

Abstract
Germline pathogenic variants in two genes encoding the lysine-specific histone methyltransferase genes SETD1A and SETD2 are associated with neurodevelopmental disorders (NDDs) characterized by developmental delay and congenital anomalies. The SETD1A and SETD2 gene products play a critical role in chromatin-mediated regulation of gene expression. Specific methylation episignatures have been detected for a range of chromatin gene-related NDDs and have impacted clinical practice by improving the interpretation of variant pathogenicity. To investigate if SETD1A and/or SETD2-related NDDs are associated with a detectable episignature, we undertook targeted genome-wide methylation profiling of > 2 M CpGs using a next-generation sequencing-based assay. A comparison of methylation profiles in patients with SETD1A variants (n = 6) did not reveal evidence of a strong methylation episignature. A review of the clinical and genetic features of the SETD2 patient group revealed that, as reported previously, there were phenotypic differences between patients with truncating mutations (n = 4, Luscan-Lumish syndrome; MIM:616831) and those with missense codon 1740 variants [p.Arg1740Trp (n = 4) and p.Arg1740Gln (n = 2)]. Both SETD2 subgroups demonstrated a methylation episignature, which was characterized by hypomethylation and hypermethylation events, respectively. Within the codon 1740 subgroup, both the methylation changes and clinical phenotype were more severe in those with p.Arg1740Trp variants. We also noted that two of 10 cases with a SETD2-NDD had developed a neoplasm. These findings reveal novel epigenotype–genotype–phenotype correlations in SETD2-NDDs and predict a gain-of-function mechanism for SETD2 codon 1740 pathogenic variants.
Introduction
Precise epigenetic regulation of gene expression is critical for normal human development (1). In the past two decades, increasing numbers of developmental disorders have been found to result from pathogenic variants in genes with important roles in chromatin structure and/or function or in DNA methylation (2–5). These epigenetic developmental disorders were initially delineated by clinicians who recognized a specific clinical phenotype often consisting of combinations of neurodevelopmental delay, congenital defects and characteristic facial dysmorphisms. With the advent of high-throughput genome-wide sequencing technologies, there has been a marked expansion in the number of neurodevelopmental disorders that are known to result from variants in epigenetic regulators (6–8). In addition, it has become clear that (a) specific clinical syndromes may result from pathogenic variants in multiple genes and (b) the phenotypic spectrum of some epigenetic disorders is wider than originally proposed, for instance, some isolated neurodevelopmental disorders may result from variants in genes previously linked to a specific syndrome (7,9–12). These developments can complicate the interpretation of variants of uncertain significance (VUS) in NDD genes, such that the absence of a classical phenotype may not be enough to exclude pathogenicity. Recently, it has been recognized that many epigenetic developmental disorders are associated with altered methylation profiles (episignatures) in peripheral blood (3). This observation has opened new approaches to aid VUS interpretation and investigate genotype–phenotype correlations and, to date, episignature alterations have been described in >50 distinct epigenetic disorders (13).
Enzymatic modification of amino acids within histone tails is a critical process for epigenetic regulation. Histone lysine methyltransferases may be divided into two classes: those containing the SET domain and those that lack the SET domain (14). Genes encoding SET domain containing lysine-specific histone methyltransferases include genes encoding histone H3 lysine 4 (KMT2) methyltransferases (KMTs), Nuclear receptor binding SET Domain protein 1 (NSD1) and SET genes (e.g. SETD1A, SETD1B and SETD2). Pathogenic variants in KMT2 genes are associated with epigenetic neurodevelopmental disorders including Wiedemann-Steiner syndrome (MIM:605130; KMT2A), childhood dystonia 28 (DYT-KMT2B, MIM:617284; KMT2B), Kleefstra syndrome type 2 (MIM:617768; KMT2C), Kabuki syndrome type 1 (MIM:147920; KMT2D) and O’Donnell-Luria-Rodan syndrome (MIM:618512, KMT2E) that have overlapping but distinct phenotypes and distinct episignatures (2,5,15–22). Heterozygous pathogenic variants in NSD1 are associated with Sotos syndrome (MIM:117550) which is characterized by pre- and post-natal overgrowth, macrocephaly, facial dysmorphisms, developmental delay and tumor susceptibility (20,21). Pathogenic variants in SETD1A have been reported in association with early onset epilepsy, neurodevelopmental delay and an increased risk of schizophrenia (MIM:618832; MIM:619056), and variants in SETD1B (MIM:619000), SETD2 and SETD5 (MIM:616761) have also been associated with NDDs (22–26). The target lysines may differ between different SET-domain-containing proteins, for instance, H3K4 (histone 3 lysine 4) for SETD1A and H3K36 (histone 3 lysine 36) for SETD2 and NSD1 (27). Interestingly, SETD2 variants have been associated with a range of clinical phenotypes. Germline heterozygous SETD2 pathogenic variants were first described in association with a Sotos-like congenital overgrowth disorder associated with macrocephaly, intellectual disability, autism and obesity [known as Luscan-Lumish syndrome (MIM:616831)] (7,28–31). More recently a recurrent missense substitution [c.5218C > T (p.Arg1740Trp/R1740W)] was described in association with phenotypes of global developmental delay, failure to thrive and feeding difficulties, microcephaly (now known as Rabin-Pappas syndrome; MIM:620155) that was distinct from Luscan-Lumish syndrome plus a milder phenotype relating to a c.5219G > A (p.Arg1740Gln/R1740Q) variant (25).
To gain further insights into genotype–phenotype correlations and potential epigenotype alterations in SETD1A and SETD2-related neurodevelopmental disorders, we undertook genome-wide methylation profiling studies of ~2 M CpGs in these conditions and assessed the methylation episignatures of these disorders.
Results
Genotype–phenotype correlations
The details of the germline variants in SETD1A (n = 6) and SETD2 (n = 10) are listed in Tables 1 and 2. Three variants were recurrent: SETD1A [c.4582-2_4582-1del (n = 3 individuals), SETD2 (c.5219G > A (R1740Q, n = 2) and SETD2 c.5218C > T (R1740W, n = 4)]. The frequency of the clinical features in individuals with SETD2 variants is summarized in Table 2. The patient group with SETD2 variants was subdivided into those three subgroups; based on previously published genotype–phenotype correlations from Rabin et al. (25): four individuals with truncating variants (SETD2-LLS-PX subgroup) and two groups with missense substitutions at codon 1740; Type 1 (R1740W, n = 4) and Type 2 (R1740Q, n = 2). As described previously, all individuals with SETD2 LoF variants displayed macrocephaly and mild to moderate intellectual disability. In contrast, those with codon 1740 missense substitutions demonstrated microcephaly rather than macrocephaly and microcephaly, severe intellectual disability, congenital anomalies (renal, cardiac and central nervous system) and a vascular retinopathy were present in Type 1 (R1740W) subgroup (see Table 2). A further feature of the SETD2-cohort was the occurrence of tumors in two of 10 individuals. A patient with a SETD2 LOF variant developed multiple brain stem gliomas (SETD2-LLS-P2) from age 6 years and a patient (SETD2-R1740W-P4) with Type 1 (R1740W) variant with a metastatic high-grade chondroblastic osteosarcoma in her right proximal tibia and right mandible age 15 years. It was not possible to say which was the primary lesion and following a decision for palliative treatment, she died shortly after presentation. In addition, a further patient (SETD2-R1740W-P2) was previously reported to have had a hypothalamic hamartoma (25). The frequency of the clinical features in individuals with SETD1A variants is summarized in Table 2.
Table 1.
SETD2 and SETD1A variant details
| Sample ID | Variant | Age/sex | Previously published | ACMG-AMP criteria |
|---|---|---|---|---|
| SETD2-R1740Q-P1 | c.5219G > A p.(Arg1740Gln) de novo missense |
12/F | PM1, PM2, PM6, PP3 | |
| SETD2-R1740Q-P2 | c.5219G > A p.(Arg1740Gln) de novo missense |
5/M | “ | |
| SETD2-R1740W-P1 | c.5218C > T p.(Arg1740Trp) de novo missense |
0.5/M | Patient 7 (25) | PM1, PM2, PM6, PP3 |
| SETD2-R1740W-P2 | c.5218C > T p.(Arg1740Trp) de novo missense |
10/M | Patient 11 (25) | “ |
| SETD2-R1740W-P3 | c.5218C > T p.(Arg1740Trp) de novo missense |
10/F | Patient 9 (25) | “ |
| SETD2-R1740W-P4 | c.5218C > T p.(Arg1740Trp) de novo missense |
12/F | Patient 4 (25) | “ |
| SETD2-LLS-P1 | c.4438_4441del p.(Val1480fs) de novo frameshift |
12/M | PVS1, PM2, PM6, PP3 | |
| SETD2-LLS-P2 | c.1647_1667delinsTG p.(Ser550AspfsTer23) de novo frameshift |
4.5/M | Case 1 (30) | PVS1, PM2, PM6, PP3 |
| SETD2-LLS-P3 | c.513del p.(Pro172fs) de novo frameshift |
2/M | PVS1, PM2, PM6, PP3 | |
| SETD2-LLS-P4 | c.4457_4460delAGAA p.(Lys1486ArgfsTer28) de novo frameshift |
20/F | PVS1, PM2, PM6, PP3 | |
| SETD1A-P1 | c.4582-2_4582-1del de novo splice acceptor |
3/M | “ | |
| SETD1A-P2 | c.4582-2_4582-1del de novo splice acceptor |
17/F | “ | |
| SETD1A-P3 | c.4582-2_4582-1del de novo splice acceptor |
13/F | “ | |
| SETD1A-P4 | c.4711C > T p.(Arg1571Ter) Nonsense |
3/F | PVS1, PM2, PP3 | |
| SETD1A-P5 | c.2289dup p.Val764Serfs*61 de novo frameshift |
7/F | PVS1, PM2, PM6, PP3 | |
| SETD1A-P6 | Whole gene deletion CNV (852 kb loss of 41 genes) |
12/F | PVS1, PM2, PP3 |
Table 2.
