

Abstract
BACKGROUND
Colorectal cancer is a complex disease with high mortality rates. Over time, the treatment of metastatic colorectal cancer (mCRC) has gradually improved due to the development of modern chemotherapy and targeted therapy regimens. However, due to the inherent heterogeneity of this condition, identifying reliable predictive biomarkers for targeted therapies remains challenging. A recent promising classification system—the consensus molecular subtype (CMS) system—offers the potential to categorize mCRC patients based on their unique biological and molecular characteristics. Four distinct CMS categories have been defined: immune (CMS1), canonical (CMS2), metabolic (CMS3), and mesenchymal (CMS4). Nevertheless, there is currently no standardized protocol for accurately classifying patients into CMS categories. To address this challenge, reverse transcription polymerase chain reaction (RT-qPCR) and next-generation genomic sequencing (NGS) techniques may hold promise for precisely classifying mCRC patients into their CMSs.
AIM
To investigate if mCRC patients can be classified into CMS categories using a standardized molecular biology workflow.
METHODS
This observational study was conducted at the University of Chile Clinical Hospital and included patients with unresectable mCRC who were undergoing systemic treatment with chemotherapy and/or targeted therapy. Molecular biology techniques were employed to analyse primary tumour samples from these patients. RT-qPCR was utilized to assess the expression of genes associated with fibrosis (TGF-β and β-catenin) and cell growth pathways (c-MYC). NGS using a 25-gene panel (TumorSec) was performed to identify specific genomic mutations. The patients were then classified into one of the four CMS categories according to the clinical consensus of a Tumour Board. Informed consent was obtained from all the patients prior to their participation in this study. All techniques were conducted at University of Chile.
RESULTS
Twenty-six patients were studied with the techniques and then evaluated by the Tumour Board to determine the specific CMS. Among them, 23% (n = 6), 19% (n = 5), 31% (n = 8), and 19% (n = 5) were classified as CMS1, CMS2, CMS3, and CMS4, respectively. Additionally, 8% of patients (n = 2) could not be classified into any of the four CMS categories. The median overall survival of the total sample was 28 mo, and for CMS1, CMS2, CMS3 and CMS4 it was 11, 20, 30 and 45 mo respectively, with no statistically significant differences between groups.
CONCLUSION
A molecular biology workflow and clinical consensus analysis can be used to accurately classify mCRC patients. This classification process, which divides patients into the four CMS categories, holds significant potential for improving research strategies and targeted therapies tailored to the specific characteristics of mCRC.
Keywords: Metastatic colorectal cancer, Targeted therapy, Consensus molecular subtypes, Personalized medicine
Core Tip: Colorectal cancer is molecularly heterogeneous. Consensus molecular subtype classification sheds light on its biology, potentially guiding targeted therapy selection. However, an optimal consensus molecular subtype classification mechanism remains elusive. This workflow, which combines reverse transcription polymerase chain reaction and next-generation sequencing, introduces a novel approach for molecular patient classification. We aim to use these techniques to improve the precision of tumour subtyping.
INTRODUCTION
Colorectal cancer (CRC) exhibits high incidence and mortality rates. At the time of diagnosis, approximately 25% of patients already present with metastatic disease, while 50% of those initially diagnosed with localized stages later develop disseminated disease[1]. Recent years have seen significant advancements in systemic therapies for metastatic colorectal cancer (mCRC) patients, including diverse combination chemotherapy regimens, targeted therapy, immunotherapy, and multi-kinase inhibitors[2]. Despite these improvements, patients’ responses remain variable and unpredictable due to the molecular heterogeneity of this disease. Thus, it is imperative to identify specific mutations for a personalized treatment approach[3].
Numerous efforts have attempted to identify distinct molecular mCRC phenotypes. In 2015, bioinformatic studies revealed a promising classification system with four consensus molecular subtypes (CMS)[4]. This classification system has gained widespread clinical acceptance and is currently guiding various ongoing clinical trials[5]. The four CMS are as follows: CMS1, or immune subtype, primarily affects young patients and exhibits rapid progression and resistance to conventional therapies. This subtype may benefit from aggressive chemotherapy and, potentially, immunotherapy. CMS2, or canonical subtype, is characterized by mutations in specific pathways linked to cellular metabolism. CMS3, or metabolic subtype, is characterized by mutations in pathways responsible for cellular metabolism, with a high prevalence of KRAS pathway mutations. Finally, CMS4, or mesenchymal subtype, is associated with mutations in fibrogenesis and epithelial-mesenchymal transition pathways, leading to a poor prognosis and a higher incidence of metastasis[5]. To date, there is no established methodology for effectively classifying patients into CMS categories. However, given that each CMS is linked to distinct patterns of mutations and gene expression, it is plausible that a molecular biology workflow designed to identify specific mutations could help accurately classify patients into different CMS groups[6]. Therefore, the objective of this study was to establish a workflow for assigning mCRC patients to CMS categories using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and next-generation sequencing (NGS) techniques.
