

Abstract
Background
Diffuse gliomas represent over 80% of malignant brain tumors ranging from low-grade to aggressive high-grade lesions. Within isocitrate dehydrogenase (IDH)-mutant gliomas, there is a high variability in survival and a need to more accurately predict outcome.
Methods
To identify and characterize a predictive signature of outcome in gliomas, we utilized an integrative molecular analysis (using methylation, mRNA, copy number variation (CNV), and mutation data), analyzing a total of 729 IDH-mutant samples including a test set of 99 from University Health Network (UHN) and 2 validation cohorts including the German Cancer Research Center (DKFZ) and The Cancer Genome Atlas (TCGA).
Results
Cox regression analysis of methylation data from the UHN cohort identified CpG-based signatures that split the glioma cohort into 2 prognostic groups strongly predicting survival that were validated using 2 independent cohorts from TCGA and DKFZ (all P-values < .0001). The methylation signatures that predicted poor outcomes also exhibited high CNV instability and hypermethylation of HOX gene probes. Integrated multi-platform analyses using mRNA and methylation (iRM) showed that parallel HOX gene overexpression and simultaneous hypermethylation were significantly associated with increased mutational load, high aneuploidy, and worse survival (P-value < .0001). A 7-HOX gene signature was developed and validated using the most significantly associated HOX genes with patient outcome in both 1p/19q codeleted and non-codeleted IDHmut gliomas.
Conclusions
HOX gene methylation and expression provide important prognostic information in IDH-mutant gliomas that are not captured by current molecular diagnostics. A 7-HOX gene signature of outcome shows significant survival differences in both 1p/19q codeleted and non-codeleted IDH-mutant gliomas.
Keywords: gene expression, HOX gene, IDH-mutant gliomas, integrated molecular analysis, methylation
Key Points.
Isocitrate dehydrogenase (IDH)-mutant gliomas with both hypermethylation and overexpression of HOX genes have worse outcome
This group of gliomas exhibited elevated copy number instability and increased mutational load
A 7-HOX gene signature of outcome was developed with prognostic implications in both 1p/19q codeleted and non-codeleted IDH-mutant gliomas
Importance of the Study.
In this study, we characterized a methylation signature that is predictive of outcomes in isocitrate dehydrogenase (IDH)-mutant gliomas. The comprehensive individual and integrated RNA and methylation (iRM) analyses showed that IDH-mutant gliomas with hypermethylation and increased expression of HOX genes have significantly worse outcomes. We further identified a 7-HOX gene signature that correlated to significantly worse survival in both 1p/19q codeleted and non-codeleted IDH-mutant gliomas. This provides valuable new insights into the potential of HOX genes for determination of new molecular subtypes of glioma that are not resolvable by conventional histological means and is also independent of previous molecular subtypes. The novel prognostic subgroups based on iRM provide new insights into further molecular subtyping of IDH-mutant gliomas. These results demonstrate the importance of integrated HOX gene methylation and expression in gliomas, and a 7-HOX gene signature that can serve as a predictor of outcome in IDH-mutant gliomas.
Diffuse gliomas represent over 80% of all malignant brain tumors with highly variable survival outcomes, ranging from lower-grade tumors to high-grade aggressive malignancies.1 Recent advances in molecular characterization of diffuse gliomas identified several molecular markers with diagnostic and prognostic relevance including the mutations in isocitrate dehydrogenase (IDH) genes.2,3 Classically, IDH mutation (IDHmut) status was the main factor used for classification and prognostication of diffuse gliomas.4 IDH mutations often occur in younger patients and the presence of an IDHmut confers a distinct survival advantage compared to similar IDH wild-type (IDHwt) tumors.2,4 Similarly, within IDHmut gliomas, combined allelic loss of chromosomes 1p and 19q (1p/19q codeletion) is a significant predictor of improved chemosensitivity and overall survival.5 For the first time, molecular markers are now used in addition to histology as part of a recent World Health Organization (WHO) classification grading system to reduce inter-observer variability and provide accurate stratification.4,6 The newest WHO classification has reserved “glioblastoma (GBM)” solely for IDHwt tumors, with previously classified IDHmut GBM now referred to as IDHmut grade 4 astrocytomas.6 Subsequent analyses including homozygous deletion of the CDKN2A gene are also associated with worse prognosis in both 1p/19q codeleted and non-codeleted IDHmut gliomas, independent of histologic markers including microvascular proliferation and necrosis and astrocytomas with CDKN2A homozygous deletion are now reclassified as grade 4 IDHmut tumors.6,7 Despite these discoveries, there remains a high variability in outcome of IDHmut gliomas that cannot be explained by currently known molecular markers.8
A multi-platform pan-cancer project from The Cancer Genome Atlas (TCGA) Research Network investigated 6 different platforms including DNA methylation, mRNA expression, DNA copy number, microRNA expression, protein expression, and exome mutation.9 These platforms often complement or exceed the accuracy of tumor classification based solely on histology or morphology and their integration can help identify biologically distinct subtypes that are not resolvable by morphology alone.10 Previously, we have shown that combined mRNA and methylation analysis distinguishes different survival classes for various tumor types and provides a clinically and biologically relevant stratification based on molecular classification.11
DNA methylation alterations have widely been reported in human cancers. One recent example of this involves the HOX genes.12–15 Cancer-specific methylation patterns including global hypomethylation and gene or region-specific hypermethylation are common.16 HOX genes are a highly conserved subgroup of the homeobox gene family that regulate numerous cellular processes including apoptosis and angiogenesis.12 Aberrant HOX gene expression and methylation are associated with various malignancies, and recent research highlighted the importance of these genes as both biomarkers and potential therapeutic targets.12–15
Within the CNS (central nervous system), HOX genes play critical roles in the developing brain including cell fate determination, which aid in the establishment of functional neuronal networks and the formation of distinct neuronal subtypes.17 While HOX genes are mostly absent from healthy adult brains, the presence of HOX gene deregulation and aberrant expression have been detected in malignant brain tumors, mainly GBM.17 Multiple prior studies exploring HOX methylation and gene expression in GBM demonstrated their role in treatment resistance, tumor aggressiveness, and poor prognosis.18–20 More specifically, HOXA9 has been identified as an oncogene in GBM, with activation leading to decreased apoptosis, increased cellular proliferation, and shorter survival.21–23
Within IDHmut gliomas, DNA methylation analyses using the promoter regions in IDHmut grade 4 astrocytomas from TCGA identified and validated a glioma-CpG island methylator phenotype (G-CIMP) associated with younger age and improved outcome.24 This signature has also been extended to IDHmut glioma.25 Although unsupervised analysis of genome-wide methylation signatures can group tumors based on IDHmut and 1p/19q codeletion status, further exploration of DNA methylation subtypes and integration of DNA methylation with other genomic platforms including gene expression is needed to better understand this heterogeneous disease and to leverage these signatures as additional predictors of outcome.26
In this study, we aimed to determine a biomarker signature of outcome in IDHmut gliomas using an independent cohort combined with 2 external validation cohorts by expanding our preliminary study,27 and further characterize this predictive signature through multi-platform analysis integrating methylation, gene expression, and somatic mutation.
Materials and Methods
Study Overview
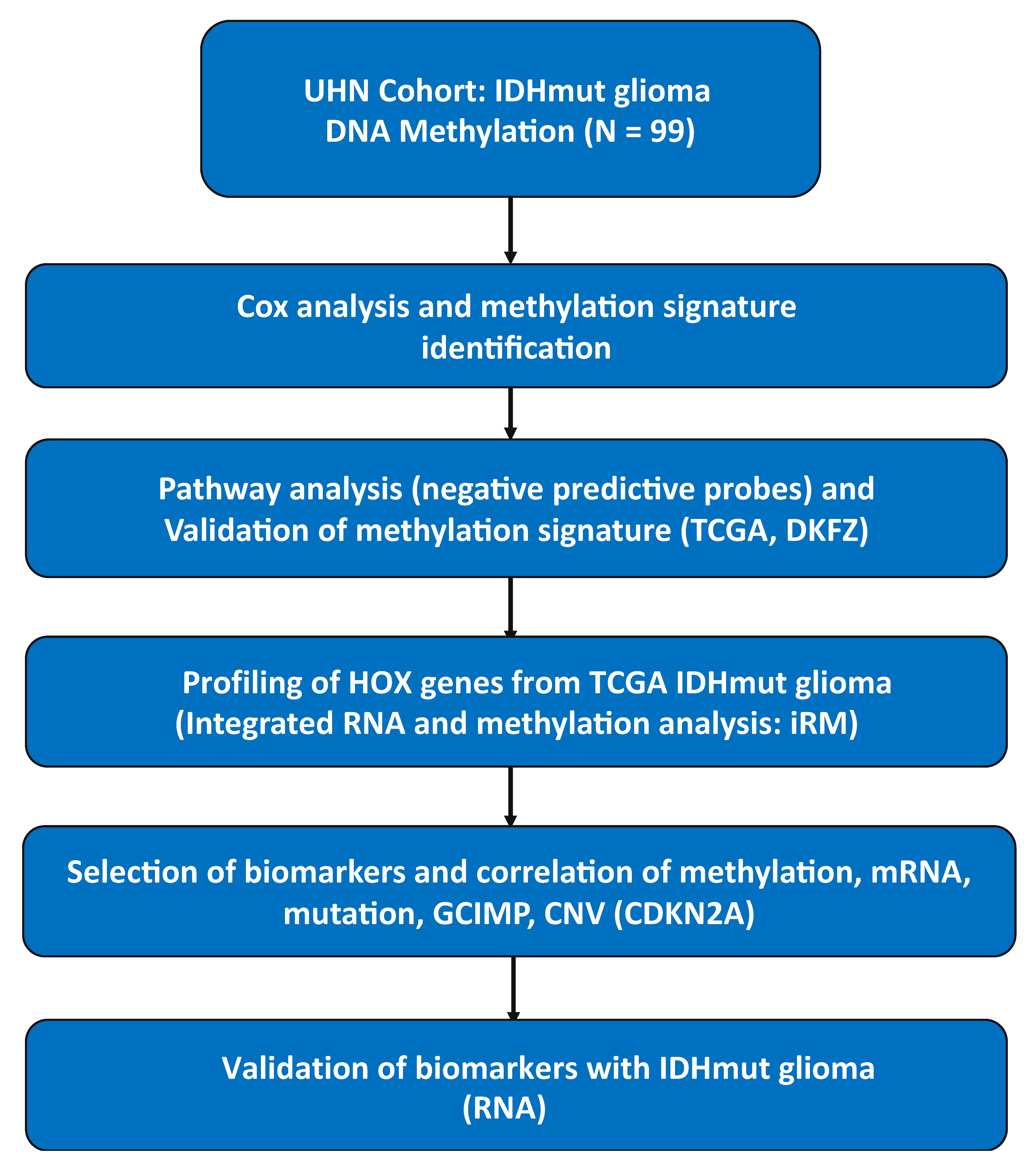
The overview and outline of the study are available in Supplementary Figure 1. Using IDHmut gliomas, we examined 3 cohorts of methylation datasets including a cohort at the University Health Network (UHN) as a training set, and 2 validations set from TCGA and German Cancer Research Center (DKFZ) totaling 729 samples: 99 from UHN, 419 from TCGA and 211 from DKFZ (Table 1).