Frequency (%) of clinical features in the three subgroups of patients with pathogenic variants in SETD2
| SETD2 variant subgroups | |||
|---|---|---|---|
| R1740Qa | R1740Wa | Loss of Function variantsa | |
| Number of patients | 2 | 4 | 4 |
| Clinical Features | |||
| Tall stature (HP:0000098) (%) | 0 | 0 | 25% |
| Obesity HP:0001513 (%) | 50 | 0 | 50 |
| Macrocephaly (HP:0000256) (%) | 0 | 0 | 100 |
| Microcephaly (HP:0000252) (%) | 0 | 100 | 0 |
| Retinopathy (HP:0000488) with retinal hemorrhage and/or detachment (%) | 0 | 100 | 0 |
| Micrognathia HP:0000347 (%) | 0 | 50 | 0 |
| Failure to thrive (HP:0001508) requiring nasogastric feeding (%) | 0 | 50 | 0 |
| Renal anomaly (HP:0000077) (cystic dysplasia or dilation renal pelvis) (%) | 0 | 100 | 0 |
| Congenital heart anomaly HP:0001627) (%) | 0 | 100 | 25 |
| Scoliosis HP:0002650 (%) | 0 | 50 | 25 |
| Severe developmental delay (HP:0012758) (%) | 0 | 100 | 0 |
| Mild/moderate developmental delay (HP:0012758) (%) | 100 | 0 | 100 |
| Seizures (HP:0001250) (%) | 0 | 50 | 0 |
| CNS structural anomaly (HP:0002011) (%) | 0 | 100 | 25 |
| Neoplasm (HP:0002664) (%) | 0 | 25 | 25 |
Detailed descriptions of the SETD2-R1740W patient phenotypes are included in Rabin et al. (25).
aR1740Q = c.5219G > A p.(Arg1740Gln). R1740W = c.5218C > T p.(Arg1740Trp). Loss of Function variants = frameshift variants.
Table 3.
Frequency (%) of clinical features in the three subgroups of patients with pathogenic variants in SETD1A
| % with clinical feature | |
|---|---|
| Number of patients | 6 |
| Clinical features | |
| Macrocephaly (HP:0000256) (absolute or relative) (%) | 83 |
| Short stature (HP:0004322) <10th centile (%) | 33 |
| Non-specific facial dysmorphisms HP:0001999 (%) | 100 |
| Joint laxity (HP:0001388) or hypermobility (%) | 50 |
| Sleep disturbance (HP:0002360) (%) | 66 |
| Brain MRI anomalies (HP:0002011) (%) | 17 |
| Mild—moderate neurodevelopmental delay (HP:0012758) (%) | 83 |
| Severe—profound neurodevelopmental delay (HP:0012758) (%) | 17 |
| Autistic spectrum features (HP:0000729) (%) | 50 |
| Seizures (HP:0001250) (%) | 17 |
| Congenital anomalies | 0 |
Epigenotype–genotype–phenotype analysis
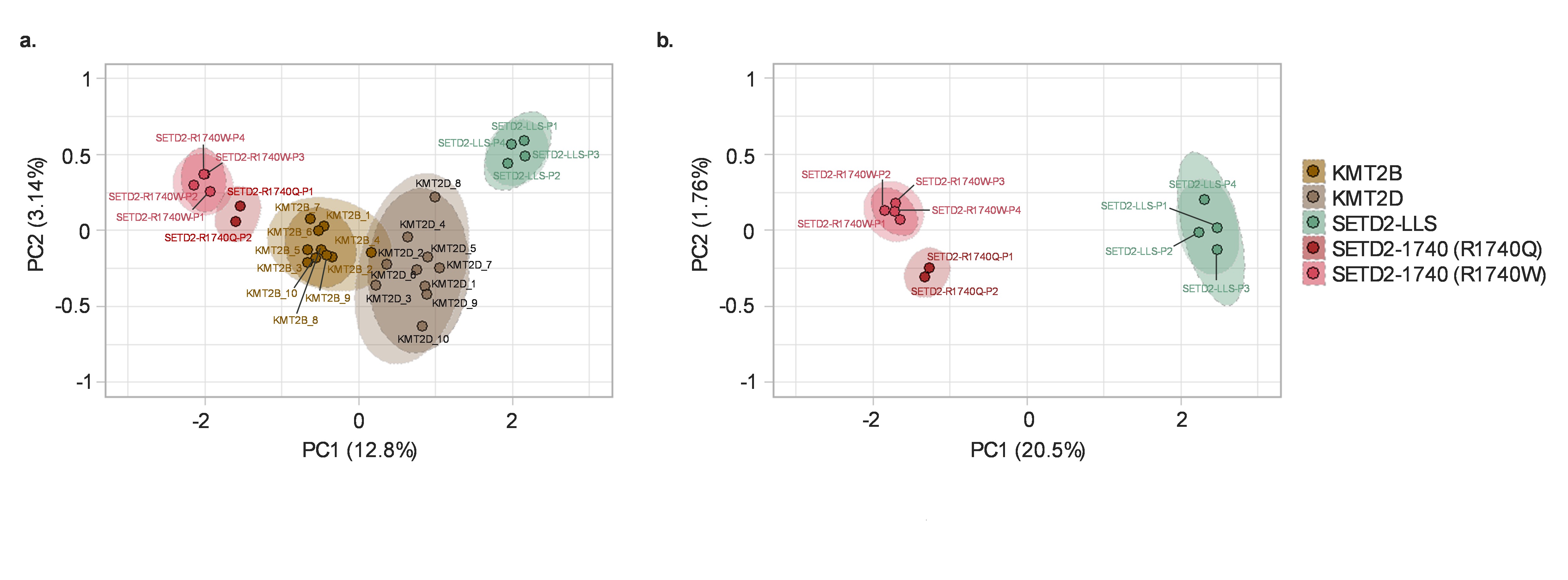
Principal component analysis (PCA) of DNA methylation profiles: The PCA (unsupervised clustering) of significant CpG sites after filtering was performed separately for individuals with SETD1A NDD and SETD2 NDD (Fig. 1). SETD1A samples with pathogenic variants (n = 6) were not distinguishable from the control cohort (Fig. 1C) and subclassifying the SETD1A NDD samples into those with the recurrent SETD1A splice acceptor variant (n = 3) and other cases with pathogenic variants (n = 3) (frameshift, stop gained and CNV) did not show any detectable difference between the two groups (Fig. 1C). Comparison of SETD2 NDD patient samples and controls separated those with loss of function variants and those with codon 1740 missense variants (Fig. 1A and B). Based on previous genotype–phenotype correlations (25), the SETD2-1740 group were subdivided into Type 1 (R1740W) and Type 2 (R1740Q) and when compared with non-1740 pathogenic variants (SETD2-LLS) each subgroup was distinct from controls (Fig. 1B). The methylation episignatures were then interrogated for individual SETD1A and SETD2 NDD individuals.
Figure 1.
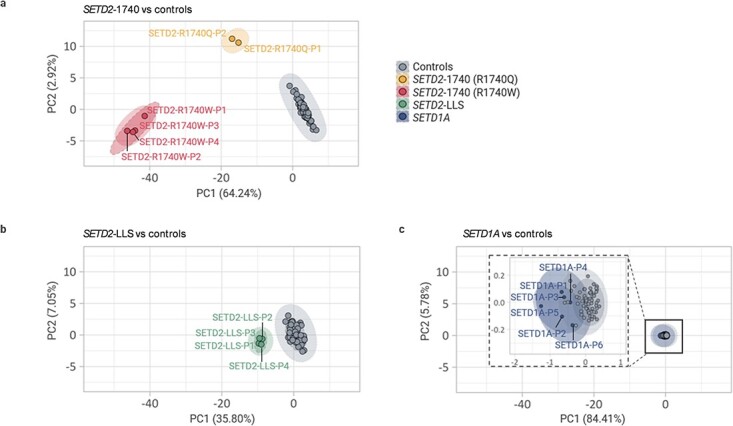
Clustering of SETD2 and SETD1A based on methylation episignatures. Unsupervised PCA clustering results for SETD1A and SETD2 group. Sample name and group annotations were applied by each group after PCA. Dotted line: group names annotation, Solid line: clustering by ‘stat_ellipse’ function in R (assumes a multivariate t-distribution; applied except for the Type 2 (R1740Q) group since this function applies only when there are more than two samples in a group). (A) There were two distinct groups in SETD2 NDD patient samples. There was a significant difference between SETD2-1740 samples and controls but Type 2 (R1740Q) cases were closer to controls than Type 1 (R1740W). (B) Despite being distinct from controls, the distance of SETD2-LLS cases were much closer to the control group than SETD2-1740 Type2 (R1740Q) cases. (C) SETD1A cases were not distinguishable from the healthy controls. There are no detectable differences between the two groups of SETD1A splice acceptor variants (SETD1A-P1, P2, P3) or pathogenic variants (SETD1A-P4, P5, P6).
SETD1A NDD episignatures: Comparison of methylation profiles in 6 individuals with a SETD1A NDD to those in 64 controls identified 7 significant differentially methylated CpG positions (DMPs) with 1 CpG island (chr12:49782966-49783193) and no DMBs detected (shown in Fig. 2). Using similar methodology, we previously found that comparison of methylation profiles in KMT2B NDD individuals (n = 10) and KMT2D associated Kabuki syndrome individuals (n = 10) to control samples (n = 29) identified 1812 and 89 significant DMPs, respectively (5). From the results of in-house laboratory healthy controls quality control, random sampling of the methylation profiles from 64 normal control samples for a range of 1 to 15 normal controls showed a mean 3.6 (range 0 to 17) significant CpGs. Therefore, there was no evidence of a strong methylation episignature in the SETD1A NDD group and, as shown in Figure 2, the hierarchical clustering was unable to clearly distinguish SETD1A from control groups.
