

Abstract
We have reported that a high-salt (4.0% NaCl) dietary intake activates mTORC1 and inhibition of this pathway with rapamycin blunts the chronic phase of salt-induced hypertension and renal injury in Dahl salt-sensitive (SS) rats. In SS rats, high-salt intake is known to increase the renal production of H2O2 by NOX4, the most abundant NOX isoform in the kidney, and the global knockout of NOX4 blunts salt-sensitivity in these rats. Here, we explored the hypothesis that elevations of H2O2 by NOX4 in high-salt fed SS rat stimulate mTORC1 for the full development of salt-induced hypertension and renal injury. Our in vitro studies found that H2O2 activates mTORC1 independent of PI3K/AKT and AMPK pathways. To determine the in vivo relevance of NOX4/H2O2/mTORC1 in the salt-induced hypertension, SS-Nox4 knockout (SSNox4−/−) rats were daily administrated with vehicle/rapamycin fed a high-salt diet for 21 days. Rapamycin treatment of SSNox4−/− rats had shown no augmented effect on the salt-induced hypertension nor upon indices of renal injury. Significant reductions of renal T lymphocyte and macrophage together with inhibition of cell proliferation were observed in rapamycin treated rats suggesting a role of mTORC1 independent of NOX4 in the proliferation of immune cell. Given the direct activation of mTORC1 by H2O2 and absence of any further protection from salt-induced hypertension in rapamycin treated SSNox4−/− rats, we conclude that NOX4-H2O2 is a major upstream activator of mTORC1 that contributes importantly to salt-induced hypertension and renal injury in the SS rat model.
Keywords: NOX4, Dahl S rats, H2O2, mTORC1, salt-sensitive
Summary
A novel role for the NOX4-H2O2 has been revealed in vivo showing it serves as an upstream regulator of mTORC1 signaling and this pathway is a downstream determinant of salt-induced hypertension and renal injury in a clinically relevant model of hypertension.
Graphical Abstract
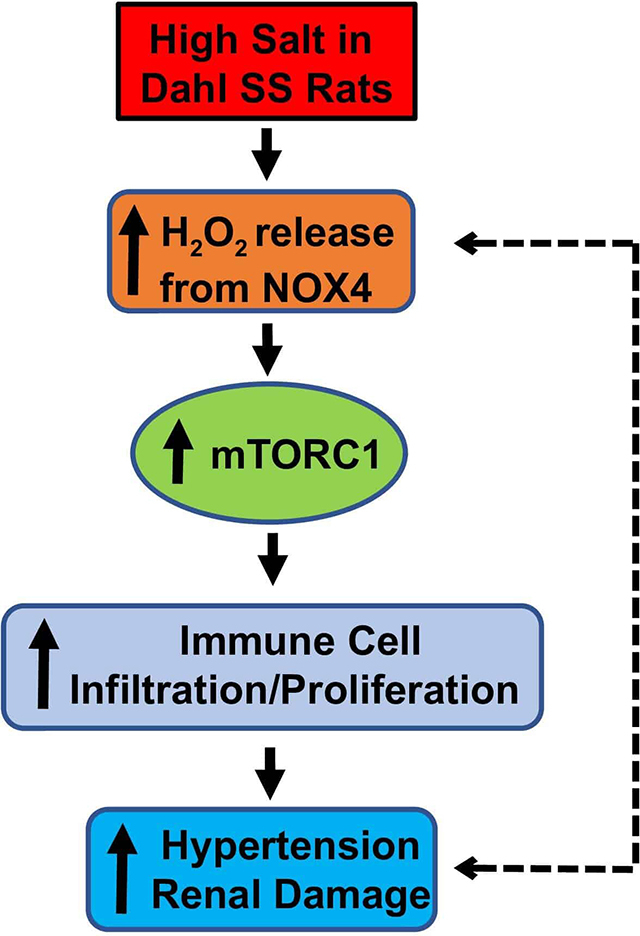
Introduction
The mTOR (mammalian target of rapamycin) is a highly conserved serine/threonine kinase which senses extracellular and intracellular signals involved in regulation of cell proliferation/growth by growth factors (insulin or insulin-like growth factors), nutrients (amino acids or glucose), and reactive oxygen species (ROS)1. mTOR forms two structurally and functionally distinct multiprotein complexes, mTORC1 and mTORC2, which correspond to two major branches within the overall signaling network. The activity of both complexes has been associated with elevation of ROS including hydrogen peroxide (H2O2) in several disease conditions2, 3. We found that chronic inhibition of mTORC1 with rapamycin blunted salt-induced hypertension4 but suppression of both complexes (mTORC1/2) with PP242 inhibitor completely abolished salt-induced hypertension in SS rats5. The connecting link between H2O2 and mTOR signaling in blood pressure salt-sensitivity is not yet fully understood.