Table 1.
Patient Characteristics of IDH-Mutant Gliomas Based on University Health Network, The Cancer Genome Atlas, and DKFZ Cohorts.27 The CDKN2A Status is for the CDKN2A Homozygous Deletion.
| Characteristics | Cohorts | ||
|---|---|---|---|
| UHN | TCGA | DKFZ | |
| N = 99 | N = 419 | N = 211 | |
| Pathological grade | |||
| 2 | 29 (29.3%) | 221 (52.7%) | 54 (25.6 %) |
| 3 | 59 (59.6%) | 190 (45.3%) | 90 (42.7%) |
| 4 | 11 (11.1%) | 7 (1.7%) | 67 (31.8%) |
| Unknown | 0 | 1 (0.2%) | 0 |
| CDKN2A status | |||
| Retained | 76 (76.8%) | 397 (94.7%) | 173 (82%) |
| Lost | 23 (23.2%) | 22 (5.3%) | 38 (18%) |
| WHO 2021 grade | |||
| 2 | 26 (26.3%) | 108 (25.8%) | 54 (25.6 %) |
| 3 | 46 (46.5%) | 232 (55.4%) | 75 (35.5%) |
| 4 | 27 (27.3%) | 29 (6.9%) | 82 (38.9%) |
| G-CIMP status | |||
| High | 47 (47.5%) | 234 (55.8%) | 185 (87.7%) |
| Low | 7 (7.1%) | 12 (2.9%) | 26 (12.3%) |
| Codel | 45 (45.4%) | 172 (41.1%) | 0 |
| Unknown | 0 | 1 (0.2%) | 0 |
| Age (at diagnosis) | |||
| Median (range) | 39 (22–73) | 39 (14–75) | 36 (15–70) |
| Unknown | 0 | 2 | 5 |
| Survival (months) | |||
| Median (range) | 90.8 (2.5–227.2) | 25.8 (0–182.3) | 49.7 (0.23–268) |
| Unknown | 0 | 1 | 0 |
UHN and DKFZ Methylation Data Processing
We profiled a total of 99 IDHmut glioma (53 astrocytomas and 46 oligodendrogliomas) samples based on Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) data (450K) which were processed and analyzed at the UHN/ Princess Margaret Genomics Center after local research ethics board approval. 27 In addition to our cohort from UHN, we also had a total of 211 IDHmut glioma samples based on 450K methylation data from DKFZ (144 astrocytomas of which 54 were grade 2 and 90 grade 3, and 67 grade 4 IDHmut astrocytomas). We used the Bioconductor package (version 3.3) for loading and processing of the methylation data. The generated methylation data was preprocessed using the Minfi package. We performed ssNoob normalization for the samples that passed quality control after excluding failed probes. Further methylation data filtering was performed at cross-reactive probes and single nucleotide polymorphisms at CpG sites using dropLociWithSnps function in the Minfi package. We obtained the methylation values of each CpG site by β value ranging from 0 (unmethylated) to 1 (fully methylated).
Copy Number Variation Obtained From Methylation
We obtained copy number variation (CNV) (including CDKN2A loss) from the 450K data using a Bioconductor-based R package called conumee. Conumee generates copy number calls and shows whole chromosome views to pre-select genes with qualitative features. The current conumee package has 450K methylation profiles of normal tissue samples to be used as a reference.
Consensus Clustering
We performed unsupervised hierarchical clustering of significant probes from Cox regression analysis. Rows are for the genes and columns are for the samples. We also used ConsensusClusterPlus Bioconductor package with Spearman correlation for the distance metric and Ward (Ward.2) for the linkage algorithm with 1000 resampling steps (epsilon = 0.8) to ensure the robustness of the putative cluster to resampling variability and stability of clustering results. Silhouette statistical analysis of subgroup hierarchical structures indicates the optimal number of clusters.
Selection of 7-HOX Genes as a Biomarker to Predict OS
We generated a table of Cox regression analysis (Supplementary Table 1) for all HOX genes of methylation and mRNA in the IDHmut glioma cohort from TCGA. We evaluated different combinations of those HOX genes systematically by analyzing each platform independently (signed average (SA)28 of methylation and gene expression) to find the overlapped significant combinations and filter out the combinations with large variations. Finally, the most significant HOX gene combinations were extracted to use as biomarkers for further integrative analysis of methylation and gene expression data using the following approaches.
Step 1.
Out of 40 HOX genes, 16 significant and common HOX genes were selected from Supplementary Table 1 based on Cox regression analysis of HOX gene expression and methylation.
Step 2.
Various combinations of HOX genes were evaluated ranging from 2 HOX genes to 16 HOX genes for both methylation and gene expression. For each combination of selected HOX genes, samples were split into 2 groups based on high-low methylation or expression values using SA method.28 Then, we performed survival analysis and calculated P-values.
Step 3.
Finally, we ranked all combinations based on P-values with the smallest values at the top. 7-HOX genes such as HOXA4, HOXA7, HOXA10, HOXA13, HOXD3, HOXD9, and HOXD10 were selected as composite biomarkers that significantly predict OS in both IDHmut methylation and mRNA cohorts.
Determination of G-CIMP Status
The G-CIMP low is characterized by the loss of DNA methylation and is associated with worse prognosis, while the opposite is true for G-CIMP high tumors.24 For the TCGA cohort, we obtained G-CIMP status from the clinical data (13 cases out of 412 IDHmut Gliomas used in our study) in Ceccarelli et al.26 For the UHN and DKFZ cohorts, we used the approach proposed by Ceccarelli et al.26
Based on the methylation of 163 probes obtained based on overlapped 27K and 450K methylation analyses that were used for classifying G-CIMP status,25 we used a random forest classifier to identify G-CIMP status.
Statistical Analysis
All statistical analyses were done using the R Bioconductor package (version 3.3). All statistical tests performed were 2-sided, with P-values less than .05 as the threshold for significance, unless otherwise stated. To investigate the clinical relevance of integrative methylation and mRNA subtypes, we conducted Kaplan–Meier survival analyses on the integrative subtypes of mRNA and methylation using the survival package in Bioconductor. A log-rank test was performed to calculate P-values.
For supervised analysis of RNAseq data, we presented the genes with adjusted P-values (FDR) of less than 0.05 and with fold-change of above 2 thresholds. Genes were ranked based on P-values with the smallest values at the top. For supervised analysis of methylation data, we used a limma-based modeling approach where FDR < 0.05 and absolute mean differences > 0.1 were considered to be significant.
We performed multivariable analysis to identify factors associated with survival after adjusting for certain parameters like tumor grade of patients in order to identify factors that can predict patient survival independent of various factors.
Results
Identification of a Methylation Signature That Predicts Outcome in IDHmut Gliomas
The UHN cohort was used as a training set to identify a methylation signature of outcome for overall survival in IDHmut gliomas using a Cox proportional hazard regression analysis of each individual probe with outcome data. A total of 6798 significant probes (P-value < .01) corresponding to 3530 genes were selected (Supplementary Table 2) for further analysis. The significant markers were grouped together as a composite to perform survival analysis.
Of the 6798 significant probes identified, a total of 5914 hypermethylated probes (87%) with hazard ratios (HR) > 1 were significantly associated with worse survival (considered negative predictors), and hypomethylation of 884 probes (13%) with HR < 1 were significantly associated with longer survival (considered positive predictors) (Supplementary Table 3).27 Using the significant probes with P-values < .01 from the Cox regression modeling, we identified a methylation signature of outcome that was significantly associated with patient survival (P-value < .0001). The negative predictive probes (signatures) dominate CpG islands (48%) compared to positive predictive probes (17%), while the expected genomic coordinates for 450K array are 31% (Supplementary Table 3).
