

Abstract
NAFLD, or metabolic dysfunction–associated steatotic liver disease, has increased in prevalence hand in hand with the rise in obesity and increased free sugars in the food supply. The causes of NAFLD are genetic in origin combined with environmental drivers of the disease phenotype. Dietary intake of added sugars has been shown to have a major role in the phenotypic onset and progression of the disease. Simple sugars are key drivers of steatosis, likely through fueling de novo lipogenesis, the conversion of excess carbohydrates into fatty acids, but also appear to upregulate lipogenic metabolism and trigger hyperinsulinemia, another driver. NAFLD carries a clinical burden as it is associated with obesity, type 2 diabetes, metabolic syndrome, and cardiovascular disease. Patient quality of life is also impacted, and there is an enormous economic burden due to healthcare use, which is likely to increase in the coming years. This review aims to discuss the role of dietary sugar in NAFLD pathogenesis, the health and economic burden, and the promising potential of sugar reduction to improve health outcomes for patients with this chronic liver disease.
INTRODUCTION
NAFLD, or metabolic dysfunction–associated steatotic liver disease,1 was initially described in the early 1800s.2 Since then, NAFLD has grown from a relatively unknown disease to a major cause of liver-related morbidity and mortality.3 NAFLD is clinically characterized by steatosis occupying > 5% of hepatocytes in the absence of alcohol consumption.4,5 The condition exists on a spectrum that ranges from simple steatosis to NASH, or metabolic dysfunction-associated steatohepatitis,1 which can progress to fibrosis and cirrhosis, predisposing patients to HCC.6 NAFLD is closely associated with other metabolic disorders such as type 2 diabetes (T2D), metabolic syndrome, polycystic ovarian syndrome, obesity, dyslipidemia, and cardiovascular disease (CVD).7–9
Population trends
In association with the obesity epidemic and the major changes in the food supply over the past few decades, the global prevalence of NAFLD has risen markedly to an estimated 25.2%. By 2040, it is projected that over half the adult population will have NAFLD.10 In the United States, the prevalence has risen to ≥ 34.0% in adults,11 and 11% in adolescents, ranging from 27% to 43% of those with childhood obesity.5 There are marked differences in NAFLD prevalence by nation, ethnicity, and race within countries. These differences are largely due to socioeconomic factors, sugar consumption, and, in part, due to the population prevalence of genetic polymorphisms underlying the susceptibility for developing NAFLD.12,13 In the United States, the highest prevalence of NAFLD is seen in those of Hispanic heritage, possibly driven by the inheritance of the most prevalent polymorphism associated with NAFLD, patatin-like phospholipase domain–containing protein three (PNPLA3).14
Healthcare burden
NAFLD and its complications cause a considerable healthcare burden worldwide. According to Younossi et al,15 the US annual direct medical costs of NAFLD are ~$103 billion, and in the European countries (Germany, France, Italy, and the United Kingdom), the annual cost is about $35 billion, with total costs highest in patients aged 45–65.15 Furthermore, in the US, the annual hospitalization rate due to NAFLD has tripled from 2007 to 2014, with a greater increase in males versus females and Latino/Hispanics versus other ethnicities.16 Recent data suggest that NAFLD will become the most common indication for liver transplantation in the near future.17 NAFLD accounts for the highest increase in disability-adjusted life years compared to other liver-related chronic diseases.18 At the individual level, the cost of medical care (due to testing, monitoring, and hospitalization) for a patient with NAFLD is estimated to be nearly twice as high compared with healthy individuals.15 In addition, there is an indirect societal impact due to absenteeism, caregiver burden, and reduced health-related quality of life.19
THE ROLE OF SUGAR IN NAFLD PATHOPHYSIOLOGY
While the causes of NAFLD are multifactorial, dietary intake of added sugars, especially fructose, has been of interest as a driver of the disease for a long time.20 The link between steatosis in the liver and fructose consumption has been attributed to Pliny, the Elder, who wrote that Marcus Apicius, the famous Roman chef, would make fatty liver (foie gras) by overfeeding geese dried figs (a rich source of fructose). The process of force-feeding geese originally comes from ancient Egypt, with depictions of the practice found in the tomb of Mereruka dated 2500 BC21 (Figure 1). Furthermore, in 1860, the German chemist Justus von Liebig also observed that dietary carbohydrates stimulated steatosis in the liver.13,22
FIGURE 1.
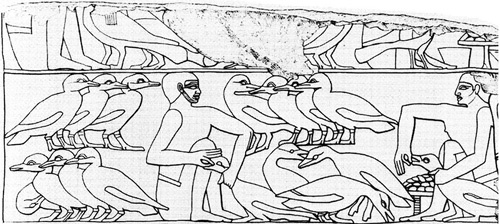
A bas relief depiction from the tomb of Mereruka, 2500 BC, illustrating the ancient Egyptian practice of overfeeding geese to produce foie gras.20
Excessive dietary sugar intake is thought to hold a major role in the onset and progression of NAFLD. The Dietary Guidelines for Americans 2020–2025 and the World Health Organization recommend that the intake of added sugars should not exceed 10% of total energy intake.23,24 This translates to ~50 gm (200 calories) of sugar on a 2000-calorie/day diet. A further reduction to less than 5% of total energy intake is recommended for additional health benefits.25 While total sugar consumption in the United States has decreased over recent years,26 current intakes remain above these guidelines, with sugar-sweetened beverages (SSB) as the top source, followed by desserts and sweet snacks.27 The average American adult and child consume about 17 teaspoons (68 gm) of added sugar per day,28 and the most common added sugars in the contemporary human diet include sucrose (table sugar) and high fructose corn syrup.29
Recent evidence suggests that the overconsumption of added dietary sugars, especially fructose, precipitates hepatic steatosis due to complex mechanisms that ultimately promote increased lipogeneses and impaired fatty acid oxidation13 (Figure 2). Conversely, reducing the consumption of free sugars (added sugars and naturally occurring sugar in fruit juice, honey, etc.) can significantly improve hepatic steatosis in both adults and children with NAFLD.33–35
FIGURE 2.
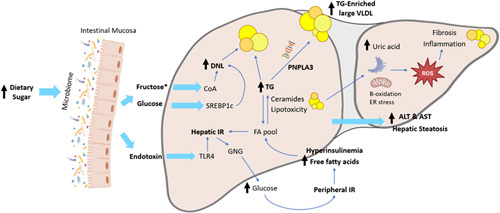
Biological mechanisms of NAFLD development by a high free sugar diet. Dietary sugars in the gut can alter the microbiome, increasing endotoxin that promotes hepatic IR.30 Monosaccharides enter the liver, and fructose is metabolized into fructose-1-P and further into acetyl-CoA, fueling DNL.12 Fructose, glucose, and insulin activate ChREBP & SREBP-1c, which transcriptionally activates genes in DNL. FA accumulation can exceed the liver’s capacity, which leads to ectopic lipid deposition and lipotoxicity. This promotes impaired mitochondrial beta-oxidation and ER stress, driving the production of ROS, hepatic IR, inflammation, and fibrosis through a complex set of mechanisms.31 Fructose metabolism also produces a drop in intracellular phosphate, resulting in increased uric acid formation, which is associated with oxidative stress and hepatic fat accumulation.32 Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ChREBP, carbohydrate-responsive element–binding protein; DNL, de novo lipogenesis; ER, endoplasmic reticulum; FA, fatty acid; GNG, gluconeogenesis; IR, insulin resistance; PNPLA3, patatin-like phospholipase domain–containing protein 3; ROS, reactive oxygen species; SREBP-1c, sterol regulatory element–binding protein 1c; TG, triglycerides; TLR4, toll-like receptor 4. Figure adapted with permission from Welsh et al., 2023.