NADPH oxidase 4 (Nox4) is the most abundant Nox isoform in the kidney and the major source of H2O26 but its role in cardiovascular and related renal injury is not well understood. There is growing evidence indicating the role of this gene in many other pathophysiological conditions. For example, transforming growth factor beta induces the upregulation of Nox4 in human aortic smooth muscle cells 7 and is also a major contributor to oxidative stress in diabetic nephropathy8. In type 1 diabetes, the activation of mTOR enhances oxidative stress via upregulation of Nox4 and Nox1 and NADPH oxidase activity9. Nox4 dependent activation of mTORC2 via AMPK is observed in idiopathic pulmonary hypertension10.
We have found that endogenously produced H2O2 from NOX4 in the kidney contributes to salt-induced hypertension in SS rats11, 12. Also, that mTORC1 is elevated in the kidney of SS fed a high-salt diet and Sprague Dawley rats receiving a non-hypertensive H2O2 infusion in the renal interstitial space for 3 days4. Together, these observations suggested that mTORC1 activation is signaled by elevations of H2O2. However, the molecular mechanism by which mTORC1 is activated by high-salt feeding in SS rats and by H2O2 in particular is largely unknown and is the focus of the present study.
Methods Summary
All supporting data used for this study are available within the article and in the online-only Data Supplement. In vitro studies were performed in normal rat kidney epithelial (NRK-52E) cell line. Experiments were performed with male SSNox4−/− rats11. Rapamycin (I.P., 1.5 mg/kg/day) was chronically administered to 10 weeks old SSNox4−/− rats fed a low-salt (0.4% NaCl) diet for 4 day and 21 day after switching to a high-salt (4.0% NaCl) diet. As we have described11, radiotelemetry catheters and transmitters were surgically implanted for recording 24 h/day arterial pressure. On the final day of the 4.0% NaCl diet period, rats were placed in a metabolic cage for a 24 h urine collection. Immunohistochemistry, Western blot, renal tubular injury and glomerular score were performed as we have described11, 13, 14. Detailed experimental methods and description of antibodies are available in the online-only Data Supplement.
Statistical Methods
Data are presented as mean values ± standard error. A two-way analysis of variance (ANOVA) for repeated measures test were used for blood pressure analysis. For multiple groups, statistical significance was analyzed by one-way ANOVA (Tukey’s post hoc multiple comparison test). A paired Student’s t test was used to compare the effect of treatments on body weight. P<0.05 was considered significant.
Results
H2O2 activated mTORC1 independent of PI3K/AKT pathway in vitro.
Figure 1A illustrates the canonical activation of mTORC1 pathway. In this study, we examined the effect of H2O2 on this pathway using a normal rat kidney epithelial (NRK-52E) cell line in the presence or absence of pharmacological inhibitors, LY294002 (PI3K inhibitor) and rapamycin (mTORC1 inhibitor). As shown in Figure 1B&C and Figure S1A, 1 μM and 5 μM of H2O2 showed significant increases of mTORC1 activity measured by functional markers S6K1T389/S6K1, pS6S235/236/S6, and 4E-BP1T37/46/4E-BP1. Higher dose of H2O2 (100 μM) exerted significant inhibition of pS6S235/236/S6 and 4E-BP1T37/46/4E-BP1. As seen in the Figure 1D and Figure S1B, activation of mTORC1 by H2O2 was found to be independent of the suppression of the protein level of negative regulators PTEN and TSC2. The level of NOX4 protein and pAMPKT172/AMPK remained unaffected with the increasing mTORC1 activity however suppression of mTORC1 with H2O2 at 100 μM significantly activated AMPK (Figure S1C&D). To further explore the mechanism, NRK-52E cells were pretreated with 0.5–20 μM of LY294002 for 30 minutes to inhibit PI3K/AKT signaling. As shown in Figure S1E. 0.5 μM LY294002 abolished PI3K dependent phosphorylation of AKT at the T308 position thereby blocking the PI3K/AKT upstream stimulatory actions on mTORC1 activity. Quantitively, as shown in Figure 1E and Figure S1F, this dose of LY294002 reduced the levels of pS6S235/236/S6 and 4E-BP1T37/46/4E-BP1 by 70–85 % which was reversed by different doses of H2O2 (1–25 μM). As shown in Figure 1F, increasing doses of H2O2 failed to reverse the rapamycin specific inhibition of pS6S235/236/S6 suggesting that H2O2 directly activates mTORC1 independent of the PI3K/AKT and AMPK pathways.