Consensus clustering was then performed based on all 6798 significant probes to determine if cluster members were associated with clinical features and patient survival (to show risk groups). Figure 1A shows that the consensus cluster based on the methylation signatures from Cox analysis (Cox proportional hazard regression analysis) using our UHN methylation cohort divided the glioma cohort into 2 prognostic subgroups.27 A Kaplan–Meier diagram depicts significant survival differences between these 2 groups (P-value < .0001) (Figure 1B).27
Figure 1.
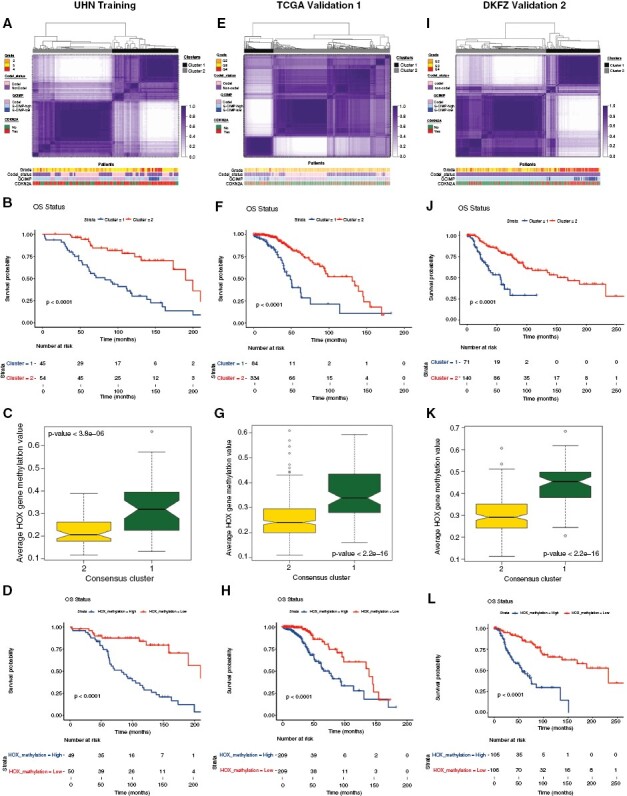
Methylome analysis of IDH-mutant gliomas identifies a distinct signature of outcome.27 (A) Consensus cluster based on the methylation signatures from Cox regression analysis of the University Health Network methylation cohort. (B) Kaplan–Meier diagram showing the survival statistics of methylation subtypes. (C) Boxplots of mean methylation values of HOX genes chosen from Cox analysis for 2 consensus cluster subtypes. The boxplot of average methylation values was calculated from the probes that overlapped with HOX genes for each sample in each consensus cluster subtype. (D) HOX gene-specific methylation patterns from the Cox analysis with significant probes show survival differences (Kaplan–Meier diagram) by median of average methylation values. (E):l Validation of global methylation signature related to patient outcome (A, B) and HOX gene-specific methylation patterns (C, D) using Kaplan–Meier survival analysis in 2 independent The Cancer Genome Atlas and DKFZ cohorts.
To determine the specific regions of these CpG sites, we next examined the location of these methylation signatures, including the promoter and gene body regions (Supplementary Table 3).27 The characterization of the methylation signature revealed that negative predictive probes were enriched within the promoter and island methylated regions compared to the positive predictive probes (Supplementary Table 3).
In particular, we observed enrichment of HOX genes as reflected in the univariable Cox hazard analysis and these significant HOX gene probes showed HR > 3. Figure 1C shows boxplots of mean methylation values of HOX genes chosen from Cox analysis (HOXA11AS, HOXA13, HOXA3, HOXA4, HOXA5, HOXA6, HOXA7, HOXB3, HOXB7, HOXB8, HOXC11, HOXC12, HOXC13, HOXC4, HOXD10, HOXD11, HOXD13, and HOXD3) for the 2 consensus cluster subgroups.27 The boxplot of average methylation values was calculated from the probes that overlapped with HOX genes, where worse survival consensus cluster subtype showed significantly elevated HOX average methylation values. HOX genes demonstrated significantly increased methylation values (hypermethylation) in the consensus cluster and were associated with worse survival (P-value < 0.0001).
Based on the significant HOX gene probes from the Cox analysis, SAs were performed28 and all glioma samples were then split into 2 groups by median. Consistently, these HOX methylation signatures also showed significant survival differences where hypermethylation of these probes was also associated with worse survival (negative predictors with HR > 1, P-value < .0001) (Figure 1D).27Figure 1D shows HOX gene-specific methylation patterns from the Cox analysis where significant probes demonstrated survival differences (Kaplan–Meier diagram) by median of average methylation values. These results demonstrate that HOX gene methylation patterns are associated with significant survival differences in IDHmut glioma and HOX hypermethylation is associated with worse survival in IDHmut glioma.
Validation of the Methylation Signature Using 2 Independent Cohorts
To ensure the consistency of our methylation signature and avoid bias, 2 independent rounds of validation were performed using TCGA and DKFZ cohorts using the same methylation signature/ CpGs (all 6798 significant probes) obtained from above UHN test cohort (Figure 1E to I).27 Based on the identified same methylation signature of outcome in IDHmut glioma and consensus clustering using the methylation signature from the UHN cohort, IDHmut glioma cohorts from both TCGA and DKFZ were again split into 2 prognostic groups. The survival analyses of TCGA and DKFZ methylation datasets validated the patient outcome-related methylation signatures (P-value < .0001 for both cohorts). Overall, both independent validation cohorts (Figure 1E and I) consistently showed that the methylation signatures predicting negative outcomes were associated with higher histologic grade, G-CIMP low status, and high copy number instability (specifically CDKN2A loss).27
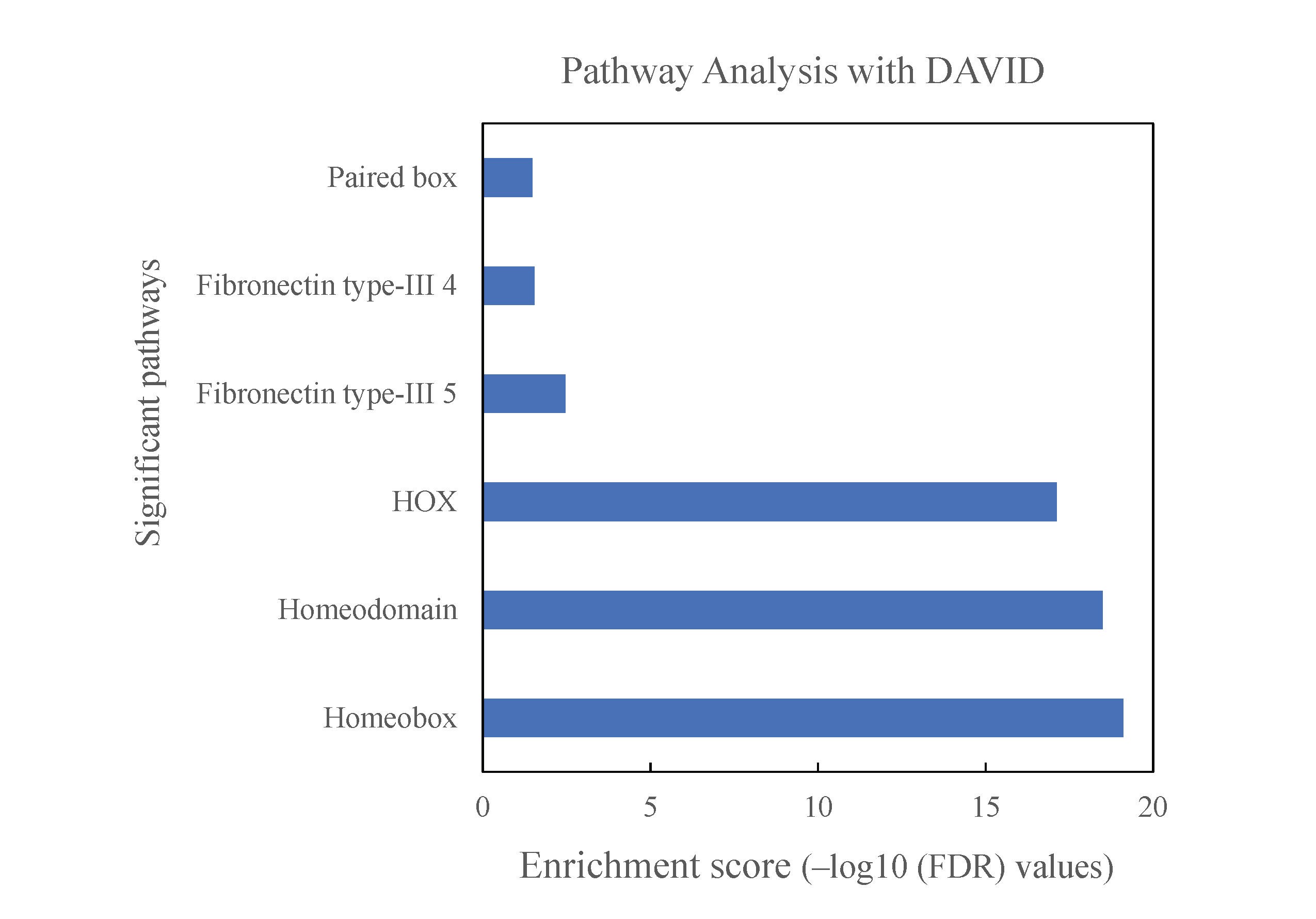
Multivariable analysis (Table 2) after adjusting for clinically relevant factors including grade, copy number (CDKN2A status), G-CIMP status, and 1p/19q codeletion status showed prognostic factors associated with survival in high versus low HOX groups (P-value = .000027, P-value = .009, P-value = .00025, respectively for UHN, TCGA, DKFZ cohorts). Next, pathway analysis and gene ontology were performed using the genes with negative outcomes that overlapped with significantly hypermethylated probe sets (HR > 1 and P-value < .01) using DAVID (the database for annotation, visualization, and integrated discovery). Gene ontology showed that the methylation signature was enriched for both homeobox genes and its subset of HOX genes (Supplementary Figure 3. HOX genes are also reflected in the univariable Cox hazard analysis in the UHN cohort, where high methylation of several CpG sites at HOX genes were correlated with worse outcome in IDHmut glioma. HOX gene details from the Cox analysis including chromosome location and number of significant probes are included in Supplementary Table 4.