Sugar and digestive mechanisms
It is important to note that although fructose and glucose share the same molecular formula (C6H12O6), they are absorbed and metabolized differently in the gut.36 Glucose is transported from the intestinal lumen into the enterocytes through an energy-requiring process mediated by the sodium-glucose cotransporter 1, whereas fructose is absorbed through a facilitated passive transport mechanism by GLUT5 on the apical border of enterocytes.37,38 At the basolateral membrane, GLUT2 facilitates the passive absorption of both glucose and fructose into the circulation.36 The small intestine plays a key role in dietary fructose metabolism. Studies in mice have shown that within the enterocyte, the majority of fructose is converted to glucose and other organic acids. Thus, the small intestine acts as a shield, protecting the liver from the lipogenic effects of fructose. However, excessive fructose intake can overwhelm intestinal fructose absorption, which is transported to the liver through the portal vein.36,39 A recent study measured fructose absorption/metabolism in 9 children with biopsy-proven NAFLD compared with 6 obese and 9 lean non-NAFLD controls, ages 8–18 years. The subjects with NAFLD demonstrated increased absorption and exaggerated metabolic response (elevated serum glucose, insulin, and uric acid) to fructose administration compared to lean children, while obese children without NAFLD had an intermediate response.40
Sugars and gut microbial mechanisms
The human gut microbiome plays a significant role in human health and disease.41 The liver is directly linked to the intestines through the portal vein and is a major site for the detoxification of products coming through the portal blood, including microbial metabolites.42 In the digestive tract, excessive dietary sugars can alter resident microbial diversity, promoting dysbiosis, increased gut permeability,43 and elevated circulating endotoxin (lipopolysaccharide).30 In both adults and children with NAFLD, plasma endotoxin levels are elevated, suggesting either a failure in endotoxin removal or an increase in production that surpasses the liver's cleanup mechanisms.30,44
In healthy physiology, endotoxins circulate in the bloodstream at low concentrations,45 and the majority are cleared by the liver (~80%).46,47 Although the mechanisms are still being investigated, recent evidence in animal models revealed that LPS disappears rapidly from the circulation (half-life of 2–4 min) and is scavenged primarily by the liver sinusoidal endothelial cells, which possess a high endocytic ability, and to a lesser extent by the Kupffer cells.48 In patients with NAFLD, the function of the Kupffer cells is impaired, which may result in disturbed hepatic clearance and increased levels of circulating LPS, leading to accelerated liver injury.49 Endotoxemia activates the innate immune system and stimulates hepatic toll-like receptor 4, which induces inflammasomes and proinflammatory cytokines, promoting hepatic inflammation and insulin resistance (IR) (Figure 2).50,51 In children with NAFLD, administration of high fructose beverages caused postprandial rises in endotoxin.44 Studies in animal models have shown that high-sugar diets increase the relative abundance of LPS-producing Proteobacteria in the gut while simultaneously decreasing the abundance of Bacteroidetes spp., some of which are considered protective against the effects of endotoxin.52–54 A recent human study revealed that a diet supplemented with high-fructose syrup significantly altered microbial composition, notably reducing the abundance of the genus Ruminococcus, known for its beneficial butyrate-producing bacteria.55 Furthermore, high doses of dietary fructose can overwhelm intestinal fructose absorption and clearance, causing fructose to spill over to the colon.36,39 In the colon, fructose generates the short-chain fatty acid acetate through microbial fermentation.56 Acetate can enter the portal circulation and be converted to acetyl-CoA by acetyl-CoA synthetase in the liver, potentially providing more carbon sources for de novo lipogenesis.42
Sugars and hepatic mechanisms
At the center of NAFLD pathogenesis, there exists an imbalance between hepatic lipid accumulation and removal, which is driven by inappropriately increased de novo lipogenesis. In hepatocytes, the monosaccharides glucose and fructose drive de novo lipogenesis through transcriptional activation of sterol regulatory element–binding protein-1c (SREBP-1c) and carbohydrate-responsive element–binding protein (ChREBP), which results in increased hepatic steatosis and directly affects insulin signaling and lipotoxicity.31,57 These monosaccharides also directly fuel de novo lipogenesis by providing necessary substrates for fatty acid and triglyceride synthesis, including acetyl-CoA and glycerol.58,59 Tracer experiments have shown that the contribution of de novo lipogenesis to intrahepatic lipid stores in NAFLD patients is ~26%, with ~59% derived from circulating fatty acids and ~15% from dietary fats.60 Additionally, a study comparing patients with NAFLD to healthy controls without steatosis revealed that de novo lipogenesis was 3-fold greater in NAFLD subjects.61 Interestingly, in individuals with obesity and NAFLD, Smith et al found a much larger contribution of de novo lipogenesis (~40%) to intrahepatic triglyceride formation, suggesting that elevated de novo lipogenesis is a distinct feature of NAFLD pathophysiology.62 They also demonstrated that increases in circulating insulin stimulate hepatic de novo lipogenesis in individuals with NAFLD.62 Further, they explored the concept that insulin mechanisms in the liver diverge, whereas the insulin receptors for glucose metabolism are resistant, leading to increased glycemia; the lipogenic mechanisms are intact and are overstimulated by excess insulin action lipogenesis.62–64 On the other hand, a study by Ter Horst et al revealed that patients with NAFLD and ce novo lipogenesis due to increased availability of lipogenic substrates like dietary fructose, while glucose-stimulated/insulin action lipogenesis was attenuated. However, the authors also stated that residual insulin-driven expression of lipogenic enzymes through SREBP-1c activation likely still plays a role. (Figure 3).65 Furthermore, insulin-resistant livers fail to suppress the activation of the transcription factor FoxO1, which upregulates 2 genes required for gluconeogenesis, phosphoenolpyruvate carboxykinase and glucose 6-phosphatase, leading to hyperglycemia.66
FIGURE 3.
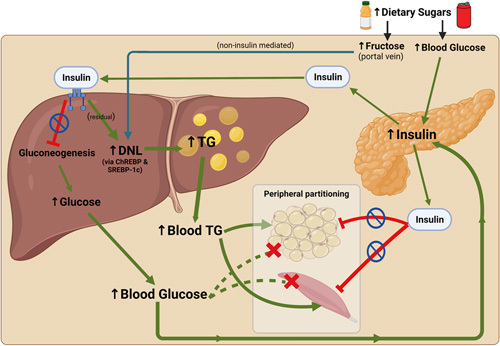
Mediators of hepatic de novo lipogenesis in obese individuals with NAFLD & insulin resistance. Dietary glucose stimulates insulin secretion from pancreatic beta cells. With hepatic IR, insulin fails to suppress hepatic gluconeogenesis; however, stimulation of DNL continues through residual activation of SREBP-1c. Dietary fructose is transported through the hepatic portal vein directly to the liver, where it stimulates de novo lipogenesis through non-insulin–mediated activation of ChREBP. Additionally, IR in adipose and muscle tissue prevents peripheral glucose uptake. Overall, these processes promote hyperglycemia and hyperlipidemia, driving NAFLD progression.65–67 Abbreviations: ChREBP, carbohydrate-responsive element–binding protein; DNL, de novo lipogenesis; IR, insulin resistance; SREBP-1c, sterol regulatory element–binding transcription factor 1; TG, triglycerides. Figure created using Biorender.
