

Abstract
The myocyte enhancer factor 2 C (MEF2C) gene encodes a transcription factor important for neurogenesis and synapse development and contains common variants associated with intelligence (IQ) and educational attainment (EA). Here, we took gene expression data from the mouse cortex of a Mef2c mouse model with a heterozygous DNA binding-deficient mutation of Mef2c (Mef2c-het) and combined these data with MEF2C ChIP-seq data from cortical neurons and single-cell data from the mouse brain. This enabled us to create a set of genes that were differentially regulated in Mef2c-het mice, represented direct target genes of MEF2C and had elevated in expression in cortical neurons. We found this gene-set to be enriched for genes containing common genetic variation associated with IQ and EA. Genes within this gene-set that were down-regulated, i.e. have reduced expression in Mef2c-het mice versus controls, were specifically significantly enriched for both EA and IQ associated genes. These down-regulated genes were enriched for functionality in the adenylyl cyclase signalling system, which is known to positively regulate synaptic transmission and has been linked to learning and memory. Within the adenylyl cyclase signalling system, three genes regulated by MEF2C, CRHR1, RGS6, and GABRG3, are associated at genome-wide significant levels with IQ and/or EA. Our results indicate that genetic variation in MEF2C and its direct target genes within cortical neurons contribute to variance in cognition within the general population, and the molecular mechanisms involved include the adenylyl cyclase signalling system’s role in synaptic function.
Keywords: MEF2C, cognition, RNA-seq, ChIP-seq, adenylyl cyclase
Introduction
The myocyte enhancer factor 2 C (MEF2C) gene encodes a transcription factor protein that regulates gene expression in the brain across the lifespan from neurodevelopment, including neurogenesis and synapse development, to adulthood [1]. Microdeletions on chromosome 5q14.3 encompassing MEF2C plus truncating and missense mutations within the gene have been described as causal for MEF2C haploinsufficiency syndrome (MCHS) where typical clinical characteristics include severe global developmental delay with absent speech, limited walking, seizures, stereotypic movements and features of autism spectrum disorder (ASD) [2]. MEF2C also contains common variants associated with schizophrenia (SCZ) [3], intelligence (IQ) [4], and educational attainment (EA) [5].
Multiple studies have used mouse models of Mef2c to investigate its role in behaviour, cognition, and its influence on the structure and function of the brain and constituent cell types, in order to identify molecular mechanisms that may underpin human phenotypes associated with MEF2C. As constitutive Mef2c null mice have early lethality [6], models have been based on conditional knockout (cKO) of the gene. Homozygous postnatal deletion of Mef2c in the brain resulted in a significant increase in spine numbers in the hippocampus, plus alterations in locomotor activity and motor coordination deficits [7]. Homozygous embryonic deletion of Mef2c in the forebrain resulted in impairment in hippocampal-dependent learning and memory, with Mef2c having a mediating role in synapse formation during activity-dependent refinement of synaptic connectivity [8]. Homozygous embryonic deletion of Mef2c in cortical and hippocampal excitatory neurons reduced cortical network activity with an imbalance of excitatory and inhibitory synaptic transmission [9]. These Mef2c cKO mice exhibited impairments in multiple behavioural phenotypes, e.g. fear learning and memory, multiple social behaviours, socially-motivated ultrasonic vocalizations, reward-related behaviours, and repetitive motor behaviours [9]. As Mef2c is a transcription factor, the latter study also used RNA sequencing (RNA-seq) to study the effect of Mef2c cKO on gene expression in the cortex. Differentially expressed genes (DEGs) were enriched for genes involved in neuron differentiation and development (up-regulated genes) and synaptic transmission and ion transport (down-regulated genes), and were enriched for ASD risk genes [9]. In a subsequent study, we reported that these Mef2c cKO DEGs were also enriched for common genetic variation associated with SCZ, IQ, and EA, as well as rare de novo mutations (DNMs) reported in ASD and intellectual disability (ID) [10].
A weakness in the models described above in terms of their construct validity for human phenotypes caused by mutation of the MEF2C gene is that they are based on homozygous deletion of Mef2c, whereas individuals affected by MCHS have a heterozygous (Het) mutation. To address this, Harrington and colleagues generated a global heterozygous Mef2c mutant mouse lacking exon 2, which encodes a large portion of the DNA-binding MADS/MEF2 domains that are often the sites for MCHS mutations [11]. These DNA-binding deficient Mef2c-Het mice were found to exhibit many of the symptoms associated with MCHS in humans, including disturbances in social interaction and communication, hyperactivity, repetitive behaviours, and reduced pain sensitivity [11]. At a biological level, these mutant mice displayed deficits in presynaptic and postsynaptic glutamatergic excitatory transmission in the somatosensory cortex. RNA-seq was performed on the whole cortex of 35–40 day postnatal Mef2c-het and control mice, capturing hundreds of genes differentially expressed as a result of the mutation. These genes were found to be significantly enriched for ASD risk genes and genes expressed in excitatory neurons [11].
Mef2c is expressed in both neurons and microglia. The Mef2c-het mutation exhibited cell type specific behavioural effects. The mutation solely in forebrain excitatory neurons reproduced most of the global effects except for social deficits, whereas the mutation in microglia reproduced most of the global effects except for anxiety-like behaviours. The mutation solely in parvalbumin-positive GABAergic interneurons and cerebellar Purkinje cells showed no effects at all. Overall, this suggested that MEF2C regulates typical brain development and function through multiple cell types [11].
Here, we sought to use gene expression data from the Mef2c-het model to explore further the contribution of both common and rare genetic variation within the set of genes regulated by Mef2c to neurodevelopmental disorders and cognitive phenotypes in a cell type specific manner. We integrated gene expression data with chromatin immunoprecipitation sequencing (ChIP-seq) data for MEF2C [12] and single cell RNA-seq (scRNA-seq) data [13] to identify dysregulated genes in cortical and microglial cells and direct transcriptional targets of MEF2C in cortical neurons. This allowed for the cell type specific analysis of the set of genes and biological processes that were directly dysregulated as a consequence of Mef2c mutation.