Figure 2.

Methylation episignatures for SETD1A samples. SETD1A NDD did not exhibit a prominent methylation episignature. The hierarchical clustering was not able to separate SETD1A from control groups. A comparison of methylation profiles in 6 patients with a SETD1A NDD to those in 64 controls. Methylation analysis detected 7 significant differentially methylated CpG positions with 1 CpG island (chr12:49782966-49783 193) and no DMBs were detected.
The methylation profiling of SETD1A samples (n = 3) with the recurrent splice-acceptor variant were then compared with normal controls but this identified only 17 significant DMPs with 2 DMBs and 1 CpG island (shown in Supplementary Material, Fig. S1). The small number of significant DMBs found in the SETD1A cohort was consistent with no strong methylation episignature. As shown in Figure 2B, the hierarchical clustering was unable to clearly distinguish SETD1A from control groups. A CpG island (GRCh37:Chr12:49782966-49783193) that passed the filter is associated with the SPATS2 (spermatogenesis associated serine-rich 2) gene, but no other genes or ClinGen diseases have been identified in this region as of yet.
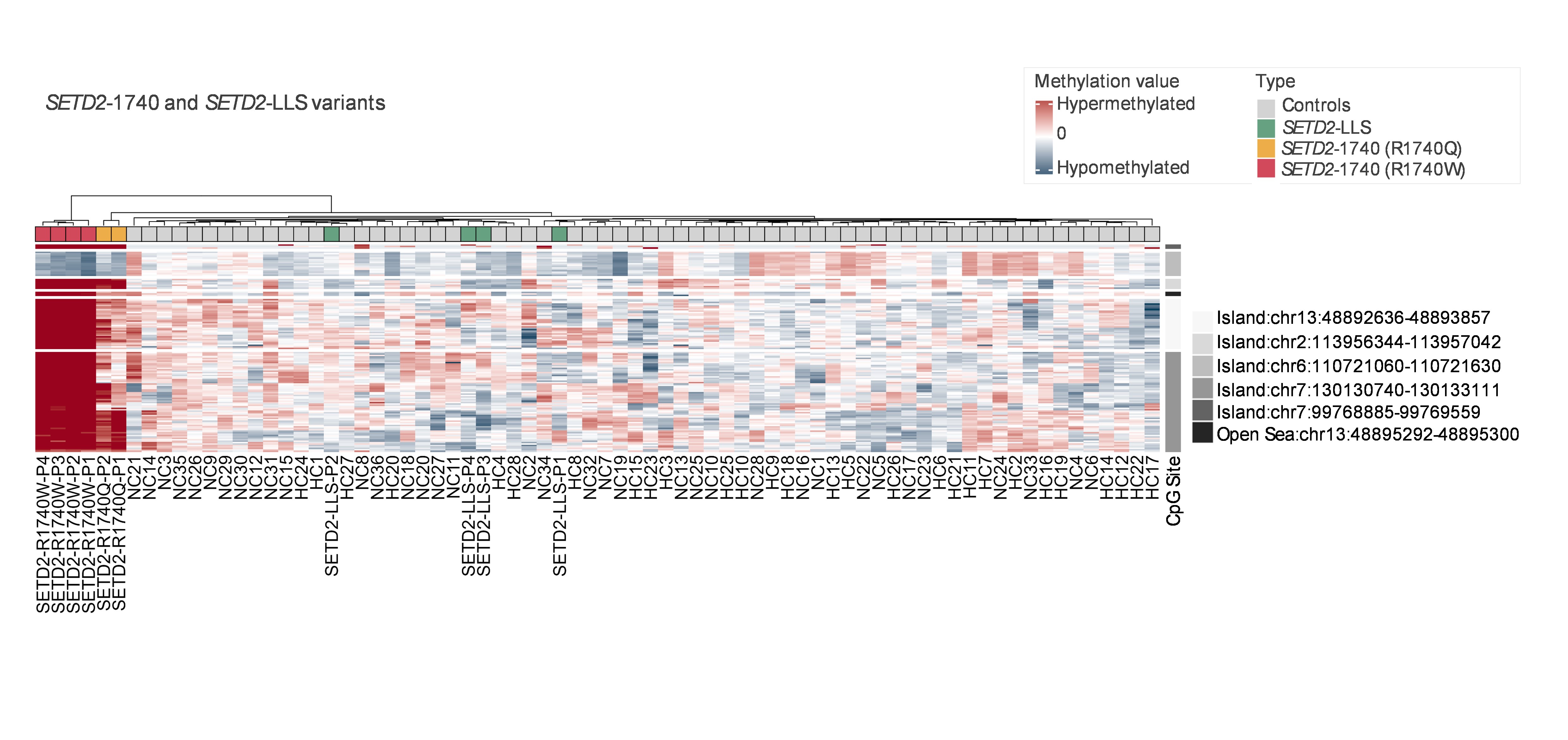
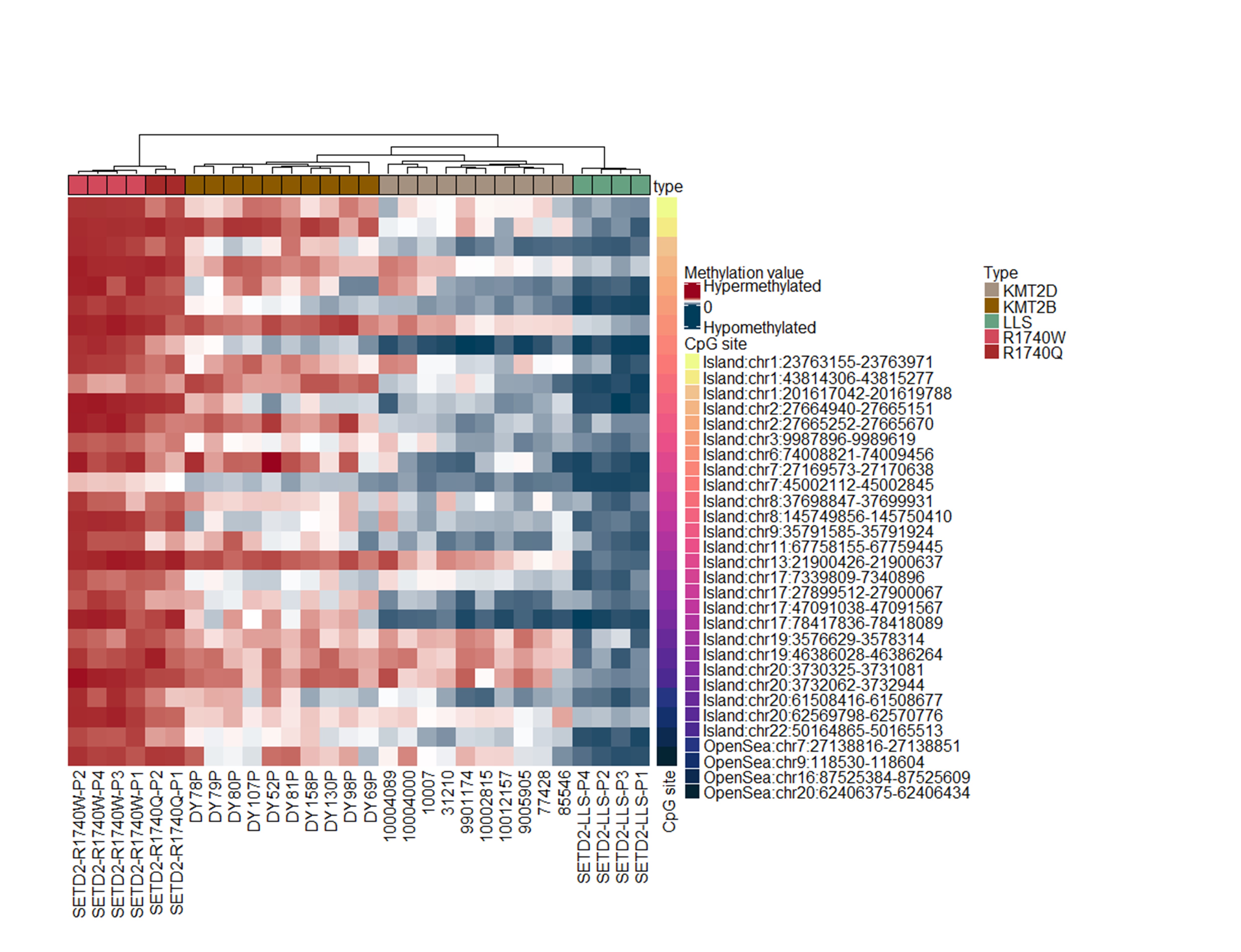
SETD2-1740 and SETD2-LLS NDD cohorts: In contrast to the SETD1A NDD individuals, a combined analysis of the methylation profiles in 10 individuals with SETD2 variants to controls (n = 64) identified 135 significant DMPs (17 hypomethylated and 118 hypermethylated for SETD2-1740, 81 hypomethylated and 54 hypermethylated for SETD2-LLS) with 1 DMB and 5 CpG islands (Supplementary Material, Fig. S2). Furthermore, when the methylation profiles of SETD2-1740 samples and non-SETD2-1740 samples were analyzed separately, there were clearer and distinct, methylation episignatures in both groups (Figs 3 and 4). Methylation episignatures for non-SETD2-1740 (LoF) variant individuals (n = 4) displayed 778 DMPs (767 hypomethylated and 11 hypermethylated), including 34 CpG Islands and 8 DMBs (Fig. 3).
Figure 3.

