

SUMMARY
We characterized a prospective endometrial carcinoma (EC) cohort containing 138 tumors and 20 enriched normal tissues using 10 different omics platforms. Targeted quantitation of two peptides can predict antigen processing and presentation machinery activity, and may inform patient selection for immunotherapy. Association analysis between MYC activity and metformin treatment in both patients and cell lines suggests a potential role for metformin treatment in non-diabetic patients with elevated MYC activity. PIK3R1 in-frame indels are associated with elevated AKT phosphorylation and increased sensitivity to AKT inhibitors. CTNNB1 hotspot mutations are concentrated near phosphorylation sites mediating pS45-induced degradation of β-catenin, which may render Wnt-FZD antagonists ineffective. Deep learning accurately predicts EC subtypes and mutations from histopathology images, which may be useful for rapid diagnosis. Overall, this study identified molecular and imaging markers that can be further investigated to guide patient stratification for more precise treatment of EC.
Keywords: Endometrial cancer, proteogenomics, target assays, deep learning, PIK3R1, CTNNB1, Metformin, CPTAC
eTOC Blurb
Dou et al. report the characterization of a prospective endometrial carcinoma (EC) cohort using 10 different omics platforms. They identify potential molecular and imaging markers for guiding patient stratification for more precise treatment of EC.
Graphical Abstract
INTRODUCTION
Endometrial carcinoma (EC) is the most common gynecologic malignancy in developed nations1–3. Over the past decade, the incidence of EC has steadily increased (~1% annually1,4). Curiously, EC-specific mortality has steadily worsened over the past 10 years, despite the fact that postmenopausal vaginal bleeding is a common early symptom and the majority of early stage, well-differentiated EC can be cured by hysterectomy5. Worsening EC outcomes have been attributed to the increasing incidence of aggressive EC histotypes, particularly among Black and Hispanic women5. For high-risk EC patients, adjuvant radiotherapy and cytotoxic chemotherapy have been shown to decrease the likelihood of disease recurrences and improve survival. However, failure rates associated with these treatments remain unacceptably high6. In other situations, the ability of radiotherapy or cytotoxic chemotherapy to improve outcomes for EC patients at intermediate risk of disease progression or recurrence remains much less clear. In part, this may depend on the patterns of driver mutations present7. Effective therapies capable of curing EC once widely metastatic are lacking.
Comprehensive genomic profiling by The Cancer Genome Atlas (TCGA) has led to a better understanding of key genomic drivers of EC, including the identification of four EC subtypes defined solely by their genomic signatures: POLE ultramutated, microsatellite instability hypermutated (MSI-H), copy-number low (CNV-L), and copy-number high (CNV-H)8. Associations between these subtypes and clinical outcomes are now being evaluated. Integrated genomic and proteomic characterization of EC by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) revealed additional oncogenic signaling pathways and regulatory mechanisms, and identified specific defects in antigen presentation machinery (APM) which appear important for determining clinical responses to immune checkpoint inhibition (ICI) therapy9.
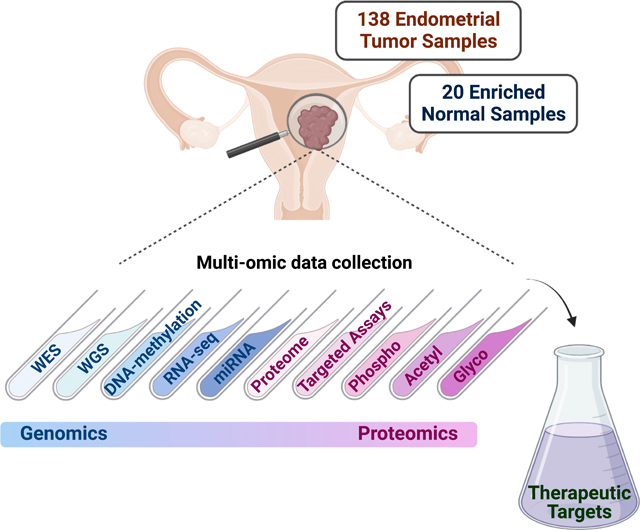
Here, we present the results of a comprehensive proteogenomic analysis of a prospectively curated set of 138 EC tumors and 20 specimens enriched for normal endometrium from healthy donors. Analysis of this independent cohort, incorporating pre-existing EC tumor and cell line cohorts, has not only confirmed published findings from our recent exploratory studies, but also provided biological insights relevant to potential therapeutic strategies. We also evaluated the utility of using machine learning for parsing clinically-relevant genomic features based solely on the analysis of histological slides routinely created for clinical evaluation.
RESULTS
Proteogenomic landscape of an independent EC cohort
We characterized the proteogenomic landscape of 138 EC tumors (including 119 endometrioid, 13 serous, 3 clear cell) with matched blood normal samples and 20 enriched normal endometrium samples from healthy donors (Table S1) using 10 omic platforms including whole-genome sequencing (WGS), whole-exome sequencing (WES), methylation array, total RNA sequencing (RNA-seq), microRNA sequencing, targeted proteomics, global proteomics, phosphoproteomics, acetylproteomics, and glycoproteomics (STAR methods). A total of 10,135 proteins and 25,300 phosphorylation, 5,556 acetylation, and 6,513 glycosylation sites were quantified (Figure 1A), using rigorous quality control metrics (Figure S1A–I). Principal component analysis (PCA) separated the tumors and normal samples based on global proteome, phosphoproteome, or glycoproteome data, but not on acetylproteomics data, with no batch effect observed in the tandem mass tag (TMT) plexes. (Figure S1E–H). Significant overlaps were observed for quantifiable global proteomics and phosphoproteomics features between the exploratory cohort9 and this independent cohort. However, more acetylation sites were quantified in this cohort due to platform improvement (Figure 1B).
Figure 1: Proteogenomic landscape of the independent EC cohort.

(A) Multi-omic data availability in the independent EC cohort. The main heatmap shows each patient per column and the side heatmap (right) shows each enriched normal sample per column.
(B) Identified and quantified proteomic features across the exploratory (Exp) and independent (Ind) cohorts.
(C) Oncoplot showing the most frequently mutated genes in the independent cohort identified by whole genome sequencing. Each column is a patient. Side table (right) compares mutation frequencies per gene for the two CPTAC cohorts and TCGA cohort.
(D) Focal-level copy number variations (CNVs) across the genome (x-axis) versus G-score (y-axis), which is the Frequency x Amplitude of the CNV. Significant amplifications are shown in red and deletions are shown in blue, with annotated peaks containing known tumor drivers.
(E) Prioritization of CNV drivers across the two CPTAC cohorts.
(F) Barplot showing pathways enriched from CNV drivers analysis (E).
(G) Scatter plot of protein level tumor/normal between the two CPTAC cohorts.
(H) Cis/trans effects of somatic mutations (y-axis) on protein expression (x-axis). See also Figure S1 and Tables S1 and S2.
Top mutated genes in this cohort included PTEN, ARID1A, PIK3CA, PIK3R1, and CTNNB1 whose mutation frequencies were similar to those in the exploratory cohort (Figure 1C). Mutation frequencies were lower in the TCGA EC cohort, due to a higher proportion of serous tumors8. The mutation frequencies were highly correlated across the three cohorts (Figure S1J). Tumors were classified into four genomic subtypes: 6 POLE, 47 MSI-H, 66 CNV-L, 16 CNV-H tumors and 3 unclassified because of missing WGS-based CNV data (Figure 1C and Figure S1K–L; Table S1). One tumor was identified as both POLE and CNV-H by genomic data, highlighting the high degree of heterogeneity in certain EC tumors that likely requires careful consideration in treatment decisions. In general, these results indicate that mutational features of this current independent cohort are similar to previous EC studies.
WGS-based CNV analysis identified significant focal amplifications and deletions. Several known tumor drivers were located within the amplified regions (e.g., SETDB1, ECT2, ATAD2, GRB7, and CCNE1) (Figure 1D; Table S2). We prioritized CNV driver genes by their correlation with mRNA and protein levels (Figure 1E) and filtered them by comparing protein levels in tumors vs enriched normal tissues, which identified 351 and 237 potential CNV drivers from this and the exploratory cohorts, respectively, of which a significant overlap of 88 genes was found between them (Figure 1E; Table S2; p<2.2e-16), and these were significantly enriched in proliferation and cell cycle related processes (Figure 1F).
We identified 1,292 up- and 1,488 down-regulated proteins when tumors were compared to enriched normal tissues (FDR < 0.01, |Log2FC| > 0.25). Significantly differentially expressed proteins were highly overlapping between the independent cohort and the exploratory cohort (Figure 1G). Interestingly, two cancer/testis antigens, PBK and KIF2C, were found to be significantly upregulated in both cohorts9. The cis- and trans-effects on the proteome of somatic driver mutations were similar across cohorts (Figure 1H). For example, CTNNB1 mutations were significantly positively correlated with higher CTNNB1 and LEF1 protein levels. We confirmed that MLH1 and HOX family genes were silenced by DNA methylation in this independent cohort (Figure S1M). circRNAs show stronger positive correlations than that between their host genes (Figure S1N) and miR-200c-3p significantly negatively correlated with QKI, a circRNA regulator, protein level (Figure S1O). These results suggest that many findings at the multi-omics level are highly consistent between the two CPTAC EC studies.
PIK3R1 in-frame indels are potential markers of AKT inhibition response
Previous studies have shown that the PI3K-AKT pathway is frequently altered in endometrioid tumors8–10 with PTEN, PIK3CA, and PIK3R1 being the most frequently mutated genes in the pathway. PTEN is mutated in ~80% of endometrioid tumors and mostly co-occurs with PIK3CA and PIK3R1 mutations, which are mutually exclusive (Figure 2A). Although PIK3CA is commonly mutated in many cancer types10, PIK3R1 mutations, especially in-frame indels of PIK3R1, are more frequent in EC (Figures 1C, S2A). Interestingly, these in-frame indels are tightly clustered at the two ends of the P85/iSH2 domain (Figure S2B), which are structurally close to each other in a region of PIK3R1 that interacts with PIK3CA (Figure 2B). Of note, this clustering pattern is not observed for truncations and missense mutations (Figure S2C–D). This suggests a potentially distinct function of PIK3R1 in-frame indels from other PIK3R1 variants.
Figure 2: PIK3R1 in-frame indels show induction of activating AKT phosphosites.

(A) PTEN, PIK3CA, and PIK3R1 mutations across independent (Ind), exploratory (Exp), and TCGA cohorts. P-values derived from Fisher’s exact test.
(B) 3D structure of PI3K complex. PIK3CA protein is colored in green, PIK3R1 protein is colored in blue, and location of PIK3R1 in-frame variants is shown in red.
(C-D) Boxplots comparing PIK3R1 mRNA (C) and protein levels (D) between PIK3R1 variants across the independent (Ind), exploratory (Exp), and TCGA cohorts. P-values derived from Student’s t-test
(E) Survival analysis of TCGA PTEN mutated EC patients harboring PIK3R1 and PIK3CA variants. P-values derived from log-rank test.
(F) Boxplots comparing AKT1-pT308 levels between PIK3R1 and PIK3CA variants in the independent cohort. P-values derived from Student’s t-test.
(G-H) Boxplots comparing TCGA RPPA data for AKT-pT308 (G) and AKT-pS473 (H) levels between PIK3R1 and PIK3CA variants. P-values derived from Student’s t-test.
(I) Western blot for AKT pT308 and pS473 in HEC-151 cells with CRISPR-Cas9 created T576 deletion.
(J) Boxplots comparing DepMap EC cell lines’ response to MK-2206. P-values derived from Student’s t-test.
(K) Schematic showing consequences of PIK3R1 in-frame variants.
Boxplots: Box portion represents Interquartile range (IQR), midline corresponds to the median, and whiskers range from the minimum (bottom) and maximum (top) variability outside the first and third quartiles (Q1 and Q3). Outliers are shown as points above whiskers. See also Figure S2 and Table S3.
To test this hypothesis, we examined cis-effects of different PIK3R1 mutations on mRNA and protein levels (Figure 2C–D). In-frame indels were associated with comparable PIK3R1 protein (p85α subunit of PI3K) levels (Figure 2D; Table S3), while truncating mutations were associated with lower protein levels compared to the WT PIK3R1 group11. Since PIK3R1 is a suppressor of AKT phosphorylation, we expected that lower levels of PIK3R1 by truncating mutations would result in activation of the PI3K-AKT pathway and worse clinical outcomes. Surprisingly, PIK3R1 in-frame indels were associated with worse survival compared to both PIK3R1 truncating and PIK3CA hotspot mutations in the TCGA cohort, despite our finding that they do not result in reduced levels of PIK3R1 protein (Figure 2E). The Both WT group (PIK3CA WT and PIK3R1 WT) had similar survival with the PIK3R1 in-frame indel group. This may be due to the fact that this cohort is significantly enriched for CNV-high ECs and contains a lower proportion of POLE-mutated cancers (Figure S2E).
We hypothesized that PIK3R1 proteins with in-frame indels abrogate the ability of PIK3R1 to suppress AKT phosphorylation, leading to elevated levels of phosphorylated AKT1. To examine this hypothesis, we first examined the relationships between AKT1 phosphorylation level and PTEN, PIK3CA, and PIK3R1 mutation status. As expected, PTEN WT samples have significantly lower AKT1-T308 phosphorylation levels than PTEN mutated samples12 (Figure 2F). Moreover, samples with in-frame indels had higher levels of AKT1-T308 phosphorylation compared with cancers carrying PIK3CA hotspot or PIK3R1 truncating mutations (Figure 2F). Since this phosphosite was not identified in the exploratory cohort, TCGA Reverse Phase Protein Array (RPPA) data was used to further confirm our hypothesis that AKT-T308 phosphorylation is higher in tumors harboring PIK3R1 in-frame indels and the lowest in PTEN WT tumors (Figure 2G). We observed the same trend for another well studied phosphorylation site, AKT-S473 (Figure 2H). Moreover, AKT protein levels were not consistently altered by PIK3CA and PIK3R1 mutations in the independent cohort or TCGA (Figure S2F–G). These results are consistent with a previous study that showed PIK3R1 in-frame indels were able to more efficiently promote AKT phosphorylation than other mutations in EC cell lines13. To further confirm the relation between PIK3R1 in-frame indels and AKT-T308 and AKT-S473 phosphorylation, we created an in-frame deletion, T576Del, in an EC cell line, HEC151, by CRISPR-Cas9 (Figure S2H). As expected, both AKT-T308 and AKT-S473 were upregulated at phosphorylation level (Figure 2I). Taken together, these results suggest that PIK3R1 in-frame indels are associated with worse outcomes in PTEN-mutated EC patients and contribute to altered PI3K-AKT pathway by increasing AKT1 phosphorylation.
Various drugs have been developed to target the PI3K-AKT pathway, but many have failed in clinical trials14,15. For example, results from a clinical trial with MK-2206, a well-established AKT inhibitor targeting AKT-S473, failed to improve survival in EC patients with PIK3CA mutated EC16. In light of our analyses described above, we next evaluated whether PIK3R1 in-frame indels may be a better marker of clinical responses to AKT inhibition than PIK3CA mutations using data from EC cell lines from DepMap17. As expected, EC cell lines with PIK3R1 in-frame indels and mutated PTEN were significantly more sensitive to MK-2206 compared to lines with PIK3CA hotspot mutation, PIK3R1 truncation, or WT PTEN (Figure 2J; Table S3). This result was also observed for two additional AKT inhibitors, GDC-0068 (NCT02465060, a phase II study of multiple cancer types including EC with inclusion criteria for tumors with AKT mutations) and GSK2110183 (Figure S2I–J; Table S3). In summary, our results suggest the PIK3R1 in-frame indels with mutated PTEN could be a potential biomarker for response to AKT inhibition (Figure 2K).
Selected Reaction Monitoring (SRM) assay accurately predicts APM status
Effective cellular APM has been reported as an important factor of response to ICI18–20 independent of tumor-mutation burden (TMB)9. By grouping tumors according to their TMB and APM levels, we confirmed the finding of high APM variation in TMB-H tumors9 (Figure S3A;Table S4). The TMB-H/APM-L group were enriched for JAK1 mutations (p = 1.05e-3, Fisher’s test) and had lower JAK-STAT, APM, and HLA protein levels (Figure S3B). TMB-H/APM-H tumors were immune hot with activated CD8+ T cells (Figure 3A). Moreover JAK1 variants were very rare in TMB-L tumors (Table S4) suggesting JAK1 mutations are associated with TMB-H tumors. This potential interaction between JAK1 mutations and TMB-H status warrants further examination in other EC cohorts.
Figure 3: Selective reaction monitoring (SRM) assay for antigen presentation machinery (APM) status.