Table 2.
Multivariable Cox Analyses of HOX Gene Clusters Within University Health Network, The Cancer Genome Atlas, DKFZ Cohorts.27 Overall Survival Characteristics Based on Grade, G-CIMP Status, CDKN2A Status. HOX gene Cluster is Obtained Based on the Signed Average of Significant HOX Methylation Probes From Cox Analysis and Split by Median
| Covariate | Multivariable Cox | |
|---|---|---|
| HR (95% CI) | P-value | |
| UHN cohort (N = 99) | ||
| HOX gene Methylation | 3.86 (1.97–7.58) | 8.76E-05 |
| Grade (3 vs.2) | 0.9 (0.4–2.01) | .77 |
| Grade (3 vs. 4) | 3.77 (1.3–11.4) | .019 |
| CODEL (non-code vs. Codel) | 1.21 (0.13–11.16) | .87 |
| G-CIMP (high vs. low) | 6.72 (1.73–26.15) | .006 |
| Age | 1.00 (0.96–1.03) | .88 |
| Gender (female vs. male) | 1.78 (0.91–3.49) | .09 |
| CDKN2A (Loss vs. no loss) | 2.45 (1.18–5.07) | .016 |
| TCGA validation cohort (N = 419) | ||
| HOX gene Methylation | 2.8 (1.6–5.1) | .0005 |
| Grade (3 vs 2) | 0.5 (0.3–0.9) | .013 |
| Grade (3 vs. 4) | 1.3 (0.2–10.2) | .79 |
| CODEL (non-code vs. Codel) | 2.0 (0.8–5.1) | .16 |
| G-CIMP (high vs. low) | 1.92E–06 (0–Inf) | .997 |
| Age | 1.0 (1.0–1.1) | 1.46E-05 |
| Gender (female vs. male) | 1.0 (0.6–1.7) | .88 |
| CDKN2A (Loss vs. no loss) | 3.1 (1.2–7.5) | .015 |
| DKFZ validation cohort (N = 211) | ||
| HOX gene Methylation | 2 (1.1–3.7) | .025 |
| Grade (3 vs. 2) | 0.4 (0.3–0.7) | .0003 |
| Grade (3 vs. 4) | 0.8 (0.5–1.5) | .48 |
| G-CIMP (high vs. low) | 1.0 (0.5–2.3) | .92 |
| Age | 1.0 (1.0–1.0) | .29 |
| Gender (female vs. male) | 0.6 (0.4–0.9) | .016 |
| CDKN2A (Loss vs. no loss) | 1.1 (0.5–2.1) | .85 |
HOX Gene Expression and Its Association With Overall Survival
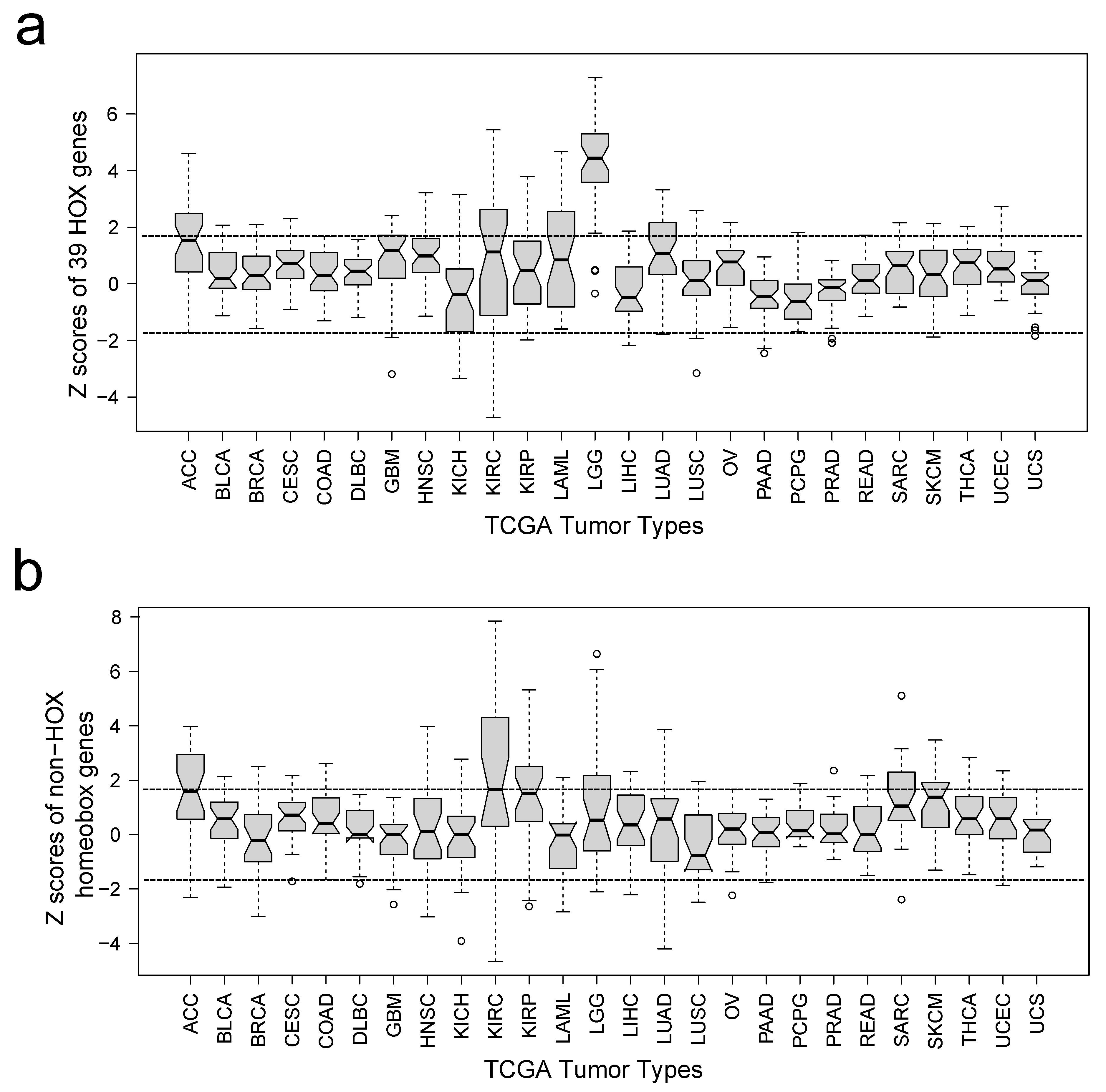
To better understand the association between HOX gene expression and methylation with patient outcomes, we first examined HOX gene expression and its correlation with overall survival using TCGA transcriptome profiles downloaded from PRECOG (PREdiction of Clinical Outcomes from Genomic Profiles).29,30 PRECOG as an analytical tool correlates CIBERSORT cell type abundance data from transcriptome profiles with overall survival outcomes across 26 tumor types from TCGA.29,30 For positive survival-associated Z scores, high expression or upregulation is associated with shorter survival. For negative survival-associated Z scores, high expression or up-regulation is associated with longer survival. Given that the pathway analysis demonstrated an enrichment for both homeobox genes and its subset of HOX genes, we performed survival-associated Z score analysis for HOX genes and non-HOX homeobox genes separately (Supplementary Figure 2A and B, respectively). Survival-associated Z score analysis for HOX genes demonstrated that HOX genes were prognostic in gliomas with a median Z score of 4.5 (P-value < .0001) but not significant in any of the other 25 tumor types (Z score of 1.95 is associated with P-value = .05). Survival-associated Z score analysis for non-HOX homeobox genes did not demonstrate any significant Z scores for any of the other 26 tumor types, including gliomas. Consequently, our subsequent analyses focused only on HOX genes and not non-HOX homeobox genes.