MATERIALS AND METHODS
Study design and participants
In this observational study conducted between 2020 and 2023, we analyzed primary tumor tissue samples from mCRC patients who were receiving systemic treatment at the University of Chile Clinical Hospital. Colon or rectal tissue samples were collected through colonoscopy or surgical procedures. The samples were processed and stored according to protocols established by the Biobank of Tissues and Fluids at the University of Chile (http://biobanco.uchile.cl/). Both formalin-fixed paraffin-embedded (FFPE) tissue biopsies and fresh neoplastic tissue (frozen without fixation) were examined.
The inclusion criteria for this study were as follows: Patients diagnosed with unresectable mCRC (colon or rectal cancer) confirmed through histological diagnosis. Undergoing treatment at the University of Chile Clinical Hospital. Receiving systemic therapy in accordance with international clinical guidelines (National Comprehensive Cancer Network[7] and European Society of Medical Oncology)[8]. Treatment regimens included chemotherapy (FOLFOX, CAPOX, or FOLFIRI) and targeted therapy (bevacizumab, aflibercept, cetuximab, panitumumab, regorafenib, and TAS102). Chemotherapy and targeted therapy regimens were selected by the physicians on a case-by-case basis.
The exclusion criteria for this study were as follows: Patients who underwent the removal of metastases (metastasectomy) before enrollment. Any comorbidity leading to a life expectancy of less than six months. Inability to maintain clinical follow-up.
RT-qPCR
The expression of TGF-β, β-catenin, and c-MYC was investigated as follows: RNA was extracted from FFPE tissue using the RecoverAllTM Total Nucleic Acid Isolation Kit for FFPE (Invitrogen). Subsequently, the concentration of each RNA sample was determined using the Quant-iTTM RiboGreenTM RNA Reagent Kit (Invitrogen) on a Cytation 3 instrument (BioTek). RNA (1000) ng was then used to prepare cDNA with the AffinityScript qPCR cDNA Synthesis Kit (Agilent) according to the manufacturer’s instructions. Amplifications by qPCR (real-time PCR) was conducted in triplicate using the Brilliant II SYBR Green qPCR Master Mix kit (Agilent) on an Eco Real-time PCR System (Illumina). The following cycling conditions were used: an initial denaturation step at 95ºC for 10 min, then 40 cycles of amplification (each cycle is 10 s at 95ºC, 30 s at 60ºC and 15s at 72°C). A melting curve for each primer ensured amplification of a single product. Finally, six FFPE non-tumour tissue samples treated in the same manner as the FFPE tumour tissues from each patient were included as controls. The relative expression was calculated using the ΔΔCt method[9] and normalized using expression levels of reference genes: B2M, PPIA, and RPLP0. Table 1 presents a summary of the primers used to conduct the RT-qPCR experiments[10-15].
Table 1.
Primers employed for reverse transcription-quantitative polymerase chain reaction experiments to determine the expression of β-catenin, c-Myc and TFG-β, and the genes used as reference genes
|
Name
|
Primer
|
Sequence
|
Product Lenght
|
Ref.
|
| TGF-β | Forward | 5’- TACCTGAACCCGTGTTGCTCTC-3’ | 122 | [10] |
| Reverse | 5’- GTTGCTGAGGTATCGCCAGGA-3’ | |||
| β-catenin | Forward | 5’- CACAAGCAGAGTGCTGAAGGTG-3’ | 146 | [11] |
| Reverse | 5’- GATTCCTGAGAGTCCAAAGACAG-3’ | |||
| c-MYC | Forward | 5’-GCCACGTCTCCACACATCAG-3’ | 132 | [12] |
| Reverse | 5’-TGGTGCATTTTCGGTTGTTG-3’ | |||
| B2M | Forward | 5’-GTGCTCGCGCTACTCTCTC-3’ | 150 | [13] |
| Reverse | 5’-GTCAACTTCAATGTCGGAT-3’ | |||
| PPIA | Forward | 5’-GCAAATGCTGGACCCAACACAAAT-3’ | 174 | [14] |
| Reverse | 5’-AATGGTGATCTTCTTGCTGGTCTTG-3’ | |||
| RPLP0 | Forward | 5’-GCAATGTTGCCAGTGTCTG-3’ | 142 | [15] |
| Reverse | 5’-GCCTTGACCTTTTCAGCAA-3’ |
NGS
The presence of genomic mutations was assessed using a 25-gene panel (TumorSec) as described by our team[16]. The RecoverAllTM Total Nucleic Acid Isolation Kit for FFPE was utilized to extract genomic deoxyribonucleic acid (DNA) from FFPE samples. Briefly, samples were incubated with 1 mL of Histo-Clear at 50°C for 3 min to remove paraffin. The supernatant was then removed, followed by two ethanol washes, and the residual ethanol was evaporated using a SpeedVAC (Thermo Scientific). The samples were then incubated overnight in a digestion solution containing proteases. The next day, the samples were incubated at 80°C for 15 min and an isolation additive was added and centrifuged. Subsequently, the supernatant was transferred to a filter column and centrifuged to isolate the RNA, which was then treated with DNase. The column contained the DNA, which was subsequently treated with RNase. The DNA and RNA were washed with wash buffers and eluted in elution buffer in separate tubes.
