

Abstract
Background
Resistance to existing therapies is a significant challenge in improving outcomes for glioblastoma (GBM) patients. Metabolic plasticity has emerged as an important contributor to therapy resistance, including radiation therapy (RT). Here, we investigated how GBM cells reprogram their glucose metabolism in response to RT to promote radiation resistance.
Methods
Effects of radiation on glucose metabolism of human GBM specimens were examined in vitro and in vivo with the use of metabolic and enzymatic assays, targeted metabolomics, and FDG-PET. Radiosensitization potential of interfering with M2 isoform of pyruvate kinase (PKM2) activity was tested via gliomasphere formation assays and in vivo human GBM models.
Results
Here, we show that RT induces increased glucose utilization by GBM cells, and this is accompanied with translocation of GLUT3 transporters to the cell membrane. Irradiated GBM cells route glucose carbons through the pentose phosphate pathway (PPP) to harness the antioxidant power of the PPP and support survival after radiation. This response is regulated in part by the PKM2. Activators of PKM2 can antagonize the radiation-induced rewiring of glucose metabolism and radiosensitize GBM cells in vitro and in vivo.
Conclusions
These findings open the possibility that interventions designed to target cancer-specific regulators of metabolic plasticity, such as PKM2, rather than specific metabolic pathways, have the potential to improve the radiotherapeutic outcomes in GBM patients.
Keywords: glioblastoma, metabolism, plasticity, PPP, radiation resistance
Key Points.
- GBM rewires glucose metabolism to resist oxidative stress induced by radiation.
- GLUT3 translocates to the cell surface for more efficient glucose uptake after radiation.
- PKM2 regulates glucose flux through the antioxidant PPP following radiation.
Importance of the Study.
Glioblastomas (GBM) are aggressive brain tumors with dismal prognosis that is in part due to a remarkable resistance to modern therapies, including radiation therapy (RT). This study shows that GBM respond to radiation by rewiring glucose metabolism in a way that promotes survival during RT. Following RT, glioblastomas upregulate glucose consumption by increasing GLUT3 at the cell surface. Glucose-derived metabolites are preferentially routed through the antioxidant PPP, thus optimizing for survival during the lethal oxidative stress induced by radiation. PKM2, is identified as one of the main molecular regulators of the increased flux through the PPP. Small molecule activators of PKM2 show promise for significantly enhancing the therapeutic benefit of RT and improving overall outcomes in this deadly disease.
More than two-thirds of adults diagnosed with glioblastoma (GBM) will die within 2 years of diagnosis, making GBM one of the most lethal of cancers. Post-surgical radiation therapy (RT) combined with temozolomide1, and more recently the addition of alternating electrical tumor treating fields2 are currently the only treatment approaches that significantly improve overall survival, over surgery alone. Yet, GBM tumors inevitably recur, largely within the radiation field3. Overcoming radiation resistance of these tumors is one of the major remaining frontiers in radiation oncology that, if resolved, could potentially dramatically improve outcomes in this disease.
Amongst the recognized drivers of the remarkable radioresistance in GBM is tumor metabolism and its potential for promoting resistance to oxidative stress, such as generated during RT. Disparate oncogenic mutations often converge on canonical metabolic pathways that fulfill the enhanced biosynthetic demands during cancer growth, making altered metabolism a recognized hallmark of cancer4, including GBM5. Aerobic glucose catabolism via glycolysis (aerobic glycolysis) is thought to provide an advantage to cancer cells by enhancing the pool of glycolytic intermediates that serve as input for various biosynthetic pathways branching from glycolysis. This partially explains the “Warburg effect” observed in most tumors6,7. Relevant to RT, glycolytic branches, such as the pentose phosphate pathway (PPP) are also critical contributors to the cell’s pool of reducing equivalents that maintain a balanced redox status and can thus promote survival under oxidative stress conditions8.
The last decade brought the recognition that tumors do not have a fixed, albeit aberrant, metabolic profile but rather exhibit remarkable metabolic plasticity that allows for rapid, on-demand reprogramming in an ever-changing tumor microenvironment. We and others have provided evidence for inherent metabolic plasticity in GBM cells9,10 that presumably provides survival advantages and contributes to therapeutic resistance. The impact of radiation on tumor metabolism, and the molecular regulators involved in metabolic reprogramming during RT remain largely uninvestigated. Here we aim to narrow this knowledge gap as it applies to GBM radiotherapy.
The M2 isoform of pyruvate kinase (PKM2) is a tumor-specific, candidate mediator of GBM metabolic plasticity. PKM2 acts as a “metabolic switch” to promote rewiring of metabolic fluxes in a context-dependent manner by rapidly shifting between different oligomeric states11. Under proliferating conditions, growth factor signaling inhibits the enzymatic activity of PKM212. This creates a bottleneck in glycolysis making upstream glycolytic intermediates available for anabolic pathways, such as the PPP, that support tumor growth. In addition, the PPP contributes to the maintenance of cellular redox balance via the production of NADPH, promoting survival under oxidative stress conditions13 but the therapeutic implications in RT-resistant cancers remain unknown.
Here, we show that irradiated GBM cells rewire their glucose metabolism to drive a metabolic antioxidant response via the PPP that promotes resistance to the oxidative stress induced by radiation, and that PKM2 is a major regulator of this response. Evidence that PKM2 is overexpressed in GBM tumors, while normal brain tissue expresses the constitutively active PKM114,15, points to PKM2 as a potentially actionable therapeutic target for combining with RT. Here, we provide initial evidence that supports this approach.
Materials and Methods
Cell Culture
Primary human glioblastoma lines, GBM217, GBM374, and GBM382 were established at UCLA as described in Laks et al.16. Patient-derived xenografts (GBM38)17 were provided by Dr. Jann Sarkaria (Mayo Clinic). See Supplementary Materials and Methods for more details.