Figure 1:

A, in the canonical mTOR pathway, ligand IGF binds to the receptor tyrosine kinase (RTK), which activates phosphatidylinositol-3-kinase (PI3K) which in turn phosphorylates phosphatidylinositol 4,5-biphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). Phosphatase and tensin homolog (PTEN) dephosphorylate PIP3 acting as a key negative regulator of PI3K signaling. PIP3 activates PDK1 which phosphorylates protein kinase B (PKB/AKT) at T308 thereby phosphorylating the negative regulator tuberous sclerosis complex 1 (TSC1)-TSC2 to activate mTORC1. mTORC1 is rapamycin sensitive and thought to facilitate many of its downstream effects through S6 kinase 1 (S6K1), ribosomal S6, and translational inhibitor 4E-binding protein 1 (4E-BP1). Energy stress activates AMPK (AMP-activated protein kinase) which represses mTORC1. LY294002 and rapamycin inhibit the PI3K/AKT and mTORC1, respectively. In vitro experiment, NRK-52E cells were serum deprived overnight and treated with H2O2 for 30 min or pretreated with LY294002 or rapamycin for 30 min before incubating with increasing concentration of H2O2 for 30 min. B-D, effect of increasing concentrations of H2O2 on pS6S235/236/S6, p4E-BP1T37/46/4E-BP1, and PTEN/β-ACTIN from three different experiments. E, pretreatment with LY294002 inhibited pS6S235/236/S6 and addition of H2O2 reversed pS6S235/236/S6 in pretreated cells. F. H2O2 failed to reverse pS6S235/236/S6 in rapamycin pretreated cells. Quantification of Western blots performed using Image Lab 5.2. Representative Western blots are shown below each graph. Analysis of variance (ANOVA) with Tukey’s post hoc test was used for multiple comparisons. * P<0.05, ns; non-significant.
Elimination of NOX4-H2O2 in SS rat reduced mTORC1 activity in response to high-salt diet.
The results of in vitro studies prompted us to propose a non-canonical activation of mTORC1 by NOX4-H2O2 in the kidney of SS rats fed a high-salt diet (Figure 1A). Consistent with our in vitro findings, neither the cortical and medullary protein levels of PTEN (the first upstream negative regulator of mTORC1), nor its downstream effector substrate pAKTT308/AKT controlled by PDK1 kinase differed in SS and SSNox4−/− rats fed a 4.0% NaCl diet for 21 day (Figure 2A&B). Also seen in Figure 2C, TSC2 protein which is a second upstream negative regulator of mTORC1 remained unchanged in these tissues. As expected, pS6K1T389/S6K1 and pS6S235/236/S6 which are downstream substrates of mTORC1 were reduced in the cortex and medulla of SSNox4−/− rats compared to SS rats (Figure 2D&E). These responses are a consequence of the high-salt diet since no significant differences of pS6S235/236/S6 and p4E-BP1T37/46/4E-BP1 were observed in the renal cortex and medulla of SS and SSNox4−/− rats maintained on 0.4% NaCl (low-salt) diet since weaning (Figure 2F and Figure S2).
Figure 2:

Western blot of cortical and medullary tissues obtained from SS (n=6) and SSNox4−/− (n=5 or 6) rats fed a 4.0% NaCl (A-E) or 0.4% NaCl (F) diet for 21 days. Shown here is the membrane staining of PTEN, pAKTT308, AKT, TSC2, pS6K1T389, S6K1, pS6S235/236, S6 and GAPDH. Unpaired t test was used to compare the strain difference. * P<0.05.
These data indicate that NOX4 produced H2O2 is not essential for the maintenance of mTORC1 activity with a low-salt diet intake but played a critical role in the dysregulation of this pathway with a high-salt diet in SS rats. The most relevant and novel finding of this study is NOX4 generated H2O2 serves an upstream regulator of mTORC1 activity in rats maintained on 4.0% NaCl and in vitro observation obtained in the NRK-52E cell line.
Rapamycin failed to augment protection from salt-induced hypertension in SSNox4−/− rats.
We have found that salt-induced hypertension of rapamycin treated SS rats mimics the phenotypes observed in SSNox4−/−11. This suggested that NOX4 generated H2O2 most likely activated mTORC1 and that reduction of mTORC1 activity with rapamycin produce a similar phenotype as that found in SSNox4−/− rat fed a high-salt diet. If this hypothesis was true, further inhibition of mTORC1 with rapamycin in SSNox4−/− in which renal mTORC1 activity was also found to be low would not be expected to augment additional protection from salt-induced hypertension. This indeed was found to be the case in the present studies s summarized in Figure 3A in which no statistical differences of mean arterial pressure (MAP) were observed between rapamycin and vehicle treated rats throughout the low-salt and high-salt diet period. Historical data from our laboratory4 of vehicle treated SS rats were plotted in the graph to show the effect of elimination of Nox4 on MAP.
