

Abstract
Genome-wide association studies (GWAS) have linked hundreds of thousands of sequence variants in the human genome to common traits and diseases. However, translating this knowledge into a mechanistic understanding of disease-relevant biology remains challenging, largely because such variants are predominantly in non-protein-coding sequences that still lack functional annotation at cell-type resolution. Recent advances in single-cell epigenomics assays have enabled the generation of cell type, sub-type, and state-resolved maps of the epigenome in heterogeneous human tissues. These maps have facilitated cell type-specific annotation of candidate cis-regulatory elements and their gene targets in the human genome, enhancing our ability to interpret the genetic basis of common traits and diseases.
INTRODUCTION
How the human genome dictates individual phenotypic traits and propensity to disease is one of the fundamental questions in biology1. Common traits and diseases have been linked to sequence variation at thousands of loci through genome-wide association studies [G] (GWAS); however, over 90% of disease-associated variants map outside of protein-coding sequences and remain largely unannotated to date2,3. Growing evidence suggests that a substantial fraction of these variants may affect binding of transcription factors to cis-regulatory elements [G] (CREs), thereby altering target gene expression in specific cell types3–7. Yet, identifying such regulatory variants and delineating their functional roles has been difficult.
Epigenome profiling has proved extremely valuable in delineating CREs in the human genome6,8–10. The epigenome refers to the covalent modifications of nucleic acids and histone proteins that make up the chromosomes in the nucleus11–13. An expanded definition of the epigenome also includes features such as chromatin accessibility and 3D genome conformation. Owing to the actions of sequence-specific transcription factors and chromatin binding proteins, the epigenomic landscape of a cell is dynamically established during development in a cell-type and sequence-context dependent manner. Activation of CREs is frequently accompanied by epigenetic changes, including increased chromatin accessibility, DNA hypomethylation, or the presence of certain histone modifications. The strong correlation between CRE activity and specific epigenetic features allows such regions to be mapped across different tissues and cell types using epigenome profiling techniques that combine biochemical assays with high-throughput sequencing6,14,15. Indeed, the Encyclopedia of DNA elements (ENCODE)8, Roadmap Epigenomics Project6, and the International Human Epigenome Consortium15, among others, have carried out large-scale epigenome profiling studies to annotate millions of candidate cis-regulatory elements [G] (cCREs)3,5–7,14,16–19.
With these catalogs of cCREs, it was shown that disease-associated non-coding variants are strongly enriched in cCREs in a tissue- or cell-type dependent manner, supporting the notion that disease-associated non-coding variants likely act by disrupting gene expression in cell types or tissues relevant to disease pathogenesis and phenotypic traits5–7. However, existing catalogs of cCREs have several key limitations that prevent a more comprehensive understanding of risk variant function. Conventional epigenomic assays profile cells in ‘bulk’ to produce population average measurements, and therefore the profiles of individual cell types are obscured in assays performed on heterogeneous primary tissue. Furthermore, rare cell types that make up a small proportion of the tissue will not be well represented in epigenomic profiles of bulk tissue. While conventional bulk assays can be applied to cell populations that have been purified, for example using fluorescence-activated cell sorting [G] (FACS), these assays have not yet been applied to all human cell types due to a scarcity of suitable marker genes and antibody reagents. In addition, bulk methods require established markers to capture the epigenome of discrete cell states within human cell types, and they do not capture continuous heterogeneity in the epigenome that describes transitions between cell states and cell types, for example during development or in response to environmental stimuli or disease. Finally, the target genes of cCRE activity in individual cell types remain largely unknown. Together these barriers have limited our ability to comprehensively interpret the function of non-coding variants and determine how they contribute to common traits and disease.
Recent advances in single-cell technology (for consistency and simplicity, we will use single-cell as a label irrespective of whether cells or nuclei are used as input material) have enabled epigenomic profiling of individual cells or nuclei in a highly parallel fashion in a single experiment13,20. In particular, methods have been developed to assay single-cell chromatin accessibility21–23, histone modifications24–33, transcription factor binding24,30,34,35, 3D chromatin conformation [G]36, and DNA methylation37,38 and its derivatives39–41 across a range of platforms utilizing droplet-, micro-well-based or combinatorial barcoding schemes13. Data from these assays29,42–46 not only allow deconvolution of individual cell types from mixed populations but also make it possible to profile the epigenome of constituent cell types. From these cell-type-resolved epigenome profiles, cCREs active in human cell types, target genes and transcriptional regulators of cCRE activity, and heterogeneity in their activity within a cell type can all be characterized, which can then be used to improve annotation of disease-associated noncoding variants13. By comparison, single-cell profiling of gene expression can also deconvolute common and rare cell types and sub-types from mixed populations, at both higher resolution and reduced cost, but lacks the information necessary to localize cCREs and annotate non-coding variants.
Here, we review recently generated cCREs atlases derived from single-cell epigenome assays of human tissues and highlight the utility of these maps in interpreting genetic variants affecting common traits and disease through identifying relevant cell types, cell sub-types, and cell states, prioritizing causal disease variants in cell type-specific cCREs, determining the cell type-specific function of disease variants, and linking disease variants to their target genes.
CIS-REGULATORY ELEMENT ATLASES
Atlases of cCREs derived from single-cell epigenomic assays have recently been generated for many primary human cells, tissues, organoids, and cell lines47–77 (Table 1). These studies have primarily profiled accessible chromatin, as these techniques are currently the most widely available, although studies for other molecular modalities are emerging (Box 1, reviewed in13). Initial single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) studies were performed in small numbers of cells, which limited the ability to detect cell populations and novel cCREs. More recently, higher throughput assays have been developed, such as single-cell combinatorial indexing (sci)ATAC-seq and droplet-based single-cell (sc)ATAC-seq, which allow accessible chromatin to be mapped in thousands to millions of cells.
Table 1: Single-cell epigenomic studies on human tissues and annotation of disease associated variants.
The listed studies showcase the diverse human tissue types profiled using single cell epigenomic assays, including the number of cells and modalities, to generate cCRE catalogs and analyze disease associated variants.
| Single-cell modality | Method | # cells | Summary of catalog | Genetic enrichment tests | Variant prioritization | Variant functional effects | Variant target gene links | Reference |
|---|---|---|---|---|---|---|---|---|
| Pan-tissue | ||||||||
| Chromatin accessibility | sciATAC-seq | 615,998 | 890k cCREs in 111 cell types from 30 adult tissues, and 1.15M cCREs in 222 cell types from 45 adult and fetal tissues when combined with Domcke et al 2020 | S-LDSC using cell type cCREs | Credible set variants identified in statistical fine-mapping overlapping cell type cCREs | - | ABC links in each cell type | 55 |
| Chromatin accessibility | sciATAC-seq3 | 790,957 | 1.05M cCREs in 124 cell types across 15 fetal tissues | S-LDSC using cell type cCREs | - | Allelic imbalance mapping of cell type cCREs | Cicero co-accessibility in cell type cCREs | 58 |
| Artery | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 7,009 | 329k cCREs in 5 cell types in atherosclerotic lesions | Custom enrichment test based on permuted background sites | Variants in LD with index variants overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs | 50 |
| Chromatin accessibility | 10x scATAC-seq | 28,316 | 324k cCREs in 14 coronary artery cell types | S-LDSC using cell type cCREs | Variants in LD with index variants overlapping cell type cCREs | Chromatin QTL mapping of cell type cCREs | Cicero co-accessibility in cell type cCREs, integrated cCRE and gene expression co-activity | 71 |
| Brain | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 70,361 | 359k cCRES in 24 isocortex, hippocampus, striatum, and substantia nigra cell types | S-LDSC using cell type cCREs | Variants in LD with genome-wide significant variants overlapping cell type cCREs | Predicted effects on cell type cCREs using SVMs | Cicero co-accessibility in cell type cCREs | 51 |
| Chromatin accessibility | 10x scATAC-seq | 130,418 | cCREs in 7 prefrontal cortex cell types from control and late-stage Alzheimer’s disease samples | S-LDSC using cell type cCREs, gchromVAR in single microglia cells | Credible set variants from previous statistical fine-mapping overlapping cytokine-responsive cCREs | - | Cicero co-accessibility in cell type cCREs, integrated cCRE and gene expression co-activity | 52 |
| Chromatin accessibility | 10x scATAC-seq | 31,304 | 658K cCREs in prefrontal cortex developmental cell types | - | - | Predicted effects on cell type cCREs using BPNet | Integrated cCRE-gene expression co-activity | 60 |
| Chromatin accessibility | 10x scATAC-seq | 45,205 | cREs in 11 frontal cortex cell types from supranuclear palsy, corticobasal degeneration, and control donors | Custom enrichment tests | - | - | - | 62 |
| Chromatin accessibility | 10x scATAC-seq | 87,339 | 253k cCREs in 12 major prefrontal cortex cell types | No genetic analyses performed | 65 | |||
| Chromatin accessibility | 10x scATAC-seq, | 80,827 | cCREs in 31 cell types from stem cell-derived cerebral organoids | No genetic analyses performed | 72 | |||
| SHARE-seq | ||||||||
| DNA methylation | snmC-seq | 2,784 | 498k differentially methylated regions in 21 prefrontal cortex neuronal subtypes | No genetic analyses performed | 46 | |||
| DNA methylation, 3D interactions | sn-m3C-seq | 4,200 | 37k chromatin interactions in 14 prefrontal cortex cell types | No genetic analyses performed | 75 | |||
| Chromatin accessibility, DNA methylation, 3D interactions | snmCAT-seq, sn-m3C-seq, snmC-seq, snmC-seq2, sciATAC-seq | 27,587 | cCREs and DMRs in 63 cell types and sub-types from frontal cortex | S-LDSC using cell type cCREs and DMRs | - | - | - | 68 |
| Colon | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 447,829 | cCREs in 34 colon cell types from normal, polyps, and colorectal cancer donors | No genetic analyses performed | 61 | |||
| Heart | ||||||||
| Chromatin accessibility | sciATAC-seq | 79,515 | 287k cCREs in 12 cell types from multiple regions of the adult human heart. | S-LDSC using cell type cCREs | Credible set variants in statistical fine-mapping overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs | 49 |
| Chromatin accessibility | 10x scATAC-seq | 46,086 | cCREs in 8 heart cell types from control and myocardial infarction donors | No genetic analyses performed | 66 | |||
| Hematopoietic | ||||||||
| Chromatin accessibility | dsciATAC-seq | 136,463 | cCREs in 15 cell types in resting and stimulated bone marrow | - | Credible set variants in previous statistical fine-mapping overlapping differential cell type cCREs | - | - | 77 |
| Chromatin accessibility | 10x scATAC-seq | 35,038 | cCREs in 26 cell types from bone marrow and peripheral blood | No genetic analyses performed | 76 | |||
| Chromatin accessibility | 10x scATAC-seq | 63,882 | 571k cCREs in 31 peripheral blood cell types and sub-types | No genetic analyses performed | 54 | |||
| Chromatin accessibility | sciATAC-seq, 10x scATAC-seq | 131,554 | 448k cCREs in 28 pancreas and peripheral blood cell types and sub-types. | S-LDSC using cell type cCREs; fgwas using cell type-specific cCREs | Credible set variants from statistical fine-mapping overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs | 47 |
| Kidney | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 27,034 | 215k cCREs in 15 kidney cell types | No genetic analyses performed | 79 | |||
| Chromatin accessibility | 10x scATAC-seq | 50,986 | cCREs in 11 kidney cell types in control and polycystic kidney disease donors | No genetic analyses performed | 80 | |||
| Chromatin accessibility | 10x scATAC-seq | 68,456 | cCREs in 20 kidney cell types in control and diabetic kidney disease donors | S-LDSC using cell type cCREs and differentially accessible cCREs | - | Allelic imbalance mapping of single cells | Integrated cCRE-gene expression co-activity | 74 |
| Chromatin accessibility | 10x scATAC-seq | 57,229 | cCREs in 12 kidney cell types | S-LDSC using cell type cCREs, gchromVAR in single cells | Variants from GWAS overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs; Priority scoring of genes based on multiple different datasets | 67 |
| Chromatin accessibility | 10x scATAC | 9,460 | cCREs in 20 kidney cell types | No genetic analyses performed | 73 | |||
| Lung | ||||||||
| Chromatin accessibility | sciATAC-seq | 90,980 | 398k cCREs in 19 lung cell types | - | Credible set variants from previous statistical fine-mapping overlapping cell type cCREs | Predicted effects on cell type cCREs using deltaSVM | Cicero co-accessibility in cell type cCREs | 53 |
| Chromatin accessibility | scTHS-seq | 40,427 | 427k cCREs in 20 lung cell types | gchromVAR using single cell profiles | - | - | - | 63 |
| Pancreas | ||||||||
| Chromatin accessibility | sciATAC-seq | 1,456 | cCREs in 3 pancreatic islet cell types | Enrichment of cell type cCREs using GREGOR and fGWAS | Credible set variants from previous statistical fine-mapping overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs | 57 |
| Chromatin accessibility | sciATAC-seq | 15,298 | 229k cCREs in 12 pancreatic islet cell types | S-LDSC using cell type cCREs; fGWAS using cell type-specific cCREs, PolyTest using single cell profiles | Credible set variants from previous statistical fine-mapping overlapping cell type cCREs | Predicted effects on cell type cCREs using deltaSVM | Cicero co-accessibility in cell type cCREs | 48 |
| Chromatin accessibility | sciATAC-seq, 10x scATAC-seq | 131,554 | 448k cCREs in 28 pancreas and peripheral blood cell types | S-LDSC using cell type cCREs; fgwas using cell type-specific cCREs | Credible set variants from statistical fine-mapping overlapping cell type cCREs | - | Cicero co-accessibility in cell type cCREs | 47 |
| Chromatin accessibility | 10x scATAC-seq | 7,829 | cCREs in 7 cell types from cytokine-treated and untreated pancreatic islets | - | Credible set variants from previous statistical fine-mapping overlapping cytokine-responsive cCREs | - | Co-accessibility in cytokine-stimuated and baseline cell types | 69 |
| Chromatin accessibility | 10x scATAC-seq | 12,473 | cCREs in 9 pancreatic islet cell types | S-LDSC using cell type cCREs linked to a target gene | Variants in LD with index variants overlapping cell type cCREs | - | - | 70 |
| Retina | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 102,802 | cCREs in 8 cell types from fetal retina and stem cell-derived retinal organoids | No genetic analyses performed | 64 | |||
| Skeletal muscle | ||||||||
| Chromatin accessibility | 10x scATAC-seq | 5,053 | cCREs in 7 skeletal muscle cell types | S-LDSC of 404 traits using cell type cCREs | Variants identified in previous statistical fine-mapping overlapping cell type cCREs | Predicted effects on cell type cCREs using SVMs | Cicero co-accessibility in cell type cCREs | 56 |
| scMultiome | ||||||||
Box 1: Current status of techniques for generating single cell epigenomic atlases.