Characterization and Integration of All HOX Gene Methylation and HOX Gene Expression
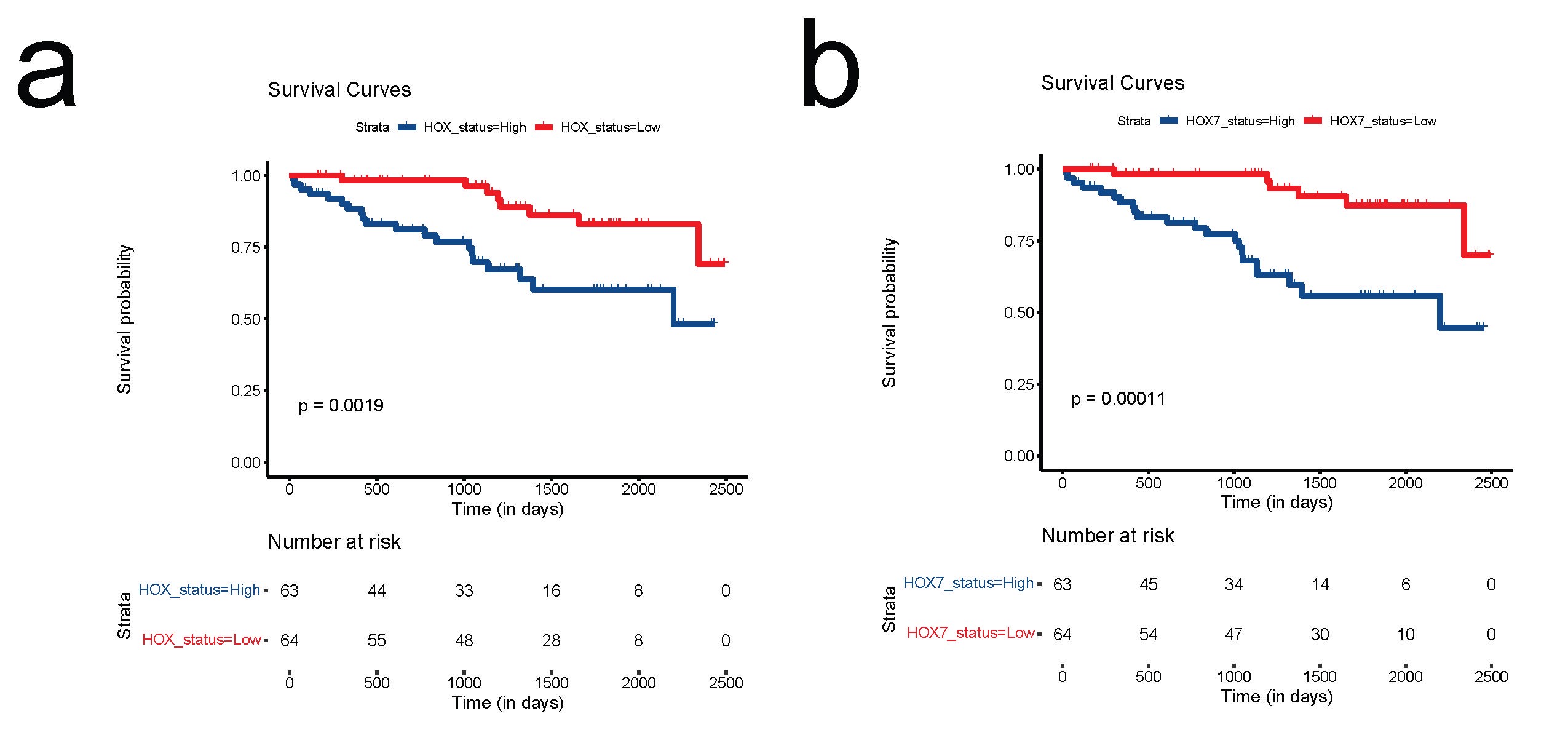
To examine whether multi-omics integration is a better prognostic score, 412 IDHmut glioma samples from TCGA with matched methylation, mRNA, CNV, and mutational datasets were used to identify subtypes with unique molecular signatures. Based on the average methylation levels of all CpGs mapped to each HOX gene, we calculated SA methylation values for each sample and split IDHmut gliomas into 2 groups consisting of hypermethylation and hypomethylation by median. Overall survival based on HOX gene methylation patterns showed that hypermethylation of HOX genes correlates with statistically significantly worse outcomes in IDHmut glioma (P-value = .0023, HR = 2[1.3–3.5]) (Figure 2A). Similarly, HOX gene expression was also split into 2 groups consisting of high and low expression groups which showed that overexpression of HOX genes was also associated with a statistically significantly worse survival (P-value < .0001, HR = 3[1.8–5.1]) (Figure 2B).
Figure 2.
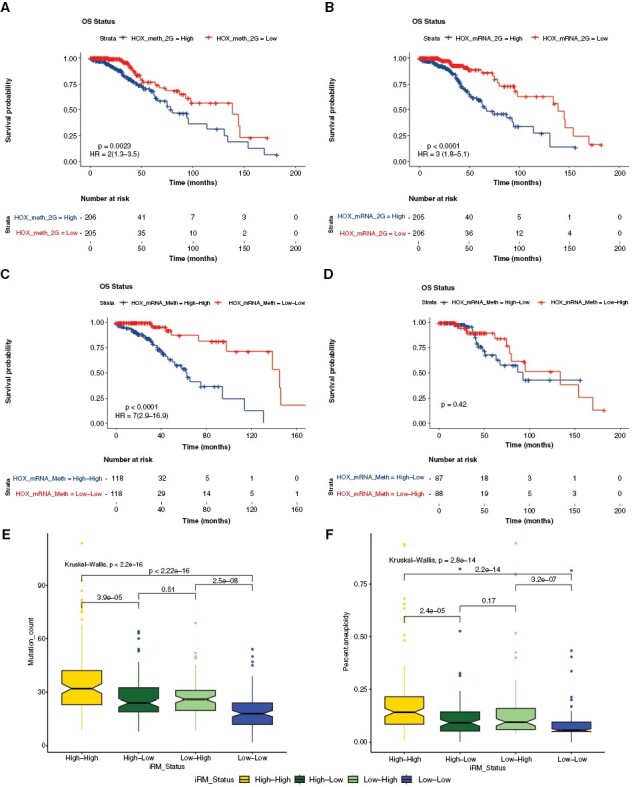
Characterization and integration of all HOX gene methylation and gene expression. HOX gene-specific methylation and gene expression patterns in similar directionality are related to patient survival. (A) Kaplan–Meier diagram showing overall survival based on HOX gene methylation pattern (hypermethylation vs. hypomethylation) with hazard ratios (95% Confidence Interval: CI). (B) Kaplan–Meier survival analysis based on expression of HOX genes with hazard ratios (95% CI). (C) Kaplan–Meier diagram with the survival statistics of integrative RNA-methylation (iRM) subtypes of similar directionality of HOX gene methylation and HOX gene expression (iRM high and iRM low). (D) Kaplan–Meier diagram with survival statistics of iRM subtypes of opposite directionality of HOX gene methylation and HOX gene expression. (E) Boxplot showing the association of these integrative subtypes with mutational rate. (F) Boxplot showing the association of these integrative subtypes with aneuploidy.
Following single platform-based HOX gene expression and methylation levels, an integrated multi-platform-based HOX gene subtype was created by combining individual subtypes (high and low-risk groups) from HOX gene expression and methylation separately by taking the median values. The integrated RNA and methylation approach (iRM) that groups them based on High and Low status split gliomas into 4 subtypes based on the directionality of HOX gene-based methylation and gene expression analysis (high methylation-high expression, low methylation-low expression, low methylation-high expression, and high methylation-low expression). Interestingly, the 2 integrated subtypes of similar directionality: High methylation-high expression (iRM high) and low methylation-low expression (iRM low) produced highly prognostic groups that predicted statistically significant survival (P-value < .0001, HR = 7[2.9–16.9]) (Figure 2C). The iRM (HR = 7) analysis of HOX genes performed better than individual HOX gene expression or methylation analysis with HR of 2 and 3, respectively (Figure 2). The 2 integrated subtypes of opposite directionality: Low methylation-high expression and high methylation-low expression, showed no survival differences (Figure 2D). Additional boxplots demonstrating the association of iRM with mutational rate (Figure 2E) and aneuploidy (Figure 2F) showed increased mutational and aneuploidy counts for the iRM high group and decreased counts for the iRM low group. This finding demonstrates a high degree of correlation between integrative subtypes and somatic mutations, with the iRM high group associated with worse survival having increased mutational load and aneuploidy, and the iRM low group associated with better survival having decreased mutational load and aneuploidy. Individually, both methylation and expression clearly separated high and low-risk groups, while both hypermethylation and overexpression of HOX genes in similar directionality were correlated with significantly worse outcomes in these tumors (P-value < .0001, HR = 7[2.9–16.9]).
Supplementary Table 1 shows Cox regression analysis of individual HOX gene methylation and expression in IDHmut glioma including methylation of CpG sites at gene promoter and gene body (averaged probe values for each HOX gene), where majority of HOX genes have high HR and significant P-values (P-value < .001). Only iRM cohort of the same directionality (both iRM low and high) showed significantly increased HR and significant P-values (P-value < .001) for majority of HOX gene expression and methylation analyzed (Supplementary Table 1), while for the iRM cohort of opposite directionality, significance was lost for majority of HOX gene expression and methylation (Supplementary Table 1). Although methylation of some HOX genes was significantly oppositely correlated with HOX gene expression in 35% (13/37) of them (P-value < .05), significant correlation was lost for all those HOX genes in iRM cohort of same directionality (Supplementary Table 5).
7-HOX Gene Biomarker Signature Predicts Outcome in Glioma
To identify the most significant outcome predictors, candidate HOX gene biomarkers were selected from the iRM high group that were significantly associated with patient outcome using TCGA IDHmut LGG samples. A separate Cox analysis of outcome for both HOX methylation and HOX gene expression was conducted for all HOX gene methylation and gene expression. A total of 84% of HOX gene methylation HRs were significantly associated with survival (31/37, P-value < .05, HR > 8), and 75% of HOX gene expression HRs were significantly associated with survival (30/40, P-value < .05, HR > 1) (Supplementary Table 1).