Irradiation and Treatments
Cells were irradiated at room temperature using an experimental X-ray irradiator (Gulmay Medical Inc, Suwanee, GA) at a dose rate of 7.1702 Gy/min. Unless otherwise stated, cells were treated with 10 μM of the PKM2 activator, TEPP46 (MedChem Express) 3 h prior to irradiation. 2-Deoxy-D-Glucose, 2DG (Sigma) was used at a dose of 5 mM in glucose-free complete GBM media, 2 h prior to irradiation. GSH-MEE (Sigma) was used at 5 mM, 5 minutes prior to irradiation. See Supplementary Materials and Methods for more details.
Gliomasphere Formation Assay
Gliomasphere-forming capacity was determined by plating cells in GBM media, into 96-well non-treated plates, at a range of cell densities appropriate for the different cell lines and doses of radiation. The surviving fraction was determined as previously described18. See Supplementary Materials and Methods for more details.
Glucose Uptake, Lactate Production, and ROS Detection
G6PDH, 6PGD, and PKM2 Activity Assays
In Vitro Metabolomics
For metabolite extraction and measurement methods See Supplementary Materials and Methods.
Immunofluorescence and Immunocytochemistry
Protein Extraction, Cell Fractionation, Western Blotting, RNA Extraction, and Real-time Quantitative Polymerase Chain Reaction
Subcutaneous Tumor Models in Mice
GBM374 cells were suspended in a solution of 1:1 mixture of Matrigel® basement membrane matrix (Corning) and DMEM medium and 50 000 cells were injected subcutaneously in the hind legs of NSG. For details on treatments see Supplementary Materials and Methods.
Intracranial Tumor Models: Survival Studies
GBM cells were implanted into the right striatum of the brains of mice using a stereotactic device. For more details on implantation methods, treatments, and IHC staining of brain tissue see Supplementary Materials and Methods.
In Vivo Stable Isotope Tracing
For details see Supplementary Materials and Methods.
In Vivo Micro-PET/Micro-computed Tomography Imaging
Male NSG mice were implanted subcutaneously with GBM374 gliomaspheres. Once the tumors were palpable, mice underwent micro-PET imaging immediately followed by micro-computed tomography imaging in the Genisys8 scanner (Sofie Biosciences). For details see Supplementary Materials and Methods.
Brain Penetration of PKM2-Activator
For sample preparation and detection see Supplementary Materials and Methods.
Statistics
Unless otherwise stated, statistical significance was determined by performing 2-sided t tests or 2-way ANOVA using Prism v9.3.0. P values were considered significant below 0.05 (*P < .05; **P < .01, ***P < .001, and ****P < .0001). All experiments were independently repeated at least 3 times.
See Supplementary Materials and Methods for statistical analysis of Subcutaneous tumor model and Intracranial model.
Data Availability
Raw data for the metabolomics study were generated at the UCLA Metabolomics center and at the University of Michigan Metabolomics core. Derived data supporting the findings of this study are available from the corresponding author upon request. Other data generated in this study are presented within the article and its supplementary files.
Ethics
For in vivo experiments, all animal procedures were approved by the UCLA Institutional Animal Care and Use Committee and by the University Committee on Use and Care of Animals at the University of Michigan.
Results
Radiation induces an enhanced “Warburg-like” metabolic phenotype in GBM
To determine the effect of radiation on glucose utilization, we used patient-derived, treatment-naïve GBM specimens with defined genetic features and mutation status (see Materials and Methods), that were propagated as gliomaspheres (gs) shown to maintain the phenotype of originating tumors19. The fluorescent analogue of glucose, 2NBDG was used as a surrogate for “glucose uptake”. The in vitro basal glucose uptake (Supplementary Figure 1A) reflected the relative growth rates of the different gliomasphere specimens (Supplementary Figure 1B, C). Adherent (ad) cultures, established by growing gliomaspheres in the presence of serum, took up more 2NBDG relative to gliomaspheres (Supplementary Figure 1D). All cultures significantly increased 2NBDG uptake following radiation by as much as 2-fold, in a radiation dose-dependent manner (Figure 1A–C, Supplementary Figure 1E, F), despite the expected slower growth rate (Supplementary Figure 1G) and smaller spheroid sizes of irradiated gliomaspheres (see Figure 3F). To determine if the enhanced glucose uptake persists beyond the first 72 h following radiation, we measured 2NBDG uptake in sequential generations of gliomaspheres (Supplementary Figure 1H). Glucose uptake remained elevated for up to 2 weeks following the initial radiation exposure with a single dose of 8 Gy (Figure 1D and Supplementary Figure 1I).
Figure 1.
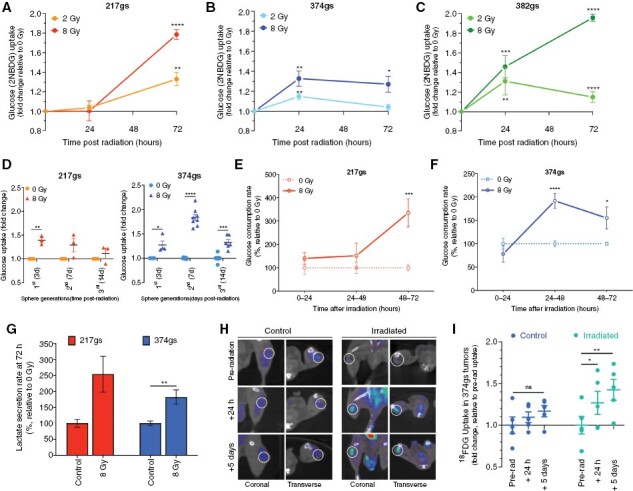
Radiation increases glucose consumption in GBM. (A–D) Glucose uptake measured via flow cytometry (2NBDG assay) in GBM217 (A), GBM374 (B) and GBM382 (C) gliomaspheres (gs). Unpaired t-tests, n = 3. (D) Glucose uptake after sequential generations of GBM217gs and GBM374gs following radiation. Unpaired t-tests, n = 3–4. (E–F) Glucose consumption rate determined via YSI biochemistry analyzer, in GBM217gs (E) and GBM374gs (F). Paired t-tests, n = 3. (G) Lactate secretion rate per cell determined via YSI. Paired t-tests, n = 3. (H–I) Glucose uptake measured via PET scan in tumor-bearing mice infused intravenously with 18F-FDG. Tumors outlined with a white circle on representative images (H). FDG uptake levels. Paired t-tests, n = 4 (I).