Quantification and quality analysis: The purity and quantity of DNA and RNA were determined by measuring absorbance at 260/280 nm with the PicoGreen assay (Quant-iTTM PicoGreen® dsDNA, Invitrogen) and the Quant-iTTM RiboGreenTM RNA Reagent Kit, respectively, on a Cytation 3 instrument (Biotek). Additionally, DNA quality analysis was conducted by measuring fragment size with the HS Genomic DNA Analysis Kit (DNF-488) (Agilent) on a Fragment Analyzer instrument (Agilent). As the extraction of genomic DNA from FFPE samples often results in low yields and degradation ranging from more than 1000 bp to less than 200 bp, fragments less than 200 bp were not used for library preparation due to excessive degradation. To ensure adequate DNA quantity, a minimum of four, 6-μm FFPE sections per patient were used for sequencing. Moreover, each sample needed to contain more than 20% tumour content.
Library preparation: The KAPA HyperPlus Library Preparation Kit (Kapa Biosystems) was utilized to prepare DNA libraries. Library sizes and concentrations were verified for quality control purposes. The 260/280 nm ratio was measured with Cytation equipment and quantification was carried out using the Quant-iTTM PicoGreenTM dsDNA Assay Kit. Furthermore, library sizes were visualized using the HS NGS Analysis Kit in a Fragment Analyzer instrument.
NGS: NGS was conducted following a protocol previously published by our team[9]. For sequencing, an equimolar pool of libraries (4 nM) was prepared, diluted, and denatured to achieve a final concentration of 9.4–9.5 pM according to guidelines in the "MiSeq System Denature and Dilute Libraries Guide" (Illumina). Paired-end sequencing (300 cycles) was performed using the Illumina MiSeq System (MiSeq Reagent Kits v2). Finally, bioinformatics analysis was conducted.
Classification of patients into CMS categories
Given the absence of a singular marker that differentiates each of the four CMS categories on its own, we developed a comprehensive protocol involving analysis by a Tumour Board consisting of experts in Molecular and Medical Oncology. Each case was individually assessed and the CMS was determined based on the criteria defined by Guinney et al[4]. The Tumour Board relied on patients’ clinical characteristics, mismatch repair (MMR) expression, and RT-qPCR and NGS results. Each patient’s CMS was determined by consensus among all committee members. Patients for whom a CMS consensus could not be reached were considered unclassifiable.
The Tumour Board employed the following criteria to classify each patient into one of the four CMS categories. It is important to note that none of these elements individually serve as a specific CMS marker; instead, classifications were based on the combination of multiple elements and reached through tumour board consensus. CMS1: presence of BRAF mutation; MMR protein deficiency; low TGF-β, β-catenin, and c-MYC mRNA expression; and absence of APC or KRAS mutations. CMS2 and CMS3: presence of APC mutation; absence of BRAF mutation (with a predominance of KRAS mutations in CMS3); MMR-proficient; low TGF-β and β-catenin mRNA expression; and high c-MYC mRNA expression. CMS4: MMR-proficient; high expression of TGF-β and β-catenin mRNA; low expression of c-MYC mRNA; and presence of non-categorical mutations identified through NGS[6].
Ethics
All procedures conducted in this study were in full compliance with the ethical standards set by the Institutional and National Research Committee, as well as the principles outlined in the 1964 Declaration of Helsinki and its subsequent amendments. Ethical approval for this study was obtained from the Ethics Committee of the University of Chile Clinical Hospital and Faculty of Medicine prior to beginning the research. Informed written consent was obtained from all patients before their participation in the trial.
Statistics
Results are presented as the number and percentage of total patients included in this study. To determine the appropriate sample size, we considered the estimated prevalence of each mCRC CMS. According to previous work[4], the expected prevalence of each CMS is approximately 20%–25%. A sample size of 25 patients was deemed sufficient to analyze the prevalence and distribution of the different CMS categories. Indeed, prior research has utilized sample sizes of 20–30 patients; thus, a sample size of 25 patients is consistent with the literature. For the overall survival analysis of the studied patients, log-rank test was conducted using GraphPad Prism 10.0 software.
RESULTS
Between 2020 and 2023, a total of 26 patients with unresectable mCRC undergoing systemic treatment at the University of Chile Clinical Hospital were included in this study. Table 2 presents the demographic and clinical characteristics of the patients, including age, gender, primary tumour site, and the presence or absence of MMR proteins. Each patient is identified with a number from 1–26.
Table 2.