Results
Generation of MEF2C gene-sets
The original set of DEGs from the Mef2c-het model contained 476 mouse genes (false discovery rate [FDR] < 0.05) [11] When converted to human orthologues for analysis using human genetic data, this “DEGs_All” gene-set contained 460 genes (Supplementary Table 1). By integrating the Mef2c-het gene expression data with ChIP-seq data [12] and scRNA-seq data [13], we generated a set of 139 DEGs (Supplementary Table 2) that are direct transcriptional targets of MEF2C and highly expressed in cortical neurons. We refer to this gene-set as “DEGs_Direct_Neuron”. As ChIP-seq data was not available for microglial cells, our set of 222 DEGs that are expressed in this cell type in the cortex, called “DEGs_Microglia” was just based on integration of the Mef2c-het gene expression data with scRNA-seq data (Supplementary Table 3).
Analysis of MEF2C gene-sets using GWAS data
Gene-set analysis was performed to test the DEGs_All, DEGs_Direct_Neuron and DEGs_Microglia gene-sets for enrichment of common genetic variation associated with SCZ, IQ, and EA using genome-wide association study (GWAS) data. The DEGs_All and DEGs_Microglia gene-sets were not found to be significantly enriched for genes associated with any of these phenotypes. However, the DEGs_Direct_Neuron gene-set was found to be significantly enriched for genes associated with IQ (P = 1.98 × 10−4) and EA (P = 6.77 × 10−6) (Fig. 1A; Supplementary Table 4). In terms of individual genes within DEGs_Direct_Neuron, 16 genes were significantly associated with EA and 12 genes were associated with IQ (with 9 genes associated with both IQ and EA) after multiple testing correction (Supplementary Table 2).
Figure 1.
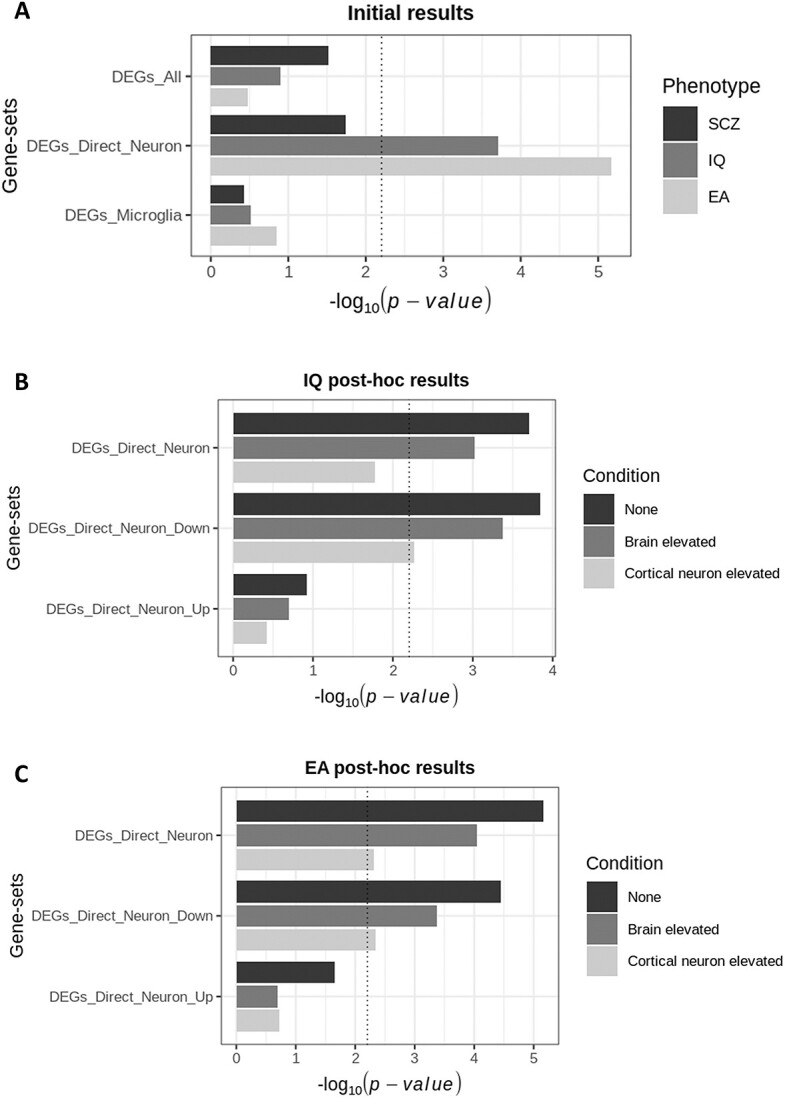
Results from MAGMA gene-set analysis of MEF2C gene-sets using GWAS data. (A) Results from the initial analysis of MEF2C gene-sets using GWAS data for SCZ, IQ and EA. The DEGs_Direct_Neuron gene-set was significantly enriched for common genetic variation associated with IQ and EA. (B, C) Post-hoc results based on these two significant results observed in the initial analysis. DEGs_Direct_Neuron was used to create two more gene-sets based on whether genes were up or down regulated upon MEF2C disruption. Each of the three gene-sets were tested for enrichment of common genetic variation associated with IQ (B) and EA (C), conditioning on brain elevated genes and cortical neuron elevated genes. The vertical lines show the significance threshold using the Bonferroni method to correct for the nine initial tests (P = 0.0056). Full results are detailed in Supplementary Table 4.
To investigate if the results for IQ and EA were more specifically due to genes negatively or positively regulated by Mef2c, we split the DEGs_Direct_Neuron gene-set into DEGs that were up- or down-regulated in Mef2c-Het mice versus controls, creating DEGs_Direct_Neuron_Up (n = 61 genes) and DEGs_Direct_Neuron_Down (n = 78 genes), respectively (Supplementary Table 2). Gene-set analysis results indicated that the enrichment signal is stronger and more significant for down-regulated DEGs in Mef2c-Het mice compared to up-regulated DEGs (Fig. 1B [IQ results] and Fig. 1C [EA results]).
Genes associated with cognition are known to be enriched in multiple brain regions, including the cortex, as well as in various neuronal cell types [4]. Therefore, it is possible that the enrichments detected here could be due to direct target genes of MEF2C in cortical neurons representing a subset of genes with elevated expression in the brain or more specifically cortical neurons. Conditioning the gene-set analysis on brain elevated genes accounted for little of the detected enrichments for IQ and EA, with negligible effect on the P values (Fig. 1B and C; Supplementary Table 4). Conditioning on genes with elevated expression in cortical neurons (n = 4114; listed in Supplementary Table 5) partially accounted for the enrichment of the DEGs_Direct_Neuron gene-set for genes associated with IQ and EA, although the enrichment for EA genes within this gene-set remained significant below the original multiple testing correction P value threshold (P = 0.00486 for EA; Fig. 1C). The enrichments for the DEGs_Direct_Neuron_Down gene-set also remained significant after conditioning on genes with elevated expression in cortical neurons (P = 0.00548 for IQ; P = 0.00448 for EA) (Fig. 1B and C).