(A) Heatmap of immune subgroups and related pathways derived from ssGSEA pathway scores using protein level as input for the independent cohort. Each column corresponds to individual samples in the main heatmap and the mean score for each immune subgroup in the side (right) panel. Each row represents a pathway.
(B-C) Histograms showing the frequency of correlations between SRM peptides from the same genes (B) and SRM peptides with TMT-based protein levels (C) for the independent cohort.
(D-E) Heatmaps showing SRM-based peptide quantitation of JAK-STAT and selected APM proteins in the exploratory (D) and independent (E) cohorts. Columns correspond to individual samples and rows represent peptides. JAK1 mutations enriched for TMB-H/APM-L groups indicated by asterisks (* p < 0.05, ** p < 0.01). P-values determined by Fisher’s exact test.
(F) ROC curves showing model performances of classifiers using two peptides, PSMB10-LPFTALGSGQDAALAVLEDR and PSMB9-VSAGEAVVNR.
(G) ROC curves showing model performances of the ORFlog classifier on the independent cohort, comparing top N peptides used per model.
Unlike TMB which is already used as a clinical biomarker21,22, there are no clinical biomarkers to determine APM status. The previous approach for computing APM scores relies on proteome-wide measurements, and is not suitable as a diagnostic assay9,23. To develop targeted proteomic assays, we selected a panel comprising 17 JAK-STAT and APM proteins, and 51 other immune related proteins, including two peptides for each protein (Table S4, Figure S3C). In both exploratory and independent cohorts, we observed a strong correlation between peptides originating from the same protein, as well as between the targeted assay and TMT measurements, particularly for proteins with relatively higher levels (Figures 3B–C, S3D–E). The targeted assays verified downregulation of APM proteins that were previously identified as downregulated in the TMB-H/APM-L group through global proteomics (Figures 3D–E). By utilizing these peptide measurements, we constructed machine learning models that accurately predicted the APM status, achieving an AUC of 0.961 using only two peptides: PSMB9−VSAGEAVVNR and PSMB10−LPFTALGSGQDAALAVLEDR (Figures 3F–G, S3F–J). We applied this SRM assay to immunotherapy-treated MSI-H EC tumors (10 sensitive and 2 resistant) (Table S4). The Immunotherapy-sensitive group had higher peptide levels than the TMB-H/APM-L group (Figure S3K–L), without reaching statistical significance, possibly as a result of the small cohort size. Therefore, this simple SRM assay should be further validated to evaluate its suitabillity for predicting APM status in a clinical setting.
MYC activity is the target and potential biomarker for metformin treatment
Obesity and type 2 diabetes (T2D) are well recognized risk factors for EC24–26. In a recent metaanalysis, metformin treatment was shown to improve overall survival of EC patients with T2D27. A number of mechanisms by which metformin potentially improves EC outcomes include its ability to modulate autophagy28, inhibit mitochondrial respiration29, modify epigenetic signatures30, and directly suppress cell growth31,32. However, the mechanisms by which metformin impacts EC and potentially improves patient outcomes remain unclear.
To examine molecular determinants underlying metformin response, we integrated clinical phenotypes with multi-omics data. RNA-seq-based pathway analysis showed significantly lower proliferation-related MYC activity in metformin-treated patients (Figures 4A, S4A; Table S1). A similar relationship was observed where MYC protein level decreased when Ishikawa EC cell lines were treated with metformin33. We also analyzed responses of 22 EC cell lines (8 sensitive, 14 insensitive) from DepMap17 (Figure S4B) and found that metformin-sensitive cell lines had significantly higher MYC activity than resistant ones (Figure 4B). Collectively, we interpreted these observations to indicate that MYC potentially serves as a biomarker for metformin responses in EC. To further evaluate this hypothesis, we sought to determine whether downregulation of MYC activity could be a direct consequence of metformin treatment34. We found MYC activity to be downregulated in several CMap signatures of metformin-treated cell lines (Figure 4C; Table S5). Analyzing two metformin-sensitive and two insensitive EC cell lines from DepMap (Figure S4B), we observed higher MYC protein levels in the metformin-sensitive cell lines (Figure 4D), but not mRNA levels (Figure S4C). Use of a previously validated reporter assay confirmed higher levels of MYC activity in the two cell lines with greater MYC protein levels (Figure S4D). Cell lines with higher MYC activity were more sensitive to metformin treatment (Figure 4E). Moreover, mRNA expression of MYC and multiple MYC targets were suppressed in response to metformin treatment (Figure S4E; Table S5). In general, decreases in putative MYC-regulated transcripts occurred earlier and to a greater degree in the two cell lines with greatest sensitivity to metformin. These experiments demonstrate that metformin treatment significantly decreases MYC activity, with a trend toward increased sensitivity in cell lines which express high levels of MYC. These results suggest that an important mechanism by which metformin decreases EC growth is by directly downregulating MYC activity.
Figure 4: Metformin may target MYC in EC.

(A) MYC Targets V2 enrichment plots from pathway analysis comparing metformin-treated versus untreated patients with Type2 Diabetes (T2D) in the independent cohort.
(B) MYC Targets V2 enrichment plot from pathway analysis comparing metformin-sensitive versus insensitive EC cell lines from DepMap.
(C) Volcano plot of MYC Targets V2 pathway scores (x-axis) versus -log10(FDR) (y-axis) for CMAP metformin treatment signatures.
(D) Western blot showing MYC expression in four EC cell lines.
(E) Dose-response curves of EC cell lines treated with metformin at increasing concentrations (x-axis).
(F-G) Survival analysis of TCGA MSI-H (F) and CNV-L (G) tumors with high and low MYC activity. (H) Scatter plot of MYC activity (y-axis) versus MYC IHC score (x-axis). P-values derived from log-rank test.
(I) Heatmap of all endometrioid tumors in the independent cohort, sorted by MYC activity (top panel) and grouped by diabetes and metformin treatment status. Side boxplots (right) compare MYC activity (top) and BMI (bottom) across the diabetes/treatment groups. MYC IHC scores (third panel from top) are shown from samples with IHC data available. P-values derived from Student’s t-test (boxplots) and Spearman correlation (left panel).
Boxplots: Box portion represents Interquartile range (IQR), midline corresponds to the median, and whiskers range from the minimum (bottom) and maximum (top) variability outside the first and third quartiles (Q1 and Q3). Outliers are shown as points above whiskers.
Next, we evaluated the relationship between T2D, EC outcomes, MYC expression and metformin exposure. We found EC patients with T2D whose tumors were characterized by high levels of MYC protein have significantly worse overall survival (OS) (Figure S4F) than MYC-low patients (Table S5). Interestingly, high levels of MYC were also associated with worse OS in both MSI-H and CNV-L subsets of EC (Figure 4F–G), but not in serous tumors (Figure S4G). Although the MYC protein is not quantifiable by global proteomics, we found MYC activity to be significantly higher in MYC-high tumors in TCGA RPPA data (Figure S4H). MYC activity also significantly correlated with MYC Immunohistochemistry (IHC) score (Figure 4H; Table S5), but not MYC mRNA levels (Figure S4I). Therefore, we used inferred MYC activity as a readout of MYC protein level to further examine relationships between MYC and EC genotypes/phenotypes. We divided patients into 3 groups, non-diabetic, diabetic untreated, and diabetic treated, and sorted patients by their MYC activity from high to low (Figure 4I). As predicted by our earlier results, metformin-treated tumors had significantly lower levels of MYC activity when compared to untreated tumors from T2D patients (p = 4.49e-2, t test). A similar trend was observed when we re-evaluated our earlier exploratory cohort (Figure 4I and S4J). Of note, we found that MYC activity in ECs from non-diabetic patients was similar or even higher than those observed in diabetic patients not previously treated with metformin (Figure 4I and S4J). We also found that MYC activity was negatively correlated with BMI in this cohort (p = 1.26e-2, R = −0.24).
Collectively, these observations suggest that high levels of MYC activity may be a biomarker for identifying EC patients most likely to benefit from metformin treatment, including non-diabetic and non-obese patients.
Comprehensive pathway analysis reveals EC tumors separate by high and low transporter activity
Calculating the activation score for 8,615 pathways using EC protein levels (Table S6), revealed the transporters axis to be the most significant subtype across EC tumors (Figure S5A–B). The transporter axis is composed of transmembrane proteins including ATP-binding cassette transporters, ATPases, ion channels and solute carriers, all of which mediate exchange of ions, metabolites and nutrients between the intra- and extracellular space. EC cancers with high transporter scores featured elevated levels of transporters, biosynthetic and metabolic pathways for lipids and glycans, synthesis of fatty acyl-CoAs, insulin, triglyceride, steroid and cholesterol, and downregulation of proinflammatory cytokine signaling responsible for activation of CD4+ and CD8+ T lymphocytes. The transporter-high tumors were associated with a higher CTNNB1 mutation frequency (Figure S5A, p = 0.004), resulting in stabilization of β-catenin and transcriptional activation of target genes involved in glycosylation and in molecular exchange required for lipid metabolic reprogramming and cancer progression. Among numerous β-catenin transcriptional targets were immune checkpoints35,36 contributing to an immunosuppressive environment.
CTNNB1 hotspot mutations inhibit DKK-induced degradation
Hotspot mutations in exon 3 of CTNNB1 (β-catenin) (Figure S5C), potential drivers of tumorigenesis37, were significantly enriched in CNV-L tumors (Figure 5A, S5D–E) and with a higher frequency in CNV-L than in TMB-H tumors (Figure S5F). CTNNB1 hotspot mutations led to significant upregulation of Wnt-β-catenin signaling at both RNA and protein levels and downregulation of several immune related pathways (Figure 5B), and these were also true across the whole cohort (Table S6). Wnt signaling proteins including CTNNB1 and LEF1 were upregulated in CTNNB1 hotspot-mutated tumors despite DKK4, an inhibitor of the Wnt pathway38, being the most significantly upregulated protein (Figure 5C, Table S6). Because CTNNB1 hotspot mutations occur at key phosphorylation sites or neighboring amino acids, they may block phosphorylation-dependent β-catenin degradation39. Consistent with this hypothesis, S45 phosphorylation was significantly downregulated in hotspot-mutated tumors, whereas phosphosites distant from the hotspot mutations were mostly upregulated (Figure 5D, Table S6). We also found significantly upregulated cell proliferation and transporter activity and downregulated total immune score in tumors with CTNNB1 hotspot mutations, consistent with existing knowledge37.
Figure 5: CTNNB1 hotspot mutations block DKK induced degradation.

(A) Mosaic plot showing distribution of CTNNB1 mutations across CNV-L tumors versus all other tumors in the independent cohort. P-value determined from Chi-Square Test.
(B) Scatter plot of pathway Normalized Enrichment Scores (NES) comparing CTNNB1 hotspot mutant versus WT CNV-L tumors at the protein (x-axis) and RNA (y-axis) levels in the independent cohort. Points with FDR < 0.01 at both protein and RNA levels are colored in red, RNA only are in green, protein only are in blue, and neither are in gray.
(C) Volcano plot of protein log2 fold change (x-axis) between CTNNB1 hotspot and WT CNV-L tumors versus −log10 FDR (y-axis) determined by Student’s T-test. Points with FDR < 0.01 and log2 fold change < −0.5 or > 0.5 are shown in blue and red, respectively.
(D) Heatmap showing mRNA, protein, and phosphoprotein values for CTNNB1, LEF1 protein, MYC activity score, Immune Score, and Transporters Score across CNV-L tumors with and without CTNNB1 hotspot mutations. Side panel showing boxplots (right) compares mutants versus WT tumors. P-values determined by Wilcoxon Rank Sum Test.
(E) Schematic depicting proposed downstream implications of hotspot mutations in CTNNB1.
(F) ROC curves of Lasso regression models predicting CTNNB1 hotspot mutation status using exploratory protein data as training and independent protein data as testing. Models vary by which samples (all tumors or just CNV-L tumors) and which proteins (all proteins or only Wnt- β-catenin pathway proteins) were used.
(G) Venn diagram showing top 10 proteins selected by regression analysis per model.
Boxplots: Box portion represents Interquartile range (IQR), midline corresponds to the median, and whiskers range from the minimum (bottom) and maximum (top) variability outside the first and third quartiles (Q1 and Q3). Outliers are shown as points above whiskers.
CTNNB1 hotspot mutations are associated with higher risk of recurrence40,41 and therefore useful for making treatment decisions. Currently, sequencing is used to identify these mutations, but sequencing is not always available or covered by insurance for low-grade tumors42,43, revealing an unmet need for a more accessible diagnostic tool. To screen for potential protein biomarkers, we used proteomic data from the exploratory cohort to train regression models and tested their performance using the independent cohort. The best performing model used CNV-L tumors and Wnt proteins (AUC = 0.99) (Figure 5F–G, Table S6). Using IHC for CTNNB1, LEF1, and MYC, we validated elevated protein levels and MYC activity in hotspot-mutated tumors. Since EC tumors derive from the epithelium, membrane CTNNB1 stains positive in all cells. However, nuclear CTNNB1 was significantly higher in CNV-L hotspot-mutated tumors compared to WT (Figure S5G), implying a higher occurrence of oncogenic activity in these tumors.
Together, our data suggest that CTNNB1 hotspot mutations block phosphorylation induced β-catenin degradation and may render Wnt-FZD antagonists ineffective for ECs with the hotspot mutations (Figure 5E), and that protein-based assays, such as IHC, can be used to detect hotspot mutation status, suggesting possibilities beyond bulk sequencing for clinical testing.
Deep learning models using histopathology slides predict subtypes and mutation status
The CNV-H molecular subtype of EC, which includes some high-grade endometrioid (Figure S6A) and all histologically serous (Figure S6B) cases, has the worst patient outcomes8. In contrast, POLE tumors, typically ultra-mutated with favorable outcomes, are not currently identifiable by human pathologists without sequencing the tumors (Figure S6C). Here, we trained convolutional neural network models on H&E stained slides from TCGA and CPTAC exploratory cohorts to predict the molecular subtypes, histological subtypes, and common mutations of EC44, and tested them on the independent cohort (Figure 6A). We qualitatively confirmed that our models were able to successfully distinguish CNV-H and non-CNV-H samples (Figure 6B–C). Notably, a rare dual CNV-H and POLE tumor was also picked out by the imaging model, which aligned to both molecular subtypes (Figure S6D). tSNE plots showing CNV-H and POLE predictions of this sample show separation of tiles based on their subtype prediction, highlighting the heterogeneity in this tumor. Using similar tSNE plots for samples with either CNV-H or POLE subtype, but not both, the model does not predict high CNV-H scores for POLE tiles (not shown). The highest AUROC obtained from our POLE model was 0.925, which was higher than the 0.89 previously reported44. We then tested and re-trained with an independent cohort from NYU, which resulted in AUROCs ranging 0.6–0.8 (Figure 6A, Table S7).
Figure 6: Deep learning models successfully classify molecular features.

(A) Barplots showing the mean AUROC per model from internal training data split tests (trained on TCGA and exploratory cohorts) and independent tests (tested on independent cohort plus NYU cohort for POLE predictions). Bar color is determined by AUROC value coming from internal or independent tests, and outlines denote if the top performing model architecture comes from the internal or independent test.
(B-C) tSNE plots where each point is a tile, colored by predicted CNV-H score (B) and true CNV-H label (C).
(D) Distribution of chromosome 1q copy number status across all tumors in the independent cohort, grouped by genomic and histologic subtypes.
(E) Boxplots of xCell immune scores (y-axis) comparing tumors with 1q gain versus no gain (xaxis). P-values determined by Wilcoxon Rank Sum Test.
(F) Volcano plot of differentially expressed glycopeptides in tumors with 1q gain versus no gain. X-axis shows log2 fold change and the y-axis shows -log10 FDR, determined by Student’s T-test.
(G) Heatmap of PARP1 multi-omic levels in samples with and without 1q gain.
(H) Boxplots of olaparib (PARP-inhibitor) response in DepMap EC cell lines with and without PARP1 amplification. P-values determined by Wilcoxon Rank Sum Test.
Boxplots: Box portion represents Interquartile range (IQR), midline corresponds to the median, and whiskers range from the minimum (bottom) and maximum (top) variability outside the first and third quartiles (Q1 and Q3). Outliers are shown as points above whiskers.
