

Abstract
Background
Glioblastoma (GB) is incurable at present without established treatment options for recurrent disease. In this phase I first-in-human clinical trial we investigated safety and feasibility of adoptive transfer of clonal chimeric antigen receptor (CAR)-NK cells (NK-92/5.28.z) targeting HER2, which is expressed at elevated levels by a subset of glioblastomas.
Methods
Nine patients with recurrent HER2-positive GB were treated with single doses of 1 × 107, 3 × 107, or 1 × 108 irradiated CAR-NK cells injected into the margins of the surgical cavity during relapse surgery. Imaging at baseline and follow-up, peripheral blood lymphocyte phenotyping and analyses of the immune architecture by multiplex immunohistochemistry and spatial digital profiling were performed.
Results
There were no dose-limiting toxicities, and none of the patients developed a cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome. Five patients showed stable disease after relapse surgery and CAR-NK injection that lasted 7 to 37 weeks. Four patients had progressive disease. Pseudoprogression was found at injection sites in 2 patients, suggestive of a treatment-induced immune response. For all patients, median progression-free survival was 7 weeks, and median overall survival was 31 weeks. Furthermore, the level of CD8+ T-cell infiltration in recurrent tumor tissue prior to CAR-NK cell injection positively correlated with time to progression.
Conclusions
Intracranial injection of HER2-targeted CAR-NK cells is feasible and safe in patients with recurrent GB. 1 × 108 NK-92/5.28.z cells was determined as the maximum feasible dose for a subsequent expansion cohort with repetitive local injections of CAR-NK cells.
Keywords: adoptive immunotherapy, CAR, NK cells, glioblastoma, HER2, phase I first, in, human clinical trial
Key Points.
First clinical trial to explore adoptive chimeric antigen receptor (CAR)-NK cell therapy for glioblastoma patients.
Intracranial injection of up to 108 CAR-NK cells was safe.
None of the patients developed a cytokine release syndrome or ICANS.
Importance of the Study.
This phase I first-in-human clinical trial with nine patients with recurrent HER2-positive glioblastoma investigated intracranial injection of a single dose of 1 × 107, 3 × 107, or 1 × 108 HER2-targeted CAR-NK cells during relapse surgery. There were no dose-limiting toxicities, and none of the patients developed a cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome. Best responses were stable disease in five patients, four patients had progressive disease. Imaging revealed pseudoprogression at injection sites in two patients, suggestive of a treatment-induced immune response. Progression-free survival in both of these patients was 37 weeks, with overall survival of 98 and 135 weeks, respectively. For all patients, the level of CD8+ T-cell infiltration in recurrent tumor tissue prior to CAR-NK cell injection positively correlated with time to progression. These results demonstrate that intracranial injection of HER2-targeted CAR-NK cells is feasible and safe, encouraging further evaluation of this approach.
Glioblastoma (GB) is the most common malignant primary brain tumor in adults. It still carries a dismal prognosis with median overall survival (OS) of less than 1 year in non-selected cohorts.1 Despite multimodal therapy, recurrence is almost universally observed, with available experimental therapies typically not able to extend median survival in the relapse situation beyond 6–8 months.2 One interesting outlier is the work of Cloughesy et al. where neoadjuvant treatment with the anti-PD-1 checkpoint inhibitor pembrolizumab as compared to adjuvant pembrolizumab treatment prolonged median overall survival from 6.3 to 13.2 months.3 Nevertheless, there is still an urgent need for more effective treatments.
Recent clinical trials with chimeric antigen receptor (CAR)-engineered autologous T-cells demonstrated feasibility and safety of this approach in recurrent GB, with signs of clinical activity and transient responses observed in some of the patients.4–8 In contrast to T-cells, natural killer (NK) cells lack an antigen-specific T-cell receptor and can therefore be applied for adoptive immunotherapy in an allogeneic setting without a relevant risk of inducing graft-versus-host disease.9,10 This advantage extends to CAR-engineered NK cells, with CD19-targeted allogeneic CAR-NK cells shown to achieve high response rates in lymphoma and leukemia patients in an initial clinical trial,11 without complications such as cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS) frequently associated with CAR-T-cell therapies.12 In addition, NK cells exhibit natural cytotoxicity, which is triggered by activating receptors like NK group 2D (NKG2D) and natural cytotoxicity receptors, and can complement CAR-mediated target cell lysis. This may reduce the risk of tumor immune escape due to the selection of antigen-loss variants observed in some cases after CAR-T-cell therapy.13
The concept of CAR-NK cells as an off-the-shelf therapeutic for adoptive cancer therapy was initially established with the continuously expanding and clinically usable NK cell line NK-92, and later extended to healthy donor-derived peripheral blood NK cells, cord blood NK cells, and NK cells differentiated from CAR-engineered induced pluripotent stem cells (iPSC).14–17 By lentiviral transduction, we previously generated a GMP-compliant and molecularly and functionally well-characterized NK-92 single-cell clone (NK-92/5.28.z) that carries a second-generation CAR with CD28 costimulatory and CD3ζ signaling domains, targeting the tumor-associated HER2 (ErbB2) antigen.18,19 While this receptor tyrosine kinase is not expressed in adult healthy brain tissue,20 HER2 protein overexpression was found in a large proportion of primary GB tumors, and was correlated with impaired survival.21–23 In preclinical studies, NK-92/5.28.z cells displayed high and selective cytotoxicity against HER2-positive GB cell lines and primary GB stem cell cultures that was retained under hypoxic conditions and in the presence of high concentrations of immunosuppressive TGF-β, which are typical for the immunosuppressive GB tumor microenvironment.23 In orthotopic GB mouse xenograft models, intratumoral administration of the CAR-NK cells delayed tumor development and markedly extended survival. In syngeneic immunocompetent GB mouse models, such direct antitumor effects of NK-92/5.28.z cells were accompanied by CAR-NK-mediated activation of durable endogenous antitumor immunity, resulting in cures and protection against tumor rechallenge at distant sites.23–25 While limiting in vitro activity of the CAR-NK cells to a few days, radiation with 10 Gy as included as a safety measure for clinical application of NK-92 cells did not decrease efficacy of NK-92/5.28.z cells in vivo.19,23
Based on these data, we designed the CAR2BRAIN phase I first-in-human clinical trial to determine feasibility and safety of local injection of HER2-specific NK-92/5.28.z CAR-NK cells in patients with recurrent GB, and to interrogate the tumor immune architecture for biomarkers predictive of efficacy. Here, we report results of the dose-escalation cohort.
Materials and Methods
Study Design and Patient Information
Written informed consent was obtained from all patients prior to trial screening procedures and enrollment. We applied a modified 3 + 3 dose-escalation scheme with 3 dose levels, differing from the classical 3 + 3 dose-escalation scheme in determining maximum feasible dose after just 3 patients at the highest dose level in case none of these patients developed dose-limiting toxicity (DLT). This procedure was justified by the subsequently planned expansion cohort of at least 6 additional patients with repetitive injections at the same dose level. Consequently, the possible patient number ranged between 9 and 18. An independent “Data and Safety Monitoring Board” (DSMB) allocated the adverse events (AEs) to their plausible causes (“study drug-related,” “injection-related,” “device-related,” “disease-related” or “unknown”), each with a probability (“certain,” “probable,” “possible,” “unlikely,” or “unrelated”). DLTs were defined as all “study drug-related” AEs of common terminology criteria for adverse events (CTCAE) grade ≥3, for which the DSMB noted a possible, probable, or certain causal relationship. The study was approved by the Goethe University Hospital Ethics Committee, Frankfurt, Germany (269/17) and performed in accordance with the principles of Good Clinical Practice, the Declaration of Helsinki, and all applicable clinical trial and advanced therapy medicinal product regulations. The trial was registered before study initiation at clinicaltrials.gov (NCT03383978), the German Clinical Trial Register (DRKS; www.drks.de; DRKS00013333), and the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT number 2016-000225-39). An interim analysis was performed upon completion of the dose-escalation cohort, which is presented herein with complete follow-up of the patients.
