriers,47 suggesting that ApoE4 is specifically associated with Aβ-related cortical tau accumulation, but not to the initial entorhinal emergence of tau. This result pattern also provides an explanation for why ApoE4 carriers without Aβ pathology exhibit tau in the medial temporal lobe but seldom beyond this region and do not develop pure tauopathies.24,25
Our second main finding revealed that ApoE4 was not only associated with accelerated tau accumulation through higher Aβ levels, but also had possible synergistic effects with Aβ on tau spreading, congruent with cross-sectional evidence of higher tau PET at given level of Aβ in ApoE4 carriers.43 Specifically, we demonstrated that the rate of Aβ-related tau accumulation was moderated by ApoE4 across regions vulnerable to early-stage tau aggregation and spread (ie, in regions strongly connected to tau epicenters Q1-Q2).3,16,29 These results are congruent with a biphasic AD pathophysiological framework, which proposes an Aβ-dependent then a later Aβ-independent tau accumulation phase,30 supported by mouse model evidence demonstrating that Aβ-targeting antibodies reduced early but not later tau changes.48 ApoE4-related tau trajectories diverged at relatively low levels of around 12 to 15 centiloids consistently across both samples, suggesting that ApoE4 may influence tau accumulation before patients are Aβ-PET positive using commonly applied thresholds.44,45 This finding highlights the need for earlier centiloid-gauged therapeutic windows for ApoE4 carriers to effectively intervene in Aβ-related tau spreading.27
A key question is how Aβ facilitates tau spreading despite spatial incongruity and how ApoE4 modulates this process. Recent network connectivity research suggests that the onset of transneuronal tau spreading is subject to remote connectivity changes induced by the emergence of Aβ in regions connected to the entorhinal cortex49 (where tau typically emerges first). This process may be accelerated in Apoe4 carriers who, according to electrophysiological50 and functional magnetic resonance imaging (fMRI)51 evidence, exhibit hyperconnectivity in primary Aβ-harboring regions compared to noncarriers. Remote Aβ-induced hyperconnectivity to tau epicenters may facilitate its spread to the rest of the cortex in ApoE4 carriers. It is unclear why ApoE4 carriers exhibit increased neuronal hyperconnectivity, but recent neuroinflammation research reveals that ApoE4-related Aβ plaques are structurally less compact52 and trigger a greater inflammatory response,53 which may amplify its pathological effects. Further, recent work in transgenic mouse models has found that ApoE4 is associated with earlier Aβ seeding and a stronger Aβ-induced astrogliosis,54 and recent work in humans has shown that markers of astrocyte abnormality (ie, glial fibrillary acidic protein) are linked to a stronger association between Aβ and tau biomarkers.55 Therefore, ApoE4 may be associated with an earlier Aβ-induced astrogliosis, triggering earlier-onset tau spreading; however, this remains to be specifically tested.
Our exploratory analysis, in which we used simulated interventions across several Aβ-defined therapeutic windows and boundaries, illustrated that, compared to noncarriers, fewer ApoE4 carriers were required to detect intervention-related tau attenuation at lower Aβ levels. This analysis translates this study’s major findings into an interventional context by conveying how the rate and timing of Aβ-related tau accumulation and chosen readout region may impact clinical trial design and, consequently, meeting surrogate end points. Q1 exhibited the largest intergroup differences, possibly reflecting its sensitivity to capturing individualized tau PET accumulation.3 Importantly, this analysis is calculated according to observed tau accumulation rates and not specific to anti-Aβ intervention and thus does not consider when anti-Aβ treatment can no longer attenuate tau accumulation, which we predict would occur in line with the centiloid thresholds established in the previous analysis.3
We argue, as others have, that the current rigid approach of dichotomizing participants according to preestablished Aβ PET thresholds without considering patient-specific risk factors begs reconsideration.32 There are reports of Aβ-PET negative individuals with AD-like tau pathology56,57 who may exemplify patients who enter the AD spectrum while formally defined as Aβ-negative owing to accelerated tau spreading at lower Aβ levels. This concept may clarify why empirical research has identified increased medial temporal lobe tau in Aβ-negative ApoE4 carriers compared to noncarriers24,58 by proposing this increased tau to be Aβ related but at subthreshold levels. A novel aspect of the present study is including moderately cognitively impaired Aβ-negative patients, a group that inevitably includes patients without AD; however, this is statistically and conceptually necessary to detect the subthreshold Aβ-related tau spreading in individuals with AD.