Figure 3.
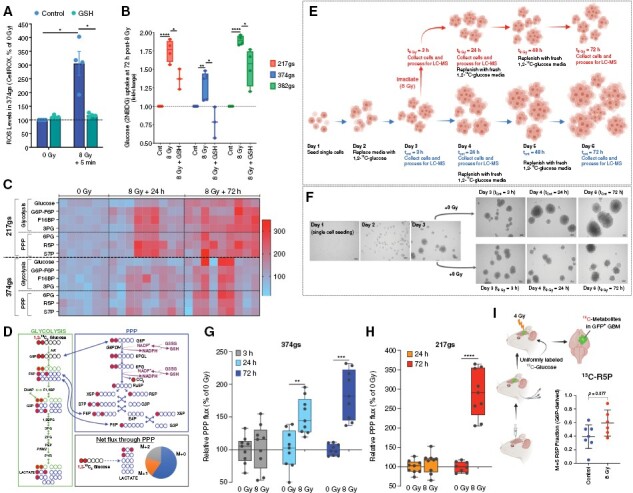
Irradiated gliomaspheres enhance flux through the PPP. (A) ROS levels determined with CellROX 5 min after radiation. Unpaired t-tests, n = 4. (B) Glucose uptake via 2NBDG, with 5 mM GSH added prior to radiation. Unpaired t-tests, n = 4. (C) Heatmap of relative ion counts for glycolytic and PPP metabolite levels. Pyruvate levels were not detectable. (D) 1,2-13C2 glucose flux and labeled carbon distribution through glycolysis (left panel) and the PPP (top right panel). Net flux through the PPP results in 3 M+0 lactates, one M+1 lactate and one M+2 lactate. (E) Protocol for glucose flux measurement. (F) Images of GBM374gs growth (10×). (G–H) Relative PPP flux was determined via glucose tracing experiments, using M+1 and M+2 lactate levels and glucose uptake rates in GBM217gs and GBM374gs treated as depicted in Figure 3E. Two-way ANOVA, n = 3. (I) Tissues were harvested from patient-derived GBM38 intracranial tumors, then assessed for 13C labeling by LC-MS. Fractions of ribose 5-phosphate (R5P) produced directly from the oxidative arm of the PPP were determined by dividing the M+5 fraction of R5P by the M+6 fraction of G6P. Unpaired t-tests, n = 6–7.
To validate the observations obtained with the 2NBDG method, we used biochemistry analyzers (YSI) as a complementary method that allows for a more direct, time-resolved, per cell basis measure of extracellular glucose concentration in growth media without disturbance to cell growth. This method confirmed the enhanced uptake of glucose by gliomaspheres with similar time kinetics following radiation (Figure 1E, F).
In general, cancer cells, including GBMs, convert most of the glucose carbons to lactate via glycolysis, even when oxygen is readily available (Warburg effect6). Similarly, irradiated gliomaspheres secrete more lactate (Figure 1G), suggesting that at least some of the “extra” glucose taken up following radiation is being converted to lactate.
To determine whether the enhanced glucose utilization following radiation also occurs in vivo, we used the glucose analogue [18F]fluorodeoxyglucose ([18F]-FDG), which allows for imaging of glucose uptake in vivo via PET. Consistent with the in vitro observations, subcutaneous GBM tumors took up significantly more glucose at 24 h and 5 days following radiation (Figure 1H, I, Supplementary Figure 1J, K).
Radiation induces translocation of glucose transporter 3 (GLUT3) to the cell membrane
To probe the mechanism behind the increase in glucose uptake by irradiated gliomaspheres we analyzed levels of the main glucose transporters (GLUT) and hexokinases (HK) in GBM cells (Figure 2A). Only HK2 gene expression levels increased following radiation (Figure 2A). At the protein level, GLUT1 levels were extremely low compared to GLUT3 and did not change following radiation (Supplementary Figure 2A). While GLUT3 was abundant (Supplementary Figure 2A) its total protein levels decreased following radiation (Figure 2B), reflecting the decreased gene expression levels (Figure 2A). Although gene expression levels of HK2, the predominant isoform in GBM, increased following radiation (Figure 2A), this did not correlate with increased protein levels (Figure 2C, Supplementary Figure 2B). Therefore, total protein levels of GLUTs or HKs did not explain the increase in glucose uptake following radiation.
Figure 2.
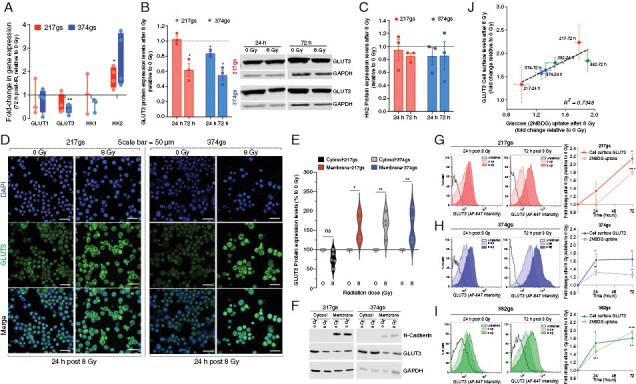
Radiation induces translocation of glucose transporters to the cell membrane. (A) GLUT1/3 and HK1/2 gene expression levels. Unpaired t-tests, n = 3. (B) GLUT3 total protein levels. Unpaired t-tests, n = 3. (C) HK2 total protein levels. Unpaired t-tests, n = 3. (D) Cell surface GLUT3 protein levels via confocal microscopy at 40×. Scale bar = 50 µm. (E–F) Cytosolic and membrane levels of GLUT3 protein at 24 h post-radiation. Two-way ANOVA, n = 3. (G–I) Cell surface GLUT3 protein levels via flow cytometry (left histogram panels) quantified and overlayed with 2NBDG uptake levels from Figure 1A–C (right panels). Unpaired t-tests, n = 3. (J) Correlation between fold change in GLUT3 protein levels at the cell surface and fold change in glucose uptake after radiation.