Preparation of NK-92.5.28.z CAR-NK Cells
NK-92/5.28.z cells stably express a CAR comprising a HER2-specific single-chain fragment variable (scFv) antibody, a CD8α hinge region, and CD28 and CD3ζ signaling domains, previously shown to be superior in NK-92 cells to a CD3ζ-only or a composite CD137-CD3ζ CAR with respect to cytotoxic activity or stability of cell surface expression, respectively.18 The CAR-NK cells were grown from a GMP-compliant and qualified master cell bank generated from a single clone of lentivirally transduced NK-92 cells.19 Individual patient dosages were expanded from a maintenance culture, irradiated at 10 Gy, washed and provided as fresh cell suspension reconstituted in GMP grade single donor human serum containing 100 U/mL IL-2 (Proleukin; Novartis Pharma, Nürnberg, Germany). Manufacturing and release testing was timed to allow same-day injections of the cells.
CAR-NK Cell Dosing
Dosing was based on previous experience from the ASPECT study and intracranial injection of NK-92/5.28.z cells in orthotopic mouse GB models.23,26 The CAR-NK cells were delivered to the operating room at a cell density of 5 × 107/mL, and were then diluted directly prior to injection in single donor human serum containing 100 U/mL IL-2 to a density of 0.5 × 107/mL or 1.5 × 107/mL, or kept at 5 × 107/mL as required for the respective dose level. During relapse surgery and after resection of the tumor mass, NK-92/5.28.z cells were injected into the wall of the resection cavity in a total volume of 2 mL divided into 20–40 portions of each 50–100 μL, aiming to avoid eloquent areas of the brain. We initially used hypodermic 25 G needles, and subsequently switched to blunt 25 G needles to reduce bleeding risk. While the injected volume remained to the largest part intraparenchymal, we did observe some reflux into the resection cavity, which was left there and not aspirated if possible. Otherwise, the relapse surgery was performed according to local standard therapy procedures. As part of this routine, all patients intraoperatively received high-dose intravenous dexamethasone (20–40 mg) to prevent local swelling, with rapid postoperative tapering over approximately 3 days conditional on the postoperative edema.
MR and O-(2-[18F]fluoroethyl)-L-tyrosine -PET Imaging
All patients received Magnetic resonance imaging (MRI) examinations within 72 hours after surgery, and 4, 12, and 24 weeks thereafter, and at additional time points in case of clinical deterioration. MRI examinations were conducted using a 3 Tesla MRI scanner (Skyra; Siemens, Erlangen, Germany). A standardized imaging protocol was employed, containing a T2-weighted sequence, diffusion-weighted imaging, susceptibility-weighted imaging, a fluid-attenuated inversion recovery (FLAIR) sequence, T1-weighted sequences before and after the administration of a gadolinium-based contrast agent and dynamic susceptibility contrast perfusion-weighted imaging (see also Supplementary Table S1). Positron emission tomography (PET) imaging in patient CB011 was performed using the radiolabeled amino acid O-(2-[18F]fluoroethyl)-L-tyrosine (FET), as described previously.27 Briefly, after overnight fasting the patient underwent a dynamic PET scan from 0 to 50 minutes post-injection of 3 MBq of FET per kg of body weight on an ECAT Exact HR+ PET scanner (Siemens Healthineers, Munich, Germany) in 3-dimensional mode (32 rings; axial field of view, 15.5 cm). FET uptake in the tissue was expressed as standardized uptake values and analyzed by regions of interest on the summed PET data from 20 to 40 minutes post-injection.
Multispectral Imaging
Formalin-fixed and paraffin-embedded primary and recurrent glioblastoma sections were stained using the Opal Polaris 6-color kit (NEL861001KT; Akoya Biosciences, Marlborough, MA) based on the tyramide signal amplification immunostaining technique. Multiplex stainings targeting human CD3, CD8α, von Willebrand Factor, CD163 and Iba-1, and/or human CD3, CD8α, CD4, programmed cell death protein (PD)-1, FoxP3, and Ki-67 (see Supplementary Table S2) were performed on a LabSat Research Automated Staining Instrument (Lunaphore Technologies SA, Tolochenaz, Switzerland). DAPI was used to detect all nuclei. Multiplex stainings using the antibody panels depicted in Supplementary Table S2 were acquired on a Vectra Polaris instrument (Akoya Biosciences) using MOTiF technology, which provided unmixed whole slide scans in a streamlined workflow within 6–15 minutes at 0.5 µm/pixel, depending on section size. Post-CAR-NK treatment specimens were available from subsequent relapse surgeries of two patients. Another post-treatment specimen was available from an autopsy, which however yielded a poor staining outcome due to autolytic processes. Whole slide multispectral image analysis was performed with (1) Phenochart, version 1.0.12, a whole slide scan viewer, (2) InForm Image Analysis Software (Akoya Biosciences), which was used for spectral unmixing and batch analysis, and (3) HALO software (Indica Labs, Albuquerque, NM), which was used for fusing of batched images to a multispectrally unmixed whole slide image and further analysis.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism version 8.4.3 and 9.3.1 (GraphPad Software, San Diego, CA), as well as JMP 16 software (SAS, Heidelberg, Germany).
Other Materials and Methods may be found in Supplementary Material.
Results
Patient Characteristics and Treatment
Clinical characteristics, prior treatments, and molecular pathology results of all patients assigned to receive CAR-NK cells are given in Table 1. Key inclusion criterion was a recurrent HER2-positive GB as determined by immunohistochemistry in tumor tissue from the most recent surgery (exemplarily shown in Supplementary Figure S1), with relapse surgery already planned based on clinical routine practice and multidisciplinary tumor board recommendation independent from study participation. Required prior treatments were at least standard of care for glioblastoma, including radiotherapy and temozolomide chemotherapy. Minimum age for participation was 18 years, with a minimum Karnofsky performance score of 50%. Main exclusion criteria were pretreatment with anti-angiogenic therapy in the last four weeks prior to study entry, coagulation disorder, and active autoimmune disease. Patients had to be off other antineoplastic therapy for 2 weeks prior to study entry, but temozolomide was allowed up to 48 hours before CAR-NK injection (not applicable for the patients reported herein). At the time of inclusion, dexamethasone up to a total dose of 4 mg per day was allowed if medically indicated. In total, 101 patients suffering from recurrent glioblastoma were assessed for study eligibility. Of these, 90 patients were excluded due to too low (56 patients) or inconclusive HER2 scores (3 patients), or other inclusion/exclusion criteria (22 patients). Nine patients declined study participation. Ultimately, 11 patients were included in the dose-escalation cohort of the study (Figure 1). Two patients were not treated due to withdrawal of consent or product not being released, respectively. Nine patients in three cohorts received single doses of 1 × 107, 3 × 107, or 1 × 108 CAR-NK cells (Table 1), with the applied cell products all conforming to the predetermined release criteria (Supplementary Table S3).