Strengths and Limitations
A strength of the present study is the independent validation in the Avid-A05 sample, which conveyed overall congruent results. Nevertheless, cognitively normal ApoE4 carriers are underrepresented in Avid-A05, leading to slightly more advanced tau levels (Figure 1), which may explain why mediation and interaction effects tended to be skewed slightly toward later Braak stages. Furthermore, the use of individualized connectivity-based tau spreading stages can more sensitively capture spatial heterogeneity in tau accumulation, therefore better gauging the extent of tau spreading compared to Braak stage–specific readouts.59 Nevertheless, several caveats should be considered when interpreting our results. First, unspecific flortaucipir off target is commonplace,60 particularly in the hippocampus and basal ganglia—hence their exclusion from our analysis. However, we cannot confirm that confounding was not introduced from off-target binding elsewhere. Excluded regions, such as the hippocampus, may be particularly informative about early-stage tau spreading,61,62 which unfortunately we cannot explore until larger data with second-generation tau PET tracers become available to us. Second, tau accumulation modeling across connected regions relies on the accurate mapping of tau PET to resting state functional magnetic resonance imaging-assessed functional connectivity, which, owing to distant multisynaptic connections,63,64 cannot be structurally confirmed owing to current methodological shortcomings.65 Third, this project’s focus is purely pathophysiological and has exclusively drawn conclusions from a surrogate end point (ie, tau accumulation) but, as future clinical progression is associated with tau severity, we believe our findings are likely to translate into clinical outcomes. Accordingly, we demonstrate that longitudinal MMSE scores, converted to annual MMSE change rates, align with tau PET increases and predict tau PET changes (eTable 4 in Supplement 1). It should be mentioned that ADNI and Avid-A05 have slightly different clinical diagnostic criteria for mild cognitive impairment and cognitively normal designations; however, we believe this did not lead to incongruous clinical classifications between the 2 cohorts owing to the rigorous expert clinical judgment in both protocols. Moreover, we emphasize the exploratory nature of the sample size estimation analysis, which was not replicated in Avid-A05 due to insufficient data throughout the centiloid spectrum and overly dispersed tau PET SUVR change rates influencing power predictions. Additionally, this project has reported P values uncorrected for multiple comparisons to reduce type II error, which we believe to be statistically appropriate given the hypothesis-driven and cross-validation approach.39,66 Additionally, this study would generally benefit from longer tau PET follow-up times, particularly at early and potentially slower tau spreading stages, and the inclusion of more diverse cohorts, since ApoE4 may have different effects across races.67
Conclusions
In conclusion, we demonstrate independently validated evidence that ApoE4 was associated with accelerated and earlier Aβ-related tau spreading, which may drive faster clinical AD progression in ApoE4 carriers. Our findings have implications for trial design by illustrating that ApoE4 carriers may require earlier intervention to effectively attenuate tau spreading and associated clinical deterioration. Moreover, our results motivate further research into Aβ thresholds that determine clinical trial inclusion according to patient-specific characteristics, such as ApoE4, so that AD progression can be targeted in time to prevent tau spreading.
eAppendix
eTable 1. Demographics and Clinical data stratified by ApoE carriage
eTable 2. Regression model ANOVA & Akaike information criterion
eTable 3. Mediation Results controlled for clinical diagnosis
eTable 4. Interaction effects estimated by linear regression controlled for clinical diagnosis
eTable 5. Demographic and Clinical data for dementia subjects stratified by ApoE4 carriage
eTable 6. Interaction effects estimated by linear regression in CN, MCI & Dementia subjects
eTable 7. Effect of Annual tau-SUVR change on MMSE change
eFigure 1. Group-average tau-PET SUVRs in ADNI at baseline
eFigure 2. Required sample sizes to detect simulated intervention effects with tau change as an endpoint
eFigure 3. Scatterplots illustrating the interaction effect between ApoE4 status and centiloid on the annual rate of tau SUVR change in CN, MCI & Dementia subjects
Data sharing statement
References