Clinical characteristics, overall survival, and reverse transcription-quantitative polymerase chain reaction results of the n = 26 patients included in the final analysis
|
Patient number
|
Age
|
Gender
|
Site of primary cancer
|
Overall survival (mo)
|
Miss-match repair proteins expression
|
β-catenin expression (RT-qPCR) (relative expression with respect to reference gene average)
|
c-MYC expression (RT-qPCR) (relative expression with respect to reference gene average)
|
TGF-β expression (RT-qPCR) (relative expression with respect to reference gene average)
|
CMS
|
| 1 | 69 | Male | Sigmoid | 5 | Proficient | 0.185 | 2.864 | 0.201 | CMS2 |
| 2 | 85 | Female | Right colon | 31 | Proficient | 0.100 | 0.352 | 0.169 | CMS1 |
| 3 | 68 | Female | Rectal and sigmoid | 12 | Proficient | 0.042 | 0.384 | 0.076 | CMS3 |
| 4 | 57 | Male | Rectal and sigmoid | 34 | Proficient | 2.684 | 18.817 | 9.778 | CMS3 |
| 5 | 45 | Female | Transverse | 40 | Proficient | 1.812 | 19.445 | 5.231 | CMS2 |
| 6 | 62 | Male | Rectum | 28 | Proficient | 0.010 | 4.401 | 0.973 | CMS3 |
| 7 | 54 | Male | Rectum | 20 | Proficient | 0.301 | 3.234 | 1.433 | CMS2 |
| 8 | 55 | Male | Sigmoid | 53 | Proficient | 0.080 | 1.870 | 11.718 | CMS4 |
| 9 | 73 | Male | Sigmoid | 62 | Proficient | 0.038 | 0.645 | 0.461 | CMS1 |
| 10 | 79 | Male | Rectum | 40 | Proficient | 0.121 | 2.080 | 3.513 | CMS3 |
| 11 | 56 | Female | Right colon | 29 | Proficient | 0.235 | 3.799 | 14.700 | CMS4 |
| 12 | 66 | Female | Right colon | 10 | Proficient | 0.351 | 6.004 | 76.116 | CMS4 |
| 13 | 53 | Male | Sigmoid | 52 | Proficient | 0.233 | 3.863 | 2.688 | CMS4 |
| 14 | 75 | Male | Sigmoid | 35 | Proficient | 0.089 | 0.760 | 0.205 | CMS3 |
| 15 | 63 | Male | Right colon | 32 | Proficient | 0.089 | 0.760 | 0.466 | CMS3 |
| 16 | 48 | Female | Sigmoid | 28 | Proficient | 0.038 | 1.110 | 0.498 | CMS3 |
| 17 | 53 | Female | Rectum | 20 | Proficient | 0.084 | 1.124 | 0.801 | Not classifiable |
| 18 | 71 | Female | Right colon | 12 | Proficient | 0.083 | 1.540 | 0.897 | CMS1 |
| 19 | 61 | Female | Sigmoid | 45 | Proficient | 0.106 | 9.208 | 6.820 | CMS4 |
| 20 | 71 | Male | Rectum | 10 | Proficient | 0.013 | 0.855 | 0.065 | CMS3 |
| 21 | 49 | Female | Sigmoid | 6 | Deficient | 0.063 | 2.968 | 1.871 | CMS1 |
| 22 | 74 | Male | Right colon | 11 | Deficient | 0.047 | 0.552 | 0.249 | CMS1 |
| 23 | 65 | Female | Rectum | 39 | Proficient | 0.059 | 0.828 | 0.084 | Not classifiable |
| 24 | 59 | Female | Sigmoid | 8 | Proficient | 0.045 | 1.324 | 0.152 | CMS2 |
| 25 | 54 | Male | Sigmoid | 5 | Deficient | 0.036 | 0.543 | 0.127 | CMS1 |
| 26 | 69 | Male | Sigmoid | 22 | Proficient | 0.192 | 5.025 | 2.654 | CMS2 |
Each patient is individually identified in the first column on the left with a sequential number ranging from 1 to 26. Additionally, the consensus molecular subtype assigned based on the Tumour Board analysis is provided. RT-qPCR: reverse transcription-quantitative polymerase chain reaction; TGF-β: Transforming growth factor beta; CMS: consensus molecular subtype.
Molecular studies
Table 2 illustrates the results of an RT-qPCR-based gene expression analysis of TGF-β, β-catenin, and c-MYC in each of the patients studied. It is observed that the expression of these three genes is heterogeneous among patients. Table 3 provides a comprehensive overview of the mutations identified with the 25-gene TumorSec panel. The most frequently observed mutations were in KRAS, TP53 and ARID1A. All observed mutations were single nucleotide variants (SNVs) and two patients possessed deletions.
Table 3.
