

Abstract
Background
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG), also known as non-structural maintenance of chromosomes condensin I complex subunit G, is mitosis-related protein that widely existed in eukaryotic cells. Increasing evidence has demonstrated that aberrant NCAPG expression was strongly associated with various tumors. However, little is known about the function and mechanism of NCAPG in glioblastoma (GBM).
Methods
The expression and prognostic value of NCAPG were detected in the clinical databases and tumor samples. The function effects of NCAPG downregulation or overexpression were evaluated in GBM cell proliferation, migration, invasion, and self-renewal in vitro and in tumor growth in vivo. The molecular mechanism of NCAPG was researched.
Results
We identified that NCAPG was upregulated in GBM and associated with poor prognosis. Loss of NCAPG suppressed the progression of GBM cells in vitro and prolonged survival in mouse models of GBM in vivo. Mechanistically, we revealed that NCAPG positively regulated E2F transcription factor 1 (E2F1) pathway activity. By directly interacting with Poly (ADP-ribose) polymerase 1, a co-activator of E2F1, and facilitating the PARP1-E2F1 interaction to activate E2F1 target gene expression. Intriguingly, we also discovered that NCAPG functioned as a downstream target of E2F1, which was proved by the ChIP and Dual-Luciferase results. Comprehensive data mining and immunocytochemistry analysis revealed that NCAPG expression was positively associated with the PARP1/E2F1 signaling axis.
Conclusions
Our findings indicate that NCAPG promotes GBM progression by facilitating PARP1-mediated E2F1 transactivation, suggesting that NCAPG is a potential target for anticancer therapy.
Keywords: E2F1, glioblastoma (GBM), NCAPG, PARP1, tumor progression
Graphical Abstract
Graphical Abstract.
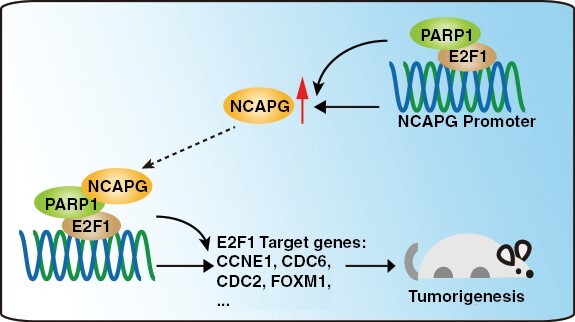
Key Points.
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) promotes glioblastoma progression by facilitating Poly (ADP-ribose) polymerase 1 (PARP1)-mediated E2F transcription factor 1 (E2F1) transactivation.
E2F1 directly activates the transcription of NCAPG.
The NCAPG/PARP1/E2F1 molecular axis is critical for glioblastoma progression.
Importance of the Study.
Our results revealed a novel tumor-promotive role of nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) for its recruitment of Poly (ADP-ribose) polymerase 1 (PARP1) to facilitate the PARP1- E2F transcription factor 1 (E2F1) interaction, resulting in the transactivation of E2F1 and upregulation of NCAPG and other oncogenic genes correlated with glioblastoma progression. These findings indicated the critical role of NCAPG/PARP1/E2F1 molecular axis in glioblastoma progression and should be target for tumor therapy.
Glioblastoma (GBM), the most commonly occurring cancer in malignant brain tumors, is ranked as grade IV glioma, and its 5-year case fatality rate ranks only after pancreatic and lung cancer.1 Up to now, the recurrence of the infiltrated glioma cannot be avoided.2 The recurrent GBM has less sensitivity to any therapy. Although numerous mechanisms and therapies including immune checkpoint blockade, chimeric antigen receptor-modified T therapy, and oncolytic virotherapy, still most patients die of the disease within 2 years.3–5 Fortunately, interest in glioblastoma continues to grow as more and more detailed and helpful mechanisms are studied and described.
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) plays a pivotal role in chromosome structural stabilization during mitosis and meiosis.6 A high expression level of NCAPG indicates poor prognosis and low survival rate.7 In addition, in the mechanisms we already know, NCAPG was involved in many pathways, including promoting cell proliferation through activating the PI3K pathway, resisting breast cancer to trastuzumab through activating tyrosine-protein kinase Src/Signal transducer and activator of transcription 3 pathway, and regulating the cellular mitosis through G2/mitotic-specific cyclin B1 (CCNB1) overexpression.8–10 Due to the high expression in various tumors, NCAPG is expected to be an emerging cancer marker and a promising target for tumor treatment.
E2F transcription factor 1 (E2F1) is 1 of the 8 members (E2F1-8) of the E2F family. E2F1 is best known for its roles in regulating gene expression, including cell cycle progression,11 apoptosis under DNA damage and serum deprivation,12 and metastasis.13 One of the most important mechanisms is the interaction between E2F1 and its binding proteins, including dimerization proteins (DP), p107 (RBL1), p130 (RBL2), and pRB (RB1).14 E2F1 promotes transcription by recruiting the transcriptional activator, when interacting with DP, whereas, E2F1 transcriptional activity is inhibited when interacting with retinoblastoma family proteins (RBL1, RBL2, and RB1).15 Additionally, E2F1 is also modified by Poly (ADP-ribose) polymerase 1 (PARP1), a nuclear enzyme that plays a pivotal role in genomic stability and chromatin remodeling.16 It has been reported that PARP1 interacted with the DNA-binding domain of E2F1 and then facilitated E2F1 transactivation in a PARylation-independent manner.17 Interestingly, the interaction between PARP1 and E2F1 could be enhanced by some factors, for example, MZF1-AS1.17 In tumor cells, the E2F1 signaling pathway was hyperactivated by PARP1 and promoted cancer progression.18 Hence, targeting signaling pathways linked to E2F1 would be a promising therapeutic option in GBM.
In this study, we found that NCAPG bound PARP1 to enhance the PARP1/E2F1 interaction, resulting in E2F1 transactivation. In addition, we also found NCAPG to be an undiscovered downstream gene of E2F1. Therefore, our studies revealed a crucial role of NCAPG in E2F1 signaling and may offer a promising target for the treatment of GBM patients.
Materials and Methods
Animal Studies
All animal experiments were approved by the Institutional Animal Care and Use Committee of Southwest University. 4-week-old female non-obese diabetic/severe combined immunodeficiency disease mice were randomized into indicated groups (each group n = 12). 5 × 105 cells were transplanted into the right frontal lobe of each mouse by a microinjector. The living status of the mice was recorded. At the end of the experiment, all mice were sacrificed and brains were taken out and H&E staining was performed.