Single cell techniques have been developed in recent years to probe diverse epigenomic layers including chromatin accessibility, DNA methylation, histone modifications and chromatin interactions13.
Chromatin accessibility assays (as single modality and, to a lesser degree, as part of multi-omic workflows with transcriptome profiling) are widely employed to assess the gene regulatory landscape at the single cell level47–74,76,77,79,80. Reasons for their widespread use include robust protocols across scales to enable studies from thousands to millions of cells and availability of commercial solutions such as from 10x Genomics.
Robust plate-based single cell DNA methylation profiling techniques have also been used for human tissue profiling46,68,75. However, its use is limited by the high per cell cost, relatively low throughput without automation and lack of commercial solutions, but a very recent high-throughput combinatorial barcoding approach with lower per cell cost might help to overcome some of these hurdles168.
Chromatin interaction profiling has not yet been adopted widely for human tissue profiling due to low throughout, high cost, data sparsity and lack of commercial solutions68,75.
Very recently, multiple proof-of-principle studies showed histone modification profiling in single cells with and without transcriptomes from the same cell13,32,33. These workflows are complex and often require custom reagents not readily available for purchasing. We presume that continued optimization will lead to robust, scalable and widely applicable protocols amenable to human tissue profiling.
One of the primary advantages of single-cell analyses is the ability to map the epigenome of both abundant and rare cell populations, the latter of which are frequently under-represented in existing catalogs that have been derived using bulk assays. For example, droplet-based scATAC-seq analysis of 63,882 cells from peripheral blood and bone marrow defined 31 immune cell types, which included relatively rare cell types such as basophils, demonstrating that rare cell types could be identified from heterogeneous archival tissue54. Application of sciATAC-seq to 15,298 pancreatic islet cells from three donors, identified 228,873 cCREs in 12 cell-types , including cCREs in less common cell types that had not been included in previous catalogs based on bulk assays, such as delta and gamma cells48. Profiling of 615,998 cells from 30 distinct adult human tissues from multiple donors using sciATAC-seq identified 111 cell types, including rare cell types whose chromatin profiles did not strongly correlate with any existing bulk chromatin profiles in ENCODE, such as enterochromaffin cells (0.06% of total nuclei profiled in the intestine) or Paneth cells (0.045% of total nuclei profiled in the intestine)55. Hybrid approaches that first perform cell sorting of broad cell populations followed by single-cell analyses may also help map the epigenome of rare sub-types, but such approaches would only apply to cell populations with known markers and reliable antibodies.
Single-cell epigenome assays also identify more cCREs than bulk assays, owing to their improved ability to detect cell-type specific cCREs. For example, of 571,400 total cCREs identified in peripheral blood and bone marrow using scATAC-seq, 20.4% were specific to a single cell type54 and this cell-type specificity would be missed by bulk assays. Similarly, nearly half of the 359,022 cCREs identified by scATAC-seq of multiple brain regions were found only in a single cell type, at least partly explaining why 66% of these CREs were not identified in bulk ATAC-seq data from the same tissue. By contrast, the single-cell data recovered 89% of the cCREs detected in bulk data in addition to identifying a higher overall number of CREs51. A study that generated sciATAC-seq data for adult human tissues55 and combined it with fetal sciATAC-seq data from another study58 identified 1.15 million cCREs, of which 34.8% and 51% were not present in previous bulk-derived cCRE catalogs in EpiMap3 and ENCODE14, respectively, although it should be noted that inclusion in these catalogs required that two distinct epigenetic signatures be present and they therefore represent a subset of data from these resources. Furthermore, we determined that 10% of cCREs were not identified in a survey of accessible chromatin across 733 samples using DNase I hypersensitivity assays despite this study profiling 5-fold more samples78.
Similar to scRNA-seq data, clustering analysis of single-cell epigenomic data can be used to resolve distinct sub-types and states within specific cell types, information that cannot be obtained from bulk data even from sorted cell populations. This approach has identified 30 different neuronal sub-types with distinct epigenomic profiles in the brain51, seven sub-types of fibroblasts with distinct tissue-of-origin dependent epigenomic profiles55 and, in pancreatic islets, multiple clusters of endocrine beta, alpha, and delta cells that represent distinct cellular states48. It has also identified differential representation of astrocyte and microglia sub-types in prefrontal cortex from control and Alzheimer’s disease (AD) samples52. We note that defining cell sub-types and states can also be performed from single-cell expression data and thus may be preferred in applications where this is the primary goal. In several studies48,52, the chromatin accessibility profiles of cells were also ordered on pseudo-time trajectories to reveal continuous epigenomic heterogeneity within cell types, which could help infer gene regulatory networks (GRNs) driving cell state transitions. Multiple studies have integrated single cell epigenomic profiles with single cell gene expression data (reviewed in ref13) to define GRNs that link transcription factors to their downstream target gene sets60,65,72. Single-cell epigenomic profiling has also provided insight into transcriptional programs and molecular mechanisms underlying lung63 or brain development65,72 as well as changes in pathologies including kidney diseases67,74,79,80, AD52 and colorectal cancer61.
PRIORITIZING DISEASE VARIANTS AND CELLS
The additional information provided by single-cell assays collectively enables new insight into the biological function of risk loci identified in GWAS. These insights come primarily from: the expanded repertoire of active cCREs, and greater knowledge about the cell types in which they are active, generated by single-cell studies and to which methods originally developed for genetic analyses of conventional bulk data can be applied; and the identification of continuous patterns of heterogeneity within a cell type, such as state transitions or developmental trajectories, to which novel methods being actively developed for genetic analyses of single-cell profiles can be applied.
Identifying cell types enriched for trait or disease association
Catalogs of cCREs identified from single-cell epigenomic assays can be used in genetic enrichment analyses to infer the cell types involved in common complex traits and diseases (Figure 1a). Such studies have primarily used cCREs derived from ‘pseudo-bulk’ profiles of each cell type47–49,51,55,59 and applied enrichment methods previously developed in the context of conventional bulk data. These studies build on the substantial number of studies that have performed genetic enrichment analyses using catalogs of cCREs derived using conventional bulk assays3,5–7,14,78,81, and which have identified many cell types involved in common complex diseases.
Figure 1: Identifying common trait-enriched and disease-enriched cell types and sub-types.
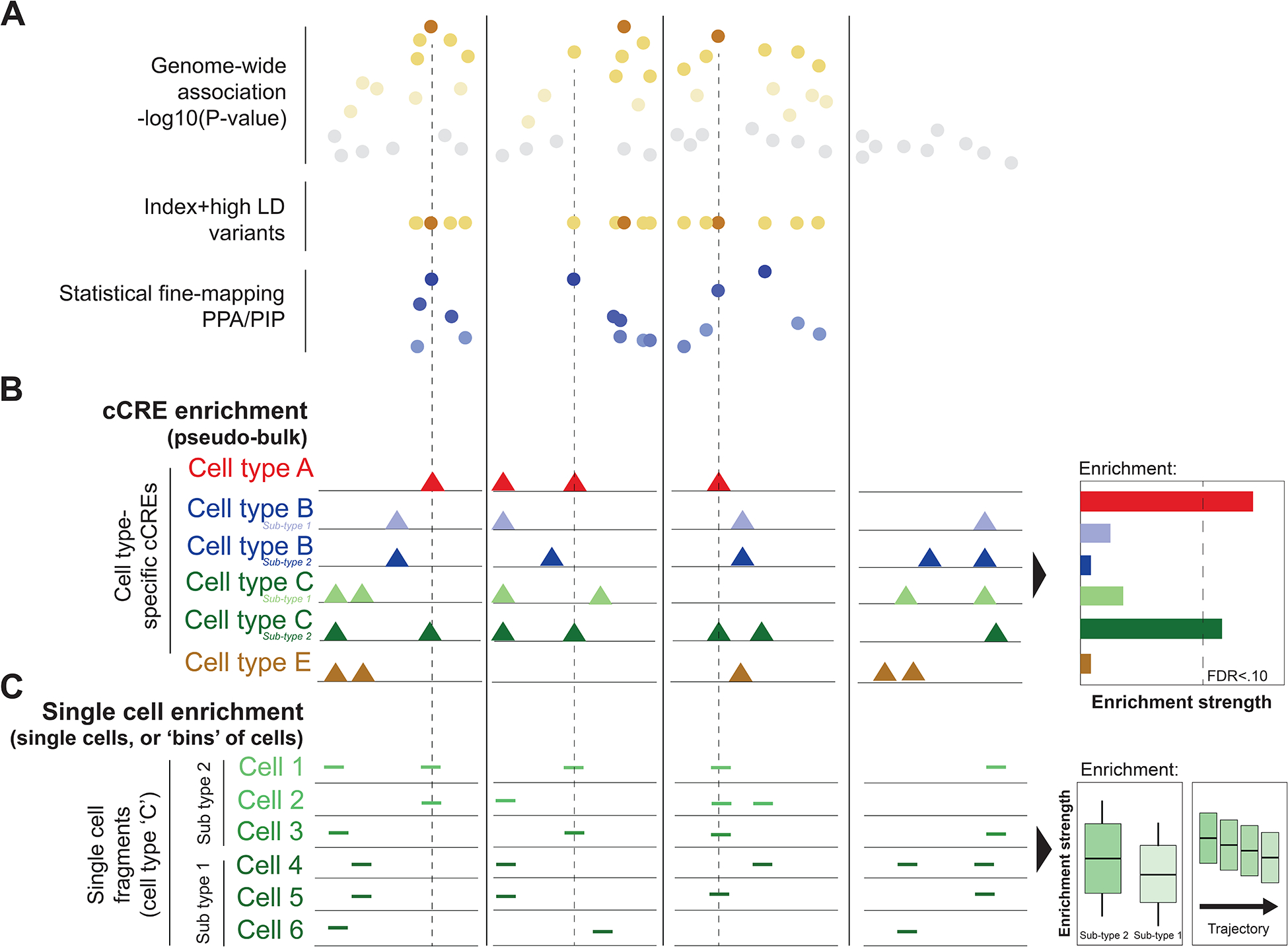
Overview of approaches for identifying disease-enriched cell types and states that have been used in single-cell epigenomics studies. a, Enrichment analysis methods can make use of genome-wide summary statistics (top), variants in linkage disequilibrium (LD) with the ‘index’ (or ‘sentinel’) variant at known loci (middle), or posterior probabilities from statistical fine-mapping at known loci (bottom). In the example, locus 1, 2 and 3 are disease-associated loci, whereas locus 4 shows no association (and therefore is not included in the middle and bottom panels). Each circle represents a genetic variant. Darker colors represent stronger association and grey denotes little to no association. b, cCREs (coloured triangles) defined from cell type ‘pseudo-bulk’ epigenomic profiles can be used in enrichment analyses to identify disease-enriched cell types and sub-types. The methods used to test pseudo-bulk cCREs for genetic enrichment are primarily co-opted from previous studies of conventional ‘bulk’ genomics. In this example, there is significant enrichment for cell type A and sub-type 2 of cell type C but not for other cell types or sub-types. The dashed lines highlight disease-associated variants overlapping cCREs in the enriched cell types and sub-types at loci 1, 2 and 3 but not locus 4, which contains no disease-associated variants. c, Epigenomic profiles such as accessible chromatin fragments (represented by horizontal lines) of each cell can be used to identify enrichment in individual cells or ‘bins’ of closely related cells, between different cell sub-types, and across gradients of cells ordered along a trajectory. In the example, sub-type 2 cells show stronger enrichment than sub-type 1 and there is a decreasing gradient of enrichment moving from sub-type 2 to sub-type 1 cells. The dashed lines highlight associated variants overlapping accessible chromatin reads in cells in sub-type 2 at loci 1, 2 and 3 and not locus 4.