Interestingly, when these 2 groups of HOX genes were compared, a total of 25 genes had significant HRs > 1 for both methylation and gene expression. Various combinations of HOX genes were then tested ranging from 2 to 16 HOX genes based on the SA method for iRM groups. Interestingly, 7-HOX genes (HOXA4, HOXA7, HOXA10, HOXA13, HOXD3, HOXD9, and HOXD10 located on chromosomes 2 and 7) exhibiting high methylation and expression can be used as biomarkers to determine the prognosis in IDHmut glioma. iRM high versus iRM low groups of these 7-HOX genes showed a significant p-value and increased HR corresponding to worse outcomes in the iRM high group compared to the iRM low group while opposite directionality of mRNA and methylation was not significant (Figure 3A).
Figure 3.
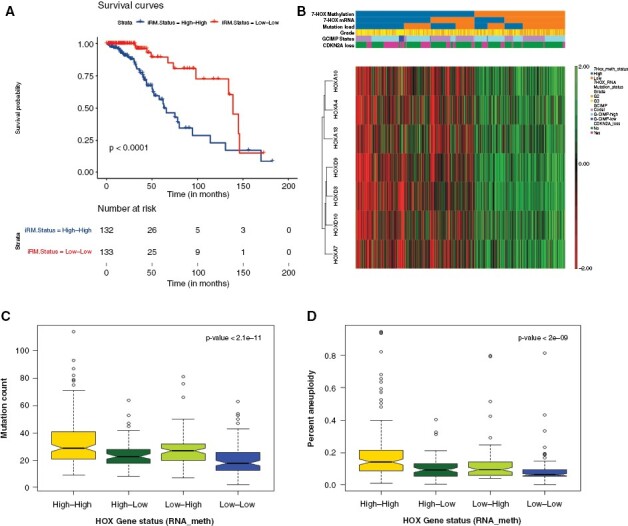
7-HOX genes as biomarkers of outcome in IDH-mutant gliomas. (A) Kaplan–Meier diagram for integrated RNA and methylation analysis of 7-HOX gene signature (signed average of HOX gene methylation and HOX gene expression). (B) Unsupervised Hierarchical clustering and dendrogram for IDHmut glioma types for mRNA and methylation, where heatmap is only for the methylation data for 7-HOX genes. (C) Boxplot showing the associations of these integrative subtypes with mutational rate. (D) Boxplot showing the associations of these integrative subtypes with aneuploidy.
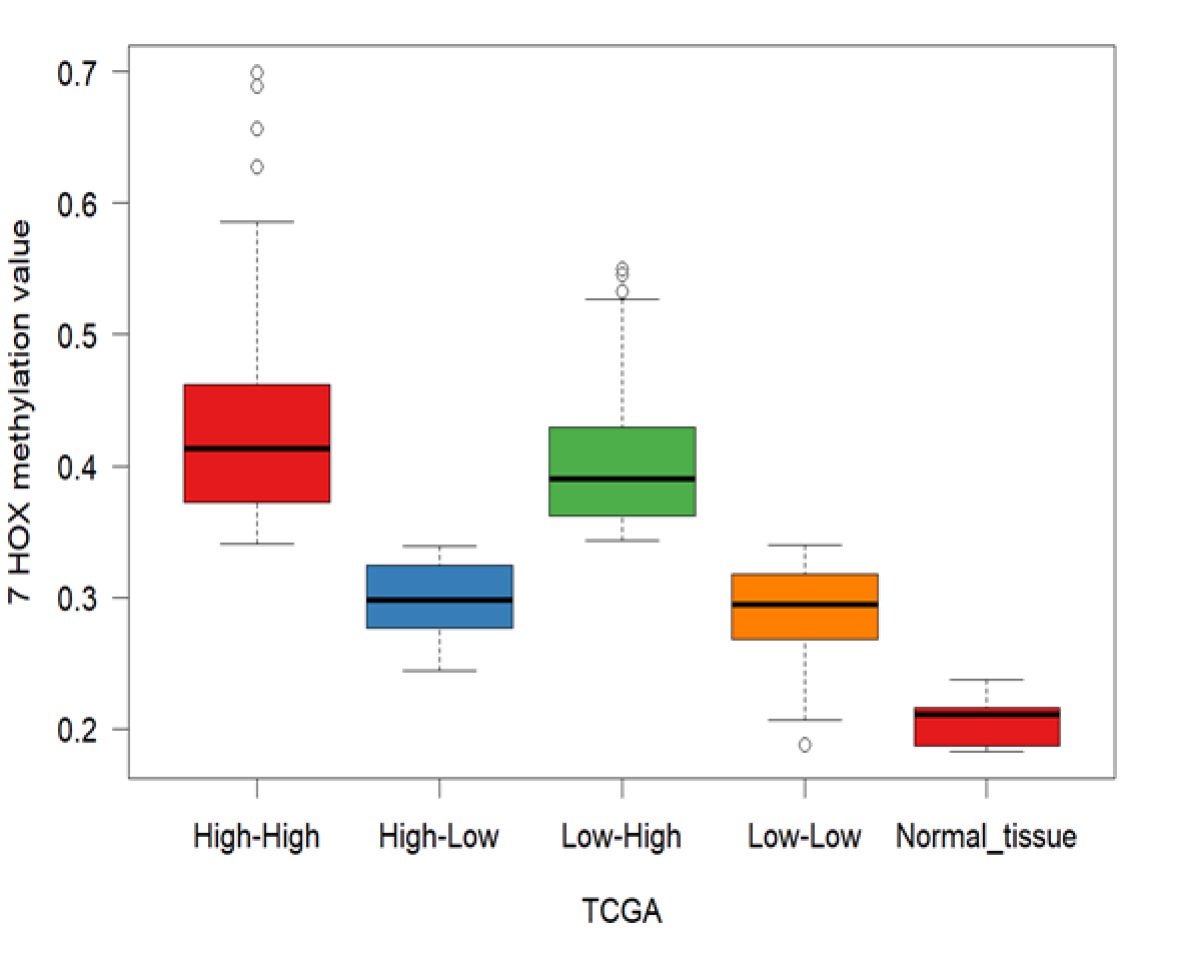
We next validated the prognostic significance of our 7-HOX gene signature using hierarchical clustering methylation data. This was used to identify the association of methylation subtypes with mRNA subtypes, mutational load, and other clinical factors including grade, CDKN2A copy number alterations and G-CIMP status. Our data show a strong association of hypermethylation with overexpression of the 7-HOX genes (P-value < .001 based on the chi-squared test) and higher mutational burden (P-value < .001 based on the chi-squared test) (Figure 3B), where the heatmap is based on the methylation data of the 7-HOX genes. Tumor mutational burden has recently been shown to be associated with poor outcome in diffuse gliomas using a multi-omics approach.31 In addition, the iRM high group also exhibited high copy number instability and a significant proportion of G-CIMP low tumors. In fact, G-CIMP low status constituted a subset (12/147 = 8%) of the iRM high group, where a total of 92% (12/13) of the G-CIMP low samples were present in the iRM high group (Figure 3B). Furthermore, we show that the iRM high group of the 7-HOX genes is associated with higher mutational burden and higher aneuploidy (Figure 3C and D).