We then explored the possibility that glucose transporters translocate to the cell membrane more efficiently following radiation, thus providing irradiated GBM cells with a greater capacity to take up glucose. Immunofluorescence analysis revealed increased levels of GLUT3 on the cell surface following radiation, in both gliomaspheres (Figure 2D) and adherent cultures (Supplementary Figure 2C), whereas GLUT1 cell surface levels remained relatively similar (Supplementary Figure 2D). The radiation-induced increase in cell surface levels of GLUT3 was also confirmed via cell fractionation methods (Figure 2E, F) and quantitatively via flow cytometry (Figure 2G–I) and closely mirrored the kinetics of glucose uptake by gliomaspheres (Figure 2G–I, right hand panels). The fold change in 2NBDG uptake induced by radiation linearly correlated with the fold change in cell surface levels of GLUT3 (Figure 2J), providing a plausible explanation for the increase in glucose uptake by irradiated gliomaspheres (Figure 1).
Radiation rewires glucose metabolism to enhance PPP activity
We hypothesized that the increase in glucose uptake by irradiated gliomaspheres is a response to the oxidative stress induced by radiation. To test this, we irradiated gliomaspheres in the presence of a potent antioxidant, reduced glutathione (GSH), provided in the form of glutathione reduced ethyl ester (GSH-MEE), a membrane/lipid permeable derivative of GSH, at concentrations that prevent the accumulation of radiation-induced reactive oxygen species (ROS) (Figure 3A, Supplementary Figure 3A). GSH partially or fully prevented the increase in glucose uptake (Figure 3B), pointing to an adaptive role for glucose in mitigating the oxidative stress induced by radiation.
To understand the fate of glucose carbons in irradiated gliomaspheres, we measured metabolite levels via LC-MS using untargeted and targeted metabolomics. Untargeted analysis revealed that total abundance levels of glucose and metabolites in upper glycolysis, such as phosphorylated glucose/fructose-6-phosphate (G6P/F6P), fructose-1,6-bisphosphate (F1,6BP), and 3-phosphoglycerate (3PG) increased following radiation (Figure 3C and Supplementary Figure 3B, C). G6P serves as the branching point of glycolysis into the oxidative (ox) arm of the PPP (oxPPP), while F6P, F1,6BP, and 3PG can reversibly feed into the non-oxidative (non-ox) PPP arm (Figure 3D). Analysis of abundance levels of PPP metabolites, such as 6-phosphogluconate (6PG) and ribulose-5-phosphate (R5P) in the oxPPP, and sedoheptulose 7-phosphate (S7P) in the non-oxPPP revealed increased abundance in irradiated gliomaspheres (Figure 3C and Supplementary Figure 3B, C).
Abundance of metabolite levels is the net result of metabolite production and consumption and does not inform on metabolic flux. Therefore, to better understand the flux of glucose carbons following radiation, we performed 1,2-13C2-glucose tracing experiments and analyzed the 13C-labeled metabolite levels via LC-MS to distinguish glucose carbons routed through the PPP from those oxidized strictly via glycolysis20 (Figure 3D, E). As depicted in Figure 3D, HK converts 1,2-13C2-glucose to 1,2-13C2-G6P, which can either be metabolized through glycolysis or through the PPP. When metabolized through glycolysis, the downstream metabolites will maintain 2 13C-labeled carbons (M+2). When 1,2-13C2-G6P is shunted through the PPP, an oxidative decarboxylation reaction removes the 13C label on the first position of glucose, generating singly 13C-labeled metabolites (M+1), some of which feed back into the glycolysis pathway via the non-oxPPP (Figure 3D). We used the difference in lactate 13C-labeling (Supplementary Figure 3D, E) normalized by the rate of glucose consumption (as determined in Figure 1E, F) to calculate absolute flux of glucose carbons through the PPP. Despite the heterogeneity in sphere sizes and growth rates (Figure 3E, F), targeted metabolomics analysis revealed a clear increase in PPP flux in irradiated gliomaspheres that becomes significant by 24 h following radiation and is sustained for up to at least 72 h, the last time point analyzed (Figure 3G, H).
The 1,2-13C2-glucose tracer is less adequate for determining PPP flux in vivo due to label mixing in other tissues and the circulatory system that would produce many different, unexpected forms of labeled glucose. Therefore, we used a uniformly labeled 13C-glucose tracer to infuse a mouse intracranial model of patient-derived gliomaspheres (Figure 3I). We observed that the G6P-derived fraction of R5P was increased in irradiated tumors, indicative of enhanced PPP activity (Figure 3I and Supplementary Figure 3F).
PKM2 enables the radiation-induced increase in PPP activity to harness its antioxidant power
The enzymatic activity of the first rate-limiting enzyme in the PPP, glucose-6-phosphate dehydrogenase (G6PDH), or the second enzyme, 6-phosphogluconate dehydrogenase (6PGD) were not affected by radiation, although oxidative stress induced by hydrogen peroxide (H2O2) significantly downregulates G6PDH activity (Supplementary Figure 4A, B). This indicated that the radiation-induced shunting of glucose carbons through the PPP (Figure 3G, H) cannot be explained by alterations in PPP enzyme activity. Others have shown that acute oxidative stress induced via means other than radiation, inhibits the enzymatic activity of the last rate-limiting enzyme in glycolysis, PKM2 and that this supports antioxidant production by promoting glucose oxidation via the PPP13. Since the increase in glucose uptake by irradiated gliomaspheres (Figure 1) is accompanied by increased PPP flux (Figure 3G–I) and this seems to be a response to oxidative stress generated by radiation (Figure 3B), we considered the possibility that oxidative stress induced by radiation inhibits PKM2 activity, resulting in increased PPP flux.