Mutations identified in the n = 26 patients included in the final analysis through massive genomic sequencing using the TumorSec panel
|
Patient number
|
Mutation
|
Mutation variant classification
|
Affected protein
|
Variant type
|
| 1 | TSC2 | Missense | p.R1729C | SNV |
| TP53 | Missense | p.R175H | SNV | |
| 2 | KRAS | Missense | p.G12C | SNV |
| 3 | KRAS | Missense | p.G12V | SNV |
| TP53 | Missense | p.R175H | SNV | |
| 4 | KRAS | Missense | p.Q61H | SNV |
| PIK3CA | Missense | p.E545G | SNV | |
| 5 | TP53 | Missense | p.P152L | SNV |
| 6 | KRAS | Missense | p.G12D | SNV |
| 7 | BRCA2 | Missense | p.K584E | SNV |
| ARID1A | Nonsense | p.Q1584 | SNV | |
| 8 | KRAS | Missense | p.N116H | SNV |
| TP53 | Missense | p.R175H | SNV | |
| PIK3CA | Missense | p.H1047R | SNV | |
| BRAF | Missense | p.N581Y | SNV | |
| 9 | BRCA2 | Frameshift (deletion) | p.N863Ifs11 | SNV |
| ARID1A | Frameshift (deletion) | p.P1326Rfs155 | SNV | |
| PIK3CA | Missense | p.H1047R | SNV | |
| 10 | PTEN | Nonsense | p.Y225 | SNV |
| KRAS | Missense | p.G12C | SNV | |
| TP53 | Frameshift (insertion) | p.Q317Pfs20 | SNV | |
| 11 | KRAS | Missense | p.Q61H | SNV |
| 12 | KRAS | Missense | p.G12D | SNV |
| TP53 | Missense | p.R280K | SNV | |
| 13 | TP53 | Missense | p.R273H | SNV |
| 14 | KRAS | Missense | p.G12D | SNV |
| TP53 | Missense | p.P278L | SNV | |
| 15 | KRAS | Missense | p.K117N | SNV |
| TP53 | Missense | p.R282W | SNV | |
| 16 | KRAS | Missense | p.G12D | SNV |
| TP53 | Frameshift (deletion) | p.S260Qfs3 | Deletion | |
| 17 | KRAS | Missense | p.Q61L | SNV |
| BRCA2 | Missense | p.S3147Y | SNV | |
| TP53 | Missense | p.R249G | SNV | |
| 18 | KRAS | Missense | p.G12C | SNV |
| ARID1A | Frameshift (deletion) | p.Q611Hfs7 | Deletion | |
| 19 | TP53 | Missense | p.Y220C | SNV |
| 20 | KRAS | Missense | p.A59G | SNV |
| KRAS | Missense | p.G12D | SNV | |
| TP53 | Missense | p.H214R | SNV | |
| 21 | NRAS | Missense | p.Q61R | SNV |
| ARID1A | Frameshift (deletion) | p.K1072Nfs21 | SNV | |
| 22 | TP53 | Missense | p.R273C | SNV |
| 23 | TP53 | Nonsense | p.E51 | SNV |
| ARID1A | Frameshift (deletion) | p.Q372Sfs19 | SNV | |
| 24 | TP53 | Missense | p.R248W | SNV |
| PIK3CA | Missense | p.E545K | SNV | |
| 25 | PTEN | Nonsense | p.Q149 | SNV |
| KRAS | Missense | p.G13D | SNV | |
| TSC2 | Missense | p.R1713C | SNV | |
| TP53 | Missense | p.R273C | SNV | |
| TP53 | Missense | p.R158H | SNV | |
| ARID1A | Nonsense | p.R1335 | SNV | |
| 26 | BRCA2 | Missense | p.E3002K | SNV |
| TP53 | Missense | p.C176Y | SNV |
SNV: Single nucleotide variant.
Classification of patients into CMS categories
Out of the 26 patients analyzed, a specific CMS could be identified for 24 patients (92%) by clinical consensus by the Tumour Board. Two patients (8%) were found to be unclassifiable. Figure 1 illustrates the distribution of patients across the four CMS categories. Specifically, 23% (n = 6), 19% (n = 5), 31% (n = 8), and 19% (n = 5) were classified as CMS1, CMS2, CMS3 and CMS4, respectively. Remarkably, the percentage of patients classified into each CMS category closely aligns with findings reported by Guinney et al[4]. The median overall survival of the total sample was 28 mo (Figure 2A), and for CMS1, CMS2, CMS3 and CMS4 it was 11, 20, 30 and 45 mo respectively, with no statistically significant differences between groups (Figure 2B).
Figure 1.
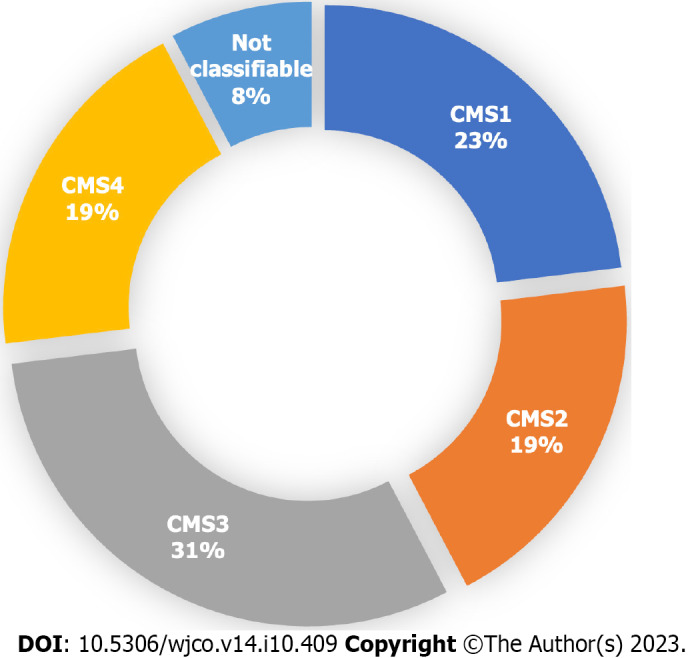
Proportion of patients in each consensus molecular subtype after analysis by the Tumour Board among the 26 patients included on the final analysis. A specific consensus molecular subtype (CMS) was successfully identified in 24 out of the 26 patients. CMS1 n = 6. CMS2 n = 5. CMS3 n = 8. CMS4 n = 5. Not classifiable n = 2. Each patient underwent an individual assessment by the Tumour Board, and a consensus was reached to determine their molecular subtype. Classification was based on clinical and histological characteristics, as well as the results of RT-qPCR (β-catenin, c-MYC and TGF- β) and NGS (TumorSec panel). CMS: Consensus molecular subtypes.
