

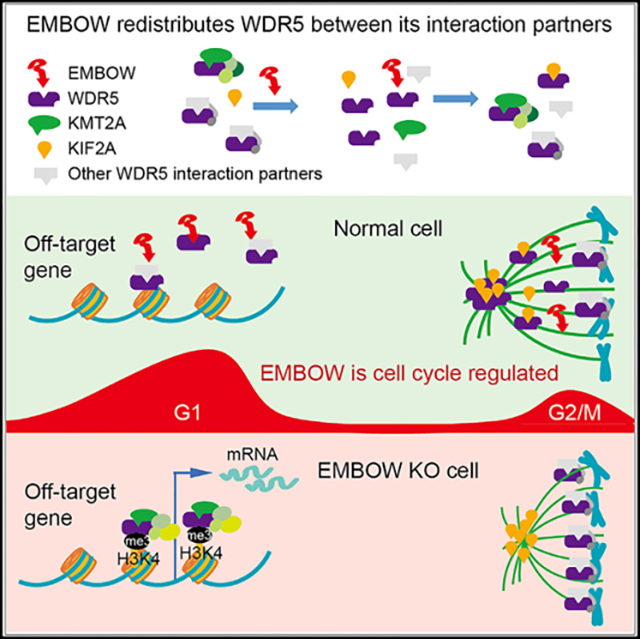
SUMMARY
The conserved WD40-repeat protein WDR5 interacts with multiple proteins both inside and outside the nucleus. However, it is currently unclear whether and how the distribution of WDR5 between complexes is regulated. Here, we show that an unannotated microprotein EMBOW (endogenous microprotein binder of WDR5) dually encoded in the human SCRIB gene interacts with WDR5 and regulates its binding to multiple interaction partners, including KMT2A and KIF2A. EMBOW is cell cycle regulated, with two expression maxima at late G1 phase and G2/M phase. Loss of EMBOW decreases WDR5 interaction with KIF2A, aberrantly shortens mitotic spindle length, prolongs G2/M phase, and delays cell proliferation. In contrast, loss of EMBOW increases WDR5 interaction with KMT2A, leading to WDR5 binding to off-target genes, erroneously increasing H3K4me3 levels, and activating transcription of these genes. Together, these results implicate EMBOW as a regulator of WDR5 that regulates its interactions and prevents its off-target binding in multiple contexts.
Graphical Abstract
In brief
Chen et al. show that human SCRIB dually encodes an unannotated, cell-cycle-regulated microprotein, EMBOW. EMBOW interacts with WDR5 and regulates its binding to multiple interaction partners, including KMT2A and KIF2A, safeguarding WDR5 on-target binding on chromatin and the mitotic spindle during the cell cycle.
INTRODUCTION
WDR5 is a conserved WD40-repeat protein that functions as a scaffold for the KMT2 complexes to deposit histone H3 lysine 4 (H3K4) di- and tri-methylation (me2,3).1 WDR5 also plays important roles in multiple complexes in the nucleus and cytoplasm of the cell, such as recruiting the anaphase-promoting complex to rapidly reactivate specific gene promoters upon exit from mitosis2 and interacting with KIF2A to ensure faithful spindle assembly during mitosis.3 WDR5 also recruits MYC to chromatin to drive tumorigenesis,4 promotes the epithelial-to-mesenchymal transition,5 and is overexpressed in cancer, including leukemia,6 colon cancer,7 and bladder cancer.8 Pharmacological inhibition or loss of WDR5 decreases cancer cell proliferation,8–11 suggesting that WDR5 is a potential anti-cancer drug target. WDR5 interacts with most protein partners via two distinct binding sites at opposing faces of its beta-barrel fold, and pharmacological targeting efforts have generally focused on one of these binding sites, termed the WIN site, with the goal of disrupting its interaction with KMT2 proteins.12 However, WIN-site inhibitors may not predominantly act by changing H3K4me3 levels, suggesting that their mechanism of action is incompletely understood.9,13 In order to develop safe and effective therapeutic strategies to target WDR5, it is essential to obtain a complete understanding of the functional complexes of WDR5, how its complexes are regulated, and which of these interactions are disrupted by WIN-site inhibitors.
Thousands of unannotated small open reading frames (smORFs; defined here as ORFs of fewer than 150 codons) are translated in mammalian cells.14 These ORFs are found in 5′ and 3′ untranslated regions (UTRs) of mRNA (upstream and downstream ORFs, uORFs and dORFs, respectively), in frame-shifted ORFs overlapping annotated protein-coding sequences (alternative ORFs [alt-ORFs]), and in noncoding RNAs.15 Genome annotation consortia excluded smORFs because of their short lengths, initiation from non-AUG start codons, out-of-frame overlap with annotated protein coding sequences, and/or limited homology to protein domains of known function.16,17 Hundreds of mammalian smORF-encoded polypeptides (SEPs; also termed micropeptides, small proteins, and microproteins) affect cell proliferation in CRISPR knockout (KO) screens,18,19 but only several dozen have been defined at the molecular level. Even fewer uORF-encoded microproteins have been characterized, perhaps because of long-standing evidence that uORFs act in cis to downregulate translation of the downstream protein.20 However, uORF-encoded microproteins can also function in trans, including MP31, which regulates lactate metabolism in glioblastoma21; alt-LAMA3, which functions in ribosomal RNA biogenesis22; and mouse Gm9999, which encodes two polypeptides, Kastor and Polluks, that regulate spermatogenesis.23 Some uORF microproteins bind to the downstream protein, leading their dual coding transcripts to be compared with bacterial operons.18 Therefore, uORFs have the potential to encode bioactive microproteins.
The human SCRIB gene encodes a homolog of Drosophila Scribble, which is a PDZ domain-containing protein involved in cell polarity and organization of multiprotein complexes.24 The 5′ UTR of the human SCRIB mRNA contains an uORF that partially overlaps the SCRIB coding sequence in an alternative reading frame; this uORF is translated in several human cancer cell lines.25,26 Although the SCRIB uORF downregulates the translation of SCRIB in cis,26 it is currently unknown whether the uORF-encoded microprotein also functions in trans. Here, we demonstrate that the human SCRIB uORF is translated to produce a cell-cycle-regulated microprotein that binds to the WIN site of WDR5. We provide evidence that changing levels of this microprotein, which we propose to name EMBOW, or endogenous microprotein binder of WDR5, alters the association of WDR5 with multiple interaction partners. We focus on two of them, KIF2A and KMT2A, because the molecular and cellular outcomes of these interactions have been previously well characterized. We show that high EMBOW levels constrain WDR5 association with KMT2A, preventing H3K4me3 deposition at off-target genes; high EMBOW also results in increased WDR5 binding to KIF2A, promoting correct mitotic spindle assembly. Finally, EMBOW regulates cell-cycle progression via its interaction with WDR5. EMBOW therefore reconfigures the WDR5 WIN-site interactome and is involved in regulating the multifunctionality of WDR5.
RESULTS
SCRIB dually encodes an unannotated, conserved nuclear microprotein
A microprotein mapping uniquely to an alternative reading frame of human SCRIB transcript variant 1 (tv1; Figure 1A) was previously detected in K562, MOLT4, HeLa, MCF-7, A549, and HCT-116 cells.25,26 This peptide, previously termed oSCRIB, maps to an uORF initiating at an AUG start codon and extending into the SCRIB coding sequence by 284 nt in the −1 reading frame, which encodes a microprotein of 120 amino acids with completely different sequence from SCRIB because of the alternative reading frame. We propose to rename it EMBOW based on its cellular role. In order to determine how broadly EMBOW is expressed, we additionally profiled HT1080 cells and the primary melanoma cell line YUZEST27 using proteogenomic technology28 and detected a tryptic peptide uniquely mapping to EMBOW in both samples (Figures 1B and 1C). These results, combined with prior studies, are consistent with ubiquitous expression of EMBOW in immortalized and primary human cells from varied tissues of origin and in cancer.
Figure 1. SCRIB dually encodes an unannotated nuclear microprotein.
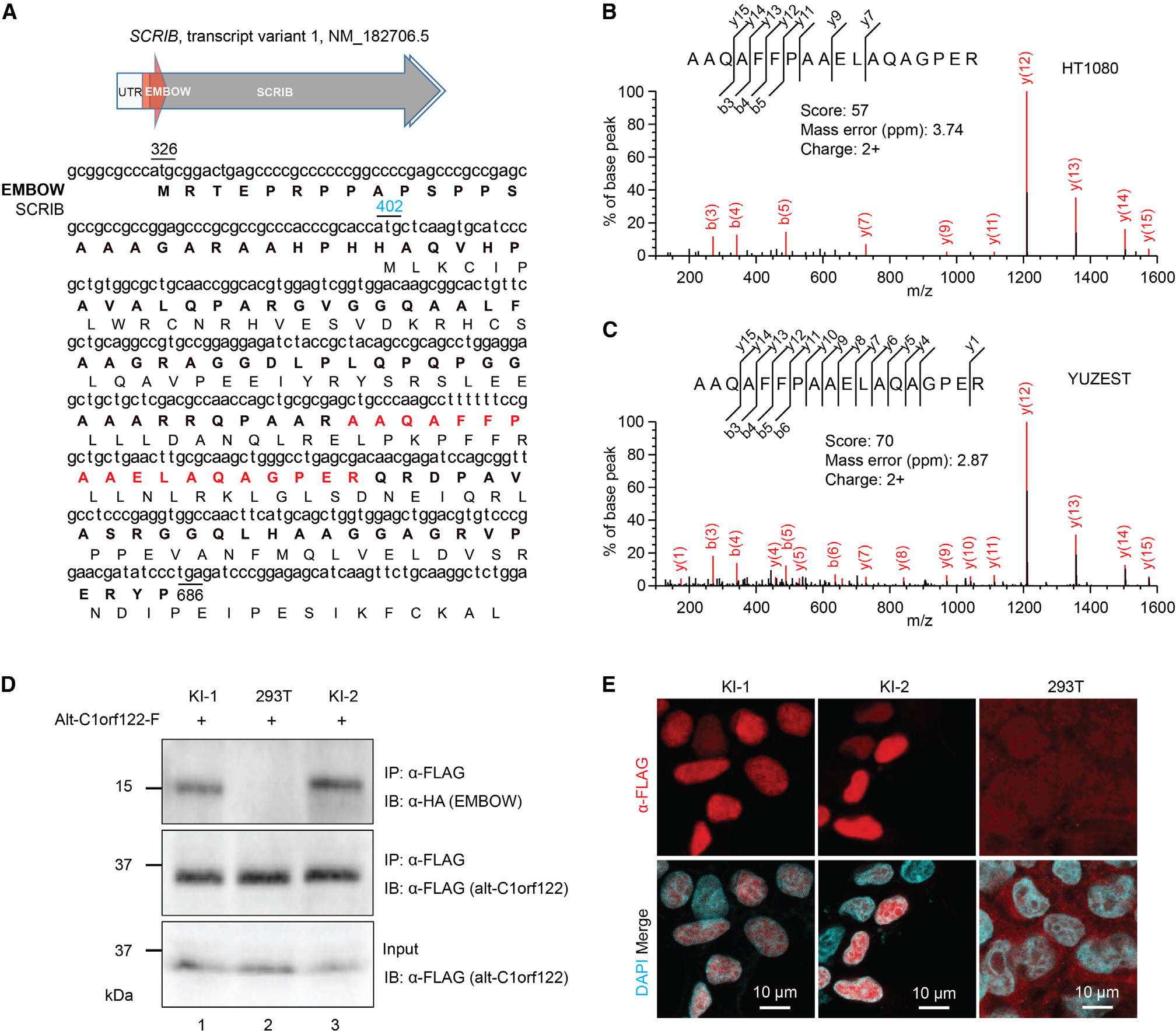
(A) Top: schematic representation of human SCRIB tv1: light gray arrow, 5′ and 3′ UTR; dark gray arrow, SCRIB coding sequence; red arrow, smORF encoding EMBOW. Bottom: the cDNA sequence of human SCRIB tv1 is shown with the protein sequences of EMBOW (bold) and SCRIB indicated below. The start codons of EMBOW (black) and SCRIB (blue) and the stop codon of EMBOW (black) are numbered. Highlighted in red is the tryptic peptide of EMBOW detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
(B and C) MS/MS spectra of EMBOW tryptic peptide detected in HT1080 and primary cultured melanoma cells (YUZEST). Mascot score, precursor mass error and precursor charge state are presented.
(D) Control (293T) or EMBOW-FLAG-HA knockin (KI) HEK293T cells were transiently transfected with a plasmid encoding alt-C1orf122-FLAG,25 serving as an FLAG-IP positive control, and IPs were performed followed by immunoblotting (IB). Cell lysates (4%) before IP (input) were used as the loading controls. Data are representative of three biological replicates.
(E) Immunostaining of two independent EMBOW-FLAG-HA KI cell lines (KI-1 and KI-2) and HEK293T cells (293T) as a negative control with anti-FLAG (red) and DAPI (cyan). Scale bars, 10 μm. Data are representative of three biological replicates.