1q amplification leads to increased glycoprotein levels and may be a biomarker for PARP-inhibition treatment
Increased CNV, a hallmark of cancer, is correlated with poorer outcomes, increased immune evasion, and lowered response to immunotherapies45,46. While the CNV-H subtype shows the highest concentration of CNV (Figure S1K), there are patterns of chromosome-specific alterations that occur across all EC subtypes, such as gains of 1q, the most common arm level CNV in EC (Figure 6D, Table S2). Tumors with 1q gain have a significantly lowered overall immune score, CD8+ naive T cell score, myeloid dendritic cell score, and microenvironment score (Figure 6E, Table S2). Using TCGA data, we saw a significant decrease in disease-free survival among tumors with 1q gain (Figure S6G, p=2.9e-3). Interestingly, pathways associated with protein glycosylation were significantly enriched in samples with 1q gain (Figure S6E). Using glycoproteomics data, we observed upregulation of glycopeptides for polymeric immunoglobulin receptor (PIGR) and other glycosylated proteins in tumors with 1q gain (Figure 6F). Focusing on glycosyltransferases or glycosidases encoded on 1q, we found 7 proteins involved in glycosylation (Figure S6F) including poly [ADP-ribose] polymerase 1 (PARP1), a key protein involved in DNA damage repair47. We observed increased PARP1 copy number, RNA, protein, phosphorylation, and acetylation levels, not only in tumors with 1q gain, but also in tumors with focal amplifications of PARP1 (Figure 6G). To test if there is any correlation between PARP1 amplification and drug response to PARP inhibitors (PARP-i), we used DepMap and observed that EC cell lines with PARP1 amplification showed increased sensitivity to the PARP-i olaparib (p=0.04) (Figure 6H). Sensitivity to niraparib and talazoparib, two other PARP-i in clinical use, also appeared to be greater in samples with PARP1 gain, although these differences were not statistically significant (Figure S6H). Gain of PARP1 may be an evolutionary advantage for EC tumors and could be a possible biomarker for treatment with PARP-i.
Multi-omic clustering uncovers a CTNNB1 hotspot mutation enriched CNV-L subgroup and a 1q gain MSI-H subgroup
Multi-omic clustering identified four clusters based on CNV, mRNA, protein, phosphosite, and acetylation site levels, which loosely follow the traditional genomic subtypes (Figure 7A). All CNV-H tumors fall into Cluster 1 with significant enrichment of TP53-mutated tumors (p=4.54E-06) and a proliferation signature (Figure 7B, Table S7). Clusters 2 and 3 were enriched for the CNV-L subtype (p=0.0006, p=0.001), with Cluster 3 characterized by CTNNB1 hotspot mutations (p=4.53E-07), arm 1q gain (p=7.39E-05), upregulated transporter activity (p=0.005) and downregulated immune pathways. Cluster 4 was enriched for MSI-H (p=1.70E-08) and TMB-H, and includes a majority of POLE tumors. Clusters 2 and 3 have complementary pathway signatures with Cluster 2 showing upregulation of immune-related pathways. Cluster 4 has enhanced activity in cell-cycle, metabolic and DNA damage related pathways in addition to mTOR and TNFα signaling. Comparison of only MSI-H samples among three clusters reveals an enrichment of 1q gain in Cluster 3 (p=0.002) and enrichment of ELMSAN1 mutation in Cluster 4 (p=7.05E-05), suggesting subgroups within the MSI-H subtype.
Figure 7: Multi-omic and glycoproteomic NMF clustering separates samples into 4 clusters.

(A) Heatmap of all tumors in the independent cohort, separated by multi-omic NMF clusters. Panels show histologic subtypes, histologic grade, genomic subtypes, APM class, transporter status, mutation status of selected genes, 1q copy number status, immune score, and corresponding glyco-NMF cluster assignment.
(B) Heatmap of mean ssGSEA pathway enrichments per cluster.
(C) Heatmap of glycopeptide levels for all independent cohort tumors, separated by glyco-peptide derived NMF clusters. Side panel (left) annotates types of glycans.
(D) Dot plots of glyco-enzyme levels between tumor versus normal across glyco-clusters, separated by glyco-enzyme function: Precursor (left), Trimming (middle), and Capping (right). Red denotes positive log2 fold change (higher in tumor) while blue indicates negative log2 fold change (higher in normal). Size of dots is determined by -log10 p-value, which comes from Student’s T-test.
(E) Dot plot comparing tumor and normal samples’ glycosylated kinases in the PI3K-AKT pathway by glycans (x-axis) and corresponding peptide (y-axis). Red denotes positive log2 fold change (higher in tumor) while blue indicates negative log2 fold change (higher in normal). Size of dots is determined by -log10 p-value, which comes from Student’s T-test.
See also Figure S7.
Glycopeptides and the corresponding unmodified protein levels are significantly different between tumor and normal samples
Altered glycosylation in tumors is known to be associated with increased metastatic potential and immune evasion48. In our data, oligomannose (HM) occupies 19% of N-linked glycopeptides, and ~70% of the rest were complex glycans with fucose (Fuc) (26%), sialic acids (Sia) (17%), or both (27%) (Figure S7A). Comparing tumor and normal samples, we observed 121 upregulated and 296 downregulated glycopeptides (Figure S7B), and ~80% of the glycopeptides involved in the lysosome pathway were upregulated, while ~60% of glycopeptides involved in the PI3K-AKT pathway were downregulated. The change in levels between tumor and normal samples of N-linked glycopeptides and the corresponding unmodified protein levels were correlated (r=0.71) (Figure S7C–D). Interestingly, three of the downregulated genes involved in trimming were MAN1C1 for trimming mannose and MGAT4C and MGAT5B for branching with GlcNAc, which may result in reduced synthesis of complex-type N-linked oligosaccharides, especially for bisected tetra-antennary complex N-glycans.
NMF clustering of glycopeptides groups tumors into four subtypes that are distinct from the multi-omics clusters
Unsupervised NMF clustering of glycopeptides levels49 divides the tumors into four subtypes (Figure 7C). Clusters 1 and 3 were mostly associated with the complex glycans Sia and Fuc, Cluster 2 with Sia, and Cluster 4 with HM. Chromosome 1q gain was most frequent in Cluster 1, TP53 mutations were frequent in Clusters 2 and 4, and CTNNB1 mutations were rare in Cluster 2. The levels of glycopeptides enriched in each cluster and the corresponding protein correlated with each other (Figure S7E). Proteins associated with oligosaccharide precursor synthesis were downregulated in all four clusters (Figure 7D). The glyco-clusters were, in general, not related to genomic subtypes except for a slight enrichment of the CNV-L group in Cluster 3 (Figure S7F), providing a complementary subtyping of EC tumors.
Focusing on the PI3K-AKT pathway differences between tumor and normal samples (Figure 7E), we observed 15 glycopeptides from 7 kinases. Most of these (12/15) were upregulated in tumors, including multiple glycosylation sites from FLT1. A significantly downregulated oligomannose structure was observed on EGFR, which indicates that glycosylation of EGFR could be altered in tumor samples in the trimming step, which may influence the function of EGFR in tumors.
DISCUSSION
This study is a comprehensive proteogenomic investigation of 138 prospectively collected EC tumors and 20 enriched normal endometrium samples, and provides a valuable community resource that can be used for both hypothesis generation and testing. Findings initially reported in Dou et al.9 were confirmed in this independent cohort, including the functional validation of the effects of frequently observed gene mutations, proteomic markers of clinical and genomic tumor subgroups, and the impact of defects in antigen presentation on patterns of immune infiltrates in TMB-H tumors. The concordance between the two studies was remarkably high, demonstrating the robustness of our proteogenomic strategy, including its sample collection protocols and analytical methods. Of note, the correlation between measured transcript and protein levels was in the range typically seen for tumors (median correlation across all genes of 0.48), once again demonstrating that, unsurprisingly, transcript levels are often not accurate for predicting protein levels for a given gene, as transcripts and protein can have largely different degradation rates and regulatory mechanisms.
Our current study expands our previous work in EC in several ways, including the exploration of additional data types: glycoproteomics, contributing further insights into EC tumor biology, and targeted mass spectrometry-based assays, which provide a path to the development of clinical assays.
ICI therapy provides a promising treatment option for many tumor types including EC. High TMB has been shown to predict response to ICI treatment in some cases, and pembrolizumab has been approved by the FDA to treat MSI-H cancers. However, high failure rates are observed among EC patients triaged to ICI therapy by currently available biomarkers. For example, recent prospective studies evaluating the clinical efficacy of dostarlimab in EC have demonstrated response rates no higher than 45% in MSI-H ECs50. Our previous study indicated that defects in antigen presentation may serve as a key rate-limiting factor which prevents variant peptide antigens from being processed and presented properly, and renders the host immune system unable to mount a tumoricidal response9. To address this potential clinical need, we developed an SRM assay for two peptides that can be used to predict APM status with high accuracy (AUC=0.96). Given its high accuracy, we believe this assay is amenable to being validated for use in clinical laboratories.
A second key finding of our current study is that MYC activity can potentially be used as a biomarker for triaging EC patients to metformin treatment. Over the past decade, a multitude of clinical studies have evaluated metformin as a strategy for EC prevention and treatment. For reasons that remain unclear, outcomes of these studies, which have typically focused on evaluating the impact of metformin in patients impacted by T2D and obesity, remain conflicted51–53. We found that high levels of MYC activity are associated with worse survival for both CNV-L and MSI-H ECs. Despite controversy whether sufficient circulating levels of metformin can be achieved clinically to biologically impact a cancer, our data clearly indicate that real-world metformin treatment results in lower levels of MYC activity in EC patients with T2D across both our current and previous EC cohorts. Lastly, we have uncovered an association between MYC activity in EC and patient BMI, with higher levels of MYC activity observed in non-obese patients. Thus, the response of ECs to metformin may be tempered by a complex relationship between MYC activity and BMI. In the future, it will be important to further parse these relationships as well as to directly test our therapeutic hypothesis that high MYC activity can serve as a biomarker for metformin response even in non-diabetic EC patients. Given the relatively limited correlation (cor = 0.38) we observed between MYC activity and MYC IHC score, this may require development of an alternative diagnostic.
Despite rapid clinical uptake of somatic tumor sequencing, traditional histology, being faster and cheaper, is still widely used to determine cancer treatments. In part, this is because the cost of genomic technologies has largely limited their use to advanced stage and metastatic disease, e.g., most insurances do not cover sequencing of low-grade EC. We therefore explored the possibility that features derived from traditional histopathology images could be used to predict the molecular features of a tumor. The high degree of accuracy achieved with our algorithm as a predictive model for some EC subtypes and mutations, specifically POLE-mutated EC, is promising but needs to be tested on a larger cohort.
Our study demonstrates the ability of proteogenomic analysis to increase our understanding of EC tumor biology and to generate new hypotheses. We have highlighted a few examples of integrative analysis across the omics data modalities that can provide insights with potential clinical applications.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to the Lead Contact, David Fenyö (david@fenyolab.org).
Materials availability
This study did not generate new unique reagents.
DATA AND CODE AVAILABILITY
Clinical and proteomic (raw MS files and processed data files) data reported in this paper, including both exploratory and independent CPTAC datasets, can be accessed via the Proteomic Data Commons (PDC) at: https://pdc.cancer.gov/ (PDC000125, PDC000126, PDC000226). Genomic and transcriptomic data files for both CPTAC cohorts (phs001287) and TCGA cohort (phs000178) can be accessed via Genomic Data Commons (GDC) at: https://portal.gdc.cancer.gov/projects/CPTAC-3. The DepMap datasets can be accessed via the DepMap portal: https://depmap.org/portal/. Processed CPTAC data for both cohorts used in this publication can also be found in the PDC, the Python package called ‘cptac’ (https://pypi.org/project/cptac/, install via pip) to allow programmatic access and LinkedOmics via http://www.linkedomics.org/data_download/CPTAC-UCEC-independent/ 64. Histological and radiological images for both CPTAC cohorts (10.7937/k9/tcia.2018.3r3juisw) and TCGA cohort (10.7937/k9/tcia.2016.gkj0zwac) can be accessed via Imaging Data Commons (IDC) at https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ucec, and The Cancer Imaging Archive at https://wiki.cancerimagingarchive.net/pages/viewpage.action?pageId=33948263. Deidentified digitized H&E slides from NYU reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Patient Selection
The tumor, normal tissue, and whole blood samples used in this manuscript were prospectively collected between May 2016 and May 2019 for the CPTAC program from 5 different countries. Informed consent was collected for each patient and all patients consented to donate their specimens to CPTAC. Each sample collection site has an internally approved IRB. None of the sites use any central IRB and none are involved in trials. The average age of the cohort was 64 yrs. Biospecimens were collected from newly diagnosed patients with endometrial cancer (EC) who were undergoing surgical resection and had received no prior treatment for their disease, including chemotherapy or radiotherapy, and were collected independent of grade or stage. EC cases were graded using the FIGO (International Federation of Gynecology and Obstetrics) system or the American Joint Committee on Cancer TNM staging system, which are functionally identical.
Sample Collection
The CPTAC Biospecimen Core Resource (BCR) at the Pathology and Biorepository Core of the Van Andel Research Institute in Grand Rapids, Michigan manufactured and distributed biospecimen kits to the Tissue Source Sites (TSS) located in the US, Europe, and Asia. Each kit contained a set of pre-manufactured labels for unique tracking of every specimen respective to TSS location, disease, and sample type, used to track the specimens through the BCR to the CPTAC proteomic and genomic characterization centers. Tissue specimens averaging 302 mg were snap-frozen by the TSS within a 30 min cold ischemic time (CIT) (CIT average = 17 min) and an adjacent segment was formalin-fixed paraffin-embedded (FFPE) and H&E stained by the TSS for quality assessment to meet the CPTAC EC requirements. Routinely, several tissue segments for each case were collected. Tissues were flash frozen in liquid nitrogen (LN2) then transferred to a liquid nitrogen freezer for storage until approval for shipment to the BCR. Specimens were shipped using a cryoport that maintained an average temperature of under −140°C to the BCR with a time and temperature tracker to monitor the shipment. Receipt of specimens at the BCR included a physical inspection and review of the time and temperature tracker data for specimen integrity, followed by barcode entry into a biospecimen tracking database. Specimens were again placed in storage at LN2 temperatures until further processing. Acceptable EC tumor tissue segments were determined by TSS pathologists based on the percent viable tumor nuclei (> 80%), total cellularity (> 50%), and necrosis (< 20%). Segments received at the BCR were verified by BCR and Leidos Biomedical Research (LBR) pathologists and the percent of total area of tumor in the segment was also documented. Additionally, disease-specific working group pathology experts reviewed the morphology to clarify or standardize specific disease classifications and correlation to the proteomic and genomic data. Specimens selected for the discovery set were determined on the maximal percent in the pathology criteria and best weight. Specimens were pulled from the biorepository using an LN2 cryocart to maintain specimen integrity and then cryopulverized. The cryopulverized specimen was divided into aliquots for DNA (30 mg) and RNA (30 mg) isolation and proteomics (50 mg) for molecular characterization. Nucleic acids were isolated and stored at −80°C until further processing and distribution; cryopulverized protein material was returned to the LN2 freezer until distribution. Shipment of the cryopulverized segments used cryoports for distribution to the proteomic characterization centers and shipment of the nucleic acids used dry ice shippers for distribution to the genomic characterization centers; a shipment manifest accompanied all distributions for the receipt and integrity inspection of the specimens at the destination. The DNA sequencing was performed at the Broad Institute, Cambridge, MA and RNA sequencing was performed at the University of North Carolina, Chapel Hill, NC. Material for proteomic analyses was sent to the Proteomic Characterization Center (PCC) at Pacific Northwest National Laboratory (PNNL), Richland, Washington.
Enriched normal sample collection
20 enriched normal endometrium tissue samples were collected according to CPTAC’s Standard Operating Procedure (SOP) for collection and preservation of research specimens. Enriched normal samples were obtained from healthy patients with an indication for surgery due to the presence of a non-cancerous lesion (hyperplasia, endometrial polyps, myoma) and served as an independent cohort of normal endometrial samples. The collected fragments were excised carefully to avoid any contamination of myometrium and other stromal components. After excision, the normal tissue samples were snap-frozen within a 30 min cold ischemic time, and next flash-frozen in liquid nitrogen, and transferred to a liquid nitrogen freezer for storage until approval for shipment to BCR. An adjacent segment of each sample was formalin-fixed paraffin-embedded and H&E stained to perform quality assessment. To meet CPTAC pathology acceptance criteria, collected normal tissue samples had to include at least 80% of endometrial cells. Thus, enriched normal tissue means that the normal tissue includes at least 80% of endometrial cells.