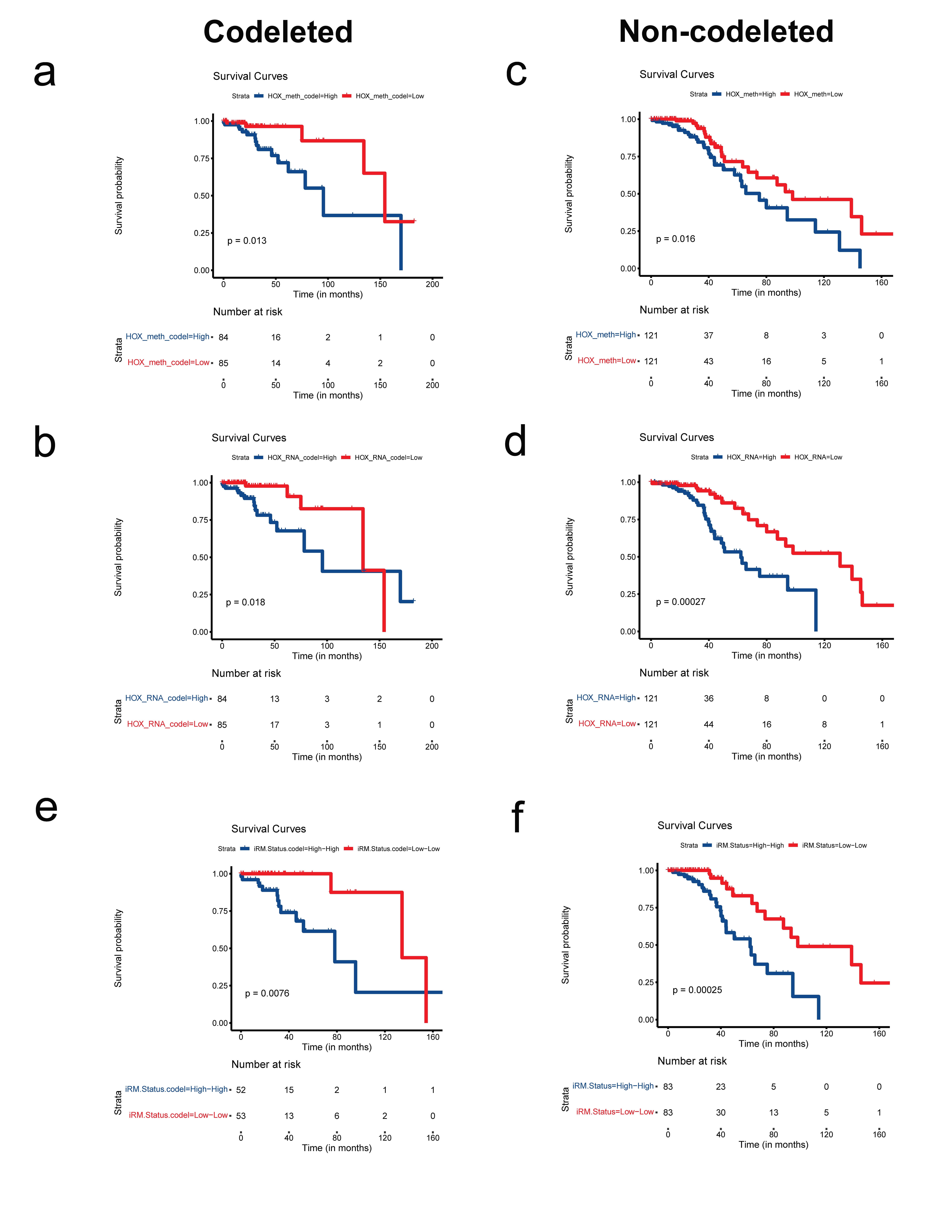
Given the robustness of the 7-HOX gene signature in determining overall IDHmut glioma prognosis, we then applied our 7-HOX gene signature to subtypes of IDHmut glioma such as 1p/19q codeleted and non-codeleted gliomas for further risk stratification. To examine this, we separated IDHmut glioma based on 1p/19q codeletion status with matched methylation and mRNA datasets that includes 1p/19q codeleted gliomas without G-CIMP low tumors and non-codeleted gliomas with G-CIMP tumors. Similar to our findings using IDHmut glioma we show that in both subtypes of IDHmut glioma, HOX gene overexpression, and hypermethylation predicts worse survival for both codeleted and non-codeleted glioma (Figure 4A to D). Consistently iRM high and iRM low tumors also exhibit significant survival differences for both codeleted and non-codeleted subtypes of IDHmut glioma with P-values of .001 and .0059, respectively (Figure 4E and F). Again, opposite direction of methylation and expression (high methylation/low expression and low methylation/high expression groups) shows no survival differences (not shown here).
Figure 4.
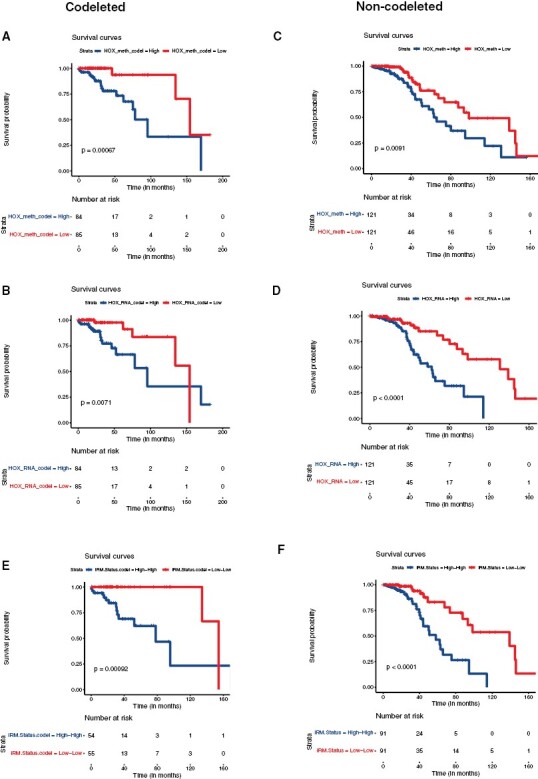
Applicability of 7-HOX gene signature in the subtypes of IDHmut glioma with 1p/19q codeletion and without 1p/19q codeletion: (A) Overall survival shown using Kaplan–Meier diagram based on HOX gene methylation pattern (hypermethylation vs hypomethylation) in 1p/19q codeleted IDHmut glioma. (B) Kaplan–Meier survival analysis based on expression of HOX genes in 1p/19q codeleted IDHmut glioma. (C) Overall survival was shown using Kaplan–Meier diagram based on HOX gene methylation pattern (hypermethylation vs. hypomethylation) in non-codeleted IDHmut LGG. (D) Kaplan–Meier survival analysis based on expression of HOX genes in non-codeleted IDHmut glioma. (E) Kaplan–Meier diagram with the survival statistics of integrative RNA-methylation (iRM) subtypes of similar directionality of HOX gene methylation and HOX gene expression (iRM high and iRM low) in codeleted IDHmut glioma. (F) Kaplan–Meier diagram with the survival statistics of integrative RNA-methylation (iRM) subtypes of similar directionality of HOX gene methylation and HOX gene expression (iRM high and iRM low) in non-codeleted IDHmut glioma.
Discussion
Traditional classification of diffuse gliomas as defined by WHO is based on histopathological grading.6,32 Molecular characterization of these tumors has further classified gliomas based on IDH mutation status and 1p/19q codeletion status, and these molecular features are correlated with patient survival.8,26,43
Despite this, there is still a wide range of clinical outcomes in patients with IDHmut gliomas that is not accounted for by current clinical and pathological parameters. We postulated that methylation signatures might help to better understand the variability in outcome of IDHmut gliomas and found that there was a negative correlation between outcome and G-CIMP low, together with high copy number instability. Most importantly, this signature was enriched for HOX genes, where HOX gene hypermethylation was associated with worse survival in IDHmut gliomas. Integrated analyses of RNA and methylation data (iRM) identified HOX gene hypermethylation and overexpression were associated with significantly worse outcomes in IDHmut gliomas, with increased mutational burden and aneuploidy.
Furthermore, we found that a subset of 7-HOX genes located at chromosomes 2 and 7 (HOXA4, HOXA7, HOXA10, HOXA13, HOXD3, HOXD9, and HOXD10), exhibiting hypermethylation and overexpression in the iRM high group, can serve as potential biomarkers predicting patient survival in both 1p/19q codeleted and non-codeleted IDHmut gliomas.
HOX genes are an evolutionarily conserved family of genes, in vertebrates there are a total of 39 HOX genes divided into 4 separate clusters, A through D, located on chromosomes 7, 17, 12, and 2, respectively.33 While aberrant HOX gene expression and methylation have been reported in the development and progression of multiple cancers,12 the majority of research to date exploring HOX genes in gliomas is limited to GBM.17–23 We sought to identify novel signatures of outcome in IDHmut gliomas using a multi-platform integrated approach, and independently found a high enrichment of HOX genes in the negatively prognostic subgroups in all 3 study cohorts. While we observed aberrant methylation and expression of HOX genes from all 4 clusters, our final prognostic 7-HOX gene signature in IDHmut gliomas includes HOX genes from clusters A and D, located on chromosomes 7p14 and 2q31, respectively.12 Both of these HOX gene clusters are located at topological associating domain (TAD) boundaries, in which chromatin interactions are highly favored.12 Deletion or hypermethylation of specific CTCF-binding sites at these TAD boundaries can promote dysregulation of HOX gene expression.12,34 Unlike HOX clusters A and D, HOX clusters B and C are not associated with a TAD boundary, and although they have also been reported to be dysregulated in GBM, may represent a different mechanism of dysregulation.12 These findings may account for the observed enrichment of HOXA and HOXD clusters in our final signature.
We also unexpectedly found that the HOX genes associated with worse prognosis displayed seemingly paradoxical hypermethylation and overexpression. While it is generally accepted that hypermethylation of gene promoters leads to downregulation of gene expression,35,36 several recent publications describe a paradoxical relationship between DNA hypermethylation and upregulation of gene expression and its associated clinical significance.13,37–40 A recent review showed some putative mechanisms of gene expression regulation and hypermethylation at different gene clusters for different cancers.41 Interestingly, they reported positive association of promoter methylation and corresponding gene expression of GATA4, HOXD12, ESR1, TWIST1, and MGMT.41 Similarly, we found that HOXD12 hypermethylation and gene expression were correlated with poor patient survival in IDHmut glioma. Within gliomas, a recent paper also explored this concept of paradoxical hypermethylation and overexpression of genes using integrated analyses of 70 adult gliomas and found that the majority of transcriptional alterations resulted from DNA methylation-independent mechanisms, including altered methylation at histone H3 trimethylation at lysine 27 (H3K27me3).42 Within GBM, HOX clusters demonstrate a drastically reduced level of H3K27me3 versus normal brain, providing evidence that this loss may be an important mechanism of aberrant expression.42 Additional studies have identified multiple genes in GBM with significant methylation of their main CpG islands/ promoter with the use of an alternative promoter, including the gene HOXA10, identified in our 7-HOX gene signature, and HOXC11.15,21,42 These mechanisms are not specific to GBM, and may serve as mechanisms to explain our findings of non-canonical hypermethylation and overexpression in IDHmut gliomas.