A recent study of 40 adolescent boys aged 11–16 years with NAFLD, undergoing an 8-week feeding protocol, demonstrated that the rate of de novo lipogenesis decreased from 25% to 17% in parallel to decreases in alanine aminotransferase and hepatic steatosis, consistent with the theory that de novo lipogenesis is a critical metabolic function linking dietary sugars and NAFLD.67
Unlike glucose, which is metabolized by most cells in the human body, fructose is metabolized primarily by the liver.68 Fructose metabolism is considered a major contributor to de novo lipogenesis in that it bypasses the rate-limiting enzyme of glycolysis (phosphofructokinase-1) and is rapidly phosphorylated by fructokinase C (the principal isoform of fructokinase in the liver) to fructose-1-phosphate without any negative feedback control.69 For this reason, fructose is considered more lipogenic than glucose. This reaction also requires ATP consumption, producing a drop in intracellular phosphate, ultimately resulting in purine nucleotide turnover and elevated uric acid formation (Figure 2).70,71 The decrease in ATP can induce oxidative stress and mitochondrial dysfunction.72 A study by Lanaspa et al demonstrated that the generation of mitochondrial oxidative stress from uric acid induces hepatic steatosis in HepG2 cells exposed to fructose.72 Furthermore, Choi et al confirmed that uric acid has direct effects on hepatic steatosis through the induction of endoplasmic reticulum stress and activation of SREBP-1c. SREBP-1c stimulates lipogenesis through the activation of lipogenic enzymes expressed in the liver, including acetyl-CoA carboxylase 1, fatty acid synthase, and stearoyl-CoA desaturase-1.32
The fatty acids accumulating in the liver can be esterified to form triglycerides and subsequently stored as lipid droplets or exported as VLDL particles into circulation.73 The storage of fatty acids in lipid droplets is thought to play a protective role in the disease process74 since excess fatty acid buildup can lead to the production of lipotoxic intermediates such as diacylglycerols, lysophosphatidylcholine species, ceramides, free cholesterol, and bile acids.75 These hepatotoxic lipids promote endoplasmic reticulum stress, mitochondrial dysfunction, and activation of NADPH oxidase, leading to downstream production of reactive oxygen species.57,76 These processes drive hepatic IR and inflammation, promoting disease progression to NASH and fibrosis (Figure 2). Additionally, hepatic glucotoxicity, which refers to the toxic effects of excess sugar intake on the liver, and hyperglycemia can disrupt insulin signaling.75 Hepatic IR enhances gluconeogenesis and represses insulin-dependent glycogen synthesis, leading to a vicious cycle of hyperglycemia and dysmetabolism, eventually causing hepatocyte injury and death.75,77,78
Furthermore, the secretion of triglyceride-enriched VLDL from the liver is increased in absolute terms in NAFLD; however, this increase does not match the overwhelming accumulation of lipids in the hepatocytes.79 The structure of VLDL comprises a triglyceride-enriched core with a monolayer of phospholipids that integrate proteins, such as apolipoprotein B-100, which are required for proper delivery and uptake of lipids to tissues. Impaired synthesis of apolipoprotein B-100 has been demonstrated in subjects with NASH, which may be related to disturbed redox balance and hyperinsulinemia.80,81 These processes lead to reduced VLDL assembly and excretion, resulting in hepatic lipid accumulation.57
Sugars and extrahepatic mechanisms
Overconsumption of added sugars can cause adaptations beyond the liver in the brain, adipose tissue, skeletal muscle, and pancreas. These extrahepatic and central responses to high sugar intake produce downstream changes that can indirectly contribute to hepatic steatosis. Added sugar intake triggers neuroadaptations in the mesolimbic reward pathway, promoting hedonic caloric intake above energy needs, leading to increased adiposity.82 Appetite and satiety levels are influenced by the type of sugar consumed. Glucose intake stimulates insulin secretion from the pancreatic beta cells, whereas fructose has a negligible impact on circulating insulin levels. Leptin (the satiety hormone) is triggered by insulin-mediated glucose metabolism, while fructose bypasses leptin secretion and attenuates the suppression of ghrelin (the hunger hormone), increasing appetite.83 Shapiro et al demonstrated that high fructose intake also induced leptin resistance in rats.84 These changes can eventually lead to obesity and peripheral insulin resistance, promoting hepatic steatosis since hyperinsulinemia triggers adipose tissue lipolysis, causing an increase in free fatty acid delivery to the liver.57 Additionally, insulin resistance in skeletal muscle can promote increased hepatic de novo lipogenesis by diverting ingested glucose away from muscle glycogen synthesis toward the liver for hepatic triglyceride synthesis.85–87
Sugars and genetic mechanisms
While the addition of high levels of simple sugars to the diet is a relatively modern phenomenon, the genetic underpinnings driving biological responses to this diet variant are not. There are a number of genetic polymorphisms linked to NAFLD in humans, but one of the most common appears to further explain the sugar–NAFLD relationship. The single nucleotide polymorphism in the PNPLA3 gene (I148 M variant, rs738409), which encodes a 481 amino acid protein called adiponutrin, is strongly associated with NAFLD in the setting of increased insulin resistance and body mass index.88,89 This polymorphism promotes hepatic steatosis by inhibiting the activity of lipases on the surface of lipid droplets, reducing triglyceride mobilization.90 Individuals who carry this variant are more susceptible to increased hepatic fat accumulation when sugar consumption is high.91 In the United States, the highest frequency of PNPLA3 rs738409 was found among those of Latino/Hispanic origin (0.49) compared to European ancestry (0.23) and African ancestry (0.17).88 A nutrigenetic analysis in Latino/Hispanic children demonstrated that triglyceride accumulation in the liver was dependent on sugar intake in those with the homozygous GG substitution of the PNPLA3 genotype.91 This evidence suggests that added sugar consumption may exacerbate the effects of this polymorphism by increasing intrahepatic lipid volume in the setting of a decreased ability to mobilize and remove lipids. Little is known about the impact of dietary sugars on other genes, including the glucokinase regulatory gene, which regulates de novo lipogenesis by controlling hepatocyte glucose influx, and the transmembrane 6-superfamily member 2, which regulates VLDL secretion.92 Therefore, more research is needed to investigate the interaction between dietary sugars and these genetic polymorphisms in NAFLD pathogenesis.13
THE BURDEN OF DIETARY SUGARS
Globally, the food industry in high-income countries has become saturated with appetizing and potentially addictive products containing added sugars.93,94 This movement is now increasingly apparent in low-income and middle-income countries.95 In food production, sugar is not only used as a caloric sweetener but also as a bulking and browning agent, humectant, texture modifier, fermentation substrate, and preservative.96 As a result, added sugars are a ubiquitous component of processed foods and beverages in the food supply.97 With the myriad of synonyms for sugar used on nutrition labels, manufacturers disguise the total sugar content in food products, making efforts to reduce consumption challenging.98
The biology of sugar preferences
Human sensory systems have evolved to detect and prefer the once rare, calorie-rich, sweet-tasting foods.99,100 When food availability was scarce, sweet foods were likely a vital energy source. Unfortunately, this biological preference makes humans vulnerable to today’s food environment, which is abundant in processed foods rich in refined sugars.101 Fructose is particularly problematic since it is the sweetest tasting natural sugar, 1.2–2.0 times sweeter than table sugar.102 The consumption of fructose has increased by 30% in the last 40 years and by 500% over the last century due to the increased consumption of processed foods and SSB.103
Recent studies in animal models and humans have shown that added sugars have habit-forming properties similar to alcohol, tobacco, cocaine, and caffeine.104,105 Excessive consumption of sugars can stimulate the reward pathways in the brain through an intense dopamine release.82 According to DiNicolantonio et al, chronic sugar intake can lead to “dopamine deficiency” in the brain due to the downregulation of the dopamine D2 receptors. Over time, this can lead to withdrawals, promoting the drive for perpetual sugar intake and addiction.106
People of all ages are susceptible to the pervasiveness of added sugar. Today, an American child as early as 2 years of age is more likely to consume a sugar-sweetened product than a fruit or vegetable.107 Given that food preferences are established in early childhood, this may drive lifelong diet preferences.108 The distinction between fruits, which naturally contain glucose and fructose, and processed foods with added sugar should be noted. Despite their sweet taste, fruits contain relatively low amounts of simple sugars compared to processed foods and SSB. Additionally, whole fruits contain fiber and antioxidants, which may protect against the harmful metabolic effects of sugar consumption.109
Hepatic consequences of added sugars
Excessive added sugar intake, especially fructose, is linked with metabolic abnormalities that can cause NAFLD, which can progress to advanced liver disease.110,111 In children and adolescents, fructose has a dose-dependent correlation with NAFLD onset and development of fibrosis.13 A recent meta-analysis revealed a significant positive association between higher consumption of SSB and the risk of NAFLD.112 According to Geidl-Flueck et al, even modest consumption of added sugars from beverages sweetened with fructose or sucrose over several weeks led to increased hepatic fatty acid synthesis in healthy men.113 Additionally, a systematic review of 7 studies including 4639 subjects showed that SSB consumers had a 53% increased risk of developing NAFLD compared with nonconsumers.114 In a group of otherwise healthy, overweight adults, the consumption of SSB for 6 months significantly increased hepatic steatosis, skeletal muscle fat, and visceral fat.115 This suggests that the daily consumption of SSB leads to an increased risk of metabolic and CVDs, including NAFLD. While earlier human studies are confounded by the fact that fructose was provided in the context of overfeeding and weight gain, more recent studies show that the effects of dietary fructose are independent of caloric intake and body mass index.116–118
Extrahepatic consequences of added sugar
With excessive sugar consumption, NAFLD typically occurs in the setting of obesity, dysglycemia, atherogenic dyslipidemia, and hypertension.8 Eventually, extrahepatic manifestations of NAFLD can occur, including chronic kidney disease and CVD,119 the leading cause of death among NAFLD patients.120 Additionally, NAFLD may play a role in the onset and progression of T2D and metabolic syndrome rather than just being an outcome of these conditions.121,122 A recent meta-analysis revealed that NAFLD was associated with about a 2-fold increase in the risk of incident T2D and metabolic syndrome over a median follow-up of 5 years.123
HOPE FOR THE FUTURE: THE BENEFITS OF SUGAR REDUCTION FOR NAFLD