Methylation episignatures for SETD2-LLS samples. The methylation episignatures for SETD2-LLS LoF variant patients (n = 4) displayed 778 DMPs (767 hypomethylated and 11 hypermethylated), including 34 CpG Islands and 8 DMBs. (A) Hierarchical clustering on the top annotation bar revealed that 4 SETD2-LLS patients have remarkable hypomethylated profiles that are distinguishable from control samples. (B) The majority of DMPs detected as significant are CpG islands (91.13%), whereas the rest of the DMPs (69 DMPs within 8 DMBs) are located in the Open Sea area (i.e. the rest of the genomic regions except Shelf, Shore or CpG Islands). (C) Normalized methylation values were used to determine whether methylated DMPs gained or lost methylation. The horizontal line (red) indicates a confidence interval of 3 standard deviations (3SD). As a result, all four patients (all of whom are frameshift variants) had similar LOM patterns of methylation. *DMB: Differentially methylated blocks *GOM: Gain of methylation *LOM: Loss of methylation.
Figure 4.

Methylation episignatures for SETD2-1740 samples. The SETD2-1740 samples (n = 6) are revealed to have extensive methylation alterations with 7566 significant DMPs (789 hypomethylated and 6777 hypermethylated) containing 281 CpG Islands and 62 DMBs. (A) Hierarchical clustering dendrogram revealed that the majority of significant DMPs in SETD2-1740 cases exhibited very clear hypermethylation patterns compared with controls. A noteworthy observation is that 4 SETD2-1740 patients with Type 1 (R1740W) are distinguishable from Type 2 cases (R1740Q) and healthy controls. (B) Of the 7566 DMPs, 94.96% of DMPs are located on CpG islands, followed by Open Sea (4.85%), Shore (0.13%), and Shelf (0.05%). (C) Gain or loss of methylated DMPs were determined by the normalized methylation values. The horizontal line (red) indicates a confidence interval of 3 standard deviations (3SD). As a result, all four Type 1 (R1740W) cases showed a more severe hypermethylation pattern than in the two Type 2 (R1740Q) cases. *DMB: Differentially methylated blocks *GOM: Gain of methylation *LOM: Loss of methylation.
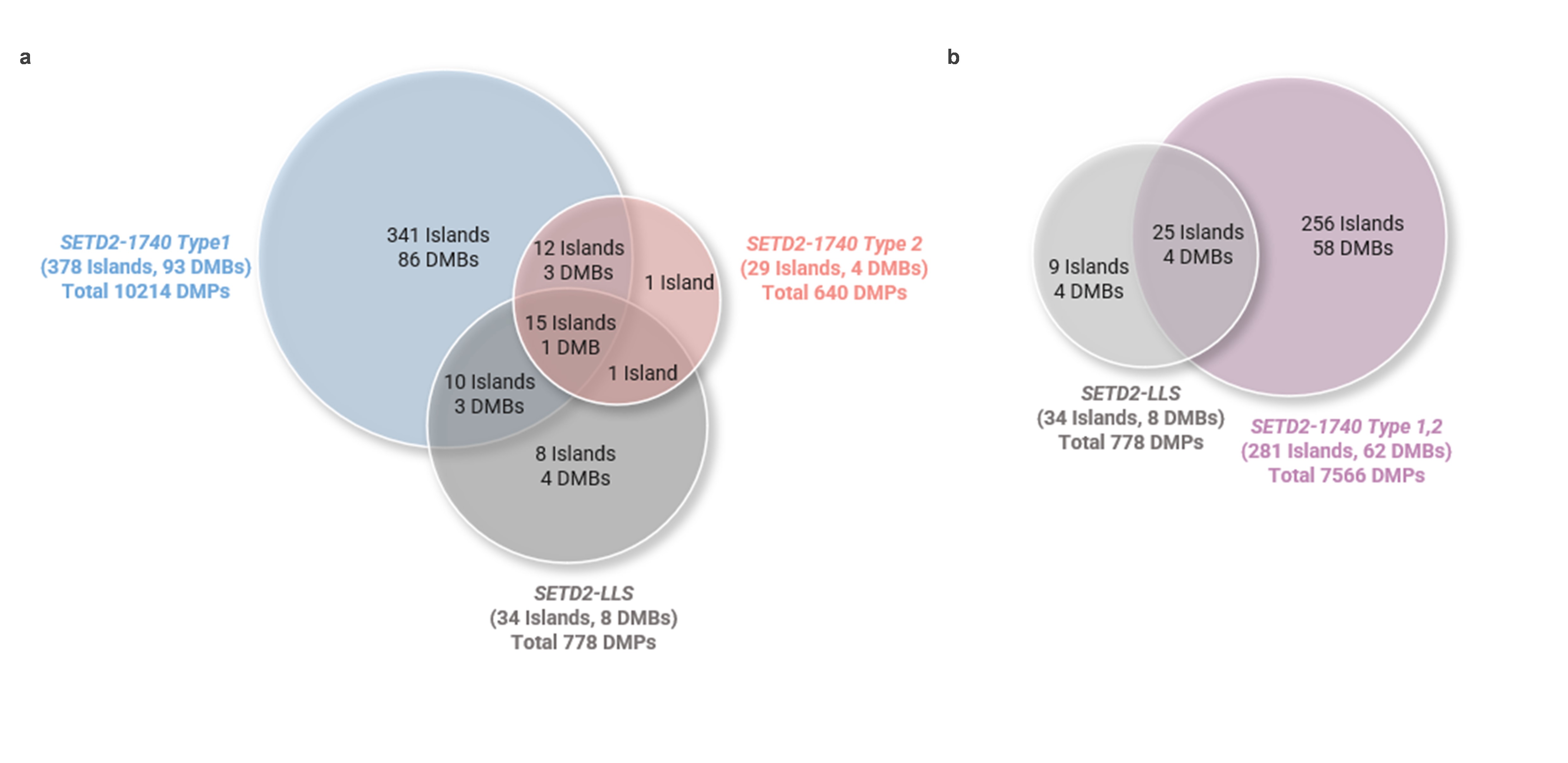
The SETD2-1740 samples (n = 6) demonstrated more extensive methylation alterations with 7566 significant DMPs (789 hypomethylated and 6777 hypermethylated) containing 281 CpG Islands and 62 DMBs (Fig. 4). Thus, there were both quantitative and qualitative differences in the methylation episignatures between SETD2 LoF variant samples and SETD2-1740 missense substitution individuals with the majority of significant DMPs in SETD2-1740 cases exhibiting hypermethylation compared with controls. Previously, it has been observed that there are phenotypic differences between Type 1 (R1740W) and Type 2 (R1740Q) cases (R1740W and R1740Q, respectively) that Type 1 (R1740W) having a more severe phenotype. Episignature results showed that all four Type 1 cases showed a more severe hypermethylation pattern than in the two Type 2 (R1740Q) cases (Fig. 4A and 4C). In addition, as alterations at individual DMPs may correlate with alterations at other DMPs, we used the caret package (Classification And Regression Training) to identify non-redundant DMPs and identified 139 DMPs for SETD2-1740 and 60 DMPs for SETD2-LLS group (see Supplementary Material, Tables S1 and S2) (32).
Comparative analysis for SETD2-1740 and SETD2-LLS cases confirmed that methylation episignatures of SETD2-LLS were less distinctive than SETD2-1740 cases (shown in Supplementary Material, Fig. S2). Of the 7566 significant DMPs in the SETD2-1740 cases, 513 DMPs (include 25 Islands and 4 DMBs) were also significant in non-SETD2-1740 (LoF variant) samples (see Supplementary Material, Fig. S3). In the SETD2-LLS group, over half of the significant CpG islands and DMBs overlapped with the SETD2-1740 group. In order to investigate if the overlapped CpGs might contribute to their certain overlapped phenotypes (whereas those that are not overlapped may provide insight into their distinct phenotypes), we examined the genes and pathways affected by methylation changes in target genes.
Gene and pathway analysis for target gene methylation alterations
The 7566 DMPs in SETD2-1740 were associated with 362 genes whereas the DMPs in non-SETD2-1740 individuals were associated with 43 genes (29 genes were common to both SETD2-1740 and non-SETD2-1740 cases) (Supplementary Material, Table S3). Given the phenotypic differences between the two subgroups of SETD2 NDD, we compared the biological pathways for genes associated with SETD2-1740 only, non-SETD2-1740 only and both patient groups (see Supplementary Material, Tables S4–S6). A clear phenotypic difference between SETD2-1740 and non-SETD2-1740 individuals is the occurrence of growth retardation in the former and frequent overgrowth in the latter. Genes implicated in growth control associated with DMPs included NRP2, LTBP3, MRPS34, RAI1, SUCLA2, IGF2BP1 and IGF1R in the SETD2-1740 cases. Among them, NRP2 and IGF2BP1 were identified in non-SETD2-1740 individuals as well. Neither of these genes has been confirmed to cause overgrowth or growth retardation in the past. However, methylation episignature results showed that both NRP2 (chr9:35791585-35791924) and IGF2BP1 (chr17:47091038-47091567) were associated with hypermethylated CpG islands in SETD2-1740 but hypomethylated CpG islands in non-SETD2-1740. Additionally, all overlapped DMPs between two SETD2 groups showed the opposite methylation profiles (Fig. 5).
Figure 5.

Methylation episignatures for overlapped CpGs in SETD2 patients. Common CpGs between SETD2-1740 and SETD2-LLS. Overlapped DMPs were identified from the data in Figure 2 (SETD2-1740 versus control) and Figure 3 (SETD2-LLS versus control) (NOT based on a comparative analysis from Supplementary Material, Fig. S3). Several overlapped DMPs are summarized in Supplementary Material, Figure S4A and SB. There are 25 CpG islands and 4 DMBs overlapped between the two groups. All DMPs show (A) hypermethylated across 6 SETD2-1740 cases [Type 2 (R1740Q) cases display slightly milder episignatures than Type 1 (R1740W)] whereas (B) all hypomethylated in 4 SETD2-LLS cases. The episignatures of both groups were completely opposite.