Figure 3:

A, Baseline of mean arterial pressure (MAP) of SSNox4−/− rat was obtained by measuring three days of continuous arterial pressure followed by four day treatment of vehicle (close circle; n=8) or rapamycin (I.P. 1.5 mg/kg, open circle; n=8) on 0.4% NaCl then switched to 4.0% NaCl diet for 21 day receiving similar treatment. B, Diurnal variation of MAP averaged in 12 h bins to correspond to the day (6am-6pm) and night (6pm- 6am) cycles of these rats is shown. Historical MAP data of vehicle treated SS rats is shown4. C, hypertrophy index (kidney weight/body weight). D, showing average body weight before and after treatment. E, average food intake over the 21 days of 4.0% NaCl diet. F, steady-state sodium excretion at day 21 of the 4.0% NaCl diet. A two-way analysis of variance (ANOVA) for repeated measures; Holm-Sidak post hoc was used for arterial pressure. Unpaired t-test was used to compare the effect of treatments. * P<0.05 and ns; non-significant.
Pressure recordings were also analyzed for effects of rapamycin on diurnal rhythms. As shown in Figure 3B and Figure 3SA&B, rapamycin treated rats exhibited almost complete abolishment of blood pressure diurnal rhythms in MAP, diastolic blood pressure (DBP) and systolic blood pressure (SBP) which were apparent even during the first day of treatment. Vehicle treated rats exhibited no changes in blood pressure diurnal rhythms. Heart rate responses (Figure 3SC) differed from those of blood pressure in that rapamycin had little effect upon heart rate diurnal rhythms but rather reduced heart rate over the initial 8 days of treatment before returning to baseline through the remainder of the study. No vehicle effects upon heart rate were apparent.
Renal hypertrophy which is commonly observed in high-salt fed SS rats remained unchanged as expressed by kidney to body weight ratio (renal hypertrophy index) in rapamycin treated versus vehicle treated SSNox4−/− rats (Figure 3C). However, as shown in Figure 3D, rapamycin treated SSNox4−/− rats exhibited nearly no body weight gain over the 21 day of the 4.0% NaCl diet compared to a significant gain of weight which occurred in vehicle treated SSNox4−/− rats. Average total food intake did not differ significantly in rapamycin and vehicle treated SSNox4−/− rats during 4.0% NaCl diet period (Figure 3E). Also shown in Figure 3E and Figure 3SF, steady-state levels of Na+ and K+ excretion rate on day 21 of 4.0% NaCl did not differ significantly in rapamycin and vehicle treated SSNox4−/− rats further indicating rats were eating similar amounts of food.
Rapamycin didn’t augment reduction of kidney injury in SSNox4−/−.
As summarized in Figure 4A–C and Figure S4A, rapamycin treatment of SSNox4−/− rats did not result in significant reduction of kidney injury as evidenced by the albumin-to-creatinine ratios (ACR), protein-to-creatinine ratios (PCR), medullary cast positive area, and glomerular injury scores. Similarly as shown in Figure 4D–F and Figure S4B–D, the expression level of renal fibrosis related genes (Col1a1, α-Sma), the kidney injury marker Kim-1, the oxidative stress related genes (p67, Nox2) and the biomarker S-nitrotyrosine remained unaffected in renal cortex and medulla of rapamycin treated SSNox4−/− compared with vehicle treated SSNox4−/− rats. This protection from renal injury that we have previously demonstrated with rapamycin treated SS4 mimicked that observed in the SSNox4−/− further indicating that NOX4 generated H2O2 is a crucial upstream activator of mTORC1 in kidney injury in SS rats.
Figure 4:

A, albumin to creatinine was determined in urine samples of SSNox4−/− rats treated with vehicle (n=7) or rapamycin (n=7) collected for 24 h on the last day of the 4.0 % NaCl. B, summary of the outer medullary tubular injury (percentage of tubular cast positive region) and representative sections of trichrome-stained kidney from vehicle (n=6) or rapamycin (n=7) treated rats. C. summary of glomerular injury. D&E, Quantitative polymerase chain reaction assessment of genes involved in renal fibrosis (Col1a1) and renal injury (Kim-1) in the cortex and medulla of vehicle (n=6) or rapamycin (n=5) treated SSNox4−/− rats. F, S-nitrotyrosine expression was determined in cortex and medulla of these rats by Western blot. Representative immunoblots of cortex and medulla are shown. Unpaired t-test was used to compare the effect of treatments. * P<0.05.
Rapamycin reduced immune cell infiltration and cell proliferation in the kidney of SSNox4−/− rats.
As shown in the Figure 5A&B, rapamycin significantly reduced renal infiltration of T lymphocytes (CD3+ cells/mm2) in the cortex of SSNox4−/− rats from 80 ± 6 to 41 ± 5 and in the medulla from 107 ± 7 to 28 ± 3 compared to vehicle treatment. Rapamycin also reduced macrophage (CD68+ cells/mm2) infiltration in the cortex and medulla of SSNox4−/− rats from 24 ± 4 to 11 ± 2 and from 32 ± 4 to 10 ± 2, respectively. Rapamycin virtually abolished the cell proliferation (Ki67+ cells/mm2) in the cortex from 132 ± 29 to 3 ± 0 and in the medulla from 157 ± 50 to 6 ± 0 compared to vehicle treated rats. Yet, as shown in Figure 5E&F, the expression of genes such as Ccl2 and Mmp9, involved in immune cell infiltration did not differ significantly in renal cortex and medulla of rapamycin and vehicle treated SSNox4−/− rats. Together, these data suggest that rapamycin is a potent inhibitor of cell proliferation which in part may be explained by the reduction of immune cell infiltration into the kidney of SSNox4−/− rats.