Lifestyle change is the first-line therapy for NAFLD.124 To prevent the development of NAFLD and its complications, it is imperative for our society to adopt interventions to support sugar reduction in the population, especially in children who are most vulnerable.125 Potential strategies include food education, the distribution of fruits and vegetables at school, and increased taxes on foods that contain any form of added sugar.125
Taxation
Sugar taxation has recently emerged as a viable strategy in many countries (eg, Mexico, Denmark, and South Africa) and US jurisdictions (eg, Berkeley, California).126–129 A recent systematic review revealed that sugar taxation is a cost-effective policy option to reduce the health and economic burden related to the overconsumption of added sugars. These savings were driven by avoided healthcare costs and tax revenue exceeding intervention costs.130 Additionally, a microsimulation model developed by Vreman et al assessed the health and economic benefits of interventions aimed at reducing the intake of added sugars and estimated that a 20% reduction in added sugar intake would significantly reduce the prevalence of hepatic steatosis, NASH, cirrhosis, HCC, obesity, T2D, and CVD. Direct medical costs and disease-attributable disability-adjusted life years would also be reduced; these effects increased proportionally when added sugar intake was reduced by 50%.131
Individual interventions
Clinical trials have demonstrated that dietary sugar restriction interventions that include meal provision and instruction are particularly effective therapeutic strategies for NAFLD. One short-term intervention study in 41 children (9–18 y) with obesity showed that dietary restriction of the free sugar fructose (~4% of total energy intake) via meal provision was associated with improvements in hepatic steatosis (from 7.2% to 3.8%), hepatic de novo lipogenesis, and insulin kinetics.35 Aligning with this, Schwimmer et al, recently conducted an 8-week, randomized, controlled intervention study in 40 adolescent boys with NAFLD (11–16 y.) to test the effect of dietary free sugar restriction on hepatic fat and found that the diet treatment group achieved a significantly greater reduction in hepatic fat (from 25% at baseline to 17% at week eight) compared to the usual control diet (21%–20%).33 In this study, meals were provided for the whole family along with individualized menu planning to restrict free sugars to less than 3% of total energy needs in the intervention diet group. A subsequent analysis of the same study participants by Cohen et al evaluated the effects of the 8-week sugar restriction on hepatic de novo lipogenesis, measured as the percentage contribution to plasma triglyceride palmitate using a 7-day metabolic labeling protocol with heavy water. Results revealed that the dietary sugar restriction significantly reduced hepatic de novo lipogenesis and fasting insulin among adolescents with NAFLD.67 Furthermore, plasma metabolomics and metagenomics were performed to investigate the systemic changes that occurred with the reduction in steatosis and de novo lipogenesis. The analyses revealed that the low free sugar diet, compared to the usual diet, was associated with metabolome and microbiome changes that may reflect biological mechanisms linking dietary sugar restriction to a therapeutic decrease in hepatic fat.132
On the other hand, a recent study by Schmidt et al, found that a dietitian-led sugar reduction intervention did not improve liver outcomes in Latino youth with obesity (11–18 y of age).133 However, this study only involved nutrition education without meal provision, and the goal for sugar restriction was only ≤ 10% of total calories.
The combination of a low-sugar diet and time-restricted feeding (16 consecutive hours of fasting and 8 hours of eating time daily) has also been shown to reduce adiposity and improve markers of liver function, dyslipidemia, and inflammation in adult patients with NAFLD.134 Another study in overweight or obese adults with NAFLD showed that a low free sugar diet intervention over 12 weeks resulted in reduced fibrosis scores and steatosis scores with improved glycemic indices and decreased concentrations of biomarkers of inflammation, triglycerides, and total cholesterol levels.135 Moreover, a study in children and adolescents (7–18 y.) with NAFLD found that a low fructose and low glycemic index/load dietary intervention over 6 months resulted in improvements in body composition and plasma markers of liver dysfunction and cardiometabolic risk.136
Further research is needed to confirm the long-term effectiveness of sugar reduction for NAFLD prevention and treatment, along with continued public health initiatives to decrease added sugars in our food system. Please see Table 1 for a detailed summary of the studies focusing on taxation and sugar restriction mentioned above.
TABLE 1.
Sugar taxation & restriction studies
| Author, Year | Study population | Study design | Intervention | Main findings |
|---|---|---|---|---|
| Sugar taxation studies with SSB consumption–focused outcomes | ||||
| Colchero et al, 2016126 | 6253 households in 53 cities in Mexico | Observational study using data from Nielsen Mexico’s Consumer Panel Services on the purchase of beverages in Mexico from January 2012 to December 2014 | Excise tax of 1 peso/L on SSB | Purchase of taxed beverages decreased by an average of 6% and at an increasing rate of up to a 12% decline by December 2014. Reductions are higher among households of low SES. |
| Falbe et al, 2016127 | Low-income neighborhoods in Berkeley vs. the comparison cities of Oakland and San Francisco, California | Repeated cross-sectional study to examine changes in pre-tax to post-tax beverage consumption based on data from an interviewer-administered beverage frequency questionnaire | Excise tax ($0.01/oz) on SSB | Consumption of SSBs decreased by 21% in Berkeley and increased by 4% in comparison cities (P = .046). Water consumption increased more in Berkeley (+ 63%) than in comparison cities (+ 19%; P < 0.01). |
| Sugar restriction studies with liver health–focused outcomes | ||||
| Schwarz et al, 2017116 | 41 nondiabetic Latino and African American children (9–18 y) with obesity and metabolic syndrome, who identified as having habitual high sugar consumption (fructose intake >50 g/d) | Convenience cohort within-subject intervention with repeated measures | Meal provision restricting fructose ingestion only to naturally occurring fructose in fruits and vegetables (~ 15 gm/d/ 4% of total kcal, for 9 d) by substituting complex carbohydrates for excess dietary fructose while maintaining a neutral energy balance | Liver fat decreased from a median of 7.2% to 3.8% (P < .001). VAT decreased from 123 cm3 to 110 cm3 (P < .001). The DNL AUC decreased from 68% to 26% (P < 0.001). Insulin kinetics improved (P < 0.001). Changes occurred irrespective of baseline liver fat. |
| Schwimmer et al, 201933 | 40 adolescent boys with biopsy-confirmed NAFLD, ages 11–16 y | Randomized, parallel assignment clinical trial without blinding |
8-week LFSD: Individualized menu planning and meal provision for the entire household to restrict free sugar intake to less than 3% of daily calories | Mean decrease in hepatic steatosis from baseline to week 8 was significantly greater for the intervention diet group (25%–17%) vs. the usual diet group (21%–20%), and the adjusted week 8 mean difference was −6.23% (P < 0.001). |
| Cohen et al., 202167 | 40 adolescent boys with biopsy-confirmed NAFLD, ages 11–16 y | Randomized, parallel assignment clinical trial without blinding |
8-week LFSD (see Schwimmer et al., 2019, 2019 above) + 7-day metabolic labeling protocol with heavy water | Hepatic DNL was significantly decreased in the treatment group (from 34.6% to 24.1%) vs. the control group (33.9%–34.6%), along with greater decreases in hepatic fat and fasting insulin. |
| Cohen et al., 2023132 | 40 adolescent boys with biopsy-confirmed NAFLD, ages 11–16 y | Randomized, parallel assignment clinical trial without blinding |
8-week LFSD (see Schwimmer et al., 2019 above) + plasma metabolomics analysis | The LFSD treatment, compared to the usual diet, was associated with differential expression of 419 metabolite features (P < 0.05), which were enriched in amino acid pathways, including methionine/cysteine and serine/glycine/alanine metabolism (P < 0.05), and lipid pathways, including omega-3 and linoleate metabolism (P < 0.05).Microbiome changes included an increase in richness at the phylum level and changes in a few genera within Firmicutes. |
| Schmidt et al, 2022133 | 105 Latino adolescents (11–18 y) with obesity (BMI ≥95th percentile for age & sex) | Parallel-design randomized controlled dietary intervention trial | A 12-week dietitian-led sugar reduction intervention, including nutrition education to promote free sugar reduction to ≤ 10% of total calorie needs | Mean free sugar intake decreased in the intervention group vs. control (11.5%–7.3% compared with 13.9%–10.7% of total energy needs, respectively; P = 0.02). There were no significant effects on liver outcomes or anthropometrics (P all > 0.10). |
| Kord-Varkaneh et al, 2023134 | 52 overweight/obese adults with NAFLD, ages 18–50 y | Randomized clinical trial | A 12-week time-restricted feeding intervention (16 h fasting/8 h feeding daily) + a low-sugar diet (< 3% total energy needs) | The time-restricted feeding intervention group reduced body fat, body weight, WC, BMI, fasting blood glucose, liver enzymes (ALT, AST, GGT), lipids (TG, TC, LDL-cholesterol), and inflammatory markers (hs-CRP and cytokeratin-18), all statistically significant vs. control (P<0.05). |
| Khodami et al, 2022134 | 43 overweight/obese adults with FibroScan-proven NAFLD, ages 18–60 y | Randomized two-arm, parallel dietary intervention | 12-week LFSD intervention (dietitian instruction to limit free sugars to <10% of total energy needs) | The LFSD intervention group compared with the usual diet control group, significantly decreased ALT, TG, TC, FBS, insulin, HOMA-IR, hs-CRP, TNF-α, and NF-kb (P < 0.05). The LFSD group also reduced fibrosis score and steatosis score, with increased QUICKI compared to the control (P < 0.05). |
| Mager et al., 2015136 | Children and adolescents with NAFLD (n = 12) and healthy controls (n = 14), ages 7–18 y | Prospective dietary intervention | Dietary education/sample menus to promote the consumption of a low fructose (< 7% energy needs) and low glycemic index (45–55)/glycemic load (< 80) (FRAGILE) diet over 6 mo | In children with NAFLD, there were significant reductions in SBP, percentage BF, and plasma concentrations of ALT (P = 0.04), Apo-B-100 (P < .001), and HOMA-IR at 3 and 6 mo (P < 0.05). Dietary reductions in fructose and GL were related to reductions in SBP (P = 0.01), ALT (P = 0.004), HOMA-IR (P = 0.03), and percentage BF. No changes in laboratory variables were observed in the healthy control group except for Apo-B-100. |
Abbreviations: ALT, alanine aminotransferase; Apo-B-100, apolipoprotein B-100; AST, aspartate aminotransferase; BF, body fat; BMI, body mass index; DNL, de novo lipogenesis; FBS, fasting blood sugar; GGT, gamma-glutamyl transferase; GL, glycemic load; HOMA-IR, Homeostatic Model Assessment for Insulin Resistance; hs-CRP, high sensitivity C-reactive protein, TG, triglycerides; LFSD, low free sugar diet; QUICKI, quantitative insulin sensitivity check index; SBP, systolic blood pressure; SSB, sugar-sweetened beverages; TC, total cholesterol; VAT, visceral adipose tissue; WC, waist circumference.