Table 1.
Patient Characteristics, Chimeric Antigen Receptor (CAR)-NK Cell Dose, Treatment Response and Patient Survival
| ID | Age | Sex | MGMT | IDH | Prior Treatments | HER2 Score at Screening (0–12) |
HER2 Score at Relapse Surgery (0–12) |
NK-92/5.28.z Cell Dose |
Best Response | PFS (Weeks) | OS (Weeks) | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CB001 | 49 | M | − | WT | S, XRT-TMZ | 6 | 6 | 1 × 107 | SD | 7 | 23 | |
| CB002 | 57 | F | − | WT | S, XRT-TMZ | 9 | 2 | n.a. | n.a. | n.a. | n.a. | product not released |
| CB003 | 60 | M | − | WT | S, XRT-TMZ, TTF | 2 | 6 | 1 × 107 | SD | 16 | 18 | |
| CB004 | 44 | F | − | WT | S, XRT-TMZ, TTF | 2 | 12 | 1 × 107 | PD | 4 | 22 | |
| CB005 | 46 | M | + | WT | S, XRT-TMZ, reS, reXRT, CCNU/TMZ, Nivo | 6 | 6 | 3 × 107 | PD | 2 | 37 | |
| CB006 | 30 | M | − | WT | S, XRT-TMZ | 8 | 3 | 3 × 107 | PD | 4 | 40 | |
| CB007 | 57 | M | + | WT | S, XRT-TMZ | 6 | 3 | 3 × 107 | PD | 3 | 31 | |
| CB008 | 59 | M | + | WT | S, XRT-TMZ | 4 | 8 | n.a. | n.a. | n.a. | n.a. | withdrawal of consent |
| CB009 | 70 | F | + | WT | S, XRT-TMZ | 2 | 6 | 1 × 108 | SD | 37 | 135 | |
| CB010 | 61 | M | − | WT | S, XRT-TMZ, reS, TTF |
2 | 9 | 1 × 108 | SD | 11 | 30 | |
| CB011 | 59 | M | + | WT | S, XRT-CCNU/TMZ | 3 | 8 | 1 × 108 | SD | 37 | 98 |
F, female; M, male; MGMT +, MGMT promotor hypermethylation; MGMT -, no MGMT promotor hypermethylation; IDH WT, IDH wild type; CCNU/TMZ, lomustine/temozolomide; reS, relapse surgery; Nivo, Nivolumab; reXRT, relapse radiotherapy; S, surgery; TMZ, temozolomide 5/28; TTF, tumor treating fields; XRT, radiotherapy; XRT-CCNU/TMZ, radiotherapy with CCNU and temozolomide (in accordance with CeTeG); XRT-TMZ, radiotherapy with concomitant and adjuvant temozolomide (in accordance with EORTC 26981); n.a., not applicable; PD, progressive disease; SD, stable disease; PFS, progression-free survival; OS, overall survival. MGMT promotor methylation and IDH status were determined with the Illumina 850k methylation array and data processing via molecularneuropathology.org.
Figure 1.
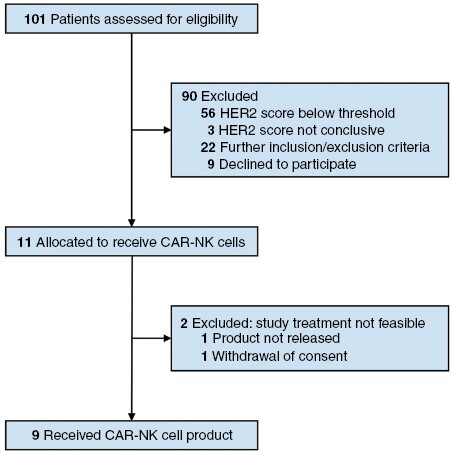
CONSORT Flow Diagram.
Safety of Intracranial CAR-NK Cell Injection
We recorded 7 severe AEs (Table 2), but none of the patients developed DLT after injection of CAR-NK cells as defined by the study protocol. Patient CB003 suffered from a postoperative meningitis CTCAE grade 3 with fever CTCAE grade 1, with a probable causal relationship to the tumor resection, and a transient elevation of liver enzymes CTCAE grade 3, which ceased after switching the antibiotic treatment from meropenem/vancomycin to linezolid/ceftriaxone. This patient reported fluctuating fevers with a maximum of 39°C, which began 9 days after surgery, and presented at our outpatient department after he additionally developed headaches beginning on day 14 after surgery. On the same day, we performed a lumbar puncture, which yielded laboratory results (leukocytes 986/µL, lactate 3.88 mmol/L, and total protein 1910 mg/L), and cerebrospinal fluid cytology (predominantly neutrophilic granulocytes) indicative of bacterial meningitis. While we were not able to specify the pathogenic bacteria by cerebrospinal fluid (CSF) culture and polymerase chain reaction (PCR), symptoms completely regressed within 24 hours after initiation of treatment with meropenem and vancomycin. Also, the CSF pathologies gradually normalized in repeat lumbar punctures. As in-depth microbiological testing did not evidence contamination of the cell product and glioblastoma relapse surgery was indicated independent from study participation, both the meningitis and the subsequent elevation of liver enzymes were assessed as unrelated to the study product and probably disease-related. The same patient with a preexisting structural epilepsy due to the glioblastoma subsequently experienced a generalized epileptic seizure CTCAE grade 2, which was disease-related. Similarly, patient CB005 who also suffered from a preexisting structural epilepsy had one disease-related epileptic seizure. In patient CB010, CTCAE grade 3 infarction and hemorrhage occurred immediately after resection with local injection of NK-92/5.28.z cells. The small and therefore not space-occupying hemorrhage was assessed as possibly related to the injection procedure by the DSMB, with an unlikely relationship to the study product. The perioperative cerebral ischemia located adjacent to the resection cavity in the access route of the tumor resection but not in the area where the intraoperative injections were performed, had a probable causal relationship to the tumor resection, which per se was not a study-specific procedure. Other AEs CTCAE ≥ grade 2 included one patient (CB001) with an increase in a preexisting perifocal edema CTCAE grade 3 resulting in a deterioration of his hemiparesis and an expressive aphasia CTCAE grade 2. Patient CB004 developed a hemiparesis CTCAE grade 3 due to tumor progression. Patient CB006 suffered from 2 focal epileptic seizures CTCAE grade 3 in the context of his preexisting structural epilepsy. As in patient CB005, we did not observe an increase in frequency or severity of seizures during study participation. None of the AEs were deemed related to the study product or procedure. Importantly, none of the patients developed a CRS or an immune effector cell-associated neurotoxicity syndrome.
Table 2.