Somatic SETD2 mutations occur in a range of human cancers, in particular clear cell renal cell carcinomas (ccRCCs). None of the DMP-associated genes in SETD2 NDD individuals were frequent targets for somatic mutations in ccRCC, but 51 DMP-associated genes have been linked to oncogenesis (details supplied in ‘NCG7.0’ tab from Supplementary Material, Tables S4–S6). Among non-SETD2-1740 individuals, 7 genes (MPL, NAV1, PRRT3, MYO1G, ELMO3, NLRP3 and KLHDC4) were classified as cancer genes from the Network of cancer genes database (http://ncg.kcl.ac.uk/query.php). Among them, MPL (chr1:43814306-43815277), NAV1 (chr1:201617042-201619788), PRRT3 (chr3:9987896-9989619) and MYO1G (chr7:45002112-45002845) were also identified in altered DMPs in SETD2-1740 individuals. MPL (proto-oncogene), NAV1 and PRRT3 are putative oncogenes implicated as candidate cancer drivers. MYO1G is a putative tumor suppressor. All overlapped genes were associated with hypermethylation in SETD2-1740 and hypomethylation in non-SETD2-1740 (see Fig. 5).
Discussion
We investigated potential methylation episignatures in individuals with germline variants in the lysine methyltransferase SETD1A and SETD2 genes using a targeted bisulfite sequencing-based approach that profiles ~2 M CpG sites across the genome. Though we did not detect evidence for a strong episignature for SETD1A, we found distinctive epigenotype–genotype–phenotype correlations for SETD2-associated NDDs. To our knowledge, investigations of the episignature of SETD1A and SETD2 NDDs have not been reported previously with an NGS-based methylation profiling strategy but a methylation array-based episignature has been reported for SETD2 (LLS) and SETD1B- related NDD (13,33). We note that in the SETD2-LLS methylation array-based episignature from Levy et al. (4,13) loss of methylation alterations predominanted (similar to that in our NGS-based SETD2-LLS methylation episignature with hypomethylated patterns) and further comparison between the episignatures is provided in Supplementary Material, Table S7. Although a variety of investigative options are available for genome-wide methylation profiling, the most commonly applied are Illumina methylation arrays that can analyze 450 000 or ~850 000 CpGs. Previously we described the use of the Illumina TruSeq Methyl Capture Library kit and next-generation sequencing (EPIC-NGS) to detect methylation episignatures for KMT2B-associated dystonia and Type 1 and KMT2D-related Kabuki syndrome, confirming the results of methylation profiles for these disorders using methylation arrays (5,34). To validate the specificity of SETD2-1740 and SETD2-LLS methylation episignatures compared with those of KMT2B-related childhood-onset dystonia and KMT2D-related Kabuki syndrome type 1 cohort, unsupervised clustering was performed and 29 significant DMBs could successfully discriminate SETD2 NDDs from other NDDs (see Supplementary Material, Figs S4 and S5 and Supplementary Material, Table S8).
More than 50 human disorders have been investigated for methylation episignatures [reviewed by Levy et al. (13)] and episignatures have been described for many of these. However, the strength and specific patterns of methylation alterations vary between disorders. Though we did not identify a robust episignature for SETD1A NDD using an approach that interrogates more than twice the number of CpGs as methylation array-based methods, there is little information regarding the relative performance of the two techniques and we cannot exclude that an episignature may have been detectable with another methodology and/or a larger number of individuals with pathogenic variants. However, we suggest that if there is a methylation episignature for SETD1A NDD then it is less pronounced for SETD2, KMT2B and KMT2D-associated disorders. In addition, it should be noted that our results relate to the methylome of peripheral blood and while this is a standard approach for the investigation of epigenetic disorders caused by pathogenic variants in chromatin-modifying genes, we cannot exclude the possibility that a SETD1A methylation episignature might vary between tissues and be more prominent in target organs such as the central nervous system (mosaic epimutations with variable levels in different tissues are well-recognized in epigenetic disorders associated with disordered genomic imprinting (35). Finally, we note that while SETD1A and SETD2 target different histone lysines (H3K4 and H3K36 respectively), KMT2B and KMT2D also act on histone 3 lysine 4 (H3K4).
In contrast to SETD1A NDD, the results for SETD2 NDD revealed clear methylation episignatures correlated with the phenotypic heterogeneity described previously in SETD2-associated NDDs. Thus, SETD2-1740 patterns of methylation differed both from controls and those with non-SETD2-1740 LoF variants in both the extent of methylation alterations (7566 and 778 significant DMPs respectively) but also the direction of methylation alterations with predominant hypermethylation in SETD2-1740 and predominant hypomethylation in non-SETD2-1740 variants. Furthermore, though the number of individuals were small, within the SETD2-1740 subgroup, there was an apparent difference between Type 1 (R1740W) and Type 2 (R1740Q) individuals with predominant hypermethylation alterations in both groups but with more pronounced alterations in Type 1 cases. In an analysis of 15 individuals with missense substitutions at codon 1740 (12 with R1740W and 3 with R1740Q), recently Rabin et al. (25) contrasted the phenotype of Type 1 (R1740W) individuals from that of 12 individuals with non-1740 variants (mostly truncating) who presented with autistic spectrum disorder (ASD) or an LLS, a Sotos-like NDD that is characterized by macrocephaly, overgrowth or obesity, ASD and variable intellectual disability but without congenital malformations of internal organs (25). In contrast, individuals in the Type 1 (R1740W) group were characterized by severe intellectual disability, CNS malformations, microcephaly, failure to thrive and multiple congenital malformations (e.g. congenital heart defects and urogenital anomalies) (25). Interestingly, individuals in Type 2 (R1740Q) group exhibited a similar, but milder, phenotype to Type 1 (R1740W) cases with intellectual disability and a small head circumference but without significant congenital anomalies. Therefore, although larger numbers of cases are required for confirmation, the more extensive methylation changes in Type 1 (R1740W) than in Type 2 (R1740Q) individuals do correlate with phenotypic severity.
In contrast to individuals diagnosed with LLS, cases with SETD2-1740 missense substitutions do not exhibit significant overgrowth. Interestingly, the methylation episignatures of LLS individuals and of other non-imprinting epigenetic disorders associated with congenital overgrowth such as Sotos syndrome and DNMT3A-overgrowth syndrome (Tatton-Brown-Rahman syndrome; MIM:615879) demonstrate predominant hypomethylation (36,37). A number of growth-related genes were associated with significantly altered DMPs in the current study e.g. the insulin-like growth factor receptor (IGF1R) gene, which was hypermethylated in the SETD2-1740 group. Germline mutations in IGF1R are associated with pre- and post-natal growth retardation and microcephaly (38) and combined investigations of the epigenetic and transcriptional effects of germline SETD2 LoF and codon 1740 mutations could provide candidate genes for aspects of SETD2-associated NDDs. Moreover, we have found that among detected genes related to hypo- or hypermethylation patterns in SETD2-1740 and SETD2-LLS group (Supplementary Material, Tables S4 and S5), several genes related to NDDs are up-regulated (3 genes including MRPS34) and down-regulated (12 genes including PLA2G6 and MBOAT7) in SETD2 knockout mice model (Setd2mNul oocyte) compared with healthy controls from GSE112835 (39) (see ‘differentially expressed genes from GSE112835’ tab in Supplementary Material, Tables S4 and S5). Our findings support that genes affected by aberrant methylation patterns could impact SETD2 functioning, thereby could affect the phenotypes, although this may not be the primary cause since it is the second hit by aberrant methylation, and functional effects of SETD2 deficiency may differ between mice and humans.
We note that two of 10 individuals with a SETD2 NDD reported here had developed a tumor (a sarcoma in one case and multiple gliomas in the other). In addition, a further patient had had a CNS hamartoma. Tumor predisposition has been associated with other chromatin gene disorders associated with congenital overgrowth such as Sotos syndrome (e.g. overall risk ~ 3% including teratoma, neuroblastoma, ganglioma, leukemia, lung cancer and glioma) and Weaver syndrome (e.g. overall risk ~ 2% including leukemia, neuroblastoma and lymphoma) that are caused by mutations in the histone methyltransferases NSD1 and EZH2, respectively (21,40). Furthermore, (a) SETD2 is frequently mutated in a range of human cancers, with the highest frequencies in mesothelioma, endometrial cancer and renal cell carcinoma but somatic SETD2 mutations also occur in sarcoma and glioblastoma multiforme (TCGA PanCancer Atlas) and (b) SETD2 has been implicated in the repair of DNA double-strand breaks (41). Therefore, though neoplasia has not been reported previously in the published literature as a feature of a SETD2 NDD, we are aware of an additional and unpublished case of a SETD2 NDD patient with osteosarcoma (J. Bernat, personal communication) and this aspect requires further investigation and clinicians looking after these individuals should be aware of a possible causal association. However, at this stage, we would not recommend a tumor surveillance program until the tumor risks and types have been confirmed and the cost-effectiveness of a surveillance protocol could be more accurately predicted. We note that neoplasia occurred in both SETD2-1740 and non-SETD2-1740 patient groups. SETD2 is a tumor suppressor gene and the most somatic driver SETD2 mutations are truncating but the c.5218C > T (R1740W) somatic variant is listed five times (out of 2938 unique samples with mutations) entries in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (accessed 5.01.2023) (the exact germline SETD2 LLS-associated loss of function mutations described here are not present in the COSMIC database but a somatic mutation producing a very similar truncated protein (p.Lys1486Argfs*29) to that in SETD2-LLS-P4 has been reported in a single renal cell carcinoma (42). Therefore, despite evidence that germline truncating and Type 1 (R1740W) mutations have different effects on developmental phenotype and epigenotype, both can be oncogenic.