Although radiation had no effect on PKM2 protein levels (Supplementary Figure 4C–E) it suppressed its enzymatic activity (Figure 4A), but this was prevented by the addition of GSH-MEE (Figure 4B and Supplementary Figure 4F), suggesting that radiation-induced oxidative stress promotes the suppression of PKM2 activity. The addition of the reducing agent dithiothreitol (DTT) to the cell lysate prior to measuring PK activity, partially restored PKM2 activity (Figure 4B and Supplementary Figure 4F), pointing to a reversible oxidizing event as the likely culprit for the suppressed PKM2 activity. TEPP46, a small molecule activator of PKM221 (Supplementary Figure 4G, H) prevented the suppression of PKM2 activity (Figure 4C) and the increase in lactate secretion following radiation (Figure 4D). Although TEPP46 had no effect on basal PPP flux, it dampened the radiation-induced increase (Figure 4E), pointing to PKM2 as a promoter of enhanced PPP flux following radiation of gliomaspheres.
Figure 4.
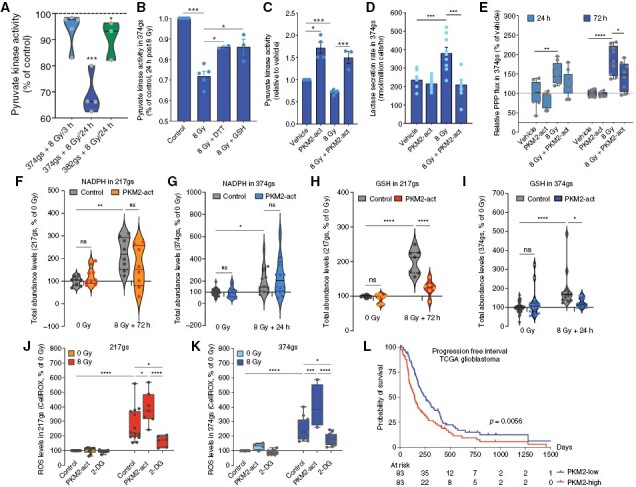
Radiation suppresses PKM2 enzymatic activity, which regulates glucose flux through the PPP. (A) Violin plots of PK activity in GBM374gs and GBM382gs after radiation. Dotted line represents the 0 Gy control. (B–C) PK activity in the presence of DTT (1 mM) or GSH (5 mM) added prior to radiation (B) or in the presence of a PKM2 activator (TEPP46) (C). Unpaired t-tests, n = 3. (D) Lactate secretion per cell via YSI in GBM374gs, 72 h post-radiation. Paired t-tests, n = 3. (E) Relative PPP flux determined from M+1 and M+2 lactate levels and glucose uptake rates in GBM374gs. Unpaired t-tests, n = 3. (F–I) Abundance levels (ion counts) of NADPH and GSH in GBM217gs and GBM374gs, measured via LC-MS. Two-way ANOVA, n = 3. (J–K) ROS levels in GBM374gs and GBM217gs via CellROX. Two-way ANOVA, n = 3. (L) Progression-free interval in PKM2-low and PKM2-high expressing GBM tumors.
The increase in PPP flux is accompanied by increased NADPH and GSH abundance levels (Supplementary Figure 4I, J), suggesting a PPP contribution to the NADPH/GSH pool following radiation. While PKM2 activators do not seem to affect NADPH levels (Figure 4F, G), pointing to other sources of NADPH production following radiation, they do reduce GSH levels (Figure 4H, I). This suggests that radiation-induced ROS levels should be exacerbated when PKM2 is activated. Indeed, TEPP46 increases ROS levels following radiation (Figure 4J, K). Alternatively, NADPH supply via the PPP can be increased by providing cells with 2-deoxyglucose (2DG). Although 2DG is a commonly used glycolysis inhibitor that cannot be shunted into the glycolysis pathway downstream of phosphoglucose isomerase, it can still be shunted into the PPP to produce NADPH22,23. 2DG dampened ROS accumulation in irradiated gliomaspheres (Figure 4J, K), likely by stimulating NADPH production via the PPP and maintaining reduced GSH pools. Analysis of PKM2 expression levels in the TCGA glioblastoma cohort revealed a strong correlation to progression-free survival (Figure 4L), highlighting an important role for PKM2 in GBM and suggesting that PKM2 might be a potential therapeutic target.
PKM2 activators sensitize GBM to radiation therapy
We evaluated the potential of pharmacological activation of PKM2 via TEPP46 in radiosensitizing GBM. Using a modified in vitro clonogenic “sphere forming assays” (SFA, See Materials and Methods) we showed that TEPP46 radiosensitizes gliomaspheres from all 3 lines (Figure 5A–C) and this effect lasts beyond the first generation of gliomaspheres (Figure 5D). Relevant to potential in vivo application, TEPP46 had no apparent effect as a single agent or as a radiosensitizer on normal human astrocytes in vitro (Figure 5E), indicating that PKM2 activators will not affect normal brain cells, which likely rely more on PKM1 for their metabolic needs14.
Figure 5.
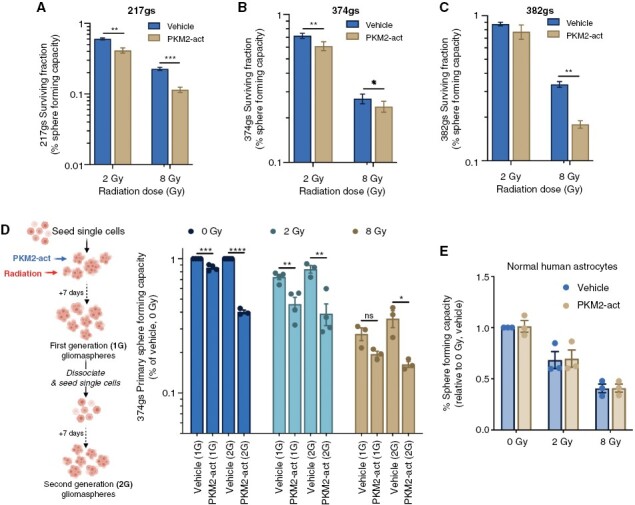
PKM2 activation radiosensitizes gliomaspheres in vitro. (A–C) Surviving fraction of gs determined via sphere forming capacity assays. Unpaired t-tests, n = 3. (D) The spheres were then dissociated and re-seeded without further treatment and allowed to form secondary spheres (second generation, 2G). Unpaired t-tests, n = 3. (E) Sphere forming capacity assay with normal human astrocytes. Unpaired t-tests, n = 3.