Adverse Events
| No. (%) of Patients (n = 9) | |||
|---|---|---|---|
| Adverse Event | Grade 3 | Grade 4 | Grade 5 |
| Possibly related | |||
| Central nervous system | |||
| Intracranial hemorrhage | 1 (11%) | 0 (0%) | 0 (0%) |
| Unrelated | |||
| Central nervous system | |||
| Ischemic stroke | 1 (11%) | 0 (0%) | 0 (0%) |
| Brain edema, localized | 1 (11%) | 0 (0%) | 0 (0%) |
| Epileptic seizure | 2 (22%) | 0 (0%) | 0 (0%) |
| Hemiparesis | 2 (22%) | 0 (0%) | 0 (0%) |
| Aphasia | 1 (11%) | 0 (0%) | 0 (0%) |
| Meningitis | 1 (11%) | 0 (0%) | 0 (0%) |
| Others | |||
| Increased liver enzymes | 1 (11%) | 0 (0%) | 0 (0%) |
All Adverse Events (AEs; including Severe Adverse Events) CTCAE grade ≥3 from all evaluable patients according to their causal relationship to the study product and study-related procedures as evaluated by the Data and Safety Monitoring Board (DSMB) of the CAR2BRAIN study.
Peripheral blood was collected at regular intervals from all patients, and extracted DNA was tested for the presence of the CAR sequence by qPCR. In 3 patients, CAR DNA was detected during the first days after intracranial NK-92/5.28.z injection (Supplementary Figure S2), indicating access of the cells to the circulation immediately following relapse surgery. The CAR sequence was not detected in any of the patients at later time points (up to day 517 in patient CB009 and day 447 in patient CB011; data not shown). Thus, there was no evidence for long-term engraftment of the irradiated NK-92/5.28.z cells. Total NK cell counts in peripheral blood of the patients remained within physiological limits (Supplementary Figure S3). Furthermore, we did not observe an elevation of troponin T levels, new electrocardiogram abnormalities, an increase of retention parameters, or any not otherwise explained liver enzyme elevations that would indicate solid organ toxicity.
Levels of a broad panel of cytokines were measured in patient sera at predefined time points, without observing an increase of pro-inflammatory factors after CAR-NK injection when compared to pretreatment values (Supplementary Figure S4). Of note, there was no elevation of IL-6 and IFNγ levels, which both play a central role in the development of CRS and ICANS. There were also no relevant changes in lymphocyte phenotypes including T-cell subtype distribution in peripheral blood after NK-92/5.28.z injection (Supplementary Figure S5). None of the patients showed induction of complement-activating antibodies to either HLA-typed lymphocytes, parental NK-92, or NK-92/5.28.z cells at week 4 after treatment. This indicates that intracranial CAR-NK treatment did not induce a humoral immune response to the cell-based product (Supplementary Table S4). Patient CB004 had preexisting anti-HLA-B7 antibodies, which did not increase at week 4 after treatment with NK-92/5.28.z.
Treatment Response
Low passage primary glioblastoma cell cultures could be established from the resected tumor tissues of 3 patients (CB005, CB006, and CB009). These GB cells retained HER2 surface expression and triggered CAR-NK cell cytotoxicity ex vivo (Supplementary Figure S6). Progression-free (PFS) and overall survival (OS) of the patients after tumor resection and CAR-NK cell injection are detailed in Figure 2A. Median PFS was 7 weeks (range 2–37 weeks). A median of 1 (range 0–3) lines of salvage therapies including re-irradiation, temozolomide, lomustine, etoposide, regorafenib, bevacizumab, nivolumab, and palbociclib were administered (Table 1). While PFS was within the range expected from historical control data for all other patients, with 37 weeks it was notably longer in 2 of the 3 patients treated at the highest dose level (CB009 and CB011). Even though permitted as per protocol, initiation of follow-up treatment while still taking part in the study was only necessary in patient CB003. Overall survival yielded a similar signal as PFS, with OS for most patients comparable to historical control data (Figure 2A). Median OS in the whole cohort was 31 weeks (range 18–135 weeks), but with patients CB009 and CB011 treated at the highest NK-92/5.28.z dose also showing an extended OS of 135 and 98 weeks after CAR-NK cell therapy, respectively. There was no correlation between HER2 score at inclusion or at relapse surgery with PFS or OS.
Figure 2.
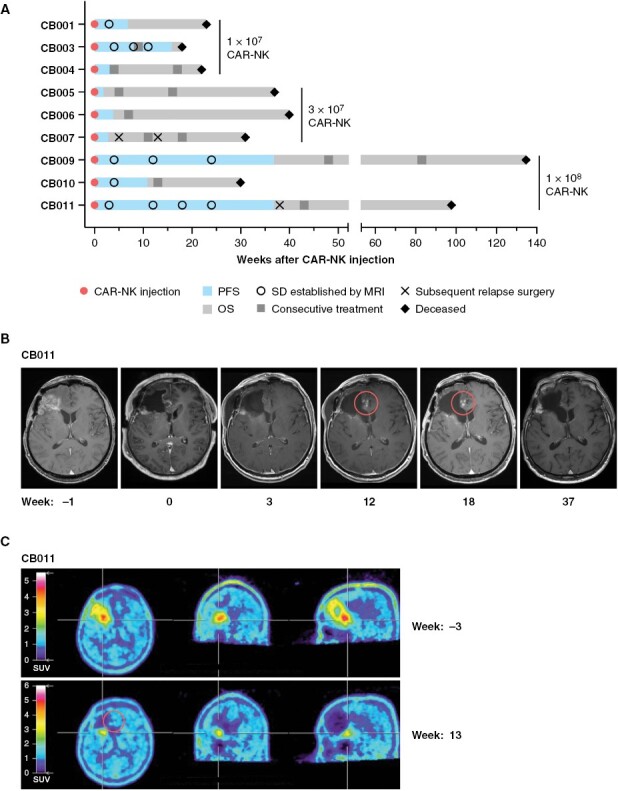
Treatment response. (A) Progression-free (PFS) and overall survival (OS) of patients treated with NK-92/5.28.z cells. Dose levels, time points of initiation of consecutive treatments, and subsequent additional relapse surgeries are indicated. Best response was stable disease (SD) for patients CB001, CB003, CB009, CB010, CB011, and progressive disease (PD) for patients CB004, CB005, CB007. (B) Magnetic resonance imaging (MRI) showing spot-like contrast enhancements in the resection margin in patient CB011 detected 12 and 18 weeks after local NK-92/5.28.z injections as a possible correlate of an induced immune reaction. (C) 18F-FET positron emission tomography indicating pseudoprogression in patient CB011.
Imaging did not yield any evidence for widespread inflammatory or encephalitic changes in the patients. Interestingly, in patient CB011 we observed transient spot-like contrast enhancements in MRI adjacent to the resection margin that appeared at week 12 after NK-92/5.28.z injection and resolved over the following weeks without further therapy (Figure 2B). Perfusion MRI did not indicate increased perfusion, and there was O-(2-[18F]fluoroethyl)-L-tyrosine enhancement in this area during positron emission tomography. The lesions were therefore classified as pseudoprogression (Figure 2C). Similar pseudoprogression was transiently observed in patient CB009 (not shown).
