

Abstract
目的
研究Polo样激酶1(PLK1)抑制剂在奥希替尼耐药的非小细胞肺癌(NSCLC)细胞中的作用及其合用奥希替尼的抗肿瘤效果。
方法
利用药物浓度递增法构建对奥希替尼耐药的NCI-H1975细胞模型;在耐药细胞上合用肿瘤经典通路抑制剂库化合物与奥希替尼,筛选与奥希替尼存在协同作用的化合物;利用基因集富集分析考察奥希替尼耐药通路;利用磺酰罗丹明B染色法考察PLK1抑制剂对奥希替尼耐药细胞的抑制作用及其合用奥希替尼的抗肿瘤作用。
结果
成功建立奥希替尼耐药细胞模型(耐药指数=43.45)。PLK1抑制剂GSK 461364和BI 2536与奥希替尼存在协同作用,与敏感细胞比较,奥希替尼耐药细胞中PLK1调控通路和细胞周期通路显著激活,且在接受奥希替尼治疗的表皮生长因子受体突变NSCLC患者队列中,PLK1信使RNA水平与患者的无进展生存期呈负相关(R=-0.62,P<0.05),证实NSCLC细胞中PLK1过度激活可能导致细胞对奥希替尼耐药。进一步体外实验发现,PLK1抑制剂伏拉塞替和GSK 461364对奥希替尼耐药细胞的半抑制浓度小于敏感细胞,相较于单用奥希替尼,PLK1抑制剂与奥希替尼合用可显著增强对耐药细胞的增殖抑制作用。
结论
PLK1抑制剂与奥希替尼合用对奥希替尼耐药的NSCLC细胞的抑制作用更强,有可能用于奥希替尼耐药患者的干预和治疗。
Keywords: 非小细胞肺癌, NCI-H1975细胞, 奥希替尼, 耐药性, Polo样激酶抑制剂, 联合用药, 生物信息学
Abstract
Objective
To investigate the effects of PLK1 inhibitors on osimertinib-resistant non-small cell lung carcinoma (NSCLC) cells and the anti-tumor effect combined with osimertinib.
Methods
An osimertinib resistant NCI-H1975 cell line was induced by exposure to gradually increasing drug concentrations. Osimertinib-resistant cells were co-treated with compounds from classical tumor pathway inhibitor library and osimertinib to screen for compounds with synergistic effects with osimertinib. The Gene Set Enrichment Analysis (GSEA) was used to investigate the activated signaling pathways in osimertinib-resistant cells; sulforhodamine B (SRB) staining was used to investigate the effect of PLK1 inhibitors on osimertinib-resistant cells and the synergistic effect of PLK1 inhibitors combined with osimertinib.
Results
Osimertinib-resistance in NCI-H1975 cell (resistance index=43.45) was successfully established. The PLK1 inhibitors GSK 461364 and BI 2536 had synergistic effect with osimertinib. Compared with osimertinib-sensitive cells, PLK1 regulatory pathway and cell cycle pathway were significantly activated in osimertinib-resistant cells. In NSCLC patients with epidermal growth factor receptor mutations treated with osimertinib, PLK1 mRNA levels were negatively correlated with progression free survival of patients (R=-0.62, P<0.05), indicating that excessive activation of PLK1 in NSCLC cells may cause cell resistant to osimertinib. Further in vitro experiments showed that IC50 of PLK1 inhibitors BI 6727 and GSK 461364 in osimertinib-resistant cells were lower than those in sensitive ones. Compared with the mono treatment of osimertinib, PLK1 inhibitors combined with osimertinib behaved significantly stronger effect on the proliferation of osimertinib-resistant cells.
Conclusion
PLK1 inhibitors have a synergistic effect with osimertinib on osimertinib-resistant NSCLC cells which indicates that they may have potential clinical value in the treatment of NSCLC patients with osimertinib resistance.