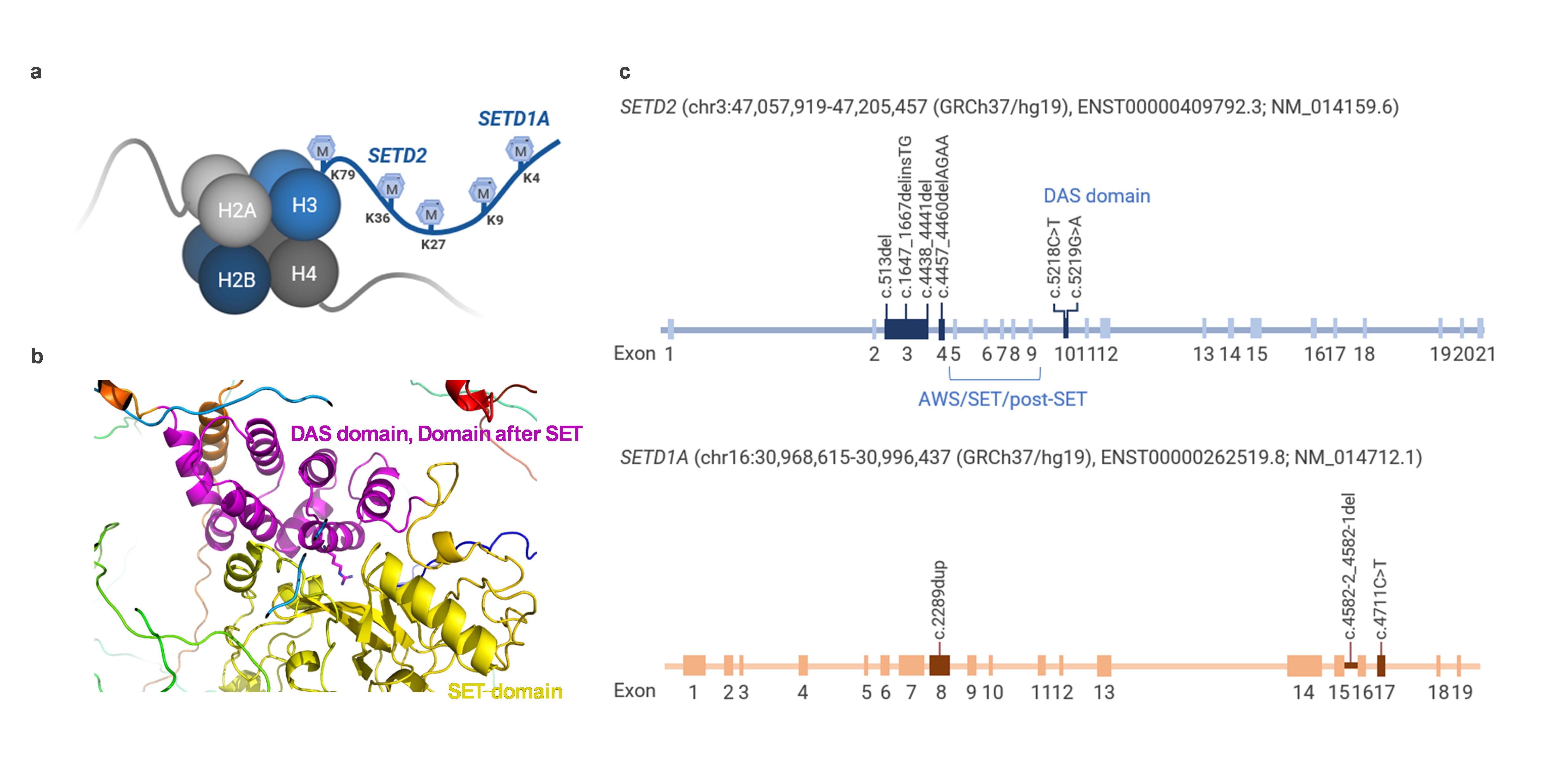
Codon 1740 is not located in any known functional domain of the SETD2 human protein, and maps to an all α-helix domain which is C-terminally adjacent to the post-SET domain (referred to as DAS domain, domain after SET) (Fig. 6A). Though the DAS domain does not map to a specific functional domain, it is highly conserved across multiple species (including Danio rerio and Xenopus tropicalis) suggesting functional importance. The AlphaFold model of SETD2 (AF-Q9BYW2-F1) shows that the DAS domain can fold as a globular domain, further confirming its functional importance (Fig. 6B) (43). A further DALI search shows that the DAS domain structurally resembles the conserved region of chromatin remodeling factor Iws1 (44,45). The Iws1 conserved region is formed by a single HEAT repeat followed by two ARM repeats and has been shown to interact with an N-terminal region of Spt6 (Fig. 6C). This suggests that the DAS domain could act a protein–protein interaction module. Furthermore, from the AlphaFold model, Arg1740 is on the surface of the DAS domain, so the mutants, including R1740W and R1740Q, possibly disrupt the interaction network of the DAS domain, but not disrupt its overall folding (Fig. 6B). The observed differences in epigenotypes between SETD2 truncating mutations SETD2-1740 missense substitutions would be consistent with a gain-of-function effect in the latter. Though the precise mechanisms of the effects of Type 1 (R1740W) and Type 2 (R1740Q) substitutions on SETD2 function remain to be elucidated, the location within the DAS domain in a region predicted to function as a binding site for an unidentified protein provides a hypothesis that could be tested in further studies, as could whether the differences in observed phenotype severity for Type 1 (R1740W) and Type 2 (R1740Q) substitutions were correlated with differential effects on protein binding and gain-of-function effects.
Figure 6.
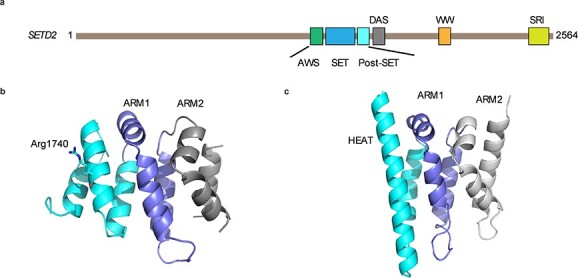
SETD2 codon 1740 structural predictions. (A) Domain architecture of human SETD2. (B) AlphaFold model of SETD2 DAS domain. (C) The structure of Iws1 conserved domain (PDB IDn2XPL).
In summary, we investigated genotype and epigenotype features of SETD1A- and SETD2-associated NDDs. We found evidence that SETD2-associated NDD may be associated with tumor susceptibility and, though we did not identify a clear methylation episignature for SETD1A-NDD, for SETD2-associated NDDs we identified methylation alterations that correlated with the clinical heterogeneity of SETD2 codon 1740 and non-codon 1740 LoF mutations. These observations illustrate how methylation profiling and the detection of specific methylation episignature can provide insights into potential mechanisms of disease that might provide insights into possible approaches for the development of therapeutic interventions.
Materials and Methods
Study cohort
Genomic DNA from patients with germline SETD1A (n = 6) or SETD2 (n = 10) variants were investigated. All variants were categorized as likely pathogenic or pathogenic according to ACMG-AMP criteria (https://wintervar.wglab.org/) (see Table 1). Details of patient ages at DNA sampling and sex are presented in Table 1. Locations of the SETD2 (GRCh37/hg19, ENST00000409792.3; NM_014159.6) and SETD1A (GRCh37/hg19, ENST00000262519.8; NM_014712.1) variants on the respective proteins are shown in Supplementary Material, Fig. S6. For the individuals with a SETD1A-associated variant (1 male and 5 females) the mean age at DNA sampling was 9.2 years, whereas it was 8.8 years in the SETD2-NDD group (6 males and 4 females). Five SETD2-NDD patients had been described previously (SETD2-R1740W-P4, SETD2-R1740W-P1 SETD2-R1740W-P3 and SETD2-R1740W-P2 were patients 4, 7, 9 and 11, respectively, in Rabin et al. (25) and SETD2-LLS-P2 was Case 1 in van Rij et al. (30). Methylation profiling results from the SETD1A- and SETD2-NDD DNA samples were compared with those from 64 healthy control subjects (age range from 0 to 40 with 31 males and 33 females) (29 of whom were included in a previous publication) (5). All 64 controls were examined, and no significant age-related episignature was detected among them. Additionally, age, gender, and batch effects were corrected during differential methylation analysis. The study was performed in accordance with the Helsinki Declaration and written informed consent was obtained from parents/guardians of the patients and blood samples were collected under local study ethical approval. The study was approved by South Birmingham Research Ethics Committee.
Molecular studies
DNA was extracted from whole blood by standard methods. DNA samples were quantified using the Qubit™ dsDNA BR Assay Kit (Invitrogen, ThermoFisher). DNA samples were sequenced on an Illumina NextSeq 2000 using a TruSeq® Methyl Capture EPIC kit. Bisulfite conversion, library preparation, target enrichment and sequencing were performed at the Stratified Medicine Core Laboratory, Department of Medical Genetics of the University of Cambridge, as described previously (5).
Data processing and bioinformatic analysis
The sequencing data were extracted in a FASTQ format. As a first step in filtering, sequencing reads with Phred score ≤ 30 were removed and adapter trimming steps were performed using Trim-Galore software (www.bioinformatics.babraham.ac.uk/projects/trim galore/). The trimmed sequences were subjected to FASTQC to ensure quality control. The sequenced reads were then aligned to the Genome Reference Consortium Human Build 37 (GRCh37) using the Bismark software v0.17.0 (www.bioinformatics.babraham.ac.uk/projects/bismark/) (46). Following alignment, PCR duplicates were removed using the ‘deduplicate bismark’ option. As a final step, methylation values (.bismarkcov format) were extracted using the ‘bismark methylation extractor’ function.