SUGAR REDUCTION AND PRECISION NUTRITION
Dietary recommendations for obesity issued over the past decade have aimed to mitigate disease progression using a one-size-fits-all approach.137,138 This strategy has shown only moderate success, and some recommended strategies for generalized weight loss may not be the most effective in the setting of elevated hepatic fat and alanine aminotransferase.139–145 Recent studies have demonstrated that personalized nutrition interventions tailored to individuals or subgroups of individuals are more effective than conventional dietary advice.146 Precision nutrition (aka personalized nutrition) utilizes information on individual characteristics or phenotypes of disease to develop targeted nutritional recommendations.147,148
NAFLD is a heterogeneous condition with a broad spectrum of clinical manifestations, pathogenesis, and response to treatment.149–151 This heterogeneity is thought to arise from multiple factors, including sex, hormonal status, genetics, gut microbiota composition, other comorbidities, and certain exposures, including diet and physical activity.150 Carrillo-Larco et al identified 3 phenotypes in adults with NAFLD using a machine-learning approach. The majority of subjects fell into the average NAFLD phenotype, whereas 6% and 10% of the remaining subjects fell into phenotypes characterized by (1) high levels of anthropometrics, systolic blood pressure, and glucose (highest all-cause mortality), or (2) high levels of liver biomarkers and cholesterol, respectively.152 Furthermore, recent studies have demonstrated that diet and lifestyle modifications, including sugar reduction, are more effective in decreasing intrahepatic fat in NAFLD patients who are carriers of the PNPLA3 I148M gene polymorphism versus noncarriers.153,154 A tailored dietary approach that accounts for differences in clinical, genetic, and metabolic characteristics between responders and nonresponders to nutrition therapies for NAFLD, including sugar reduction, may be more effective than traditional lifestyle recommendations. Indeed, Zelber-Sagi et al suggested that current dietary regimens for NAFLD, such as low carbohydrate or reduced refined sugar interventions, could be improved by tailoring nutrition recommendations to meet individual preferences and goals, therefore improving long-term adherence and health outcomes.138 Although precision nutrition interventions have shown promise in several chronic disease populations,147,155 there is limited research in patients with NAFLD. Further research is needed to determine subphenotypes of NAFLD and tailor nutrition advice to individuals or subgroups of similar individuals with NAFLD to improve health outcomes.
CONCLUSION
In conclusion, NAFLD is the most common cause of chronic liver disease and is strongly associated with the metabolic conditions of insulin resistance, T2D, and obesity. The increasing prevalence of NAFLD is linked to the rise of sugar consumption; therefore, dietary strategies incorporating restriction may provide an effective disease prevention and treatment solution. While the role of dietary sugar in NAFLD pathogenesis is still being elucidated, current consumption levels surpass the World Health Organization's guideline of no more than 10% of total energy intake. Given the health and economic impact of NAFLD, it's crucial to reduce free sugar intake to alleviate the current burden and prevent future obesity-related comorbidities.
Acknowledgments
FUNDING INFORMATION
Supported by the National Institutes of Health (NIH) (grant numbers R01NR019083 and R01DK125701).
CONFLICTS OF INTEREST
Miriam B. Vos serves as a consultant to Boehringer Ingelheim, Novo Nordisk, Eli Lilly, Intercept, Takeda and Alberio. She has stock or stock options in Thiogenesis and Tern Pharmaceuticals. Her institution has received research grants (or in kind research services) from Target Real World Evidence, Quest, Labcorp, and Sonic Incytes Medical Corp. The remaining authors have no conflicts to report.
Footnotes
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; Apo-B-100, apolipoprotein B-100; BF, body fat; BMI, body mass index; ChREBP, carbohydrate-responsive element–binding protein; CVD, cardiovascular disease; DNL, de novo lipogenesis; ER, endoplasmic reticulum; FA, fatty acid; FAS, fatty acid synthase; FBS, fasting blood sugar; GNG, gluconeogenesis; GL, glycemic load; HOMA-IR, Homeostatic Model Assessment for insulin resistance; hs-CRP, high sensitivity C-reactive protein; IR, insulin resistance; LFSD, low free sugar diet; MetS, metabolic syndrome; PNPLA3, patatin-like phospholipase domain–containing protein 3; QUICKI, quantitative insulin sensitivity check index; ROS, reactive oxygen species; SBP, systolic blood pressure; SREBP-1c, sterol regulatory element–binding protein 1c; SSB, sugar-sweetened beverages; TC, total cholesterol, TG, triglycerides; T2D, type 2 diabetes; TLR4, toll-like receptor 4; VAT, visceral adipose tissue; WC, waist circumference.
Contributor Information
Helaina E. Huneault, Email: hhuneau@emory.edu.
Ana Ramirez Tovar, Email: ana.maria.ramirez.tovar@emory.edu.
Cristian Sanchez-Torres, Email: cristian.julian.sanchez-torres@emory.edu.
Jean A. Welsh, Email: jwelsh1@emory.edu.
Miriam B. Vos, Email: mvos@emory.edu.