Keywords: Non-small cell lung carcinoma, NCI-H1975 cells, Osimertinib, Drug resistance, Polo-like kinase1 inhibitors, Drug combination, Bioinformatics
NSCLC包括肺腺癌和鳞状细胞癌,占肺癌的80%~85%,是全球癌症相关死亡的主要原因之一[1]。约50%的亚洲NSCLC患者中检测到EGFR激活突变[2],这些患者采用EGFR-TKI治疗后存活率及临床结局显著改善[3]。作为第三代EGFR-TKI,奥希替尼对EGFR敏感性激活突变以及T790M耐药突变细胞的抑制率更高[4],是EGFR-T790M突变所致耐药的NSCLC患者的一线治疗药物[5]。然而,尽管治疗初期患者对奥希替尼的响应率良好,但仍不可避免会出现获得性耐药[6]。因此,探寻奥希替尼的耐药机制、开发奥希替尼耐药的治疗策略对NSCLC患者的临床治疗具有重要意义。
PLK1属于丝/苏氨酸蛋白激酶的Polo亚家族,在人体具有有丝分裂活性的细胞和组织中高表达,参与调控细胞周期进展、分化和存活等多种细胞过程[7]。PLK1在多种人类恶性肿瘤中高表达并与患者的不良预后相关[8],有研究报道PLK1驱动NSCLC细胞的上皮-间充质转化从而促进NSCLC转移[9]。近年来,研究者开发出多款PLK1小分子抑制剂,这些小分子抑制剂在肿瘤临床前治疗研究中展现出良好的活性,但因为剂量限制性毒副作用及脱靶效应在临床试验中表现欠佳[10]。
本研究在成功构建奥希替尼耐药的NSCLC细胞株基础上,筛选出对奥希替尼耐药细胞有抑制作用的PLK1抑制剂,并考察了PLK1抑制剂与奥希替尼的协同治疗效果,旨在为NSCLC的奥希替尼耐药问题提供新的解决方案。
1. 材料与方法
1.1. 主要试剂和仪器
人NSCLC细胞株NCI-H1975购自中国科学院典型培养物保藏委员会细胞库;NCI-H1975敏感株与耐药株源自GSE146850队列(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146850),PC-9敏感株与PC-9耐药株源自GSE153183队列(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153183)。肿瘤经典通路抑制剂化合物库(L3500)为美国Selleck公司产品;奥希替尼、PLK1抑制剂为美国MedChemExpress公司产品;RPMI-1640培养基、胎牛血清为美国Thermo Fisher公司产品;胰蛋白酶为美国Invitrogen 公司产品;Tris-base为德国BioFroxx公司产品;SRB为美国Sigma-Aldrich公司产品。
细胞培养箱与生物安全柜为美国Thermo Electron公司产品;普通倒置显微镜为日本Leica公司产品;多功能酶标仪为美国Thermo Fisher Scientific公司产品。
1.2. 细胞培养
NCI-H1975细胞在添加了10%胎牛血清、100 U/mL青霉素和100 μg/mL链霉素的RPMI-1640培养基中培养,所有细胞均置于培养箱(37 ℃、5%二氧化碳)中,待细胞生长至合适密度时进行传代培养,经0.05%胰蛋白酶消化传代,所有实验细胞均传代不超过十次。
1.3. 药物浓度递增法构建耐药株
取汇合度60%~80%的人NSCLC细胞株NCI-H1975(即药物敏感细胞株,记为H1975敏感细胞),在培养基加入1 nmol/L的奥希替尼,培养24 h后观察细胞状态。若细胞大量死亡则弃含药培养基并用磷酸盐缓冲液清洗,更换不含药物培养基继续培养,待细胞生长速度至对数生长期,消化传代后再次加入1 nmol/L奥希替尼培养24 h,及时观察细胞状态,若细胞仍大量死亡则继续做撤药处理,待细胞增殖至正常状态后,重复上述循环,直至当细胞在1 nmol/L 奥希替尼培养条件下稳定增殖并连续传代三次以上。按20 nmol/L的药物浓度差提升奥希替尼浓度,继续培养24 h。若细胞增殖速度异常缓慢或大量死亡,则更换为原含1 nmol/L奥希替尼的培养基,待其恢复至正常增殖速度后再次更换为筛选浓度培养基。重复上述循环,直至细胞在该筛选药物浓度培养条件下稳定增殖并连续传代三次以上。期间需在不同时间段测量细胞对于奥希替尼的IC50以确定耐药指数,耐药指数=耐药细胞IC50/亲本细胞IC50。耐药株构建时间共计7个月,最终细胞维持培养浓度为1 μmol/L,耐药株记为H1975耐药细胞。