Correlation of Disease Course after CAR-NK Therapy with CD8+ T-Cell Infiltration
From patients CB007 and CB011 post-treatment tumor tissue was available following subsequent relapse surgeries. Patient CB005 consented to have an autopsy performed. We did not observe loss of HER2 antigen expression in post-treatment tumors as a conceivable escape mechanism, as HER2 expression was largely stable before and after CAR-NK cell therapy (HER2 scores of 3/12 and 4/12 for CB007; 8/12 and 6/12 for CB011). While the autopsy tissue from patient CB005 was HER2-negative (HER2 score before CAR-NK cell therapy: 6/12), beginning autolytic alterations were impeding evaluability. All available patient samples (primary, recurrent, and post CAR-NK therapy tumor tissues) were investigated using multiplex immunofluorescence panels that included CD3 (T-cells), CD8 (cytotoxic T lymphocytes), CD4/FoxP3 (regulatory T-cells), CD163 (myeloid cells), and von Willebrand factor (vasculature) (Figure 3A). Thereby, a higher proportion of intratumoral CD8+ T-cells in the recurrent but not the primary tumor tissue correlated with better PFS and OS (Figure 3B). Applying the median of 1.50% of total cells in the recurrent tumor samples being CD8+ T-cells as a cutoff value suggested a different probability of survival after CAR-NK therapy for the resulting patient groups, with a median PFS of 4 weeks (4 patients; range 3–7 weeks) for the group with <1.50% CD8+ T-cells (range 0.15%–0.60%) and 16 weeks (5 patients; range 2–37 weeks) for the group with ≥1.50% CD8+ T-cells (range 1.50%–6.40%) (P = .0528, Log-rank test) (Figure 3C). Median OS after NK-92/5.28.z treatment in these patient groups was 27 weeks (range 22–40 weeks) and 37 weeks (range 18–135 weeks), respectively (P = .3023; Log-rank test). Nevertheless, evaluation of a larger patient cohort would be needed to confirm this trend.
Figure 3.
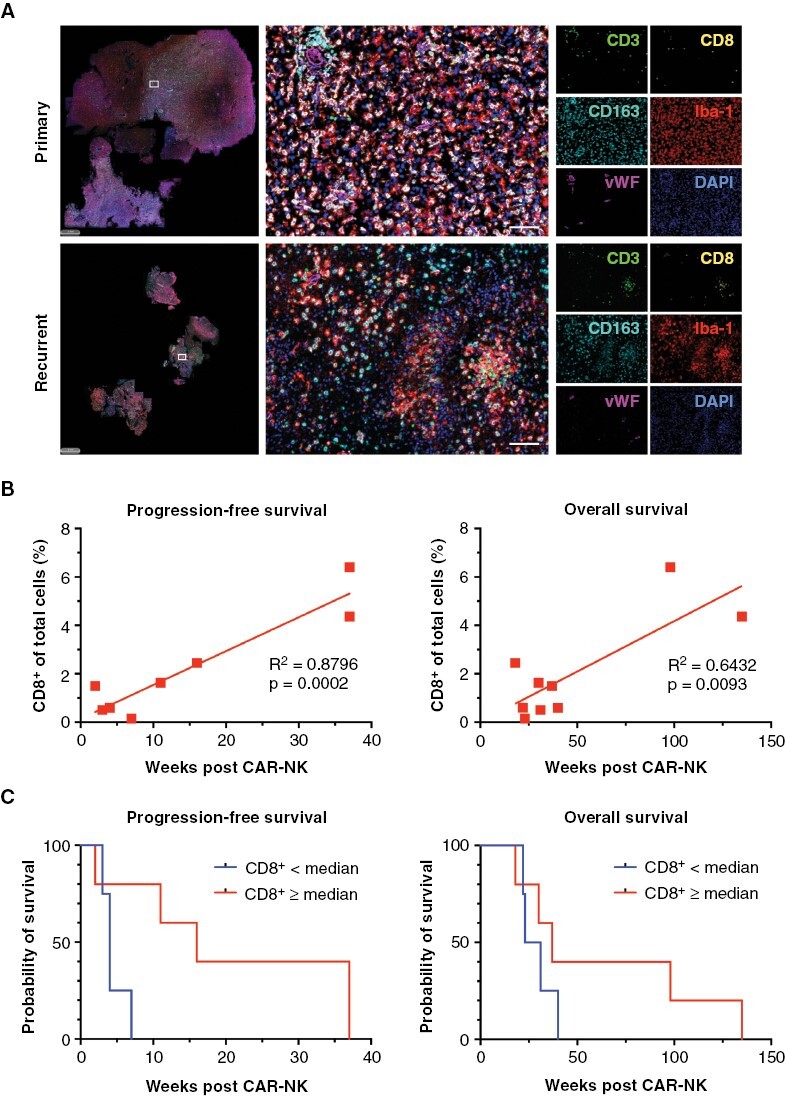
Spatial analysis of the tumor microenvironment and correlation with progression-free and overall survival following chimeric antigen receptor (CAR)-NK therapy. (A) Composite images showing Opal 6-color multiplex immunofluorescence stainings for CD3 (green), CD8 (yellow), Iba-1 (red), CD163 (cyan) and von Willebrand Factor (vWF, magenta) to identify T lymphocytes, myeloid, and endothelial cells. DAPI was used to detect nuclei (blue). Individual images of primary (treatment-naïve, upper panel) and recurrent (post radiation and chemotherapy with temozolomide, lower panel) glioma tissues of patient CB001 are shown. Scale bars: 100 µm. (B) Simple linear regression analysis revealing an increase in the proportion of CD8+ T-cells in recurrent tumor tissues prior to CAR-NK therapy as a significant predictor for progression-free (P = .0002, R2 = 0.8796) and overall survival (P = .0093, R2 = 0.6432). (C) Kaplan–Meier survival analysis post CAR-NK therapy based on CD8+ T-cell frequency in recurrent tumor tissues. Statistical significance was assessed using the Log-rank test.
Of note, patients CB009 and CB011 who achieved an extended PFS and OS after NK-92/5.28.z injection, also showed the most substantial infiltration with CD8+ T-cells in their recurrent tumors with 4.36% and 6.40%, respectively (Figure 3B). We also noticed changes in the proportions of infiltrating immune cells between recurrent tumor tissues obtained before and after CAR-NK therapy in patients CB007 and CB011, where a subsequent relapse surgery was performed (Figure 4). Thereby, primary tumor tissue of patient CB007, recurrent tumor tissue collected immediately before CAR-NK injection and upon a second relapse early after CAR-NK injection that was resected at week 5, all displayed low levels of CD8+ T-cell infiltration, but with the proportion of PD-1+ cells within the CD8+ T-cell population steadily increasing from 30% (primary tumor) to 83% (second recurrence) (Figure 4A). The recurrent tumor of patient CB011 that developed relatively late after CAR-NK treatment (PFS of 37 weeks) and was resected at week 38 displayed markedly reduced CD8+ T-cell counts when compared to the value at the time point of adoptive cell therapy (Figure 4B). Nevertheless, the PD-1+ subpopulation of CD8+ T-cells remained low in this patient even upon second recurrence.
Figure 4.