REFERENCES
- 1. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023. doi: 10.1097/HEP.0000000000000520. [DOI] [PubMed] [Google Scholar]
- 2. Schaffner F, Thaler H. Nonalcoholic fatty liver disease. Prog Liver Dis. 1986;8:283–298. [PubMed] [Google Scholar]
- 3. Huh Y, Cho YJ, Nam GE. Recent epidemiology and risk factors of nonalcoholic fatty liver disease. Journal of obesity & metabolic syndrome. 2022;31:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, et al. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988-1994. Am J Epidemiol. 2013;178:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr. 2013;162:496–500.e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muthiah MD, Cheng Han N, Sanyal AJ. A clinical overview of non-alcoholic fatty liver disease: A guide to diagnosis, the clinical features, and complications-What the non-specialist needs to know. Diabetes Obes Metab. 2022;24(Suppl 2):3–14. [DOI] [PubMed] [Google Scholar]
- 7. Lim S, Kim J-W, Targher G. Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends in Endocrinology & Metabolism. 2021;32:500–514. [DOI] [PubMed] [Google Scholar]
- 8. Glass LM, Hunt CM, Fuchs M, Su GL. Comorbidities and nonalcoholic fatty liver disease: The Chicken, the Egg, or Both?. Federal practitioner : for the health care professionals of the VA, DoD, and PHS. 2019;36:64–71. [PMC free article] [PubMed] [Google Scholar]
- 9. Mohammad Maysara A, Muhammad Talal S, Firas B, Yasser A-K, Yamen E, Srinivasan D, et al. Association of non-alcoholic fatty liver disease and polycystic ovarian syndrome. BMJ Open Gastroenterology. 2020;7:e000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Le MH, Yeo YH, Zou B, Barnet S, Henry L, Cheung R, et al. Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical bayesian approach. Clin Mol Hepatol. 2022;28:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noureddin M, Ntanios F, Malhotra D, Hoover K, Emir B, McLeod E, et al. Predicting NAFLD prevalence in the United States using National Health and Nutrition Examination Survey 2017–2018 transient elastography data and application of machine learning. Hepatology Communications. 2022;6:1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sulaiman SA, Dorairaj V, Adrus MNH. Genetic polymorphisms and diversity in nonalcoholic fatty liver disease (NAFLD): A mini review. Biomedicines. 2022;11:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69:2672–2682. [DOI] [PubMed] [Google Scholar]
- 15. Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016;64:1577–1586. [DOI] [PubMed] [Google Scholar]
- 16. Adejumo AC, Samuel GO, Adegbala OM, Adejumo KL, Ojelabi O, Akanbi O, et al. Prevalence, trends, outcomes, and disparities in hospitalizations for nonalcoholic fatty liver disease in the United States. Anna Gastroenterol. 2019;32:504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Esteban JPG, Asgharpour A. Evaluation of liver transplant candidates with non-alcoholic steatohepatitis. Trans Gastroenterol Hepatol. 2022;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allen AM, Lazarus JV, Younossi ZM. Healthcare and socioeconomic costs of NAFLD: A global framework to navigate the uncertainties. J Hepatol. 2023;79:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Younossi ZM, Tampi R, Priyadarshini M, Nader F, Younossi IM, Racila A. Burden of illness and economic model for patients with nonalcoholic steatohepatitis in the United States. Hepatology. 2019;69:564–572. [DOI] [PubMed] [Google Scholar]
- 20. Best CH, Hartroft WS, Lucas CC, Ridout JH. Liver damage produced by feeding alcohol or sugar and its prevention by choline. British Med Jl. 1949;2:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oriental Division NYPL A bas relief in necropolis. In. Public Domain 2500 BC.
- 22. Schwarz J-M, Clearfield M, Mulligan K. Conversion of sugar to fat: Is hepatic de novo lipogenesis leading to metabolic syndrome and associated chronic diseases? J Osteopathic Med. 2017;117:520–527. [DOI] [PubMed] [Google Scholar]
- 23. Organization WH. WHO calls on countries to reduce sugars intake among adults and children. 2023.
- 24. 2020-2025 DGfA. Make Every Bite Count With the Dietary Guidelines. 2023.
- 25. Organization WH. Guideline: Sugars intake for adults and children. 2020.
- 26. Welsh JA, Sharma AJ, Grellinger L, Vos MB. Consumption of added sugars is decreasing in the United States. Am J Clin Nutr. 2011;94:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ricciuto L, Fulgoni VL, Gaine PC, Scott MO, DiFrancesco L. Sources of added sugars intake among the U.S. population: Analysis by selected sociodemographic factors using the National Health and Nutrition Examination Survey 2011–18. Front Nutr. 2021;8::687643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prevention CfDCa. Get the Facts: Added Sugars. 2023.
- 29. Rippe JM, Sievenpiper JL, Lê K-A, White JS, Clemens R, Angelopoulos TJ. What is the appropriate upper limit for added sugars consumption? Nutr Rev. 2016;75:18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm. 2010;7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferré P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes, Obesity & Metab. 2010;12:83–92. [DOI] [PubMed] [Google Scholar]
- 32. Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab Invest. 2014;94:1114–1125. [DOI] [PubMed] [Google Scholar]
- 33. Schwimmer JB, Ugalde-Nicalo P, Welsh JA, Angeles JE, Cordero M, Harlow KE, et al. Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: A Randomized Clinical Trial. JAMA. 2019;321:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lustig RH, Mulligan K, Noworolski SM, Tai VW, Wen MJ, Erkin-Cakmak A, et al. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring). 2016;24:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwarz J-M, Noworolski SM, Erkin-Cakmak A, Korn NJ, Wen MJ, Tai VW, et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology. 2017;153:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merino B, Fernández-Díaz CM, Cózar-Castellano I, Perdomo G. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients. 2019;12:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferraris RP, Choe JY, Patel CR. Intestinal absorption of fructose. Annu Rev Nutr. 2018;38:41–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Douard V, Ferraris RP. The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol. 2013;591:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018;27:351–361.e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sullivan J, Le M, Pan Z, Rivard C, Love-Osborne K, Robbins K, et al. Oral fructose absorption in obese children with non-alcoholic fatty liver disease: Fructose absorption in paediatric NAFLD. Pediatr Obesity. 2014;10:188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang B, Yao M, Lv L, Ling Z, Li L. The human microbiota in health and disease. Engineering. 2017;3:71–82. [Google Scholar]
- 42. Park G, Jung S, Wellen KE, Jang C. The interaction between the gut microbiota and dietary carbohydrates in nonalcoholic fatty liver disease. Exper & Molecular Med. 2021;53:809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hrncir T, Hrncirova L, Kverka M, Hromadka R, Machova V, Trckova E, et al. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms. 2021;9:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin R, Willment A, Patel SS, Sun X, Song M, Mannery YO, et al. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int J Hepatol. 2014;2014:560620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Erlanson-Albertsson C, Stenkula KG. The importance of food for endotoxemia and an inflammatory response. Int J Mol Sci. 2021;22:9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Freudenberg MA, Galanos C. Bacterial lipopolysaccharides: Structure, metabolism and mechanisms of action. Int Rev Immunol. 1990;6:207–221. [DOI] [PubMed] [Google Scholar]
- 47. Mathison JC, Ulevitch RJ. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. J Immunol. 1979;123:2133–2143. [PubMed] [Google Scholar]
- 48. Yao Z, Mates JM, Cheplowitz AM, Hammer LP, Maiseyeu A, Phillips GS, et al. Blood-borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J Immunol. 2016;197:2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. 2009;51:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nature Rev Gastroenterol & Hepatol. 2016;13:412–425. [DOI] [PubMed] [Google Scholar]
- 51. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61:1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Do MH, Lee E, Oh MJ, Kim Y, Park HY. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients. 2018;10:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khan S, Waliullah S, Godfrey V, Khan MAW, Ramachandran RA, Cantarel BL, et al. Dietary simple sugars alter microbial ecology in the gut and promote colitis in mice. Sci Trans Med. 2020;12:eaay6218. [DOI] [PubMed] [Google Scholar]