Methylation analysis with RnBeads package: Raw methylation beta-values and annotation information of CpG sites (Open Sea, Shelf, Shore and CpG Island), including the P-value of each CpG position were extracted using the Bioconductor RnBeads R package (https://rnbeads.org/). The RnBeads package was implemented in R software (available on R version 3.6.3). The Bismarkcov files were directly loaded into RnBeads as BED files. As part of the loading procedure, regions with X, Y chromosomes and CpGs that fall near a SNP were removed. The sequencing coverage threshold was set at 10X. CpG sites with exceptionally high coverage outliers and missing values present in more than one sample were removed during the filtering process. Differential methylation analysis option (by ‘limma’ method) was used for extracting beta-values and annotation (mean difference between groups, a P-value of each site) information (47,48). When computing differentially methylated CpG sites, PCA was performed to assess any batch effect (age, gender, batch) and identify significant outliers. If a significant batch effect was detected, the target variables were adjusted by surrogate variable analysis (SVA) using the sva package. Then, age, batch, and gender were considered as covariates when assessing differentially methylated regions by the limma method using the RnBeads ‘covariate.adjustment.columns’ option. Moreover, cell type heterogeneity is widely recognized as a source of confounding in DNA methylation profiling studies, thus an adjustment step using the Houseman method (49) was included in the pipeline to minimize the effects of variations in blood composition. Under the limma package, RnBeads offers cell type adjustment, which were enabled with the ‘differential.adjustment.celltype’ and ‘inference.reference.methylome.column’ = ‘Celltype’ options. By incorporating estimates of cell type proportions into the differential methylation analysis, differences in cell type proportions were adjusted, and any potential bias related to differences in cell type composition between samples could be eliminated, thus increasing the accuracy of the results.
As a result, three different output files were obtained: (1) mean difference between control and disease groups, (2) site P-value obtained from a two-sided Welch t-test or alternatively from linear models employed in the limma package and (3) combined P-value for CpG Islands using a generalization of Fisher’s method. The obtained data were used for the further differentially methylated block (DMB) and methylation episignature analysis. A summary of the sequencing coverage and sequencing reads is provided in Supplementary Material, Table S9. Data is available on request from the authors (subject to patient consent).
Detection and visualization of methylation episignatures: Only CpG sites (or CpG islands) with a neighboring CpG count greater than 5 kb were selected for further analysis. Neighboring CpGs located in CpG sites (Open Sea, Shore and Shelf) were then combined together and assigned as ‘DMB’. DMBs were combined on the basis of their functional similarity. Each DMB has a size ranging from 5 to 200 kb. On the other hand, CpG islands are determined by the Ensembl genome browser (http://www.ensembl.org) (48). Based on the discussed threshold, significant DMBs from SETD2 and SETD1A cohorts were selected for methylation signature analysis. As a first filtering step, DMBs that contained fewer than three EPIC-NGS target regions were filtered out. After that, only DMBs (including CpG Islands) with an adjusted P-value lower than 0.05 (false discovery rate < 0.05) and a methylation difference between controls and diseases group of more than 20% were considered significant for genome-wide CpG site methylation analysis. Moreover, genes associated with significant DMBs and CpG islands, including OMIM/Morbid association, were annotated using the DECIPHER browser (https://www.deciphergenomics.org/).
A heatmap plot of methylation profiles was generated by plotting normalized beta-values (normalized methylation value by mean control value [(β-valuesample − β-valuecontrol_mean)/σ]. It was created with the ComplexHeatmap package (version 2.2.0). Hierarchical clustering was performed using a complete-linkage method by the ‘hclust’ option, and this unsupervised clustering result was then displayed as a dendrogram above the top annotation bar in the heatmap. A scatter plot provided detailed information about methylation patterns. More specifically, by comparing the methylation beta-values of individuals with the mean values of ±3 standard deviation confidence interval of healthy individuals, a significant gain or loss of methylation could be identified. A clustering plot was created using a PCA algorithm based on pre-processed beta-values (not normalized) in order to remove potential bias from normalized data. PCA (unsupervised clustering) was performed with the ‘prcomp’ option in R (v.3.6.3).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the patients and families for their participation in this study and Dr Moira Blyth for help with patient ascertainment. The views expressed are those of the authors and not necessarily those of the NHS or Department of Health. We acknowledge support from the NIHR UK Rare Genetic Disease Research Consortium. The University of Cambridge has received salary support (E.R.M.) from the NHS in the East of England through the Clinical Academic Reserve. The views expressed are those of the authors and not necessarily those of the NHS or Department of Health. We acknowledge support from the NIHR UK Rare Genetic Disease Research Consortium.
Conflict of Interest statement. Dr Menzies reports personal consultancy fees from Mendelian Ltd, a rare disease digital health company, outside the submitted work. The other authors have no competing interests to declare.
Contributor Information
Sunwoo Lee, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK.
Lara Menzies, Department of Clinical Genetics, Great Ormond Street Hospital, London WC1N 3JH, UK.
Eleanor Hay, Department of Clinical Genetics, Great Ormond Street Hospital, London WC1N 3JH, UK.
Eguzkine Ochoa, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK.
France Docquier, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK; Stratified Medicine Core Laboratory NGS Hub, Department of Medical Genetics, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Fay Rodger, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK; Stratified Medicine Core Laboratory NGS Hub, Department of Medical Genetics, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Charu Deshpande, Manchester Centre for Genomic Medicine, Manchester University Hospitals NHS Foundation Trust, Saint Mary’s Hospital, Manchester, UK.
Nicola C Foulds, Wessex Clinical Genetics Services, University Hospital Southampton NHS Foundation Trust, Southampton, UK.
Sébastien Jacquemont, CHU Sainte-Justine Research Centre, Montreal, Quebec, Canada; Department of Pediatrics, University of Montreal, Montreal, Quebec, Canada.
Khadije Jizi, CHU Sainte-Justine Research Centre, Montreal, Quebec, Canada.
Henriette Kiep, Department of Neuropediatrics, University Hospital for Children and Adolescents, Leipzig, Germany.
Alison Kraus, Yorkshire Regional Genetics Service, Chapel Allerton Hospital, Leeds, UK.
Katharina Löhner, Department of Genetics, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Patrick J Morrison, Patrick G Johnston Centre for Cancer Research and Cell Biology, Queens University Belfast, Belfast, UK.
Bernt Popp, Institute of Human Genetics, University of Leipzig Medical Center, Leipzig, Germany; Center of Functional Genomics, Berlin Institute of Health at Charité, Universitätsmedizin Berlin, Berlin, Germany.
Ruth Richardson, Northern Genetics Service, The Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Arie van Haeringen, Department of Clinical Genetics, Leiden University Hospital, Leiden, The Netherlands.
Ezequiel Martin, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK; Stratified Medicine Core Laboratory NGS Hub, Department of Medical Genetics, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Ana Toribio, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK; Stratified Medicine Core Laboratory NGS Hub, Department of Medical Genetics, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Fudong Li, MOE Key Laboratory for Cellular Dynamics, The School of Life Sciences, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230026, China.
Wendy D Jones, Department of Clinical Genetics, Great Ormond Street Hospital, London WC1N 3JH, UK.
Francis H Sansbury, All Wales Medical Genomics Service, NHS Wales Cardiff and Vale University Health Board and Institute of Medical Genetics, University Hospital of Wales, Heath Park, Cardiff, UK.
Eamonn R Maher, Department of Medical Genetics, University of Cambridge, Cambridge CB2 0QQ, UK.
Funding
NIHR Cambridge Biomedical Research Centre (BRC-1215-20014 and NIHR203312); Rosetrees Trust (to E.O., S.L. and E.R.M.).
Ethical conduct of research
This study was performed in accordance with the Helsinki Declaration and written informed consent was obtained from parents/guardians of the patients. Blood samples were collected under local study ethical approvals. The study was approved by South Birmingham Research Ethics Committee.