Cells
Established GBM cell lines (LN-229, U-118 MG, and A172), HEK293FT, and normal astroglia cells (SVGP12) were purchased from the American Type Culture Collection (ATCC, USA). Two patient-derived GBM cell lines (GBM-3 and GBM6) were obtained from Dr. Hao W (Daping Hospital). Cells were cultured as previously described.19
Transfection
Indicated plasmids were transferred into the cells by Lipofectamine 2000 according to the manufacturer’s instructions. The shRNA sequences were listed in Supplementary Table I.
Quantitative PCR
TRIzoL reagent was used to extract total RNA as described by the manufacturer’s instructions. The reverse transcription and the detection methods were described previously.20 The primer pairs were shown in Supplementary Table II.
Western Blot, Immunoprecipitation, and LC-MS-MS Assay
For western blot and Co-IP assay, the detailed procedures were described in the previous article.21
For the LC-MS-MS proteomics analysis, the proteins were harvested by the Co-IP experiments and the gel of WB was stained by Fast Silver Stain Kit. The silver-stained gel was sent to the Applied Protein Technology, Shanghai for proteomics analysis.
MTT and BrdU Assay
Methylthiazolyldiphenyl-tetrazolium bromide (MTT) and BrdU assays were performed as previously described.19
Sphere Formation Assay
Spheres from GBM cells were seeded into the ultra-low attachment 24-well plate and cultured with serum-free medium supplemented with rh-EGF (20 ng/mL) and rh-bFGF (20 ng/mL). The number of neurospheres in each well was detected.
Migration, and Invasion Assay
Migration and invasion assays were performed as previously described.19
Dual-Luciferase Reporter Gene Assay
pGL3-basic, phRL-TK, and other indicated plasmids were co-transfected into the 293FT cells, cells were lysed after 48 hours by cell lysis buffer. The procedure of Dual-Luciferase Reporter Gene Assay was followed by Dual-Luciferase Reporter Gene Assay Kit (Yeasen).
Chromatin Immunoprecipitation and Re-ChIP
Cells were cross-linked by 1% formaldehyde at 37°C and terminated by glycine solution. Cells were lysed by sodium dodecyl sulfate-lysis buffer. The solution was incubated with indicated antibody at 4°C overnight, and then incubated with Protein A + G agarose for 1 hour. The next steps were followed by the protocol of the ChIP Assay Kit (Beyotime). The primer pairs were shown in Supplementary Table II.
GST-Pulldown
Indicated plasmids were transformed into the Rosetta DE3. Isopropyl β-D-thiogalactoside was used to induce the cells to express indicated protein expression. Glutathione S-transferase (GST)-tagged protein was pre-combined with GST purified resin, and the lysate containing His tagged protein was slowly passed through the GST resin bound to the GST-tagged protein, after three times of PBS buffer washing, the proteins were eluted. Western blot assay was used to determine whether they interacted with each other.
Duo-Link PLA Assay
Prior to initiation, cells were deposited on slides and pretreated for fixation, restoration, and permeability. The slides were incubated with indicated antibodies mixture at 4°C overnight after blocking by Duo-link blocking buffer. The following steps were followed by the manufacturer’s protocol.
Gene Set Enrichment Analysis
gene set enrichment analysis (GSEA) was performed using the Broad Institute GSEA version 4.0.3 software. The Chinese Glioma Genome Atlas (CGGA) database and The Cancer Genome Atlas (TCGA) database respectively downloaded from the Chinese Glioma Genome Atlas (http://www.cgga.org.cn/) and UCSC Xena (https://xena.ucsc.edu/public/).
Patient Data Analysis and Patient Tumor Tissues
Patient and gene expression data were downloaded from the gene expression profiling interactive analysis (GEPIA), GlioVis, TCGA, CGGA, and R2 databases (http://r2.amc.nlhttp://r2platform.com). GPS-Prot, String, and Gene MANIA were used to draw the grid of protein interactions. Cistrome DB database were used to analyze the ChIP-sequencing data. Tumor samples and prior approval were gained from the department of neurosurgery, Daping hospital, Chongqing, China. Tumor tissue micro-arrays were obtained from Bioaitech Co., Ltd. (Henan, China). All the patients provided written informed consent to participate. Tissue analysis was approved by the ethics committee of Southwest University of China.
Statistical Analysis
All experiments were carried out in triplicates and all graphs were obtained using Graphpad Prism 8 and IBM SPSS Statistics software were used to process the data and presented as Mean ± SD. Kaplan Meier method was used to draw the survival curve, and Cox Regression Analysis was used to evaluate the correlation of indicated proteins expression. Student’s t-Test was used to evaluate the significance, and the results were considered significant as P < .05.
Results
NCAPG Is Overexpressed and Promotes Tumor Progression in GBM
NCAPG was reported to act as a crucial protein that promoted numerous cancer progressions,7 which was consistent with the analysis of the GEPIA database (Figure 1A). To explore the role of NCAPG in GBM, the GEPIA, TCGA, Rembrandt, and Gravendeel databases were analyzed and showed that NCAPG expression was significantly higher in GBM samples (Figure 1B-E). Additionally, we observed that NCAPG expression was positively correlated with the malignancy of glioma (Supplementary Figure 1A–D). Furthermore, high NCAPG expression was linked to poor prognosis of glioma patients in the CGGA, TCGA, Frejie, and Phillips datasets (Figure 1F, G and Supplementary Figure 2). To further research the biological role of NCAPG, CGGA database were used to identify the characteristics of glioma that were mediated by NCAPG expression. We discovered that NCAPG expression was closely linked to age, tumor grade, TCGA subtype, isocitrate dehydrogenase mutation, and the 1p19q Codeletion status in glioma (Supplementary Table III; Supplementary Figure 1E and 1F). To further validate the role of NCAPG in GBM, we detect NCAPG expression in human GBM samples by immunohistochemical staining and western blot analysis. The expression of NCAPG was clearly higher in GBM tissues than the normal brain tissues (Figure 1H and 1I). Consistently, NCAPG expression profile was detected and indicated that NCAPG expression was significantly increased in GBM cell lines and GBM parent cells (GBM-3) (Figure 1J and 1K). Taken together, these results indicated that high NCAPG expression predicted poor survival of GBM patients.
Figure 1.