1.4. 肿瘤经典通路抑制剂库筛选与奥希替尼具有协同作用的化合物
将肿瘤经典通路抑制剂库中化合物(0.1 μmol/L)与奥希替尼(0.5 μmol/L)联合用于H1975耐药株中,使用SRB染色检测并计算细胞株的增殖抑制率,以单用奥希替尼(0.5 μmol/L)的细胞株增殖抑制率为对照,筛选出肿瘤经典通路抑制剂库中与奥希替尼存在协同作用的化合物。
1.5. SRB染色法考察细胞存活率
H1975敏感细胞和H1975耐药细胞分别以3.0×104个/孔的细胞密度接种于96孔板中(100 μL/孔),于细胞培养箱中培养24 h后,给予梯度浓度的相应化合物孵育72 h。孵育结束弃去上清液,每孔加入100 μL 10%三氯乙酸溶液固定,弃培养液,用双蒸水洗涤后烘干。每孔加入100 μL SRB染色液(用1%冰醋酸配置,每500 mL加入1.5~2.0 g SRB粉末),室温放置20 min,用1%冰醋酸洗涤干净后烘干。每孔加入100 μL Tris-base碱溶液溶解结合到细胞内的SRB染色液,置于室温摇床20 min后,检测515 nm波长处的吸光度值并计算细胞存活率。
1.6. 差异基因富集分析考察耐药株中PLK1信号通路的变化情况
选取GSE146850队列中H1975奥希替尼耐药株与敏感株以及GSE153183队列中PC-9耐药株与敏感株的转录组数据,使用R软件包limma 3.42.2对数据进行比较并计算差异基因,筛选差异基因标准为P<0.05且log2FC的绝对值大于1。对分析得到的差异基因采用R软件包clusterProfiler 4.0进行基因集富集分析,评估差异基因中相关信号通路的富集情况。
1.7. 采用NSCLC患者队列数据分析PLK1表达水平与患者无进展生存期的相关性
选取11例接受过奥希替尼治疗后的EGFR突变的NSCLC患者的RNA测序数据与生存随访数据(https://github.com/aleighbrown/pwgs_snakemake),采用皮尔逊相关性分析考察患者PLK1 mRNA表达水平与患者无进展生存期的相关性。
1.8. 统计学方法
采用RStudio(R软件包ggplot2)、Excel和Graphpad Prism 8软件进行相关实验数据的分析和绘图。所有数据均以均数±标准差(±s)表示,组间差异比较采用双侧student’s t检验,P<0.05表示差异具有统计学意义。
2. 结果
2.1. 奥希替尼耐药株构建成功
SRB染色结果见图1。H1975敏感细胞多呈多边形,细胞轮廓明显,易成团生长;H1975耐药细胞多呈梭形,轮廓不规则,分散程度较高。处理72 h后奥希替尼对H1975耐药细胞的IC50显著高于H1975敏感细胞(分别为1550.00和35.67 nmol/L,P<0.05),耐药指数为43.45,见图2。表明H1975耐药株构建成功。
图1. 奥希替尼敏感和耐药H1975细胞显微镜下形态.

A:敏感细胞多呈多边形,细胞轮廓明显,易成团生长;B:耐药细胞多呈梭形,轮廓不规则,分散程度较高. 标尺=200 μm.
图2. 奥希替尼处理72 h后敏感和耐药H1975细胞的生存曲线.

2.2. 筛选出与奥希替尼具有协同作用的肿瘤经典通路抑制剂
SRB染色结果显示,实验浓度下单用奥希替尼对H1975耐药细胞的增殖抑制率为35.80%;而将肿瘤经典通路抑制剂库中化合物(0.1 μmol/L)与奥希替尼(0.5 μmol/L)联合作用72 h,抑制率前十的化合物见表1。其中合用抑制率排名前二的化合物为PLK1抑制剂GSK 461364和BI 2536,与奥希替尼合用后对H1975耐药株的抑制率分别达到90.10%和87.90%。表明PLK1抑制剂GSK 461364、BI 2536与奥希替尼合用能够显著抑制奥希替尼耐药细胞的增殖,提示奥希替尼的耐药现象可能与NSCLC中PLK1异常激活相关。
表1.