Figure 5:

A-D, quantification of T lymphocyte (CD3+) and macrophage (CD68+) cells per mm2 of kidneys of vehicle (n=7) or rapamycin (n=7) treated SSNox4−/− rats. (A&C) CD3+ cells/mm2 and (B&D) CD68+ cells/mm2 in the renal cortex and medulla and representative kidney sections illustrating the immunohistochemical localization of T lymphocyte and macrophage cells in the kidney of these rats. Scale bar 50 μM. E&F, Quantitative polymerase chain reaction assessment of Ccl2 and Mmp9 in the cortex and medulla of vehicle (n=6) or rapamycin (n=5) treated SSNox4−/− rats. Unpaired t-test was used to compare the effect of treatments. * P<0.05.
Rapamycin reduced mTORC1 activity in SSNox4−/− rats.
Renal cortical and medullary mTORC1 and mTORC2 activities were determined in vehicle or rapamycin treated SSNox4−/− rats by Western blot. As shown in Figure 6A&B, rapamycin significantly reduced the cortical mTORC1 activity with the same tendency in the medullary tissue compared with vehicle treated rats. As we found previously4, rapamycin had no inhibitory effect on the functional marker of mTORC2 activity (pAKTS473/AKT) in cortex and medulla of kidney of these rats as well (Figure 6C&D).
Figure 6:

A&B, pS6S235/236 and S6 were measured in the cortex and medulla of SSNox4−/− rats treated with vehicle (n=6) or rapamycin (n=5) by Western blot. Corresponding representative immunoblots are shown below each graph. C&D, measurement and quantification of pAKTS473 and AKT and corresponding representative immunoblots are shown below each graph. GAPDH is a loading control. Unpaired t test was used to compare treatments. * P<0.05 and ns; non-significant.
Discussion
H2O2 is an upstream regulator of mTORC1 which does not require PI3K/AKT and AMPK pathways.
Previous in vitro studies have failed to clarify the effects of oxidative stress and H2O2 upon activation of mTORC12, 3. There have been conflicting reports showing that mTOR is alternatively activated or inhibited by oxidative stress, depending on cell type and oxidant level of the cell15–17. Stimulation of S6K activity by H2O2 treatment was observed using mouse epidermal JB6 cells with responses being dose- and time-dependent2, 3. In contrast, H2O2 was found to impair mTORC1 activity in a human lung epithelial cell line A549 and normal bovine aortic smooth cells17. The results of the present study reconcile these conflicting observations. First, a biphasic response of H2O2 was observed in normal rat kidney epithelial cells (Figure 1) where mTORC1 was strongly activated by physiologically relevant concentrations of H2O2 but suppressed by supra-physiological concentrations. Second, H2O2 was found to directly activate mTORC1 independent of PI3K/AKT and AMPK pathways in this cell type. This observation contradicts the report that H2O2 dependent mitogenic signaling in cultured type II pneumocyte cells activates mTOR-kinase via the PI3K/AKT pathway 23, perhaps a cell specific difference.
The in vivo effects of endogenously produced H2O2 upon mTORC1 activity have not been previously examined. Evidence indicates that the major reactive oxygen species released by NOX4 is H2O2 18. Studies in our laboratory have demonstrated that intracellular H2O2 in medullary thick ascending limb below detectable limits by fluorescence in SSNox4−/− rats12. The SSNox4−/− rat model was therefore of great utility in examining the relationship between NOX4-H2O2 and mTORC1 signaling. Supporting the conclusion that H2O2 mediated activation of mTORC1 is independent of the canonical mTOR pathway (Figure 2) which was shown by the absence of differences in levels of PTEN, pAKTT308 and TSC2 expression between SS rats (which exhibit high kidney levels of H2O2 in response to high-salt intake) and SSNox4−/− rats. This further indicates that the actions of H2O2 are upstream of mTORC1 since downstream effector components such as pS6K1 and pS6 were reduced in the kidneys of the SSNox4−/− compared to SS rats. It is interesting to note that pS6 and p4E-BP1 (functional markers of mTORC1 activity) were similar in both SS and SSNox4−/− rats fed a 0.4% NaCl since weaning. This is perhaps to be expected since mTORC1 normally senses and integrates several signaling inputs including insulin/IGF1, amino acids to maintain renal homeostasis1, 19 but becomes dysregulated during many pathophysiological conditions such as diabetes20, cancer21, and hypertension4, 5 22. Together, our observations suggest that the basal physiologic levels of mTORC1 activity are maintained independent of NOX4-H2O2 for normal renal function but become dysregulated by NOX4 generated H2O2 in SS rats fed a high-salt diet.