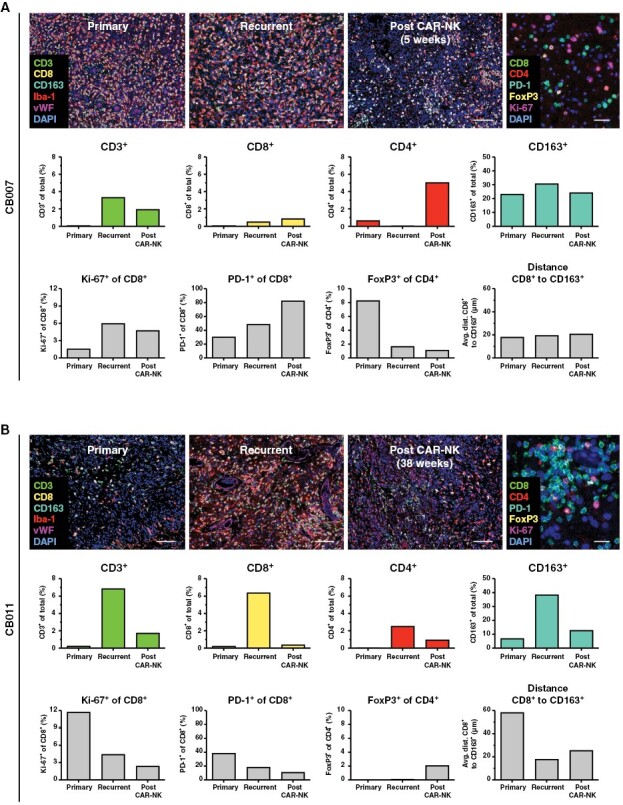
Spatial analysis of immune cell subpopulations in the course of disease progression. Multiplex immunofluorescence images of regions of interests selected within whole slide scans derived from primary, recurrent and post chimeric antigen receptor (CAR)-NK therapy tumor tissues of patients CB007 (A) and CB011 (B) are displayed. Two different staining panels to detect CD3, CD8, CD163, Iba-1, vWF, DAPI (mixed panel; scale bars: 100 µm), and CD8, CD4, PD-1, FoxP3, Ki-67, DAPI (lymphoid panel; scale bars: 50 µm) were employed. Staining with the lymphoid panel is shown for recurrent tumor tissue as an example.
Whole exome sequencing was performed, and tumor mutational burden (TMB) and microsatellite instability (MSI) scores were determined for all of the 21 available patient samples (initial tumor and relapse tumor prior to CAR-NK cell injection from all 9 patients, subsequent relapse tumor from patients CB007 and CB011, autopsy tissue from patient CB005). Both TMB and MSI were in a low range, with the exception of a recurrent tumor in one patient (CB005; Supplementary Table S5). Neither TMB nor MSI scores predicted disease course after therapy. To characterize the cellular composition of the tumor microenvironment on an epigenetic level, large-scale DNA methylation analysis, followed by reference-based deconvolution to determine the proportions of infiltrating CD14+ monocytes, CD56+ NK cells, CD8+ and CD4+ effector T-cells, regulatory T-cells, CD19+ B cells, eosinophils, and neutrophils was performed. Principal component analysis neither revealed a clustering according to best response (SD vs. PD) following CAR-NK treatment, nor the type of recurrent tumor sample (pre or post CAR-NK injection) (Supplementary Figure S7).
Discussion
The dose-escalation cohort of the CAR2BRAIN study reported here demonstrates feasibility and safety of local intracerebral injections of up to 1 × 108 HER2-CAR NK cells. Neither neurotoxic nor systemic side effects of NK-92/5.28.z therapy were apparent. This is in accordance with the favorable safety profile of CAR-NK cells in the systemic treatment of leukemia and lymphoma.11
Pseudoprogression in patients CB009 and CB011 observed in the injected area several weeks after CAR-NK treatment at the highest dose level may represent a signal of immune activation by the experimental therapy, encouraging further evaluation of this approach. Furthermore, it is intriguing to note that for all patients the extent of CD8+ T-cell infiltration in pretreatment tumor tissue correlated with PFS and OS. Whether this represents an independent predictive parameter as suggested by a recent report,28 or is related to outcome after CAR-NK cell therapy remains to be determined.
HER2 is expressed in a substantial proportion of glioblastoma patients and constitutes a relevant target for immunotherapy.5,7,23 The HER2 screening method employed here allowed rapid evaluation of a large number of patients, with a good correlation between the screening performed on archival tissue and the results from the tissue resected during the study intervention. Indeed, low passage glioblastoma cell cultures obtained from resected tumor tissues of 3 patients at the time point of CAR-NK injection revealed continued HER2 surface expression of the patient-derived cells and sensitivity to HER2-specific NK-92/5.28.z cells ex vivo.
Broadening the experience from earlier trials with systemic application of NK-92 cells,29–34 our study extends the evidence for safe and potentially effective therapies based on this cell line to intracerebral injections of a HER2-CAR-engineered permanent cell clone. After generation and extensive pharmacological characterization and biosafety testing, CAR-engineered variants such as NK-92/5.28.z can be stored as cell banks for unlimited time, providing a well-defined product readily available on demand.19,24 The efficient workflow established in this study, in contrast to current CAR-T-cell therapies, reduces the time between treatment decision and administration of the cells to 1 week, without the necessity to rely on the patient’s own immune cells. NK-92 cells have a unique phenotype with the absence of most inhibitory killer cell immunoglobulin-like receptors and high expression of activating natural cytotoxicity receptors. In contrast to primary NK cells, they can be continuously expanded without changing their phenotype, facilitating the generation of stable off-the-shelf cell products that include genetically engineered variants.24,29,35 Disadvantages of NK-92 cells include the need to irradiate them for safety reasons before injection or infusion into patients, which requires repeated treatments for prolonged activity as currently done in the expansion cohort of our trial. In contrast, CAR-NK cells generated from primary NK cells of healthy donors do not require irradiation and can therefore engraft for several months if they are additionally engineered to produce IL-15.11,16 Still, the more limited ex vivo expansion potential of primary NK cells and the generally low efficiency of gene transfer makes it more difficult to generate the cell numbers needed for clinical application, in particular when avoiding feeder-cell based expansion.36,37 Features desirable for CAR effectors that distinguish NK cells from T lymphocytes include the lack of a T-cell receptor of unknown specificity, the NK cells’ endogenous natural cytotoxicity, and the apparently reduced risk for severe side effects such as CRS and ICANS.11,38 NK cells may also provide more efficient immune activation through crosstalk with bystander immune cells like dendritic cells.39
This is the first full report of a CAR-NK cell-based intervention in a solid tumor indication. Specific challenges for CAR-NK cell treatment of glioblastoma include choosing an optimal route of application, and overcoming the hypoxic and immunosuppressive tumor microenvironment.40 Concerning the latter, we found in preclinical experiments that NK-92/5.28.z cells are highly resistant to hypoxia, and retain their specific cell-killing activity even at an oxygen concentration as low as 0.1% and at high concentrations of TGF-β.23 Limitations of our study include the necessity to use for safety reasons irradiated NK-92/5.28.z cells. Nevertheless, upon repeated intracranial injection, irradiated and non-irradiated NK-92/5.28.z cells were similarly effective against orthotopic glioblastoma tumors in xenograft models.23 In immunocompetent mice, in addition, endogenous antitumor immunity based on IgG antibodies and T-cells was induced by the CAR-NK treatment, which was critical to achieve cures.23,24 This indicates (1) the CAR-NK cells’ high cytotoxic activity irrespective of prior irradiation, and (2) their general ability to mobilize an endogenous immune response against the tumor.