ICI treated sample collection
Medical records were abstracted to identify uterine cancer patients who had received treatment with ICI therapy. Pre-ICI FFPE tumor specimens were retrieved from those with archived tissue stored at UAB. Medical records were analyzed to obtain basic demographic data, histology, stage, treatments used, and best response to ICI therapy. Best response to therapy was collected by reviewing all imaging reports obtained on therapy. Complete response was defined as no radiographic evidence of residual disease; partial response was defined as any reduction in lesion size by > 1 cm; stable disease was defined as no growth or reduction of lesions > 1 cm; progressive disease was defined as growth of lesions > 1 cm or oncologist’s determination of clinical progression.
Cell lines
The EC cell line, HEC-151, was purchased from JCRB cell bank. Cells were cultivated using MEM containing 10% FBS and 1% penicillin–streptomycin in a 37 °C incubator with 5% CO2. Cell lines were routinely tested and confirmed negative for Mycoplasma contamination by Lonza Mycoalert Mycoplasma Detection Kit.
EC cell lines, HEC265, HEC-108, HEC-251, were obtained from either the JCRB Cell Bank. EC cell line, HEC1A, was obtained from the American Type Culture Collection. Cells were cultured in MEM media supplemented with 15% FBS. Identity of each cell line was verified by a PCR-based method for Short Tandem Repeat (STR) profiling prior to receipt. Once thawed, cell lines were cultured for fewer than 20 passages and regularly screening for mycoplasma infection by two independent methods: MycoStrip (Rep-mys-10; Invivogen) and RT-PCR (Venor GeM, Cat MP00025–1KT; Sigma-Millipore).
METHOD DETAILS
Sample Processing for Genomic DNA and Total RNA Extraction
Our study sampled a single site of the primary tumor from surgical resections, due to the internal requirement to process a minimum of 125 mg of tumor issue and 50 mg of adjacent normal tissue. DNA and RNA were extracted from tumor and adjacent normal specimens in a co-isolation protocol using QIAGEN’s QIAsymphony DNA Mini Kit and QIAsymphony RNA Kit. Genomic DNA was also isolated from peripheral blood (3–5 mL) to serve as matched normal reference material. The Qubit dsDNA BR Assay Kit was used with the Qubit 2.0 Fluorometer to determine the concentration of dsDNA in an aqueous solution. Any sample that passed quality control and produced enough DNA yield to go through various genomic assays was sent for genomic characterization. RNA quality was quantified using both the NanoDrop 8000 and quality assessed using Agilent Bioanalyzer. A sample that passed RNA quality control and had a minimum RIN (RNA integrity number) score of 7 was subjected to RNA sequencing. Identity match for germline, normal adjacent tissue, and tumor tissue was assayed at the BCR using the Illumina Infinium QC array. This beadchip contains 15,949 markers designed to prioritize sample tracking, quality control, and stratification.
Whole Exome Sequencing
Whole Exome Sequencing Methods
Library Construction
Library construction was performed as described in Fisher et al., with the following modifications: initial genomic DNA input into shearing was reduced from 3 μg to 20–250 ng in 5 0μL of solution. For adapter ligation, Illumina paired end adapters were replaced with palindromic forked adapters, purchased from Integrated DNA Technologies, with unique dual-indexed molecular barcode sequences to facilitate downstream pooling. Kapa HyperPrep reagents in 96-reaction kit format were used for end repair/A- tailing, adapter ligation, and library enrichment PCR. In addition, during the post-enrichment SPRI cleanup, elution volume was reduced to 30 μL to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted.
In-solution hybrid selection
After library construction, libraries are pooled into groups of up to 96 samples. Hybridization and capture were performed using the relevant components of Illumina’s Nextera Exome Kit and following the manufacturer’s suggested protocol, with the following exceptions: first, all libraries within a library construction plate were pooled prior to hybridization. Second, the Midi plate from Illumina’s Nextera Exome Kit was replaced with a skirted PCR plate to facilitate automation. All hybridization and capture steps were automated on the Agilent Bravo liquid handling system.
Preparation of libraries for cluster amplification and sequencing
After post-capture enrichment, library pools were quantified using qPCR (automated assay on the Agilent Bravo), using a kit purchased from KAPA Biosystems with probes specific to the ends of the adapters. Based on qPCR quantification, libraries were normalized to 2nM.
Cluster amplification and sequencing
Cluster amplification of DNA libraries was performed according to the manufacturer’s protocol (Illumina) using exclusion amplification chemistry and flowcells. Flowcells were sequenced utilizing Sequencing-by-Synthesis chemistry. The flow cells are then analyzed using RTA v.2.7.3 or later. Each pool of whole exome libraries was sequenced on paired 76 cycle runs with two 8 cycle index reads across the number of lanes needed to meet coverage for all libraries in the pool. Pooled libraries were run on HiSeq4000 paired end runs to achieve a minimum of 150x on target coverage per each sample library. The raw Illumina sequence data were demultiplexed and converted to fastq files, adapter and low-quality sequences were trimmed. The raw reads were mapped to the HG38 human reference genome. The validated bams were used for downstream analysis and variant calling.
PCR-Free Whole Genome Sequencing
Whole Genome Sequencing, PCR-Free
Preparation of libraries for cluster amplification and sequencing
An aliquot of genomic DNA (350 ng in 50 μL) is used as the input into DNA fragmentation (aka shearing). Shearing is performed acoustically using a Covaris focused-ultrasonicator, targeting 385bp fragments. Following fragmentation, additional size selection is performed using a SPRI cleanup. Library preparation is performed using a commercially available kit provided by KAPA Biosystems (KAPA Hyper Prep without amplification module) and with palindromic forked adapters with unique 8-base index sequences embedded within the adapter (purchased from IDT). Following sample preparation, libraries are quantified using quantitative PCR (kit purchased from KAPA Biosystems), with probes specific to the ends of the adapters. This assay is automated using Agilent’s Bravo liquid handling platform. Based on qPCR quantification, libraries are normalized to 1.7 nM and pooled into 24-plexes.
Cluster amplification and sequencing (HiSeq X)
Sample pools are combined with HiSeqX Cluster Amp Regents EPX1, EPX2 and EPX3 into single wells on a strip tube using the Hamilton Starlet Liquid Handling system. Cluster amplification of the templates is performed according to the manufacturer’s protocol (Illumina) with the Illumina cBot. Flow Cells are sequenced to a minimum of 15x on HiSeqX utilizing sequencing-by-synthesis kits to produce 151bp paired-end reads. Output from Illumina software is processed by the Picard data-processing pipeline to yield BAM files containing demultiplexed, aggregated aligned reads.
All sample information tracking is performed by automated LIMS messaging.
Illumina Infinium MethylationEPIC BeadChip Array
The MethylationEPIC array uses an 8-sample version of the Illumina Beadchip capturing >850,000 methylation sites per sample. The EPIC array includes sample plating, bisulfite conversion, and methylation array processing. After scanning, the data was processed through an automated genotype calling pipeline. Data generated consists of raw idats and a sample sheet
RNA Sequencing.
Total RNA Sequencing
Indexed cDNA sequencing libraries were prepared from the RNA samples using the TruSeq Stranded RNA Sample Preparation Kit and bar-coded with individual tags. Quality control was performed at every step, and the libraries were quantified. Indexed libraries were prepared and run on HiSeq4000 paired end 75 base pairs to generate a minimum of 120 million reads per sample library with a minimum of 90% mapped reads. The raw Illumina sequence data were demultiplexed and converted to fastq files, and adapter and low-quality sequences were trimmed for downstream analysis.
miRNA Sequencing
Indexed small RNA sequencing libraries were prepared from the RNA samples using the TruSeq Small Total RNA Sample Prep Kit and bar-coded with individual tags. Quality control was performed at every step, and the libraries were quantified. Indexed libraries were prepared and loaded on the Hiseq4000 to generate a minimum of 10 million reads per library with a minimum of 90% reads mapped. The raw Illumina sequence data were demultiplexed and converted to fastq files for downstream analysis.
MS Sample Processing and Data Collection
Protein extraction and Lys-C/trypsin tandem digestion
Approximately 50 mg of each of the pulverized OCT-embedded uterine tumor and normal tissues were homogenized separately in 200 μL of lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA, 2 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF, 10 mM NaF, 1:100 v/v Sigma phosphatase inhibitor cocktail 2, 1:100 v/v Sigma phosphatase inhibitor cocktail 3, 20 μM PUGNAc, and 5 mM sodium butyrate). Lysates were precleared by centrifugation at 20,000 g for 10 min at 4 °C and protein concentrations were determined by BCA assay (ThermoFisher Scientific) and adjusted to 8 μg/μL with lysis buffer. Proteins were reduced with 5 mM dithiothreitol for 1 h at 37 °C, and subsequently alkylated with 10 mM iodoacetamide for 45 min at 25°C in the dark. Samples were diluted 1:4 with 50 mM Tris, pH 8.0 and digested with Lys-C (Wako) at 1:50 enzyme-to-substrate ratio sequencing grade modified trypsin (Promega, V5117) at 1:50 enzyme-to-substrate ratio. After 2 h of digestion at 25 °C, an aliquot of the same amount of sequencing grade modified trypsin (Promega, V5117) was added to the samples and further incubated at 25 °C overnight (~14 h). The digested samples were then acidified with 100% formic acid to 1% of the final concentration of formic acid. Tryptic peptides were desalted on C18 SPE (Waters tC18 SepPak, WAT054925) and dried using Speed-Vac.
TMT-11 labeling
Desalted peptides were labeled with 11-plex TMT reagents using conditions modified from the manufacturer’s instructions (ThermoFisher Scientific). Peptides (400 μg) from each of the tumors were dissolved in 80 μL of 50 mM HEPES, pH 8.5 solution, and mixed with 400 μg of TMT reagent that was dissolved freshly in 20 μL of anhydrous acetonitrile. Channel 126 was used for labeling the internal reference sample (pooled from all tumor and normal samples in the CPTAC UCEC Exploratory cohort – Cell. 2020, 180(4):729–748) throughout the sample analysis. After 1 h incubation at RT each sample was diluted with 60 μL 50 mM HEPES pH 8.5, 20% acetonitrile. Quench of the reaction was achieved by adding 12 μL of 5% hydroxylamine and incubation for 15 min at RT. Peptides labeled by different TMT reagents were then mixed, dried using Speed-Vac, reconstituted with 3% acetonitrile, 0.1% formic acid and were desalted on tC18 SepPak SPE columns.
Fractionation
Approximately 3.5 mg of 11-plex TMT labeled sample was separated on a reversed-phase Agilent Zorbax 300 Extend-C18 column (250 mm × 4.6 mm column containing 3.5-μm particles) using Agilent 1200 HPLC System. Solvent A was 5 mM ammonium formate, pH 10, 2% acetonitrile and solvent B was 5 mM ammonium formate, pH 10, 90% acetonitrile. The flow rate was 1 mL/min and the injection volume was 900 μL. The LC gradient started with a linear increase of solvent B to 16% in 6 min, then linearly increased to 40% B in 70 min, 4 min to 44% B, 5 min to 60% B and another 14 of 60% solvent B. A total of 96 fractions were collected into a 96 well plate throughout the LC gradient. These fractions were concatenated into 24 fractions by combining 4 fractions that are 24 fractions apart (i.e., combining fractions #1, #25, #49, and #73; #2, #26, #50, and #74; and so on). For proteome analysis, 5% of each concatenated fraction was dried down and resuspended in 2% acetonitrile, 0.1% formic acid to a peptide concentration of 0.1 mg/mL for LC-MS/MS analysis. The rest of the fractions (95%) were further concatenated into 12 fractions (i.e., by combining fractions #1 and #13; #3 and #15; and so on), dried down, and subjected to immobilized metal affinity chromatography (IMAC) for phosphopeptide enrichment.
Phosphopeptide enrichment using IMAC
Fe3+-NTA-agarose beads were freshly prepared using the Ni-NTA Superflow agarose beads (QIAGEN, #30410) for phosphopeptide enrichment. For each of the 12 fractions, peptides were reconstituted in 500 μL IMAC binding/wash buffer (80% acetonitrile, 0.1% trifluoroacetic acid) and incubated with 20 μL of the 50% bead suspension for 30 min at RT. After incubation, the beads were washed 2 times each with 50 μL of wash buffer and once with 50 μL of 1% formic acid on the stage tip packed with 2 discs of Empore C18 material (Empore Octadecyl C18, 47 mm; Supleco, 66883-U). Phosphopeptides were eluted from the beads on C18 using 70 μL of Elution Buffer (500 mM K2HPO4, pH 7.0). 50% acetonitrile, 0.1% formic acid was used for elution of phosphopeptides from the C18 stage tips. Samples were dried using Speed-Vac, and later reconstituted with 10 μL of 3% acetonitrile, 0.1% formic acid for LC-MS/MS analysis.
Immunoaffinity purification of acetylated peptides
Tryptic peptides from the flow-through of IMAC were combined into four samples follow concatenation scheme by combining 3 fractions that were 4 fractions apart (i.e., combining fractions #1, #5 and #9 as a new fraction) and dried down using Speed-Vac. The dried peptides were reconstituted in 1.4 mL of the immunoaffinity purification (IAP) buffer (50 mM MOPS/NaOH pH 7.2, 10 mM Na2HPO4 and 50 mM NaCl). After dissolving the peptide, the pH of the peptide solution was checked using pH indicator paper. The antibody beads from PTMScan® Acetyl-Lysine Motif [Ac-K] Kit (Cell Signaling, #13416) were freshly prepared. Briefly, the antibody beads were centrifuged at 2,000 x g for 30 sec and all buffers from the beads were removed; the antibody beads were then washed with 1 mL of IAP buffer for four times and finally resuspend in 40 μL of IAP buffer. For each fraction, half of the antibody in each tube was transferred to the peptide solution and incubated on a rotator overnight at 4 °C. After removing the supernatant, the reacted beads were washed with 1 mL of PBS buffer five times. For the elution of acetylated peptides, the antibody beads were incubated 2 times each with 50 μL of 0.15% TFA at room temperature for 10 min. The eluted peptides were transferred to the stage tip packed with two discs of Empore C18 material. The C18 stage tips were washed by 1% formic acid and 50% acetonitrile, and 0.1% formic acid was used for elution of peptides from the C18 stage tips. The eluted peptides were dried using Speed-Vac, and reconstituted with 13 μL of 2% acetonitrile, 0.1% formic acid contained 0.01% DDM (n-Dodecyl β-D-maltoside) right before the LC-MS/MS analysis.
The acetylated peptides prepared by IP from the IMAC flow-through may very well miss those peptides that are both phosphorylated and acetylated. Splitting the samples for independent IP and IMAC may improve the chance of recovering such peptides, assuming having both PTMs on the same peptide does not impact the affinity of either the IP or IMAC process. However, acetylated peptides are estimated to be 10 times lower in abundance than the phosphopeptides, hence much larger input may be needed to recover the dual-modified peptides. Given the extremely low stoichiometry of these dual-modified peptides and the sample size limitations, it was not pursued in this work.
Enrichment of glycopeptides
Peptides from the flow-through of acetylated peptide enrichment were desalted on the reversed phase C18 SPE column (Waters). The glycopeptides were enriched with OASIS MAX solid-phase extraction (Waters). The MAX cartridge was conditioned with 3×1 mL of ACN, then 3×1 mL of 100 mM triethylammonium acetate buffer, followed by 3×1 mL of water, and finally 3×1 mL of 95% ACN / 1% TFA. The peptides were reconstituted in 95% ACN / 1% TFA and loaded twice. The cartridge was washed with 4×1 mL of 95% ACN / 1% TFA to remove non-glycosylated peptides. The glycopeptide fraction was eluted with 50% ACN / 0.1% FA, dried down, and reconstituted in 3% ACN / 0.1% FA prior to ESI-LC-MS/MS analysis.
LC-MS/MS data acquisition
Fractionated samples prepared for global proteome, phosphoproteome, and acetylome analysis were separated using a nanoACQUITY UPLC system (Waters) by reversed-phase HPLC. The analytical column was manufactured in-house using ReproSil-Pur 120 C18-AQ 1.9 μm stationary phase (Dr. Maisch GmbH) and slurry packed into a 25-cm length of 360 μm o.d. x 75 μm i.d. fused silica picofrit capillary tubing (New Objective). The analytical column was heated to 50 °C using an AgileSLEEVE column heater (Analytical Sales and Services). The analytical column was equilibrated to 98% Mobile Phase A (MP A, 0.1% formic acid/3% acetonitrile) and 2% Mobile Phase B (MP B, 0.1% formic acid/90% acetonitrile) and maintained at a constant column flow of 200 nL/min. The sample was injected into a 5-μL loop placed in-line with the analytical column which initiated the gradient profile (min:%MP B): 0:2, 1:6, 85:30, 94:60, 95:90, 100:90, 101:50, 110:50 (for global proteome and phosphoproteome analysis); 0:2, 1:6, 235:30, 244:60, 245:90, 250:90, 251:50, 260:50 (for acetylome analysis). The column was allowed to equilibrate at start conditions for 30 minutes between analytical runs.