Figure 2.

Classification of patients into consensus molecular subtype categories. A: Kaplan-Meier Curve with overall survival (OS) of the n = 26 patients included on the final analysis. mOS = 28 mo; B: Kaplan-Meier curve which shows OS of patients based on their molecular subtype classification. The median overall survival times were 11, 20, 30, and 45 mo for CMS1, CMS2, CMS3, and CMS4, respectively. There were no statistically significant differences observed among the studied groups (P = 0.0968).
DISCUSSION
The objective of the workflow outlined in this manuscript was to develop an RT-qPCR- and NGS-based method by which to classify mCRC patients into CMS categories. Our results demonstrate that it is possible to classify mCRC patients into a specific CMS in approximately 90% of the cases.
To date, there are no validated tools from prospective studies for classifying patients into the four CMS categories. Although genomic platforms such as ColotypeR[17] and CMSCaller[18] have been utilized, they have not significantly impacted clinical practice. Our findings present an alternative protocol for patient classification, leveraging a 25-gene panel (TumorSec) and a three-gene RT-qPCR panel (TGF-β, β-catenin, and c-MYC). The selected genes play vital roles in the epithelial-mesenchymal transition, particularly TGF-β and β-catenin, which are specific to CMS4 (fibrotic)[19]. Additionally, c-MYC was chosen due to its utility for identifying CMS2 (metabolic)[20]. However, distinguishing between CMS2 and CMS3 remains challenging as they share genetic signatures and patterns of gene expression.
The relevance of classifying mCRC patients into CMS categories must be contextualized. Thus far, the selection of targeted therapies and the design of clinical studies have primarily relied on the identification of KRAS, NRAS, and BRAF mutations and MMR expression analyses[7-8]. However, incorporating knowledge of the CMS categories can offer significant advantages in both aspects. First, it can enhance the selection of targeted therapies, enabling a more personalized approach. Additionally, a better understanding of the CMS categories can lead to improved clinical study design, allowing for more tailored and effective treatments for patients with specific CMS profiles[6]. For instance, CMS1, characterized by high lymphocytic infiltration and a worse prognosis, may benefit from aggressive therapeutic strategies such as combination triplet chemotherapy (FOLFOXIRI) and anti-angiogenic agents[21]. Monodrug immunotherapy could also be beneficial for these patients given their high frequency of microsatellite instability-high tumours as demonstrated in the KEYNOTE177 study[22]. Considering the high prevalence of BRAF mutations, future studies should examine the efficacy of BRAF inhibitors for these patients[23]. CMS2 and CMS3 share significant features and may respond to similar agents. For example, they may show sensitivity to anti-EGFR therapy, especially in CMS2 cases[24]. However, CMS3 patients frequently develop KRAS mutations, primarily in exon 2, leading to constitutive activation of the mitogen-associated protein kinase pathway, associated with a poorer prognosis and response to standard treatment[25]. CMS4, which carries the worst prognosis, calls for the development of new strategies targeting the epithelial-mesenchymal transition or the TGF-β pathway. CMS4 tumours also show better response to irinotecan-based treatments or anti-angiogenic agents such as bevacizumab[26].
It is important to note that the classification of CMS can also predict the prognosis of patients with mCRC[4]. While this study documented the overall survival of patients, there were no significant differences between groups, likely due to the low number of patients in each CMS category. Therefore, it cannot be established whether patients with different CMSs have different prognoses.
The principal innovation of this exploratory study lies in the establishment of a protocol for the classification of mCRC patients into CMS through RT-qPCR (TGF-β, β-catenin, and c-MYC) and a 25-gene NGS panel (TumorSec). Our results demonstrates that this combined approach has the potential to classify patients with mCRC into one of the four CMS categories in over 90% of cases. As there is currently no gold-standard for conducting this clinical-molecular classification, this approach may represent a significant advancement in the development of an optimal technique that could become the standard for these purposes. In the future, it is important to further explore CMS categories and incorporate this knowledge into clinical practice. While this protocol proposes a CMS classification scheme, prospective and large-scale studies are imperative to assessing whether this methodology truly influences therapeutic decisions for patients[5] and for validating the clinical utility of CMS categories[6].
CONCLUSION
In conclusion, we successfully classified mCRC patients into CMS categories using an RT-qPCR and NGS-based workflow. This approach opens avenues for tailoring therapies according to CMS subtypes, potentially leading to improved patient outcomes.
ARTICLE HIGHLIGHTS
Research background
Colorectal cancer is a heterogeneous disease; therefore, it is crucial to progress towards a molecular consensus classification in order to predict prognosis and therapy response.
Research motivation
The primary motivation is to progress towards a consensus molecular classification of metastatic colorectal cancer patients, to better guide targeted therapy.
Research objectives
The aim of this study is to classify a sample of metastatic colorectal cancer patients into consensus molecular subtypes using a reverse transcription -quantitative polymerase chain reaction polymerase chain reaction (RT-qPCR) and next-generation genomic sequencing (NGS) protocol.