- 54. Satokari R. High intake of sugar and the balance between pro- and anti-inflammatory gut bacteria. Nutrients. 2020;12:1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beisner J, Gonzalez-Granda A, Basrai M, Damms-Machado A, Bischoff SC. Fructose-induced intestinal microbiota shift following two types of short-term high-fructose dietary phases. Nutrients. 2020;12:3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Geng Y, Faber KN, de Meijer VE, Blokzijl H, Moshage H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol Int. 2021;15:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jin ES, Sherry AD, Malloy CR. Metabolism of glycerol, glucose, and lactate in the citric acid cycle prior to incorporation into hepatic acylglycerols. J Biol Chem. 2013;288:14488–14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Federico A, Rosato V, Masarone M, Torre P, Dallio M, Romeo M, et al. The Role of fructose in non-alcoholic steatohepatitis: Old relationship and new insights. Nutrients. 2021;13:1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130:1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brown MS, Goldstein JL. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008;7:95–96. [DOI] [PubMed] [Google Scholar]
- 65. Ter Horst KW, Vatner DF, Zhang D, Cline GW, Ackermans MT, Nederveen AJ, et al. Hepatic insulin resistance is not pathway selective in humans with nonalcoholic fatty liver disease. Diabetes Care. 2021;44:489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cohen CC, Li KW, Alazraki AL, Beysen C, Carrier CA, Cleeton RL, et al. Dietary sugar restriction reduces hepatic de novo lipogenesis in adolescent boys with fatty liver disease. J Clin Invest. 2021;131:e150996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr. 1993;58:754s–765s. [DOI] [PubMed] [Google Scholar]
- 69. Mäenpää PH, Raivio KO, Kekomäki MP. Liver adenine nucleotides: Fructose-induced depletion and its effect on protein synthesis. Science. 1968;161:1253–1254. [DOI] [PubMed] [Google Scholar]
- 70. Smith CM, Rovamo LM, Raivio KO. Fructose-induced adenine nucleotide catabolism in isolated rat hepatocytes. Can J Biochem. 1977;55:1237–1240. [DOI] [PubMed] [Google Scholar]
- 71. van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J. 1977;162:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287:40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75:3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65:1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Free Radical Biol Med. 2020;152:116–141. [DOI] [PubMed] [Google Scholar]
- 77. Palma R, Pronio A, Romeo M, Scognamiglio F, Ventriglia L, Ormando VM, Lamazza A, et al. The role of insulin resistance in fueling NAFLD pathogenesis: From molecular mechanisms to clinical implications. J Clin Med. 2022;11:3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sekizkardes H, Chung ST, Chacko S, Haymond MW, Startzell M, Walter M, et al. Free fatty acid processing diverges in human pathologic insulin resistance conditions. J Clin Invest. 2020;130:3592–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fabbrini E, Tiemann Luecking C, Love-Gregory L, Okunade AL, Yoshino M, Fraterrigo G, et al. Physiological mechanisms of weight gain-induced steatosis in people with obesity. Gastroenterology. 2016;150:79–81.e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Konishi K, Miyake T, Furukawa S, Senba H, Kanzaki S, Nakaguchi H, et al. Advanced fibrosis of non-alcoholic steatohepatitis affects the significance of lipoprotein(a) as a cardiovascular risk factor. Atherosclerosis. 2020;299:32–37. [DOI] [PubMed] [Google Scholar]
- 82. Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev. 2008;32:20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Teff KL, Elliott SS, Tschöp M, Kieffer TJ, Rader D, Heiman M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89:2963–2972. [DOI] [PubMed] [Google Scholar]
- 84. Shapiro A, Mu W, Roncal C, Cheng K-Y, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. American journal of physiology. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1370–R1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Flannery C, Dufour S, Rabøl R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes. 2012;61:2711–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018;27:22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Goyal NP, Schwimmer JB. The genetics of pediatric nonalcoholic fatty liver disease. Clin Liver Dis. 2018;22:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci USA. 2019;116:9521–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Davis JN, Le KA, Walker RW, Vikman S, Spruijt-Metz D, Weigensberg MJ, et al. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr. 2010;92:1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Moore JB. From sugar to liver fat and public health: Systems biology driven studies in understanding non-alcoholic fatty liver disease pathogenesis. Proc Nutr Soc. 2019;78:290–304. [DOI] [PubMed] [Google Scholar]
- 93. Popkin BM, Hawkes C. Sweetening of the global diet, particularly beverages: Patterns, trends, and policy responses. Lancet Diabetes Endocrinol. 2016;4:174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Popkin BM. Global nutrition dynamics: The world is shifting rapidly toward a diet linked with noncommunicable diseases. Am J Clin Nutr. 2006;84:289–298. [DOI] [PubMed] [Google Scholar]
- 95. Baker P, Machado P, Santos T, Sievert K, Backholer K, Hadjikakou M, et al. Ultra-processed foods and the nutrition transition: Global, regional and national trends, food systems transformations and political economy drivers. Obesity Rev. 2020;21:e13126. [DOI] [PubMed] [Google Scholar]
- 96. Koivistoinen P, Hyvönen L. The use of sugar in foods. Int Dent J. 1985;35:175–179. [PubMed] [Google Scholar]
- 97. Murphy SP, Johnson RK. The scientific basis of recent US guidance on sugars intake. Am J Clin Nutr. 2003;78:827S–833S. [DOI] [PubMed] [Google Scholar]
- 98. Francisco UoCS. Hidden in Plain Sight. 2023.
- 99. Russell C, Baker P, Grimes C, Lindberg R, Lawrence MA. Global trends in added sugars and non-nutritive sweetener use in the packaged food supply: Drivers and implications for public health. Public Health Nutr. 2023;26:952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Beauchamp GK. Why do we like sweet taste: A bitter tale? Physiol Behav. 2016;164:432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Drewnowski A, Mennella JA, Johnson SL, Bellisle F. Sweetness and food preference. J Nutr. 2012;142:1142S–1148S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Singh RS, Chauhan K, Singh RP. Enzymatic approaches for the synthesis of high fructose syrup Gahlawat SK, Salar RK, Siwach P, Duhan JS, Kumar S, Kaur P. Plant Biotechnology: Recent Advancements and Developments. Singapore: Springer Singapore; 2017:189–211. [Google Scholar]
- 103. Vancells Lujan P, Viñas Esmel E, Sacanella, Meseguer E. Overview of non-alcoholic fatty liver disease (NAFLD) and the role of sugary food consumption and other dietary components in its development. Nutrients. 2021;13:1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ahmed SH, Guillem K, Vandaele Y. Sugar addiction: Pushing the drug-sugar analogy to the limit. Curr Opin Clin Nutr Metab Care. 2013;16:434–439. [DOI] [PubMed] [Google Scholar]
- 105. Lustig RH. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J Am Diet Assoc. 2010;110:1307–1321. [DOI] [PubMed] [Google Scholar]
- 106. DiNicolantonio JJ, O’Keefe JH, Wilson WL. Sugar addiction: Is it real? A narrative review. British Journal of Sports Medicine. 2018;52:910. [DOI] [PubMed] [Google Scholar]
- 107. Mennella JA, Bobowski NK, Reed DR. The development of sweet taste: From biology to hedonics. Rev Endocr Metab Disord. 2016;17:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. De Cosmi V, Scaglioni S, Agostoni C. Early taste experiences and later food choices. Nutrients. 2017;9:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tajima R, Kimura T, Enomoto A, Saito A, Kobayashi S, Masuda K, et al. No association between fruits or vegetables and non-alcoholic fatty liver disease in middle-aged men and women. Nutrition. 2019;61:119–124. [DOI] [PubMed] [Google Scholar]
- 110. Younossi ZM. Non-alcoholic fatty liver disease – A global public health perspective. J Hepatol. 2019;70:531–544. [DOI] [PubMed] [Google Scholar]
- 111. Stefan N, Häring H-U, Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diab Endocrinol. 2019;7:313–324. [DOI] [PubMed] [Google Scholar]
- 112. Chen H, Wang J, Li Z, Lam CWK, Xiao Y, Wu Q, et al. Consumption of sugar-sweetened beverages has a dose-dependent effect on the risk of non-alcoholic fatty liver disease: An updated systematic review and dose-response meta-analysis. Int J Environ Res Public Health. 2019;16:2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Geidl-Flueck B, Hochuli M, Németh Á, Eberl A, Derron N, Köfeler HC, et al. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: A randomized controlled trial. J Hepatol. 2021;75:46–54. [DOI] [PubMed] [Google Scholar]
- 114. Wijarnpreecha K, Thongprayoon C, Edmonds PJ, Cheungpasitporn W. Associations of sugar- and artificially sweetened soda with nonalcoholic fatty liver disease: A systematic review and meta-analysis. Qjm. 2016;109:461–466. [DOI] [PubMed] [Google Scholar]
- 115. Maersk M, Belza A, Stødkilde-Jørgensen H, Ringgaard S, Chabanova E, Thomsen H, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am J Clin Nutr. 2012;95:283–289. [DOI] [PubMed] [Google Scholar]