与奥希替尼合用72 h对耐药H1975细胞抑制率排名前十的肿瘤经典通路抑制剂及其靶点
| 序 号 | 药物名称 | 合用抑制率(%) | 靶 点 |
|---|---|---|---|
| 1 | GSK 461364 | 90.10 | PLK1 |
| 2 | BI 2536 | 87.90 | PLK1和BRD4 |
| 3 | Barasertib | 85.63 | Aurora B |
| 4 | GSK 1070916 | 85.50 | Aurora B/C |
| 5 | Sapanisertib | 85.22 | mTOR1/2 |
| 6 | BAY-876 | 84.75 | GLUT1 |
| 7 | LY2090314 | 84.63 | GSK-3 |
| 8 | Berzosertib | 82.76 | ATR |
| 9 | Vistusertib | 79.82 | mTOR1/2 |
| 10 | Dabrafenib Mesylate | 79.50 | Raf |
PLK:Polo样激酶;BRD:溴结构域蛋白;Aurora B/C:极光激酶B/C;mTOR:哺乳动物雷帕霉素靶蛋白;GLUT:葡萄糖转运体;GSK:糖原合酶激酶;ATR:共济失调毛细血管扩张和Rad3相关激酶;Raf:快速加速纤维肉瘤蛋白.
2.3. 奥希替尼耐药可能与PLK1通路激活有关
奥希替尼耐药株(H1975耐药细胞与PC-9耐药细胞)中PLK1调控通路和细胞周期通路被显著激活(P<0.01),见图3。同时,在接受奥希替尼治疗的EGFR突变NSCLC患者队列中,奥希替尼治疗后患者的PLK1 mRNA水平与无进展生存期呈负相关(R=-0.62,P<0.05),见图4。结果提示,NSCLC细胞中PLK1过度激活可能导致细胞对奥希替尼耐药。
图3. 奥希替尼耐药株与亲本敏感株中PLK1调控通路和细胞周期通路相关基因富集结果.

A:H1975细胞PLK1调控通路;B:H1975细胞周期通路;C:PC-9细胞PLK1调控通路;D:PC-9细胞周期通路. PLK:Polo样激酶.
图4. 接受奥希替尼治疗的EGFR突变非小细胞癌患者中PLK1 mRNA水平与无进展生存期的相关性.

EGFR:表皮生长因子受体;PLK:Polo样激酶.
2.4. 奥希替尼耐药细胞对PLK1抑制剂更敏感
采用PLK1单靶点抑制剂伏拉塞替(BI 6727)和GSK 461364进行细胞增殖抑制实验。SRB染色结果显示,伏拉塞替和GSK 461364作用72 h后,H1975耐药细胞的IC50(分别为19.36和8.24 nmol/L)显著低于H1975敏感细胞(分别为37.83和28.54 nmol/L,均P<0.05),见图5,表明奥希替尼耐药细胞对PLK1抑制剂更敏感。
图5. 伏拉塞替或GSK 461364作用后奥希替尼敏感和耐药H1975细胞的生存曲线.

GSK:糖原合酶激酶;PLK:Polo样激酶.
2.5. PLK1抑制剂与奥希替尼合用可增强对耐药细胞的增殖抑制作用
根据上述结果,选择对H1975耐药细胞增殖抑制率约在30%的PLK1抑制剂浓度与奥希替尼合用。伏拉塞替(12.5 nmol/L)和GSK 461364(6.25 nmol/L)分别与奥希替尼联合作用于H1975耐药细胞72 h后,H1975耐药细胞的存活率较单用奥希替尼组显著降低(均P<0.05,图6),表明PLK1抑制剂与奥希替尼合用可增强对耐药细胞的增殖抑制作用,提示合用PLK1抑制剂与奥希替尼可能是解决临床NSCLC奥希替尼耐药的新途径。
图6. 伏拉塞替或GSK 461364联合奥希替尼作用后H1975耐药细胞的生存曲线.