A commonly-used method for genetic enrichment analyses is stratified linkage disequilibrium score regression [G] (S-LDSC)82, which determines whether variants genome-wide linked to a cCRE annotation have enriched trait heritability. S-LDSC is designed to test for enrichment of an annotation using variants genome-wide, such as all cCREs active in a given cell type. Different enrichment methods may be more appropriate for other analyses, such as when analyzing a small subset of cCREs (for example, cell type-specific cCREs or stimulus-responsive cCREs) or using only variants at known disease loci. Orthogonal methods, such as fGWAS (functional GWAS), which were also originally developed in the context of bulk data, have been applied in single-cell epigenome studies in these contexts3,83–86 (Figure 1b).
Application of genetic enrichment methods to cCREs derived from single-cell epigenomics assays has identified cell types enriched for a wide range of common complex traits and diseases, as well as disease endophenotypes47–52,55,56,58. For example, 32 of 34 UK biobank phenotypes showed enriched heritability in cCREs for at least one fetal cell type58 and 3,220 enriched trait-cell type pairs were found across 222 adult and fetal cell types55. This study implicated a larger number of links between cell types and common complex disease risk than previous bulk-derived catalogs based on accessible chromatin14,78, which is typical of single-cell enrichment analyses and is likely due to the greater number of cell types tested and the larger number of available genetic association studies. However, it is worth noting that studies performing S-LDSC analyses often correct for multiple tests using family-wise error rate, where false discovery rates of 10% or even higher are considered significant. With increasing numbers of trait-cell type pairs tested, a greater number of false positives will be identified and a more stringent threshold may be appropriate.
In many cases, these enrichments mirror previously reported findings for cell types already present in catalogs derived using conventional bulk assays. For example, the prominent role for microglia in AD risk is supported by enrichment analysis of both scATAC-seq and bulk ATAC-seq data51,52,87,88. In other cases, single-cell studies have highlighted previously unknown relationships between cell types and complex phenotypes. In one study, 160 of the 450 traits and diseases with at least one enriched cell type had cell type enrichments not reported in previous studies, including for cell types not annotated in previous bulk catalogs55. For example, type 2 diabetes (T2D) risk variants were enriched for pancreatic delta and gamma cell cCREs, thyroid stimulating hormone (TSH) level-associated variants were strongly and specifically enriched for thyroid follicular cell cCREs, and chronic obstructive pulmonary disorder (COPD)-associated variants were enriched for stromal smooth muscle cell cCREs.
Despite the usefulness of these analyses, there are several considerations when interpreting results from cCRE catalogs derived from single cell epigenomics data that do not apply to catalogs based on bulk assays. For rarer cell types, particularly for catalogs where smaller numbers of cells are profiled, the peak calls may not be saturated, and therefore these cell types will have fewer cCREs and reduced power in enrichment analyses compared to more common cell types. Moreover, cell types may not represent completely ‘pure’ populations, as misclassification of cells and technical issues such as doublets may result in artefactual peak calls.
Nevertheless, single-cell epigenome studies have collectively demonstrated that expanded atlases of cCREs derived from pseudo-bulk profiles of individual cell types, used in combination with previously developed genetic enrichment methods, can provide insight into cell types involved in common complex disease.
Defining cell sub-types and states enriched for disease risk
One key advantage of single-cell epigenomics over conventional bulk assays is the ability to resolve cCREs active in specific cell sub-types or cell states that may not have been previously known or may not yet have been characterized using cell sorting approaches. Genetic enrichment methods, as described above, can also be applied to cCREs derived from pseudo-bulk profiles of sub-types and states within a cell type to identify enrichments for complex traits and disease (Figure 1b).
A primary example is the brain, which has an incredible diversity of cell types and sub-types, many of which have not been well characterized by cell sorting- or isolation-based methods and likely play distinct roles in complex traits and disease. For example, variants associated with schizophrenia were found to be enriched in cCREs active in specific specialized excitatory neuronal sub-types derived from scATAC-seq of multiple brain regions, including BNDF+ and BNDF− excitatory neurons, striatopallidal medium spiny neurons, and cortical intratelencephalic neurons51,68. Analysis of an integrated single-cell map of 31 cortical cell types and sub-types also identified highly specific neuronal sub-type cCRE enrichment patterns for other traits, such as bipolar disease and autism68. Single-cell epigenomics studies have also described distinct sub-types of the same cell type that are present in different tissues. The seven fibroblast sub-types identified across human tissues in the study mentioned above showed distinct patterns of enrichment for different common traits and diseases; for example, cardiac fibroblasts showed sub-type-specific enrichment for variants associated with myocardial fractal dimension whereas epithelial fibroblasts showed sub-type-specific enrichment for variants associated with balding55.
Single-cell epigenome studies have also revealed enrichment of genetic association for cCREs active in discrete cellular states defined within a cell type, such as those that perform different cellular functions or that change during disease or in response to stimulation. As noted above, variants associated with AD risk were enriched for different sub-types of microglia, and the same microglial sub-types had differential representation in AD compared to non-diseased samples52. In another study, variants associated with fasting glucose level (a measure of insulin secretion) were specifically enriched in cCREs predicted to be active in a beta cell state enriched for insulin secretion-related processes compared to other beta cell states48. In both cases, the cellular states had not been previously documented, and therefore had no available profiles from previous studies using bulk assays.
One consideration for these analyses is that the resolution of cell sub-types and states is based on computational clustering of single-cell profiles. Novel sub-types and states reported in these studies therefore may not in every case reflect biologically meaningful heterogeneity. Additional validation experiments, such as cell sorting based on newly-identified sub-type or state markers followed by characterization using functional assays, could confirm that these cells represent distinct functional units but are typically not performed in single-cell studies.
Overall, these findings demonstrate that single-cell epigenome assays can provide insight into which cell sub-types and states are involved in complex traits and diseases.
Identifying disease-relevant heterogeneity within a cell type
Unlike conventional bulk assays, single-cell epigenomics enables moving beyond discrete definitions of cell sub-types or states to continuous representations of cells within a cell type. Several novel approaches have been developed that can leverage the profiles of individual cells, or ‘bins’ of closely-related cells, instead of pseudo-bulk profiles to identify disease-enriched cell subsets, state transitions or cellular trajectories (Figure 1c).
The method g-chromVAR was developed to determine whether fine-mapped GWAS variants are enriched for cell type-specific peak profiles compared to a matched background and can be applied to bulk as well as individual cell profiles84. Application of this method to bone marrow scATAC-seq profiles revealed that monocyte-associated and platelet count-associated variants were enriched for cells from different sub-populations of common myeloid progenitors. Furthermore, variants associated with platelet counts had increasing enrichment among single cells ordered along a developmental trajectory from stem cells to megakaryocytes, which are precursors to platelet cells, but not on trajectories to erythroid and lymphoid cells. A previously developed method, PolyTest85, was used to test disease-associated variants for enrichment of single-cell profiles in pancreatic islets, and trajectory analysis revealed that fasting glucose-associated variants were increasingly enriched on a path towards beta cells in an insulin secretion-related state.
One consideration for enrichment analyses using single cells is that the profiles of individual cells are very sparse, and there is thus limited information available for each cell. Approaches to circumvent this issue include binning cells with similar profiles and creating pseudo-bulk aggregates per bin, or imputing the epigenomic profiles of each cell based on profiles of similar cells89. These approaches can provide richer profiles for each cell and facilitate genetic enrichment analyses.
By leveraging the epigenomic profiles of individual cells, single-cell epigenomics provides additional insight into dynamic processes that occur within a cell type, such as disease trajectories or state transitions, and how they contribute to risk of common complex disease.
Prioritizing variants causal for disease association signals
Complex trait-associated and disease-associated loci often contain many associated variants that are not directly causal owing to linkage disequilibrium [G] (LD), as well as independent signals that each have distinct causal variants. Multiple strategies can be used to define sets of candidate causal variants at disease loci, including basic approaches such as identifying all variants in high LD (LD variants [G]) with the index variant [G] , and statistical fine-mapping [G] approaches that assign causal probabilities (posterior probability of association [G] (PPA), or posterior inclusion probability [G] (PIP)) to variants at each signal90–94. The outcome of statistical fine-mapping is typically credible sets [G] of variants that explain the majority of the PPA and/or PIP and represent the most likely candidate variants for the signal. Compared to LD-based approaches, in which all linked candidate variants are considered equal, statistical fine-mapping has the advantage of ascribing varying levels of confidence to each variant, but with the drawback that these methods are typically more sensitive to genotyping and imputation errors.
Statistical fine-mapping or LD-based analyses often result in multiple candidate causal variants for a given signal, and in these cases epigenomic data can be used to prioritize candidates. Catalogs of cCREs derived from conventional bulk assays have been used to annotate candidate causal variants at disease association signals in specific cell types3,7,78,88,95, which has provided substantial insight into the mechanisms of association signals for many diseases. Single-cell epigenomics studies have improved the ability to prioritize candidate variants at disease-associated loci over previous catalogs in multiple ways (Figure 2a,b).
Figure 2: Prioritizing candidate causal variants at disease-associated loci.
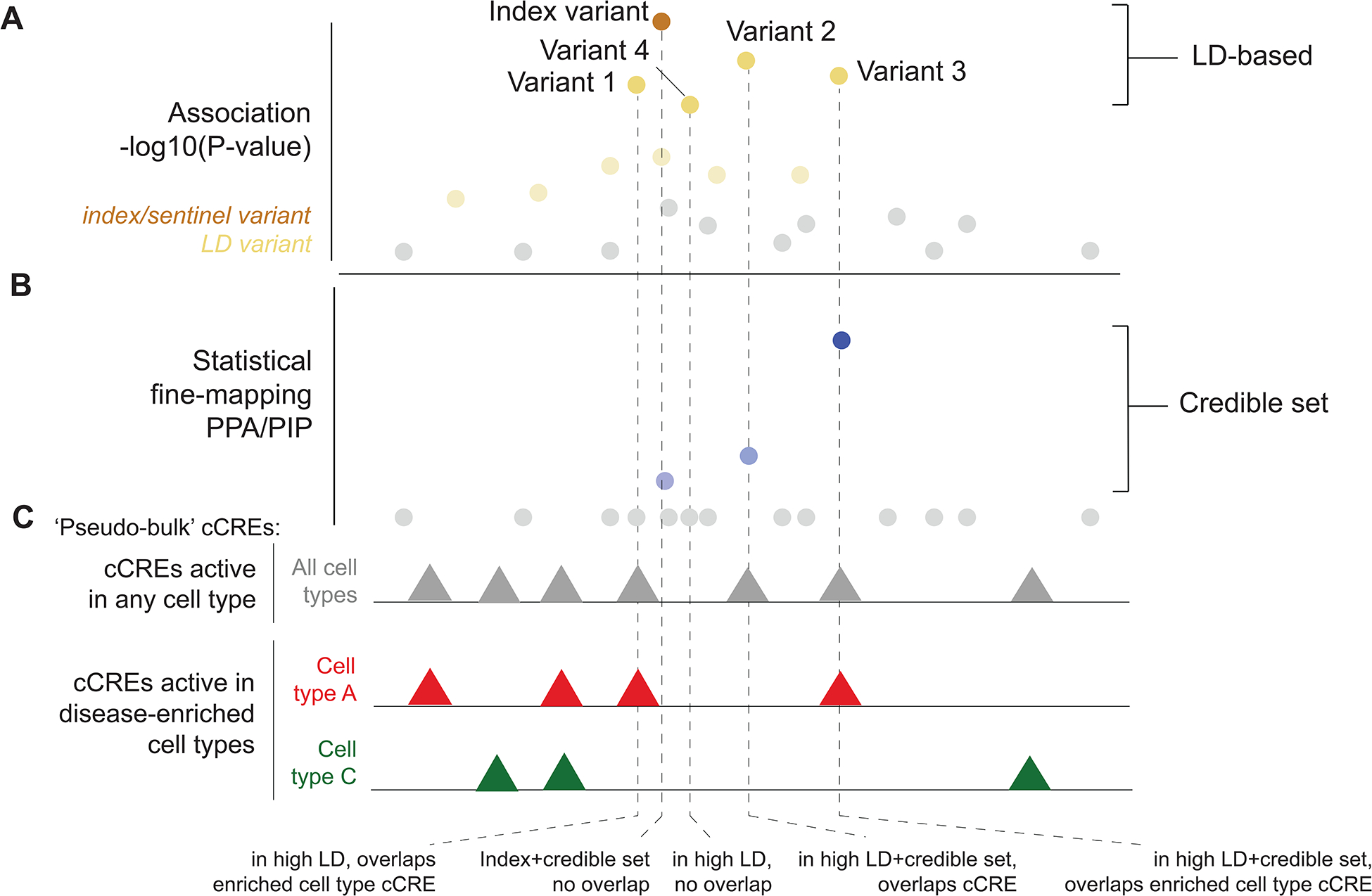
Overview of approaches for prioritizing variants causal for disease-associated loci used in single-cell epigenomics studies. a, Variant association data at a locus, where the most strongly associated ‘index’ variant and variants in linkage disequilibrium (LD) with the index variant are highlighted. In an LD-based approach, variants in high LD (typically r2>.8, although not specified in this example) with the index variant are prioritized as causal candidates, in this case variants 1–4. b, Statistical fine mapping derives the posterior probability that variants at a locus are causal for a risk signal. In a ‘credible set’ approach, variants that cumulatively explain a certain amount of the posterior probability (for example, 99% or 95%) of a risk signal from fine-mapping are prioritized as candidates. In this example, the index variant and variants 3 and 4 are in the credible set and thus prioritized by fine-mapping. c, cCREs identified using pseudo-bulk profiles of each cell type mapping to a locus are used to further prioritize among candidate variants. Studies have considered either cCREs active in any cell type, or just cCREs active in disease-enriched cell types. When prioritizing variants based on overlap with cCREs (indicated by dashed lines), variants 1, 3 and 4 overlap cCREs active in any cell type and variant 1 and 4 overlap cCREs active in a disease-enriched cell type. In this example, the index variant itself does not overlap any cCREs. Note that in these examples the predicted mechanism is that the causal variant affects cCRE activity, but in reality, variants can affect other regulatory mechanisms such as RNA splicing or stability.