Research methods
Patients with unresectable metastatic colorectal cancer who were undergoing systemic treatment with chemotherapy and/or targeted therapy. Molecular biology techniques were employed to analyse primary tumour samples from these patients. RT-qPCR was utilized to assess the expression of genes associated with fibrosis (TGF-β and β-catenin) and cell growth pathways. NGS using a 25-gene panel (TumorSec) was performed to identify specific genomic mutations. The patients were then classified into one of the four CMS categories according to the clinical consensus of a Tumour Board.
Research results
n = 26 metastatic colorectal cancer patients analyzed. 23% (n = 6), 19% (n = 5), 31% (n = 8), and 19% (n = 5) were classified as CMS1, CMS2, CMS3, and CMS4, respectively. Additionally, 8% of patients (n = 2) could not be classified into any of the four CMS categories.
Research conclusions
It is possible to classify patients with metastatic colorectal cancer into consensus molecular subtypes through RT-qPCR and NGS techniques.
Research perspectives
Prospective studies are needed to determine if this classification is useful and if it has an impact on predicting the survival of patients with metastatic colorectal cancer.
Footnotes
Institutional review board statement: The study was reviewed and approved by the Medical Oncology Supervisor of Bradford Hill Clinical Research Center.
Informed consent statement: All study participants, or their legal guardian, provided informed written consent prior to study enrollment.
Conflict-of-interest statement: All the authors declare have not conflict of interest to declare.
STROBE statement: The authors have read the STROBE Statement—checklist of items, and the manuscript was prepared and revised according to the STROBE Statement—checklist of items.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: July 25, 2023
First decision: September 13, 2023
Article in press: October 8, 2023
Specialty type: Oncology
Country/Territory of origin: Chile
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Gu GL, China; Li L, China S-Editor: Liu JH L-Editor: A P-Editor: Zhang XD
Contributor Information
Jaime González-Montero, Bradford Hill Clinical Research Center, Santiago 8420383, Chile; Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile. jagonzalez@ug.uchile.cl.
Mauricio Burotto, Bradford Hill Clinical Research Center, Santiago 8420383, Chile.
Guillermo Valenzuela, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Debora Mateluna, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Florencia Buen-Abad, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Jessica Toro, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Olga Barajas, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Katherine Marcelain, Basic and Clinical Oncology Department, University of Chile, Santiago 8380453, Chile.
Data sharing statement
No additional data are available.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Alese OB, Wu C, Chapin WJ, Ulanja MB, Zheng-Lin B, Amankwah M, Eads J. Update on Emerging Therapies for Advanced Colorectal Cancer. Am Soc Clin Oncol Educ Book. 2023;43:e389574. doi: 10.1200/EDBK_389574. [DOI] [PubMed] [Google Scholar]
- 3.Sagaert X, Vanstapel A, Verbeek S. Tumor Heterogeneity in Colorectal Cancer: What Do We Know So Far? Pathobiology. 2018;85:72–84. doi: 10.1159/000486721. [DOI] [PubMed] [Google Scholar]
- 4.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ten Hoorn S, de Back TR, Sommeijer DW, Vermeulen L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. J Natl Cancer Inst. 2022;114:503–516. doi: 10.1093/jnci/djab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valenzuela G, Canepa J, Simonetti C, Solo de Zaldívar L, Marcelain K, González-Montero J. Consensus molecular subtypes of colorectal cancer in clinical practice: A translational approach. World J Clin Oncol. 2021;12:1000–1008. doi: 10.5306/wjco.v12.i11.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D, Engstrom PF, Garrido-Laguna I, Grem JL, Grothey A, Hochster HS, Hoffe S, Hunt S, Kamel A, Kirilcuk N, Krishnamurthi S, Messersmith WA, Meyerhardt J, Miller ED, Mulcahy MF, Murphy JD, Nurkin S, Saltz L, Sharma S, Shibata D, Skibber JM, Sofocleous CT, Stoffel EM, Stotsky-Himelfarb E, Willett CG, Wuthrick E, Gregory KM, Freedman-Cass DA. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J Natl Compr Canc Netw. 2018;16:359–369. doi: 10.6004/jnccn.2018.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson A, Bodoky G, Ciardiello F, D'Hoore A, Diaz-Rubio E, Douillard JY, Ducreux M, Falcone A, Grothey A, Gruenberger T, Haustermans K, Heinemann V, Hoff P, Köhne CH, Labianca R, Laurent-Puig P, Ma B, Maughan T, Muro K, Normanno N, Österlund P, Oyen WJ, Papamichael D, Pentheroudakis G, Pfeiffer P, Price TJ, Punt C, Ricke J, Roth A, Salazar R, Scheithauer W, Schmoll HJ, Tabernero J, Taïeb J, Tejpar S, Wasan H, Yoshino T, Zaanan A, Arnold D. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 9.