See also Figure S1.
To determine whether EMBOW is endogenously expressed from the SCRIB locus in HEK293T cells, we generated two independent knockin (KI) HEK293T cell lines with an FLAG-HA tag appended to the 3′ end of the EMBOW smORF. FLAG-immunoprecipitation (IP) followed by immunoblotting revealed a 15-kDa anti-HA immunoreactive band in the KI cells, which was absent from control cells, indicating endogenous expression of EMBOW (Figure 1D). Consistent with its interaction with WDR5 (vide infra), immunofluorescence demonstrated that endogenous EMBOW localizes to the nucleus of KI cells during interphase (Figure 1E).
ClustalW alignment of murine, bovine, pig, and primate SCRIB mRNAs revealed putative EMBOW homologs that, despite truncating mutations at the C terminus in some organisms, exhibit sequence similarity at the N termini (Figure S1A). A translated SCRIB uORF may therefore be conserved in mammals, and the N terminus of microprotein could be important. To determine whether mouse EMBOW can be translated, an expression vector containing the 5′ UTR of mouse SCRIB tv1 through the predicted stop codon of mouse EMBOW was constructed with an FLAG epitope tag and GFP appended to the C terminus of the protein. As shown in Figure S1B, overexpressed mouse EMBOW exhibited nucleocytoplasmic immunofluorescence in 3T3 cells, indicating that EMBOW has the potential to be translated in mice.
EMBOW directly interacts with WDR5 via the WIN site
Because microproteins bind to other proteins,29 we aimed to identify protein(s) associated with EMBOW. We performed co-immunoprecipitation (coIP) with nuclear lysates from HEK293T cells transiently overexpressing EMBOW-FLAG-HA and parental HEK293T cells as a control, followed by label-free quantitative proteomics with Maxquant.30 WDR5 was 6-fold more enriched by EMBOW coIP than any other protein (Figure 2A). We further examined the top 10 proteins enriched in the EMBOW coIP and found among this list three ribonucleoprotein or ribosomal proteins, as well as four actin- or tubulin-binding proteins (Table S1), which are, or are associated with, nonspecific proteins commonly detected in affinity purification,31 so we did not pursue them further. We therefore focused on the association of WDR5 with EMBOW.
Figure 2. EMBOW directly interacts with WDR5 via the WIN site.
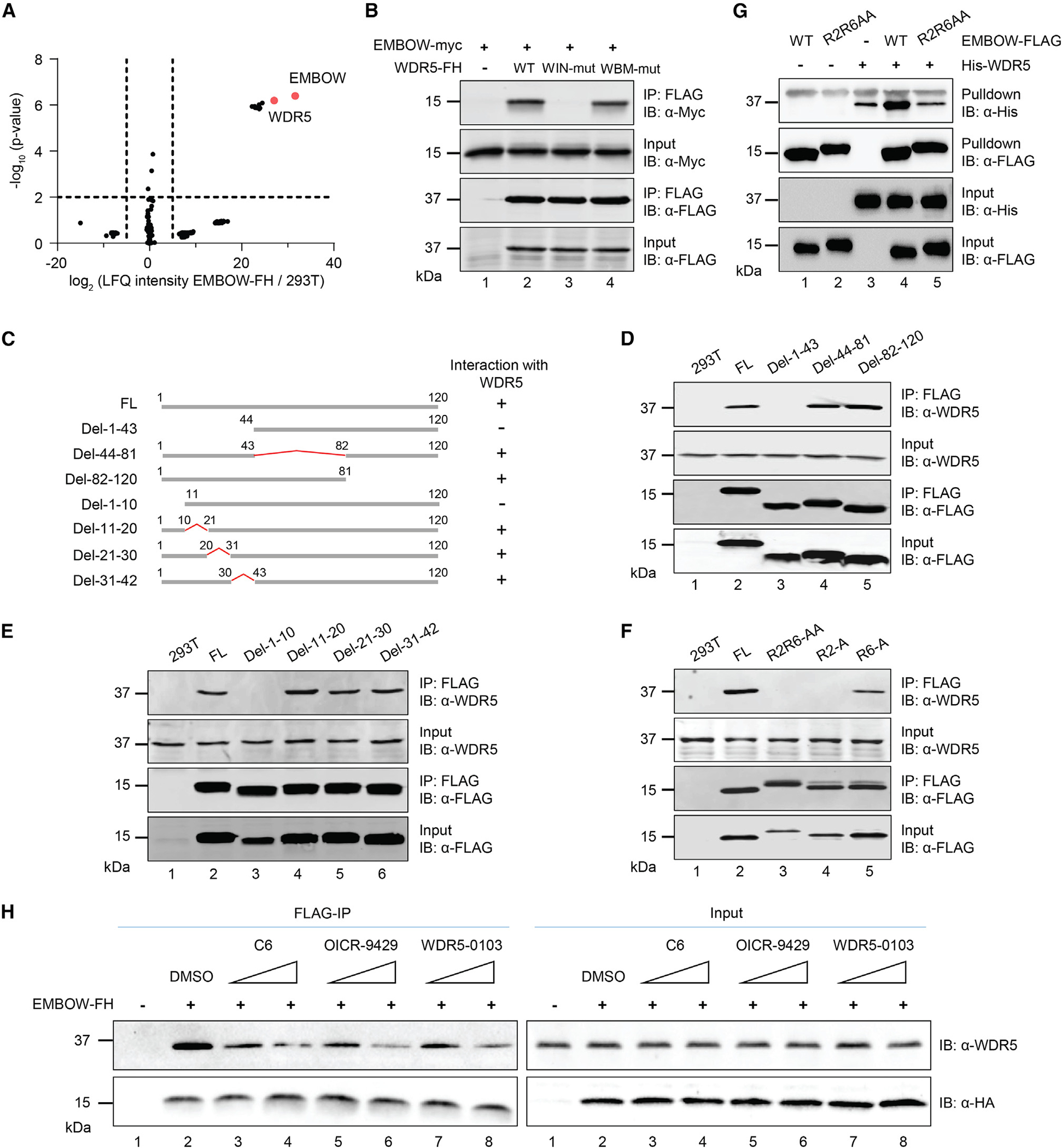
(A) Volcano plot of quantitative proteomics (N = 3 biologically independent experiments) of anti-FLAG pull-down from nuclear lysates of HEK293T cells transiently overexpressing EMBOW-FLAG-HA (EMBOW-FH) or parental (293T) HEK293T. Two-sample p value was calculated using Perseus. For complete quantitative proteomics results, see Table S4.
(B) HEK293T cells were transfected with EMBOW-myc only, or co-transfected with EMBOW-myc and wild-type (WT), or WIN site mutated (WIN-mut), or WBM site mutated WDR5-FH (WBM-mut), followed by IP and IB. Data are representative of three biological replicates.
(C) Schematic representation of full-length (FL) and deletion (del) mutant EMBOW constructs, with amino acid residues numbered above. WDR5 association status of each construct is indicated on the right.
(D–F) HEK293T cells were transfected with EMBOW full-length (FL) or mutants (listed at top), followed by IP and IB. Data are representative of three biological replicates.
(G) His-WDR5, wild-type (WT), or R2R6-to-AA mutated (R2R6AA) EMBOW-FLAG were purified from E. coli, followed by in vitro FLAG pull-down and IB. A nonspecific upper band appears in the anti-His IB channel. Data are representative of three biological replicates.
(H) HEK293T cells were transfected with EMBOW-FH or mock transfection, and FLAG-IP was performed in the absence (DMSO) or presence of WDR5 WIN-site inhibitors C6 (0.5 or 5 μM), OICR-9429 (0.5 or 5 μM), or WDR5-0103 (5 or 50 μM), followed by IB. Data are representative of three biological replicates.
WDR5 directly interacts with more than two dozen canonical proteins,32 and essentially all of these partners bind to one of two pockets on WDR5: the “WDR5-binding motif” (WBM) site, which contacts an acidic and hydrophobic residue-containing WBM motif on interacting partners, and the “WDR5-interacting” (WIN) site, which associates with arginine-containing WIN motifs on interacting partners. The canonical WIN motif is defined as [G/V]-[S/C/A]-A-R-[A/S/T], although histone H3 and PDPK1 has been reported to associate with the WIN site of WDR5 via their N-terminal sequences, ARTKQ and Ac-ARTTSQLYDAVP, respectively.33 To determine which pocket of WDR5 interacts with EMBOW, we mutated the WIN site and WBM site on WDR5 separately and immunopurified wild-type and mutated WDR5 from cells co-expressing EMBOW. As shown in Figure 2B, although wild-type and WBM-site-deficient WDR5 interacted with EMBOW, the WIN-site-deficient WDR5 did not, indicating that the WIN site is required for EMBOW binding.
We then determined which regions of EMBOW are required for its interaction with WDR5. Because EMBOW does not contain a canonical WIN motif, we started by examining the WDR5-binding capability of a series of EMBOW truncation mutants transiently overexpressed in HEK293T cells. As shown in Figures 2C–2E, deletion of the middle region or C terminus of EMBOW maintained its interaction with WDR5, whereas deletion of the N terminus abolished the interaction. Smaller deletions within the EMBOW N-terminal region localized its WDR5 interaction motif to the first 10 amino acids. We then mutated the two arginine residues within this region (R2 and R6; Figure S1A) to alanine and assessed their interaction with WDR5. As shown in Figure 2F, whereas mutation of R6 to A maintained its interaction with WDR5, mutation of R2 to A abolished the interaction, indicating the second arginine residue of EMBOW is required for its interaction with WDR5. The N terminus of EMBOW, MRTEPRPPAP, may therefore engage the WIN site of WDR5 via a similar binding mode as the N termini of PDPK1 and histone H3, which also exhibit a threonine in the third position.
Next, we wanted to determine whether mouse EMBOW interacts with WDR5. To this end, we transfected mouse EMBOW-GFP-FLAG into HEK293T cells, followed by FLAG IP and immunoblotting. As shown in Figure S1C, WDR5 co-purified with mouse EMBOW, but not with GFP-FLAG, and mutation of R2 to A on mouse EMBOW abolished the interaction. The EMBOW-WDR5 interaction therefore may be conserved in mouse.
To determine whether the interaction between EMBOW and WDR5 is direct, we purified EMBOW-FLAG and His6-WDR5 from E. coli, followed by in vitro FLAG pull-down. As shown in Figure 2G, His6-WDR5 co-purified with wild-type EMBOW, compared with FLAG beads only, and mutation of EMBOW residues R2 and R6 to A abolished the in vitro interaction, indicating that EMBOW directly interacts with WDR5 in a manner requiring residue R2.
To further confirm these results, we performed EMBOW-FLAG coIP in the presence of three different WDR5 WIN-site inhibitors,9,11,34 followed by immunoblotting. As shown in Figure 2H, all three inhibitors diminished the interaction of EMBOW with WDR5 in a dose-dependent manner, compared with DMSO vehicle, demonstrating that EMBOW interacts with WDR5 via the WIN site.
EMBOW regulates the WIN-site interactome of WDR5
To investigate the function of endogenous EMBOW, we generated EMBOW KO cells using CRISPR-Cas9. Loss of EMBOW expression was confirmed by genomic DNA PCR, followed by sequencing. The two alleles were disrupted by a 173-nt homozygous deletion, deleting the EMBOW, but not the SCRIB, start codon (Figure S2A). Consistent with specific EMBOW KO, WDR5-FLAG immunopurified endogenous EMBOW in wild-type, but not EMBOW KO, HEK293T cells (Figures S2B–S2D), and the protein level of SCRIB was not changed in EMBOW KO cells relative to wild-type cells (Figure S2E).
Given that WDR5 interacts with dozens of proteins via the WIN site, including EMBOW, we hypothesized that EMBOW binding may alter the WDR5 interactome. To test this possibility, we examined changes in WDR5-associated proteins when EMBOW is high via coIP from HEK293T cells stably expressing WDR5-FLAG and transfected with EMBOW-myc vs. empty vector. Conversely, we identified changes in WDR5-associated proteins when EMBOW is absent by conducting coIPs in parental and EMBOW KO HEK293T cells stably expressing WDR5-FLAG. We focused on proteins that were significantly differentially associated with WDR5 in opposing directions in these two experiments; proteins changing only in EMBOW overexpression or KO with statistical significance were not further considered. As shown in Figures 3A and 3B, overexpression of EMBOW increased the association of KIF2A and MRTO4 with WDR5, compared with empty vector; conversely, KIF2A and MRTO4 binding to WDR5 decreased in EMBOW KO cells, compared with HEK293T. Overexpression of EMBOW decreased the association of (1) KMT2A, the catalytic subunit of the KMT2A/MLL1 complex; (2) KANSL3 and MCRS1, two subunits of the NSL complex; and (3) other factors, including INO80 and SESN2, with WDR5. Conversely, interaction with proteins in groups 1–3 are increased upon EMBOW KO. Importantly, KIF2A and KMT2A directly interact with WDR5 via its WIN site,32 suggesting that EMBOW association with WDR5 specifically alters its WIN site, not WBM site, interactome. Consistent with this notion, the interaction between WDR5 and MYC, which binds to the WBM site,4 is not altered by EMBOW overexpression (Figure S3A). We note that several known WDR5 WIN-site interaction partners, including histone H3 and PDPK1, are not detected or are not significantly different in both of our proteomics datasets, but it cannot be concluded that EMBOW levels do not affect their interaction with WDR5. For example, it is possible that these proteins are not quantifiable with our shotgun proteomics protocol. Furthermore, exclusion of proteins changing in only one experiment (overexpression or KO) may have led to additional false negatives because of the enrichment and significance cutoffs applied. Nonetheless, our observations are consistent with perturbation of the WDR5 interactome by changing EMBOW levels.