Atlases derived from single-cell epigenome assays provide a larger repertoire of cCREs in which candidate variants can be annotated. For example, a scATAC-seq study of different brain regions identified nearly double the number of cCREs than bulk assays, and many of the cCREs identified only in scATAC-seq data were cell-type specific. Using LD-based candidate variants defined for AD or Parkinson’s disease (PD), 80% of AD and PD loci had at least one variant overlapping cCREs in one or more brain cell types derived from scATAC-seq, including in cCREs not identified in bulk data51. In another study, variants associated with type 2 diabetes (T2D) and fasting insulin level overlapped cCREs identified by scATAC-seq of skeletal muscle56. A single fine-mapped variant at the ARL15 locus overlapped a cCRE active in mesenchymal stem cells, suggesting that this variant is likely to underlie the association signal56; this cCRE was absent from previous bulk catalogs14,78. However, although single-cell studies broadly generate larger catalogs of cCREs, reduced detection of cCREs in rare cell types –particularly in studies profiling fewer cells– likely leads to some candidate variants not being annotated in these cell types.
In other studies, new insight into the cell types and states enriched for complex trait or disease association allowed for refined prioritization of candidate causal variants for these traits. For example, genetic variants associated with TSH levels were strongly and specifically enriched in cCREs active in follicular cells55, which were not profiled in previous catalogs using conventional bulk assays. Candidate variants for TSH level identified by statistical fine-mapping could be further prioritized based on overlap with follicular cell cCREs.
As complex diseases are typically caused by dysfunction in multiple cell types and processes, it is often necessary to further consider which cell type(s) are relevant to disease risk at a particular locus (Figure 2c). Determining the association of risk variants at a locus with relevant endophenotypes can help provide clues about the cell types of action, and variants can then be prioritized based on overlap with cCREs active in endophenotype-relevant cell types. Single-cell epigenomics has extended this concept by enabling insight into novel cell states within a cell type relevant to disease risk at a locus based on endophenotype association. As mentioned above, variants associated with fasting glucose, an endophenotype of T2D related to insulin secretion, were specifically enriched in an insulin secretion-related beta cell state48. This information would enable variants associated with T2D that are also associated with fasting glucose level to be prioritized based on overlap with insulin secretion state-specific cCREs.
It is typically reported that over 90% of common complex disease signals are located in non-coding regions of the genome2,3,96–99. Across studies that have annotated disease signals using catalogs of cCREs from single-cell ATAC-seq47,48,51,55, between ~60–80% of signals per trait have any candidate variant in a cell type-specific cCRE. These numbers are likely biased upwards as poorly fine-mapped signals will have many non-causal variants. When considering higher probability variants only (PPA>.10), we estimate that ~40–60% of signals per trait are annotated with a cCRE. Therefore, a non-trivial fraction of disease signals remains unannotated by catalogs of cCREs from single-cell studies. Some or all of these signals may be unannotated because additional cCREs remain to be identified, for example in cell types or cellular states not yet profiled or because they are not well captured by assaying accessible chromatin. However, some of the unannotated signals may reflect other modes of gene regulation mediated by non-coding sequence, such as splicing, translation, or RNA stability.
Finally, prioritizing candidate causal variants based on overlap with cCREs active in different disease-relevant cell types can be a subjective process. Thus, functionally-informed fine-mapping [G] (FIFM) methods have been developed that formally incorporate enrichments from epigenomic annotations as weights or priors of variants in fine-mapping directly83,100–102. When applied to conventional bulk datasets, these methods have been shown to reduce credible set sizes up to several fold compared to standard fine-mapping approaches that do not consider functional annotations83,100,103,104. However, they have not yet been applied in the context of cCREs identified in single-cell epigenomics studies.
Prioritizing disease-relevant cells with single cell expression
It is worth noting that substantial insight into cell type-specific processes involved in the genetic basis of common complex diseases has also come from gene expression data, and cell type-specific expression maps have been generated from single cell gene expression data for many human tissues105–108. Compared to single-cell epigenomics, single-cell gene expression atlases tend to have greater clustering resolution, and this increased resolution enables analysis of a richer set of cell types, sub-types, and states. Multiple methods have been developed that leverage cell type-specific expression profiles to identify cell types and sub-types enriched for trait and disease heritability109–111. These methods typically assign variants to cell types indirectly, for example based on proximity of the variant to a gene with cell type-specific expression. Incorporating cell type-specific cCREs defined from single-cell epigenomics data may therefore help further enhance these methods.
MAPPING FUNCTIONAL RISK VARIANTS
Disease associated variants overlapping cCREs in a relevant cell type may not necessarily affect the activity of the cCRE, and it is therefore necessary to distinguish between candidate variants with functional effects on cCRE activity and benign variants with no effect. Furthermore, functional variants may lie outside of the cCRE they affect, and these variants would not be prioritized based on simple cCRE overlap as described in the previous section.
Data generated using conventional bulk assays have been extensively used to map genetic effects on cell type-specific regulatory element activity and identify functional disease associated variants95,112–116. Many of these same approaches have been adopted in single-cell epigenome studies by using pseudo-bulk profiles of each cell type to identify functional non-coding variants (Figure 3). Here too, new insights gained from these analyses are currently largely owing to the larger set of cell types, sub-types, and states profiled in single-cell epigenomics versus bulk studies.
Figure 3: Identifying disease variants affecting cell type regulatory activity.
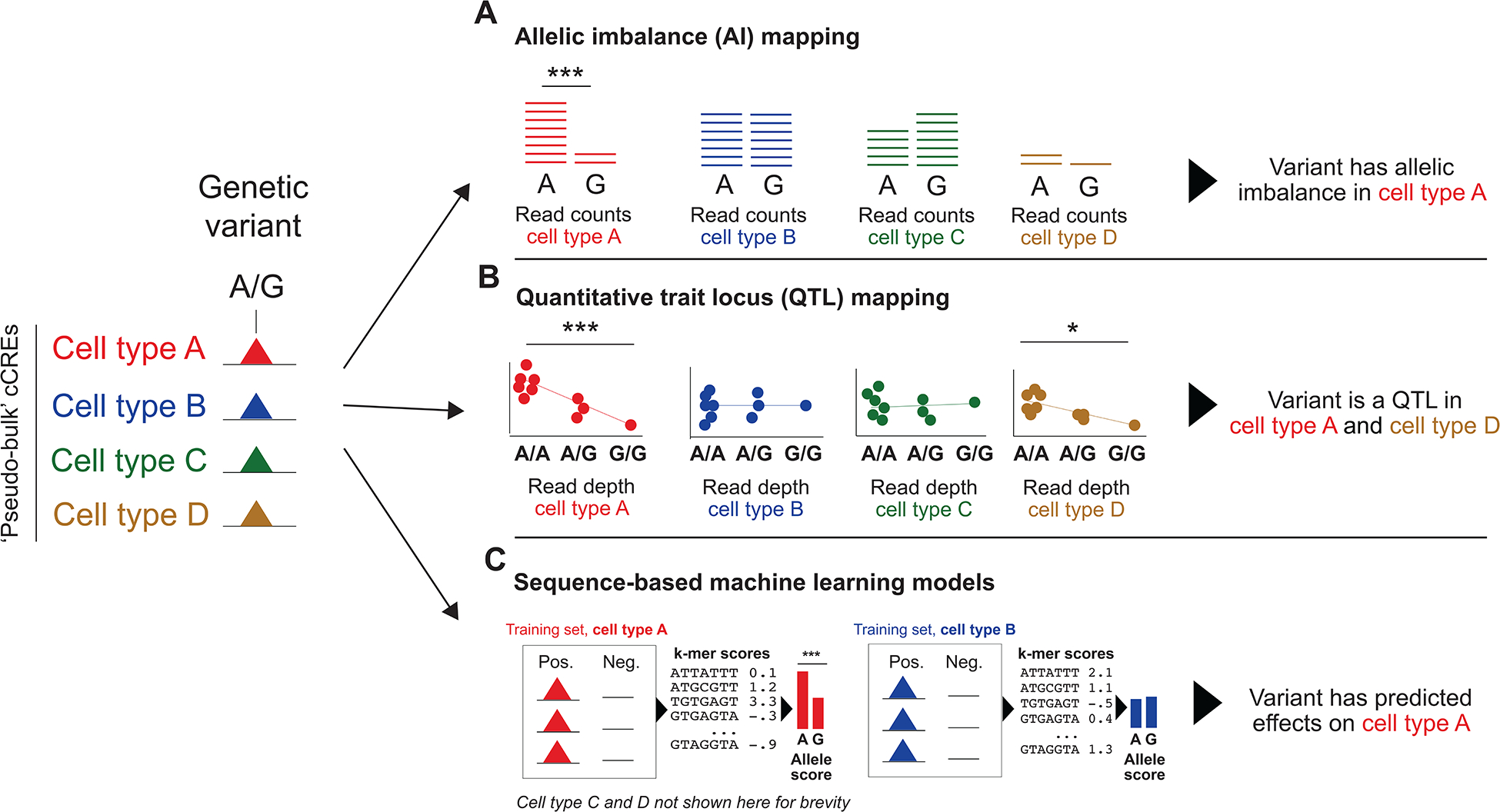
Overview of approaches for determining the functional effects of disease variants in specific cell types using single-cell epigenomics. All examples in this figure use the same genetic variant with alleles A and G overlapping a cCRE derived from pseudo-bulk profiles of cell types A, B, C, and D. Many of the methods currently used to determine variant effects on cCRE activity using pseudo-bulk profiles have been co-opted from previous studies using conventional bulk genomics. a, In allelic imbalance (AI) mapping, read counts for variant alleles in heterozygous samples are compared for each cell type. In this example, the variant has significant imbalance in allele read counts for cell type A but not for cell types B, C and D. b, In quantitative trait locus (QTL) mapping, the cell type read depth for a cCRE is compared across samples with different genotypes. In this example, the variant is a QTL in cell types A and D but not in cell types B and C. c, In sequence-based machine learning using gapped k-mers, a model is trained using cCREs active and not active in each cell type and sequence k-mers are scored based on this model. In this example the models for each cell type are trained separately, although many models can jointly consider data from many cell types. The k-mer scores for the sequence around each variant allele are compared to identify variants with large allelic differences. In this example, the genetic variant has significant allelic differences in cell type A and therefore is predicted to affect cell type A function, but does not have corresponding allelic differences in cell types B-D.
Mapping genetic effects on cell type-specific cCRE activity
One approach used to identify functional variants is allelic imbalance [G] (AI) mapping, whereby a heterozygous variant in a cCRE is assessed for differences in sequence read counts obtained from an epigenomic assay (Figure 3a). Functional variants are more likely to show imbalance, that is one of the alleles would have significantly higher counts than the other. Single-cell studies have primarily performed AI mapping using pseudo-bulk profiles comprised of reads from cells of just that cell type, although can also be performed using single cell profiles74. While AI mapping can be performed using just a few samples, the sparsity of single-cell profiles can severely limit the set of variants that can be effectively tested per sample. Furthermore, due to substantial differences in cell type abundance, and therefore available read coverage, rare cell types will have reduced sensitivity to detect significant AI effects compared to more common cell types. Therefore, the value of these analyses for interpreting disease associated variants may currently be limited to specific subset of variants with sufficient coverage in particular cell types. One advantage of single-cell data over bulk data in AI mapping, however, is that as each cell represents its own library, the number of reads that can cover a variant position is much higher.