In vivo, we determined the effect of combining TEPP46 with radiation in subcutaneous (sub-c) and orthotopic xenograft models of human GBM. In the 374gs sub-c model, TEPP46 alone was initially more efficient at controlling tumor growth than fractionated radiation (Figure 6A). However, combining TEPP46 with radiation resulted in sustained tumor control over time and in some cases, tumor regression (Figure 6A and Supplementary Figure 5A, B). Linear mixed-effects modeling identified significant negative effects of the combination regimen on tumor growth rate (Supplementary Figure 5C, D). The tumor average maximum growth rates, as estimated by smoothing spline for vehicle, radiation, TEPP46 or combined treatment were 0.18, 0.14, 0.17, and 0.07, respectively. TEPP46 had no apparent effect on the weights of the mice, supporting a favorable overall toxicity profile (Supplementary Figure 5E).
Figure 6.
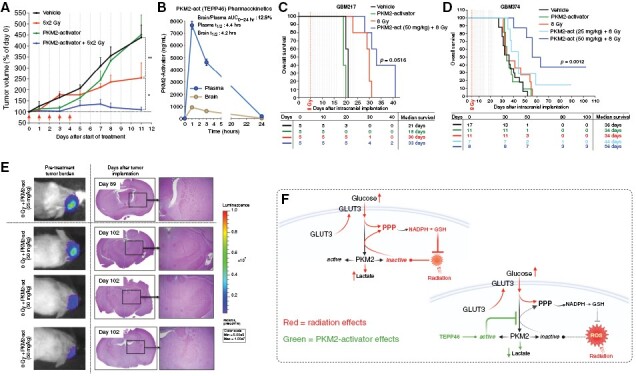
PKM2 activation radiosensitizes GBM tumors in vivo. (A) Tumor growth of GBM374 subcutaneous tumors, treated with 50 mg/kg TEPP46 (dotted vertical lines) and 2 Gy fractions of radiation ( arrows). Statistical analyses in the M&M section. (B) Pharmacokinetic analysis of TEPP46 levels in the plasma and the brain of TEPP46-treated (50 mg/kg), tumor-free NSG mice, determined by LC-MS. (C–D) Overall survival of mice bearing GBM374 and GBM217 intracranial tumors, via KM analysis (D), with endpoint as ‘time to euthanasia’ (time from tumor implantation to euthanasia). Dotted vertical lines: TEPP46 treatment days. The difference in survival among these treatment groups was determined via log-rank test (P = .001). Using Cox PH regression analysis, the difference in overall survival among treatment groups of GBM374 was also determined via LRT. See M&M for details. The table below indicates the number of mice still alive and well on a given day. (E) Luminescence images of long-term survivors’ initial tumor burden, and hematoxylin/eosin IHC staining of brains at the time of euthanasia. Right panels show zoomed-in images (4×) of the presumed tumor location. (F) Radiation increases glucose uptake in gliomaspheres, via increased membrane localization of GLUT3. Radiation also suppresses PKM2 activity, leading to a bottleneck in glycolytic flux and accumulation of glycolytic intermediates, promoting flux through the PPP, and the generation of antioxidants that have the potential to neutralize the oxidative stress induced by radiation. The use of PKM2-activators can prevent the suppression of PKM2 activity by radiation, thus antagonizing the antioxidant function of the PPP.
Previous studies show that TEPP46 penetrates the blood brain barrier24, therefore we evaluated the therapeutic potential of TEPP46 in orthotopic GBM models with different median survival times: 217gs, median survival ~3 weeks and 374gs, median survival ~5 weeks. Our own evaluation of the in vivo pharmacokinetics of TEPP46 administered intraperitoneally showed that the brain-to-plasma area under the curve (0–24 h) for TEPP46 in non-tumor bearing mice is ~13%, with a plasma and brain half-life of around 4 h (Figure 6B). As a single agent, TEPP46 had no impact on the overall survival for either model however, the combination treatment had a significant radiosensitizing effect, but only at the higher dose of 50 mg/kg of TEPP46 (Figure 6C, D). This combination treatment resulted in the greatest estimated median survival of 33 and 56 days for GBM217 and GBM374, respectively, and the lowest hazard ratio among all the groups, with 8 Gy as the reference group (Figure 6C, D, Supplementary Figure 5F, G). Notably, we observed long-term survivors with no detectable tumors in both TEPP46 doses in the GBM374 model (Figure 6E, Supplementary Figure 5H). These results suggest that the lower dosing of TEPP46 might not be reaching therapeutic concentrations in the brain. Despite this apparent limitation of TEPP46 availability in vivo, 3 out of 8 mice survived tumor-free for over 3 months, in the slower-growing GBM374 model (Figure 6C and E and Supplementary Figure 5H, I).
Discussion
The reconfiguration of carbohydrate metabolism as a regulated response to cellular oxidative stress has been recognized for over 2 decades, including the oxidative stress-induced rerouting of glucose carbons into the oxPPP25,26. Here, we report that GBM cells retain a remarkable degree of metabolic plasticity that supports their survival during oxidative stress induced by RT. They consume additional glucose following irradiation (Figure 1), akin to our observations in breast cancer27. Early studies have reported similar observations of a rapid rise in glucose uptake during RT, via [18F]-FDG-PET imaging of mouse and human tumors28,29, pointing to a more general response of tumors treated with RT. Antioxidants prevent the increase in glucose uptake (Figure 3A, B), pointing to the radiation-induced oxidative stress as the likely ‘signal’ for this response, in line with reports of a ROS-dependent increase in tumor glucose demand following radiation of mammary carcinomas30.