- 1.Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971-982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogel JW, Iturria-Medina Y, Strandberg OT, et al. ; Alzheimer’s Disease Neuroimaging Initiative; Swedish BioFinder Study . Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat Commun. 2020;11(1):2612. doi: 10.1038/s41467-020-15701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franzmeier N, Dewenter A, Frontzkowski L, et al. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci Adv. 2020;6(48):eabd1327. doi: 10.1126/sciadv.abd1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franzmeier N, Neitzel J, Rubinski A, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat Commun. 2020;11(1):347. doi: 10.1038/s41467-019-14159-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cope TE, Rittman T, Borchert RJ, et al. Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain. 2018;141(2):550-567. doi: 10.1093/brain/awx347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife. 2019;8:e49132. doi: 10.7554/eLife.49132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119-128. doi: 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535-562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biel D, Brendel M, Rubinski A, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res Ther. 2021;13(1):137. doi: 10.1186/s13195-021-00880-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ossenkoppele R, Smith R, Mattsson-Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78(8):961-971. doi: 10.1001/jamaneurol.2021.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020;12(524):eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biel D, Luan Y, Brendel M, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Combining tau-PET and fMRI meta-analyses for patient-centered prediction of cognitive decline in Alzheimer’s disease. Alzheimers Res Ther. 2022;14(1):166. doi: 10.1186/s13195-022-01105-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buckley RF, Mormino EC, Rabin JS, et al. Sex differences in the association of global amyloid and regional tau deposition measured by positron emission tomography in clinically normal older adults. JAMA Neurol. 2019;76(5):542-551. doi: 10.1001/jamaneurol.2018.4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buckley RF, Scott MR, Jacobs HIL, et al. Sex mediates relationships between regional tau pathology and cognitive decline. Ann Neurol. 2020;88(5):921-932. doi: 10.1002/ana.25878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabin JS, Yang HS, Schultz AP, et al. Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol. 2019;85(2):272-279. doi: 10.1002/ana.25399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steward A, Biel D, Brendel M, et al. Functional network segregation is associated with attenuated tau spreading in Alzheimer’s disease. Alzheimers Dement.2023;19(5):2034-2046. doi: 10.1002/alz.061626 [DOI] [PubMed] [Google Scholar]
- 17.Franzmeier N, Ossenkoppele R, Brendel M, et al. The BIN1 rs744373 Alzheimer’s disease risk SNP is associated with faster Abeta-associated tau accumulation and cognitive decline. Alzheimers Dement. 2022;18(1):103-115. doi: 10.1002/alz.055113 [DOI] [PubMed] [Google Scholar]
- 18.Franzmeier N, Rubinski A, Neitzel J, Ewers M; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat Commun. 2019;10(1):1766. doi: 10.1038/s41467-019-09564-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):140. doi: 10.1186/s13195-020-00710-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hersi M, Irvine B, Gupta P, Gomes J, Birkett N, Krewski D. Risk factors associated with the onset and progression of Alzheimer’s disease: a systematic review of the evidence. Neurotoxicology. 2017;61:143-187. doi: 10.1016/j.neuro.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 21.Benson GS, Bauer C, Hausner L, et al. Don’t forget about tau: the effects of ApoE4 genotype on Alzheimer’s disease cerebrospinal fluid biomarkers in subjects with mild cognitive impairment-data from the Dementia Competence Network. J Neural Transm (Vienna). 2022;129(5-6):477-486. doi: 10.1007/s00702-022-02461-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong YJ, Kim CM, Lee JH, Sepulcre J. Correlations between APOE4 allele and regional amyloid and tau burdens in cognitively normal older individuals. Sci Rep. 2022;12(1):14307. doi: 10.1038/s41598-022-18325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montal V, Diez I, Kim CM, et al. Network tau spreading is vulnerable to the expression gradients of APOE and glutamatergic-related genes. Sci Transl Med. 2022;14(655):eabn7273. doi: 10.1126/scitranslmed.abn7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging. 2016;37:19-25. doi: 10.1016/j.neurobiolaging.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755-766. doi: 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sando SB, Melquist S, Cannon A, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 2008;8:9. doi: 10.1186/1471-2377-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9-21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 28.Ward A, Crean S, Mercaldi CJ, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38(1):1-17. doi: 10.1159/000334607 [DOI] [PubMed] [Google Scholar]
- 29.Pichet Binette A, Franzmeier N, Spotorno N, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer’s disease. Nat Commun. 2022;13(1):6635. doi: 10.1038/s41467-022-34129-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol. 2011;68(8):1062-1064. doi: 10.1001/archneurol.2011.70 [DOI] [PubMed] [Google Scholar]