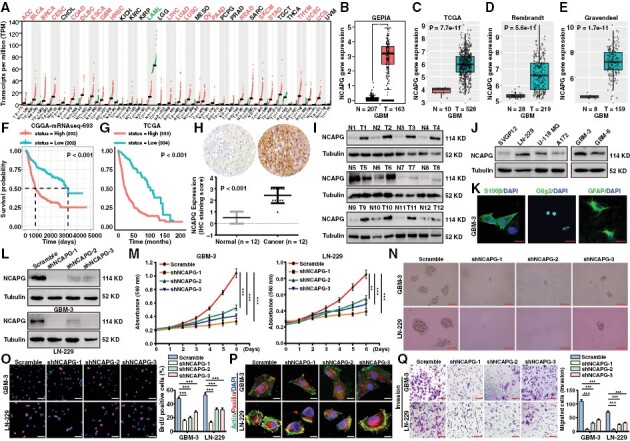
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) is overexpressed and promotes tumor progression in glioblastoma (GBM). (A) Box plot of NCAPG expression in normal (N) and tumor tissues (T) of various cancers was analyzed by the GEPIA database. (B-E) Box plot of NCAPG expression in normal and GBM tissues was analyzed by GEPIA, TCGA, Rembrandt, and Gravendeel database respectively. (F, G) Kaplan–Meier analysis of progression-free survival using data from the CGGA and TCGA database. (H) Representative immunohistochemical assays of NCAPG expression in human GBM tumors and adjacent normal brain tissues (12 pairs) (I) Western blot analysis of NCAPG protein expression in adjacent normal brain tissues (Normal, N) and GBM tissues (Tumor, T). (J) NCAPG protein expression in GBM cell lines and normal cells (SVGP12). (K) The immunofluorescence detection of the biomarkers including S100β, Olig2, and GFAP of GBM cells. (L) The efficiency of NCAPG shRNA was evaluated by western blot assay. (M) Growth curves of GBM cells. (N) Representative images of GBM spheres. (O) Representative images of BrdU assays for GBM cells. (P) The immunofluorescence staining with anti-Paxillin and actin of GBM cells. (Q) Transwell experiments were performed to detect the invasion ability of GBM cells. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
To investigate the biological function played by NCAPG, we silenced NCAPG in GBM cells (Figure 1L). MTT experiments showed that NCAPG downregulation inhibited GBM cell proliferation (Figure 1M). In addition, NCAPG depletion also retarded the sphere formation of GBM-3 and LN-229 cells (Figure 1N). Then, BrdU incorporation assays demonstrated that loss of NCAPG significantly reduced the DNA synthesis of GBM cells (Figure 1O). Paxillin was responsible for the adhesion and metastasis of tumor cells, often located at the end of the actin filaments. The immunofluorescence assays were performed and showed that loss of NCAPG showed few paxillin adhesion spots than the control group (Figure 1P). Additionally, NCAPG depletion suppressed the migration and invasion of GBM cells (Figure 1Q and Supplementary Figure 3A). Notably, the expression of metastatic proteins such as Snail, Vimentin, and N-cadherin was significantly reduced, and E-cadherin was upregulated in NCAPG-knockdown groups, whereas there had no significant change in paxillin and β-actin expression after NCAPG downregulation in GBM cells (Supplementary Figure 3B).
To further confirm that the effects of NCAPG depletion on tumor progression of GBM cells were not an off-target or viral effect, we recovered NCAPG expression after shNCAPG#1 interference by overexpression of a full-length NCAPG vector that is resistant to shNCAPG#1 targeting (Supplementary Figure 4A and 4B). Then, MTT and BrdU experiments showed that NCAPG overexpression in NCAPG-knockdown cells almost rescued the proliferation of GBM cells (Supplementary Figure 4C and 4D). Besides, restoring the expression of NCAPG in NCAPG-knockdown cells remarkably enhanced the migration and invasion of GBM cells (Supplementary Figure 4E and 4F).
Collectively, these data clearly demonstrated that NCAPG was overexpressed in GBM and linked to poor survival. Loss of NCAPG inhibited GBM cell proliferation and migration rates, suggesting that NCAPG played a critical role in GBM progression.
NCAPG Promotes the Development of GBM Through Activation of the E2F1 Pathway
To unveil the underlying mechanisms whereby NCAPG regulates GBM cell development, GSEA analysis was performed and indicated that the E2F family targets, G2/M checkpoint, mitotic spindle, and MYC proto-oncogene targets showed an obvious positive correlation with NCAPG expression in GBM (Figure 2A). E2F family targets drew our attention because E2F family has been reported to play a critical role in promoting GBM progression.22 Further GSEA analysis on the E2F family targets showed that high expression of NCAPG positively regulated the E2F1 signaling pathway (Figure 2B and 2C). To confirm the[se findings, we detected the downstream targets of E2F1 signaling through western blot and quantitative polymerase chain reaction (qPCR) experiments after NCAPG depletion. As shown in Figure 2D and 2E, the protein and mRNA levels of CDC2, CDC6, CCNE1, CCNA2, CCNB1, CDC25A, and FOXM1 were significantly reduced in the NCAPG-knockdown GBM cells, especially the shNCAPG-1# group. Therefore, we speculate that NCAPG might affect the binding affinity of E2F1 to its downstream gene promoters. Indeed, ChIP analysis demonstrated that silencing of NCAPG reduced the E2F1 enrichment on the promoter of E2F1 target genes in GBM cells (Figure 2F). Moreover, knockdown or overexpression of NCAPG reduced or enhanced E2F1 transactivation, respectively (Figure 2G and 2H). To further validate our hypothesis regarding the correlation between NCAPG and E2F1 pathway, we overexpressed E2F1 expression on the NCAPG-knockdown cells. Overexpression of E2F1 rescued the expression of E2F1 downstream proteins, proliferation, migration, and invasion of NCAPG-knockdown cells (Figure 2I-K, and Supplementary Figure 5). Additionally, we knocked down E2F1 expression on the NCAPG-overexpression GBM cells using the shE2F1-1 sequence (Supplementary Figure 6A). The results indicated that loss of E2F1 remarkably weakened the facilitated effects of NCAPG overexpression on the E2F1 downstream targets and the development of GBM cells (Figure 2L, and Supplementary Figure 6B-6E). Notably, knockdown or overexpression of NCAPG altered E2F1 activity, but not its expression. Taken together, these findings suggested that NCAPG promotes the development of GBM by increasing the expression of E2F1 target genes.
Figure 2.