Treatment in glioblastoma patients was limited to a single application in the dose-escalation part of our study due to regulatory restraints. Hence, the primary endpoint of the CAR2BRAIN phase I trial was to establish safety of single injections, which may not be sufficient for pronounced antitumor efficacy and effective immune activation. While in in vitro assays cytotoxicity of NK-92/5.28.z cells remained unaffected by high concentrations of corticosteroids (Supplementary Figure S8), intraoperative dexamethasone treatment may still have affected indirect effects of the CAR-NK cells mediated through crosstalk with endogenous immune cells. We chose local administration of the single dose distributed as multiple injections into the wall of the resection cavity to establish safety by ensuring contact of CAR-NK cells with both, remaining tumor cells and normal neural tissue. In support of this, encouraging signals for the potential of local application of cellular therapies in gliomas have also been obtained by studies applying CAR-T-cells into the resection cavity or through ventricular catheters.4,7,8 The latter study also provided evidence for immune activation following therapy. It is noteworthy to mention the possibility of inducing neutralizing antibodies to NK-92 cells or the CAR construct. No such antibodies were detected in the dose-escalation part, although they may develop after repetitive injections. Strengths of our study include the tight and complete neuroradiological follow-up, uncontaminated by salvage therapies up to the first progression after CAR-NK cell therapy in 8 out of 9 patients, and the availability of tissue from recurrent tumors after CAR-NK cell therapy in 2 patients, enabling detailed immunological profiling of recurrent tumors. For complete follow-up, after tumor progression and end of study participation, all patients were followed until they deceased.
The data obtained will be instrumental for the further development of local CAR-NK cell therapies. While our preclinical data in immunocompetent mouse models have demonstrated that activation of an endogenous immune response by NK-92/5.28.z cells is pivotal for efficient antitumor activity and protection from tumor rechallenge,23 it will be crucial to elucidate preexisting properties of the glioblastoma cells and the immune cell architecture in the tumor microenvironment that may promote this synergistic effect also in human patients. Following regulatory requirements, we irradiated the NK-92/5.28.z cells before injection into glioblastoma patients as it is currently done due to safety reasons for all NK-92-based therapeutics. This limits the time interval of their potential therapeutic activity. Hence, in the ongoing expansion cohort patients receive in addition to the first injection during relapse surgery up to 12 additional weekly injections of NK-92/5.28.z through an implanted catheter, intended to prolong exposure of remaining tumor cells to active CAR-NK cells. Preclinical work of our group has shown that co-treatment with an anti-PD-1 immune checkpoint inhibitor can invigorate immune responses induced by NK-92/5.28.z treatment, providing lasting tumor control even in more advanced tumor stages.41,42 To exploit this mechanism, we have amended the study protocol of the CAR2BRAIN trial and added an additional cohort of glioblastoma patients who will, following completion of the currently recruiting expansion cohort with repetitive injections of NK-92/5.28.z cells through an implanted Rickham reservoir, receive both, local CAR-NK cell injections into the resection cavity and systemic anti-PD-1 antibody to interrogate safety and antitumor activity of this combination.
Supplementary Material
Acknowledgments
We thank the patients and their families for their contributions to the study. We are indebted to Stephen Gottschalk and Nabil Ahmed for generous advice on the study protocol, and to Erhard Seifried and Red Cross Blood Donation Service Baden-Württemberg-Hessen for providing the infrastructure for GMP production of the study therapeutic. We thank Sina Hehn for her assistance in developing and programming the eCRF forms, Stefanie Jordan and Daniel P. Brucker for PBMC isolation, and Tim R. Fenton for contributing the reference sheets for tumor deconvolution. We gratefully acknowledge NantKwest for providing NK-92 cells from their proprietary master cell bank which we used as a basis for the generation of the CAR-engineered NK-92/5.28.z cell clone.
Contributor Information
Michael C Burger, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Marie-Therese Forster, Department of Neurosurgery, Goethe University Hospital, Frankfurt, Germany.
Annette Romanski, Institute for Transfusion Medicine and Immunohematology, Goethe University, Frankfurt and Red Cross Blood Donation Service Baden-Württemberg-Hessen, Frankfurt, Germany.
Florian Straßheimer, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Jadranka Macas, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Pia S Zeiner, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Eike Steidl, Institute of Neuroradiology, Goethe University Hospital, Frankfurt, Germany.
Stefanie Herkt, Institute for Transfusion Medicine and Immunohematology, Goethe University, Frankfurt and Red Cross Blood Donation Service Baden-Württemberg-Hessen, Frankfurt, Germany.
Katharina J Weber, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany; University Cancer Center (UCT), Goethe University Hospital, Frankfurt, Germany.
Jonathan Schupp, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Jennifer H Lun, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Maja I Strecker, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Karolin Wlotzka, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Pinar Cakmak, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Corinna Opitz, Institute for Transfusion Medicine, German Red Cross Blood Donation Service North-East and Medical Faculty Carl Gustav Carus, TU Dresden, Dresden, Germany.
Rosemol George, German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; Department of Radiotherapy and Oncology, Goethe University Hospital, Frankfurt, Germany.
Iris C Mildenberger, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany.
Paulina Nowakowska, Institute for Transfusion Medicine and Immunohematology, Goethe University, Frankfurt and Red Cross Blood Donation Service Baden-Württemberg-Hessen, Frankfurt, Germany.
Congcong Zhang, Georg-Speyer-Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany.
Jasmin Röder, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Georg-Speyer-Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany.
Elvira Müller, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Kristina Ihrig, University Cancer Center (UCT), Goethe University Hospital, Frankfurt, Germany.
Karl-Josef Langen, Research Center Jülich, Institute of Neuroscience and Medicine, Jülich, Germany; Department of Nuclear Medicine, University Hospital Aachen, Aachen, Germany.
Michael A Rieger, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Medicine II, Hematology/Oncology, Goethe University Hospital, Frankfurt, Germany.
Eva Herrmann, Institute for Biostatistics and Mathematical Modelling, Goethe University, Frankfurt, Germany.
Halvard Bonig, Institute for Transfusion Medicine and Immunohematology, Goethe University, Frankfurt and Red Cross Blood Donation Service Baden-Württemberg-Hessen, Frankfurt, Germany.
Patrick N Harter, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Yvonne Reiss, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Elke Hattingen, Institute of Neuroradiology, Goethe University Hospital, Frankfurt, Germany.
Franz Rödel, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; Department of Radiotherapy and Oncology, Goethe University Hospital, Frankfurt, Germany.
Karl H Plate, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; Institute of Neurology (Edinger Institute), Goethe University Hospital, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany.
Torsten Tonn, Institute for Transfusion Medicine, German Red Cross Blood Donation Service North-East and Medical Faculty Carl Gustav Carus, TU Dresden, Dresden, Germany; German Cancer Consortium (DKTK), Partner Site Dresden, Dresden, Germany.
Christian Senft, Department of Neurosurgery, Goethe University Hospital, Frankfurt, Germany.
Joachim P Steinbach, Dr. Senckenberg Institute of Neurooncology, Goethe University Hospital, Frankfurt, Germany; Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany.
Winfried S Wels, Frankfurt Cancer Institute (FCI), Goethe University, Frankfurt, Germany; German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Frankfurt, Germany; Georg-Speyer-Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany.
Funding
This work was supported in part by grants from the German Federal Ministry of Education and Research (BMBF) (Cluster für individualisierte Immunintervention, Ci3; FKZ 131A009), a research grant from NantKwest, grants from LOEWE Center for Cell and Gene Therapy Frankfurt (CGT) (II L 5-518/17.004 2013, 2016) and LOEWE Center Frankfurt Cancer Institute (FCI) (III L 5-519/03/03.001 - [0015]), both funded by the Hessian Ministry of Higher Education, Research and the Arts (HMWK), German Cancer Consortium (DKTK) (Joint Funding Upgrade project CAR2BRAIN), ForTra gGmbH für Forschungstransfer of Else Kröner-Fresenius-Stiftung (2020_EKTP21) and institutional funds of the Institute for Neurooncology and Georg-Speyer-Haus. Georg-Speyer-Haus is funded jointly by the German Federal Ministry of Health and Hessian Ministry of Higher Education. MCB obtained grants from intramural funding from Goethe University (Frankfurter Forschungsförderung: Nachwuchsforscher program 2018, Clinician Scientist program 2020). KJW was supported by the Mildred Scheel Career Center Frankfurt, funded by Deutsche Krebshilfe.