When genetic variants were tested for allelic imbalance using accessible chromatin profiles of cell types derived from sciATAC-seq of fetal brain and liver, a set of 586 variants with significant allelic imbalance was identified, several of which were candidate causal variants at complex trait association signals58. However, due to the limited proportion of disease-associated variants tested and the limitations of allelic imbalance mapping in testing just variants within cCREs directly, more systematic prioritization of variants causal for disease association signals was not possible.
An alternative approach is quantitative trait locus (QTL) mapping [G], which models epigenomic profiles as a function of variant genotype across samples (Figure 3b). QTL mapping requires larger sample sizes compared to AI yet can be applied to all variants at a locus, which enables variants causally underlying associations to be identified, including those outside of cCRE boundaries. The results of QTL mapping, but not AI mapping, can also be formally compared to disease risk signals using colocalization analyses117 to identify variants affecting both the epigenome and disease risk. Conversely, AI mapping may be preferred in studies with low sample numbers where QTL mapping is not feasible or when detecting sample-specific effects such as rare or private variants118. As with AI, QTL mapping in single-cell epigenome studies has to date been performed using pseudo-bulk profiles collapsed for each cell type, although these studies can also be performed at the individual cell level119,120.
QTL mapping of chromatin accessibility in 14 immune cell types and sub-types derived from scATAC-seq of 10 peripheral blood samples with RASQUAL121 (which considers allelic imbalance as part of QTL mapping122) expanded the number of immune cell types and sub-types with chromatin accessibility QTL (caQTL) profiles, although these findings are based on one non-peer reviewed pre-print. Previous studies using conventional bulk assays of sorted immune cell populations have generated profiles for only a handful of cell types and sub-types112,113,115,116,123. Immune cell type caQTLs annotated 512 fine-mapped variants for 16 immune traits and diseases, including those with cell type- and state-specific effects. Another study used RASQUAL to map caQTLs in four major cell types derived from scATAC-seq of 41 coronary artery samples, none of which had available caQTLs from previous studies, and annotated variants associated with coronary artery disease (CAD) risk71.
As with AI mapping, a current limitation of single-cell studies is that the number of caQTLs identified per cell type varies greatly depending on cell type abundance, as less abundant cell types have lower read depth and thus reduced power to map QTLs. For example, rare cell types in peripheral blood such as megakaryocytes had almost no caQTLs identified while common cell types such as monocytes had thousands of caQTLs121. In addition, both studies prioritized trait- and disease-associated variants based on simple overlap with caQTLs, although fine-mapping of caQTLs and colocalization analysis is needed to formally establish shared trait and caQTL signals.
Allelic imbalance and QTL mapping analyses using data from single-cell epigenomics assays will undoubtedly increase in number and quality as studies profile larger numbers of samples. The value of single-cell epigenomics in this context, in addition to simply offering a more efficient route to profiling cell types from a heterogeneous tissue in large sample numbers compared to sorting- or isolation-based approaches, is the ability to map genetic effects on molecular phenotypes obscured from bulk assays such as state transitions and trajectories. Conversely, the substantial disparity in AI and/or QTL detection across cell types due to differences in cell abundance may effectively limit these approaches at present to just the most common cell types.
Similarly, expression quantitative trait loci (eQTLs) are increasingly being mapped in specific cell types from single-cell gene expression profiles directly124,125. Other studies have used single cell gene expression as a ‘reference’ to deconvolute eQTLs identified in ‘bulk’ tissue into individual cell types with cell interaction analyses that link cell-type enrichment to genotype126,127. The sample sizes of single cell eQTL studies have been substantially larger than for caQTL studies to date, which enables mapping genetic effects on phenotypes within cell types such as on state transitions and trajectories120,124,125. Colocalizing eQTLs with disease association signals can then reveal cell type-specific mechanisms of disease risk for those signals, including affected gene(s)118. Cell type-specific cCREs can enhance eQTL analyses, for example, by improving fine-mapping of variants causal for eQTL signals.
Data from single-cell epigenomics assays can also be used to deconvolute caQTLs identified in bulk tissue, as has been done for expression QTLs (eQTLs)126,127. These approaches offer a potentially exciting route for increasing sample sizes in QTL mapping of specific cell types, as existing bulk tissue datasets can be re-utilized, or new datasets more cheaply generated. The extent to which cell type-specific caQTLs identified with in silico cell interaction methods recapitulate caQTLs mapped in each cell type is currently unknown, although cell interaction eQTL studies have shown high concordance with eQTLs mapped in each cell type directly127.
Predicting variant effects on cell type-specific cCRE activity
Allelic imbalance and QTL mapping require variants to be present in the tested samples, which limits their application among low frequency and rare variants. An alternative strategy is to use machine learning to create sequence-based models of cCRE activity in each cell type, and then apply these models to predict the function of variant alleles (Figure 3c). These models learn the sequence grammar that underlies why certain regions of the genome are active, which can then be used to predict the regulatory activity of new regions as well as predict the effects of variant alleles. Numerous sequence-based machine learning models [G] have been developed and applied to cCREs from conventional bulk assays to model cell type-specific regulatory element activity and variant function128–131.
Many single-cell accessible chromatin studies to date have used gapped k-mer support vector machines (SVM) to model genomic sequence of cCREs active in a cell type47,48,51,53,56,59 and predict variants with functional activity in each cell type130,132. This approach was originally developed for and applied to conventional bulk data, and the primary benefit of single-cell epigenomic assays in this context is therefore the ability to model cell types and cell states that have not been profiled in previous catalogs. For example, a study that predicted variant effects on cCRE activity in brain cell types, many of which had not been previously profiled, using three different methods identified 100 candidate causal variants with predicted function at 34 AD and Parkinson’s disease loci51. Another study used deltaSVM to predict variants affecting endocrine cell type- and state-specific cCREs, and variants with predicted effects on cell states were enriched for sequence motifs of TFs defining cell state identity48. Variants with cell type- and state-specific effects were under-represented in bulk QTLs, suggesting that these variants have muted effects in QTL studies of bulk tissue.
Predictions of variant effects in each cell type are particularly useful in interpreting the function of less common and rare variants, which are impractical to test with QTL and AI mapping without very large sample sizes. A neural network, BPNet, was used to model cell type-specific cCRE activity in the developing cerebral cortex at base-pair resolution, which was then applied to predict variants with functional effects in each cortex cell type. Rare de novo variants with predicted effects on the activity of cCREs in specific cell types during corticogenesis were enriched in autism cases compared to controls60. By comparison, there was no corresponding enrichment among de novo variants in cCREs without considering functional predictions, suggesting that the enrichment is obscured by the inclusion of many non-functional variants.
At present, there has been limited independent validation of variant effects on cell type-specific regulatory activity predicted using machine learning models in single-cell studies. In few cases, studies have compared predicted variant effect to effects estimated using AI mapping of cell type-specific profiles48. In addition, individual variants with predicted effects have been validated using luciferase gene reporters and electrophoretic mobility shift assays (EMSAs), as well as base-specific editing of human cell lines and stem cell models48. However, using data from systematic validation experiments, such as high-throughput reporter assays and genome editing screens, will be valuable in helping to calibrate machine learning models trained on cell type-specific cCREs.
More recent machine learning methods can model the epigenomic profiles of individual cells133. Over time these methods can likely be used to predict genetic effects on molecular phenotypes obscured from conventional bulk assays such as developmental trajectories or cell state transitions.
LINKING RISK VARIANTS TO TARGET GENES
Disease associated variants primarily map in cCREs that are distal to promoters, and linking distal cCREs to the genes they affect is a major challenge. Conventional assays of 3D chromatin interactions in whole tissues, sorted primary cells70,88, or cell lines have annotated target genes based on physical proximity to cCREs. Approaches to define target genes using correlative analyses across different cell types and tissues, such as in ENCODE and EpiMap3,5, provide predictions that are not inherently specific to a cell type. The activity-by-contact [G] (ABC) method links distal cCREs to gene promoters by combining both chromatin accessibility and chromatin contact data4, and has been applied to epigenomic profiles from conventional bulk assays of many cell types. However, there is limited overlap in the links detected using these different target gene prediction methods, none of which have been definitively validated and established as the standard, and there is still a need for improved maps of target gene predictions in each cell type. Single-cell epigenomics provides multiple avenues to link distal cCREs to their target genes in individual cell types.
Linking variants to target genes using single-cell correlative approaches
Bioinformatic approaches can be used to leverage single-cell chromatin accessibility data to identify distal cCREs correlated with the activity of gene promoter cCREs across individual cells (Figure 4a). For example, Cicero identifies pairs of cCREs that are co-accessible [G] across bins of related cells within a cell type (or, alternatively, using cells across different cell types) using a graphical Least Absolute Shrinkage and Selection Operator (LASSO) model134. Co-accessibility has been extensively used to link distal cCREs to putative target genes in specific cell types47,48,50–53,56,57,67,79. Moreover, co-accessible links have been shown to be enriched for 3D chromatin interactions from bulk promoter capture Hi-C (pcHi-C) data from the same tissue or cell type48,57, supporting that co-accessible links capture true target gene relationships, and many co-accessible links are cell type-specific . For example, 2.82 million co-accessible links were detected between cCREs in brain cell types using single-cell chromatin accessibility data, 20% of which were identified as physical interactions in bulk HiChIP assays from each brain region51. Moreover, more than 90% of candidate variants for AD and PD in a brain cell type cCRE were linked to a gene promoter using co-accessibility data.
Figure 4: Linking disease variants to putative target genes.
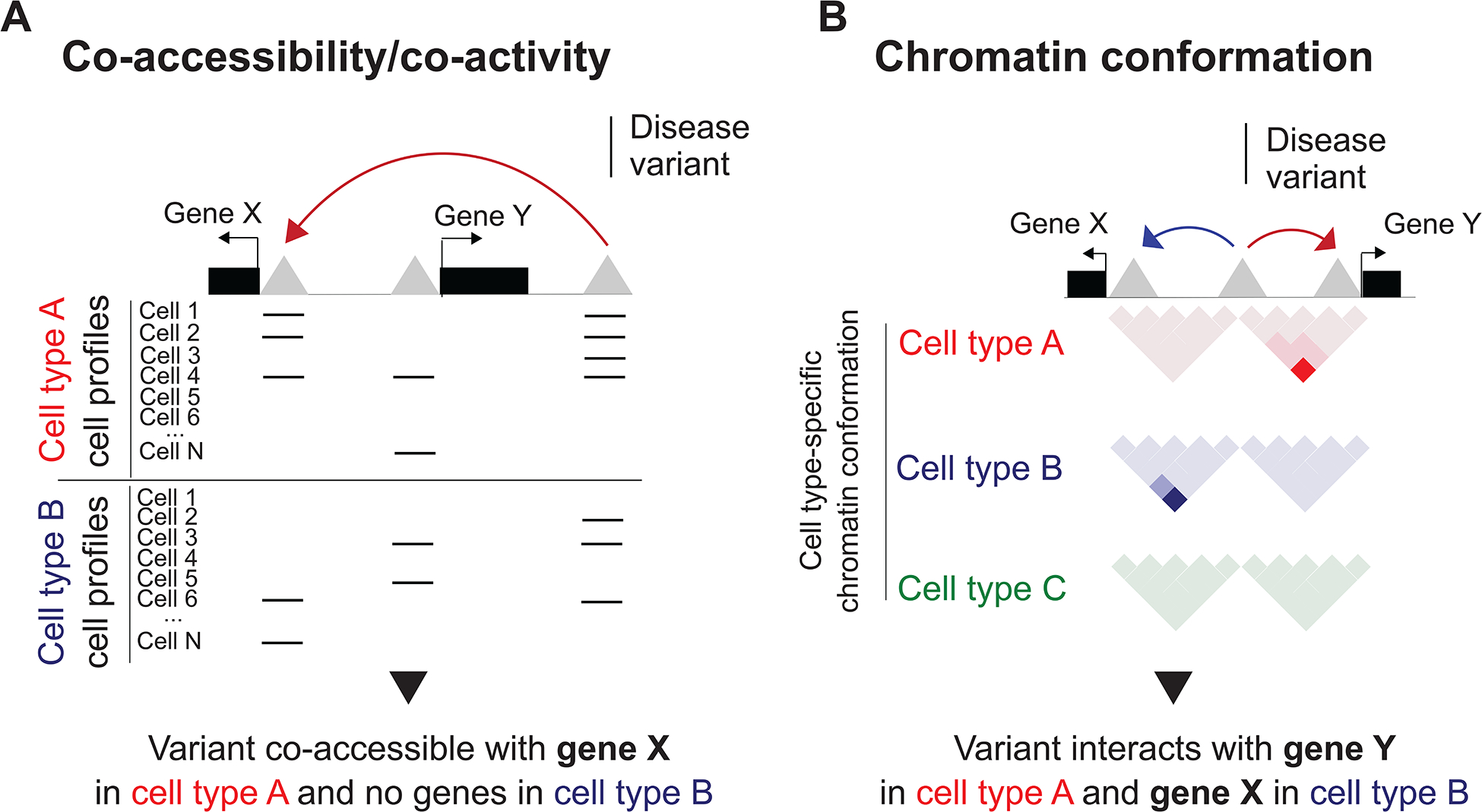
Overview of approaches for assigning disease variants to their target genes in specific cell types using single-cell epigenomics. a, Co-accessibility or co-activity of distal cCREs and promoter cCREs (both represented by grey triangles) across single-cells within a cell type using single-cell accessible chromatin data. In this example, a distal cCRE harboring a disease variant is accessible (represented by horizontal lines) in the same cells as the promoter cCRE of gene X but not gene Y in cell type A, indicating that gene X is the target gene of the distal cCRE in this cell type. By contrast, the distal cCRE is not co-accessible with either of the promoter cCREs in cell type B, suggesting it does not regulate either gene X or gene Y in this cell type b, Chromatin contacts between distal cCREs and promoter cCREs in specific cell types using single-cell 3D chromatin interaction data. In this example, a distal cCRE harboring a disease variant physically interacts with the promoter of gene Y in cell type A and with the promoter of gene X in cell type B. In cell type C, the distal cCRE does not interact with either of the genes.