Rapamycin treatment of SS rats mimics the protection of salt-induced hypertension in SSNox4−/− rats.
Over the past two decades, significant progress has been made in understanding the complexity of the mTORC1 regulation and its role in various forms of cancer but there has been limited progress in cardiovascular diseases. Rapamycin is a potent and specific inhibitor of mTORC1 and its effects have been studied in two distinct rat models of hypertension the spontaneously hypertensive rat (SHR)22 and SS rats4. In the heart, Nox4 and mTORC1 activity have been linked by findings that Nox4 induced cardiac fibrosis and hypertrophy through activation of AKT/mTOR and NFkB signaling pathways in mice23.
The present study shows that NOX4 derived H2O2 is an important determinant of mTORC1 activation and required for full development of salt-induced hypertension and progressive renal injury in SS rats. Rapamycin treatment of SS rats mimics these responses as illustrated in Figure S5A showing that BP of high-salt diet fed SS rats treated with rapamycin was nearly identical to that of SSNox4−/− rats. The reductions of renal injury in rapamycin treated SS rats to levels similar to the vehicle treated SSNox4−/− rats indicate a role of NOX4-H2O2 in the regulation of other pathways involved in renal injury (Figure S5B&C). Both SS and SSNox4−/− exhibited an initial rise of MAP of similar magnitude when rapamycin was administered during low-salt control period. The present study does not shed light on the mechanism(s) responsible for this increase but over observations of the effects of rapamycin on BP diurnal rhythms suggest that this compound has central actions on the autonomous nervous system. The major finding of the present study was that MAP rose to similar levels with high-salt feeding in SSNox4−/− vehicle and rapamycin treated rats. This supports our conclusion that the reduction of salt-induced hypertension in SS rats with knockout of NOX4 (SSNox4−/−) maybe in large part a consequence of reduced mTORC1 activity.
Inhibition of mTORC1 with rapamycin reduced renal immune cell by inhibiting cell proliferation independent of NOX4.
It is well recognized that in experimental and human hypertension, lymphocytes and macrophages home to regions of renal injury. In SS rats, inflammation contributes to the later phase of hypertension and renal injury24 and several studies have shown that although suppression of renal inflammation in SS rats does not affect the initial rise in BP during the first 7–10 day after switching to a high-salt diet, the progression to the malignant phase of hypertension is prevented4, 25. Nox4 is recognized to be important in innate26 and adaptive27 immunity but has not been previously examined with regard to the inflammatory related renal injury observed in SS rats. We have shown that rapamycin treatment of SS rats which reduced mTORC1 activity was associated with a reduced renal infiltration of T lymphocyte and macrophage and inhibition of cell proliferation4. And yet, in SSNox4−/− rats in which mTORC1 activity is already low compared with rapamycin treated SS rat4 resulted in yet a further reduction of T lymphocyte and macrophage infiltration vis- a-vis complete inhibition of cell proliferation (Figure 5SD&E) indicating that these rapamycin actions were independent of NOX4–H2O2 but dependent on inhibition of cell proliferation. This cannot be explained by a reduction in renal perfusion pressure which was very similar in rapamycin treated SS rats as in SSNox4−/− rats.
Effect of rapamycin on the expression of known genes involved in salt-induced hypertension in SS rats.
We have reported that the protective effect from salt-induced hypertension exhibited by SSNox4−/− rats is attributed in part to the parallel reduction of oxidative stress via Nox2 and its cytosolic subunits p47, p67 and others11. There is plethora of data showing that the benefits of rapamycin in many pathologies are related to transcriptional changes in ROS related genes. Increased levels of NOX2 in the heart of adult offspring from diabetic mothers is the result of AKT/mTOR activity and rapamycin treatment reduced NOX2 protein in cardiomyocytes28. Rapamycin reduces the expression of Mmp9 in Glioblastoma cells by modulation of NFkB and PKC-α signaling29. Effects of mTORC1 activity on chemokine gene expression of Ccl2 in macrophages has been reported30 and of Col131 and α-Sma32 in fibroblasts derived from urethral scar tissue and nasal polyps, respectively. It is interesting to note that many of these genes have also been implicated to be directly or indirectly involved in BP salt-sensitivity in SS rats11. Given these observations, it appears that the effects of rapamycin are similar at both the genomic and functional level to those observed in SSNox4−/− rats. This would explain why rapamycin did not show any augmentation in the protection from salt-induced hypertension in SSNox4−/− rats.