Our study is not without limitations. First, the integration of RNA and methylation (iRM) in our study was exploratory, and based on the TCGA dataset. Additional investigations with matched RNA and methylation analyses from the same samples are warranted in order to further validate our findings. Second, future wet bench studies are necessary in order to provide rationale behind our observations and establish underlying mechanisms that have not yet been investigated thoroughly for gliomas, and more specifically IDH-mutant gliomas.
In summary, using an integrative multi-platform approach, we observed a significant correlation between HOX gene hypermethylation and overexpression (iRM high) with worse outcomes in IDHmut glioma, including both 1p/19q non-codeleted and codeleted gliomas. The iRM high group was also associated with high mutational load and high aneuploidy. From the iRM group, we developed and validated a predictive 7-HOX gene signature of outcome in IDHmut gliomas. These results illustrate the strength of combining and integrating different platforms and data types (methylation, mRNA, and mutations) where complementary and dependent integration can provide a better understanding of co-regulated genes and probes with similar profiles.11 The integration of mRNA and methylation of HOX genes that distinguishes survival provides a novel clinically and biologically relevant stratification for IDHmut gliomas that could lead to a better molecular diagnosis and management of these patients. Future directions include further exploration of these predictive HOX genes, including mechanistic studies to determine the relationship between aberrant HOX gene methylation and expression in IDHmut glioma development and progression. Future targeted HOX-directed therapies may aid in the search for new treatment options in patients with gliomas.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Yasin Mamatjan, Princess Margaret Cancer Center and MacFeeters-Hamilton Center for Neuro-Oncology Research, University Health Network, Toronto, Ontario, Canada; Faculty of Science, Thompson Rivers University, Kamloops, British Columbia, Canada.
Mathew R Voisin, Princess Margaret Cancer Center and MacFeeters-Hamilton Center for Neuro-Oncology Research, University Health Network, Toronto, Ontario, Canada.
Farshad Nassiri, Princess Margaret Cancer Center and MacFeeters-Hamilton Center for Neuro-Oncology Research, University Health Network, Toronto, Ontario, Canada.
Fabio Y Moraes, Department of Oncology, Queens University, Kingston, Ontario, Canada.
Severa Bunda, Princess Margaret Cancer Center and MacFeeters-Hamilton Center for Neuro-Oncology Research, University Health Network, Toronto, Ontario, Canada.
Jonathan So, Department of Medicine, Dana-Farber Cancer Institute, Boston, Massachusetts, USA.
Mira Salih, Mount Sinai Hospital, New York, New York, USA; Department of Neuro-Oncology/Neurosurgery, Saitama Medical University International Medical Center, Hidaka, Japan.
Mitsuaki Shirahata, Department of Neuro-Oncology/Neurosurgery, Saitama Medical University International Medical Center, Hidaka, Japan.
Takahiro Ono, Department of Neurosurgery, Akita University Graduate School of Medicine, Akita, Japan.
Hiroaki Shimizu, Department of Neurosurgery, Akita University Graduate School of Medicine, Akita, Japan.
Daniel Schrimpf, Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Andreas von Deimling, Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Kenneth D Aldape, Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, USA.
Gelareh Zadeh, Princess Margaret Cancer Center and MacFeeters-Hamilton Center for Neuro-Oncology Research, University Health Network, Toronto, Ontario, Canada.
Author Contributions
YM, GZ, and KA designed the research project and interpreted the results. YM performed all data analysis. YM and MV wrote the manuscript. MV prepared figures in Illustrator. FN, KA, AD, FM, FN, SB, JS, MS, HS, TO, DS, and GZ contributed to the discussions and editing of the manuscript. All authors approved the manuscript.
Conflict of interest statement
The authors declare no personal or financial competing interests.
Funding
This work was supported by the Canadian Institutes of Health Research Project Grant.
Data Availability
Previously unpublished raw methylation datasets (idat files) from University Health Network (UHN) have been deposited to the European Genome-Phenome Archive with the dataset identifier of EGAS00001006961 (https://wwwdev.ebi.ac.uk/ega/studies/EGAS00001006961). The previously published methylation data from the German Cancer Research Center (DKFZ) can be accessed from GEO with the dataset identifier of GSE90496. The Cancer Genome Atlas (TCGA) dataset was open access.
References
- 1. Wen PY, Kesari S.. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 2. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whitfield BT, Huse JT.. Classification of adult-type diffuse gliomas: Impact of the World Health Organization 2021 update. Brain Pathol. 2022;32(4):e13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473–1479. [DOI] [PubMed] [Google Scholar]
- 6. Louis DN, Perry A, Reifenberger G, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Appay R, Dehais C, Maurage CA, et al. ; POLA Network. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019;21(12):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen AL, Holmen SL, Colman H.. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoadley KA, Yau C, Wolf DM, et al. ; Cancer Genome Atlas Research Network. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weinstein JN, Collisson EA, Mills GB, et al. ; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mamatjan Y, Agnihotri S, Goldenberg A, et al. Molecular signatures for tumor classification: An analysis of the cancer genome atlas data. J Mol Diagn. 2017;19(6):881–891. [DOI] [PubMed] [Google Scholar]
- 12. Shah N, Sukumar S.. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–371. [DOI] [PubMed] [Google Scholar]
- 13. Duan R, Han L, Wang Q, et al. HOXA13 is a potential GBM diagnostic marker and promotes glioma invasion by activating the Wnt and TGF-beta pathways. Oncotarget. 2015;6(29):27778–27793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cimino PJ, Kim Y, Wu HJ, et al. Increased HOXA5 expression provides a selective advantage for gain of whole chromosome 7 in IDH wild-type glioblastoma. Genes Dev. 2018;32(7-8):512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurscheid S, Bady P, Sciuscio D, et al. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome Biol. 2015;16(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones PA, Baylin SB.. The epigenomics of cancer. Cell. 2007;128(4):683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonçalves CS, Le Boiteux E, Arnaud P, Costa BM.. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell Mol Life Sci. 2020;77(19):3797–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26(18):3015–3024. [DOI] [PubMed] [Google Scholar]
- 19. Gaspar N, Marshall L, Perryman L, et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010;70(22):9243–9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Boiteux E, Court F, Guichet PO, et al. Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage. Mol Oncol. 2021;15(8):1995–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonçalves CS, Xavier-Magalhães A, Martins EP, et al. A novel molecular link between HOXA9 and WNT6 in glioblastoma identifies a subgroup of patients with particular poor prognosis. Mol Oncol. 2020;14(6):1224–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pojo M, Gonçalves CS, Xavier-Magalhães A, et al. A transcriptomic signature mediated by HOXA9 promotes human glioblastoma initiation, aggressiveness and resistance to temozolomide. Oncotarget. 2015;6(10):7657–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Costa BM, Smith JS, Chen Y, et al. Reversing HOXA9 oncogene activation by PI3K inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res. 2010;70(2):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noushmehr H, Weisenberger DJ, Diefes K, et al. ; Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malta TM, de Souza CF, Sabedot TS, et al. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro Oncol. 2018;20(5):608–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mamatjan Y, et al. . Identification of the prognostic signatures for isocitrate dehydrogenase mutant glioma. In: 2022 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB); Ottawa, ON, Canada; 2022:1–7. doi: 10.1109/CIBCB55180.2022.9863027 [DOI] [Google Scholar]
- 28. Gendoo DM, Ratanasirigulchai N, Schroder MS, et al. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016;32(7):1097–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L, Ge J, Lan Y, et al. Tumor mutational burden is associated with poor outcomes in diffuse glioma. BMC Cancer. 2020;20(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215–225; discussion 226. discussion 226219. [DOI] [PubMed] [Google Scholar]
- 33. Quinonez SC, Innis JW.. Human HOX gene disorders. Mol Genet Metab. 2014;111(1):4–15. [DOI] [PubMed] [Google Scholar]
- 34. Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Witte T, Plass C, Gerhauser C.. Pan-cancer patterns of DNA methylation. Genome Med. 2014;6(8):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esteller M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene. 2002;21(35):5427–5440. [DOI] [PubMed] [Google Scholar]
- 37. Spainhour JC, Lim HS, Yi SV, Qiu P.. Correlation patterns between DNA methylation and gene expression in the cancer genome atlas. Cancer Inform. 2019;18:1176935119828776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bahar Halpern K, Vana T, Walker MD.. Paradoxical role of DNA methylation in activation of FoxA2 gene expression during endoderm development. J Biol Chem. 2014;289(34):23882–23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia N, Wang J, Li Q, et al. DNA methylation promotes paired box 2 expression via myeloid zinc finger 1 in endometrial cancer. Oncotarget. 2016;7(51):84785–84797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee DD, Leao R, Komosa M, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129(4):1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith J, Sen S, Weeks RJ, Eccles MR, Chatterjee A.. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer. 2020;6(5):392–406. [DOI] [PubMed] [Google Scholar]
- 42. Court F, Le Boiteux E, Fogli A, et al. Transcriptional alterations in glioma result primarily from DNA methylation independent mechanisms. Genome Res. 2019;29(10):1605–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reuss DE, Mamatjan Y, Schrimpf D, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: A grading problem for WHO. Acta Neuropathol. 2015;129(6):867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Previously unpublished raw methylation datasets (idat files) from University Health Network (UHN) have been deposited to the European Genome-Phenome Archive with the dataset identifier of EGAS00001006961 (https://wwwdev.ebi.ac.uk/ega/studies/EGAS00001006961). The previously published methylation data from the German Cancer Research Center (DKFZ) can be accessed from GEO with the dataset identifier of GSE90496. The Cancer Genome Atlas (TCGA) dataset was open access.