There is known to be an overlap between Fragile X mental retardation protein (FMRP) and MEF2C target genes. However, conditioning on FMRP target genes had negligible effect on the enrichments detected here (Supplementary Table 6). To examine if the enrichments we detected for IQ and EA are a property of polygenic phenotypes in general, we obtained GWAS summary statistics for five other phenotypes and tested the MEF2C gene-sets for enrichment in each one. These GWAS included child-onset psychiatric disorders, other brain-related disorders, a non-brain related disease, and height. Our MEF2C gene-sets were not enriched for genes associated with any of these five phenotypes (Supplementary Table 7).
Analysis of MEF2C gene-sets using data on De novo mutations
To investigate the contribution of rare variants in the MEF2C gene-sets to SCZ, ASD, ID, and developmental disorders (DD), we tested if the gene-sets were enriched for synonymous (syn), missense (mis) and loss of function (lof) DNMs that have been reported in trio-based exome sequencing studies of these disorders. The expected number of each mutational class of DNM for each gene is estimated using a mutational background model that takes into account sequence context and gene size [14]. The observed versus expected number of DNMs within each gene-set is compared to the observed versus expected number of DNMs in all genes outside of each gene-set using a two-sample Poisson rate ratio test. The DEGs_Direct_Neuron gene-set was overrepresented for genes containing missense and loss of function DNMs reported for ID/DD at nominal significance levels. Similar to our analysis of the GWAS data above, the enrichments for mis and lof DNMs in ID/DD were stronger in the down-regulated DEGs compared to the up-regulated DEGs. However, none of the P values survive multiple testing corrections (P < 0.004; Supplementary Table 8).
Functional annotation analysis of gene-sets
We performed gene ontology (GO) enrichment analysis to investigate the functionality of the DEGs_Direct_Neuron_Down gene-set that was enriched for IQ and EA associated genes. Four of the top five enriched GO terms from an initial analysis were related to synaptic signalling (Fig. 2A; Supplementary Table 9). However, similar to our gene-set analysis above, we wanted to account for the potentially confounding effect of the origin of this gene-set, i.e. all genes in this gene-set have elevated expression in cortical neurons. Therefore, we performed a competitive GO analysis of the DEGs_Direct_Neuron_Down gene-set using all genes with elevated expression in cortical neurons as the background gene-set. The previously identified GO terms related to synaptic signalling remained significant but were no longer among the top terms. Instead, the top four terms are all related to adenylyl cyclase signalling activity (Fig. 2B; Supplementary Table 10). Adenylyl cyclase is an enzyme that synthesizes cyclic adenosine monophosphate (cAMP) from ATP [15]. cAMP subsequently regulates a number of cellular processes, including synaptic transmission [16] and has been shown to be important for learning and memory [17–19]. The top two GO terms were “activation of adenylate cyclase activity” (P = 1.1 × 10−4; Q = 0.031) and “G protein-coupled receptor signalling pathway” (P = 1.6 × 10−4; Q = 0.029). G protein-coupled receptors, upon binding of a ligand, activate the adenylyl cyclase enzyme [20]. The third most significant term was “corticotropin-releasing hormone (CRH) receptor activity” (P = 3.6 × 10−4; Q = 0.018). CRH receptors are a family of G protein-coupled receptors that bind corticotropin-releasing hormone and lead to activation of adenylyl cyclase and thereby increased levels of cAMP [21]. The fourth most significant term was “corticotrophin-releasing factor (CRF) receptor activity” (P = 3.6 × 10−4; Q = 0.011). The CRF receptor is also a G protein-coupled receptor that when activated leads to elevated cAMP levels.
Figure 2.

Gene ontology over-representation analysis results for DEGs_Direct_Neuron_Down. Top 10 most significant GO terms resulting from hypergeometric tests using all genes as the background gene-set (A) and using all genes with elevated expression in cortical neurons as the background gene-set (B). Full results are detailed in Supplementary Tables 9 and 10.
A total of 14 genes from the gene-set map to at least one of these four terms (see Supplementary Table 11 for their P values from the IQ and EA GWAS and a short summary of their function). Corticotropin releasing hormone receptor 1 (CRHR1) maps to all four GO terms and was individually significant in both the IQ and EA GWASs at genome-wide significant levels (P = 5.5 × 10−7 for IQ; P = 1.6 × 10−11 for EA). CRHR1 is the main CRH receptor in the brain and genetic variants in this gene have also been found to be significantly associated with several phenotypes including major depressive episode, generalized anxiety disorder, antidepressant response, stress and panic [22]. Two other genes within these GO terms were genome-wide significant for EA; regulator of g protein signalling 6 (RGS6; P = 9.9 × 10−8) and gamma-aminobutyric acid type a receptor subunit gamma 3 (GABRG3; P = 1.4 × 10−6). A meta-analysis conducted by the Cross-Disorder Group of the Psychiatric Genomic Consortium identified a SNP in RGS6 as one of 23 genetic loci with pleiotropic effects on ≥ four out of the eight studies psychiatric disorders [23]. GABRG6 was identified to contain SNPs significantly associated with ASD using a family-based association study and was enriched for rare variants observed in ASD patients compared to controls [24].
The latest EA GWAS results [25] lists multiple independent genome-wide significant SNPs at each of CRHR1, RGS6 and GABRG3. We reviewed the Genotype-Tissue Expression (GTEx) project data for these SNPs to link associated alleles with effects on gene expression using expression quantitative trait loci (eQTLs). At CRHR1, the T allele of the most associated SNP (rs60814418) is associated with both lower EA and reduced expression of CRHR1 in two brain regions following GTEx experiment wide-correction (Caudate (basal ganglia), P = 6.43 × 10−5; Hippocampus, P = 5.01 × 10−5). At RGS6, none of the genome-wide significant SNPs are eQTLs in brain regions. However, the A allele of the most associated SNP (rs12897542) is associated with both lower EA and reduced expression of RGS6 in heart tissue following GTEx experiment wide-correction (P = 4.3 × 10−7). Finally for GABRG3, the C allele of the most associated SNP (rs891793) is associated with both lower EA and but increased expression of GABRG3 in two brain regions following GTEx experiment wide-correction (Cerebellar Hemisphere, P = 3.4 × 10−6; Cerebellum, P = 3.71 × 10−6).