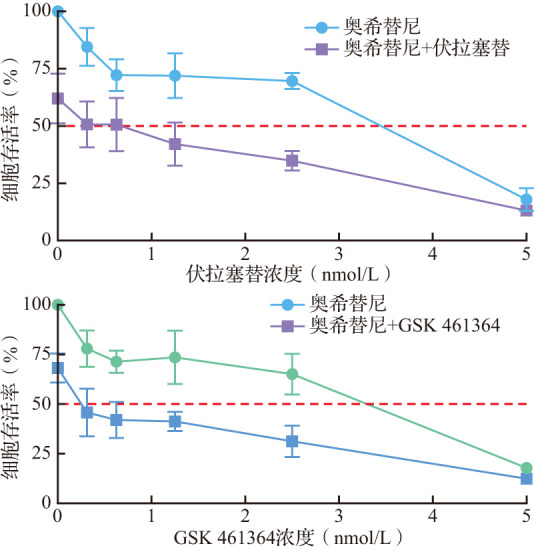
PLK:Polo样激酶;GSK:糖原合酶激酶.
3. 讨论
NSCLC是世界上最为常见的癌症相关死亡原因之一,全球每年死亡人数超过8万人[11]。约50%的亚洲NSCLC患者和11%~16%的西方国家NSCLC患者携带EGFR突变[2]。数据显示,携带EGFR敏感突变的患者获益于EGFR-TKI,其客观缓解率与无进展生存期有效改善[12]。
然而,作为携带EGFR突变的NSCLC患者的标准一线治疗手段,第一代与第二代EGFR-TKI如吉非替尼和阿法替尼等虽然在患者中显示出良好的临床收益,但多数患者在治疗后产生获得性耐药[13]。为了解决第一、二代EGFR-TKI耐药问题,研究者开发了第三代EGFR-TKI奥希替尼用于对EGFR-TKI获得性耐药的T790M阳性NSCLC患者[14]。奥希替尼在T790M驱动的第一代EGFR-TKI获得性耐药患者中显示出优越的疗效及更低的皮肤和胃肠道毒性[15]。然而与早期EGFR-TKI类似,接受奥希替尼治疗的患者依然不可避免地出现获得性耐药。现有研究揭露的耐药机制可分为EGFR依赖性耐药和EGFR非依赖性耐药[16]。EGFR依赖性耐药机制主要包括EGFR三级突变如EGFR-C797、EGFR-L718和EGFR-G719突变等类型,阻碍奥希替尼与EGFR之间形成共价结合,从而赋予患者耐药性[17]。EGFR非依赖性耐药机制主要包括旁路途径激活、组织学转化和致癌融合等[18]。目前,c-MET扩增是奥希替尼最常见的EGFR非依赖性耐药机制,占奥希替尼进展患者的5%~24%,c-MET通过与配体肝细胞生长因子结合,导致受体磷酸化并激活EGFR下游信号通路的旁路导致奥希替尼耐药[19];EGFR下游信号通路如PI3K-AKT、RAS-MAPK-ERK、JAK-STAT等异常激活也会促进肿瘤发生、增殖、迁移、侵袭和对治疗的抵抗[20];除此之外,含卷曲螺旋结构域6与RET融合等致癌融合突变也会赋予患者奥希替尼耐药性[21]。尽管在表征奥希替尼的部分分子耐药性方面已经取得了进展,但由于耐药性的异质性和复杂性,仍有30%~50%患者的耐药机制仍未明确[22]。
联合其他分子靶向药物是一种解决或延缓奥希替尼耐药的治疗策略。目前,奥希替尼联合c-MET抑制剂沃利替尼、MEK抑制剂司美替尼或RET抑制剂普拉替尼等多种分子靶向药物的临床试验显示出良好的肿瘤治疗作用与可控的毒副作用[23-25]。基于此,本研究首先成功构建了奥希替尼耐药株,并将肿瘤经典通路抑制剂库中的化合物与奥希替尼合用,旨在筛选出能有效克服奥希替尼耐药的药物组合。结果显示,在所有化合物中,PLK1抑制剂GSK 461364和BI 2536与奥希替尼合用后对奥希替尼耐药株的增殖抑制率最高。结合基因集富集分析结果,奥希替尼耐药株样本中PLK1调控通路和细胞周期通路显著激活,表明NSCLC细胞对奥希替尼耐药可能与PLK1过度激活有关,提示联合使用PLK1抑制剂与奥希替尼可能是临床上解决NSCLC患者奥希替尼耐药的新途径。接着,本研究在细胞水平考察了奥希替尼敏感和耐药细胞对PLK1抑制剂的敏感性,结果显示奥希替尼耐药细胞对PLK1抑制剂更加敏感,且PLK1抑制剂与奥希替尼合用对奥希替尼耐药细胞的增殖有较好的抑制作用。
PLK1作为丝/苏氨酸蛋白激酶的Polo亚家族成员,在有丝分裂和维持基因组稳定性方面发挥至关重要的作用,调控细胞周期进展、分化和存活等多种细胞过程[7]。相较于靶向微管等传统抗有丝分裂药物,靶向有丝分裂调节激酶PLK1可降低其在正常细胞中的微管抑制而产生的毒副作用[26]。作为具有癌症治疗潜力的靶点,近年来,研究者开发了多种PLK1抑制剂并进入临床试验。本文所使用的PLK1抑制剂伏拉塞替以及GSK 461364均为ATP竞争性抑制剂,其中伏拉塞替为二氢蝶酮衍生物类化合物,与PLK1的激酶结构域R57、L59和C133等残基通过氢键相互作用[27];GSK 461364为噻吩衍生类化合物,其对PLK1具有高度选择性[28]。目前,研究进展最快的PLK1抑制剂伏拉塞替在多组临床试验中表现出优异的抗肿瘤活性及安全性,被美国食品药品监督管理局称为治疗急性髓性白血病的“突破性疗法”[29]。然而,目前开发的PLK1抑制剂大多靶向PLK1的ATP结合位点,PLK家族ATP结合口袋的高度保守性使得多数化合物不可避免地面临脱靶效应,单药治疗时抑制剂的非特异性活性常导致剂量限制性毒性[30]。实体肿瘤对单一PLK1抑制剂的反应有限[31]。目前尚未有一款成熟的PLK1抑制剂进入临床。
本研究初步揭示了PLK1可能是解决临床奥希替尼耐药的有效靶点。关于合用PLK1抑制剂与奥希替尼增强对耐药细胞抗肿瘤作用的具体机制,有研究揭示了PLK1抑制剂伏拉塞替与厄洛替尼合用可促进肿瘤细胞的DNA损伤以及G2/M停滞并引发细胞凋亡[32]。除此之外,在EGFR-TKI获得性耐药的NSCLC患者肿瘤标本中有丝分裂调控激酶Aurora B表达增加且明显激活[33-34],而PLK1通过在Thr236位点上磷酸化Aurora B调控其激酶活性[35-36]。推测抑制PLK1可能通过进一步抑制Aurora B激酶活性从而增强奥希替尼对耐药细胞的增殖抑制作用。
综上所述,本研究通过体外筛选、细胞水平评估和生物信息学分析证实,PLK1的异常激活可能是奥希替尼耐药的新机制,PLK1抑制剂与奥希替尼合用对奥希替尼耐药细胞的抗肿瘤作用更强,可为奥希替尼耐药的NSCLC患者的临床治疗提供了新的思路。
Acknowledgments
研究得到国家自然科学基金(82104193)和浙江省自然科学基金(LY22H310001)支持
Acknowledgments
This work was supported by National Natural Science Foundation of China (82104193) and Natural Science Foundation of Zhejiang Province (LY22H310001)
[缩略语]