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 10.Salmani A, Mohammadi M, Farid Hosseini R, Tavakol Afshari J, Fouladvand A, Dehnavi S, Khoshkhooi M, Jabbari Azad F. A significant increase in expression of FOXP3 and IL-17 genes in patients with allergic rhinitis underwent accelerated rush immunotherapy. Iran J Basic Med Sci. 2019;22:989–996. doi: 10.22038/ijbms.2019.32979.7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan L, Shen H, Huang H, Yang R, Yao L. Impairment of Wnt/β-catenin signaling in blood cells of patients with severe cavitary pulmonary tuberculosis. PLoS One. 2017;12:e0172549. doi: 10.1371/journal.pone.0172549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu S, Yang Q, Yang JH, Du Z, Zhang G. Identification of suitable reference genes for investigating gene expression in human gallbladder carcinoma using reverse transcription quantitative polymerase chain reaction. Mol Med Rep. 2015;11:2967–2974. doi: 10.3892/mmr.2014.3008. [DOI] [PubMed] [Google Scholar]
- 13.Kheirelseid EA, Chang KH, Newell J, Kerin MJ, Miller N. Identification of endogenous control genes for normalisation of real-time quantitative PCR data in colorectal cancer. BMC Mol Biol. 2010;11:12. doi: 10.1186/1471-2199-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dydensborg AB, Herring E, Auclair J, Tremblay E, Beaulieu JF. Normalizing genes for quantitative RT-PCR in differentiating human intestinal epithelial cells and adenocarcinomas of the colon. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1067–G1074. doi: 10.1152/ajpgi.00234.2005. [DOI] [PubMed] [Google Scholar]
- 15.Varela T, Laizé V, Conceição N, Caldeira P, Marreiros A, Guerreiro H, Cancela ML. Expression of DUSP4 transcript variants as a potential biomarker for colorectal cancer. Biomark Med. 2020;14:639–650. doi: 10.2217/bmm-2019-0369. [DOI] [PubMed] [Google Scholar]
- 16.Salvo M, González-Feliú E, Toro J, Gallegos I, Maureira I, Miranda-González N, Barajas O, Bustamante E, Ahumada M, Colombo A, Armisén R, Villamán C, Ibañez C, Bravo ML, Sanhueza V, Spencer ML, de Toro G, Morales E, Bizama C, García P, Carrasco AM, Gutiérrez L, Bermejo JL, Verdugo RA, Marcelain K. Validation of an NGS Panel Designed for Detection of Actionable Mutations in Tumors Common in Latin America. J Pers Med. 2021;11 doi: 10.3390/jpm11090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buechler SA, Badve SS, Gokmen-Polar Y, Herring E, Ludwig K, Hummon A. ColotypeR: A tool to classify colon cancers by consensus molecular subtype and subtype-specific risk of recurrence. J Clin Oncol. 2018;36 Suppl 4:632–632. [Google Scholar]
- 18.Eide PW, Bruun J, Lothe RA, Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep. 2017;7:16618. doi: 10.1038/s41598-017-16747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;10:5. doi: 10.4103/1477-3163.78111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadanandam A, Wang X, de Sousa E Melo F, Gray JW, Vermeulen L, Hanahan D, Medema JP. Reconciliation of classification systems defining molecular subtypes of colorectal cancer: interrelationships and clinical implications. Cell Cycle. 2014;13:353–357. doi: 10.4161/cc.27769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cremolini C, Antoniotti C, Lonardi S, Bergamo F, Cortesi E, Tomasello G, Moretto R, Ronzoni M, Racca P, Loupakis F, Zaniboni A, Tonini G, Buonadonna A, Marmorino F, Allegrini G, Granetto C, Masi G, Zagonel V, Sensi E, Fontanini G, Boni L, Falcone A. Primary tumor sidedness and benefit from FOLFOXIRI plus bevacizumab as initial therapy for metastatic colorectal cancer. Retrospective analysis of the TRIBE trial by GONO. Ann Oncol. 2018;29:1528–1534. doi: 10.1093/annonc/mdy140. [DOI] [PubMed] [Google Scholar]
- 22.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 23.Molina-Cerrillo J, San Román M, Pozas J, Alonso-Gordoa T, Pozas M, Conde E, Rosas M, Grande E, García-Bermejo ML, Carrato A. BRAF Mutated Colorectal Cancer: New Treatment Approaches. Cancers (Basel) 2020;12 doi: 10.3390/cancers12061571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ten Hoorn S, Sommeijer DW, Elliott F, Fisher D, de Back TR, Trinh A, Koens L, Maughan T, Seligmann J, Seymour MT, Quirke P, Adams R, Richman SD, Punt CJA, Vermeulen L. Molecular subtype-specific efficacy of anti-EGFR therapy in colorectal cancer is dependent on the chemotherapy backbone. Br J Cancer. 2021;125:1080–1088. doi: 10.1038/s41416-021-01477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD, Taniere P, Willcox BE, Guinney J, Middleton GW. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin Cancer Res. 2018;24:224–233. doi: 10.1158/1078-0432.CCR-17-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters NA, Constantinides A, Ubink I, van Kuik J, Bloemendal HJ, van Dodewaard JM, Brink MA, Schwartz TP, Lolkema MPJK, Lacle MM, Moons LM, Geesing J, van Grevenstein WMU, Roodhart JML, Koopman M, Elias SG, Borel Rinkes IHM, Kranenburg O. Consensus molecular subtype 4 (CMS4)-targeted therapy in primary colon cancer: A proof-of-concept study. Front Oncol. 2022;12:969855. doi: 10.3389/fonc.2022.969855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.