Discussion
We have combined data from a genetic mouse model and GWAS to investigate the network of genes regulated by the transcription factor MEF2C, a causative gene for a rare neurodevelopmental disorder with associated cognitive deficits and a contributory gene to variance in cognitive ability in the general population. We sought to leverage gene expression data from the Mef2c-het mouse model to investigate if the set of genes regulated by MEF2C are also loci where common genetic variation influences cognitive ability, and from there identify the biological functions that may be involved. We report that direct target genes of MEF2C that have elevated expression in cortical neurons and are differentially expressed as a consequence of heterozygous mutation of MEF2C in the Mef2c-het mouse model are significantly enriched for genes associated with IQ and EA. MEF2C acts as both a transcriptional activator and repressor with cell type specific functionality. MEF2C’s activator function is more prominent in excitatory neurons and its repressor function more prominent in microglia [11]. It can be inferred that genes downregulated in Mef2c-het mice are activated or positively regulated by MEF2C under normal conditions, whereas genes upregulated in Mef2c-het mice are normally repressed or negatively regulated by MEF2C. Our observation that IQ- and EA-associated genes were concentrated in the neuronal genes downregulated in Mef2c-het mice suggests that variation in genes regulated by MEF2C that influences cognitive function in the general population resides in genes that are ordinarily activated by MEF2C and expressed in neurons rather than microglia. Functional annotation of these genes point to a role for adenylate cyclase activity in cognition where CRHR1, RGS6, and GABRG3 are functionally involved and are individually associated with cognitive phenotypes by GWAS.
The adenylyl cyclases (ACs) catalyze the production of cAMP, the ubiquitous second messenger in a crucial signalling pathway for learning and memory in both invertebrates and vertebrates. Characterization of D. melanogaster memory mutants revealed that several are due to defects in cAMP signalling. These include rutabaga, which encodes a Ca2+-stimulated adenylyl cyclase [26], amnesiac, which encodes an adenylyl cyclase–activating peptide [27] and a gene that encodes a subunit of the cAMP-dependent protein kinase A (PKA), the main cAMP target [28]. In another model for learning and memory, the mechanosensory neurons of Aplysia californica, cAMP/PKA signalling contributes to short- and long-lasting forms of synaptic plasticity, learning, and memory [29]. Gene disruption studies in mice have also demonstrated that adenylyl cyclase activity is required for hippocampus-dependent memory formation. For example, reduction of PKA activity in transgenic mice expressing a dominant-negative R subunit, R (AB), causes defects in late-phase long-term potentiation (L-LTP), spatial memory, and long-term contextual fear conditioning [30]. Mice lacking the Ca2+/CaM-stimulated adenylyl cyclase (AC1) display presynaptic and postsynaptic functional defects at thalamocortical synapses and have impaired spatial memory [17,31]. Furthermore, mice that lack both type 1 and 8 adenylyl cyclases (AC1 and AC8) ablate L-LTP and long-term memory (LTM) for contextual and passive avoidance learning [32]. It is hypothesized that L-LTP and LTM are both dependent upon a Ca2+ signal generated through activation of NMDA receptors. NMDA receptors trigger various signal transduction pathways, including the Erk/MAPK and PKA pathways, by stimulating the activity of adenylyl cyclases. These pathways activate CREB transcriptional pathways, resulting in the expression of genes essential for L-LTP and LTM [33]. It has also been demonstrated that learning in the bar-pressing task increased adenylyl cyclase activity [34], and that learning in an inhibitory avoidance learning task increased cAMP content [35]. However, recent studies indicate that increases in cAMP signalling may not necessarily lead to memory enhancement. In D. melanogaster, expression of constitutively active Gsα, a general activator of adenylyl cyclases, disrupts learning and memory [36]. Gene ablation of Gi, a general inhibitor of adenylyl cyclases, disrupts hippocampus-dependent memory in mice, emphasizing that continuous increases in adenylyl cyclase activity in the brain might have a negative impact on memory [37]. These findings show that memory formation depends upon a balance between mechanisms for increasing and decreasing cAMP.
An imbalance in excitatory and inhibitory synapses is thought to contribute to many neuropsychiatric phenotypes, including SCZ, for which a deficit in cognition is a core symptom. Many studies have observed reduced dendritic spine density (used to measure excitatory synapses) in postmortem brain samples from SCZ patients (reviewed in [38]). Overexpression of Mef2c in prefrontal projection neurons of mice resulted in improved cognitive performance of these mice. Interestingly, overexpression of adenylyl cyclase subtype 1 (AC1) in the forebrain of mice also enhances memory formation [39]. It was shown that when KCl concentrations are high enough to elicit membrane depolarization in cerebellar granular neurons, the cAMP-PKA pathway stimulates MEF2-dependent DNA-binding activity and gene expression by direct phosphorylation to promote neuronal survival [40]. Further studies would be needed to investigate if these effects are mediated by the alteration of similar molecular pathways. Nevertheless, these studies, as well as our findings implicate MEF2C and the adenyl cyclase signalling pathway as being dysregulated in cognition.
Reviewing individual associated genes, CRH functions via two different G protein-coupled receptors (GPCRs), CRHR1 and CRHR2. CRHR1 mediates adrenocorticotropic hormone (ACTH) release in the anterior pituitary in response to CRH [41]. The binding of CRH to CRHR1 induces adenylate cyclase activity, which raises cAMP levels in anterior pituitary corticotrophs. Through the cAMP/PKA pathway, CRH is responsible for both enhanced proopiomelanocortin (POMC) transcription and ACTH release [42]. Studies have shown that stress hormones (particularly CRH) mediate morphological alterations of hippocampus spines and synapses via CRHR1 activation [43–45].