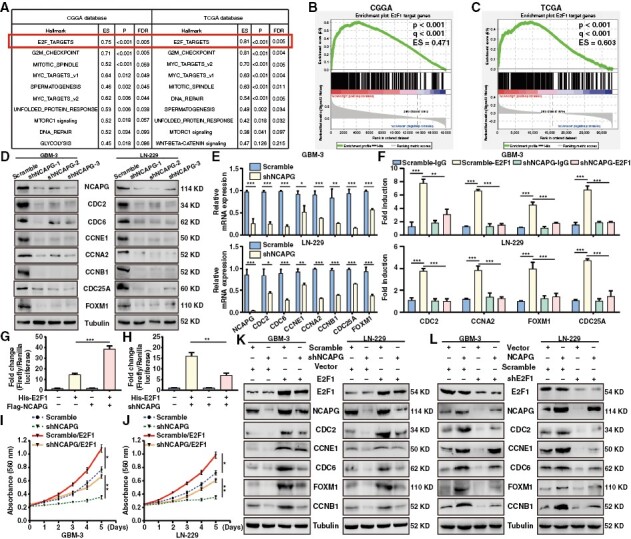
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) promotes the development of glioblastoma (GBM) through activation of the E2F transcription factor 1 (E2F1) pathway. (A) GSEA analysis of the data from CGGA and TCGA database (B, C) GSEA analysis of the correlation between NCAPG expression and E2F1 signaling pathway. (D, E) The protein and mRNA expression of E2F1 downstream targets were examined. (F) ChIP analysis of E2F1 enrichment at its target promoters. (G, H) The Dual-Luciferase reporting gene assay was used to evaluate the promoter activity of p14 in GBM cells. (I, J) MTT assays were performed after overexpression of E2F1 in NCAPG-knockdown GBM cells. (K) The expression of NCAPG, E2F1, and its downstream proteins were detected in NCAPG-knockdown GBM cells by overexpression of E2F1. (L) The expression of NCAPG, E2F1, and its downstream proteins were detected in NCAPG-overexpression GBM cells by silencing of E2F1. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
NCAPG Forms a Complex with PARP1 In Vitro and In Vivo
To research whether NCAPG directly binds E2F1 to its target gene promotes, a GST pull-down assay was performed and showed there was no direct interaction between His-tagged NCAPG and GST-tagged E2F1 protein (Supplementary Figure 7). Up to date, many transcription coactivators of E2F1 have been gradually reported, such as DNA topoisomerase IIbeta binding protein 1 (TopBP1),23 lncRNA-SLC16A1-AS1,24 and PARP1.17 To identify the putative protein partners of NCAPG in regulating E2F1 transactivation, we performed a co-immunoprecipitation (Co-IP) assay with NCAPG antibody and then analyzed by liquid chromatography-tandem mass spectrometry in GBM cells (Figure 3A). We were particularly intrigued potential importance of PARP1, a crucial co-activator of E2F1. Co-IP experiments demonstrated an interaction between NCAPG and PARP1 in GBM-3 and LN-229 cells (Figure 3B). Additionally, the GST pull-down and in situ assays also indicated that PARP1 was a direct protein partner of NCAPG (Figure 3C and 3D). Consistently, the GPS-Prot database, Gene MANIA database, and STRING Interaction Network also predicted this interaction (Figure 3E and Supplementary Figure 8). To identify the region of NCAPG and PARP1 involved in binding, a series of NCAPG and PARP1 deletion mutants were constructed and used in the Co-IP experiments. As shown in Figure 3F and 3G, the DNA Bing Domain of PARP1 and the Armadillo-like (ARM) domain of NCAPG were required for the NCAPG/PARP1 interaction. Collectively, these data demonstrated that NCAPG is directly bound to PARP1 in GBM cells.
Figure 3.
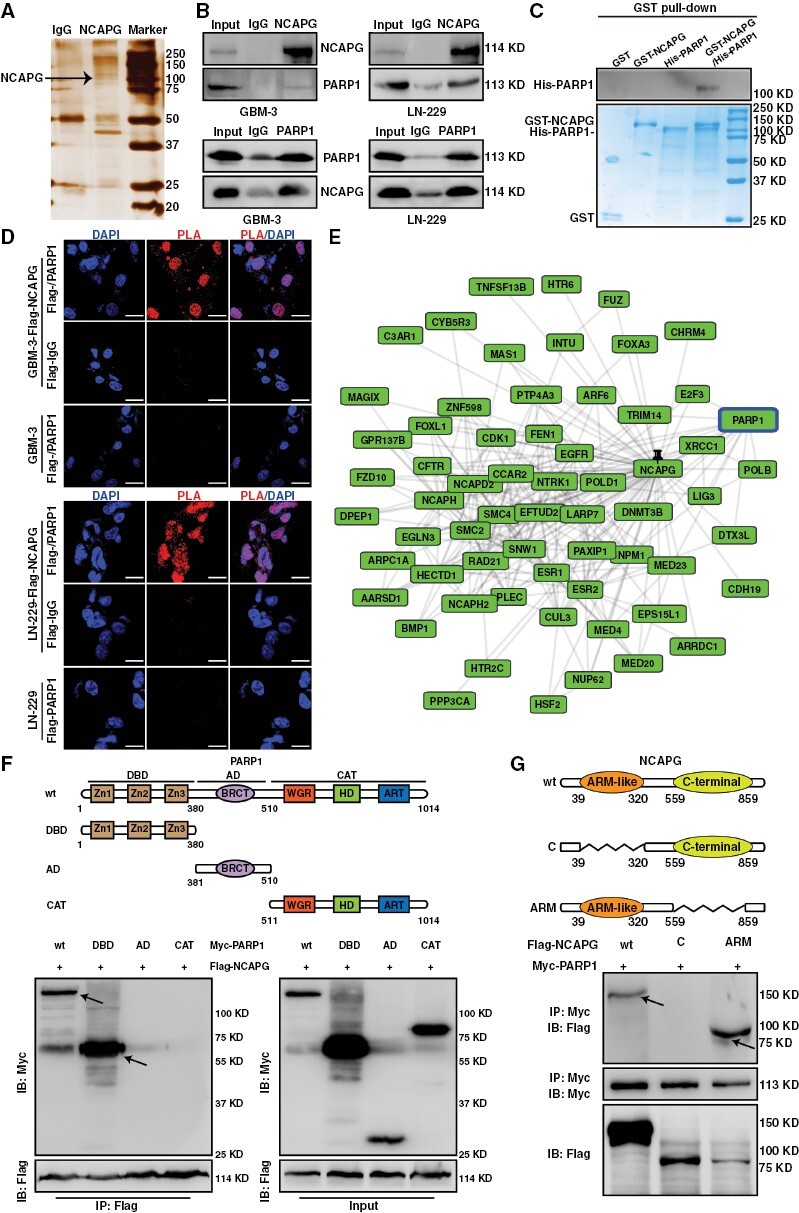
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) forms a complex with Poly (ADP-ribose) polymerase 1 (PARP1) in vitro and in vivo. (A) Mass spectrometry (MS) analysis of indicated proteins in silver staining demonstrating proteins pulled down by NCAPG from glioblastoma (GBM)-3 cells. (B) Co-IP and western blot assays show endogenous interaction between NCAPG and PARP1 in GBM cells. (C) GST pull-down assay indicating the direct interaction of PARP1 and NCAPG. (D) Duo-link proximity ligation assay (PLA) assay indicating the direct interaction between NCAPG and PARP1 within GBM cells. (E) The interaction prediction of NCAPG and PARP1 was observed in the GPS-Prot database. (F, G) The truncation and domain deletion of PARP1 and NCAPG vectors were used to determine the interaction domain. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
NCAPG Promotes GBM Progression Via PARP1-Mediated E2F1 Transactivation
Since NCAPG interacted with PARP1 and mediated E2F1 downstream genes, we hypothesized that NCAPG plays as a recruitment factor linking PARP1 and E2F1. The ChIP-re-ChIP assays were performed in Flag-NCAPG overexpression cells to explore the coregulated targets and found that CDC2, CCNA2, and CDC25A promoters were pulled down by PARP1 and Flag antibodies (Figure 4A). Additionally, in situ, assays and Co-IP assays were conducted and demonstrated that NCAPG knockdown significantly weakened the interaction between PARP1 and E2F1 (Figure 4B and 4C). Heale et al. reported that Condensin I (including NCAPG) could interact with PARP1 in interphase, and their interaction could be induced by DNA damage.25 Here, we found that NCAPG overexpression remarkably promoted the PARP1/E2F1 association after Camptothecin (CPT) treatment, which generated single-strand breaks (Supplementary Figure 9A). Additionally, we carried out serum-starvation and re-feeding experiments in LN-229 cell lines, and conducted the Duo-Link experiments and found that NCAPG promoted the interaction between PARP1 and E2F1 mainly in S phase (Supplementary Figure 9B-D). Of note, the ARM domain of NCAPG was indispensable