Our findings show that radiation promotes a net outward translocation of GLUT3 to the cell’s surface accounting for the changes in glucose transport following radiation (Figure 6F). Although the mechanism of radiation-induced translocation of GLUT3 remains to be determined, the involvement of GLUT3 in facilitating increased glucose consumption by irradiated GBM cells is not surprising. GLUT3 has a higher affinity for glucose than GLUT1, 2- or 4- and ~5-fold greater transport capacity than GLUT1 or 431, ensuring efficient glucose uptake. GLUT3 is critical to the survival of GBM stem cells in low glucose32 and subpopulations of GBM are GLUT3-addicted33. While GLUT1 and GLUT3 are the 2 main transporters in cerebral glucose metabolism, only GLUT3 levels predict for poor GBM survival32,33.
Our findings show that the radiation-induced increase in PPP flux is regulated, in part, by the suppression of PKM2 activity in gliomaspheres, due to an oxidative event (Figure 4A, B). Oxidized, kinase-inactive PKM2 blocks pyruvate production, making upstream glycolytic intermediates available for the PPP, one of the main sources of cellular NADPH13. PKM2 activators reduce flux of glucose carbons through the PPP (Figure 4E), exacerbates ROS levels (Figure 4J, K) and radiosensitize GBM (Figures 5 and 6). Together, these findings posit that enhanced carbon flux through the PPP is a PKM2-regulated, pro-survival metabolic response in irradiated GBM (Figure 6F). Although not investigated here, another potential benefit from enhanced PPP flux is the generation of nucleotide precursors for DNA repair34, a possibility that warrants further investigation.
Although seemingly contradictory, our findings are in line with others showing radiosensitization of GBM cells by knocking down PKM2 expression35,36. PKM2 can readily switch between an enzymatically active-tetramer and an inactive-dimer, altering glycolytic flux. The dimers can also translocate to the nucleus where they moonlight as regulators of DNA repair, as is demonstrated by the Sizemore and Wu studies35,36. Knocking down PKM2 would abrogate both the dimeric and tetrameric forms of PKM2, therefore abrogating both of its functions as a regulator of glycolytic flux in the cytosol and a regulator of DNA repair in the nucleus, whereas our approach is to alter the balance between the 2 conformations using the small molecule activator TEPP46, that could have greater clinical relevance.
Clearly, normal cells are also capable of reconfiguring glycolytic flux as a way to mitigate oxidative stress22,25,26 and redox-sensitive metabolic enzymes, such as GAPDH can regulate this response in normal cells37. Therefore, identifying cancer-specific regulators of redox metabolic plasticity for therapeutic targeting is crucial to the success of this approach. PKM2 is overexpressed in GBM15,24, making it an attractive target of “metabolic plasticity’ for GBM radiosensitization. Small molecule activators of PKM2 show promise in slowing tumor growth in xenograft models21,38 prompting clinical trial testing as single agents (Clinicaltrials.gov, NCT04328740, recruiting), yet to be combined with other anti-cancer therapies, including RT. The development of PKM2 imaging agents in brain tumors15,39,40, provides an additional opportunity for coupling treatment with target imaging to optimize potential for benefit. Despite critical differences, many parallels exist between the metabolic regulation of rapidly proliferating cancer cells and activated immune effector cells41. Evidence exists, although none in tumor models, suggesting that activation of PKM2 hampers immune cell activation42,43. Therefore, PKM2 targeting needs further validation in the context of an intact immune system to fully support its radiotherapeutic potential.
Supplementary Material
Acknowledgments
We thank Min Li and Mansoureh Eghbali, Department of Anesthesiology at UCLA, for their support in acquiring confocal images. We thank Mikayla Tamboline for her help with the micro-PET/CT imaging experiments in the UCLA Crump Preclinical Imaging Technology Center.
Contributor Information
Justine Bailleul, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Yangjingyi Ruan, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Lobna Abdulrahman, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Andrew J Scott, Department of Radiation Oncology, University of Michigan, Ann Arbor, Michigan, USA; Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan, USA.
Taha Yazal, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
David Sung, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Keunseok Park, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, California, USA.
Hanna Hoang, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Juan Nathaniel, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Fang-I Chu, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Daisy Palomera, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Anahita Sehgal, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Jonathan E Tsang, Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
David A Nathanson, Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Shili Xu, Jonsson Comprehensive Cancer Center, University of California, Los Angeles, California, USA; Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, California, USA; Crump Institute for Molecular Imaging, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
Junyoung O Park, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, California, USA.
Johanna ten Hoeve, Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Kruttika Bhat, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Nathan Qi, Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, Michigan, USA.
Harley I Kornblum, Jonsson Comprehensive Cancer Center, University of California, Los Angeles, California, USA; Neuropsychiatric Institute–Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, California, USA.
Dorthe Schaue, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
William H McBride, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA.
Costas A Lyssiotis, Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, Michigan, USA; Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan, USA.
Daniel R Wahl, Department of Radiation Oncology, University of Michigan, Ann Arbor, Michigan, USA; Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan, USA.
Erina Vlashi, Department of Radiation Oncology, David Geffen School of Medicine, University of California, Los Angeles, California, USA; Jonsson Comprehensive Cancer Center, University of California, Los Angeles, California, USA.
Funding
Andrew Scott was supported by F32CA260735. Dan Wahl was supported by NCI K08CA234416 (DRW), R37CA258346 (DRW), Cancer Center Support Grant P30CA46592, Damon Runyon Cancer Foundation, Ben and Catherine Ivy Foundation and the Sontag Foundation. David Nathanson and Harley Kornblum were supported by UCLA SPORE in Brain Cancer (P50 CA211015). Erina Vlashi was supported by NCI CA251872 (EV), UCLA SPORE in Brain Cancer (P50 CA211015) (EV), Jonsson Comprehensive Cancer Center at UCLA Seed Grant and the UCLA Clinical and Translational Science Institute grant UL1TR001881. The micro-PET/CT imaging was supported by the Cancer Center Support Grant (2P30CA016042-44).
Conflict of interest statement
The authors declare no conflict of interest.
Authorship statement
see Supplementary Material.