RGS6 is a member of the R7 subfamily of RGS proteins and functions as a GTPase-activating protein for Gα and Gβγ subunits of G protein-coupled receptors (GPCRs), thereby play a critical role in regulating the duration and magnitude of signalling initiated by GPCRs [46,47]. One class of GPCRs regulated by RGS6 are serotonin 1A (5-HT1A) receptors in the cortical and hippocampal neurons and mediate the antidepressant and anxiolytic effects of serotonin. RGS6 influences anxiety and depression in rodents by inhibiting the 5-HT1A heteroreceptor—adenylyl cyclase signalling axis at postsynaptic sites. By direct activation of adenylyl cyclase signalling, RGS6 facilitates cAMP accumulation and subsequent activation of PKA and CREB, which contribute to rodent anxiety and depression-related behaviours [48]. RGS6 is also essential for adult maintenance of dopaminergic (DA) neurons in the ventral substantia nigra (SN) where RGS6 functions as a critical survival factor for SN DA neurons, and its loss results in their late-age degeneration [49]. RGS6 suppresses D2-autoreceptor signalling in substantia nigra compacta (SNc) DA neurons to promote proper DA homeostasis and neurotransmission and prevent abnormal α-synuclein accumulation. By inhibiting SNc D2-autoreceptor-Gαi/o signalling, RGS6 promotes DA packaging/release by preventing vesicular DA transporter downregulation and DA transporter upregulation. In addition, RGS6 inhibition of D2-autoreceptor-Gαi/o signalling promotes adenylyl cyclase signalling, increasing DA synthesis and suppressing α-synuclein expression [50].
The multimeric transmembrane GABA receptor exists as hetero-pentameric (five different subunits: alpha, beta, gamma, delta and rho) ligand-gated ion channels and conducts chloride ions following activation by GABA, which results in neuronal hyperpolarization and inhibition of neuronal signalling [51,52]. The protein encoded by GABRG3 is a gamma subunit of GABA receptor. Most of the cytogenetic abnormalities associated with various neurodevelopmental disorders like Prader–Willi syndrome and Angelman syndrome occur as result of genetic lesions in the chromosome 15, particularly the 15q11-13 locus. This region contains a number of GABA receptor subunit genes including GABRG3 [53], making them important candidates for various neurodevelopmental disorders. These receptors have received considerable attention because decreased GABA receptor density is observed in the hippocampus of autistic patients, and suppressed GABAergic inhibition has been implicated in ASD’s aetiology [54,55]. An imbalance of the inhibitory GABAergic pathways leads to overstimulation in the brain and an inability to filter out excess stimuli from environmental and intrinsic resources [55].
Harrington et al. [11] reported that MEF2C DEGs, mainly the downregulated genes, were enriched in genes associated with ASD risk. In contrast, we did not observe significant enrichment of DNMs observed in ASD cases in our gene-sets. We did observe up to a 3-fold excess of loss of function DNMs in ASD cases in our gene-sets. However, we conservatively tested this against the rate of DNMs in ASD cases in all other genes in the genome, which is elevated above expectations based on natural rates of mutation. As a result, for ASD cases, the rate of DNM in MEF2C-regulated genes is not significantly higher than the rate in non-MEF2C-regulated genes.
Our study has several limitations. Firstly, we were unable to source ChIP-seq data for MEF2C from microglial cells, thereby inhibiting us from identifying and analysing direct targets of MEF2C in that cell type. Secondly, the RNA-seq data available just captured gene expression from whole cortex in control and Mef2c-het mice at postnatal day 35–40. Therefore, we did not have the opportunity to investigate the impact of MEF2C disruption on gene expression at different developmental timepoints, in different brain regions or at the level of single cell data. Thirdly, our study of eQTLs at CRHR1, RGS6, and GABRG3 was limited to tissues from adult samples so again we were unable to study the effect of GWAS-associated SNPs at these genes on their expression at different developmental timepoints or with single cell resolution.
In conclusion, we have found that positively regulated, direct target genes of MEF2C that are elevated in expression in cortical neurons are enriched in genes containing common genetic variation associated with IQ and EA. These genes are enriched for functionality in the adenylyl cyclase signalling system, which is known to positively regulate synaptic transmission. Other studies have found that adenylyl cyclase is required for appropriate synaptic function and its deficits lead to impaired learning and memory. We further implicate the master regulator, MEF2C, as being involved in this system. Further studies are required to understand the exact molecular mechanisms by which MEF2C functions in this system and to investigate the potential of targeting this system in cortical neurons to treat, for example, the cognitive symptoms of SCZ and ASD and associated neuropsychiatric disorders that exhibit cognitive deficits.
Materials and Methods
Ethics statement
Data were directly downloaded from published studies and no additional ethics approval was needed. Each study is referenced and details on ethics approval are available in each manuscript.
Generation of MEF2C gene-sets
Integrated RNA- and ChIP-seq analysis
Differential expression data was obtained from a published RNA-sequencing study [11]. Raw binding data was obtained from a ChIP-sequencing study [12] that performed Mef2c ChIP-sequencing in cortical neurons dissected from E15.5 mouse embryos. There was one replica available. ChIP-seq of input DNA in cortical neurons was used as a control. The raw files were downloaded from the Gene Expression Omnibus (GEO) database repository (GEO accession: GSE66710) using fastq-dump and fastq (Andrews, S. 2010; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was run to assess the quality. Fastp [56] was used to trim reads with a cut window size of 2. BWA [57] was used for alignment to the genome; this tool was chosen because of its high accuracy and suitability for short reads [58]. First, an index was created using the mus_musculus.grcm38.dna.primary assembly.fa data file as reference. Alignment was then run to create .sai files, followed by Bwa samse to convert the .sai files to .sam format for the single-end reads. Postprocessing was performed using samtools which involved converting the sam files to bam format, sorting the bam file, removing possible PCR duplicates and finally creating a file with mapping statistics. Peaks were called using macs2 [59] and a Q value threshold of 0.01.
Binding and Expression Target Analysis (BETA); http://cistrome.org/BETA/ [60] was used to integrate the RNA-seq and ChIP-seq data and infer direct target genes. Input files consisted of the differential expression data, which was in the format of a table with three columns (gene name, fold change and FDR), and the binding data (output of macs2), which had five columns (chrom, chromStart, chromEnd, name, score). A rank product (RP) was calculated for each gene based on 1) differential expression false discovery rate (FDR; genes with FDR < 0.1 were considered) and 2) binding potential based on the number of binding sites and how far away they are from the transcription start site. Genes with an RP < 0.01 were considered to be direct target genes.