Conflict of interest statement
CZ, TT, and WSW are named as inventors on patents and patent applications related to the study therapeutic owned by their respective academic institutions. MCB received honoraria for lectures or advisory board participation from Bristol Myers Squibb and Gilead Sciences. CS received honoraria for lectures, advisory board participation, expert testimony, or travel support from Stryker, Bayer, Brainlab, and Novocure. JPS received grants from Merck and UCB as well as honoraria for lectures, travel, or advisory board participation from Abbvie, Bristol-Myers Squibb, Medac, Roche, Novocure, and UCB. HB acknowledges speakers’ and advisory board honoraria from Boehringer-Ingelheim, Bristol Myers Squibb, Novartis, Sandoz-Hexal, and Medac as well as royalties and licensing fees from Medac, all unrelated to the work presented herein. No potential conflicts of interest were disclosed by the other authors.
Authorship statement
MCB, ICM, KHP, TT, CS, JPS, and WSW designed the study. EHe developed the statistical plan. MCB, MTF, PSZ, CS, and JPS enrolled and treated patients. MCB, MTF, PSZ, ES, KJW, EM, KI, KJL, PNH, EHa, KHP, CS, and JPS gathered clinical data, MCB, AR, FS, JM, SH, KJW, JS, JHL, MIS, KW, PC, CO, RG, ICM, PN, CZ, JR, MAR, HB, PNH, YR, FR, KHP, TT, JPS, and WSW performed experiments and acquired, analyzed and interpreted data. AR, SH, PN, HB, and TT produced the study product. All authors contributed to writing the manuscript, provided critical feedback during editing, and approved the final submitted version.
Data Availability
To comply with data privacy laws, individual participant data are not available for sharing. All other data supporting the findings of this study are available within the article, the supplementary information, or from the corresponding authors upon reasonable request.
References
- 1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS.. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 2021;23(12 suppl 2):iii1–iii105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lombardi G, De Salvo GL, Brandes AA, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019;20(1):110–119. [DOI] [PubMed] [Google Scholar]
- 3. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vitanza NA, Johnson AJ, Wilson AL, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. 2021;27(9):1544–1552. [DOI] [PubMed] [Google Scholar]
- 8. Majzner RG, Ramakrishna S, Yeom KW, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miller JS, Lanier LL.. Natural killer cells in cancer immunotherapy. Annu Rev Cancer Biol. 2019;3(1):77–103. [Google Scholar]
- 10. Maskalenko NA, Zhigarev D, Campbell KS.. Harnessing natural killer cells for cancer immunotherapy: Dispatching the first responders. Nat Rev Drug Discov. 2022;21(8):559–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Freyer CW, Porter DL.. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic malignancies. J Allergy Clin Immunol. 2020;146(5):940–948. [DOI] [PubMed] [Google Scholar]
- 13. Berger TR, Maus MV.. Mechanisms of response and resistance to CAR T cell therapies. Curr Opin Immunol. 2021;69:56–64. [DOI] [PubMed] [Google Scholar]
- 14. Uherek C, Tonn T, Uherek B, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100(4):1265–1273. [PubMed] [Google Scholar]
- 15. Imai C, Iwamoto S, Campana D.. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Hermanson DL, Moriarity BS, Kaufman DS.. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–192.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schönfeld K, Sahm C, Zhang C, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nowakowska P, Romanski A, Miller N, et al. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Press MF, Cordon-Cardo C, Slamon DJ.. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5(7):953–962. [PubMed] [Google Scholar]
- 21. Schlegel J, Stumm G, Brandle K, et al. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J Neurooncol. 1994;22(3):201–207. [DOI] [PubMed] [Google Scholar]
- 22. Liu G, Ying H, Zeng G, et al. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980–4986. [DOI] [PubMed] [Google Scholar]
- 23. Zhang C, Burger MC, Jennewein L, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108(5):djv375. [DOI] [PubMed] [Google Scholar]
- 24. Zhang C, Oberoi P, Oelsner S, et al. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burger MC, Zhang C, Harter PN, et al. CAR-engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Westphal M, Ylä-Herttuala S, Martin J, et al. ; ASPECT Study Group. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(9):823–833. [DOI] [PubMed] [Google Scholar]
- 27. Ceccon G, Lohmann P, Stoffels G, et al. Dynamic O-(2-18F-fluoroethyl)-L-tyrosine positron emission tomography differentiates brain metastasis recurrence from radiation injury after radiotherapy. Neuro Oncol. 2017;19(2):281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mauldin IS, Jo J, Wages NA, et al. Proliferating CD8(+) T cell infiltrates are associated with improved survival in glioblastoma. Cells. 2021;10(12):33783378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tonn T, Becker S, Esser R, Schwabe D, Seifried E.. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10(4):535–544. [DOI] [PubMed] [Google Scholar]
- 30. Arai S, Meagher R, Swearingen M, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy. 2008;10(6):625–632. [DOI] [PubMed] [Google Scholar]
- 31. Tonn T, Schwabe D, Klingemann HG, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15(12):1563–1570. [DOI] [PubMed] [Google Scholar]
- 32. Boyiadzis M, Agha M, Redner RL, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19(10):1225–1232. [DOI] [PubMed] [Google Scholar]
- 33. Williams BA, Law AD, Routy B, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8(51):89256–89268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tang X, Yang L, Li Z, et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 35. Klingemann H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy. 2023;25(5):451–457. [DOI] [PubMed] [Google Scholar]
- 36. Suck G, Odendahl M, Nowakowska P, et al. NK-92: an “off-the-shelf therapeutic” for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65(4):485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wagner J, Pfannenstiel V, Waldmann A, et al. A two-phase expansion protocol combining interleukin (IL)-15 and IL-21 improves natural killer cell proliferation and cytotoxicity against rhabdomyosarcoma. Front Immunol. 2017;8:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laskowski TJ, Biederstädt A, Rezvani K.. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. 2022;22(10):557–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferlazzo G, Morandi B.. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol. 2014;5:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim AR, Choi SJ, Park J, et al. Spatial immune heterogeneity of hypoxia-induced exhausted features in high-grade glioma. Oncoimmunology. 2022;11(1):2026019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strassheimer F, Strecker MI, Zhang C, et al. Synergistic effects of combination therapy of CAR-NK cells and anti-PD-1 antibody result in high efficacy against advanced stage orthotopic glioblastoma grafts in a syngeneic mouse model and induce protective anti-tumor immunity in vivo. Neuro Oncol. 2019;21(suppl 3):iii60. [Google Scholar]
- 42. Strecker MI, Wlotzka K, Strassheimer F, et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology. 2022;11(1):2127508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To comply with data privacy laws, individual participant data are not available for sharing. All other data supporting the findings of this study are available within the article, the supplementary information, or from the corresponding authors upon reasonable request.