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Fabian D, Guillermo Prieto Eibl M, Alnahhas I, et al. Treatment of glioblastoma (GBM) with the addition of tumor-treating fields (TTF): a review. Cancers. 2019;11(2):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chamberlain MC. Radiographic patterns of relapse in glioblastoma. J Neurooncol. 2011;101(2):319–323. [DOI] [PubMed] [Google Scholar]
- 4. Pavlova NN, Thompson CB.. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou W, Wahl DR.. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers. 2019;11(9):1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Warburg O. On the metabolism of carcinoma cells. Biochem Z. 1924;152:309–344. [Google Scholar]
- 7. DeBerardinis RJ, Chandel NS.. We need to talk about the Warburg effect. Nat Metab. 2020;2(2):127–129. [DOI] [PubMed] [Google Scholar]
- 8. Moreira JD, Hamraz M, Abolhassani M, et al. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites. 2016;6(4):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA. 2011;108(38):16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shibao S, Minami N, Koike N, et al. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro-oncology. 2018;20(3):343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaneton B, Gottlieb E.. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37(8):309–316. [DOI] [PubMed] [Google Scholar]
- 12. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. [DOI] [PubMed] [Google Scholar]
- 13. Anastasiou D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukherjee J, Phillips JJ, Zheng S, et al. Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PLoS One. 2013;8(2):e57610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beinat C, Patel CB, Haywood T, et al. A clinical PET imaging tracer ([(18)F]DASA-23) to monitor pyruvate kinase M2 induced glycolytic reprogramming in glioblastoma. Clin Cancer Res. 2021;27(23):6467–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laks DR, Crisman TJ, Shih MYS, et al. Large-scale assessment of the gliomasphere model system. Neuro-oncology 2016;18(10):1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou W, Yao Y, Scott AJ, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun. 2020;11(1):3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yazal T, Bailleul J, Ruan Y, et al. Radiosensitizing pancreatic cancer via effective autophagy inhibition. Mol Cancer Ther. 2021;(1):79–88. [DOI] [PubMed] [Google Scholar]
- 19. Gosa L, Ta L, Nathanson DA.. Processing of primary patient tumors and subsequent generation of primary cell lines. Methods Mol Biol. 2019;1897:425–431. [DOI] [PubMed] [Google Scholar]
- 20. Lee WN, Boros LG, Puigjaner J, et al. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am J Physiol. 1998;274(5):E843–E851. [DOI] [PubMed] [Google Scholar]
- 21. Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8(10):839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le Goffe C, Vallette G, Charrier L, et al. Metabolic control of resistance of human epithelial cells to H2O2 and NO stresses. Biochem J. 2002;364(Pt 2):349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu X, Olszewski K, Zhang Y, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol. 2020;22(4):476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Witney TH, James ML, Shen B, et al. PET imaging of tumor glycolysis downstream of hexokinase through noninvasive measurement of pyruvate kinase M2. Sci Transl Med. 2015;7(310):310ra169. [DOI] [PubMed] [Google Scholar]
- 25. Grant CM. Metabolic reconfiguration is a regulated response to oxidative stress. J Biol. 2008;7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ralser M, Wamelink MM, Kowald A, et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol. 2007;6(4):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang L, Bailleul J, Yazal T, et al. PK-M2-mediated metabolic changes in breast cancer cells induced by ionizing radiation. Breast Cancer Res Treat. 2019;178(1):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Furuta M, Hasegawa M, Hayakawa K, et al. Rapid rise in FDG uptake in an irradiated human tumour xenograft. Eur J Nucl Med. 1997;24(4):435–438. [DOI] [PubMed] [Google Scholar]
- 29. Hautzel H, Muller-Gartner HW.. Early changes in fluorine-18-FDG uptake during radiotherapy. J Nucl Med. 1997;38(9):1384–1386. [PubMed] [Google Scholar]
- 30. Zhong J, Rajaram N, Brizel DM, et al. Radiation induces aerobic glycolysis through reactive oxygen species. Radiother Oncol. 2013;106(3):390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simpson IA, Dwyer D, Malide D, et al. The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab. 2008;295(2):E242–E253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flavahan WA, Wu Q, Hitomi M, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16(10):1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cosset E, Ilmjärv S, Dutoit V, et al. Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell. 2017;32(6):856–868.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tong X, Zhao F, Thompson CB.. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr Opin Genet Dev. 2009;19(1):32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu S, Cao R, Tao B, et al. Pyruvate facilitates FACT-mediated γH2AX loading to chromatin and promotes the radiation resistance of glioblastoma. Adv Sci (Weinh). 2022;9(8):e2104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sizemore ST, Zhang M, Cho JH, et al. Pyruvate kinase M2 regulates homologous recombination-mediated DNA double-strand break repair. Cell Res. 2018;28(11):1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuehne A, Emmert H, Soehle J, et al. Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol Cell. 2015;59(3):359–371. [DOI] [PubMed] [Google Scholar]
- 38. Parnell KM, Foulks JM, Nix RN, et al. Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol Cancer Ther. 2013;12(8):1453–1460. [DOI] [PubMed] [Google Scholar]
- 39. Beinat C, Patel CB, Haywood T, et al. Human biodistribution and radiation dosimetry of [(18)F]DASA-23, a PET probe targeting pyruvate kinase M2. Eur J Nucl Med Mol Imaging. 2020;47(9):2123–2130. [DOI] [PubMed] [Google Scholar]
- 40. Wang D, Li C, Zhu Y, et al. TEPP-46-based AIE fluorescent probe for detection and bioimaging of PKM2 in living cells. Anal Chem. 2021;93(37):12682–12689. [DOI] [PubMed] [Google Scholar]
- 41. Andrejeva G, Rathmell JC.. similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017;26(1):49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palsson-McDermott EM, Curtis AM, Goel G, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015;21(2):347. [DOI] [PubMed] [Google Scholar]
- 43. Kono M, Maeda K, Stocton-Gavanescu I, et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight. 2019;4(12):e127395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for the metabolomics study were generated at the UCLA Metabolomics center and at the University of Michigan Metabolomics core. Derived data supporting the findings of this study are available from the corresponding author upon request. Other data generated in this study are presented within the article and its supplementary files.