Identification of cell-type specific genes
Single-cell RNA sequencing data was available for 690 000 individual cells (565 distinct cell populations) across 9 regions of the adult mouse brain [13] http://dropviz.org/). These data are in the format of an expression matrix, where column names are the cell types and row names are genes. Each cell in the matrix contains the number of unique molecular identifier (UMI) counts for each gene in each of the 565 cell populations. We first scaled the expression data by cell type using transcripts per million (TPM), where each UMI count was multiplied by 1 M and divided by the sum of UMI counts in that cell type. We removed genes with a sum TPM of less than 1000 across all cell types. We then used the metric of expression specificity described in Trubetskoy et al. [3] where the UMI count for each gene is divided by the total expression of that gene in all 565 cell types, resulting in values ranging from 0 to 1 for each gene. The matrix was then restricted to the cell types of interest. We were interested in genes specific to neuronal and microglial cells in the cortex, of which there are 114 and 6 cell types, respectively. The sum of expression specificity values for each gene across these cell types of interest was calculated and the median value was used as a threshold for identifying genes elevated in our cell types of interest, i.e. genes with a sum of expression specificity values across neuronal or microglial cell types greater than the median value were identified as being elevated in expression in that cell type. A background gene set was also created for both microglial and neuronal specific genes, consisting of all genes in the expression matrix with a sum of expression specificity values across that cell type greater than the same threshold as used for the test sets (median).
Conversion of MGI gene symbols to human orthologues
All gene-sets were converted to human orthologue genes (HGNC gene symbols or NCBI gene IDs) using mouse and human ensembl databases via Biomart in R.
Overlap between gene-sets
Seventy-six percent of the genes in the DEGs_Direct_Neuron gene-set are present in the DEGs_All gene-set. The unique genes in DEGs_Direct_Neuron (n = 33) are present due to different methods being used to generate these gene-sets, including different criteria (RP < 0.1 for all genes with FDR < 0.1). The less stringent FDR threshold of 0.1 was used in creation of this gene-set because we were more tolerant of differential expression false positives on account of also considering results from ChIP-seq analysis to call MEF2C target genes. We also wished to include genes with high MEF2C binding, but with slightly less significant differential gene expression.
Gene-set analysis
A gene-set analysis (GSA) is a statistical method for simultaneously analysing multiple common genetic markers in order to determine their joint effect. We performed GSA using region-based multi-marker analysis of genomic annotation (MAGMA) http://ctg.cncr.nl/software/magma [61] and summary statistics from published GWAS on SCZ (69 369 cases and 236 642 controls) [3], intelligence (IQ; n = 269 867) [4] and educational attainment (EA) n = 766 345 [5]. An analysis involves three steps. First, in the annotation step, SNPs with available GWAS results are mapped on to genes (GRCh37/hg19 start-stop coordinates ±20 kb). Second, in the gene analysis step, gene P values are computed for each GWAS dataset. This gene analysis is based on a multiple linear principal components regression model that accounts for linkage disequilibrium (LD) between SNPs in each gene, number of SNPs in each gene, inverse of the mean minor allele count of variants in each gene and the GWAS sample size. The European panel of the 1000 Genomes data was used as a reference panel for LD. Third, a competitive GSA based on the gene P values, also using a regression structure, was used to test if the genes in each gene-set were more strongly associated with either phenotype than other genes in the genome. The condition modifier was used to include cortical elevated genes as covariates in the regression model. MAGMA was chosen because it corrects for LD, gene size and gene density (potential confounders) and has significantly more power than other GSA tools [62].
The multiple testing corrected P value threshold for associated gene-sets was calculated using the Bonferroni method by dividing the standard P value threshold (0.05) by the number of tests performed (three gene-sets multiplied by three GWAS phenotypes = 9). This same threshold was used for post-hoc analysis, including conditional gene-set analysis [63]. The multiple testing corrected P value thresholds for individually associated genes was also calculated using the Bonferroni method by dividing 0.05 by the number of genes tested (19 110 for IQ and 18 388 for EA).
Analysis of De novo mutations
Enrichment of de novo mutations (DNMs) in our gene-sets was tested using the R package, denovolyzeR http://denovolyzer.org/ [64]. This package implements the mutational background model proposed in [14], which estimates the expected number of each mutational class of DNM for each gene based on sequence context and gene size. We tested for enrichment of synonymous, missense, and loss of function (included nonsense, frameshift, and splice) DNMs. Enrichment of DNMs in gene sets is tested using a two-sample Poisson rate ratio test. The background rate is the number of observed to expected DNMs in genes within the denovolyseR probability table that are outside the gene-set being tested.
The major source of SCZ trio data was from Howrigan et al. [65]. This study combined DNMs from a new sample of Taiwanese SCZ trios with previously published DNMs from other SCZ studies [66–72] to give a total of 2772 trios. We combined these data with SCZ DNMs from Rees et al. [73] number of trios [n] = 613), Wang et al. [74] (N= 45) and Ambalavanan et al. [75] (N= 17). In total, we had data on 3447 SCZ trios. ASD trio data was sourced from [76]. Intellectual disability and developmental disorder trio data was sourced from multiple studies. The first is Genovese et al. [77] which includes data on 192 trios with ID sourced from multiple studies. Hamdan et al. [78] contained data on 309 trios with ID and/or developmental delay. Chevarin et al. [79] contained data on 60 trios with marfanoid habitus and intellectual disability and finally, the Deciphering Developmental Disorders Study [80] performed exome sequencing of 4293 trios with developmental disorders. Chevarin and colleagues did not report syn DNMs, therefore we did not test for enrichment of syn DNMs for the DD/ID phenotype.
For each study, the data was downloaded from the Supplementary Information. For the Deciphering Developmental Disorders data, we filtered out DNMs with a posterior probability score < 0.00781 as they reported to do the same [80]. DNM annotations were re-coded to match the variant classes in the DenovolyzeR built-in probability table (syn [synonymous], mis [missense], non [nonsense], frameshift and splice [canonical splice site]). DNM annotations that did not fit into any of these classes were removed (inframe, start-loss, and stop-loss).
Gene ontology overrepresentation analysis
GO overrepresentation analysis was performed using the meta Database ConsensusPathDB web interface (http://cpdb.molgen.mpg.de/). This tool determines P values by a hypergeometric test using all HGNC symbols that are present in at least one GO category as background. Q values are calculated using the false discovery rate method [81].
Conflict of interest statement: None declared.
Funding
This work was supported by an Irish Research Council Government of Ireland Postgraduate Scholarship to LF (GOIPG/2018/1501).
Supplementary Material
Contributor Information
Laura Fahey, Centre for Neuroimaging, Cognition and Genomics (NICOG), School of Biological and Chemical Sciences and School of Psychology, University of Galway, University Road, Galway, H91 CF50, Ireland; Discipline of Bioinformatics, School of Mathematical and Statistical Sciences, University of Galway, University Road, Galway, H91 CF50, Ireland.
Deema Ali, Centre for Neuroimaging, Cognition and Genomics (NICOG), School of Biological and Chemical Sciences and School of Psychology, University of Galway, University Road, Galway, H91 CF50, Ireland.