非小细胞肺癌(non-small cell lung carcinoma,NSCLC);表皮生长因子受体(epidermal growth factor receptor,EGFR);EGFR激酶抑制剂(EGFR-tyrosine kinase inhibitors,EGFR-TKI);Polo样激酶(Polo-like kinase,PLK);磺酰罗丹明B(sulforhodamine B,SRB);半抑制浓度(half maximal inhibitory concentration,IC50);差异倍数(fold change,FC);糖原合酶激酶(glycogen synthase kinase,GSK);细胞间充质-上皮转化因子(cellular-mesenchymal to epithelial transition factor,c-MET);转染过程中重排(rearranged during transfection,RET);丝裂原活化的细胞外信号调节激酶(mitogen-activated extracellular signal-regulated kinase,MEK);腺苷三磷酸(adenosine triphosphate,ATP)
利益冲突声明
所有作者均声明不存在利益冲突
Conflict of Interests
The authors declare that there is no conflict of interests
参考文献(References)
- 1.CHEN P, LIU Y, WEN Y, et al. Non-small cell lung cancer in China [J]. Cancer Commun (Lond), 2022, 42(10): 937-970. 10.1002/cac2.12359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.SHI Y, AU J S, THONGPRASERT S, et al. A pro-spective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER)[J]. J Thorac Oncol, 2014, 9(2): 154-162. 10.1097/jto.0000000000000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.PARK K, TAN E H, O’BYRNE K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial[J]. Lancet Oncol, 2016, 17(5): 577-589. 10.1016/s1470-2045(16)30033-x [DOI] [PubMed] [Google Scholar]
- 4.JÄNNE P A, YANG J C, KIM D W, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer[J]. N Engl J Med, 2015, 372(18): 1689-1699. 10.1056/nejmoa1411817 [DOI] [PubMed] [Google Scholar]
- 5.RAMALINGAM S S, YANG J C, LEE C K, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer[J]. J Clin Oncol, 2018, 36(9): 841-849. 10.1200/jco.2017.74.7576 [DOI] [PubMed] [Google Scholar]
- 6.THRESS K S, PAWELETZ C P, FELIP E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M[J]. Nat Med, 2015, 21(6): 560-562. 10.1038/nm.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ANDRYSIK Z, BERNSTEIN W Z, DENG L, et al. The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus[J]. Nucleic Acids Res, 2010, 38(9): 2931-2943. 10.1093/nar/gkq011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.STREBHARDT K, ULLRICH A. Targeting polo-like kinase 1 for cancer therapy[J]. Nat Rev Cancer, 2006, 6(4): 321-330. 10.1038/nrc1841 [DOI] [PubMed] [Google Scholar]
- 9.KIM D E, SHIN S B, KIM C H, et al. PLK1-mediated phosphorylation of β-catenin enhances its stability and transcriptional activity for extracellular matrix remodeling in metastatic NSCLC[J]. Theranostics, 2023, 13(3): 1198-1216. 10.7150/thno.79318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LEE K S, BURKE TR J R, PARK J E, et al. Recent advances and new strategies in targeting Plk1 for anticancer therapy[J]. Trends Pharmacol Sci, 2015, 36(12): 858-877. 10.1016/j.tips.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.WANG M, HERBST R S, BOSHOFF C. Toward personalized treatment approaches for non-small-cell lung cancer[J]. Nat Med, 2021, 27(8): 1345-1356. 10.1038/s41591-021-01450-2 [DOI] [PubMed] [Google Scholar]