Figure 3. EMBOW regulates the WIN-site interactome of WDR5.
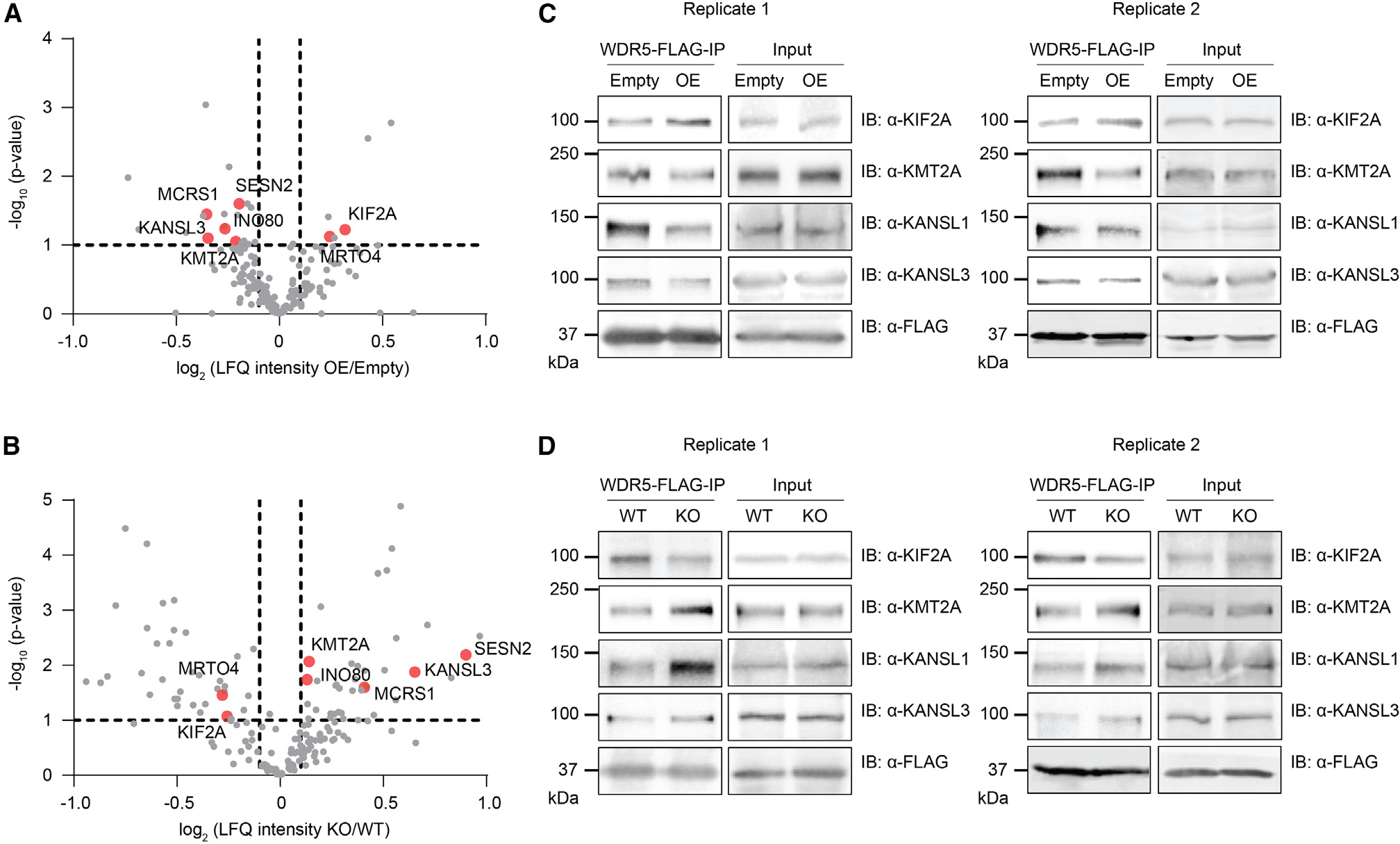
(A and B) HEK293T cells stably expressing WDR5-FLAG were transfected with EMBOW-myc (OE) or empty vector (Empty) (A), or WDR5-FLAG was stably expressed in parental (WT) or EMBOW knockout (KO) HEK293T cells (B), followed by FLAG-IP and quantitative proteomics with LFQ analysis (N = 3 biologically independent experiments). Two-sample p value was calculated using Perseus. WDR5-interacting proteins showing statistically significant (defined as −log10 (p value) ≥ 1) changes in opposite directions upon overexpression or KO of EMBOW are indicated in red, and protein names are labeled. The WDR5-interacting proteins are defined as those proteins enriched by WDR5 in HEK293T cells stably expressing WDR5-FLAG over parental HEK293T cells with log2 (fold change) ≥ 5 and −log10 (p value) ≥ 1. For complete quantitative proteomics results, see Tables S5, S6, and S7.
(C and D) WDR5-FLAG IP and immunoblotting using the four experimental samples described above. Cell lysates (4%) before IP (input) were used as loading controls. Shown are two biological replicates.
Previous reports have demonstrated that KIF2A, KMT2A, and KANSL1 are direct WDR5-interacting partners,32,33 and because our results suggest that EMBOW may regulate the distribution of WDR5 between these complexes, we chose these proteins for validation. We repeated the WDR5-FLAG coIP from cells overexpressing or lacking EMBOW and controls, followed by immunoblotting. Consistent with our proteomics results, overexpression of EMBOW increased the association of KIF2A with WDR5 and simultaneously decreased the interaction of KMT2A and KANSL1 with WDR5 (Figure 3C). Loss of EMBOW showed the opposite effect (Figure 3D). Overexpression of EMBOW also decreased the association of CXXC1, but not other KMT2 members and WRAD subunits, with WDR5 with statistical significance (Table S2). These results are consistent with partial disruption of the KMT2A complex by EMBOW overexpression and indicate that high levels of EMBOW are associated with decreased WDR5 binding to KMT2A and increased WDR5 binding to KIF2A.
We note that other WDR5-associated proteins regulated by EMBOW in our proteomics data, including SESN2, INO80, MRTO4, and MCRS1, may contribute to EMBOW-related phenotypes but require further validation.
Loss of EMBOW decreases WDR5 on the spindle pole during mitosis and shortens mitotic spindle length
WDR5 was previously reported to interact directly with KIF2A to promote proper spindle assembly during mitosis,3,35 and EMBOW regulates the interaction of WDR5 with KIF2A. We therefore wanted to determine whether EMBOW regulates WDR5 localization to the spindle. To this end, we performed WDR5 immunostaining in wild-type, EMBOW KO, rescue (KO stably expressing EMBOW), and R2A rescue (KO stably expressing R2A mutated EMBOW) metaphase HEK293T cells and calculated the ratio of WDR5 intensity associated with the spindle pole vs. spindle tubulin. As shown in Figure 4A, KO of EMBOW decreased WDR5 levels associated with the spindle pole, which can be rescued by reintroduction of wild-type EMBOW, but to a lesser extent by non-interacting R2A-mutant EMBOW (see above). However, EMBOW itself does not localize to the mitotic spindle (Figure S3B). EMBOW thus ensures correct WDR5 spindle pole localization by promoting its interaction with KIF2A.
Figure 4. Loss of EMBOW decreases WDR5 on the spindle pole, shortens spindle length, prolongs G2/M phase, and delays cell proliferation.
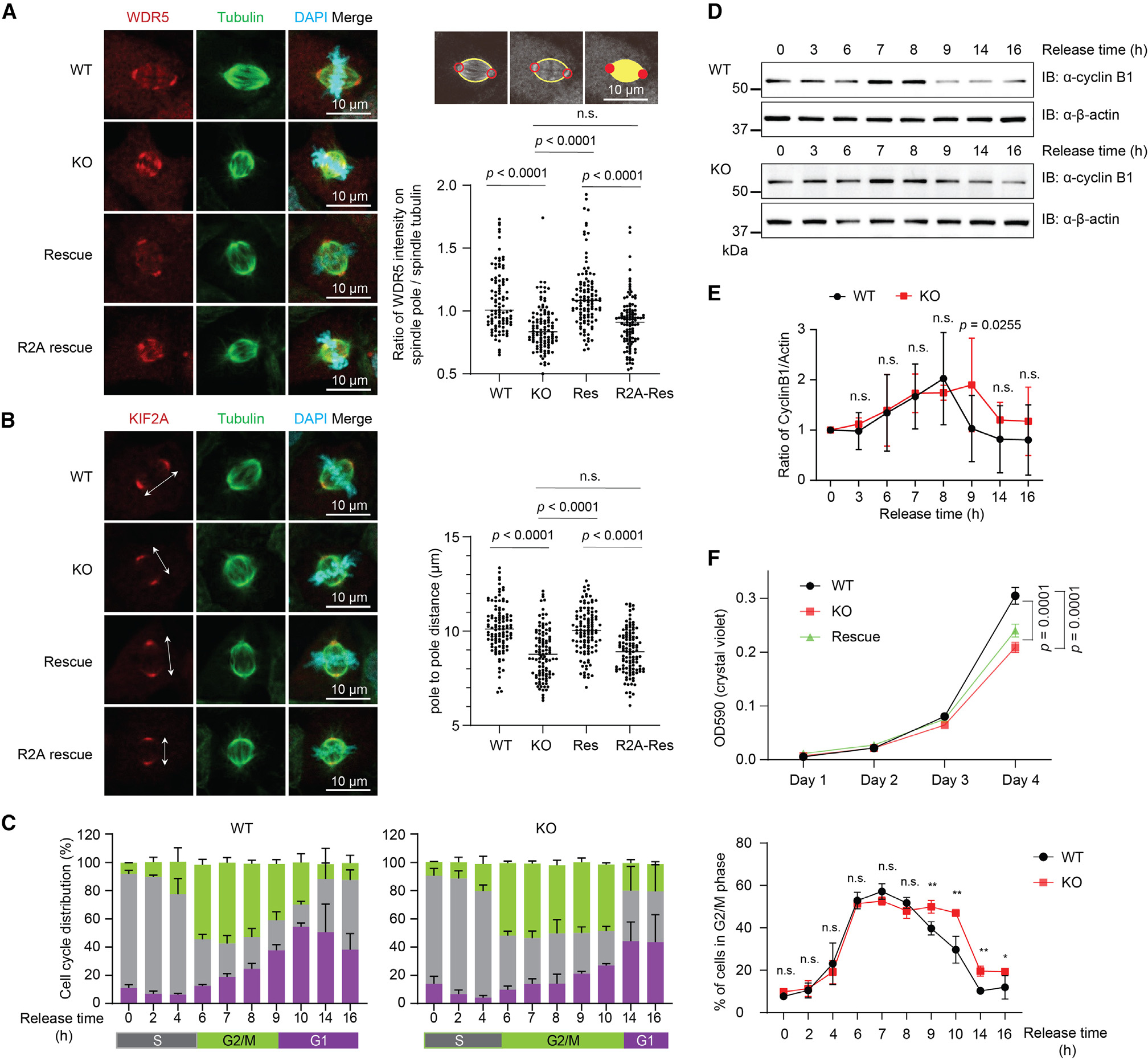
(A and B) Left: immunostaining of wild-type (WT), EMBOW KO, rescue with wild-type (rescue) or R2A-mutated (R2A rescue) EMBOW HEK293T cells with anti-WDR5 (A, red) or anti-KIF2A (B, red), anti-Tubulin (green), and DAPI (cyan). Data are representative of three biological replicates. Right: schematic representation of method to calculate the ratio of WDR5 signal colocalizing with the spindle pole and spindle tubulin are shown at the top, and quantitation of the ratio of WDR5 intensities on spindle pole and spindle tubulin in the four cell lines are shown at the bottom (A), or quantitation of the pole-to-pole distances of the four cell lines (B), totaling >100 cells for each measurement. Data represent mean values ± SEM, and significance (p value) was evaluated by one-way ANOVA (Dunnett’s test). Scale bars, 10 μm.
(C) Left: quantitation of the cell cycle of synchronized WT and EMBOW KO cells released from the G1/S boundary; release time points are indicated at the bottom. Right: percentage of WT (black) and EMBOW KO (red) cells in G2/M phase were plotted, release time points are indicated at the bottom. N = 3 biologically independent experiments. Data represent mean values ± SEM, and significance (p value) was evaluated by one-way ANOVA (Dunnett’s test). **p < 0.01. (D and E) Synchronized WT and EMBOW KO cells were released from the G1/S boundary and collected at the indicated time points, followed with immunoblotting
(D). Quantification of cyclin B1 protein changes are shown in (E). Data represent mean values ± SEM; N = 5 biologically independent experiments, and significance (p value) was evaluated by one-way ANOVA (Dunnett’s test).