MS analysis was performed using an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific). The global proteome and phosphoproteome samples were analyzed under identical conditions. Electrospray voltage (1.8 kV) was applied at a carbon composite union (Valco Instruments) coupling a 360 μm o.d. x 20 μm i.d. fused silica extension from the LC gradient pump to the analytical column and the ion transfer tube was set at 250 °C. Following a 25-min delay from the time of sample injection, Orbitrap precursor spectra (AGC 4 × 105) were collected from 350–1800 m/z for 110 min at a resolution of 60K along with data dependent Orbitrap HCD MS/MS spectra (centroid) at a resolution of 50K (AGC 1 × 105) and max ion time of 105 ms for a total duty cycle of 2 seconds. Masses selected for MS/MS were isolated (quadrupole) at a width of 0.7 m/z and fragmented using a collision energy of 30%. Peptide mode was selected for monoisotopic precursor scan and charge state screening was enabled to reject unassigned 1+, 7+, 8+, and > 8+ ions with a dynamic exclusion time of 45 seconds to discriminate against previously analyzed ions between ±10 ppm. The acetylome samples were analyzed under similar conditions except that the max ion time was 125 ms.
The TMT-labeled glycoproteome fractions were analyzed using Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). Approximately 0.5 ug of peptides were separated on an in-house packed 28 cm x 75 um diameter C18 column (1.9 um Reprosil-Pur C18-AQ beads (Dr. Maisch GmbH); Picofrit 10 um opening (New Objective)) lined up with an Easy nLC 1200 UHPLC system (Thermo Scientific). The column was heated to 50C using a column heater (Phoenix-ST). The flow rate was set at 200 nl/min. Buffer A and B were 3% ACN / 0.1% FA and 90% ACN / 0.1% FA, respectively. The peptides were separated with a 6%–30% B gradient in 84 min. Peptides were eluted from the column and nanosprayed directly into the mass spectrometer. The mass spectrometer was operated in a data-dependent mode. Parameters for glycoproteomic samples were set as follows: MS1 resolution – 60,000, mass range – 500 to 2000 m/z, RF Lens – 30%, AGC Target – 5.0e5, Max injection time – 50 ms, charge state include – 2–6, dynamic exclusion – 45 s. The cycle time was set to 2 s, and within this 2 s the most abundant ions per scan were selected for MS/MS in the orbitrap. MS2 resolution – 50,000, high-energy collision dissociation activation energy (HCD) – 35, isolation width (m/z) – 0.7, AGC Target – 1.0e5, Max injection time – 100 ms.
Construction and utilization of the Comparative Reference samples
As a quality control measure, two different types of “Comparative Reference” (“CompRef”) patient-derived xenograft (PDX) samples were generated as previously described (Li et al., 2013; Tabb et al., 2016) and used to monitor the longitudinal performance of the proteomics workflow throughout the course of this study. Briefly, the PDX tumors from established basal and luminal breast cancer intrinsic subtypes were raised subcutaneously in 8-week old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Laboratories, Bar Harbor, ME) using procedures reviewed and approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis. Xenografts were grown in multiple mice, pooled, and cryopulverized to provide a sufficient amount of uniform material for the duration of the study. Full proteome, phosphoproteome and acetylome process replicates of each of the two types of CompRef samples were prepared and analyzed as standalone 11-plex TMT experiments alongside every four TMT-11 experiments of the study samples, using the same analysis protocol as the patient samples. These interstitially analyzed CompRef samples were evaluated for depth of proteome, phosphoproteome, and acetylome coverage and for consistency in quantitative comparison between the basal and luminal models.
Genomic Data Analysis
Harmonized genome alignment
WGS, WES, RNA-Seq sequence data were harmonized by NCI Genomic Data Commons (GDC) https://gdc.cancer.gov/about-data/gdc-data-harmonization, which included alignment to GDC’s hg38 human reference genome (GRCh38.d1.vd1) and additional quality checks. All the downstream genomic processing was based on the GDC-aligned BAMs to ensure reproducibility.
Copy Number Calling
Copy number variation was detected using BIC-seq2 94, a read-depth-based CNV calling algorithm to detect somatic copy number variation (CNVs) from the WGS data of tumors. Briefly, BIC-seq2 divides genomic regions into disjoint bins and counts uniquely aligned reads in each bin. Then, it combines neighboring bins into genomic segments with similar copy numbers iteratively based on Bayesian Information Criteria (BIC), a statistical criterion measuring both the fitness and complexity of a statistical model. We used paired-sample CNV calling that takes a pair of samples as input and detects genomic regions with different copy numbers between the two samples. We used a bin size of ∼100 bp and a lambda of 3 (a smoothing parameter for CNV segmentation). We recommend calling segments as copy gain or loss when their log2 copy ratios were larger than 0.2 or smaller than −0.2, respectively (according to the BIC-seq publication).
Somatic Variant Calling
Somatic mutations were called by the Somaticwrapper pipeline v1.6 (https://github.com/ding-lab/somaticwrapper), which includes four different callers, i.e., Strelka v.295, MUTECT v1.7 71, VarScan v.2.3.8 84, and Pindel v.0.2.5 76 from WES. We kept the exonic SNVs called by any two callers among MUTECT v1.7, VarScan v.2.3.8, and Strelka v.2 and indels called by any two callers among VarScan v.2.3.8, Strelka v.2, and Pindel v.0.2.5. For the merged SNVs and indels, we applied a 14X and 8X coverage cutoff for tumor and normal, separately. We also filtered SNVs and indels by a minimal variant allele frequency (VAF) of 0.05 in tumors and a maximal VAF of 0.02 in normal samples. We filtered any SNV, which was within 10bp of an indel found in the same tumor sample. Finally, we rescued the rare mutations with VAF of [0.015, 0.05) in ccRCC driver genes based on the gene consensus list 96
Mutational Signatures
The R package SigProfilerMatrixGeneratorR (version 1.0) was used to call mutation signatures from somatic mutation data (PMID: 31470794). The maximum number of signatures was set to 10 and nmf replicate was set to 100. The activity scores of SigProfilerMatricGenetator suggested decomposed solutions were used as signature scores. Only single base substitutions signatures were included in our analysis.
DNA methylation analysis
Raw methylation idat files were downloaded from CPTAC DCC and GDC. Beta values of CpG loci were reported after functional normalization, quality check, common SNP filtering, and probe annotation using Li Ding Lab’s methylation pipeline v1.1 https://github.com/ding-lab/cptac_methylation. For downstream integrated analysis, we focused only on the methylation levels (represented as beta values) of the probes located both in the CpG island and the promoter (including 5’UTR) regions. The gene-level methylation was derived by averaging these probe-level methylation values.
Microsatellite Instability Prediction
MSI scores were calculated by MSIsensor (https://github.com/ding-lab/msisensor) and interpreted as the percentage of microsatellite sites (with deep enough sequencing coverage) that have a lesion. Samples with an MSIscore > 5 are classified as “MSI-High” and the rest will be classified as “MSS” based on a bimodal distribution of MSIscore on this cohort.
Copy Number Classification
The copy number subtypes were characterized by CNV deletion events. A sample was defined as CNV-high if more than 10% of its genome was deleted, regardless of the number of CNV-independent events.
TCGA Subtype Classification
TCGA identified four subtypes of endometrial cancer: POLE, MSI, CNV-high, and CNV-low. We replicated this subtyping for the samples in this study.
The MSI subtype consists of all samples called MSI-H, as described in the Microsatellite Instability Prediction section. To identify the POLE subtype, we looked for samples with mutations in the POLE exonuclease domain. There were 6 samples carrying exonuclease domain mutations (EDM) including 5 P286R and 1 V411L. All of them passed the criteria [CA] signature > 20% and [CG] signature < 3%. Thus, six samples were classified as POLE. Samples identified as having high CNV, as described in the Copy Number Classification section, were assigned to the CNV-high subgroup. All remaining samples not classified as MSI, POLE, or CNV-high were classified as CNV-low. The genomic subtypes of three samples, C3L-00898, C3L-02802, and C3N-02298, could not be determined due to lack of WGS-based CNV data. One sample, C3L-02557, was identified as both POLE and CNV-H.
RNA Quantification & Analysis
RNA Quantification and Circular RNA Prediction
The Hg38 reference genome and RefSeq annotations were used for the RNAseq data analysis. They were downloaded from the UCSC table browser. First, CIRI (v2.0.6) was used to call circular RNA with default parameters and BWA (version 0.7.17-r1188) was used as the mapping tool. The cutoff of supporting reads for circRNA was set to 10. Then we used a pseudo-linear transcript strategy to quantify gene and circular RNA expression 97. In brief, for each sample, linear transcripts of circular RNAs were extracted and 75bp (read length) from the 3’ end was copied to the 5’ end. The modified transcripts were called pseudo-linear transcripts. Transcripts of linear genes were also extracted and mixed with pseudo-linear transcripts. RSEM (version 1.3.1) with Bowtie2 (version 2.3.3) as the mapping tool was used to quantify gene and circular RNA expression based on the mixed transcripts. After quantification, the upper quantile method was applied for normalization. The normalized matrix was log2-transformed and separated into gene and circular RNA expression matrices.
miRNA-Seq Data Analysis miRNA-Seq FASTQ files were downloaded from GDC. We reported the mature miRNA and precursor miRNA expression in TPM (Transcripts Per Million) after adapter trimming, quality check, alignment, annotation, reads counting using Li Ding Lab’s miRNA pipeline https://github.com/ding-lab/CPTAC_miRNA. The mature miRNA expression was calculated irrespective of its gene of origin by summing the expression from its precursor miRNAs.
PROGENy Pathway Activity
The PROGENy R package was applied to the log2 transformed RSEM mRNA matrix to estimate activity of 11 cancer related pathways: EGFR, Hypoxia, JAK-STAT, MAPK, NFkB, PI3K, TGFb, TNFa, Trail, VEGF, p5377.
MS Data Interpretation
Quantification of TMT global proteomics data
LC-MS/MS analysis of the TMT11-labeled, bRPLC fractionated samples generated a total of 384 global proteomics data files. The Thermo RAW files were processed with mzRefinery to characterize and correct for any instrument calibration errors, and then with MS-GF+ v988167,72,98 to match against the RefSeq human protein sequence database downloaded on June 29, 2018 (hg38; 41,734 proteins), combined with 264 contaminants (e.g., trypsin, keratin). The partially tryptic search used a ± 10 ppm parent ion tolerance, allowed for isotopic error in precursor ion selection, and searched a decoy database composed of the forward and reversed protein sequences. MS-GF+ considered static carbamidomethylation (+57.0215 Da) on Cys residues and TMT modification (+229.1629 Da) on the peptide N-terminus and Lys residues, and dynamic oxidation (+15.9949 Da) on Met residues for searching the global proteome data.
Peptide identification stringency was set at a maximum 1% FDR at peptide level using PepQValue < 0.005 and parent ion mass deviation < 7 ppm criteria. A minimum of 6 unique peptides per 1000 amino acids of protein length was then required for achieving 1% at the protein level within the full data set.
The intensities of all 11 TMT reporter ions were extracted using MASIC software (Monroe et al., 2008). Next, PSMs passing the confidence thresholds described above were linked to the extracted reporter ion intensities by scan number. The reporter ion intensities from different scans and different bRPLC fractions corresponding to the same gene were grouped. Relative protein level was calculated as the ratio of sample level to reference level using the summed reporter ion intensities from peptides that could be uniquely mapped to a gene. The pooled reference sample was labeled with TMT 126 reagent, allowing comparison of relative protein levels across different TMT-11 plexes. The relative levels were log2 transformed and zero-centered for each gene to obtain final relative levels.
Small differences in laboratory conditions and sample handling can result in systematic, sample-specific bias in the quantification of protein levels. In order to mitigate these effects, we computed the median, log2 relative protein level for each sample and re-centered to achieve a common median of 0.
Quantification of phosphopeptides
Phosphopeptide identification for the 192 phosphoproteomics data files were performed as in the global proteome data analysis described above (e.g., peptide level FDR < 1%), with an additional dynamic phosphorylation (+79.9663 Da) on Ser, Thr, or Tyr residues. The phosphoproteome data were further processed by the Ascore algorithm (Beausoleil et al., 2006) for phosphorylation site localization, and the top-scoring sequences were reported. For phosphoproteomic datasets, the TMT-11 quantitative data were not summarized by protein but left at the phosphopeptide level. All peptides (phosphopeptides and global peptides) were labeled with TMT-11 reagent simultaneously. Separation into phospho- and non-phosphopeptides using IMAC was performed after the labeling. Thus, all the biases upstream of labeling are assumed to be identical between global and phosphoproteomic datasets. Therefore, to account for sample-specific biases in the phosphoproteome analysis, we applied the correction factors derived from median-centering the global proteomic dataset.
Quantification and analysis of acetylated peptides
Acetylated peptide identification for the 64 acetylome data files were performed as in the global proteome data analysis described above, with additional dynamic acetylation (+42.0105 Da) and carbamylation (+43.0058 Da) on Lys residues. The acetylation site localization, protein inference, and quantification of the acetylome data were performed in identical fashion as in the phosphoproteome data.
Targeted selected reaction monitoring (SRM) analysis
Stable isotope-labeled peptides
Proteotypic peptides for the target proteins were selected for targeted proteomics analysis based on the TMT identification results and well-accepted criteria 99, and the corresponding crude heavy stable isotope-labeled peptides were synthesized with 13C/15N on C-terminal lysine or arginine (New England Peptide, Gardner, MA). The heavy peptides were dissolved individually in 15% acetonitrile (ACN) and 0.1% formic acid (FA) at a concentration of 2 mM and used for creating a peptide internal standard (IS) mixture with a final concentration of 5 μMfor each peptide.
SRM assay development
The heavy peptides in the peptide mixture were evaluated for peptide response and fragmentation pattern using LC-SRM. For each peptide, transition settings were as follows: (1) precursor charges: 2, 3 and 4; (2) fragment ion charges: 1, 2 and 3; (3) ion type: y ions; and (4) m/z window: 250–1500. Transition lists were generated with optimal collision energy values by Skyline software (Version 21.2) 81. LC-SRM was then used to evaluate all heavy peptides for stability of peptide retention time, reliable heavy peptides identification, transition interferences, and endogenous peptide detectability. In the end, 3 transitions per peptide were selected for the final assay configuration for targeted quantitation of a total of 108 peptides of the 62 target proteins and 11 phosphopeptides of RB1 protein.
PRISM fractionation
High-pressure, high-resolution separations coupled with intelligent selection and multiplexing (PRISM) fractionation 100 was performed for 39 peptides to achieve higher sensitivity quantitation by SRM. A nanoACQUITY UPLC® system (Waters Corporation, Milford, MA) equipped with a reversed-phase capillary LC column (30-μm Jupiter C18 bonded particles packed in 200 μm i.d. × 50 cm capillary) was used. Separations were performed by reversed-phase LC fractionation at mobile phase flow rates of 2.2 μL/min on the binary pump systems using 10 mM ammonium formate (pH 7) in water as mobile phase A and 10 mM ammonium formate (pH 7) in 90% ACN as mobile phase B. 45 μL of sample with a peptide concentration of 0.5 μg/μL and 12 fmol/μL of heavy peptide standards was loaded onto the reversed-phase capillary column and separated into 96 fractions using a 190-min gradient of (min:%B): 35:1, 37:10, 52:15, 87:25, 112:35, 125:45, 150:90, 156:1. The eluent was automatically deposited every minute and concatenated onto 6 vials. Prior to peptide fraction collection, 20 μL of 0.1% FA was added to each well of the 96-well plate to avoid the loss of peptides. All the elute were then dried under vacuum.
Phosphopeptides enrichment by IMAC
The in-house-made IMAC tip was capped in a tip-end with a 20-μm polypropylene frits disk followed by packing with Ni-NTA silica resin (QIAGEN, Hilden, Germany). First, Ni2+ ions were removed by adding 50 mM EDTA in 1 M NaCl. The tip was then activated with 100 mM FeCl3 and equilibrated with 1% (v/v) acetic acid at pH 3.0 prior to sample loading. 1 μL of 4000 fmol/μL of each of the crude IS phosphopeptides spiked into 200 μL of 1 μg/μL tryptic peptides or into in 0.1% (v/v) trifluoroacetic acid, 80% ACN were subjected to IMAC. Followed by 1% (v/v) trifluoroacetic acid, 80% ACN, and 1% (v/v) acetic acid washing steps, the bound phosphopeptides were eluted by 200 mM NH4H2PO4 onto the activated desalting SDB-XC StageTips for desalting and directly eluted to sample vials of LC-SRM then dried under vacuum.