Gary Donohoe, Centre for Neuroimaging, Cognition and Genomics (NICOG), School of Biological and Chemical Sciences and School of Psychology, University of Galway, University Road, Galway, H91 CF50, Ireland.
Pilib Ó Broin, Discipline of Bioinformatics, School of Mathematical and Statistical Sciences, University of Galway, University Road, Galway, H91 CF50, Ireland.
Derek W Morris, Centre for Neuroimaging, Cognition and Genomics (NICOG), School of Biological and Chemical Sciences and School of Psychology, University of Galway, University Road, Galway, H91 CF50, Ireland.
References
- 1. Assali A., Harrington A.J and Cowan CW. Emerging roles for MEF2 in brain development and mental disorders. Curr Opin Neurobiol 2019;59:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vrečar I, Innes J, Jones E. et al. Further clinical delineation of the MEF2C haploinsufficiency syndrome: report on new cases and literature review of severe neurodevelopmental disorders presenting with seizures, absent speech, and involuntary movements. J Pediatr Genet 2017;06:129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trubetskoy V, Pardiñas AF, Qi T. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022;604:502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savage JE, Jansen PR, Stringer S. et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 2018;50:912–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee JJ, Wedow R, Okbay A. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 2018;50:1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phan D, Rasmussen TL, Nakagawa O. et al. BOP, a regulator of right ventricular heart development, is a direct transcriptional target of MEF2C in the developing heart. Development 2005;132:2669–78. [DOI] [PubMed] [Google Scholar]
- 7. Adachi M, Lin P-Y, Pranav H. et al. Postnatal loss of Mef2c results in dissociation of effects on synapse number and learning and memory. Biol Psychiatry 2016;80:140–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barbosa AC, Kim M-S, Ertunc M. et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A 2008;105:9391–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harrington AJ, Raissi A, Rajkovich K. et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 2016;5:e20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cosgrove D, Whitton L, Fahey L. et al. Genes influenced by MEF2C contribute to neurodevelopmental disease via gene expression changes that affect multiple types of cortical excitatory neurons. Hum Mol Genet 2021;30:961–70. [DOI] [PubMed] [Google Scholar]
- 11. Harrington AJ, Bridges CM, Berto S. et al. MEF2C Hypofunction in neuronal and neuroimmune populations produces MEF2C haploinsufficiency syndrome-like behaviors in mice. Biol Psychiatry 2020;88:488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Telese F, Ma Q, Perez PM. et al. LRP8-Reelin-regulated neuronal enhancer signature underlying learning and memory formation. Neuron 2015;86:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saunders A, Macosko EZ, Wysoker A. et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Samocha KE, Robinson EB, Sanders SJ. et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 2014;46:944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taussig R. Adenylyl cyclases. In William J. Lennarz and M. Daniel Lane (eds.) Encyclopedia of Biological Chemistry. Elsevier, pp. 42–46. [Google Scholar]
- 16. Bailey CP, Nicholls RE, Zhang X. et al. (2008) Gαi2 inhibition of adenylate cyclase regulates presynaptic activity and unmasks cGMP-dependent long-term depression at schaffer collateral-CA1 hippocampal synapses. Learn Mem 2013;15:261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu ZL, Thomas SA, Villacres EC. et al. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci 1995;92:220–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zars T, Fischer M, Schulz R. et al. Localization of a short-term memory in drosophila. Science (1979) 2000;288:672–75. [DOI] [PubMed] [Google Scholar]
- 19. Martel G, Millard A, Jaffard R. et al. Stimulation of hippocampal adenylyl cyclase activity dissociates memory consolidation processes for response and place learning. Learn Mem 2006;13:342–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ritter SL and Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol 2009;10:819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grammatopoulos DK. Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction. Br J Pharmacol 2012;166:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramoz N, Hoertel N, Nobile B. et al. Corticotropin releasing hormone receptor CRHR1 gene is associated with tianeptine antidepressant response in a large sample of outpatients from real-life settings. Transl Psychiatry 2020;10:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cross-Disorder Group of the Psychiatric Genomics Consortium, Electronic address: plee0@mgh.harvard.edu and Cross-Disorder Group of the Psychiatric Genomics Consortium . Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019;179:1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang L, Li J, Shuang M. et al. Association study and mutation sequencing of genes on chromosome 15q11-q13 identified GABRG3 as a susceptibility gene for autism in Chinese Han population. Transl Psychiatry 2018;8:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Okbay A, Wu Y, Wang N. et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat Genet 2022;54:437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Livingstone MS, Sziber PP and Quinn WG. Loss of calcium/calmodulin responsiveness in adenylate cyclase of rutabaga, a Drosophila learning mutant. Cell 1984;37:205–15. [DOI] [PubMed] [Google Scholar]
- 27. Feany MB and Quinn WG. A neuropeptide gene defined by the Drosophila memory mutant amnesiac. Science 1995;268:869–73. [DOI] [PubMed] [Google Scholar]
- 28. Skoulakis EM, Kalderon D and Davis RL. Preferential expression in mushroom bodies of the catalytic subunit of protein kinase A and its role in learning and memory. Neuron 1993;11:197–208. [DOI] [PubMed] [Google Scholar]
- 29. Castellucci VF, Nairn A, Greengard P. et al. Inhibitor of adenosine 3′:5′-monophosphate-dependent protein kinase blocks presynaptic facilitation in Aplysia. J Neurosci 1982;2:1673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abel T, Nguyen PV, Barad M. et al. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 1997;88:615–26. [DOI] [PubMed] [Google Scholar]
- 31. Iwasato T, Inan M, Kanki H. et al. Cortical adenylyl cyclase 1 is required for thalamocortical synapse maturation and aspects of layer IV barrel development. J Neurosci 2008;28:5931–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong ST, Athos J, Figueroa XA. et al. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 1999;23:787–98. [DOI] [PubMed] [Google Scholar]
- 33. Ferguson GD and Storm DR. Why calcium-stimulated adenylyl cyclases? Phys Ther 2004;19:271–76. [DOI] [PubMed] [Google Scholar]
- 34. Guillou J-L, Micheau J and Jaffard R. The opposite effects on cysteamine on the acquisition of two different tasks in mice are associated with bidirectional testing-induced changes in hippocampal adenylyl cyclase activity. Behav Neurosci 1998;112:900–8. [DOI] [PubMed] [Google Scholar]
- 35. Bernabeu R, Bevilaqua L, Ardenghi P. et al. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci 1997;94:7041–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Connolly JB, Roberts IJ.H, Armstrong JD. et al. Associative learning disrupted by impaired Gs signalling in drosophila mushroom bodies. Science 1996;274:2104–7. [DOI] [PubMed] [Google Scholar]