(F) Growth curve of WT, EMBOW KO, and rescue cells at the indicated number of days (N = 4 biologically independent experiments). Data represent mean values ± SEM; significance (p value) was evaluated via two-way ANOVA (Dunnett’s test).
n.s., not significant; OD590, optical density at 590 nm. See also Figures S3 and S4.
To determine whether EMBOW loss affects spindle assembly, we quantified mitotic spindle length by measuring the interpolar distance via KIF2A immunostaining. As shown in Figure 4B, KO of EMBOW shortened the spindle length relative to wild-type cells, which can be partially rescued by reintroduction of wild-type, but not R2A-mutant, EMBOW. This effect is not due to off-target effects of Cas9 editing, because a cell line expressing Cas9 and a guide RNA targeting luciferase do not exhibit the same spindle shortening (Figure S4A). These results suggest that EMBOW is required for spindle assembly via its interaction with WDR5.
Loss of EMBOW prolongs G2/M phase and delays cell proliferation
Defective spindle assembly leads to chromosome misalignment, prolonged mitosis, and genomic instability.3,35,36 WDR5 has additionally been reported to regulate the cell cycle via its interaction with additional WIN-site-binding proteins such as PDPK1.33 We therefore hypothesized that EMBOW could be involved in the cell cycle and cell proliferation by controlling the interaction of WDR5 with KIF2A and/or other cell-cycle regulators. To test this hypothesis, we synchronized wild-type and EMBOW KO HEK293T cells to the G1/S boundary, followed by release and flow cytometry analysis at time points corresponding to specific cell-cycle phases. As shown in Figure 4C, EMBOW KO cells exhibited a prolonged G2/M phase and an increased S phase population compared with wild-type cells. To confirm the observation that G2/M phase is prolonged upon EMBOW KO, we immunoblotted the synchronized cells to determine cyclin B1 protein levels, a marker of G2/M phase.37 As shown in Figures 4D and 4E, cyclin B1 increased from the 6- to 7-h time point in both wild-type and EMBOW KO cells, and the cyclin B1 signal decreased at the 9-h time point in wild-type cells, but not until the 14-h time point in EMBOW KO cells, consistent with a prolonged G2/M phase in EMBOW KO cells. The prolonged G2/M phase was partially rescued by reintroduction of EMBOW (Figure S3C). Consistent with these results, loss of EMBOW decreased cell proliferation, which can be partially rescued by reintroduction of EMBOW (Figure 4F). Loss of EMBOW therefore prolongs G2/M phase and delays cell proliferation via its interaction with WDR5. We did not further investigate the apparent increase in S phase population in EMBOW KO cells, which will require future experimental validation.
Given prior reports that WDR5 regulates multiple processes related to the cell cycle, including spindle assembly, transcription, and signaling,2,3,33,38 identifying the mechanism by which EMBOW loss prolongs G2/M phase will require further study. We note that, because the MYC-WDR5 interaction is not altered by EMBOW (see above), it is unlikely that EMBOW-dependent cell-cycle alterations result from the WDR5-MYC interaction.
Loss of EMBOW increases WDR5 and H3K4me3 levels of de novo genes
WDR5 is a component of methyltransferase KMT2 complexes, which catalyze histone H3K4me2/3.1 EMBOW regulates the interaction of WDR5 with KMT2A, which is the catalytic subunit of the KMT2A complex. We therefore asked whether EMBOW controls the chromatin association of WDR5 and, consequently, H3K4me3 levels. To this end, we performed chromatin IP followed by deep sequencing (ChIP-seq) in wild-type, EMBOW KO, and stable EMBOW-overexpressing HEK293T cells. As shown in Figures 5A and 5B, WDR5 predominantly localizes to promoter regions, in line with prior reports,38 whereas EMBOW signal is low and is not significantly enriched in promoter regions, suggesting it is unlikely that EMBOW interacts directly or stably with chromatin.
Figure 5. Loss of EMBOW does not change WDR5 or H3K4me3 levels at WDR5 on-target genes.
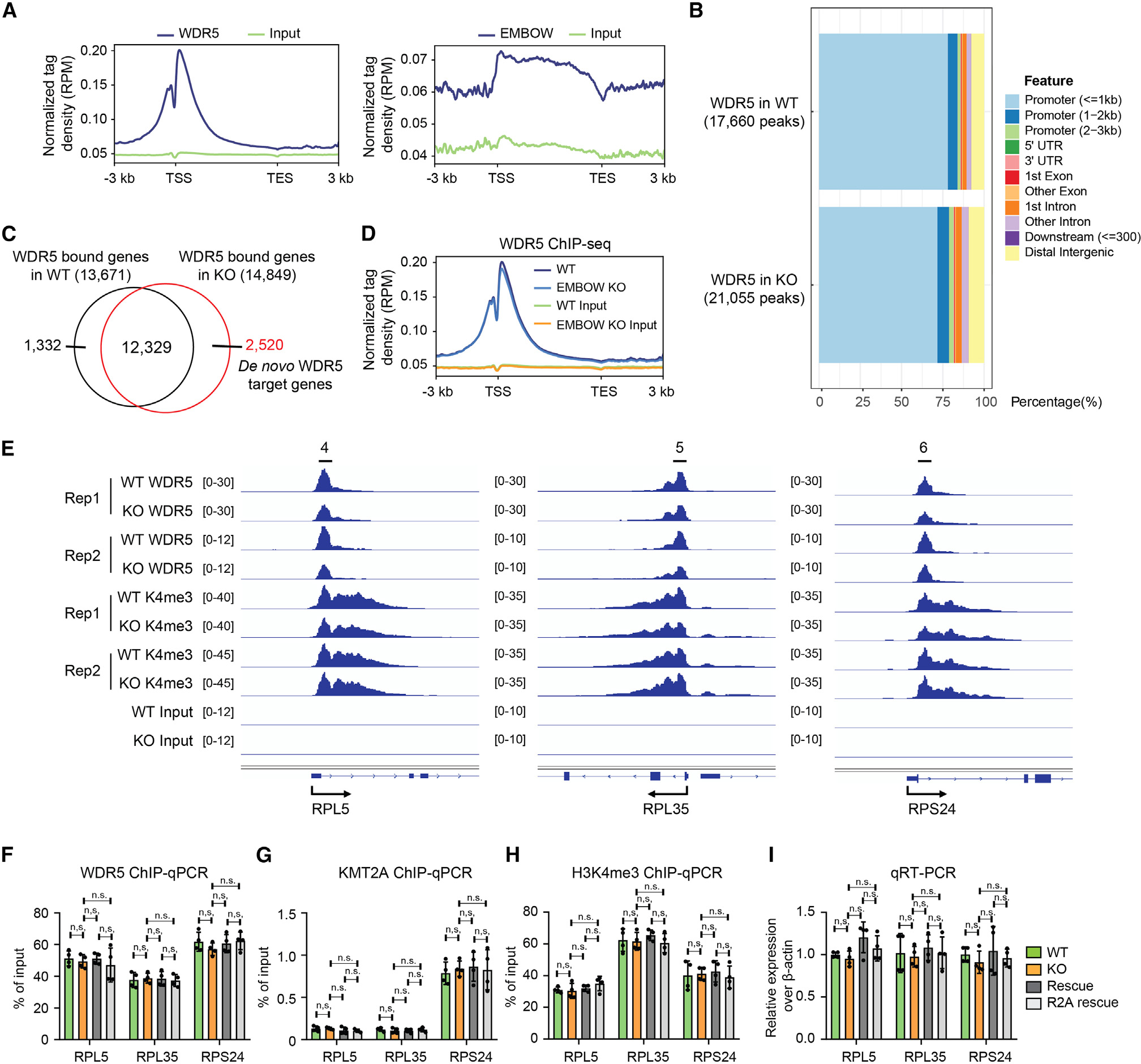
(A) Profile of WDR5 (left) and EMBOW (right) ChIP-seq signal at all UCSC (The University of California, Santa Cruz) annotated genes in HEK293T cells.
(B) The genomic distribution of WDR5 ChIP-seq peaks in wild-type (top) or EMBOW KO (bottom) cells.
(C) Venn diagram of WDR5-bound genes in WT or EMBOW KO cells. The WDR5-bound genes are defined as those genes with a WDR5 peak within ±1-kb distance of the gene’s transcription start site (TSS) with a 3-kb flanking distance.
(D) Profile of WDR5 binding on WDR5 on-target genes in WT and EMBOW KO cells.
(E–I) WDR5 binding and H3K4me3 levels at three WDR5 target ribosomal genes upon EMBOW KO, shown by ChIP-seq snapshots (E), and confirmed by ChIP-qPCR with an anti-WDR5 antibody (F), an anti-KMT2A antibody (G), or an anti-H3K4me3 antibody (H). Quantitative RT-PCR (qRT-PCR) results of the three ribosomal genes are shown in (I). Data represent mean values ± SEM; N = 4 biologically independent samples. Significance (p value) was evaluated with one-way ANOVA (Dunnett’s test).
n.s., not significant; Rep, replicate; RPM, reads per million reads.
In EMBOW KO cells, we detected 14,849 genes exhibiting WDR5 peaks, 2,520 of which we term “de novo WDR5 target genes,” which were off-target sites not occupied by WDR5 in wild-type cells (Figure 5C). We refer to genes occupied by WDR5 in wild-type cells as “on-target genes,” which, as a class, exhibited little to no changes in WDR5 occupancy upon EMBOW KO (Figure 5D). To validate the on-target gene ChIP-seq data, we performed conventional ChIP-qPCR at three ribosomal genes (Figure 5E), which were previously reported to be WDR5 targets.38 WDR5 binds to the promoter region of the three ribosomal genes in wild-type cells, and loss of EMBOW did not change WDR5 binding to these genes, consistent with WDR5 ChIP-seq data (Figure 5F). Loss of EMBOW did not change the levels of KMT2A or H3K4me3 at the promoters of these genes, nor the transcription of the three ribosomal genes (Figures 5G–5I). We speculate that WDR5 on-target genes are saturated by WDR5 in wild-type cells, and thus loss of EMBOW cannot further increase WDR5 binding to these sites.
We then examined de novo WDR5 target genes, which exhibited increased WDR5 occupancy in KO cells. As shown in Figure 6A, WDR5 signal was low in the promoter region of de novo WDR5 target genes in wild-type cells, and loss of EMBOW increased WDR5 binding to these regions. To determine whether EMBOW regulates H3K4me3 levels, we performed H3K4me3 ChIP-seq in wild-type and EMBOW KO cells. As shown in Figure 6B, loss of EMBOW increased H3K4me3 levels at the promoter region of de novo WDR5 target genes. To validate the ChIP-seq data, we performed conventional ChIP-qPCR at three representative de novo WDR5 target genes in wild-type, EMBOW KO, rescue, and R2A rescue HEK293T cells (Figure 6C). As shown in Figure 6D, loss of EMBOW increased WDR5 binding to the promoter region of the three de novo target genes, which can be partially rescued by reintroduction of wild-type EMBOW, and to a lesser extent by R2A-mutant EMBOW. Similar results were observed for KMT2A (Figure 6E). Loss of EMBOW also increased H3K4me3 levels at the promoter region of the three de novo target genes as expected, which can be partially rescued by reintroduction of wild-type, but not R2A-mutant EMBOW (Figure 6F). H3K4me3 has been reported to be a marker of transcriptionally active promoters.39 We therefore performed quantitative RT-PCR (qRT-PCR) to determine whether EMBOW regulates the transcription of the three de novo target genes. As shown in Figure 6G, loss of EMBOW increased the transcription of the three de novo target genes, which can be partially rescued by reintroduction of wild-type, but not R2A-mutant, EMBOW. This result is not due to off-target Cas9 activity, because a cell line expressing Cas9 and a guide RNA targeting luciferase exhibits no change in off-target gene H3K4me3 levels or expression (Figures S4B and S4C). We speculate that the “de novo” genes are not substantially bound by WDR5 or transcribed in normal cells, but that loss of EMBOW enables aberrant WDR5 binding and activation of these promoters. Taken together, our results could be consistent with a model in which transient association of endogenous EMBOW with WDR5 limits its binding to KMT2 complexes in the nucleus, ensuring that the activating complex binds to and methylates only high-affinity, on-target genes and preventing superstoichiometric KMT2 complex formation and association with off-target genes.
Figure 6. Loss of EMBOW increases WDR5 and H3K4me3 levels of de novo genes.