LC-SRM data acquisition
The digested tissue samples were reconstituted in 2% ACN/0.1% FA and spiked with 50 fmol/μL heavy peptides for a final concentration of 0.25 μg/μL, and 2 μL of the resulting samples were analyzed by LC-SRM using a Waters nanoACQUITY UPLC system coupled to a Thermo Scientific TSQ Altis triple quadrupole mass spectrometer (Thermo Scientific, Waltham, MA). A 100 μm i.d. × 10 cm, BEH 1.7-μm C18 capillary column (Waters) was operated at a temperature of 44 °C. The mobile phases were (A) 0.1% FA in water and (B) 0.1% FA in ACN. The peptide samples were separated at a flow rate of 400 nL/min using a 110-min gradient profile as follows (min:%B): 7:1, 9:6, 40:13, 70:22, 80:40, 85:95, 93:50, 94:95 and 95:1. Data were acquired in timescheduled SRM mode (retention time window: 15 min). PRISM collected fractions were reconstituted in 20 μL 2% ACN/0.1% FA and 9 μL were loaded onto the column and separated at a flow rate of 400 nL/min using a 70-min gradient profile as follows (min:%B): 11:0.5, 14: 5, 30:20, 37:25, 48:60, 31:95, 54:0.5. Data were acquired in unscheduled SRM mode. For enriched phosphopeptide samples, all the eluent from IMAC was dissolved in 12 μL and 9 μL were loaded onto the column and separated at a flow rate of 400 nL/min using a 42-min gradient profile as follows (min:%B): 11:0.5, 13: 5, 18:20, 23:25, 53:95, 36:0.5. Data were acquired in unscheduled SRM mode. The parameters of the triple quadruple instrument were set with 0.7 fwhm Q1 and Q3 resolution, and 1.2-s cycle time.
SRM data analysis
SRM data were analyzed using the Skyline software (Version 21.2) 81. The total peak area ratios of endogenous light peptides and their heavy isotope-labeled internal standards (i.e., L/H peak area ratios) were exported for quantitation. Peak detection and integration were carried out according to two criteria: (1) same retention time and (2) similar peak area ratios for the transitions. All data were manually inspected to ensure correct retention time, peak detection and accurate integration. All SRM results including the assay characterization data are organized as Skyline files on the Panorama server 101 and can be accessed via https://panoramaweb.org/CPTAC_UCEC.url (the account for reviewer is: Email: panorama+reviewer134@proteinms.net; Password: ALZsnvJc).
Quantification and analysis of glycopeptides
Mass spectrometry data from each individual cohort was downloaded from the DCC website. The data sets were annotated following their metadata information. MzML files were searched using the GPQuest search engine version 2.1 (Hu et al., 2018; Mertins et al., 2018) against a reported glycopeptide database reported in Glycositeatlas (Sun et al, 2019 ) appended with an equal number of decoy sequences and an N-linked glycan database, which was collected from the public database of GlycomeDB (Ranzinger et al., 2011) (http://www.glycome-db.org). The post-processing, statistical summarization, and reporting were done using MS-PyCloud (Li Chen, 2018, biorxiv). The consensus spectral library for each glycopeptide was constructed by using SpectraST and data visualization was done by OmicsOne (Hu et. al, 2021). Below are the individual search settings for glycoproteome analysis, including the necessary setting to account for individual experimental settings.
Searching N-linked glycopeptides using GPQuest. Prior to the database search, ProteoWizard 3.0 was used to convert the .RAW files to .mzML files with the “centroid all scans” option selected. The MS/MS spectra containing the oxonium ions (m/z 204.0966) in the top 10 abundant peaks after removing TMT reporter ions were considered as the potential glycopeptide candidates. Isotope error was set to (−1/0/1/2) for all searches. Cysteine carbamidomethylation (+57.0215) was specified as fixed modification. Methionine oxidation (+15.9949), and protein N-terminal acetylation (+42.01060) were specified as variable modifications. The minimum number of peaks used was set to 15, and the maximum set to 100. The precursor ion tolerance was set to 10 ppm and the fragment tolerance was set to 20 ppm. Lysine TMT labeling (+229.1629), and peptide N-terminal TMT labeling (+229.1629) were specified as fixed modifications. The best hits of all glycopeptide-spectrum matchings (GPSMs) were ranked by the Morpheus scores (Wenger and Coon, 2013) in descending order, in which those with FDR < 1% and covering > 10% total intensity of each tandem spectrum were reserved as qualified identifications.
Post-processing using MS-PyCloud. Glycopeptide-spectrum matchings (GPSMs) were filtered based on a user-defined PSM-level false discovery rate (FDR) cutoff and significant GPSMs from all sets of each cohort were grouped to infer the represented proteins parsimoniously using a bipartite graph analysis algorithm adopted in many protein inference tools 102–104. The final FDRs are then estimated at N-glycopeptide-levels using the reversed decoy search. For isobaric labeled data, the TMT reporter ion intensities are extracted from the mzML file for MS2 scans corresponding to the identified glycopeptides. For TMT data, the TMT reagent lot correction factors were used to adjust the reported TMT intensities for interference between TMT channels. Log2 ratios are calculated at GPSM-level relative to the user-specified reference channel and are then rolled up by the median value to N-glycopeptide-level (intact glycopeptide enriched). Normalized log2 ratio matrices are generated using median normalization (MD norm), and median normalization plus median absolute deviation scaling (MD norm + MAD scaling). Normalised levels are generated from the log2 ratio matrices by summing the log2 ratios with the median log2 value of the reference channel summed MS2 intensities across all sets for each N-glycopeptide using the same approach used in the proteome and phosphoproteome processing. Additional details regarding these steps can be found in 105,106.
IHC analysis
Immunohistochemical staining was performed on formalin-fixed paraffin-embedded 4-μm tumor sections obtained from 52 UCEC patients. Slides and appropriate positive and negative controls were then stained with primary and secondary antibodies using the automated platform, Dako Autostainer Link 48. The following primary antibodies were used: anti-c-MYC (1:100; rabbit monoclonal; clone Y69; cat. no. 790–4628; Ventana), anti-CTNNB1 (1:1000; rabbit polyclonal; cat. no. HPA029159; Atlas Antibodies), anti-LEF1 (pre-diluted, ready-to-use formulation; rabbit monoclonal; clone ZR336; cat. no. ZC-Z2642RL, Zeta Corporation). An EnVision FLEX kit (cat. no. K800221–2; Dako, Agilent Technologies, Inc.) was used to visualize antibody binding. Subsequently, IHC slides were quantified manually by H-score on a scale of 0 to 300 taking into account the percentage of positive tumor cells and staining intensity. Slides were also digitally scanned with automated ScanScope AT Turbo slide scanner (Aperio/Leica Microsystems) using 20X magnification and digital images were saved as.svs.
CRISPR editing materials and methods
Cell line
The endometrial carcinoma cell line HEC-151 was purchased from JCRB cell bank. Cells were cultivated using MEM containing 10% FBS and 1% penicillin–streptomycin in a 37 °C incubator with 5% CO2. Cell lines were routinely tested and confirmed negative for Mycoplasma contamination by Lonza Mycoalert Mycoplasma Detection Kit.
PIK3R1T576del editing
The SpCas9 2NLS nuclease and the CRISPRevolution sgRNA EZ kit were ordered from Synthego corporation. The sequence of the gRNA is: 5’ GGUCUCUCGUCUUUCUCAGC. The Homology-directed repair (HDR) donor ssODN (PAGE purified) and PCR primers was ordered from Sigma-Aldrich. The sequence of ssODN is: 5’ caattattcatgtataggattccatttcaaatacttacATCAAGTATTGGTCTCTtTTcCTgAGCTGGATAAGGTCT GGTTTAATGCTGTTCATACGTTTGTCAATTTCTCGATACT. The PCR primers used for screening edited alleles are: 5’ CCAGCTcAGgAAaAGACC (PIK3R1-T576del-F2) and 5’ gcaatcaccaattattcatg (PIK3R1-R4). The primers used to amplify the edited region for Sanger sequencing and deep sequencing are: 5’ AGAAGACTTGAAGAAGCAGG (PIK3R1-1) and 5’ aactcatcctgaattgtagc (PIK3R1-2).
2×105 HEC-151 cells resuspended in 5μl resuspension buffer (in Neon™ Transfection System 10 μL Kit, ThermoFisher) were mixed with sgRNA (90pmol), Cas9 (10pmol), and ssODN (60pmol) in 7μl resuspension buffer. 10μl of the resulting mixture was electroporated using Neon Transfection System (ThermoFisher) with the setting of 1400 volts, 10ms, and 4 pulses. Cells were then transferred into one well in a 24-well-dish and recovered in full medium for 2 days. Cells were then seeded in a 96-well-dish with 10~20 cells/well. When cells reached confluency, a screening PCR was conducted by using 10% of the cells from each well in a direct PCR assay using primers specifically amplifying the edited allele and Terra™ PCR Direct Red Dye Premix (TaKaRa). The remaining cells were expanded for further analyses later. After a positive sub-pool was identified, a Sanger sequencing and Deep sequencing further confirmed the editing. For these two assays, the 172bp fragment spanning the edited region was amplified by PCR using the cells from the positive wells, primers amplifying the 172bp spanning the edited region, and Terra™ PCR Direct Red Dye Premix. The purified PCR product was sent to Azenta Life Sciences for Sanger sequencing and Amplicon-EZ Next Generation sequencing, respectively.
Western blotting
Western blotting has been previously described 107 with a few modifications. Cell lysate protein (30μg) was loaded into each well of a 4–15% Mini-Protein TGX Precast gel (Bio-Rad). The first antibodies used were rabbit anti–phospho-AKT (Thr308) (Cell Signaling Technology, RRIRAB_329825, 1:500), rabbit anti-phospho-AKT (Ser473) (Cell Signaling Technology, RRID:AB 329825, 1:1000 dilution), rabbit anti-AKT (Cell Signaling Technology, RRID:AB_329827, 1:1000 dilution), and mouse anti-β-actin (Sigma-Aldrich, RRID:AB_476744, 1:5000 dilution).
In vitro Metformin sensitivity assays
Established UCEC cell lines were obtained from either the JCRB Cell Bank (HEC265, HEC-108, HEC-251) or the American Type Culture Collection (HEC1A) and cultured in MEM media supplemented with 15% FBS. Identity of each cell line was verified by a PCR-based method for Short Tandem Repeat (STR) profiling prior to receipt. Once thawed, cell lines were cultured for fewer than 20 passages and regularly screening for mycoplasma infection by two independent methods: MycoStrip (Rep-mys-10; Invivogen) and RT-PCR (Venor GeM, Cat MP00025–1KT; Sigma-Millipore). To assess metformin sensitivity, all cell lines were serum starved for 24 hours before being aliquoted in 96-well plates and incubated in MEM supplemented with 5% FBS and metformin at specified doses. Metformin (PHR 1085, Sigma Millipore) was prepared fresh in MEM media for each use. Cell counts were quantified using Cell Titer96 reagent (Promega; Cat# G3580) as previously described (Delaney MA, et al, 2017).
Gene expression was evaluated using validated Taqman assays specific for c-myc (Hs00153408_m1) as well as putative myc-regulated targets identified by our interrogation of DEPMAP data: NOP2 (Hs00999660_m1), NOP16 (Hs00212622_m1), NOLC-1 (Hs01102319_g1); WDR74 (Hs00276510_m1), PES1 (Hs00362795_g1), IMP4 (Hs00369187), GRWD1 (Hs00230365_m1) and TBRG4 (Hs01056260_g1). Total RNA was prepared from flash frozen pellets of washed cells using the MirVANA kit (AM1561; Fisher Scientific) as previously described (Delaney MA, et al, 2017). Relative levels of transcript were quantified by the delta-deltaCT method (Livak KJ and Schmittgen TD, 2001).
Western blotting was used to assess levels of expression for proteins of interest as described (Delaney MA, et al, 2017) with the following modifications. Thirty micrograms of cell lysate was loaded into each well of a 4–12% polyacrylamide gel (Invitrogen) and transferred to PVDF membranes. Membranes were then incubated with primary antibody for c-myc (Cat#E5Q6W, Cell Signaling; 1:1000 dilution;). Secondary HRP-conjugated anti-rabbit antibody (Cat#7974S; Cell Signaling) was used at 1:3000 dilution. Immunoreactivity was visualized using ECL Western PICO (Fisher Scientific). Specimen-specific immunoreactivity for GADPH (Cat#G9545; Sigma-Millipore; 1:3000 dilution), was used as a loading control. Results are reported as levels of luminescence normalized to loading control for each lane of interest. Results are reported as relative levels of expression normalized to baseline/controls.
Both a previously validated E-box luciferase reporter vector (pGL2, pGL2-E4) and CMV-beta-gal vector were obtained from Dr. Rosalie Sears (Oregon Health and Science University) and used to evaluate c-myc transcriptional activity in vitro as described (Hurlin, PJ, et al, 1997). In brief, 3 × 105 cells from each UCEC cell line were co-transfected with either 200 ng of pGL2 (non-expressing control) or pGL2-E4 and 100 ng b-galactosidase reporter plasmid using lipofectamine 2000 (Cat#: 11668027, ThermoFisher) in Opti-MEM media (Cat#31985062, ThermoFisher). After 5 minutes, media bathing transfected cells were replaced and cells incubated for 24 hours. Each transfected culture was divided and used to measure either luciferase (E1500, Promega) or b-galactosidase activity (E2000, Promega) using a manufacturer recommended protocol. After subtracting background, results are reported as luciferase activity normalized to b-galactosidase activity for each cell pellet.
For all studies, statistical significance was assessed using Student’s T-test, with significance set at p<0.05.
Other proteogenomic analyses
Mutation impact on the proteome
To assess the impact of somatic mutations on protein and phosphorylation levels, we performed cis/trans analysis as detailed in https://github.com/PayneLab/GBM_confirmatory. Briefly, samples were segregated into wildtype or mutation for a specific gene. Cis analysis then performed a Wilcoxon rank sum test to determine whether the samples with a mutation had a significantly different protein level for the mutated gene. Trans analysis was similar, with samples separated into wildtype and mutated. Then all proteins were tested to see if a significantly different level was observed (with a BH corrected FDR of 5%). See the GitHub repository for explicit code used in the manuscript.
APM subgroup prediction
The R package caret (v6.0.88) 86 was used to train and predict APM status using SRM data. There are 35 TMB high samples in the Exploratory cohort with 33 features in total. Among them, there are 20 positive samples (TMB_H/APM_H) and 15 negative samples (TMB_H/APM_L). The buil-tin backwards feature selection method, recursive feature elimination, was used to sort the 33 features based on their importance with 10-fold cross-validation and 5 times repeating. After that, 10 different classic machine learning methods with default parameters were used to train the classification model on the Exploratory cohort by adding peptides one at a time, starting with the peptide with the highest feature importance. These methods are AdaBoost, Bayesian Generalized Linear Model (Bayesglm), Logistic Model Trees (LMT), Boosted Logistic Regression (LogitBoost), Penalized Multinomial Regression (Multinom), Oblique Random Forest (ORFlog), Penalized Logistic Regression (plr), Random Forest (ranger, rf), and Regularized Random Forest (RRF). Among them, LMT, LogitBoost and plr are three different versions of logistic regression. ORFlog, parRF, ranger, rf and RRF are five different versions of random forest. Then, the trained models were applied to the independent cohort and AUC values were used to evaluate the performances of these methods.
Inferred Immune, MYC, APM, and Wnt pathway scores
All scores were inferred by single sample gene set enrichment analysis (ssGSEA) method from the GSVA R package 62,108. The immune signatures are from 109, MYC hallmark gene sets were used as MYC signature, and the KEGG antigen processing and presentation pathway gene set is used as the APM signature. The KEGG Wnt Signaling Pathway gene set was used to analyze the Wnt pathway signatures for our CTNNB1-mutated and WT tumors. mRNA expression was used to infer MYC and immune scores and protein level was used to infer the APM score.
CTNNB1 hotspot mutation prediction
The glm.net package in R was used to train and test lasso regression models to classify hotspot CTNNB1 mutations. Protein data from the exploratory CPTAC UCEC cohort (Dou et al. Cell 2020) was used for training and the current independent dataset was used as testing. Four lasso regression models were run using (1) all samples and all available proteins, (2) CNV-low samples only and all available proteins, (3) all samples and Wnt-β-catenin signaling genes (MSigDB), and (4) CNV-low samples and Wnt-β-catenin signaling genes. Before training the models, feature selection was performed using a two-sided t-test to determine top 10 genes from either all available proteins or Wnt-β-catenin signaling geneset. The top ten genes were then used in training and testing models to classify hotspot mutation or WT tumors. ROC curves were plotted using the pROC package in R (https://cran.r-project.org/web/packages/pROC/index.html).