for the PARP1-E2F1 interaction (Figure 4D). Furthermore, we detected the function of each domain of NCAPG on E2F1 transcriptional activity. As is shown in Figure 4E, NCAPG overexpression could increase E2F1 transactivation except C-terminal domain. Importantly, NCAPG-induced E2F1 transactivation could be abolished by silencing of PARP1 (shPARP1-1#) (Figure 4E and Supplementary Figure 10). Next, we sought to detect the function of NCAPG domain for E2F1 enrichment on target gene promoters. ChIP assays showed that deletion of ARM domain lost its recruitment function and reduced the binding affinity of E2F1 to its target promoters (Figure 4F). Similarly, deletion of ARM domain weakened the promotive effects of NCAPG overexpression on the E2F1 target gene expression and cell growth, suggesting that ARM domain was essential for the transactivation of E2F1 (Figure 4G-4I). These data support a critical role of NCAPG in facilitating PARP1-mediated E2F1 transactivation.
Figure 4.
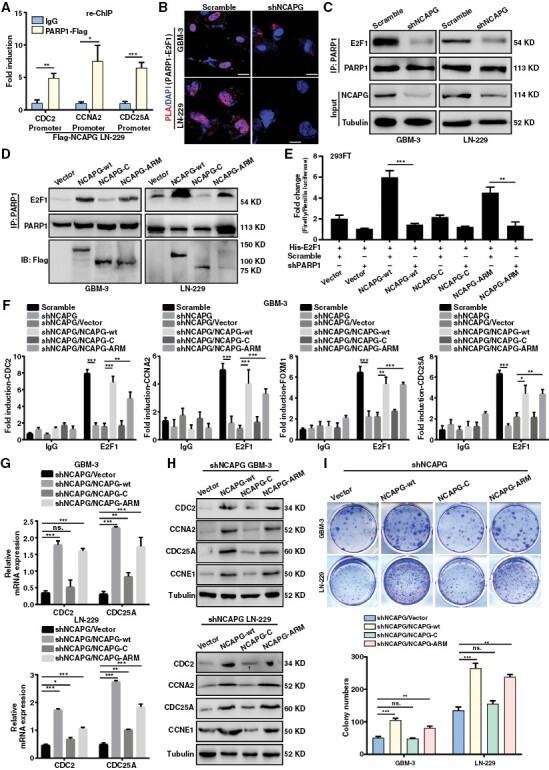
Nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG) promotes glioblastoma (GBM) progression via Poly (ADP-ribose) polymerase 1 (PARP1)-mediated E2F transcription factor 1 (E2F1) transactivation. (A) ChIP-re-ChIP assay indicating the E2F1 target promoters were co-regulated by NCAPG and PARP1 in LN-229/Flag-NCAPG cells. (B) Duo-link PLA assay was performed to detect the effect of NCAPG knockdown on the interaction between PARP1 and E2F1 in GBM cells. (C) Co-IP assay was used to determine the binding affinity of PARP1 and E2F1 protein caused by NCAPG knockdown. (D) Co-IP assay was used to determine the function of NCAPG domain for facilitating the interaction of PARP1 and E2F1. (E) The Dual-Luciferase reporting gene assay was used to determine the function of NCAPG domain on E2F1 activity. (F) ChIP assays were performed to detect the function of NCAPG domain on the enrichment of E2F1 to its target promoters. (G, H) The mRNA and protein expression of E2F1 downstream targets were examined in GBM cells to detect the function of NCAPG domain on E2F1 target genes. (I) Colony formation assay was used to detect the function of NCAPG domain on cell proliferation of GBM cells. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
E2F1 Directly Activates the Transcription of NCAPG
Given the observation that the NCAPG expression levels were altered by downregulation or overexpression of E2F1 in GBM cells (Figure 2K and 2L). Consistent with these findings, western blot and qPCR experiments demonstrated that the protein and mRNA expression of NCAPG was positively regulated by E2F1 (Figure 5A and 5B). Additionally, we analyzed RNA-seq data collected from the clinical database and found a remarkable correlation between E2F1 and NCAPG expression (Figure 5C). Therefore, we speculated that E2F1 might transcriptionally mediate NCAPG expression. Notably, Dual-Luciferase reporter analysis revealed a positive regulation of E2F1 on the NCAPG promoter (Figure 5D). According to the JASPAR dataset, we identified 2 potential binding sites of E2F1 on the NCAPG promoter (Figure 5E). Besides, we downloaded the ChIP-sequencing data of E2F1 from the Cistrome DB database and made the data visualization through IGV software. Supporting our assumption, we found that E2F1 bound to the promoter of NCAPG, and this binding commonly existed in different cancers (Figure 5F). To further confirm the enrichment of E2F1 on NCAPG promoter, ChIP analysis was conducted and showed that E2F1 bound to the P5 region of NCAPG promoter, which was consistent with the above data from the JASPAR dataset and Cistrome DB database (Figure 5G). To determine whether E2F1 directly regulated NCAPG transcription, the luciferase reporter vectors were constructed and then used to explore the effects caused by E2F1 knockdown. We found that loss of E2F1 significantly inhibited the activity of wild NCAPG promoter, but not mutant promoter (Figure 5H). To determine whether PARP1 participated in the modulation of E2F1 on NCAPG expression, we detected the promoter activity of NCAPG by silencing PARP1. We found that PARP1 knockdown abolished the increased NCAPG promoter activity induced by overexpression of E2F1 (Figure 5I). Similarly, loss of PARP1 reduced the enrichment of E2F1 on NCAPG promoter in GBM cells (Figure 5J). Lastly, PARP1 depletion clearly reduced the mRNA and protein level of NCAPG (Figure 5K, 5L and Supplementary Figure 11). However, NCAPG expression and the interaction of NCAPG/PARP1 or PARP1/E2F1 did not change after treatment with the PARP1 inhibitor, suggesting that E2F1 promoted NCAPG transcription in a PARylation-independent manner (Supplementary Figure 12). Collectively, these data demonstrated that E2F1 directly regulated the transcription of NCAPG in GBM cells.