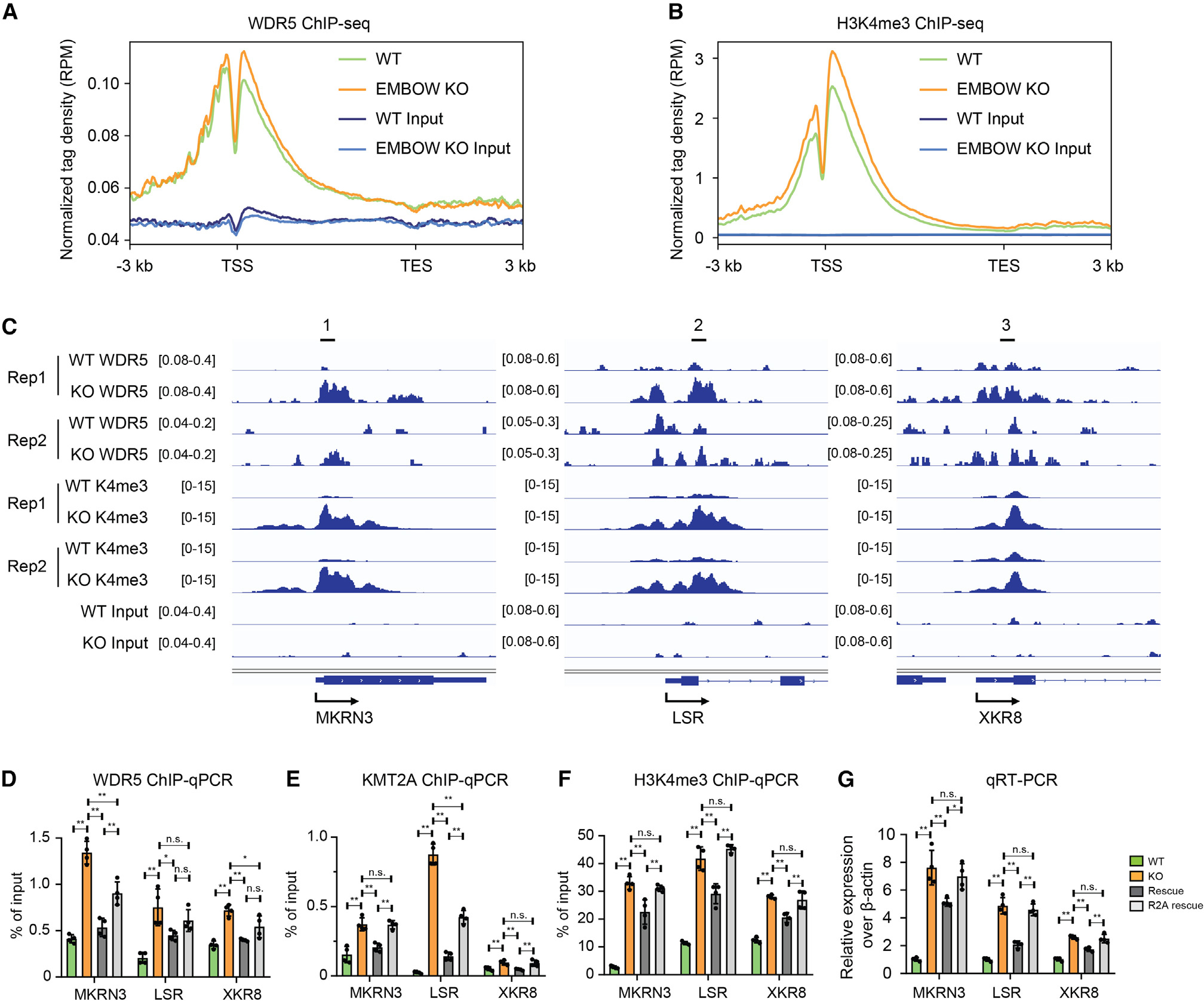
(A and B) Profile of WDR5 binding (A) or histone modification of H3K4me3 (B) on de novo WDR5 target genes in WT and EMBOW KO HEK293T cells.
(C–G) WDR5 binding and H3K4me3 levels at three de novo WDR5 target genes upon EMBOW KO shown by ChIP-seq snapshots (C) and confirmed by ChIP-qPCR with an anti-WDR5 antibody (D), an anti-KMT2A antibody (E), or an anti-H3K4me3 antibody (F). qRT-PCR results of the three target genes are shown in (G). Data represent mean values ± SEM; N = 4 biologically independent samples. Significance (p value) was evaluated with one-way ANOVA (Dunnett’s test).
**p < 0.01, *p < 0.05. Rep, replicate. See also Figure S4.
EMBOW expression is cell cycle regulated
Having demonstrated that EMBOW is involved in WDR5 functions in spindle assembly and regulation of transcription, which occur in different phases of the cell cycle,40,41 we asked whether EMBOW is itself cell cycle regulated. We synchronized EMBOW-FLAG-HA KI HEK293T cells to G2/M phase or the G1/S boundary, then collected the cells at time points corresponding to specific cell-cycle phases, followed by FLAG-IP and immunoblotting. As shown in Figure S5A, EMBOW levels increased at late G1, then decreased at the G1/S boundary; subsequently, EMBOW levels recovered to a lower level during G2/M. Similar results were observed for HEK293T cells stably expressing EMBOW (Figure S5B). SCRIB did not change during the cell cycle (Figure S5C), suggesting that the expression of these two proteins is decoupled, despite their synthesis from the same mRNA. WDR5 and KIF2A also did not change during the cell cycle, whereas the protein level of KMT2A increased at the G1/S boundary, consistent with a published report42 (Figure S5C). These results indicate that EMBOW expression is cell cycle regulated, with a global maximum at late G1 phase and a smaller increase at G2/M.
DISCUSSION
In this work, we have demonstrated that human SCRIB gene dually encodes cell-cycle-regulated EMBOW, which binds to the WIN site of WDR5 to regulate its distribution between interaction partners, ensuring correct targeting of WDR5 at different stages of the cell cycle and preventing mislocalization at gene promoters or the mitotic spindle. Furthermore, WIN-site inhibitors dissociate the WDR5-EMBOW complex, and it will be of interest in the future to test whether this contributes to the anti-proliferative mechanism of these molecules.
EMBOW was previously termed oSCRIB, and the uORF that encodes it was shown to downregulate the translation of SCRIB.26 Herein, we demonstrate that EMBOW also acts on WDR5 in trans. Multiple recent papers support the idea that two proteins coordinately expressed from bicistronic/dual coding human genes (e.g., an uORF microprotein and the downstream canonical protein) can function in related pathways, analogous to co-encoded cistrons within bacterial operons.18,43 We therefore hypothesize that the two proteins that are dually encoded in the SCRIB locus, EMBOW and SCRIB, might have related functions. SCRIB is a context-dependent promoter or inhibitor of tumor progression44,45 that regulates cell polarization signaling pathways by binding to PDZ ligands.24,46 It is interesting that WDR5 has also been reported to function in cell polarity.47,48 It is therefore possible that EMBOW also controls cell polarization via its interaction with WDR5. Alternatively, it is more broadly possible that EMBOW and SCRIB have similar effects on cell proliferation despite acting on different pathways.
The requirement of the EMBOW-WDR5 interaction for chromatin function and spindle assembly regulation and partial-to-complete rescue with EMBOW reintroduction, coupled with a lack of observed change in SCRIB protein levels in EMBOW KO cells, suggest that EMBOW is directly responsible, at least in part, for the observed effects on WDR5 interactions and function in this study. However, although we focused on the effect of EMBOW on WDR5’s association with well-characterized partners KMT2A and KIF2A, additional proteins such as KANSL1, MRTO4, INO80, and SESN2 also change in their association with WDR5 depending on EMBOW levels (Figures 3A and 3B). Future work will therefore be required to determine whether EMBOW regulates additional WDR5 activities associated with these factors. In addition, as noted above, it is possible that our quantitative proteomic analysis missed other WDR5 interaction partners that are affected by EMBOW; follow-up work will therefore be required to determine the full extent to which EMBOW affects the WDR5 interactome and whether additional WDR5 interaction partners are involved in chromatin, spindle, and cell-cycle phenotypes associated with EMBOW KO.
Our results suggest that one role of EMBOW is to promote correct localization of WDR5 at the mitotic spindle. With respect to spindle assembly, a prior study reported that WDR5 deletion resulted in elongation of the mitotic spindle. In this work, we find that EMBOW loss leads to a shortened mitotic spindle—a different outcome from WDR5 KO. We propose that this difference arises because WDR5 is still present but mislocalized in EMBOW KO cells. With analogy to examples in which loss-of-function mutations or pharmacological inhibitors do not phenocopy gene silencing/KO,49,50 it may be reasonable that the phenotypes associated with EMBOW vs. WDR5 KO are different, while supporting a role for the EMBOW-WDR5 interaction in spindle assembly.
With regard to EMBOW-mediated regulation of the WDR5-KMT2A interaction in chromatin, our data suggest that EMBOW KO increases WDR5 binding, methylation, and transcription of off-target genes that are not normally occupied by KMT2 complexes in wild-type cells, without altering activation of on-target genes previously reported to be bound by KMT2, such as ribosomal protein genes. On-target genes are highly occupied by WDR5 in both wild-type and EMBOW KO cells. One possible explanation for these observations is that the association of WDR5 with KMT2A is tuned by EMBOW in wild-type cells during G1 phase such that on-target genes are maximally occupied, but EMBOW dissociates excess KMT2 complexes to prevent binding to off-target genes, which speculatively could have lower affinity for KMT2. In EMBOW KO, we hypothesize that excess WDR5-KMT2A interactions occur, leading to deposition at off-target genes and aberrant activation; however, because on-target genes are already maximally bound by WDR5 complexes, EMBOW KO cannot change their activation state further.
The molecular mechanism by which EMBOW exerts its effect on the WDR5 interactome is currently unclear. Furthermore, given that KMT2A colocalizes with WDR5 both in chromatin and on the spindle pole,3 how can apparent global disruption of its association with WDR5 by EMBOW promote spindle assembly? One proposal that is consistent with our observations and prior studies relies on a transient association between EMBOW and WDR5 that, at high EMBOW concentrations, dissociates WDR5 from all of its WIN-site complexes. Because EMBOW does not remain stably bound to WDR5 (consistent with the absence of EMBOW signal from chromatin and the mitotic spindle), WDR5 would then be free to redistribute between all available interaction partners. The outcomes of this WDR5 redistribution would depend on which interaction partners are accessible during a given burst of EMBOW expression during the cell cycle. For example, during G2/M phase, when the nuclear membrane breaks down, transient EMBOW expression and binding could promote dynamic relocalization of WDR5 from preexisting chromatin-associated complexes to proteins associated with the mitotic spindle. This hypothesis is speculative, and many other mechanisms of action for EMBOW are possible, including the possibility that EMBOW-dependent changes in cell-cycle progression alter the apparent WDR5 interactome indirectly.
Overall, although the mechanism(s) by which EMBOW regulates WDR5 function and cell-cycle progression remains to be elucidated, it is clear that EMBOW is part of the WDR5 WIN-site interactome, and that its loss alters WDR5-protein interactions, localization, and function. It may therefore be important to consider EMBOW in future studies of WDR5 multifunctionality and WIN-site inhibition.
Limitations of the study
We reported putative conservation of EMBOW expression and function from mouse to human, but we do not have direct evidence for endogenous expression of EMBOW in any organism other than human. Furthermore, non-GFP-tagged mouse EMBOW fusions were not detectable, possibly suggesting that mouse EMBOW protein is unstable when overexpressed. Given that in-frame stop codons have been lost from mouse to human, resulting in elongation of EMBOW (Figure S1A), evidence of conservation should be interpreted with caution.
We utilized shotgun proteomics to analyze changes in the WDR5 interactome resulting from manipulation of EMBOW expression. This method is relatively insensitive to low-abundance, hydrophobic, or insoluble proteins,51 and thus some WDR5 interaction partners may have escaped detection. In addition, we considered only the subset of WDR5 binders that were oppositely regulated by changes in EMBOW levels in our data for functional validation, further focusing on KMT2A and KIF2A. It is therefore likely that EMBOW could regulate additional WDR5 interactions beyond those examined in this study.
Finally, additional studies will be required to support our hypothesis that EMBOW redistributes WDR5 between its interaction partners. Our hypothesis relies on a low-affinity interaction between EMBOW and WDR5, as well as transiently high EMBOW expression levels sufficient to displace higher-affinity WDR5-binding partners in cells. Direct quantitation of these biophysical parameters would be required to test this model. In addition, quantitation of the distribution of WDR5 between complexes throughout the cell cycle using size exclusion chromatography in EMBOW-overexpressing or KO cells could provide additional support. Furthermore, although we demonstrated that EMBOW KO increases H3K4me3 levels at some loci, we do not demonstrate that EMBOW inhibits KMT2 complex activity. Further studies, including in vitro enzymatic assays, will be needed to address this point.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sarah A. Slavoff (sarah.slavoff@yale.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Proteomics data have been deposited under accession PRIDE: PXD036699 to the PRIDE repository and are publicly available as of the date of publication. ChIP-seq data were deposited in the NCBI under GEO: GSE213209 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
HEK 293T and 3T3 cells were purchased from ATCC, and cultured in DMEM (Corning, 10-013-CV) with 10% FBS (Sigma, F0392) and 1% penicillin/streptomycin (VWR, 97063-708) in a 5% CO2 atmosphere at 37°C.