- 37. Pineda VV, Athos JI, Wang H. et al. Removal of Giα1 constraints on adenylyl cyclase in the hippocampus enhances LTP and impairs memory formation. Neuron 2004;41:153–63. [DOI] [PubMed] [Google Scholar]
- 38. Obi-Nagata K, Temma Y and Hayashi-Takagi A. Synaptic functions and their disruption in schizophrenia: from clinical evidence to synaptic optogenetics in an animal model. Proc Jpn Acad Ser B Phys Biol Sci 2019;95:179–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H, Ferguson GD, Pineda VV. et al. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 2004;7:635–42. [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Tang X, Li M. et al. Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase A Signaling pathway in neuronal survival. J Biol Chem 2005;280:16705–16713. [DOI] [PubMed] [Google Scholar]
- 41. Risbrough VB and Stein MB. Role of corticotropin releasing factor in anxiety disorders: A translational research perspective. Horm Behav 2006;50:550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soto-Rivera CL and Majzoub JA. Adrenocorticotrophin. In: Shlomo Melmed (ed) The Pituitary. Elsevier, pp. 47–83. [Google Scholar]
- 43. Chen Y, Dube CM, Rice CJ. et al. (2008) Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci 2017;28:2903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maras PM and Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci 2012;35:315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andres AL, Regev L, Phi L. et al. NMDA receptor activation and Calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J Neurosci 2013;33:16945–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dohlman HG and Thorner J. RGS proteins and signalling by heterotrimeric G proteins. J Biol Chem 1997;272:3871–74. [DOI] [PubMed] [Google Scholar]
- 47. Anderson GR, Posokhova E and Martemyanov KA. The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem Biophys 2009;54:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stewart A, Maity B, Wunsch AM. et al. Regulator of G‐protein signaling 6 (RGS6) promotes anxiety and depression by attenuating serotonin‐mediated activation of the 5‐HT1Areceptor‐adenylyl cyclase axis. FASEB J 2014;28:1735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bifsha P, Yang J, Fisher RA. et al. Rgs6 is required for adult maintenance of dopaminergic neurons in the ventral substantia Nigra. PLoS Genet 2014;10:e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo Z, Ahlers-Dannen KE, Spicer MM. et al. Age-dependent nigral dopaminergic neurodegeneration and α-synuclein accumulation in RGS6-deficient mice. JCI Insight 2019;5:e126769. 10.1172/jci.insight.126769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang S, Guo X, Dong X. et al. GABAA receptor subunit gene polymorphisms predict symptom-based and developmental deficits in Chinese Han children and adolescents with autistic spectrum disorders. Sci Rep 2017;7:3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Menold MM, Shao Y, Wolpert CM. et al. Association analysis of chromosome 15 GABAA receptor subunit genes in autistic disorder. J Neurogenet 2001;15:245–59. [DOI] [PubMed] [Google Scholar]
- 53. Sutcliffe JS and Nurmi EL. Genetics of childhood disorders: XLVII. Autism, part 6: duplication and inherited susceptibility of chromosome 15q11-q13 genes in autism. J Am Acad Child Adolesc Psychiatry 2003;42:253–56. [DOI] [PubMed] [Google Scholar]
- 54. Blatt GJ, Fitzgerald CM, Guptill JT. et al. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 2001;31:537–43. [DOI] [PubMed] [Google Scholar]
- 55. McCauley JL, Olson LM, Delahanty R. et al. A linkage disequilibrium map of the 1-Mb 15q12 GABAA receptor subunit cluster and association to autism. Am J Med Genet 2004;131B:51–59. [DOI] [PubMed] [Google Scholar]
- 56. Chen S, Zhou Y, Chen Y. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keel BN and Snelling WM. Comparison of Bburrows-Wheeler transform-based mapping algorithms used in high-throughput whole-genome sequencing: application to illumina data for livestock genomes. Front Genet 2018;9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y, Liu T, Meyer CA. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang S, Sun H, Ma J. et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 2013;8:2502–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leeuw CA, Mooij JM, Heskes T. et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015;11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leeuw CA, Neale BM, Heskes T. et al. The statistical properties of gene-set analysis. Nat Rev Genet 2016;17:353–64. [DOI] [PubMed] [Google Scholar]
- 63. Leeuw CA, Stringer S, Dekkers IA. et al. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat Commun 2018;9:3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ware JS, Samocha KE, Homsy J. et al. Interpreting de novo variation in human disease using denovolyzeR. Curr Protoc Hum Genet 2015;87:7.25.1–7.25.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Howrigan DP, Rose SA, Samocha KE. et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat Neurosci 2020;23:185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Girard SL, Gauthier J, Noreau A. et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet 2011;43:860–63. [DOI] [PubMed] [Google Scholar]
- 67. Xu B, Roos JL, Dexheimer P. et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet 2011;43:864–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu B, Ionita-Laza I, Roos JL. et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet 2012;44:1365–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gulsuner S, Walsh T, Watts AC. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013;154:518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guipponi M, Santoni FA, Setola V. et al. Correction: exome sequencing in 53 sporadic cases of schizophrenia identifies 18 putative candidate genes. PLoS One 2015;10:e0141630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCarthy SE, Gillis J, Kramer M. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 2014;19:652–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fromer M, Pocklington AJ, Kavanagh DH. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014;506:179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rees E, Han J, Morgan J. et al. De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nat Neurosci 2020;23:179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang Q, Li M, Yang Z. et al. Increased co-expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci Rep 2015;5:18209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ambalavanan A, Girard SL, Ahn K. et al. De novo variants in sporadic cases of childhood onset schizophrenia. Eur J Hum Genet 2016;24:944–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Satterstrom FK, Kosmicki JA, Wang J. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020;180:568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Genovese G, Fromer M, Stahl EA. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016;19:1433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hamdan FF, Srour M, Capo-Chichi J-M. et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet 2014;10:e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chevarin M, Duffourd Y, A. Barnard R. et al. Excess of de novo variants in genes involved in chromatin remodelling in patients with marfanoid habitus and intellectual disability. J Med Genet 2020;57:466–74. [DOI] [PubMed] [Google Scholar]
- 80. Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Benjamini Y and Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.