Figure 5.
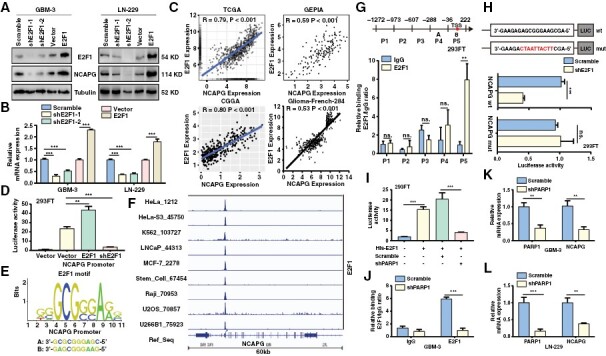
E2F transcription factor 1 (E2F1) directly activates the transcription of nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG). (A, B) The protein and mRNA expression of NCAPG and E2F1 were detected. (C) The correlation of NCAPG and E2F1 expression was analyzed. (D) The Dual-Luciferase reporting gene assay was performed to investigate the effect of E2F1 overexpression or E2F1 knockdown on NCAPG promoter activity respectively. (E) Predicted sequences of E2F1 motif in NCAPG promoter by JASPAR. (F) The ChIP-sequencing data of E2F1 were downloaded from the Cistrome DB database and visualized by IGV software. (G) ChIP analysis of the region of E2F1 enrichment at NCAPG promoter. (H) Luciferase reporter constructs containing the truncated wild NCAPG promoter or E2F1 motif mutant promoter were con-transfected with a shE2F1 or with an empty vector into 293FT cells, and luciferase activity was evaluated. (I) The Dual-Luciferase reporter gene assay demonstrates the promoter activity of NCAPG in the 293FT cells transfected with His-E2F1 and/or sh polymerase 1 (PARP1). (J) ChIP analysis of E2F1 enrichment at NCAPG promoter in glioblastoma-3 cells expression Scramble or shPARP1. (K, L) The mRNA level of NCAPG in PARP-silenced cells were determined by qPCR assay. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
The NCAPG/PARP1/E2F1 Molecular Axis Exists In Vivo
To evaluate whether NCAPG depletion affects tumor growth of GBM cells, we performed the colony formation and orthotopic implantation assays. We found that the tumor growth of GBM cells was significantly retarded after NCAPG knockdown in vitro and in vivo (Figure 6A and 6B). Of note, the overall survival of GBM cells was clearly improved by silencing of NCAPG (Figure 6C). And the effects of NCAPG knockdown on tumor growth were not related to gender (Supplementary Figure 13 A and B). Next, we evaluated NCAPG and PARP1/E2F1 signaling pathways (PARP1, E2F1, CDC6, CCNB1, and FOXM1) in microarray tissues from 47 glioma patients with cancers using IHC staining. High volume of NCAPG and PARP1/E2F1 signaling pathway was observed in most tumor specimens, and significant correlations were found between the expression of NCAPG and the gene of PARP1/E2F1 signaling pathway (Figure 6D and 6E). These findings were also proved by the TCGA and CGGA database analysis (Figure 6F). Additionally, we performed the orthotopic implantation assays and found that E2F1 overexpression significantly rescued the tumor growth of NCAPG-knockdown GBM-3 cells (Supplementary Figure S13C). And the effects of NCAPG overexpression on the expression of E2F1 and target proteins in mouse xenograft models were consistent with the effects of NCAPG overexpression on GBM cells (Supplementary Figure S13D). Moreover, we analyzed the R2 database to determine the correlation between the expression of these proteins and the malignancy of glioma. The results indicated that NCAPG was highly correlated with E2F1 and its target proteins including CDC2, CCNB1, MMP9, FOXM1, CCNE1, and CDC6 (Figure 6G). Besides, we detected the effects of NCAPG on immune microenvironment in glioma, and we found that NCAPG expression was negatively correlated with CD4+ T cell in-filtration in glioma (Supplementary Figure S14A-C). GSEA analysis indicated that NCAPG was positively associated with NF-KB signaling pathway, while was negatively correlated with the regulation of Th1 cell immune response and T cell receptor pathway (Supplementary Figure. S14D-F). Next, we performed the qPCR analysis and found that the expression level of IL4R and IL1B was clearly increased in the NCAPG-knockdown GBM cells, suggesting that NCAPG was involved in promoting immunosuppression in the glioma microenvironment. (Supplementary Figure. S14 G and H). Collectively, these results support the critical role of NCAPG in glioblastoma progression and should be targeted for tumor therapy.
Figure 6.
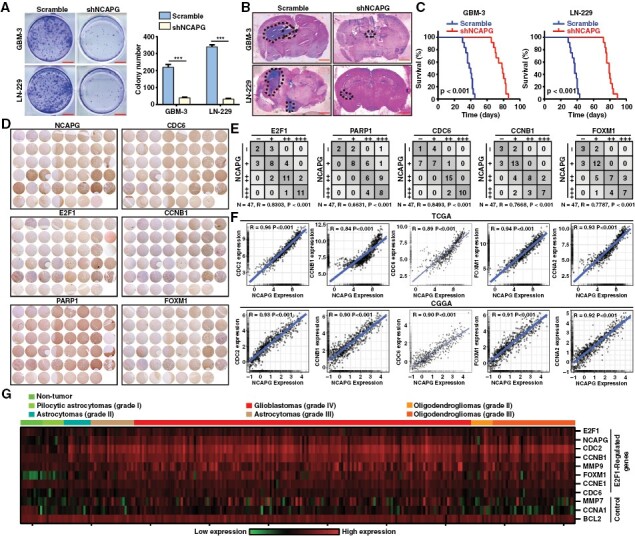
The nonstructural maintenance of chromatin condensin I complex subunit G (NCAPG)/ Poly (ADP-ribose) polymerase 1/ E2F transcription factor 1 (E2F1) molecular axis exists in vivo. (A) Colony formation assay was performed to validate the impact of NCAPG knockdown on cell growth. (B) Intracranial in situ tumorigenesis model mice were established to evaluate the effect of NCAPG downregulation on the tumor growth of glioblastoma cells. (C) The survival of the mice was recorded and the survival curve was drawn with the Kaplan–Meier method. (D, E). Tumor tissue micro-arrays were purchased and conducted by IHC staining. The expression correlation between NCAPG and PARP1, E2F1, and its downstream targets were quantified. (F) The expression correlation between NCAPG and downstream targets of E2F1 was analyzed using the data from the TCGA and CGGA databases. (G) Gene expression profile analysis on the R2 dataset using NCAPG, E2F1, E2F1-regulated genes, and other control genes. All data are shown as the means ± SD; *P < .05, **P < .01, ***P < .001.