Little is known about the link of NOX4-mTORC2 in salt-induced hypertension. Others have found that a NOX4 dependent activation of mTORC2 in pulmonary hypertension10. We have reported the combined inhibition of both mTORC1 and mTORC2 with PP242 completely prevented and reversed salt-induced hypertension in SS rats5, a far greater response than observed with specific inhibition of mTORC1 alone with rapamycin treatment4 indicating that mTORC2 contributed importantly to this remarkable protection from salt-induced hypertension in SS rats. However, until a specific inhibitor of mTORC2 is developed, the connection between NOX4 and mTORC2 can only be inferred. We cannot rule out the possibility that a similar NOX4/H2O2/mTORC2 cascade maybe operating in SS rats fed a high-salt diet.
In summary, the results of the present study show that the protective effects upon BP and renal injury observed in SSNox4−/− rats fed a high salt diet are largely a consequence of reduction of NOX4-H2O2 stimulated mTORC1 activity. H2O2 was found to effectively activate mTORC1 independent of the PI3K-AKT and AMPK pathways. Rapamycin treatment of SSNox4−/− rats resulted in no further protection of BP and kidney injury than that observed in SSNox4−/− rats. We conclude that NOX4 driven BP and kidney injury in SS rats fed a high-salt diet is a consequence of H2O2 activation of mTORC1 pathway.
Perspectives
High-salt dietary intake stimulates H2O2 production in the kidney in part through the activation of NOX4 and through increased activity of the electron transport chain in SS rat. The present study identifies a novel direct pathway whereby elevations of tissue H2O2 can stimulate mTORC1 independent of the PI3K/AKT and AMPK pathways. mTORC1 is one of the major pathways contributing to cellular energy sensing and the control of a number of important cellular functions such as mitochondrial biogenesis and function, cellular proliferation, and autophagy. The novel evidence that H2O2 can directly target mTORC1 and thereby enhances salt-induced hypertension and renal injury heightens our understanding of redox sensing and the complexity of intracellular signaling involved in the coordination of kidney energy metabolism in the development of salt-sensitive hypertension.
Supplementary Material
Novelty and Significance.
What is new?
First study to identify the importance of NOX4 in the regulation of mTORC1 pathway in salt-induced hypertension and kidney injury in SS rats.
First study showing that H2O2 can directly activate mTORC1 independent of PI3K-AKT and AMPK pathways.
First study demonstrating that a high-salt diet stimulates NOX4-H2O2 which serves as an upstream regulator of mTORC1 activity and its related downstream pathways involved in determining BP salt-sensitivity and kidney injury.
What is relevant?
Evidences of NOX4-H2O2 and mTORC1 signaling have been studied separately in the Dahl S model of hypertension. The present study integrated and revealed in vivo relevance of NOX4-H2O2 in the activation of mTORC1 in salt-induced hypertension and renal injury in SS rats.
Acknowledgements
We thank Megan Stumpf for radiotelemetry implantation and tissue collection.
Source of Funding
This work was supported by National Institute of Health, National Institute of Heart, Lung and Blood grant HL-116264 (AWC).
Footnotes
Conflicts of Interest/Disclosures
None.
References
- 1.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mtor and its downstream targets s6k1 and 4ebp1/eif4e. Genes Dev. 2002;16:1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C, Li J, Ke Q, Leonard SS, Jiang BH, Zhong XS, Costa M, Castranova V, Shi X. Ultraviolet-induced phosphorylation of p70(s6k) at thr(389) and thr(421)/ser(424) involves hydrogen peroxide and mammalian target of rapamycin but not akt and atypical protein kinase c. Cancer Res. 2002;62:5689–5697 [PubMed] [Google Scholar]
- 3.Bae GU, Seo DW, Kwon HK, Lee HY, Hong S, Lee ZW, Ha KS, Lee HW, Han JW. Hydrogen peroxide activates p70(s6k) signaling pathway. J. Biol. Chem 1999;274:32596–32602 [DOI] [PubMed] [Google Scholar]
- 4.Kumar V, Wollner C, Kurth T, Bukowy JD, Cowley AW Jr. Inhibition of mammalian target of rapamycin complex 1 attenuates salt-induced hypertension and kidney injury in dahl salt-sensitive rats. Hypertension. 2017;70:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar V, Evans LC, Kurth T, Yang C, Wollner C, Nasci V, Zheleznova NN, Bukowy J, Dayton A, Cowley AW Jr. Therapeutic suppression of mtor (mammalian target of rapamycin) signaling prevents and reverses salt-induced hypertension and kidney injury in dahl salt-sensitive rats. Hypertension. 2019;73:630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill PS, Wilcox CS. Nadph oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607 [DOI] [PubMed] [Google Scholar]
- 7.Martin-Garrido A, Brown DI, Lyle AN, Dikalova A, Seidel-Rogol B, Lassegue B, San Martin A, Griendling KK. Nadph oxidase 4 mediates tgf-beta-induced smooth muscle alpha-actin via p38mapk and serum response factor. Free Radic Biol Med. 2011;50:354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 nad(p)h oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem 2005;280:39616–39626 [DOI] [PubMed] [Google Scholar]