PANOPLY analysis
Proteomics data – including global proteome, phosphoproteome, acetylome and glycoproteome – along with RNAseq gene expression and CNV data from the V3.0 data freeze were reformatted into GCT files (https://clue.io/connectopedia/gct_format) and input to PANOPLY v1.1. PANOPLY 110 is a cloud-based platform for automated and reproducible proteogenomic data analysis implementing a wide array of algorithms.
In the first round of analysis, we focused on multi-omic clustering using all the proteomics, RNAseq and CNV data as input. Based on preliminary exploration, the number of clusters kmin was set to 4. Clustering was performed using all features from all -omes (balance_omes=false), filtered to exclude 5% of features globally with the lowest standard deviation (sd_filt=0.05 with filt_mode=global).
Once the clusters were defined, over-representation analysis was performed to determine enrichments for various selected sample annotation terms in the clusters using Fisher’s exact test on the set of samples forming the cluster core. Functional characterization of the clusters was performed using gene set enrichment analysis (GSEA) on the matrix of signed multi-omic feature weights (W). Details are available in the PANOPLY documentation Wiki (https://github.com/broadinstitute/PANOPLY/wiki).
Subsequently, the sample annotation was updated to include the multi-omic cluster ID for each sample. This updated sample annotation along with the proteogenomics datasets previously described are then used as input to a subsequent run of the PANOPLY pipeline to perform immune analysis, outlier analysis, association analysis and connectivity map analysis among others. Each analysis outputs a report summarizing results in addition to detailed plots and tables.
Neural networks for subtype and mutation predictions
Digital whole slide images (WSI) stained with hematoxylin and eosin (H&E) were obtained as SVS files. TCGA, CPTAC Exploratory (Dou et al. 2020), and CPTAC independent cohort images were obtained from the The Cancer Imaging Archive (TCIA). NYU slides were obtained from the NYU Center for Biospecimen Research & Development (CBRD, RRID:SCR_017930). Slides were scanned at either 40x (TCGA cohort) or 20x (CPTAC and NYU cohorts). Due to the size and the multi-resolution data structure of the WSI, the Python package, Openslide, was used to cut slides into 299 × 299 pixel tiles at 10x, 5x, and 2.5x equivalent magnification of the scanned WSI. 10x, 5x, and 2.5x tiles covering the same regions were then grouped into tile sets and were treated as 1 sample for multi-resolution models following the Panoptes sample preparation (Hong et al Cell Rep Med 2021, PMID: 34622237). In short, tiles were excluded if more than 40% of the pixels are of white background and irrelevant contaminants. Stain colors were normalized using the method described in Vahadane et al. 2016 (PMID: 27164577). Labels for each task were one-hot encoded at per-tile level. Multi-resolution and single-resolution convolutional neural network (CNN) imaging models were trained on TCGA and CPTAC exploratory cohorts, which were randomly split into training, validation, and testing at a per-patient ratio of 8:1:1. The CPTAC independent dataset was used for completely independent testing of the trained models.
For each task, 13 model architectures were applied to the data. These include Inception V1, V2, V3, InceptionResnet V1, V2, and Panoptes 1, 2, 3, 4 with and without a clinical variable branch (I1, I2, I3, I5, I6, X2, X1, X4, X3, X2CF, X1CF, X4CF, X3CF, respectively). Inception and InceptionResnet are established deep CNN classifiers (Szegedy et al. 2017, DOI: https://doi.org/10.1609/aaai.v31i1.11231). The Panoptes architecture is a multi-resolution InceptionResnet-based CNN architecture that takes in three tiles of the same region on each slide (Hong et al 2021, PMID: 34622237). Panoptes1 and Panoptes2 have three branches based on InceptionResnet1 and InceptionResnet1, respectively. Panoptes3 and Panoptes4 use the same architecture as Panoptes1 and 2, respectively, and contain an additional 1-by-1 convolutional layer between the concatenation of branches and the global average pooling. Models with an added clinical feature layer were fed patient age and BMI (Panoptes models + CF).
Receiver Operating Characteristic (ROC) curve, plotting true positive rate against false positive rate, and the area under the ROC curve (AUROC), were calculated using the R package PRROC (https://cran.r-project.org/web/packages/PRROC/index.html). 95% confidence intervals were calculated using a 2000 times bootstrap method by randomly sampling true labels and prediction probabilities. Mean AUROC with 95% confidence intervals were plotted for the top performing internal split test architectures per task, top performing internal test architecture for the independent test, and the top performing architecture for the independent test (Figure 6A).
The best performing models were selected based on the highest AUROC at the per-slide (patient) level. For visualization, activations of 20,000 randomly sampled tiles of the second-to-the-last layer of the test set were extracted and dimensionally reduced and plotted with tSNE for feature visualization. Example tiles were highlighted and sent to pathologists for secondary review. Selected whole slide cases from the test set were fed into the trained model and per-tile level predictions were aggregated into a heatmap layer to overlay onto the original slides for feature visualization and localization.
To correct for multiple testing, we adapted the method used by Kather et al. 2020111, and included a list of p-values and false discovery rates (FDR, with Benjamini-Hochberg correction) for top performing models at the slide level (Table S7). The p-values were obtained from one-sided ttests between the probability values of patients in the positive class and the negative class, for each corresponding model and test datasets.
To address the limited sample size for the POLE subtype, we obtained 13 positive POLE slides from the NYU Center for Biospecimen Research & Development (CBRD), as well as 78 non-POLE slides to control for batch effect. We did not obtain clinical data for NYU samples other than EC genomic subtype, and therefore could not test or train with any of the CF model architectures. We first tested the X4 architecture trained on TCGA and CPTAC exploratory on the NYU POLE positive samples and CPTAC independent samples, labeled “POLE + NYU Test”. We then trained new models with TCGA, CPTAC exploratory, and all NYU samples, and then applied to the current independent cohort for testing across 4 Panoptes architectures, labeled “POLE + NYU Train”.
Antigen Presentation Machary (APM) status models were trained using only CPTAC cohorts due to the lack of proteomic data in the TCGA dataset. APM status was derived from a proteomic based scoring metric. We therefore tested two modeling methods for the APM classifier: (1) training on the exploratory cohort and testing on independent, labeled “APM-high”, and (2) training on the combined exploratory and independent cohorts and testing on the internal split, labeled “APM-high Mixed”.
Transporters score
Determining the most distinct separation of tumors into distinct classes along a given scoring axis was completed by Inferred Pathway Activity Score (IPAS) (https://doi.org/10.1101/2020.10.19.345629). Pathway activity scores from proteome profiles of EC tumors were determined by using a collection of 8,600 pathways and cell type signatures from various databases: KEGG 112, Reactome 112,113, HMDB 114, SMPDB 114,115, Hallmark 116, WikiPathways 117, BioPlanet 118, BioCarta 119, xCell 120, Tabula Sapiens 121. Each pathway score was treated as a dimension and the tumors were ranked along the axis by increasing scoring value. At each point i of the axis, geometrically averaged P values were computed between distributions of individual protein levels in class [1,i] and class [i+1,N], (where N is a number of tumors) and determined the minimum of the geometrically averaged P value. It is assumed that the minimal value the averaged P determines the most distinct separation of given molecular profiles (tumors) along a given axis. Mann Whitney Wilcoxon test was used for computing individual P values.
To assess significance of the obtained optimal classes of tumors, 10,000 randomly formed tumor classes of the same sizes were generated. For each randomly formed tumor class, the combined P values 122,123 and the actual histograms of P values were computed and compared to combined P values and P value histograms of the optimal classes. Both the number of times combined P values of randomly selected formed tumor classes were less than the combined P values of original classes and the number of times individual P values of a certain power (eg. P < 10−6) in random classes were less than P values of the same power in original classes were obtained. The obtained numbers were divided by the number of tests (104), and significance was assessed of the obtained optimal classes 123. Mutation enrichment in a specific class (eg. Transporters) was performed by Fisher test.
SRM data batch effect correction
The ICI treated samples are formalin-fixed paraffin-embedded specimens which are different to the fresh frozen method used by CPTAC. Combat method implemented in the R package ez.combat was used to do the batch effect correction with default parameters 87. Peptides with more than 50% NAs were removed, and then remaining NAs were set to the minimum values of non-NA numbers. After batch effect correction, these values were reset to NAs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis and visualization was done with a diverse set of methods depending on which data modality was analyzed. The methods are described in the methods details section. When multiple tests were performed p-values were corrected for multiple testing using the Benjamini-Hochberg method to control for the false discovery rate.
Supplementary Material
Table S1. Genotype, phenotype, immune scores, clinical, and pathological characteristics of tumor and normal samples. Related to Figure 1.
Table S2. Mutation signatures, CNV drivers, CNV arm immune score correlations, and PARP-i drug sensitivity data. Related to Figures 1 and 6.
Table S3. PIK3R1 mutation status, AKT inhibitor response of EC cell lines, and sequencing of CRISPR-Cas9 edited cells. Related to Figure 2.
Table S4. Immune subgroup, immune scores and SRM peptide levels for ICI treated samples. Related to Figure 3.
Table S5. Inferred MYC activity in CMAP, TCGA, Exploratory and Independent cohorts, total MYC IHC scores. Related to Figure 4.
Table S6. Transporters scores and classes, CTNNB1 mutation protein fold change, and top classifying genes for hotspot mutation. Related to Figure 5.
Table S7. Deep learning models, multi-omic NMF clustering, and glycopeptide NMF clustering. Related to Figures 6 and 7.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti–phospho-AKT (S473) | Cell Signaling | Catalog: 9271 |
| anti–phospho-AKT (T308) | Cell Signaling | Catalog: 9275 |
| Anti- GAPDH | Sigma | Catalog: G9545 |
| anti-myc | Cell Signaling | Catalog: E5Q6W |
| rabbit anti-AKT | Cell Signaling | Catalog: 9272 |
| anti–β-actin | Sigma-Aldrich | Catalog: A5441 |
| HRP-conjugated anti-rabbit antibody | Cell Signaling | Catalog: 7074S |
| Biological Samples | ||
| Primary tumor and normal tissue samples | This paper | N/A |
| Patient-derived xenograft tissue samples | Washington University in St. Louis | Dou et al.9 |
| Chemicals, peptides, and recombinant proteins | ||
| 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid | Sigma | Catalog: H3375 |
| Ammonium bicarbonate | Sigma | Catalog: 9830 |
| Ammonium formate | Sigma | Catalog: 9735 |
| Aprotinin | Sigma | Catalog: A6103 |
| Calcium chloride | Sigma | Catalog: C1016 |
| Dithiothreitol | Thermo Scientific | Catalog: 20291 |
| Ethylenediaminetetraacetic acid | Sigma | Catalog: E7889 |
| Fetal Bovine Serum | Sigma | Catalog: F7524 |
| Formic acid | Sigma | Catalog: 33015 |
| Iodoacetamide | Thermo Scientific | Catalog: A3221 |
| Iron (III) chloride | Sigma | Catalog: 451649 |
| Leupeptin | Roche | Catalog: 11017101001 |
| Lipofectamine 2000 | Invitrogen | Catalog: 52785 |
| Lysyl Endopeptidase | Wako Chemicals | Catalog: 129–02541 |
| Metformin | Sigma | Catalog: PHR 1084 |
| MOPS buffer | Invitrogen | Catalog: NP0001 |
| Ni-NTA Superflow Agarose Beads | Qiagen | Catalog: 30410 |
| OptiMem Media | ThermoFisher | Catalog: 31985062 |
| Phenylmethylsulfonyl fluoride | Sigma | Catalog: 93482 |
| Phosphatase Inhibitor Cocktail 2 | Sigma | Catalog: P5726 |
| Phosphatase Inhibitor Cocktail 3 | Sigma | Catalog: P0044 |
| Polyvinylidene fluoride membrane (PVDF) | Invitrogen | Catalog: IB401002 |
| Sequencing grade modified trypsin | Promega | Catalog: V517 |
| Sodium butyrate | Sigma | Catalog: 303410 |
| Sodium chloride | Sigma | Catalog: S7653 |
| Sodium fluoride | Sigma | Catalog: S7920 |
| Urea | Sigma | Catalog: U0631 |
| Acetonitrile, Optima LC/MS | Fisher Chemical | Catalog: A955–4 |
| Water, Optima LC/MS | Fisher Chemical | Catalog: W6–4 |
| Trifluoroacetic acid | Sigma | Catalog: 302031 |
| Triethylammonium acetate buffer | Sigma | Catalog: 90358 |
| MEM | Corning | Catalog: 10–010-CV |
| Critical Commercial Assays | ||
| Beta-Galactosidase Assay System | Promega | Catalog: P2000 |
| BCA Protein Assay Kit | Thermo Scientific Pierce | Catalog: A53225 |
| Cell Titer 96 Assay Reagents | Promega | Catalog: G3580 |
| Chemiluminescence PICO Assay Kit | Thermo Fisher | Catalog: 34578 |
| Luciferase Assay System | Promega | Catalog: E1500 |
| PTMScan Acetyl-Lysine Motif [Ac-K] Kit | Cell Signaling | Catalog: 13416 |
| Reversed-phase tC18 SepPak | Waters | Catalog: WAT054925 |
| Tandem mass tags - 11plex | Thermo Scientific | Catalog: A34808 |
| Taqman Assay - MYC | Fisher Scientific | Hs00153408_m1 |
| Taqman Assay - NOP2 | Fisher Scientific | Hs00099660_m1 |
| Taqman Assay - NOP16 | Fisher Scientific | Hs00212612 _m1 |
| Taqman Assay - NOLC1 | Fisher Scientific | Hs01102319_g1 |
| Taqman Assay - WDR74 | Fisher Scientific | Hs00276510_m1 |
| Taqman Assay - PES1 | Fisher Scientific | Hs00362795_g1 |
| Taqman Assay - IMP4 | Fisher Scientific | Hs00369187_m1 |
| Taqman Assay - GRWD1 | Fisher Scientific | Hs00230365_m1 |
| Taqman Assay - TBRG4 | Fisher Scientific | Hs01056260_g1 |
| Taqman Universal Master Mix | Applied Biosystems | Catalog: 444038 |
| Oasis MAX 1 cc Vac Cartridge, 10 mg Sorbent per Cartridge | Waters | Catalog: 186004649 |
| Mycoalert Mycoplasma Detection Kit | Lonza | Catalog: LT07–318 |
| MycoStrip Test Kit | Invivogen | Catalog: Rep-mys-10 |
| MycoPlasma RT-PCR Test Kit | Venor GEM | MP00025 |
| SpCas9 nuclease 2NLS | Synthego | SpCas9 2NLS Nuclease (300 pmol) |
| CRISPRevolution sgRNA EZ kit | Synthego | Customized |
| Neon Transfection System 10 μL Kit | ThermoFisher | Catalog: MPK1025 |
| Neon Transfection System | ThermoFisher | Catalog: MPK5000 |
| Terra PCR Direct Red Dye Premix | TaKaRa | Catalog: 639270 |
| Mini-Protein TGX Precast gel | Bio-Rad | Catalog: 4561086 |
| Deposited data | ||