Discussion
Increasing evidence has demonstrated that aberrant expression of NCAPG was strongly associated with various tumors.7 Functionally, NCAPG has been found to facilitate cell proliferation, migration, and invasion by mediating the PI3K/AKT pathway in hepatocarcinoma,9 the GSK3β/β-catenin pathway in oral carcinoma,26 the TGF-β pathway in lung adenocarcinoma,27 and the p38 MAPK pathway in ovarian cancer.28 Recently, other studies showed that NCAPG was associated with tumor progression and might serve as a prognostic biomarker in glioma.29 However, the biological function and molecular mechanism of NCAPG in GBM were unclear. We found that NCAPG silencing significantly suppressed GBM progression. RNA-seq GSEA profiles revealed the positive correlation between NCAPG expression and E2F1 pathway activity. Indeed, our data indicated that silencing of NCAPG diminished the enrichment of E2F1 on its target gene promoters. Additionally, the promotive effect of NCAPG on GBM progression was abolished by inhibition of E2F1 expression. Given the crucial oncogenic roles of NCAPG in many cancers, we suspect that the NCAPG/E2F1 axis is not limited to GBM. For example, previous studies provided independent evidence that NCAPG was positively associated with CCNA2, CDC2, and FXOM1, which were downstream genes of E2F1.30–32 Of note, our results showed that knockdown or overexpression of NCAPG altered the activity of E2F1, but not its expression.
Prior studies have reported that E2F1 activity was regulated by its protein partners. For instance, TopBP1 inhibited transcriptional activity and localization of E2F1, resulting in the induction of S-phase entry and apoptosis in response to DNA damage.23 In contrast, lncRNA-SLC16A1-AS1, an E2F1-responsive lncRNA, formed an RNA-protein complex with E2F1 and promoted E2F1 activity in bladder cancer.24 Moreover, PARP1 played as a co-activator of E2F1 in facilitating the expression of target genes in a PARylation-independent manner.33 Interestingly, this PARP1-E2F1 interaction could be enhanced by lncRNA MZF1-AS1.17 Therefore, we speculate that NCAPG might regulate E2F1 transactivation by interacting with E2F1 or other proteins. As reported, NCAPG could bind to β-catenin to activate the Wnt/β-catenin pathway in colorectal cancer.34 In addition, NCAPG positively regulated ligand-dependent enhancer activation in part by interacting with HECTD1, an E3 ubiquitin ligase responsible for the recruitment of transcriptional coactivators and corepressors via its ubiquitination function.35 Here, we found that, surprisingly, NCAPG was required for E2F1 transactivation, at least in part based on its recruitment of an E2F1 co-activator, PARP1. Notably, we observed that the ARM domain of NCAPG was essential for the PARP1/NCAPG interaction and E2F1 transactivation in GBM cells. Interestingly, our evidence also discovered that E2F1 promoted the transcription of NCAPG in a PARylation-independent manner, suggesting the presence of NCAPG/PARP1/E2F1 feedback loops in GBM cells.
It has been reported that NCAPG participated in tumor progression by control of the PI3K/AKT, Wnt/β-catenin, and other pathways.9,34 These findings suggest that precisely targeting NCAPG by inhibition of NCAPG mRNA or protein levels may prolong the prognosis of tumor patients with high NCAPG expression.36 Based on our findings, the expression of NCAPG was highly correlated with the PARP1/E2F1 pathway in GBM. Targeting of NCAPG/PARP1/E2F1 signaling pathway may aid GBM therapy. Of course, E2F1 signaling pathway is an attractive target for tumor treatment. However, transcription factors are poor drug targets. Additionally, PARP1 inhibitors failed to inhibit NCAPG-facilitating GBM progression as PARP1 mediated E2F1 transactivation as well as NCAPG expression in a PARylation-independent manner. Unexpectedly, we observed that E2F1 transactivation was inhibited by blocking of NCAPG recruitment, suggesting that targeting NCAPG may lower E2F1 signaling activity. Future studies are needed to explore the biological implications of the interaction between NCAPG and PARP1 and to lay a theoretical foundation for the development of novel therapeutic avenues for malignant tumors.
In summary, our findings uncover a novel mechanism in which E2F1 facilitated the expression of NCAPG to ultimately enhance the interaction between PARP1 and E2F1, persistently augmenting the transactivation of E2F1. These findings indicate the crucial roles of NCAPG/PARP1/E2F1 axis in GBM progression.
Supplementary Material
Contributor Information
Jianbing Hou, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China; Advanced Research Center in Brain Diseases, Jinfeng Laboratory, Chongqing, China.
Pan Huang, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China.
Minghao Xu, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China.
Hao Wang, Department of Neurosurgery, Daping Hospital, The Third Military Medical University, Chongqing, China.
Yaqian Shao, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China.
Xuelian Weng, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China.
Yudong Liu, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China.
Hongbo Chang, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China; Advanced Research Center in Brain Diseases, Jinfeng Laboratory, Chongqing, China.
Li Zhang, Department of Radiology and Nuclear Medicine, The First Hospital of HeBei Medical University, Hebei Province, China.
Hongjuan Cui, State Key Laboratory of Resource Insects, Southwest University, Chongqing, China; Advanced Research Center in Brain Diseases, Jinfeng Laboratory, Chongqing, China.
Funding
This research was supported by the Natural Science Foundation of Chongqing (cstc2022ycjh-bgzxm0145 and cstc2020jcyj-msxmX0678), the Fundamental Research Funds for the Central Universities (SWU-XDZD22006 and SWU-KT22034).
Conflict of interest statement
The authors declare no conflict of interest.
Data Availability
All of the data and material in this paper are available when requested.
Authorship statement
J.H., P.H., M.X., H.W., Y.S., X.W., Y.L., H.C., Z.L., and H.C. have participated in investigation, methodology, and validation of data presented in this article. J.H., H.P., and M.X. are responsible for Formal Analysis of data. J.H., H.P., and M.X. wrote and edited this manuscript, Z.L. and H.C. read and revised this manuscript.