- 31.Meisl G, Hidari E, Allinson K, et al. In vivo rate-determining steps of tau seed accumulation in Alzheimer’s disease. Sci Adv. 2021;7(44):eabh1448. doi: 10.1126/sciadv.abh1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13(3):205-216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180-184. doi: 10.1038/ng0694-180 [DOI] [PubMed] [Google Scholar]
- 34.Navitsky M, Joshi AD, Kennedy I, et al. Standardization of amyloid quantitation with florbetapir standardized uptake value ratios to the Centiloid scale. Alzheimers Dement. 2018;14(12):1565-1571. doi: 10.1016/j.jalz.2018.06.1353 [DOI] [PubMed] [Google Scholar]
- 35.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270-279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaefer A, Kong R, Gordon EM, et al. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb Cortex. 2018;28(9):3095-3114. doi: 10.1093/cercor/bhx179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ewers M, Luan Y, Frontzkowski L, et al. ; Alzheimer’s Disease Neuroimaging Initiative and the Dominantly Inherited Alzheimer Network . Segregation of functional networks is associated with cognitive resilience in Alzheimer’s disease. Brain. 2021;144(7):2176-2185. doi: 10.1093/brain/awab112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Leon MJ, Pirraglia E, Osorio RS, et al. ; Alzheimer’s Disease Neuroimaging Initiative; National Alzheimer’s Coordinating Center . The nonlinear relationship between cerebrospinal fluid Aβ42 and tau in preclinical Alzheimer’s disease. PLoS One. 2018;13(2):e0191240. doi: 10.1371/journal.pone.0191240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong RA. When to use the Bonferroni correction. Ophthalmic Physiol Opt. 2014;34(5):502-508. doi: 10.1111/opo.12131 [DOI] [PubMed] [Google Scholar]
- 40.Fleisher AS, Chen K, Liu X, et al. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34(1):1-12. doi: 10.1016/j.neurobiolaging.2012.04.017 [DOI] [PubMed] [Google Scholar]
- 41.Maass A, Landau S, Baker SL, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage. 2017;157:448-463. doi: 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young CB, Johns E, Kennedy G, et al. ; Alzheimer’s Disease Neuroimaging Initiative; A4 Study Team . APOE effects on regional tau in preclinical Alzheimer’s disease. Mol Neurodegener. 2023;18(1):1. doi: 10.1186/s13024-022-00590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Therriault J, Benedet AL, Pascoal TA, et al. ; Alzheimer’s Disease Neuroimaging Initiative . APOEε4 potentiates the relationship between amyloid-β and tau pathologies. Mol Psychiatry. 2021;26(10):5977-5988. doi: 10.1038/s41380-020-0688-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB-PET centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement. 2019;15(2):205-216. doi: 10.1016/j.jalz.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amadoru S, Doré V, McLean CA, et al. Comparison of amyloid PET measured in centiloid units with neuropathological findings in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):22. doi: 10.1186/s13195-020-00587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weigand AJ, Bangen KJ, Thomas KR, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Is tau in the absence of amyloid on the Alzheimer’s continuum? a study of discordant PET positivity. Brain Commun. 2020;2(1):fcz046. doi: 10.1093/braincomms/fcz046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salvadó G, Grothe MJ, Groot C, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Differential associations of APOE-ε2 and APOE-ε4 alleles with PET-measured amyloid-β and tau deposition in older individuals without dementia. Eur J Nucl Med Mol Imaging. 2021;48(7):2212-2224. doi: 10.1007/s00259-021-05192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321-332. doi: 10.1016/j.neuron.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 49.Lee WJ, Brown JA, Kim HR, et al. Regional Aβ-tau interactions promote onset and acceleration of Alzheimer’s disease tau spreading. Neuron. 2022;110(12):1932-1943. e5. doi: 10.1016/j.neuron.2022.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koelewijn L, Lancaster TM, Linden D, et al. Oscillatory hyperactivity and hyperconnectivity in young APOE-ε4 carriers and hypoconnectivity in Alzheimer’s disease. Elife. 2019;8:e36011. doi: 10.7554/eLife.36011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pihlajamäki M, Sperling RA. Functional MRI assessment of task-induced deactivation of the default mode network in Alzheimer’s disease and at-risk older individuals. Behav Neurol. 2009;21(1):77-91. doi: 10.1155/2009/276384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephen TL, Cacciottolo M, Balu D, et al. APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice. Acta Neuropathol Commun. 2019;7(1):82. doi: 10.1186/s40478-019-0729-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J Neuroinflammation. 