Single-cell co-accessibility analyses have been widely adopted, in part because they require only the chromatin accessibility profiles already obtained from a single-cell ATAC-seq assay to predict target genes of cCREs. In these analyses, the accessibility of a gene promoter is used to reflect gene activity; however, promoter accessibility is often a poor proxy for gene expression. A newer set of methods identifies single-cell ‘co-activity [G] ’ between cCREs and gene expression levels (from RNA-seq data) directly135. These methods can leverage data from ‘paired’ modalities, such as gene expression and accessible chromatin, generated from the same cell to correlate cCRE and gene expression across cells. Alternatively, studies have performed computational integration of ‘single’ modality ATAC-seq and RNA-seq data (diagonal integration methods, reviewed in ref. 13), and integrated maps similarly can be used to correlate cCRE activity and gene expression across bins of closely matched cells60,61,66,136. Compared to single-cell co-accessibility, co-activity links showed stronger enrichment for 3D chromatin interactions135, suggesting they may more accurately predict target genes.
A general limitation of correlation-based approaches is that some of the links may reflect secondary effects, rather than direct cis-regulatory effects. Furthermore, the sensitivity and specificity to recover true regulatory relationships from single-cell correlation-based methods may be lower than for other target gene prediction methods137. Validation studies of target gene links derived from single-cell-correlation based studies have been limited thus far to genome or epigenome editing experiments of a handful of loci47–49. More systematic validation of target gene predictions using experimental techniques such as genome or epigenome editing followed by gene expression profiling or flow cytometry of fluorescently-labeled transcripts will be needed to better calibrate the sensitivity and specificity of these predictions.
Linking variants to target genes using 3D chromatin conformation
Chromatin conformation can also be used to link disease variants to putative target genes based on physical proximity between cis-regulatory elements (Figure 4b). The development of assays to measure 3D chromatin conformation in single cells, such as single cell Hi-C, single cell methyl-HiC or single-nucleus methyl-3C sequencing (sn-m3C-seq), enables the physical proximity between distal cCREs and target genes to be defined in each cell type138–141. Reanalysis of data from a previous study, in which 14 cell types were defined in prefrontal cortex using sn-m3C-seq, identified chromatin loops in all 14 cell types, most of which were specific to a cell type, with cell type-specific loops linked to genes with enriched expression in the same cell type141. Using these loops, 445 variants associated with common neuropsychiatric traits and disease were linked to 189 target genes, which included cell type-specific interactions, such as AD-associated variants that had astrocyte-specific interactions with the APOE promoter.
A general limitation of 3C-based approaches is that the resolution of interactions is often several kilobases or lower, restricting the ability to link single cCREs to target genes or proximal cCREs to gene promoters. Although single-cell Hi-C-based assays can define 3D chromatin conformation in specific cell types, to date they have been applied to few human tissues. Key barriers to their wider adoption are the limited availability and high costs per-cell of these technologies relative to other single-cell modalities. In particular, high costs mean that fewer cells are typically assayed in an experiment which limits the ability to profile rare cell types and heterogeneity within cell types.
Finally, methods originally developed for conventional bulk data, such as ABC, can be co-opted to predict target genes in each cell type identified in single-cell studies. For example, ABC was used to link candidate variants for complex traits and diseases to putative target genes with pseudo-bulk cell type profiles from scATAC-seq and contacts from promoter-capture HiC (pCHi-C) assays of matched bulk tissues55. On average, cell types had 726,000 total linkages between distal cCREs and genes, of which 6,800 were specific to a cell type. Predictions from ABC can likely be further improved if chromatin contact maps for each cell type are generated using single-cell Hi-C assays.
FUTURE PERSPECTIVES
There are multiple challenges ahead to further improve atlases of cCREs derived from single-cell epigenomics. These include the under-sampling of rare cell types in current single cell atlases, more robust cataloging of cell sub-types and cell states, and incomplete knowledge of the biological function of cCREs in the human genome. Below we provide our perspectives and strategies to bridge these gaps.
Improving the representation of rare cell types
The proportion of cell types obtained from single-cell assays reflects the abundance of the cell type in the tissue of origin, and profiles for rarer cell types therefore can be under-sampled and define fewer cCREs particularly when using assays that profile a low number (thousands) of cells. Wider adoption of techniques such as high-content combinatorial indexing142,143 for profiling hundreds of thousands to millions of cells per experiment will help saturate the detection of cCREs in rarer cell types. However, the lack of commercial kits and the relatively high sequencing costs associated with these assays limit their application. Alternatively, hybrid approaches that perform commercial single-cell assays on previously sorted cell populations may also be valuable in enriching for rare cell sub-types. While this approach requires additional sample preparation steps to sort or isolate cells prior to single cell assays, it may be an accessible and cost-effective solution in the short term for cell types with available markers and robust antibodies.
Improving the classification of cell sub-types and cell states
As single cell atlases continue to accumulate, the catalogs of candidate sub-types and states undoubtedly will grow rapidly. Currently, the definition of cell sub-types and states from single-cell data alone is based on computational clustering. Therefore, novel sub-groupings of cells identified within a cell type ideally requires independent molecular validation, such as immunostaining, fluorescence in situ hybridization (FISH) or targeted cell ablation experiments, to establish they indeed represent distinct functional units. Furthermore, technical limitations of single-cell assays, such as doublets in the datasets, can complicate the identification and characterization of transitional cell states and resolution of ‘pure’ populations of cells. Study designs that pool multiple samples per single-cell assay will be powerful in accurately flagging doublet cells based on sample genotype. Although these designs require laborious up-front sample preparation to process and pool multiple samples prior to single cell assay, they are already being widely applied in single cell studies of gene expression124,125.
Improving functional interpretation of disease-associated variants
There are numerous avenues moving forward through which cell type-resolved epigenomics, either using single-cell assays or bulk assays of sorted or isolated cells, can improve functional interpretation of disease-associated variants. Current studies have thus far largely provided reference maps of cell type-specific cCREs by profiling only a few samples for each tissue. Larger sample sizes will enable mapping of QTLs for epigenomic profiles of cell types and, in the case of single-cell data, state transitions and developmental trajectories that are obscured in bulk data. For single-cell assays, cost-effective studies will be facilitated by pooling samples and demultiplexing cells based on genotype144, such as in recent eQTL studies using hundreds of pooled samples in single-cell RNA-seq124,125. The value of single-cell assays compared to bulk assays of sorted cells in resolving each cell type is magnified with larger sample sizes, for which it becomes increasingly intractable to isolate all cell types and sub-types in every sample. As rare and private variants cannot be easily assayed using QTL mapping even in large sample sizes, continued improvement of machine learning methods that leverage epigenomic profiles to predict variant effects on cell type and states, as well as cell trajectories and state transitions, will also be needed. Atlases of cell type-specific cCREs are also largely lacking context across ancestries, environmental exposures and disease states; single-cell data will be particularly useful in identifying disease associated variants that affect stimulus- and disease-relevant state transitions95.
Moving forward, integrating data generated across different studies, both from single-cell and conventional bulk assays, will help to improve prioritization of disease risk mechanisms, as was recently done for target genes of disease-associated variants67,145. These efforts also should consider that genes can be affected by multiple independent disease associated variants at a locus, and that associated variants can affect more than one gene.
These efforts to interpret disease risk variants all collectively depend on biochemical assays of the epigenome, and thus additional evidence is needed to help establish the variant function. One approach is to map QTLs across additional layers such as gene expression or protein levels85,118,146, in addition to cellular, tissue and endophenotypes147,148, where colocalizing QTLs can help resolve the specific variants causal for shared disease and QTL signals149. However, a major challenge in QTL mapping is that large sample sizes are needed to have sufficient power for discovery and colocalization. The function of specific variants can also be studied by removing them from their genomic context, for example using reporter assays that can measure biological functions including transcription factor binding, enhancer, promoter, silencer, and RNA processing activity150–153. While these assays are well suited to studying large numbers of candidate variants in a single, high-throughput experiment, the extent to which reporter function recapitulates function in the native genome context is not well established. Finally, genome editing of specific variant alleles enables variants to be studied in their native context for effects on genome and cellular function154, although these experiments are currently lower throughput.
Improving functional annotation of the human genome
To date, single-cell epigenome studies have primarily profiled accessible chromatin only, largely due to the wide availability of commercial ATAC-seq assays. However, there are multiple additional layers to the epigenome including histone modifications, DNA methylation, and transcription factor binding, for which single-cell techniques are increasingly available (Box 1, reviewed in ref.13). These assays can further improve annotation of the epigenome of specific cell types by identifying classes of cCREs not captured by accessible chromatin, annotating the biological state and function of cCREs, and determining the transcriptional regulators of cCRE activity. Techniques that map multiple modalities in the same cell, such as accessible chromatin and gene expression, will also help improve cell type and sub-type definition by leveraging the higher resolution of gene expression data and enable annotation of target genes using methods to correlate cCRE activity with expression levels directly155,156. Integration of chromatin accessibility assays with assays that profile chromatin interactions (whether to define 3D chromatin interactions70 or in the context of methods such as ABC4) in single-cells or after sorting of specific populations will further improve the annotation of target genes of cCRE activity and risk variant activity in each cell type . Emerging techniques for spatial epigenomics will also be important in defining the distinct cis-regulatory programs of cell types in different spatial niches157–159.
Finally, functional screens that combine genome editing with single-cell genomics have enabled the effects of genes and cCREs on downstream regulatory programs to be understood, in both cis and trans160,161. While these screens have to date primarily used gene expression as a read-out, recent techniques such as Perturb-ATAC provide single-cell epigenomic profiles that can be used to annotate genomic elements affected by cCRE activity162. When intersected with genetic association data, the results of screens using single-cell epigenomics can help annotate gene networks through which disease-associated variants act.
Therapeutic implications of improved cCRE characterization
A new class of therapeutics are being developed using CRISPR-based genome or epigenome editing to target specific non-coding regions harboring disease risk variants154,163. For example, a recent cell therapy for sickle cell disease uses CRISPR inactivation of a disease-associated enhancer for BCL11A in patient derived hematopoietic stem cells164–166. Improved annotation and characterization of cCREs in the human genome can enhance efforts to design such therapeutics. A catalog of cCREs in rare, disease-relevant cell types and sub-types can reveal regions harboring risk variants that may represent therapeutic targets for that cell type. In addition, knowledge of cCRE activity across many cell types and sub-types can be used to prioritize cCREs highly specific to a cell type which may limit off-target effects. Furthermore, determining the direction of effect of a risk allele on cCRE activity using caQTL or AI mapping can provide insight into whether a therapeutic should activate or inactivate a given region. Finally, another area of potential therapeutic interest is designing synthetic enhancers that drive highly cell type-specific expression based on sequence-based models trained on cell type and sub-type cCREs167.