- 116. Schwarz J-M, Noworolski SM, Wen MJ, Dyachenko A, Prior JL, Weinberg ME, et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J Clin Endocrinol Metab. 2015;100:2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pollock NK, Bundy V, Kanto W, Davis CL, Bernard PJ, Zhu H, et al. Greater Fructose Consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents 1–23. J Nutr. 2012;142:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Imamura F, O’Connor L, Ye Z, Mursu J, Hayashino Y, Bhupathiraju SN, et al. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributable fraction. Bmj. 2015;351:h3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li AA, Ahmed A, Kim D. Extrahepatic manifestations of nonalcoholic fatty liver disease. Gut Liver. 2020;14:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Liu Y, Zhong G-C, Tan H-Y, Hao F-B, Hu J-J. Nonalcoholic fatty liver disease and mortality from all causes, cardiovascular disease, and cancer: A meta-analysis. Scientific Reports. 2019;9:11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tariq R, Axley P, Singal AK. Extra-hepatic manifestations of nonalcoholic fatty liver disease: A review. J Clin Exper Hepatol. 2020;10:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J Hepatol. 2018;68:335–352. [DOI] [PubMed] [Google Scholar]
- 123. Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31:936–944. [DOI] [PubMed] [Google Scholar]
- 124. Lassailly G, Caiazzo R, Pattou F, Mathurin P. Perspectives on treatment for nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1835–1848. [DOI] [PubMed] [Google Scholar]
- 125. Ribeiro A, Igual‐Perez MJ, Santos Silva E, Sokal EM. Childhood fructoholism and fructoholic liver disease. Hepatol Comm. 2019;3:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Colchero M, Popkin B, Rivera J, Ng SW. Beverage purchases from stores in Mexico under the excise tax on sugar sweetened beverages: Observational study. BMJ. 2016;352:h6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Falbe J, Thompson HR, Becker CM, Rojas N, McCulloch CE, Madsen KA. Impact of the Berkeley excise tax on sugar-sweetened beverage consumption. Am J Public Health. 2016;106:1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mytton OT, Clarke D, Rayner M. Taxing unhealthy food and drinks to improve health. Bmj. 2012;344:e2931. [DOI] [PubMed] [Google Scholar]
- 129. Paarlberg R, Mozaffarian D, Micha R. Viewpoint: Can U.S. local soda taxes continue to spread? Food Policy. 2017;71:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu S, Veugelers PJ, Liu C, Ohinmaa A. The cost effectiveness of taxation of sugary foods and beverages: A systematic review of economic evaluations. Appl Health Econ Health Policy. 2022;20:185–198. [DOI] [PubMed] [Google Scholar]
- 131. Vreman RA, Goodell AJ, Rodriguez LA, Porco TC, Lustig RH, Kahn JG. Health and economic benefits of reducing sugar intake in the USA, including effects via non-alcoholic fatty liver disease: A microsimulation model. BMJ Open. 2017;7:e013543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cohen CC, Huneault H, Accardi CJ, Jones DP, Liu K, Maner-Smith KM, et al. Metabolome × Microbiome changes associated with a diet-induced reduction in hepatic fat among adolescent boys. Metabolites. 2023;13:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schmidt KA, Jones RB, Rios C, Corona Y, Berger PK, Plows JF, et al. Clinical Intervention to reduce dietary sugar does not impact liver fat in latino youth, regardless of PNPLA3 genotype: A randomized controlled trial. J Nutr. 2022;152:1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kord-Varkaneh H, Salehi-Sahlabadi A, Tinsley GM, Santos HO, Hekmatdoost A. Effects of time-restricted feeding (16/8) combined with a low-sugar diet on the management of non-alcoholic fatty liver disease: A randomized controlled trial. Nutrition. 2023;105:111847. [DOI] [PubMed] [Google Scholar]
- 135. Khodami B, Hatami B, Yari Z, Alavian SM, Sadeghi A, Varkaneh HK, et al. Effects of a low free sugar diet on the management of nonalcoholic fatty liver disease: A randomized clinical trial. Europ J Clin Nutr. 2022;76:987–994. [DOI] [PubMed] [Google Scholar]
- 136. Mager DR, Iñiguez IR, Gilmour S, Yap J. The effect of a low fructose and low glycemic index/load (FRAGILE) dietary intervention on indices of liver function, cardiometabolic risk factors, and body composition in children and adolescents with nonalcoholic fatty liver disease (NAFLD). JPEN J Parenter Enteral Nutr. 2015;39:73–84. [DOI] [PubMed] [Google Scholar]
- 137. Kargulewicz A, Stankowiak-Kulpa H, Grzymisławski M. Dietary recommendations for patients with nonalcoholic fatty liver disease. Przeglad Gastroenterol. 2014;9:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zelber-Sagi S, Grinshpan LS, Ivancovsky-Wajcman D, Goldenshluger A, Gepner Y. One size does not fit all; practical, personal tailoring of the diet to NAFLD patients. Liver Int. 2022;42:1731–1750. [DOI] [PubMed] [Google Scholar]
- 139. Grønbæk H, Lange A, Birkebæk NH, Holland-Fischer P, Solvig J, Hørlyck A, et al. Effect of a 10-week weight loss camp on fatty liver disease and insulin sensitivity in obese Danish children. J Pediatr Gastroenterol Nutr. 2012;54:223–228. [DOI] [PubMed] [Google Scholar]
- 140. Ramon-Krauel M, Salsberg SL, Ebbeling CB, Voss SD, Mulkern RV, Apura MM, et al. A low-glycemic-load versus low-fat diet in the treatment of fatty liver in obese children. Child Obes. 2013;9:252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Nobili V, Marcellini M, Devito R, Ciampalini P, Piemonte F, Comparcola D, et al. NAFLD in children: A prospective clinical-pathological study and effect of lifestyle advice. Hepatology. 2006;44:458–465. [DOI] [PubMed] [Google Scholar]
- 142. Reinehr T, Schmidt C, Toschke AM, Andler W. Lifestyle intervention in obese children with non-alcoholic fatty liver disease: 2-year follow-up study. Arch Dis Child. 2009;94:437–442. [DOI] [PubMed] [Google Scholar]
- 143. Pozzato C, Verduci E, Scaglioni S, Radaelli G, Salvioni M, Rovere A, et al. Liver fat change in obese children after a 1-year nutrition-behavior intervention. J Pediatr Gastroenterol Nutr. 2010;51:331–335. [DOI] [PubMed] [Google Scholar]
- 144. Koot BGP, van der Baan-Slootweg OH, Vinke S, Bohte AE, Tamminga-Smeulders CLJ, Jansen PLM, et al. Intensive lifestyle treatment for non-alcoholic fatty liver disease in children with severe obesity: Inpatient versus ambulatory treatment. Int J Obes. 2016;40:51–57. [DOI] [PubMed] [Google Scholar]
- 145. Draijer L, Benninga M, Koot B. Pediatric NAFLD: An overview and recent developments in diagnostics and treatment. Expert Rev Gastroenterol Hepatol. 2019;13:447–461. [DOI] [PubMed] [Google Scholar]
- 146. Celis-Morales C, Livingstone KM, Marsaux CF, Macready AL, Fallaize R, O’Donovan CB, et al. Effect of personalized nutrition on health-related behaviour change: Evidence from the Food4Me European randomized controlled trial. Int J Epidemiol. 2017;46:578–588. [DOI] [PubMed] [Google Scholar]
- 147. de Toro-Martín J, Arsenault BJ, Després JP, Vohl MC. Precision nutrition: A review of personalized nutritional approaches for the prevention and management of metabolic syndrome. Nutrients. 2017;9:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. DDDDBerciano S, Figueiredo J, Brisbois TD, Alford S, Koecher K, Eckhouse S, et al. Precision nutrition: Maintaining scientific integrity while realizing market potential. Front Nutri. 2022;98:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Pal P, Palui R, Ray S. Heterogeneity of non-alcoholic fatty liver disease: Implications for clinical practice and research activity. World J Hepatol. 2021;13:1584–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Arrese M, Arab JP, Barrera F, Kaufmann B, Valenti L, Feldstein AE. Insights into nonalcoholic fatty-liver disease heterogeneity. Semin Liver Dis. 2021;41:421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lonardo A. Separating the apples from the oranges: From NAFLD heterogeneity to personalized medicine. Explor Med. 2021;2:435–442. [Google Scholar]
- 152. Carrillo-Larco RM, Guzman-Vilca WC, Castillo-Cara M, Alvizuri-Gómez C, Alqahtani S, Garcia-Larsen V. Phenotypes of non-alcoholic fatty liver disease (NAFLD) and all-cause mortality: Unsupervised machine learning analysis of NHANES III. BMJ Open. 2022;12:e067203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Shen J, Wong GL-H, Chan HL-Y, Chan RS-M, Chan H-Y, Chu WC-W, et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2015;30:139–146. [DOI] [PubMed] [Google Scholar]
- 154. Sevastianova K, Kotronen A, Gastaldelli A, Perttilä J, Hakkarainen A, Lundbom J, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss–induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94:104–111. [DOI] [PubMed] [Google Scholar]
- 155. Tuncay C, Ergoren MC. A systematic review of precision nutrition and Mediterranean Diet: A personalized nutrition approaches for prevention and management of obesity related disorders. Clin Nutr ESPEN. 2020;38:61–64. [DOI] [PubMed] [Google Scholar]