METHOD DETAILS
Cloning and genetic constructs
The 5′UTR of human SCRIB transcript variant 1 (NM_182706.5) through the stop codon of EMBOW was synthesized by GenScript with an FLAG epitope tag appended to the 3′ end of EMBOW coding sequence and was then subcloned into pcDNA3.1 for transient transfection or pLJM1 for stable cell line generation. The WDR5 cDNA clone was purchased from Addgene (a gift from Debu Chakravarti, Northwestern University, USA), and subcloned into pcDNA3.1 for transient transfection or pLJM1 for stable cell line generation. The mutations (R2A, R6A, and R2R6-AA) of EMBOW and WIN site mutation (F133A) or WBM site mutation (L240K)33 of WDR5 with epitope tag(s) were generated by ligating the PCR products into pcDNA3.1 vector.
Cell culture, lentivirus production and stable cell line generation
HEK 293T and 3T3 cells were purchased from ATCC, and for this study were grown from early-passage stocks. Cell culture, lentivirus production and stable cell line generation were performed exactly as previously described.58 Briefly, to produce lentiviruses, HEK 293T cells were cotransfected using polyethyleneimine (Polysciences, 23966) with expression construct in pLJM1 along with pMD2.G and psPAX2, and growth medium was replaced after 7–8 h. 48 h after transfection, medium containing viruses was collected and filtered through a 0.45-μm filter, and infection was performed by mixing with two volumes of fresh medium containing suspended HEK 293T cells. Twenty-four hours after infection, the growth medium was replaced. Forty-eight hours after infection, stable cells were selected with 4 μg/mL puromycin for 2 days. Early stocks of stable cell lines were established after selection. Stable cell lines were released from puromycin for 2 days before use in experiments.
Immunofluorescence
HEK 293T or 3T3 cells were plated on glass coverslips pre-treated with fibronectin (Millipore, 341635) following manufactures’ guidelines. For non-synchronized cells, cells were cultured overnight to 70–80% confluency in 12-well plate, fixed in 10% formalin for 15 min at room temperature (RT), washed with PBS once, then permeabilized with PBS containing 0.2% (v/v) Triton X-100. For spindle immunostaining, the cells were synchronized to G1/S boundary using double thymidine blocking, then released to G2/M phase, and fixed in 10% formalin for 15 min at RT, washed with PBS once, then permeabilized with prechilled methanol at −20°C for 20 min. The permeabilized cells were incubated with primary antibodies overnight at 4°C. After washing twice with PBS, the cells were incubated with secondary antibodies and DAPI for 1 h at RT, washed with PBS and mounted with Mowiol (Sigma, 81381) before viewing.
Confocal imaging was performed on a Leica SP8 LS confocal microscope (Leica Application Suite X, version 3.5.2.18963). The spindle lengths were measured as the pole-to-pole distances of anti-KIF2A immunofluorescence signal using ImageJ (version ImageJ2). For quantification of WDR5 intensity, the spindle tubulin region was defined by anti-Tubulin immunofluorescence signal, and the spindle pole region was defined by circles at the spindle pole, as shown in Figure 4A. The mean intensity ratios of WDR5 on spindle pole and spindle tubulin were measured using ImageJ (version ImageJ2).
Immunoprecipitation and proteomics
For identification of EMBOW interacting partners, parental HEK 293T cells or HEK 293T cells transiently transfected with EMBOW-FH using polyethyleneimine were grown to 80–90% confluency (~48 h post-transfection) in 15-cm dishes. Cells were collected and suspended in 1 mL of nuclear isolation buffer (10 mM HEPES-KOH pH 7.4, 100 mM KCl and 5 mM MgCl2 with 0.5% NP40 and Roche Complete protease inhibitor cocktail tablets (Roche, 11873580001)), and incubated on ice for 10 min, followed by centrifugation at 3,000 g, 4°C, 3 min. The nuclear pellets were resuspended in 1 mL lysis buffer (Tris-buffered saline (TBS) with 1% Triton X-100 and Roche Complete protease inhibitor cocktail), followed with sonication and immunoprecipitation as previously described.59 For WDR5-FLAG co-IP proteomics, HEK 293T cells stably expressing WDR5-FLAG were transfected with pcDNA3.1 empty vector or EMBOW-myc, or WDR5-FLAG was stably expressed in wild-type or EMBOW KO HEK 293T cells. Cells were growth to 80–90% confluency in 15-cm dishes, collected and suspended in 1 mL lysis buffer, followed with sonication and immunoprecipitation as described above. After the final wash, elution was in 40 μL of 3 × FLAG peptide (Sigma, F4799), at a final concentration of 100 μg/mL in lysis buffer at 4°C for 1 h. The eluted proteins were subjected to SDS-PAGE separation prior to LC-MS/MS analysis as previously described.58
Briefly, gel slices containing entire lanes were digested with trypsin at 37°C for 14–16 h. The resulting peptide mixtures were extracted from the gel, dried, subjected to ethyl acetate extraction to remove residual detergent, de-salted with peptide cleanup C18 spin column (Agilent Technologies, 5188-2750), then resuspended in 35 μL 0.1% formic acid (FA), followed by centrifugation at 21,130 g, 4°C, 30 min. A 5 μL aliquot of each sample was injected onto a pre-packed column attached to a nanoAcquity UPLC (Waters) in-line with a Thermo Scientific Q Exactive Plus Hybrid QuadrupoleOrbitrap mass spectrometer (Thermo Scientific) and a 130-min gradient was used to further separate the peptide mixtures as follows (solvent A: 0.1% FA; solvent B: acetonitrile (ACN) with 0.1% FA): Isocratic flow was maintained at 0.1 μL/min at 1% B for 40 min, followed by linear gradients from 1% B to 6% B over 2 min, 6% B to 24% B over 48 min, 24% B to 48% B over 5 min, 48% B to 80% B over 5 min. Isocratic flow at 80% B was maintained for 5 min, followed by a gradient from 80% B to 1% B over 5 min, and isocratic flow at 1% B was maintained for 10 min. The full MS was collected over the mass range of 300–1,700 m/z with a resolution of 70,000 and the automatic gain control (AGC) target was set as 3 × 106. MS/MS data was collected using a top 10 high-collisional energy dissociation method in data-dependent mode with a normalized collision energy of 27.0 eV and a 1.6 m/z isolation window. MS/MS resolution was 17,500 and dynamic exclusion was 90 s.
For identification of alt- and microproteins, ProteoWizard MS Convert was used for peak picking and files were analyzed using Mascot Daemon (version 2.5.0.1). Oxidation of methionine and N-terminal acetylation were set as variable modifications, and a previously reported three-frame translation of assembled transcripts from HEK 293T mRNA-seq was used as the database exactly as previously described.15 For co-IP proteomics searches and quantitative analysis, files were analyzed using MaxQuant, oxidation of methionine and N-terminal acetylation were set as variable modifications, and human UniProt plus EMBOW was used as the database for searching. For all analysis, a mass deviation of 20 p.p.m. was set for MS1 peaks, and 0.02 Da was set as maximum allowed MS/MS peaks with a maximum of two missed cleavages. Maximum false discovery rates (FDR) were set to 1% both on peptide and protein levels. Minimum required peptide length was five amino acids. Protein quantitation was accomplished by calculating the LFQ intensity ratio of EMBOW-FLAG or WDR5-FLAG pulldown to corresponding negative control samples using MaxQuant (version 1.6.8.0) with standard parameters. p values (two-sample t test) were calculated using Perseus (version 1.5.8.5) with standard parameters.
Generation of EMBOW knock-out (KO) and knock-in (KI) cell lines
EMBOW KO and FLAG-HA KI HEK 293T cells were generated using CRISPR-Cas9. Guide RNAs (gRNAs) were designed with the guide design tools from https://portals.broadinstitute.org/gppx/crispick/public and http://crispor.tefor.net/ to target the SCRIB genomic region (gRNA1: 5′-CAGTCCGAGCGTTCCGAGCG-3’; gRNA2: 5′-GCGGACTGAGCCCCGCCCCC-3’ for KO, and gRNA 5′-TGAGCGACAACGAGATCCAG-3′ for KI). Double-stranded DNA oligonucleotides corresponding to the gRNAs were inserted into pSpCas9(BB)-2A-GFP vector (Addgene, as a gift from F. Zhang, MIT).
For generation of KO cells, an equal mixture of the two gRNA plasmids was transfected into HEK 293T cells using polyethyleneimine, and GFP-positive cells were sorted with flow cytometry. Loss of EMBOW expression was confirmed by genomic DNA PCR, followed with sequencing, and quantitative proteomics, as shown in Figure S2. In the EMBOW KO cell line used in this study, the two alleles were disrupted by a 173-nt homozygous deletion, deleting the EMBOW start codon, but not the SCRIB start codon.
For generation of KI cells, a donor plasmid containing 300 bp homology left-arm and 300 bp homology right-arm sequence around the stop codon of EMBOW, which are separated with FLAG-HA tag and BamHI/NotI restriction sites, were synthesized by GenScript, and a DNA sequence containing the pGK promoter and blasticidin resistance gene was subcloned into the donor plasmid at the BamHI and NotI sites. An equal mixture of the gRNA and donor plasmids were transfected into HEK 293T cells using polyethyleneimine, and blascidin selection was performed 3 days post-transfection. EMBOW-FLAG-HA KI cells were confirmed by genomic DNA PCR and sequencing.
Cell cycle synchronization
For cell cycle synchronization using double thymidine (DOT scientific, DST18050), 5×105 HEK 293T cells were seeded in 6-well plates and treated with 2 mM thymidine for 16 h, followed with two PBS washes, and incubated with fresh medium for 8 h before the second 2 mM thymidine block for 14 h, following a previously published protocol.60 Cells were washed with PBS twice, then incubated with fresh medium to release from the G1/S boundary, then either fixed in 10% formalin for immunostaining, harvested for western blotting, or trypsinized and harvested for flow cytometry analysis (FACS) at specific time points after release.
For cell cycle synchronization using thymidine and nocodazole (Sigma, M1404), 5×105 HEK 293T cells were seeded in 6-well plates and treated with 2 mM thymidine for 18 h, followed by two PBS washes, and incubated with fresh medium for 3 h before the second 100 ng/mL nocodazole block for 13 h following a previously published protocol.61 Cells were washed with PBS twice, then incubated with fresh medium to release from G2/M phase, and fixed in 10% formalin for immunostaining or harvested for western blotting.
Flow cytometry analysis (FACS)
Synchronized cells were trypsinized and harvested, and after one PBS wash, cells were fixed in 70% cold ethanol for 30 min at 4°C or overnight at −20°C. After one PBS wash, the fixed cells were incubated with staining buffer (20 μg/mL RNase A (Thermo Fisher, EN0531) and 50 μg/mL Propidium Iodide (PI) in PBS (G Biosciences, 786–1273)) at 37°C for 30 min, followed by one PBS washing, and resuspended in 300 μL PBS. The cells were passed through a 40-μm Nylon Mesh (Fisherbrand, 22363547) before analysis with an S1000 Flow Cytometer from Stratedigm. At least 10,000 cells were counted and PI signal quantified using FlowJo software (version 10.8.1).
Crystal violet staining
4×104 HEK 293T cells were seeded in 12-well plates in triplicate, and fixed in 10% formalin for 15 min at RT every 24 h. After washing with ddH2O twice, cells were stained with 0.1% crystal violet in methanol for 30 min at RT in the dark, followed with three ddH2O washes and dried. The cells were then immersed in 1 mL 10% acetic acid with shaking for 20 min. Then 20 μL of the solution was combined with 80 μL ddH2O in a 96-well plate, and the optical density at 590 nm was monitored with Synergy HT.
ChIP-seq and data analysis
Wild-type, EMBOW KO, or rescue HEK 293T cells, or HEK 293T cells stably expressing EMBOW-FLAG-HA were grown to 80–90% confluency in 15-cm dishes. For H3K4me3 ChIP, the cells were washed twice with cold PBS, collected, aliquoted into five 1.5-mL tubes per 15-cm dish and flash frozen for later use. For FLAG, WDR5 and KMT2A ChIP, the cells were washed with 10 mL PBS once, then incubated with 12 mL 1% (v/v) formaldehyde (Sigma, 252549) in PBS for 10 min at RT for cross-linking, and quenched by adding 0.6 mL 2.5 M glycine in PBS at RT for 5 min. Cells were washed twice with cold PBS, collected, aliquoted into five 1.5-mL tubes per 15-cm dish and flash frozen for later use.