- 12.RAMALINGAM S S, VANSTEENKISTE J, PLAN-CHARD D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC[J]. N Engl J Med, 2020, 382(1): 41-50. 10.1056/nejmoa1913662 [DOI] [PubMed] [Google Scholar]
- 13.WESTOVER D, ZUGAZAGOITIA J, CHO B C, et al. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors[J]. Ann Oncol, 2018, 29(suppl_1): i10-i19. 10.1093/annonc/mdx703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GREIG S L. Osimertinib: first global approval[J]. Drugs, 2016, 76(2): 263-273. 10.1007/s40265-015-0533-4 [DOI] [PubMed] [Google Scholar]
- 15.SORIA J C, OHE Y, VANSTEENKISTE J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer[J]. N Engl J Med, 2018, 378(2): 113-125. 10.1056/nejmoa1713137 [DOI] [PubMed] [Google Scholar]
- 16.PASSARO A, JÄNNE P A, MOK T, et al. Overcoming therapy resistance in EGFR-mutant lung cancer[J]. Nat Cancer, 2021, 2(4): 377-391. 10.1038/s43018-021-00195-8 [DOI] [PubMed] [Google Scholar]
- 17.ZALAQUETT Z, CATHERINE RITA HACHEM M, KASSIS Y, et al. Acquired resistance mechanisms to osimertinib: the constant battle[J]. Cancer Treat Rev, 2023, 116: 102557. 10.1016/j.ctrv.2023.102557 [DOI] [PubMed] [Google Scholar]
- 18.COOPER A J, SEQUIST L V, LIN J J. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management[J]. Nat Rev Clin Oncol, 2022, 19(8): 499-514. 10.1038/s41571-022-00639-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.OXNARD G R, HU Y, MILEHAM K F, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib[J]. JAMA Oncol, 2018, 4(11): 1527-1534. 10.1001/jamaoncol.2018.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.NAGANO T, TACHIHARA M, NISHIMURA Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy[J]. Cells, 2018, 7(11): 212. 10.3390/cells7110212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.OFFIN M, SOMWAR R, REKHTMAN N, et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers[J]. JCO Precis Oncol, 2018, 2: PO.18. 00126. 10.1200/po.18.00126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.FU K, XIE F, WANG F, et al. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance[J]. J Hematol Oncol, 2022, 15(1): 173. 10.1186/s13045-022-01391-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.SEQUIST L V, HAN J Y, AHN M J, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study[J]. Lancet Oncol, 2020, 21(3): 373-386. 10.1016/s1470-2045(19)30785-5 [DOI] [PubMed] [Google Scholar]