2014;11(1):111. doi: 10.1186/1742-2094-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu CC, Zhao N, Fu Y, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96(5):1024-1032.e3. doi: 10.1016/j.neuron.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bellaver B, Povala G, Ferreira PCL, et al. Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer’s disease. Nat Med. 2023;29(7):1775-1781. doi: 10.1038/s41591-023-02380-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weigand AJ, Edwards LE, Thomas KR, Bangen KJ, Bondi MW; Alzheimer’s Disease Neuroimaging Initiative . Comprehensive characterization of elevated tau PET signal in the absence of amyloid-beta. Brain Commun. 2022;4(6):fcac272. doi: 10.1093/braincomms/fcac272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ. Subthreshold amyloid predicts tau deposition in aging. J Neurosci. 2018;38(19):4482-4489. doi: 10.1523/JNEUROSCI.0485-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Therriault J, Benedet AL, Pascoal TA, et al. Association of apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 2020;77(4):470-479. doi: 10.1001/jamaneurol.2019.4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leuzy A, Binette AP, Vogel JW, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Comparison of group-level and individualized brain regions for measuring change in longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2023;80(6):614-623. doi: 10.1001/jamaneurol.2023.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lemoine L, Leuzy A, Chiotis K, Rodriguez-Vieitez E, Nordberg A. Tau positron emission tomography imaging in tauopathies: the added hurdle of off-target binding. Alzheimers Dement (Amst). 2018;10:232-236. doi: 10.1016/j.dadm.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lace G, Savva GM, Forster G, et al. ; MRC-CFAS . Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain. 2009;132(Pt 5):1324-1334. doi: 10.1093/brain/awp059 [DOI] [PubMed] [Google Scholar]
- 62.Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6(1):85. doi: 10.1186/1750-1326-6-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Honey CJ, Sporns O, Cammoun L, et al. Predicting human resting-state functional connectivity from structural connectivity. Proc Natl Acad Sci U S A. 2009;106(6):2035-2040. doi: 10.1073/pnas.0811168106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grandjean J, Zerbi V, Balsters JH, Wenderoth N, Rudin M. Structural basis of large-scale functional connectivity in the mouse. J Neurosci. 2017;37(34):8092-8101. doi: 10.1523/JNEUROSCI.0438-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abhinav K, Yeh F-C, Pathak S, et al. Advanced diffusion MRI fiber tracking in neurosurgical and neurodegenerative disorders and neuroanatomical studies: a review. Biochim Biophys Acta. 2014;1842(11):2286-2297. doi: 10.1016/j.bbadis.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 66.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1(1):43-46. doi: 10.1097/00001648-199001000-00010 [DOI] [PubMed] [Google Scholar]
- 67.Naslavsky MS, Suemoto CK, Brito LA, et al. Global and local ancestry modulate APOE association with Alzheimer’s neuropathology and cognitive outcomes in an admixed sample. Mol Psychiatry. 2022;27(11):4800-4808. doi: 10.1038/s41380-022-01729-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weiner MW, Veitch DP, Aisen PS, et al. ; Alzheimer's Disease Neuroimaging Initiative . The Alzheimer's Disease Neuroimaging Initiative 3: continued innovation for clinical trial improvement. Alzheimers Dement. 2017;13(5):561-571. doi: 10.1016/j.jalz.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Analysis of 18F-AV-1451 PET imaging in cognitively healthy, MCI, and AD subjects (MCI). ClinicalTrials.gov identifier: NCT02116010. Updated September 22, 2020. Accessed October 4, 2023. https://academic.oup.com/amamanualofstyle/book/27941/chapter/207563234
- 70.Pontecorvo MJ, Devous MD Sr, Navitsky M, et al. ; 18F-AV-1451-A05 investigators . Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140(3):748-763. doi: 10.1016/j.jalz.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix
eTable 1. Demographics and Clinical data stratified by ApoE carriage
eTable 2. Regression model ANOVA & Akaike information criterion
eTable 3. Mediation Results controlled for clinical diagnosis
eTable 4. Interaction effects estimated by linear regression controlled for clinical diagnosis
eTable 5. Demographic and Clinical data for dementia subjects stratified by ApoE4 carriage
eTable 6. Interaction effects estimated by linear regression in CN, MCI & Dementia subjects
eTable 7. Effect of Annual tau-SUVR change on MMSE change
eFigure 1. Group-average tau-PET SUVRs in ADNI at baseline
eFigure 2. Required sample sizes to detect simulated intervention effects with tau change as an endpoint
eFigure 3. Scatterplots illustrating the interaction effect between ApoE4 status and centiloid on the annual rate of tau SUVR change in CN, MCI & Dementia subjects
Data sharing statement