Collected cells from one 1.5-mL tube were resuspended in 150 μL buffer 1 (15 mM Tris-HCl pH 7.5, 60 mM KCl, 5 mM MgCl2, 0.1 mM EGTA, 0.3 M sucrose), then combined with 150 μL buffer 2 (15 mM Tris-HCl pH 7.5, 60 mM KCl, 5 mM MgCl2, 0.1 mM EGTA, 0.3 M sucrose, 0.5% NP-40 (v/v), 1% NaDOC (w/vol)) and incubated on ice for 15 min, then combined with 300 μL buffer 3 (85 mM Tris-HCl pH 7.5, 3 mM MgCl2, 2 mM CaCl2, 0.3 M sucrose), and incubated at 37°C for 5 min. For H3K4me3 ChIP, 0.5 μL MNase (NEB, M0247S) was added into one tube, and for FLAG, WDR5 and KMT2A ChIP, 1.8 μL MNase was added into one tube, and incubated at 37°C for 15 min, then quenched by adding 6 μL 0.5 M EDTA-KOH pH 7.5, and 6 μL 100x Roche Complete protease inhibitor cocktail tablets (Roche, 11873580001), followed by centrifugation at 13,523 g, 4°C, 10 min for H3K4me3 ChIP, or followed by sonication (30% intensity, 5 s pulse with 25 s rest, 3 cycles, MICROSON XL 2000) on ice and centrifugation at 13,523 g, 4°C, 10 min for FLAG, WDR5 and KMT2A ChIP.
The DNA content in each supernatant was measured and normalized to 0.17 μg/μL with ChIP buffer (50 mM Tris-HCl pH 7.5, 30 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 0.05 mM EGTA, 5 mM EDTA, 0.3 M sucrose, 0.125% NP-40 (v/v), 0.25% NaDOC (w/vol)). The digested lysates were immunoprecipitated with anti-H3K4me3 antibody (1:200, Abclonal, A2357), anti-WDR5 antibody (1:300, Invitrogen, 9H25L13) or anti-KMT2A antibody (1:200, Cell Signaling Technology, 14197) overnight at 4°C, followed by incubating with Dynabeads Protein A (15 μL slurry per ug antibody, Invitrogen, 10001D) for 1 h at 4°C; or with anti-FLAG beads (10 μL slurry per 300 μL lysate, Sigma, A2220) for 1 h at 4°C. The beads were washed once with 1 mL low salt buffer (20 mM Tris-HCl pH 8.1, 150 mM NaCl, 2 mM EDTA, 0.1% SDS (w/vol), 1% Triton X-100 (v/v)), 1 mL high salt buffer (20 mM Tris-HCl pH 8.1, 500 mM NaCl, 2 mM EDTA, 0.1% SDS (w/vol), 1% Triton X-100 (v/v)), 1 mL LiCl buffer (50 mM Tris-HCl pH 8.1, 1 mM EDTA, 0.25 M LiCl, 1% NP40 (v/v), 1% NaDOC (w/vol)), 1 mL ×2 TE buffer (10 mM Tris-HCl pH 8.1, 1 mM EDTA).
For H3K4me3 ChIP, proteins were eluted by adding 200 μL elution buffer 1 (1% SDS in TE buffer). For WDR5 and KMT2A ChIP, proteins were eluted by adding 200 μL elution buffer 2 (1% SDS, 0.1 M NaHCO3 in ddH2O). And for FLAG ChIP, elution was in 100 μL of 3x FLAG peptide (Sigma, F4799), at a final concentration of 100 μg/mL in low salt buffer at 4°C for 1 h, and the elution was combined with 100 μL 2x elution buffer 2 (2% SDS, 0.2 M NaHCO3 in ddH2O). Crosslink reversal was performed for WDR5, KMT2A and FLAG ChIP by adding 8 μL 5 M NaCl, and incubated at 65°C for 4 h or overnight. For all ChIP, RNAs were removed by adding 1 μL RNase A (Thermo Fisher, EN0531), and incubated at 37°C for 1 h. Proteins were digested by adding 2 μL proteinase K (NEB, P8107S), 8 μL 1 M Tris-HCl pH 6.5, 4 μL 0.5 M EDTA, and incubated at 55°C for 2 h or overnight. The DNAs were extracted by adding 1 mL PB buffer (5 M guanidium HCl, 30% isopropanol (v/v)), and passed through a spin column for DNA (Epoch Life Science, 1910–250) following standard protocols for DNA purification. Quantitative ChIP-PCR was performed on selected genes, and the primers are listed in Table S3.
Library preparation and deep sequencing were performed by Yale Center for Genomic Analysis on an Illumina NovaSeq 6000 with paired-end (100 bp) reads. All reads were mapped to the human genome (hg38) using Bowtie2 (version 2.4.5).62 The BigWig normalized signal files were generated using bamCoverage from deepTools (version 3.5.1)55 for IGV visualization. WDR5-enriched regions were predicted with MACS2 (version 2.2.7.1).56 To detect changes in WDR5 and H3K4me3 levels in the wild-type and EMBOW KO HEK 293T cells, we calculated normalized WDR5 and H3K4me3 ChIP-seq tag densities according to the EMBOW-dependent WDR5 de novo target genes or WDR5 on-target genes using computeMatrix from deepTools, and plotted the tag densities using plotProfile from deepTools.
Protein purification and In vitro pulldown assay
Full length EMBOW and its mutants with FLAG tag at their C-termini were subcloned into pGEX-6P-1. The fusion proteins were expressed in BL21 E.coli by overnight culture at 20°C in LB medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) supplemented with 0.2 mM IPTG, purified using Glutathione Sepharose 4B and proteolyzed from beads using PreScission Protease. Full length WDR5 was subcloned into PET28a. The fusion protein was expressed in BL21 E.coli, purified using Ni-NTA agarose beads and eluted in 150 mM imidazole. The purified proteins were snap frozen and stored at −80°C, or used directly in in vitro binding assays. Protein concentrations were measured using Coomassie assay. The direct interaction between EMBOW and WDR5 was detected by in vitro pulldowns, 1 μg EMBOW-FLAG and 3 μg WDR5 protein were mixed in binding buffer (25 mM Tris-HCl, pH 7.4; 500 mM NaCl with 0.05% NP-40) and incubated for 3 h at 4°C, then the FLAG agarose beads were added to capture EMBOW-FLAG for another 3 h at 4°C. The immunoprecipitated proteins were eluted by boiling and detected by western blotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values and error bars represent the mean ± SEM. Significance was evaluated by a two-tailed t test using Microsoft Excel (version 2013) or GraphPad Prism (version 8.0.0), and a one-way or two-way ANOVA (Dunnett’s test) using GraphPad Prism (version 8.0.0). p values (t test) for proteomics were calculated using Perseus (version 1.5.8.5) with standard parameters, and −log10 (p value) ≥ 1 was considered significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| DYKDDDDK Tag (D6W5B) Rabbit mAb | Cell Signaling Technology | Cat#14793; RRID:AB_2572291 |
| HA Tag Polyclonal Antibody (SG77) | Invitrogen | Cat#71-5500; RRID:AB_2533988 |
| beta Actin Loading Control Monoclonal Antibody (BA3R) | Invitrogen | Cat#MA5-15739; RRID:AB_2537660 |
| MYC Epitope Tag Antibody Rabbit Polyclonal | Rockland | Cat#600-401-381; RRID:AB_217927 |
| Cyclin B1 Antibody | Cell Signaling Technology | Cat#4138; RRID:AB_2072132 |
| WDR5 Recombinant Rabbit Monoclonal Antibody (9H25L13) | Invitrogen | Cat#703745; RRID:AB_2848236 |
| SCRIB Rabbit pAb | ABclonal | Cat#A17450; RRID:AB_2772164 |
| KIF2A Polyclonal Antibody | Proteintech | Cat#13105-1-AP; RRID:AB_2265336 |
| MLL1 (D6G8N) Rabbit mAb (Carboxyterminal Antigen) | Cell Signaling Technology | Cat#14197; RRID:AB_2688010 |
| KANSL1 Rabbit pAb | ABclonal | Cat#A10755; RRID:AB_2758198 |
| KANSL3 Rabbit pAb | ABclonal | Cat#A8234; RRID:AB_2770036 |
| Anti-Lamin B1 antibody | Abcam | Cat#Ab133741; RRID:AB_2616597 |
| alpha Tubulin Polyclonal Antibody | Proteintech | Cat#11224-1-AP; RRID:AB_2210206 |
| His-Tag Monoclonal Antibody (1B7G5) | Proteintech | Cat#66005-1-IG; RRID:AB_11232599 |
| Anti-Rabbit IgG (H&L) [Goat] Peroxidase conjugated | Rockland | Cat#611-1302; RRID:AB_219720 |
| Anti-MOUSE IgG (H&L) (Min X Human Serum Proteins) Affinity Purified, Peroxidase Conjugated | Rockland | Cat#610-1319-0500; RRID:AB_11182792 |
| Monoclonal Anti-FLAG | Sigma-Aldrich | Cat#F1804; RRID:AB_262044 |
| WDR5 Polyclonal Antibody | Bethyl Laboratories | Cat#A302-430A; RRID:AB_1944300 |
| alpha Tubulin Monoclonal Antibody (DM1A) | Invitrogen | Cat#62204; RRID:AB_1965960 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Invitrogen | Cat#A-11008; RRID:AB_143165 |
| Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | Cat#A-21235; RRID:AB_2535804 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | Cat#A-21244; RRID:AB_2535812 |
| Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Invitrogen | Cat#A-11001; RRID:AB_2534069 |
| TriMethyl-Histone H3-K4 Rabbit pAb | ABclonal | Cat#A2357; RRID:AB_2631278 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pLJM1-EGFP | Sancak et al.52 | Addgene plasmid #19319 |
| DH5a | Thermo Fisher Scientific | Cat# K4520-1 |
| BL21 | Thermo Fisher Scientific | Cat# C6070-03 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| OICR-9429 | Selleckchem | Cat#S7833 |
| WDR5-0103 | Selleckchem | Cat#S2184 |
| WDR5 WIN site inhibitor C6 | Aho et al.9 | N/A |
| Thymidine | DOT scientific | Cat#DST18050 |
|
| ||
| Deposited data | ||
|
| ||
| Label-free quantitative proteomics of EMBOW interatome | This study | Table S4; PRIDE: PXD036699 |
| Label-free quantitative proteomics of WDR5 interactome change upon EMBOW overexpression or knockout | This study | Tables S5, S6, and S7; PRIDE: PXD036699 |
| WDR5 and histone H3K4me3 ChIP-seq | This study | Figures 5, 6, and S4; GEO: GSE213209 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| EMBOW-KO HEK 293T cell line | This study | N/A |
| EMBOW-FLAG-HA-KI HEK 293T cell line | This study | N/A |
| HEK 293T cell line | ATCC | Cat#CRL-3216 |
| 3T3 cell line | ATCC | Cat#CRL-1658 |
|
| ||
| Oligonucleotides | ||
|
| ||
| gRNA targeting SCRIB genomic for EMBOW KO: gRNA1: CAGTCCGAGCGTTCCGAGCG; gRNA2: GCGGACTGAGCCCCGCCCCC |
This study | N/A |
| gRNA targeting SCRIB genomic for EMBOW-FLAG-HA KI: TGAGCGACAACGAGATCCAG |
This study | N/A |
| Primers for ChIP- and qRT-PCR | This study | See Table S3 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pcDNA3.1-EMBOW-FLAG-HA | This study | N/A |
| pcDNA3.1-mEMBOW-GFP-FLAG | This study | N/A |
| pcDNA3.1-EMBOW-myc | This study | N/A |
| pcDNA3.1-WDR5-FLAG-HA | This study | N/A |
| pLJM1-WDR5-FLAG | This study | N/A |
| pLJM1-EMBOW-FLAG-HA | This study | N/A |
| pGEX-6P-1-EMBOW-FLAG | This study | N/A |
| pET28a-WDR5 | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism (version 8.0.0) | Dotmatics | https://www.graphpad.com |
| ImageJ (version ImageJ2) | Rueden et al.53 | https://imagej.nih.gov/ij/ |
| Bowtie2 (version 2.4.5) | Langmead and Salzberg54 | http://bowtie-bio.sourceforge.net/index.shtml |
| deepTools (version 3.5.1) | Ramirez et al.55 | https://deeptools.readthedocs.io/en/latest/content/installation.html |
| MACS2 (version 2.2.7.1) | Zhang et al.56 | https://github.com/taoliu/MACS |
| Mascot Daemon (version 2.5.0.1) | Matrix Science | http://www.matrixscience.com/ |
| MaxQuant (version 1.6.8.0) | Tyanova et al.57 | https://www.maxquant.org/ |
| Leica Application Suite X (version 3.5.2.18963) | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| FlowJo software (version 10.8.1) | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/flowjo-v10-software |
Highlights.
The transcript leader of SCRIB encodes a WDR5-binding microprotein, EMBOW
EMBOW is expressed at specific points in the cell cycle
EMBOW binding alters the WDR5 interactome
EMBOW loss dysregulates WDR5 binding to off-target genes and the mitotic spindle
ACKNOWLEDGMENTS
We thank Jessica J. Mohsen for suggesting the name EMBOW. We thank the Yale West Campus Imaging Core for support and assistance in this work. This work was supported in part by a Leukemia Research Foundation Hollis Brownstein Research Grant, a Mark Foundation for Cancer Research Emerging Leader Award, a Paul G. Allen Frontiers Group Distinguished Investigator Award, the National Institutes of Health (NIH; R01GM122984), and start-up funds from Yale University West Campus (to S.A.S.). X.C. and Y.C. were supported by the Shanghai Pujiang Program (22PJ1402600 to X.C.). X.C. was supported in part by a Rudolph J. Anderson postdoctoral fellowship from Yale University. K.J. was supported in part by a NIH Chemical Biology Training Grant (T32 GM067543). R.H. and A.B. were supported by the Melanoma Research Alliance (project number 828544) and the Yale SPORE in Skin Cancer (project number 2P50CA121974-11A1). K.H.L. was supported in part by a NIH Pathway to Independence Award (4 R00DK129712-03). Z.W. and J.Z. were supported by the National Natural Science Foundation of China (82173043 to Z.W.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113145.