- 24.EBERLEIN C A, STETSON D, MARKOVETS A A, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased depen-dence on RAS signaling in preclinical models[J]. Cancer Res, 2015, 75(12): 2489-2500. 10.1158/0008-5472.can-14-3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.PIOTROWSKA Z, ISOZAKI H, LENNERZ J K, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion[J]. Cancer Discov, 2018, 8(12): 1529-1539. 10.1158/2159-8290.cd-18-1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.BARR F A, SILLJÉ H H, NIGG E A. Polo-like kinases and the orchestration of cell division[J]. Nat Rev Mol Cell Biol, 2004, 5(6): 429-440. 10.1038/nrm1401 [DOI] [PubMed] [Google Scholar]
- 27.KOTHE M, KOHLS D, LOW S, et al. Selectivity-determining residues in Plk1[J]. Chem Biol Drug Des, 2007, 70(6): 540-546. 10.1111/j.1747-0285.2007.00594.x [DOI] [PubMed] [Google Scholar]
- 28.GILMARTIN A G, BLEAM M R, RICHTER M C, et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis[J]. Cancer Res, 2009, 69(17): 6969-6977. 10.1158/0008-5472.CAN-09-0945 [DOI] [PubMed] [Google Scholar]
- 29.VAN DEN BOSSCHE J, LARDON F, DESCHOOL-MEESTER V, et al. Spotlight on volasertib: preclinical and clinical evaluation of a promising Plk1 inhibitor[J]. Med Res Rev, 2016, 36(4): 749-786. 10.1002/med.21392 [DOI] [PubMed] [Google Scholar]
- 30.ZHANG J, ZHANG L, WANG J, et al. Polo-like kinase 1 inhibitors in human cancer therapy: development and therapeutic potential[J]. J Med Chem, 2022, 65(15): 10133-10160. 10.1021/acs.jmedchem.2c00614 [DOI] [PubMed] [Google Scholar]
- 31.MEDEMA R H, LIN C C, YANG J C. Polo-like kinase 1 inhibitors and their potential role in anti-cancer therapy, with a focus on NSCLC[J]. Clin Cancer Res, 2011, 17(20): 6459-6466. 10.1158/1078-0432.ccr-11-0541 [DOI] [PubMed] [Google Scholar]
- 32.WANG Y, SINGH R, WANG L, et al. Polo-like kinase 1 inhibition diminishes acquired resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer with T790M mutations[J]. Oncotarget, 2016, 7(30): 47998-48010. 10.18632/oncotarget.10332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.NILSSON M B, SUN H, ROBICHAUX J, et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components[J]. Sci Transl Med, 2020, 12(559): eaaz4589. 10.1126/scitranslmed.aaz4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.BERTRAN-ALAMILLO J, CATTAN V, SCHOUMA-CHER M, et al. AURKB as a target in non-small cell lung cancer with acquired resistance to anti-EGFR therapy[J]. Nat Commun, 2019, 10(1): 1812. 10.1038/s41467-019-09734-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.TANAKA K, YU H A, YANG S, et al. Targeting aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis[J]. Cancer Cell, 2021, 39(9): 1245-1261.e6. 10.1016/j.ccell.2021.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LEE H S, MIN S, JUNG Y E, et al. Spatiotemporal coordination of the RSF1-PLK1-Aurora B cascade establishes mitotic signaling platforms[J]. Nat Commun, 2021, 12(1): 5931. 10.1038/s41467-021-26220-z [DOI] [PMC free article] [PubMed] [Google Scholar]