CONCLUSIONS
Building on previous studies that used conventional bulk assays, recent developments in single-cell epigenomics have improved our ability to interpret the function of non-coding variants associated with common complex disease and traits. Atlases derived from single-cell epigenome assays applied to human primary tissues have increased the number of active cCREs annotated in the human genome at cell-type resolution and the number of cell types that have been profiled. In addition, these atlases have described cell sub-types and heterogeneous states within cell types that are largely absent from previous catalogs. This is particularly relevant to tissues such as the brain which contain an incredible diversity of cell types and sub-types that would be impractical to assay using cell sorting. Single-cell epigenomic studies have also enabled the target genes of cCREs in specific cell types to be predicted, although the optimal methods and techniques for defining target genes in each cell type is still an active area of research. In combination with disease association data, these atlases have been used to: identify cell types, sub-types, and states enriched for disease associations; prioritize risk variants in cell type cCREs; determine the functional effects of risk variants on cCRE activity; and link risk variants to putative target genes. Thus far, these studies have largely co-opted methods and concepts developed for bulk data by collapsing single-cell data down to pseudo-bulk profiles per cell type, sub-type, or state. Newer methods that leverage single-cell profiles directly are increasingly being developed that can provide insight into disease-relevant heterogeneity, state transitions, and trajectories within a cell type. Thus, single-cell epigenomics has proven to be a valuable addition to the toolbox for annotating disease-associated variants alongside other resources such as conventional bulk epigenomics, gene expression, and gene functional data.
ACKNOWLEDGEMENTS
We apologize to those authors whose work we fail to include in this review due to space constraints. Research in the Ren lab is funded by the Ludwig Institute and the National Institutes of Health (NIH) grants 1UM1HG009402, 1U19MH114831, 1U01MH121282, 1R01AG066018, R01AG067153, U01DA052769, 1UM1HG011585, RF1MH124612, 1R56AG069107, R01EY031663, 1U01HG012059, R24AG073198, RF1MH128838, R41MH128993, UM1 MH130994, and 1U54AG079758. The Center for Epigenomics was supported, in part, by the UC San Diego School of Medicine. The Gaulton lab is funded by NIH grants DK114650, DK120429, DK122607, DK105554 and HG012059.
GLOSSARY
- Activity-by-contact (ABC)
A method for predicting the target genes of a distal cCRE based on cCRE activity and chromatin contacts between the cCRE and gene promoter cCREs
- Allelic imbalance (AI) mapping
A statistical technique to identify genetic variants with allelic differences in a molecular phenotype (such as chromatin accessibility, transcription factor binding, or epigenetic marks) by comparing the number of reads directly covering each allele of a sample heterozygous for the variant
- Cis-regulatory element (CRE)
Genomic DNA sequences that regulate transcription of a gene including enhancers, promoters, and insulators. CREs can be identified using molecular markers such as chromatin accessibility, transcription factor binding, DNA methylation and histone modifications
- Candidate cis-regulatory element (cCRE)
Genomic DNA sequences with molecular hallmarks of regulatory elements such as chromatin accessibility, transcription factor binding, DNA methylation and histone modifications that have not yet been shown to regulate gene transcription
- Chromatin conformation
The nuclear organization of chromatin that enables physical proximity of genomic regions such as distal enhancers and promoters in 3D space. Chromatin conformation can be mapped using proximity ligation-based assays such as Hi-C
- Co-accessible
Describes pairs of cis-regulatory elements that have correlated accessible chromatin profiles either across samples or cell types if using bulk profiles, or across cells if using single-cell profiles. Co-accessibility can be used to predict the target genes of cCRE activity
- Co-activity
Describes cis-regulatory elements with accessible chromatin profiles that are correlated with the expression level of a gene across samples or single-cells, and can be used to predict the target genes of cCRE activity
- Credible Set
The minimum set of variants obtained from statistical or functionally-informed fine-mapping that cumulatively explains a high percentage of the total PPA (posterior probability of association) or PIP (posterior inclusion probability) at a disease signal (usually 99% or 95%). These variants are considered candidates for being causal for the signal
- Index variant
The variant at a disease associated locus showing the strongest association p-value, which is often used to designate the locus, but may not necessarily be causal for disease. In some cases, it is also called the ‘sentinel’ variant
- Fluorescence-activated cell sorting (FACS)
A technique for separating cell populations using flow cytometry based on cells labelled with fluorescent markers
- Functionally-informed fine-mapping (FIFM)
Statistical methods that integrate genetic data with functional annotations to identify independent association signals at a disease-associated locus, determine the enrichment of functional annotations for disease association, and resolve variants causal for each association signal using functional enrichments as weights or priors on variants
- Genome wide association study (GWAS)
Systematic testing of directly assayed or imputed genotypes of variants genome-wide for association to a binary (for example, case or control) or quantitative phenotype
- Linkage Disequilibrium (LD)
The non-random inheritance of variant alleles at a locus due to limited recombination events between variant positions leading to only a subset of observed haplotypes and highly correlated variant genotypes. At disease loci, many variants will show significant association due to being in LD with the true causal variant but are not directly causal themselves
- LD Variant
A variants at a disease-associated locus that has strong association p-values due to linkage disequilibrium with the index variant and which may or may not be causal for disease
- Posterior probability of association (PPA)
Probability obtained from Bayesian fine-mapping analyses that a variant is causal for a trait or disease association signal
- Posterior inclusion probability (PIP)
Probability obtained from Bayesian fine-mapping analyses that a variant is included in any causal model which relates to the evidence that it is causal for a trait or disease
- Quantitative trait locus (QTL) mapping
A statistical technique to identify genetic variants that have genotypes correlated with the levels of a molecular, cellular, tissue, or physiological phenotype such as chromatin accessibility, transcription factor binding, or epigenetic marks across different samples
- Sequence-based machine learning models
Methods that use machine learning to learn the sequence grammar underlying sets of active genomic regions (such as cCREs active in a cell type) compared to non-active regions. These machine learning models can then be used to predict whether a sequence is likely to have regulatory activity, which can be further leveraged to predict variant alleles with differences in predicted regulatory activity
- Statistical fine-mapping
Statistical methods that utilize only genetic data to identify independent association signals at a disease-associated locus as well as resolve variants causal for each association signal
- Stratified linkage disequilibrium score regression
Statistical technique to identify genomic annotations with enriched heritability for a common complex trait or disease based on linkage disequilibrium with associated variants genome-wide
- ToC blurb
In this Review, Gaulton et al. discuss how single-cell epigenomics methods generate cell type, sub-type, and state-resolved maps of candidate cis-regulatory elements (cCREs) in heterogeneous human tissues that can help interpret the genetic basis of common traits and diseases
Footnotes
Competing interests
B.R. is a shareholder and consultant of Arima Genomics, Inc., and a co-founder of Epigenome Technologies, Inc. K.J.G. is a consultant of Genentech and a shareholder in Vertex Pharmaceuticals and Neurocrine Biosciences. These relationships have been disclosed to and approved by the UCSD Independent Review Committee.
REFERENCES
- 1.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, doi: 10.1038/35057062 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012, doi: 10.1093/nar/gky1120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boix CA, James BT, Park YP, Meuleman W & Kellis M Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307, doi: 10.1038/s41586-020-03145-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nasser J et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243, doi: 10.1038/s41586-021-03446-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi: 10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195, doi: 10.1126/science.1222794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium, E. P. et al. Perspectives on ENCODE. Nature 583, 693–698, doi: 10.1038/s41586-020-2449-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorkin DU et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751, doi: 10.1038/s41586-020-2093-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Y et al. Spatiotemporal DNA methylome dynamics of the developing mouse fetus. Nature 583, 752–759, doi: 10.1038/s41586-020-2119-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allis CD & Jenuwein T The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500, doi: 10.1038/nrg.2016.59 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Cavalli G & Heard E Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499, doi: 10.1038/s41586-019-1411-0 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Preissl S, Gaulton KJ & Ren B Characterizing cis-regulatory elements using single-cell epigenomics. Nat Rev Genet, doi: 10.1038/s41576-022-00509-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Consortium, E. P. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710, doi: 10.1038/s41586-020-2493-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stunnenberg HG, International Human Epigenome, C. & Hirst M The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 167, 1145–1149, doi: 10.1016/j.cell.2016.11.007 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Shen Y et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120, doi: 10.1038/nature11243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461, doi: 10.1038/nature12787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Javierre BM et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384 e1319, doi: 10.1016/j.cell.2016.09.037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitt AD et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep 17, 2042–2059, doi: 10.1016/j.celrep.2016.10.061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stuart T & Satija R Integrative single-cell analysis. Nat Rev Genet 20, 257–272, doi: 10.1038/s41576-019-0093-7 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490, doi: 10.1038/nature14590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914, doi: 10.1126/science.aab1601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin W et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146, doi: 10.1038/nature15740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada A et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol 21, 287–296, doi: 10.1038/s41556-018-0248-3 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Wu SJ et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol 39, 819–824, doi: 10.1038/s41587-021-00865-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartosovic M, Kabbe M & Castelo-Branco G Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotechnol 39, 825–835, doi: 10.1038/s41587-021-00869-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ai S et al. Profiling chromatin states using single-cell itChIP-seq. Nat Cell Biol 21, 1164–1172, doi: 10.1038/s41556-019-0383-5 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Carter B et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat Commun 10, 3747, doi: 10.1038/s41467-019-11559-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grosselin K et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet 51, 1060–1066, doi: 10.1038/s41588-019-0424-9 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930, doi: 10.1038/s41467-019-09982-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods 16, 323–325, doi: 10.1038/s41592-019-0361-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartosovic M & Castelo-Branco G Multimodal chromatin profiling using nanobody-based single-cell CUT&Tag. Nat Biotechnol, doi: 10.1038/s41587-022-01535-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart T et al. Nanobody-tethered transposition enables multifactorial chromatin profiling at single-cell resolution. Nat Biotechnol, doi: 10.1038/s41587-022-01588-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33, 1165–1172, doi: 10.1038/nbt.3383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hainer SJ, Boskovic A, McCannell KN, Rando OJ & Fazzio TG Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319–1329 e1311, doi: 10.1016/j.cell.2019.03.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64, doi: 10.1038/nature12593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo H et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23, 2126–2135, doi: 10.1101/gr.161679.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smallwood SA et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11, 817–820, doi: 10.1038/nmeth.3035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mooijman D, Dey SS, Boisset JC, Crosetto N & van Oudenaarden A Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat Biotechnol 34, 852–856, doi: 10.1038/nbt.3598 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Zhu C et al. Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution. Cell Stem Cell 20, 720–731 e725, doi: 10.1016/j.stem.2017.02.013 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Inoue A, Suzuki T & Zhang Y Simultaneous mapping of active DNA demethylation and sister chromatid exchange in single cells. Genes Dev 31, 511–523, doi: 10.1101/gad.294843.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214, doi: 10.1016/j.cell.2015.05.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201, doi: 10.1016/j.cell.2015.04.044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preissl S et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci 21, 432–439, doi: 10.1038/s41593-018-0079-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80, doi: 10.1038/nbt.4038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo C et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604, doi: 10.1126/science.aan3351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiou J et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402, doi: 10.1038/s41586-021-03552-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiou J et al. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat Genet 53, 455–466, doi: 10.1038/s41588-021-00823-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hocker JD et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci Adv 7, doi: 10.1126/sciadv.abf1444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ord T et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ Res 129, 240–258, doi: 10.1161/CIRCRESAHA.121.318971 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet 52, 1158–1168, doi: 10.1038/s41588-020-00721-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morabito S et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155, doi: 10.1038/s41588-021-00894-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang A et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. Elife 9, doi: 10.7554/eLife.62522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936, doi: 10.1038/s41587-019-0206-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 184, 5985–6001 e5919, doi: 10.1016/j.cell.2021.10.024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; - This study generated a large, comprehensive atlas of cCREs in hundreds of adult and fetal cell types which identified many trait-cell type links and annotated fine-mapped risk variants.