REFERENCES
- 1.Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 81, 65–95. 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY, and Rape M (2020). Gene expression and cell identity controlled by anaphase-promoting complex. Nature 579, 136–140. 10.1038/s41586-020-2034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali A, Veeranki SN, Chinchole A, and Tyagi S (2017). MLL/WDR5 Complex Regulates Kif2A Localization to Ensure Chromosome Congression and Proper Spindle Assembly during Mitosis. Dev. Cell 41, 605–622.e7. 10.1016/j.devcel.2017.05.023. [DOI] [PubMed] [Google Scholar]
- 4.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, et al. (2015). Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452. 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen T, Li K, Liu Z, Liu J, Wang Y, Sun R, Li Z, Qiu B, Zhang X, Ren G, et al. (2021). WDR5 facilitates EMT and metastasis of CCA by increasing HIF-1alpha accumulation in Myc-dependent and independent pathways. Mol. Ther. 29, 2134–2150. 10.1016/j.ymthe.2021.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ge Z, Song EJ, Kawasawa YI, Li J, Dovat S, and Song C (2016). WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 7, 37740–37754. 10.18632/oncotarget.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neilsen BK, Chakraborty B, McCall JL, Frodyma DE, Sleightholm RL, Fisher KW, and Lewis RE (2018). WDR5 supports colon cancer cells by promoting methylation of H3K4 and suppressing DNA damage. BMC Cancer 18, 673. 10.1186/s12885-018-4580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Zhou Q, Xie K, Cheng L, Peng S, Xie R, Liu L, Zhang Y, Dong W, Han J, et al. (2021). Targeting WD repeat domain 5 enhances chemosensitivity and inhibits proliferation and programmed death-ligand 1 expression in bladder cancer. J. Exp. Clin. Cancer Res. 40, 203. 10.1186/s13046-021-01989-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aho ER, Wang J, Gogliotti RD, Howard GC, Phan J, Acharya P, Macdonald JD, Cheng K, Lorey SL, Lu B, et al. (2019). Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 26, 2916–2928.e13. 10.1016/j.celrep.2019.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang D, Chen X, Chen X, Qu Y, Wang Y, Yang Y, and Cheng Y(2020). WDR5 Promotes Proliferation and Correlates with Poor Prognosis in Oesophageal Squamous Cell Carcinoma. OncoTargets Ther. 13, 10525–10534. 10.2147/OTT.S234773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, Skucha A, Vittori S, Kuznetsova E, Smil D, et al. (2015). Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat. Chem. Biol. 11, 571–578. 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Xu J, Wang X, Long G, You Q, and Guo X (2021). Targeting WD Repeat-Containing Protein 5 (WDR5): A Medicinal Chemistry Perspective. J. Med. Chem. 64, 10537–10556. 10.1021/acs.jmedchem.1c00037. [DOI] [PubMed] [Google Scholar]
- 13.Siladi AJ, Wang J, Florian AC, Thomas LR, Creighton JH, Matlock BK, Flaherty DK, Lorey SL, Howard GC, Fesik SW, et al. (2022). WIN site inhibition disrupts a subset of WDR5 function. Sci. Rep. 12, 1848. 10.1038/s41598-022-05947-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orr MW, Mao Y, Storz G, and Qian SB (2020). Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res. 48, 1029–1042. 10.1093/nar/gkz734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, and Saghatelian A (2013). Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 9, 59–64. 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunet MA, Leblanc S, and Roucou X (2020). Reconsidering proteomic diversity with functional investigation of small ORFs and alternative ORFs. Exp. Cell Res. 393, 112057. 10.1016/j.yexcr.2020.112057. [DOI] [PubMed] [Google Scholar]
- 17.Cao X, and Slavoff SA (2020). Non-AUG start codons: Expanding and regulating the small and alternative ORFeome. Exp. Cell Res. 391, 111973. 10.1016/j.yexcr.2020.111973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Brunner AD, Cogan JZ, Nuñez JK, Fields AP, Adamson B, Itzhak DN, Li JY, Mann M, Leonetti MD, and Weissman JS (2020). Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146. 10.1126/science.aay0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prensner JR, Enache OM, Luria V, Krug K, Clauser KR, Dempster JM, Karger A, Wang L, Stumbraite K, Wang VM, et al. (2021). Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat. Biotechnol. 39, 697–704. 10.1038/s41587-020-00806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calvo SE, Pagliarini DJ, and Mootha VK (2009). Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA. 106, 7507–7512. 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang N, Li F, Zhang M, Zhou H, Chen Z, Ma X, Yang L, Wu X, Zhong J, Xiao F, et al. (2021). An Upstream Open Reading Frame in Phosphatase and Tensin Homolog Encodes a Circuit Breaker of Lactate Metabolism. Cell Metab. 33, 128–144.e9. 10.1016/j.cmet.2020.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Na Z, Dai X, Zheng SJ, Bryant CJ, Loh KH, Su H, Luo Y, Buhagiar AF, Cao X, Baserga SJ, et al. (2022). Mapping subcellular localizations of unannotated microproteins and alternative proteins with MicroID. Mol. Cell 82, 2900–2911.e7. 10.1016/j.molcel.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mise S, Matsumoto A, Shimada K, Hosaka T, Takahashi M, Ichihara K, Shimizu H, Shiraishi C, Saito D, Suyama M, et al. (2022). Kastor and Polluks polypeptides encoded by a single gene locus cooperatively regulate VDAC and spermatogenesis. Nat. Commun. 13, 1071. 10.1038/s41467-022-28677-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonello TT, and Peifer M (2019). Scribble: A master scaffold in polarity, adhesion, synaptogenesis, and proliferation. J. Cell Biol. 218, 742–756. 10.1083/jcb.201810103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao X, Khitun A, Na Z, Dumitrescu DG, Kubica M, Olatunji E, and Slavoff SA (2020). Comparative Proteomic Profiling of Unannotated Microproteins and Alternative Proteins in Human Cell Lines. J. Proteome Res. 19, 3418–3426. 10.1021/acs.jproteome.0c00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nomura Y, and Dohmae N (2021). Discovery of a small protein-encoding cis-regulatory overlapping gene of the tumor suppressor gene Scribble in humans. Commun. Biol. 4, 1098. 10.1038/s42003-021-02619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014. 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khitun A, and Slavoff SA (2019). Proteomic Detection and Validation of Translated Small Open Reading Frames. Curr. Protoc. Chem. Biol. 11, e77. 10.1002/cpch.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saghatelian A, and Couso JP (2015). Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 11, 909–916. 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M(2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guarnaccia AD, and Tansey WP (2018). Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med. 7, 21. 10.3390/jcm7020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guarnaccia AD, Rose KL, Wang J, Zhao B, Popay TM, Wang CE, Guerrazzi K, Hill S, Woodley CM, Hansen TJ, et al. (2021). Impact of WIN site inhibitor on the WDR5 interactome. Cell Rep. 34, 108636. 10.1016/j.celrep.2020.108636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, et al. (2013). Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 449, 151–159. 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi ZY, Ma XS, Liang QX, Zhang T, Xu ZY, Meng TG, Ouyang YC, Hou Y, Schatten H, Sun QY, and Quan S (2016). Kif2a regulates spindle organization and cell cycle progression in meiotic oocytes. Sci. Rep. 6, 38574. 10.1038/srep38574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bufe A, García Del Arco A, Hennecke M, de Jaime-Soguero A, Oster-maier M, Lin YC, Ciprianidis A, Hattemer J, Engel U, Beli P, et al. (2021). Wnt signaling recruits KIF2A to the spindle to ensure chromosome congression and alignment during mitosis. Proc. Natl. Acad. Sci. USA. 118, e2108145118. 10.1073/pnas.2108145118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong D, and Ferrell JE Jr. (2010). The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 21, 3149–3161. 10.1091/mbc.E10-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryan AF, Wang J, Howard GC, Guarnaccia AD, Woodley CM, Aho ER, Rellinger EJ, Matlock BK, Flaherty DK, Lorey SL, et al. (2020). WDR5 is a conserved regulator of protein synthesis gene expression. Nucleic Acids Res. 48, 2924–2941. 10.1093/nar/gkaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, and Roeder RG (2013). H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021–1036. 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bähler J (2005). Cell-cycle control of gene expression in budding and fission yeast. Annu. Rev. Genet. 39, 69–94. 10.1146/annurev.genet.39.110304.095808. [DOI] [PubMed] [Google Scholar]
- 41.Fischer M, Schade AE, Branigan TB, Müller GA, and DeCaprio JA (2022). Coordinating gene expression during the cell cycle. Trends Biochem. Sci. 47, 1009–1022. 10.1016/j.tibs.2022.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Cheng EHY, and Hsieh JJD (2007). Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 21, 2385–2398. 10.1101/gad.1574507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunet MA, Jacques JF, Nassari S, Tyzack GE, McGoldrick P, Zinman L, Jean S, Robertson J, Patani R, and Roucou X (2021). The FUS gene is dual-coding with both proteins contributing to FUS-mediated toxicity. EMBO Rep. 22, e50640. 10.15252/embr.202050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen H, Huang C, Wu J, Li J, Hu T, Wang Z, Zhang H, Shao Y, and Fu Z (2021). SCRIB Promotes Proliferation and Metastasis by Targeting Hippo/YAP Signalling in Colorectal Cancer. Front. Cell Dev. Biol. 9, 656359. 10.3389/fcell.2021.656359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kapil S, Sharma BK, Patil M, Elattar S, Yuan J, Hou SX, Kolhe R, and Satyanarayana A (2017). The cell polarity protein Scrib functions as a tumor suppressor in liver cancer. Oncotarget 8, 26515–26531. 10.18632/oncotarget.15713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephens R, Lim K, Portela M, Kvansakul M, Humbert PO, and Richardson HE (2018). The Scribble Cell Polarity Module in the Regulation of Cell Signaling in Tissue Development and Tumorigenesis. J. Mol. Biol. 430, 3585–3612. 10.1016/j.jmb.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 47.Wang P, Dreger M, Madrazo E, Williams CJ, Samaniego R, Hodson NW, Monroy F, Baena E, Sánchez-Mateos P, Hurlstone A, and Redondo-Muñoz J (2018). WDR5 modulates cell motility and morphology and controls nuclear changes induced by a 3D environment. Proc. Natl. Acad. Sci. USA. 115, 8581–8586. 10.1073/pnas.1719405115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni SS, Griffin JN, Date PP, Liem KF Jr., and Khokha MK(2018). WDR5 Stabilizes Actin Architecture to Promote Multiciliated Cell Formation. Dev. Cell 46, 595–610.e3. 10.1016/j.devcel.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss WA, Taylor SS, and Shokat KM (2007). Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 3, 739–744. 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson AS, Fong LG, and Young SG (2008). PCSK9 function and physiology. J. Lipid Res. 49, 1595–1599. 10.1194/jlr.cx00001-jlr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan P, D’Lima NG, and Slavoff SA (2018). Comparative Membrane Proteomics Reveals a Nonannotated E. coli Heat Shock Protein. Biochemistry 57, 56–60. 10.1021/acs.biochem.7b00864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 58.Cao X, Khitun A, Harold CM, Bryant CJ, Zheng SJ, Baserga SJ, and Slavoff SA (2022). Nascent alt-protein chemoproteomics reveals a pre-60S assembly checkpoint inhibitor. Nat. Chem. Biol. 18, 643–651. 10.1038/s41589-022-01003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao X, Khitun A, Luo Y, Na Z, Phoodokmai T, Sappakhaw K, Olatunji E, Uttamapinant C, and Slavoff SA (2021). Alt-RPL36 downregulates the PI3K-AKT-mTOR signaling pathway by interacting with TMEM24. Nat. Commun. 12, 508. 10.1038/s41467-020-20841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao X, Chen Y, Wu B, Wang X, Xue H, Yu L, Li J, Wang Y, Wang W, Xu Q, et al. (2020). Histone H4K20 Demethylation by Two hHR23 Proteins. Cell Rep. 30, 4152–4164.e6. 10.1016/j.celrep.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, and Marzluff WF (2000). Stem-loop binding protein, the protein that binds the 3’ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell Biol. 20, 4188–4198. 10.1128/MCB.20.12.4188-4198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data have been deposited under accession PRIDE: PXD036699 to the PRIDE repository and are publicly available as of the date of publication. ChIP-seq data were deposited in the NCBI under GEO: GSE213209 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.