- 9.Eid AA, Ford BM, Bhandary B, de Cassia Cavaglieri R, Block K, Barnes JL, Gorin Y, Choudhury GG, Abboud HE. Mammalian target of rapamycin regulates nox4-mediated podocyte depletion in diabetic renal injury. Diabetes. 2013;62:2935–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mtorc2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation. 2014;129:864–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley AW Jr., Yang C, Zheleznova NN, Staruschenko A, Kurth T, Rein L, Kumar V, Sadovnikov K, Dayton A, Hoffman M, Ryan RP, Skelton MM, Salehpour F, Ranji M, Geurts A. Evidence of the importance of nox4 in production of hypertension in dahl salt-sensitive rats. Hypertension. 2016;67:440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheleznova NN, Yang C, Cowley AW Jr. Role of nox4 and p67phox subunit of nox2 in ros production in response to increased tubular flow in the mtal of dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2016;311:F450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cowley AW Jr., Yang C, Kumar V, Lazar J, Jacob H, Geurts AM, Liu P, Dayton A, Kurth T, Liang M. Pappa2 is linked to salt-sensitive hypertension in dahl s rats. Physiol Genomics. 2016;48:62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang C, Stingo FC, Ahn KW, Liu P, Vannucci M, Laud PW, Skelton M, O’Connor P, Kurth T, Ryan RP, Moreno C, Tsaih SW, Patone G, Hummel O, Jacob HJ, Liang M, Cowley AW, Jr. Increased proliferative cells in the medullary thick ascending limb of the loop of henle in the dahl salt-sensitive rat. Hypertension. 2013;61:208–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K. Redox regulates mammalian target of rapamycin complex 1 (mtorc1) activity by modulating the tsc1/tsc2-rheb gtpase pathway. J Biol Chem. 2011;286:32651–32660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mtor pathway and complex. J Biol Chem. 2005;280:39505–39509 [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Kimball SR, Jefferson LS, Shenberger JS. Hydrogen peroxide impairs insulin-stimulated assembly of mtorc1. Free Radic Biol Med. 2009;46:1500–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of nox1 and nox4 in basal and angiotensin ii-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008;45:1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grahammer F, Haenisch N, Steinhardt F, Sandner L, Roerden M, Arnold F, Cordts T, Wanner N, Reichardt W, Kerjaschki D, Ruegg MA, Hall MN, Moulin P, Busch H, Boerries M, Walz G, Artunc F, Huber TB. Mtorc1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc. Natl. Acad. Sci. U. S. A 2014;111:E2817–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56:1600–1607 [DOI] [PubMed] [Google Scholar]
- 21.Guertin DA, Sabatini DM. Defining the role of mtor in cancer. Cancer Cell. 2007;12:9–22 [DOI] [PubMed] [Google Scholar]
- 22.Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, Abel ED, Symons JD, Jalili T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. Nadph oxidase 4 induces cardiac fibrosis and hypertrophy through activating akt/mtor and nfkappab signaling pathways. Circulation. 2015;131:643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am. J. Physiol. Renal Physiol 2014;307:F499–F508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: Direct interaction of tlr4 with nad(p)h oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of nf-kappa b. J Immunol 2004;173:3589–3593 [DOI] [PubMed] [Google Scholar]
- 27.Di Marco E, Gray SP, Chew P, Kennedy K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Differential effects of nox4 and nox1 on immune cell-mediated inflammation in the aortic sinus of diabetic apoe−/− mice. Clin Sci (Lond). 2016;130:1363–1374 [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Wang X, Wu Y, Lu X, Chidiac P, Wang G, Feng Q. Maternal diabetes up-regulates nox2 and enhances myocardial ischaemia/reperfusion injury in adult offspring. J. Cell. Mol. Med 2018;22:2200–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandrika G, Natesh K, Ranade D, Chugh A, Shastry P. Suppression of the invasive potential of glioblastoma cells by mtor inhibitors involves modulation of nfkappab and pkc-alpha signaling. Sci. Rep 2016;6:22455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ai D, Jiang H, Westerterp M, Murphy AJ, Wang M, Ganda A, Abramowicz S, Welch C, Almazan F, Zhu Y, Miller YI, Tall AR. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ. Res 2014;114:1576–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu D, Yin J, Huang S, Li H, Li Z, Chong T. Rapamycin inhibits the growth and collagen production of fibroblasts derived from human urethral scar tissue. Biomed Res Int. 2018;2018:7851327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko DY, Shin JM, Um JY, Kang B, Park IH, Lee HM. Rapamycin inhibits transforming growth factor beta 1 induced myofibroblast differentiation via the phosphorylated-phosphatidylinositol 3-kinase mammalian target of rapamycin signal pathways in nasal polyp-derived fibroblasts. Am J Rhinol Allergy. 2016;30:211–217 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.