References
- 1. Ostrom QT, Gittleman H, Stetson L, Virk S, Barnholtz-Sloan JS.. Epidemiology of intracranial gliomas. Prog Neurol Surg. 2018;30:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Hou J, Deng Q, Zhou J, et al. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP-mediated degradation of EGFR. Oncogene. 2017;36(8):1134–1144. [DOI] [PubMed] [Google Scholar]
- 3. Thomas AA, Brennan CW, DeAngelis LM, Omuro AM.. Emerging therapies for glioblastoma. JAMA Neurol. 2014;71(11):1437–1444. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J.. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat Rev Clin Oncol. 2013;10(1):14–26. [DOI] [PubMed] [Google Scholar]
- 5. Choi PJ, Tubbs RS, Oskouian RJ.. Emerging cellular therapies for glioblastoma multiforme. Cureus. 2018;10(3):e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao C, Gong J, Jie Y, et al. NCAPG Is a promising therapeutic target across different tumor types. Front Pharmacol. 2020;11:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai X, Gao J, Shi C, et al. The role of NCAPG in various of tumors. Biomed Pharmacother. 2022;155:113635. [DOI] [PubMed] [Google Scholar]
- 8. Zhang Q, Su R, Shan C, Gao C, Wu P.. Non-SMC condensin I complex, subunit G (NCAPG) is a novel mitotic gene required for hepatocellular cancer cell proliferation and migration. Oncol Res. 2018;26(2):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong C, Ai J, Fan Y, et al. NCAPG promotes the proliferation of hepatocellular carcinoma through PI3K/AKT signaling. Onco Targets Ther. 2019;12:8537–8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang L, Ren L, Chen H, et al. NCAPG confers trastuzumab resistance via activating SRC/STAT3 signaling pathway in HER2-positive breast cancer. Cell Death Dis. 2020;11(7):547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Song X, Herrup K.. Context-dependent functions of E2F1: Cell cycle, cell death, and DNA damage repair in cortical neurons. Mol Neurobiol. 2020;57(5):2377–2390. [DOI] [PubMed] [Google Scholar]
- 12. Biswas AK, Johnson DG.. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012;72(1):13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alla V, Engelmann D, Niemetz A, et al. E2F1 in melanoma progression and metastasis. J Natl Cancer Inst. 2010;102(2):127–133. [DOI] [PubMed] [Google Scholar]
- 14. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–2262. [DOI] [PubMed] [Google Scholar]
- 15. Frolov MV, Dyson NJ.. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117(Pt 11):2173–2181. [DOI] [PubMed] [Google Scholar]
- 16. Schiewer MJ, Knudsen KE.. Transcriptional roles of PARP1 in cancer. Mol Cancer Res. 2014;12(8):1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fang E, Wang X, Yang F, et al. Therapeutic targeting of MZF1-AS1/PARP1/E2F1 axis inhibits proline synthesis and neuroblastoma progression. Adv Sci (Weinh). 2019;6(19):1900581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iglesias P, Seoane M, Golan I, et al. PARP1 deficiency reduces tumour growth by decreasing E2F1 hyperactivation: A novel mechanism in the treatment of cancer. Cancers (Basel). 2020;12(10):2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hou J, Liu Y, Huang P, et al. RANBP10 promotes glioblastoma progression by regulating the FBXW7/c-Myc pathway. Cell Death Dis. 2021;12(11):967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang G, Zhu Q, Fu G, et al. TRIP13 promotes the cell proliferation, migration and invasion of glioblastoma through the FBXW7/c-MYC axis. Br J Cancer. 2019;121(12):1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hou J, Xu M, Gu H, et al. ZC3H15 promotes glioblastoma progression through regulating EGFR stability. Cell Death Dis. 2022;13(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giannopoulou AI, Kanakoglou DS, Piperi C.. Transcription factors with targeting potential in gliomas. Int J Mol Sci . 2022;23(7):3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu K, Lin FT, Ruppert JM, Lin WC.. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol. 2003;23(9):3287–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Logotheti S, Marquardt S, Gupta SK, et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during Bladder Cancer progression as target and co-activator of E2F1. Theranostics. 2020;10(21):9620–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heale JT, Ball AR, Schmiesing JA, et al. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol Cell. 2006;21(6):837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J, Sun S, Li J, et al. NCAPG, mediated by miR-378a-3p, regulates cell proliferation, cell cycle progression, and apoptosis of oral squamous cell carcinoma through the GSK-3beta/beta-catenin signaling. Neoplasma. 2021;68(6):1201–1211. [DOI] [PubMed] [Google Scholar]
- 27. Wu Y, Lin Y, Pan J, et al. NCAPG promotes the progression of lung adenocarcinoma via the TGF-beta signaling pathway. Cancer Cell Int. 2021;21(1):443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu H, Zou D, Ni N, et al. Overexpression of NCAPG in ovarian cancer is associated with ovarian cancer proliferation and apoptosis via p38 MAPK signaling pathway. J Ovarian Res. 2022;15(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng G, Han T, Hu X, et al. NCAPG promotes tumor progression and modulates immune cell infiltration in glioma. Front Oncol. 2022;12:770628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan Y, Jiang X, Tang L, et al. FOXM1/lncRNA TYMSOS/miR-214-3p-mediated high expression of NCAPG correlates with poor prognosis and cell proliferation in non-small cell lung carcinoma. Front Mol Biosci. 2021;8:785767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu B, Xiao Y, Li H, et al. Identification and verification of biomarker in clear cell renal cell carcinoma via bioinformatics and neural network model. Biomed Res Int. 2020;2020:6954793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H, Zheng P, Li Z, et al. NCAPG promotes the proliferation of renal clear cell carcinoma via mediating with CDK1. Dis Markers. 2022;2022:6758595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simbulan-Rosenthal CM, Rosenthal DS, Luo R, et al. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene. 2003;22(52):8460–8471. [DOI] [PubMed] [Google Scholar]
- 34. Shi Y, Ge C, Fang D, et al. NCAPG facilitates colorectal cancer cell proliferation, migration, invasion and epithelial-mesenchymal transition by activating the Wnt/beta-catenin signaling pathway. Cancer Cell Int. 2022;22(1):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W, Hu Y, Oh S, et al. Condensin I and II complexes license full estrogen receptor alpha-dependent enhancer activation. Mol Cell. 2015;59(2):188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao W, Xia T, Xu S, et al. Increased expression of NCAPG (Non-SMC condensing I complex subunit G) is associated with progression and poor prognosis of lung adenocarcinoma. Bioengineered. 2022;13(3):6113–6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data and material in this paper are available when requested.