References
- 1. Rodenhiser, D. and Mann, M. (2006) Epigenetics and human disease: translating basic biology into clinical applications. Can. Med. Assoc. J., 174, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aref-Eshghi, E., Rodenhiser, D.I., Schenkel, L.C., Lin, H., Skinner, C., Ainsworth, P., Paré, G., Hood, R.L., Bulman, D.E., Kernohan, K.D.et al. (2018) Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. Am. J. Hum. Genet., 102, 156–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sadikovic, B., Levy, M.A., Kerkhof, J., Aref-Eshghi, E., Schenkel, L., Stuart, A., McConkey, H., Henneman, P., Venema, A., Schwartz, C.E.et al. (2021) Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med., 23, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levy, M.A., Relator, R., McConkey, H., Pranckeviciene, E., Kerkhof, J., Barat-Houari, M., Bargiacchi, S., Biamino, E., Palomares Bralo, M., Cappuccio, G.et al. (2022) Functional correlation of genome-wide DNA methylation profiles in genetic neurodevelopmental disorders. Hum. Mutat., 43, 1609–1628. [DOI] [PubMed] [Google Scholar]
- 5. Lee, S., Ochoa, E., Barwick, K., Cif, L., Rodger, F., Docquier, F., Pérez-Dueñas, B., Clark, G., Martin, E., Banka, S.et al. (2022) Comparison of methylation episignatures in KMT2B- and KMT2D-related human disorders. Epigenomics, 14, 537–547. [DOI] [PubMed] [Google Scholar]
- 6. Harrison, A. and Parle-McDermott, A. (2011) DNA methylation: a timeline of methods and applications. Front. Genet., 2, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O’Roak, B.J., Vives, L., Fu, W., Egertson, J.D., Stanaway, I.B., Phelps, I.G., Carvill, G., Kumar, A., Lee, C., Ankenman, K.et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism Spectrum disorders. Science, 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haghshenas, S., Bhai, P., Aref-Eshghi, E. and Sadikovic, B. (2020) Diagnostic utility of genome-wide dna methylation analysis in mendelian neurodevelopmental disorders. Int. J. Mol. Sci., 21, 9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soden, S.E., Saunders, C.J., Willig, L.K., Farrow, E.G., Smith, L.D., Petrikin, J.E., LePichon, J.B., Miller, N.A., Thiffault, I., Dinwiddie, D.L.et al. (2014) Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med., 6, 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Srivastava, S., Love-Nichols, J.A., Dies, K.A., Ledbetter, D.H., Martin, C.L., Chung, W.K., Firth, H.V., Frazier, T., Hansen, R.L., Prock, L.et al. (2019) Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med., 21, 2413–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sztainberg, Y. and Zoghbi, H.Y. (2016) Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci., 19, 1408–1417. [DOI] [PubMed] [Google Scholar]
- 12. Ziats, C.A., Patterson, W.G. and Friez, M. (2021) Syndromic autism revisited: review of the literature and lessons learned. Pediatr. Neurol., 114, 21–25. [DOI] [PubMed] [Google Scholar]
- 13. Levy, M.A., McConkey, H., Kerkhof, J., Barat-Houari, M., Bargiacchi, S., Biamino, E., Bralo, M.P., Cappuccio, G., Ciolfi, A., Clarke, A.et al. (2022) Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv., 3, 100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang, Y. and Reinberg, D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
- 15. Ng, S.B., Bigham, A.W., Buckingham, K.J., Hannibal, M.C., McMillin, M.J., Gildersleeve, H.I., Beck, A.E., Tabor, H.K., Cooper, G.M., Mefford, H.C.et al. (2010) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet., 42, 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones, W.D., Dafou, D., McEntagart, M., Woollard, W.J., Elmslie, F.V., Holder-Espinasse, M., Irving, M., Saggar, A.K., Smithson, S., Trembath, R.C.et al. (2012) De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet., 91, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleefstra, T., Kramer, J.M., Neveling, K., Willemsen, M.H., Koemans, T.S., Vissers, L.E., Wissink-Lindhout, W., Fenckova, M., Akker, W.M., Kasri, N.N.et al. (2012) Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet., 91, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zech, M., Boesch, S., Maier, E.M., Borggraefe, I., Vill, K., Laccone, F., Pilshofer, V., Ceballos-Baumann, A., Alhaddad, B., Berutti, R.et al. (2016) Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am. J. Hum. Genet., 99, 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O’Donnell-Luria, A.H., Pais, L.S., Faundes, V., Wood, J.C., Sveden, A., Luria, V., Abou Jamra, R., Accogli, A., Amburgey, K., Anderlid, B.M.et al. (2019) Heterozygous variants in KMT2E cause a spectrum of neurodevelopmental disorders and epilepsy. Am. J. Hum. Genet., 104, 1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kurotaki, N., Imaizumi, K., Harada, N., Masuno, M., Kondoh, T., Nagai, T., Ohashi, H., Naritomi, K., Tsukahara, M., Makita, Y.et al. (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet., 30, 365–366. [DOI] [PubMed] [Google Scholar]
- 21. Tatton-Brown, K., Douglas, J., Coleman, K., Baujat, G., Cole, T.R.P., Das, S., Horn, D., Hughes, H.E., Temple, I.K., Faravelli, F.et al. (2005) Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am. J. Hum. Genet., 77, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh, T., Kurki, M.I., Curtis, D., Purcell, S.M., Crooks, L., McRae, J., Suvisaari, J., Chheda, H., Blackwood, D., Breen, G.et al. (2016) Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci., 19, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hiraide, T., Nakashima, M., Yamoto, K., Fukuda, T., Kato, M., Ikeda, H., Sugie, Y., Aoto, K., Kaname, T., Nakabayashi, K.et al. (2018) De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum. Genet., 137, 95–104. [DOI] [PubMed] [Google Scholar]
- 24. Yu, X., Yang, L., Li, J., Li, W., Li, D., Wang, R., Wu, K., Chen, W., Zhang, Y., Qiu, Z.et al. (2019) De novo and inherited SETD1A variants in early-onset epilepsy. Neurosci. Bull., 35, 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rabin, R., Radmanesh, A., Glass, I.A., Dobyns, W.B., Aldinger, K.A., Shieh, J.T., Romoser, S., Bombei, H., Dowsett, L., Trapane, P.et al. (2020) Genotype–phenotype correlation at codon 1740 of SETD2. Am. J. Med. Genet., 182, 2037–2048. [DOI] [PubMed] [Google Scholar]
- 26. Chen, F., Chen, J., Wang, H., Tang, H., Huang, L., Wang, S., Wang, X., Fang, X., Liu, J., Li, L.et al. (2021) Histone lysine methyltransferase SETD2 regulates coronary vascular development in embryonic mouse hearts. Front. Cell Dev. Biol., 9, 651655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Husmann, D. and Gozani, O. (2019) Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol., 26, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luscan, A., Laurendeau, I., Malan, V., Francannet, C., Odent, S., Giuliano, F., Lacombe, D., Touraine, R., Vidaud, M., Pasmant, E.et al. (2014) Mutations in SETD2 cause a novel overgrowth condition. J. Med. Genet., 51, 512–517. [DOI] [PubMed] [Google Scholar]
- 29. Lumish, H.S., Wynn, J., Devinsky, O. and Chung, W.K. (2015) Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J. Autism Dev. Disord., 45, 3764–3770. [DOI] [PubMed] [Google Scholar]
- 30. van Rij, M.C., Hollink, I.H.I.M., Terhal, P.A., Kant, S.G., Ruivenkamp, C., vanHaeringen, A., Kievit, J.A. and vanBelzen, M.J. (2018) Two novel cases expanding the phenotype of SETD2-related overgrowth syndrome. Am. J. Med. Genet. A, 176, 1212–1215. [DOI] [PubMed] [Google Scholar]
- 31. Marzin, P., Rondeau, S., Aldinger, K.A., Alessandri, J.L., Isidor, B., Heron, D., Keren, B., Dobyns, W.B. and Cormier-Daire, V. (2019) SETD2 related overgrowth syndrome: presentation of four new patients and review of the literature. Am. J. Med. Genet. C. Semin. Med. Genet., 181, 509–518. [DOI] [PubMed] [Google Scholar]
- 32. Butcher, D.T., Cytrynbaum, C., Turinsky, A.L., Siu, M.T., Inbar-Feigenber, M., Mendoza-Londono, R., Chitayat, D., Walker, S., Machado, J., Caluseriu, O.et al. (2017) CHARGE and kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet., 100, 773–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krzyzewska, I.M., Maas, S.M., Henneman, P., Lip, K.V.D., Venema, A., Baranano, K., Chassevent, A., Aref-Eshghi, E., Van Essen, A.J., Fukuda, T.et al. (2019) A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenetics, 11, 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ciolfi, A., Foroutan, A., Capuano, A., Pedace, L., Travaglini, L., Pizzi, S., Andreani, M., Miele, E., Invernizzi, F., Reale, C.et al. (2021) Childhood-onset dystonia-causing KMT2B variants result in a distinctive genomic hypermethylation profile. Clin. Epigenetics, 13, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monk, D., Mackay, D.J.G., Eggermann, T., Maher, E.R. and Riccio, A. (2019) Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet., 20, 235–248. [DOI] [PubMed] [Google Scholar]
- 36. Choufani, S., Cytrynbaum, C., Chung, B.H.Y., Turinsky, A.L., Grafodatskaya, D., Chen, Y.A., Cohen, A.S.A., Dupuis, L., Butcher, D.T., Siu, M.T.et al. (2015) NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun., 6, 10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeffries, A.R., Maroofian, R., Salter, C.G., Chioza, B.A., Cross, H.E., Patton, M.A., Dempster, E., Karen Temple, I., Mackay, D.J.G., Rezwan, F.I.et al. (2019) Growth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging. Genome Res., 29, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abuzzahab, M.J., Schneider, A., Goddard, A., Grigorescu, F., Lautier, C., Keller, E., Kiess, W., Klammt, J., Kratzsch, J., Osgood, D.et al. (2003) IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N. Engl. J. Med., 349, 2211–2222. [DOI] [PubMed] [Google Scholar]
- 39. Xu, Q., Xiang, Y., Wang, Q., Wang, L., Brind’Amour, J., Bogutz, A.B., Zhang, Y., Zhang, B., Yu, G.et al. (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat. Genet., 51, 855–856. [DOI] [PubMed] [Google Scholar]
- 40. Tatton-Brown, K., Rahman, N. (2013 Jul 18 [updated 2018 Aug 2]) EZH2-Related Overgrowth. In: Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. PMID: 23865096. [Google Scholar]
- 41. Carvalho, S., Vitor, A.C., Sridhara, S.C., Martins, F.B., Raposo, A.C., Desterro, J.M.P., Ferreira, J. and Almeida, F. (2014) SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. elife, 3, e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scelo, G., Riazalhosseini, Y., Greger, L., Letourneau, L., Gonzàlez-Porta, M., Wozniak, M.B., Bourgey, M., Harnden, P., Egevad, L., Jackson, S.M.et al. (2014) Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat. Commun., 5, 5135. [DOI] [PubMed] [Google Scholar]
- 43. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A.et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holm, L. (2022) Dali server: structural unification of protein families. Nucleic Acids Res., 50, W210–W215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Diebold, M.-L., Koch, M., Loeliger, E., Cura, V., Winston, F., Cavarelli, J. and Romier, C. (2010) The structure of an Iws1/Spt6 complex reveals an interaction domain conserved in TFIIS, Elongin A and Med26. EMBO J., 29, 3979–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krueger, F. and Andrews, S.R. (2011) Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Assenov, Y., Müller, F., Lutsik, P., Walter, J., Lengauer, T. and Bock, C. (2014) Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods, 11, 1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Müller, F., Scherer, M., Assenov, Y., Lutsik, P., Walter, J., Lengauer, T. and Bock, C. (2019) RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol., 20, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Houseman, E.A., Accomando, W.P., Koestler, D.C., Christensen, B.C., Marsit, C.J., Nelson, H.H., Wiencke, J.K. and Kelsey, K.T. (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics., 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.