- 56.Orchard P et al. Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits. Genome Res, doi: 10.1101/gr.268482.120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rai V et al. Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol Metab 32, 109–121, doi: 10.1016/j.molmet.2019.12.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Domcke S et al. A human cell atlas of fetal chromatin accessibility. Science 370, doi: 10.1126/science.aba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; - This study generated a comprehensive atlas of cCREs in fetal cell types using high-content combinatorial indexing and identified traits enriched for fetal cell types
- 59.Sheng X et al. Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments. Nat Genet 53, 1322–1333, doi: 10.1038/s41588-021-00909-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trevino AE et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069 e5023, doi: 10.1016/j.cell.2021.07.039 (2021). [DOI] [PubMed] [Google Scholar]; - This article describes an example of a study that leveraged deep learning of single cell epigenome profies to predict functional rare variants affecting autism risk
- 61.Becker WR et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet 54, 985–995, doi: 10.1038/s41588-022-01088-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Briel N et al. Single-nucleus chromatin accessibility profiling highlights distinct astrocyte signatures in progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol 144, 615–635, doi: 10.1007/s00401-022-02483-8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duong TE et al. A single-cell regulatory map of postnatal lung alveologenesis in humans and mice. Cell Genom 2, doi: 10.1016/j.xgen.2022.100108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finkbeiner C et al. Single-cell ATAC-seq of fetal human retina and stem-cell-derived retinal organoids shows changing chromatin landscapes during cell fate acquisition. Cell Rep 38, 110294, doi: 10.1016/j.celrep.2021.110294 (2022). [DOI] [PubMed] [Google Scholar]
- 65.Herring CA et al. Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution. Cell 185, 4428–4447 e4428, doi: 10.1016/j.cell.2022.09.039 (2022). [DOI] [PubMed] [Google Scholar]
- 66.Kuppe C et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777, doi: 10.1038/s41586-022-05060-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu H et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat Genet 54, 950–962, doi: 10.1038/s41588-022-01097-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo C et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genom 2, doi: 10.1016/j.xgen.2022.100107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benaglio P et al. Type 1 diabetes risk genes mediate pancreatic beta cell survival in response to proinflammatory cytokines,. Cell Genomics, 100214, doi: 10.1016/j.xgen.2022.100214. (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su C et al. 3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk. Cell Metab 34, 1394–1409 e1394, doi: 10.1016/j.cmet.2022.08.014 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turner AW et al. Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk. Nat Genet 54, 804–816, doi: 10.1038/s41588-022-01069-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uzquiano A et al. Proper acquisition of cell class identity in organoids allows definition of fate specification programs of the human cerebral cortex. Cell 185, 3770–3788 e3727, doi: 10.1016/j.cell.2022.09.010 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Q et al. Single-cell chromatin accessibility landscape in kidney identifies additional cell-of-origin in heterogenous papillary renal cell carcinoma. Nat Commun 13, 31, doi: 10.1038/s41467-021-27660-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson PC et al. Multimodal single cell sequencing implicates chromatin accessibility and genetic background in diabetic kidney disease progression. Nat Commun 13, 5253, doi: 10.1038/s41467-022-32972-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee DS et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat Methods 16, 999–1006, doi: 10.1038/s41592-019-0547-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Granja JM et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37, 1458–1465, doi: 10.1038/s41587-019-0332-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37, 916–924, doi: 10.1038/s41587-019-0147-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meuleman W et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251, doi: 10.1038/s41586-020-2559-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muto Y et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun 12, 2190, doi: 10.1038/s41467-021-22368-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muto Y et al. Defining cellular complexity in human autosomal dominant polycystic kidney disease by multimodal single cell analysis. Nat Commun 13, 6497, doi: 10.1038/s41467-022-34255-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trynka G et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 45, 124–130, doi: 10.1038/ng.2504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–1235, doi: 10.1038/ng.3404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pickrell JK Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet 94, 559–573, doi: 10.1016/j.ajhg.2014.03.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ulirsch JC et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet 51, 683–693, doi: 10.1038/s41588-019-0362-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; - This study reports the method g-chromVar that specifically leveraged single cell epigenome profiles in genetic enrichment analyses, which was used to understand the role of blood cell trait variants in hematopoesis
- 85.Li YI et al. RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604, doi: 10.1126/science.aad9417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schmidt EM et al. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606, doi: 10.1093/bioinformatics/btv201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hansen DV, Hanson JE & Sheng M Microglia in Alzheimer’s disease. J Cell Biol 217, 459–472, doi: 10.1083/jcb.201709069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139, doi: 10.1126/science.aay0793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Dijk D et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729 e727, doi: 10.1016/j.cell.2018.05.061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wakefield J A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet 81, 208–227, doi: 10.1086/519024 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wellcome Trust Case Control, C. et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet 44, 1294–1301, doi: 10.1038/ng.2435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gaulton KJ et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet 47, 1415–1425, doi: 10.1038/ng.3437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501, doi: 10.1093/bioinformatics/btw018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine mapping. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 82, 1273–1300, doi: 10.1111/rssb.12388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Calderon D et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat Genet 51, 1494–1505, doi: 10.1038/s41588-019-0505-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Edwards SL, Beesley J, French JD & Dunning AM Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 93, 779–797, doi: 10.1016/j.ajhg.2013.10.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hindorff LA et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106, 9362–9367, doi: 10.1073/pnas.0903103106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang F & Lupski JR Non-coding genetic variants in human disease. Hum Mol Genet 24, R102–110, doi: 10.1093/hmg/ddv259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Claussnitzer M et al. A brief history of human disease genetics. Nature 577, 179–189, doi: 10.1038/s41586-019-1879-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kichaev G et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet 10, e1004722, doi: 10.1371/journal.pgen.1004722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen W, McDonnell SK, Thibodeau SN, Tillmans LS & Schaid DJ Incorporating Functional Annotations for Fine-Mapping Causal Variants in a Bayesian Framework Using Summary Statistics. Genetics 204, 933–958, doi: 10.1534/genetics.116.188953 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y & Kellis M Joint Bayesian inference of risk variants and tissue-specific epigenomic enrichments across multiple complex human diseases. Nucleic Acids Res 44, e144, doi: 10.1093/nar/gkw627 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513, doi: 10.1038/s41588-018-0241-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van de Bunt M et al. Evaluating the Performance of Fine-Mapping Strategies at Common Variant GWAS Loci. PLoS Genet 11, e1005535, doi: 10.1371/journal.pgen.1005535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tabula Sapiens C et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896, doi: 10.1126/science.abl4896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eraslan G et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290, doi: 10.1126/science.abl4290 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Regev A et al. The Human Cell Atlas. Elife 6, doi: 10.7554/eLife.27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cao J et al. A human cell atlas of fetal gene expression. Science 370, doi: 10.1126/science.aba7721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Calderon D et al. Inferring Relevant Cell Types for Complex Traits by Using Single-Cell Gene Expression. Am J Hum Genet 101, 686–699, doi: 10.1016/j.ajhg.2017.09.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jagadeesh KA et al. Identifying disease-critical cell types and cellular processes by integrating single-cell RNA-sequencing and human genetics. Nat Genet 54, 1479–1492, doi: 10.1038/s41588-022-01187-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Watanabe K, Umicevic Mirkov M, de Leeuw CA, van den Heuvel MP & Posthuma D Genetic mapping of cell type specificity for complex traits. Nat Commun 10, 3222, doi: 10.1038/s41467-019-11181-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alasoo K et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50, 424–431, doi: 10.1038/s41588-018-0046-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bossini-Castillo L et al. Immune disease variants modulate gene expression in regulatory CD4(+) T cells. Cell Genom 2, None, doi: 10.1016/j.xgen.2022.100117 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liang D et al. Cell-type-specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. Nat Neurosci 24, 941–953, doi: 10.1038/s41593-021-00858-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robertson CC et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet 53, 962–971, doi: 10.1038/s41588-021-00880-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gate RE et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat Genet 50, 1140–1150, doi: 10.1038/s41588-018-0156-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383, doi: 10.1371/journal.pgen.1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330, doi: 10.1126/science.aaz1776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neavin D et al. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol 22, 76, doi: 10.1186/s13059-021-02293-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nathan A et al. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 606, 120–128, doi: 10.1038/s41586-022-04713-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Benaglio P et al. Mapping genetic effects on cell type-specific chromatin accessibility and annotating complex trait variants using single nucleus ATAC-seq. BioRxiv 2020.12.03.387894, doi: 10.1101/2020.12.03.387894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kumasaka N, Knights AJ & Gaffney DJ Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat Genet 48, 206–213, doi: 10.1038/ng.3467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen L et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 167, 1398–1414 e1324, doi: 10.1016/j.cell.2016.10.026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yazar S et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041, doi: 10.1126/science.abf3041 (2022). [DOI] [PubMed] [Google Scholar]
- 125.Perez RK et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376, eabf1970, doi: 10.1126/science.abf1970 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim-Hellmuth S et al. Cell type-specific genetic regulation of gene expression across human tissues. Science 369, doi: 10.1126/science.aaz8528 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aguirre-Gamboa R et al. Deconvolution of bulk blood eQTL effects into immune cell subpopulations. BMC Bioinformatics 21, 243, doi: 10.1186/s12859-020-03576-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kelley DR, Snoek J & Rinn JL Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 26, 990–999, doi: 10.1101/gr.200535.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 12, 931–934, doi: 10.1038/nmeth.3547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee D et al. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 47, 955–961, doi: 10.1038/ng.3331 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou J et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet 50, 1171–1179, doi: 10.1038/s41588-018-0160-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shrikumar A, Prakash E & Kundaje A GkmExplain: fast and accurate interpretation of nonlinear gapped k-mer SVMs. Bioinformatics 35, i173–i182, doi: 10.1093/bioinformatics/btz322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yuan H & Kelley DR scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat Methods 19, 1088–1096, doi: 10.1038/s41592-022-01562-8 (2022). [DOI] [PubMed] [Google Scholar]
- 134.Pliner HA et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 71, 858–871 e858, doi: 10.1016/j.molcel.2018.06.044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang L, Zhang J & Nie Q DIRECT-NET: An efficient method to discover cis-regulatory elements and construct regulatory networks from single-cell multiomics data. Sci Adv 8, eabl7393, doi: 10.1126/sciadv.abl7393 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Granja JM et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411, doi: 10.1038/s41588-021-00790-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dey KK et al. SNP-to-gene linking strategies reveal contributions of enhancer-related and candidate master-regulator genes to autoimmune disease. Cell Genom 2, doi: 10.1016/j.xgen.2022.100145 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang R, Zhou T & Ma J Ultrafast and interpretable single-cell 3D genome analysis with Fast-Higashi. Cell Syst 13, 798–807 e796, doi: 10.1016/j.cels.2022.09.004 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang S et al. DeepLoop robustly maps chromatin interactions from sparse allele-resolved or single-cell Hi-C data at kilobase resolution. Nat Genet 54, 1013–1025, doi: 10.1038/s41588-022-01116-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li X et al. SnapHiC2: A computationally efficient loop caller for single cell Hi-C data. Comput Struct Biotechnol J 20, 2778–2783, doi: 10.1016/j.csbj.2022.05.046 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yu M et al. SnapHiC: a computational pipeline to identify chromatin loops from single-cell Hi-C data. Nat Methods, doi: 10.1038/s41592-021-01231-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mulqueen RM et al. High-content single-cell combinatorial indexing. Nat Biotechnol, doi: 10.1038/s41587-021-00962-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu C et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods 18, 283–292, doi: 10.1038/s41592-021-01060-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94, doi: 10.1038/nbt.4042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gazal S et al. Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat Genet 54, 827–836, doi: 10.1038/s41588-022-01087-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yao C et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun 9, 3268, doi: 10.1038/s41467-018-05512-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu Y et al. Genetic architecture of 11 organ traits derived from abdominal MRI using deep learning. Elife 10, doi: 10.7554/eLife.65554 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Elliott LT et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216, doi: 10.1038/s41586-018-0571-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Giambartolomei C et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34, 2538–2545, doi: 10.1093/bioinformatics/bty147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Doni Jayavelu N, Jajodia A, Mishra A & Hawkins RD Candidate silencer elements for the human and mouse genomes. Nat Commun 11, 1061, doi: 10.1038/s41467-020-14853-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Inoue F & Ahituv N Decoding enhancers using massively parallel reporter assays. Genomics 106, 159–164, doi: 10.1016/j.ygeno.2015.06.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Patwardhan RP et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotechnol 27, 1173–1175, doi: 10.1038/nbt.1589 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cheung R et al. A Multiplexed Assay for Exon Recognition Reveals that an Unappreciated Fraction of Rare Genetic Variants Cause Large-Effect Splicing Disruptions. Mol Cell 73, 183–194 e188, doi: 10.1016/j.molcel.2018.10.037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li H et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther 5, 1, doi: 10.1038/s41392-019-0089-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhu C et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol 26, 1063–1070, doi: 10.1038/s41594-019-0323-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385, doi: 10.1126/science.aau0730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Deng Y et al. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 375, 681–686, doi: 10.1126/science.abg7216 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Deng Y et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383, doi: 10.1038/s41586-022-05094-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lu T, Ang CE & Zhuang X Spatially resolved epigenomic profiling of single cells in complex tissues. Cell 185, 4448–4464 e4417, doi: 10.1016/j.cell.2022.09.035 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866 e1817, doi: 10.1016/j.cell.2016.11.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gasperini M et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390 e319, doi: 10.1016/j.cell.2018.11.029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rubin AJ et al. Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176, 361–376 e317, doi: 10.1016/j.cell.2018.11.022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chavez M, Chen X, Finn PB & Qi LS Advances in CRISPR therapeutics. Nat Rev Nephrol 19, 9–22, doi: 10.1038/s41581-022-00636-2 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sankaran VG, Xu J & Orkin SH Transcriptional silencing of fetal hemoglobin by BCL11A. Ann N Y Acad Sci 1202, 64–68, doi: 10.1111/j.1749-6632.2010.05574.x (2010). [DOI] [PubMed] [Google Scholar]
- 165.Frangoul H et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N Engl J Med 384, 252–260, doi: 10.1056/NEJMoa2031054 (2021). [DOI] [PubMed] [Google Scholar]
- 166.Bauer DE et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342, 253–257, doi: 10.1126/science.1242088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.de Almeida BP, Reiter F, Pagani M & Stark A DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat Genet 54, 613–624, doi: 10.1038/s41588-022-01048-5 (2022). [DOI] [PubMed] [Google Scholar]
- 168.Nichols RV et al. High-throughput robust single-cell DNA methylation profiling with sciMETv2. Nat Commun 13, 7627, doi: 10.1038/s41467-022-35374-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]