| CPTAC Raw Genomic data | This paper and Dou et al.9 | dbGaP phs001287 |
| CPTAC Raw Proteomic data | This paper and Dou et al.9 | PDC000125, PDC000126, PDC000226 |
| CPTAC and TCGA Processed data tables | This paper, Dou et al.9, and The Cancer Genome Atlas et al.8 | http://www.linkedomics.org/ |
| CPTAC Image data | This paper and Dou et al.9 | 10.7937/k9/tcia.2018.3r3juisw https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ucec |
| TCGA Raw Genomic data | The Cancer Genome Atlas et al.8 | dbGaP phs000178 |
| TCGA Image data | The Cancer Genome Atlas et al.8 | 10.7937/k9/tcia.2016.gkj0zwac https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=tcga_ucec |
| CCLE DepMap | Corsello et al.17 | https://depmap.org/portal/ |
| Experimental models: Cell lines | ||
| HEC151 UCEC cell line | JCRB cell bank | JCRB1122 |
| HEC251 UCEC cell line | JCRB cell bank | JCRB1141 |
| HEC265 UCEC cell line | JCRB cell bank | JCRB1142 |
| HEC108 UCEC cell line | JCRB cell bank | JCRB1123 |
| HEC1A UCEC cell line | American Type Culture Collection | HTB-112 |
| Oligonucleotides | ||
| CRISPRevolution sgRNA: 5’GGUCUCUCGUCUUUCUCAGC | This paper, Synthego | N/A |
| Homology-directed repair (HDR) donor ssODN: 5’caattattcatgtataggattccatttcaaatac ttacATCAAGTATTGGTCTCTtTTcC TgAGCTGGATAAGGTCTGGTTTAATGCTGTTCATACGTTTGTCAATTTCTCGATACT | This paper, Sigma-Aldrich | N/A |
| PCR primers for PIK3R1 edited alleles: 5’CCAGCTcAGgAAaAGACC (PIK3R1-T576del-F2) and 5’gcaatcaccaattattcatg (PIK3R1-R4) | This paper, Sigma-Aldrich | N/A |
| Sanger sequencing primers for PIK3R1 edited alleles: 5’AGAAGACTTGAAGAAGCAGG (PIK3R1–1) and 5’aactcatcctgaattgtagc (PIK3R1–2) | This paper, Sigma-Aldrich | N/A |
| Software and Algorithms | ||
| Ascore (v1.0.6858) | Beausoleil et al.54 | https://github.com/PNNL-Comp-Mass-Spec/AScore/releases |
| BlackSheep | Blumenberg et al.55 | https://github.com/ruggleslab/blackSheep |
| Bowtie2 (v2.3.3) | Langmead et al.56 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| BWA (v0.7.17-r1188) | Li et al.57 | http://bio-bwa.sourceforge.net/ |
| CIRI (v2.0.6) | Gao et al.58 | https://sourceforge.net/projects/ciri/ |
| cptac (Python package) | Lindgren et al.59 | https://paynelab.github.io/cptac/ |
| eriscript (v0.5.5) | Benelli et al.60 | https://sites.google.com/site/bioericscript/ |
| GPQuest (v2.1) | Toghi Eshghi et al.61 | https://github.com/huizhanglabjhu/GPQuest |
| GSVA (v1.30.0) | Hänzelmann et al.62 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| INTEGRATE v(0.2.6) | Zhang et al.63 | https://sourceforge.net/p/integrate-fusion/wiki/Home/ |
| LinkedOmics | Vasaikar et al.64 | http://www.linkedomics.org |
| MASIC | Monroe et al.65 | https://github.com/PNNL-Comp-Mass-Spec/MASIC/releases |
| metaX | Wen et al.66 | https://github.com/wenbostar/metaX |
| MS-GF+ | Kim et al.67 | https://github.com/MSGFPlus/msgfplus/releases |
| MSIsensor (v0.2) | Niu et al.68 | https://github.com/ding-lab/msisensor |
| MS-PyCloud | Chen et al.69 | https://bitbucket.org/mschnau1/mspycloud/src/main/ |
| MuSiC (v0.4) | Dees et al.70 | http://gmt.genome.wustl.edu/packages/genome-music/index.html |
| Mutect (v1.1.7) | Cibulskis et al.71 | https://software.broadinstitute.org/cancer/cga/mutect |
| mzRefinery | Gibbons et al.72 | https://omics.pnl.gov/software/mzrefinery |
| Optitype | Szolek et al.73 | http://github.com/FRED-2/OptiType |
| PepQuery | Wen et al.74 | http://pepquery.org |
| Maftools (2.10.0) | Mayakonda et al.75 | https://github.com/PoisonAlien/maftools |
| Pindel (v0.2.5) | Ye et al.76 | http://gmt.genome.wustl.edu/packages/pindel/ |
| PROGENy | Schubert et al.77 | https://bioconductor.org/packages/release/bioc/html/progeny.html |
| pyQUILTS (v1.0) | Ruggles et al.78 | http://quilts.fenyolab.org |
| RSEM (v1.3.1) | Li et al.79 | https://deweylab.github.io/RSEM/ |
| SigProfilerMatrixGeneratorR (v1.0) | Tan et al.80 | https://github.com/AlexandrovLab/SigProfilerMatrixGeneratorR |
| Skyline (v 21.2) | MacLean et al.81 | https://skyline.ms/project/home/software/Skyline/begin.view |
| STAR-Fusion (v1.6.0) | Haas et al.82 | https://github.com/STAR-Fusion/STAR-Fusion/wiki |
| Strelka (v2) | Saunders et al.83 | https://github.com/Illumina/strelka |
| Varscan (v2.3.8) | Koboldt et al84 | http://varscan.sourceforge.net/ |
| XGboost (v0.81) | Chen et al.85 | https://xgboost.readthedocs.io/en/latest/ |
| Caret (v6.0.88) | Kuhn et al.86 | https://topepo.github.io/caret/ |
| ez.combat | Johnson et al.87 | https://github.com/TKoscik/ez.combat |
| Other | ||
| CPTAC Data Portal | Edwards et al.88 | https://cptac-data-portal.georgetown.edu/cptacPublic |
| CTdatabase | Chen et al.89 | http://www.cta.lncc.br |
| dbGaP | Tryka et al.90 | https://www.ncbi.nlm.nih.gov/gap/ |
| Genomic Data Commons | Grossman et al.91 | https://gdc.cancer.gov |
| RefSeq (downloaded from UCSC Genome Browser 2018/06/29) | O’Leary et al.92 | https://www.ncbi.nlm.nih.gov/refseq/; https://genome.ucsc.edu/cgibin/hgTables; RRID:SCR_003496 |
| UniProt (r2017.06, r2019.01) | UniProt Consortium93 | https://www.uniprot.org/; RRID: SCR_002380 |
| BioRender | BioRender | https://www.biorender.com/ |
HIGHLIGHTS.
APM status can be accurately predicted using a targeted proteomic assay
MYC activity is a potential biomarker for metformin treatment
PIK3R1 in-frame indels are associated with upregulated AKT1 phosphorylation
CTNNB1 exon 3 hotspot mutations block pS45 induced 𝛃-catenin degradation
ACKNOWLEDGEMENTS
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is supported by the National Cancer Institute of the National Institutes of Health under award numbers U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, U01CA214125, U24CA271012, U24CA271076, U24CA271114, and contracts from Leidos (S21-167 and 21X164Q); by NCI grant T32CA203690; by grant RR160027 from the Cancer Prevention & Research Institutes of Texas (CPRIT); and by funding from the McNair Medical Institute at The Robert and Janice McNair Foundation. B.Z. and M.E. are Cancer Prevention & Research Institutes of Texas Scholars in Cancer Research and McNair Medical Institute Scholars. The Pacific Northwest National Laboratory (PNNL) proteomics work described herein was performed in the Environmental Molecular Sciences Laboratory (grid.436923.9), a US Department of Energy (DOE) National Scientific User Facility located at PNNL in Richland, WA. PNNL is a multi-program national laboratory operated by the Battelle Memorial Institute for the DOE under contract DE-AC05-76RL01830. This work was also supported by a US Department of Defence funding DOD BC220523.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
CONSORTIA
Eunkyung An, Matthew L. Anderson, Andrzej Antczak, Meenakshi Anurag, Rebecca C. Arend, Thomas Bauer, Jasmin Bavarva, Chet Birger, Michael J. Birrer, Melissa Borucki, Wen Bu, Shuang Cai, Anna Calinawan, Steven A. Carr, Patricia Castro, Sandra Cerda, Daniel W. Chan, Lijun Chen, David Chesla, Arul M. Chinnaiyan, Rosalie K. Chu, Marcin P. Cieslik, Sandra Cottingham, Andrzej Czekański, Teresa Davoli, Deborah DeLair, Elizabeth G. Demicco, Kelly Devereaux, Saravana M. Dhanasekaran, Rajiv Dhir, Li Ding, Marcin J. Domagalski, Peter Dottino, Yongchao Dou, Bailee Dover, Brian J. Druker, Elizabeth Duffy, Nathan J. Edwards, Robert Edwards, Matthew J. Ellis, Jennifer Eschbacher, Mina Fam, David Fenyö, Brenda Fevrier-Sullivan, Thomas L. Fillmore, McKenzie Foxall, Jesse Francis, John Freymann, Stacey Gabriel, Gad Getz, Michael A. Gillette, Andrew K. Godwin, Charles A. Goldthwaite, Pamela Grady, Marina A. Gritsenko, Xiaofang Guo, Jason Hafron, Pushpa Hariharan, Catherine E. Hermann, Tara Hiltke, Barbara Hindenach, Katherine A. Hoadley, Runyu Hong, Galen Hostetter, Yingwei Hu, Jasmine Huang, Michael M. Ittmann, Scott D. Jewell, Ashlie Johnson, Isabelle Johnson, Corbin D. Jones, Marcin Jędryka, Andrea G. Kahn, Lizabeth Katsnelson, Karen A. Ketchum, Justin Kirby, Iga Kolodziejczak, Amy T. Ku, Chandan Kumar-Sinha, Paweł Kurzawa, Alexander J. Lazar, Rossana Lazcano, Toan Le, Jonathan T. Lei, Yi Li, Yuxing Liao, Tung-Shing M. Lih, Tai-Tu Lin, Tao Liu, Wenke Liu, Rita Jui-Hsien Lu, Avi Ma’ayan, Rashna Madan, D. R. Mani, Sailaja Mareedu, John A. Martignetti, Ramya P. Masand, Rafał Matkowski, Peter B. McGarvey, Wilson McKerrow, Mehdi Mesri, Francesmary Modugno, Matthew E. Monroe, Rebecca Montgomery, Jamie Moon, Ronald J. Moore, David Mutch, Michael D. Nestor, Alexey I. Nesvizhskii, Chelsea Newton, Kristen Nyce, Tatiana Omelchenko, Gilbert S. Omenn, Amanda G. Paulovich, Samuel H. Payne, Vladislav A. Petyuk, Barbara L. Pruetz, Liqun Qi, Boris Reva, Shannon Richey, Ana I. Robles, Karin D. Rodland, Henry Rodriguez, Kelly V. Ruggles, Dmitry Rykunov, Sara R. Savage, Eric E. Schadt, Athena A. Schepmoes, Tujin Shi, Zhiao Shi, Yvonne Shutack, Shilpi Singh, Michael Smith, Richard D. Smith, Jimin Tan, Darlene Tansil, Mason Taylor, Ratna R. Thangudu, Mathangi Thiagarajan, Matt Tobin, Chia-Feng Tsai, Ki Sung Um, Negin Vatanian, Joshua M. Wang, Pei Wang, Yi-Ting Wang, Alex Webster, Karl K. Weitz, Bo Wen, C. M. Williams, George D. Wilson, Maciej Wiznerowicz, Jason Wright, Yige Wu, Matthew A. Wyczalkowski, Xinpei Yi, Kakhaber Zaalishvili, Bing Zhang, Hui Zhang, Xu Zhang, Zhen Zhang, Grace Zhao, Rui Zhao
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Minihan AK, Patel AV, Flanders WD, Sauer AG, Jemal A, and Islami F. (2022). Proportion of Cancer Cases Attributable to Physical Inactivity by US State, 2013–2016. Med. Sci. Sports Exerc. 54, 417–423. [DOI] [PubMed] [Google Scholar]
- 2.Crosbie EJ, Kitson SJ, McAlpine JN, Mukhopadhyay A, Powell ME, and Singh N. (2022). Endometrial cancer. Lancet 399, 1412–1428. [DOI] [PubMed] [Google Scholar]
- 3.Zhang S, Gong T-T, Liu F-H, Jiang Y-T, Sun H, Ma X-X, Zhao Y-H, and Wu Q-J (2019). Global, Regional, and National Burden of Endometrial Cancer, 1990–2017: Results From the Global Burden of Disease Study, 2017. Front. Oncol. 9, 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Temkin SM, Kohn EC, Penberthy L, Cronin KA, Rubinsak L, Dickie LA, Minasian L, and Noone A-M (2018). Hysterectomy-corrected rates of endometrial cancer among women younger than age 50 in the United States. Cancer Causes Control 29, 427–433. [DOI] [PubMed] [Google Scholar]
- 5.Clarke MA, Devesa SS, Hammer A, and Wentzensen N. (2022). Racial and Ethnic Differences in Hysterectomy-Corrected Uterine Corpus Cancer Mortality by Stage and Histologic Subtype. JAMA Oncol 8, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khouri OR, Frey MK, Musa F, Muggia F, Lee J, Boyd L, Curtin JP, and Pothuri B. (2019). Neoadjuvant chemotherapy in patients with advanced endometrial cancer. Cancer Chemotherapy and Pharmacology 84, 281–285. 10.1007/s00280-019-03838-x. [DOI] [PubMed] [Google Scholar]
- 7.van den Heerik ASVM, Horeweg N, Nout RA, Lutgens LCHW, van der Steen-Banasik EM, Westerveld GH, van den Berg HA, Slot A, Koppe FLA, Kommoss S, et al. (2020). PORTEC-4a: international randomized trial of molecular profile-based adjuvant treatment for women with high-intermediate risk endometrial cancer. Int. J. Gynecol. Cancer 30, 2002–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindeboom RGH., Vermeulen M., Lehner B., and Supek F. (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan TL, and Cantley LC (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, and Bell DW (2011). PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 71, 4061–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song M, Bode AM, Dong Z, and Lee M-H (2019). AKT as a Therapeutic Target for Cancer. Cancer Res. 79, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 15.Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, Manzella L, and Vigneri P. (2021). AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 12, 662232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myers AP, Konstantinopoulos PA, Barry WT, Luo W, Broaddus RR, Makker V, Drapkin R, Liu J, Doyle A, Horowitz NS, et al. (2020). Phase II, 2-stage, 2-arm, PIK3CA mutation stratified trial of MK-2206 in recurrent endometrial cancer. Int. J. Cancer 147, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, et al. (2020). Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 1, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Donnell JS, Long GV, Scolyer RA, Teng MWL, and Smyth MJ (2017). Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 52, 71–81. [DOI] [PubMed] [Google Scholar]
- 19.Jenkins RW, Barbie DA, and Flaherty KT (2018). Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, He Z, Wang X, Li H, and Liu X-S (2019). Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 8. 10.7554/eLife.49020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, and Kurzrock R. (2017). Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 16, 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samstein RM., Lee C-H., Shoushtari AN., Hellmann MD., Shen R., Janjigian YY., Barron DA., Zehir A., Jordan EJ., Omuro A., et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu KH, and Broaddus RR (2020). Endometrial Cancer. N. Engl. J. Med. 383, 2053–2064. [DOI] [PubMed] [Google Scholar]
- 25.Friberg E, Orsini N, Mantzoros CS, and Wolk A. (2007). Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia 50, 1365–1374. [DOI] [PubMed] [Google Scholar]
- 26.Saed L, Varse F, Baradaran HR, Moradi Y, Khateri S, Friberg E, Khazaei Z, Gharahjeh S, Tehrani S, Sioofy-Khojine A-B, et al. (2019). The effect of diabetes on the risk of endometrial Cancer: an updated a systematic review and meta-analysis. BMC Cancer 19, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meireles CG, Pereira SA, Valadares LP, Rêgo DF, Simeoni LA, Guerra ENS, and Lofrano-Porto A. (2017). Effects of metformin on endometrial cancer: Systematic review and meta-analysis. Gynecol. Oncol. 147, 167–180. [DOI] [PubMed] [Google Scholar]
- 28.Tong X-P, Chen Y, Zhang S-Y, Xie T, Tian M, Guo M-R, Kasimu R, Ouyang L, and Wang J-H (2015). Key autophagic targets and relevant small-molecule compounds in cancer therapy. Cell Prolif. 48, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalyanaraman B, Cheng G, Hardy M, Ouari O, Lopez M, Joseph J, Zielonka J, and Dwinell MB (2018). A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol 14, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bridgeman SC, Ellison GC, Melton PE, Newsholme P, and Mamotte CDS (2018). Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 20, 1553–1562. [DOI] [PubMed] [Google Scholar]
- 31.Tseng H-W, Li S-C, and Tsai K-W (2019). Metformin Treatment Suppresses Melanoma Cell Growth and Motility Through Modulation of microRNA Expression. Cancers 11. 10.3390/cancers11020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue J, Li L, Li N, Li F, Qin X, Li T, and Liu M. (2019). Metformin suppresses cancer cell growth in endometrial carcinoma by inhibiting PD-L1. Eur. J. Pharmacol. 859, 172541. [DOI] [PubMed] [Google Scholar]
- 33.Wallbillich JJ, Josyula S, Saini U, Zingarelli RA, Dorayappan KDP, Riley MK, Wanner RA, Cohn DE, and Selvendiran K. (2017). High Glucose-Mediated STAT3 Activation in Endometrial Cancer Is Inhibited by Metformin: Therapeutic Implications for Endometrial Cancer. PLoS One 12, e0170318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamb J., Crawford ED., Peck D., Modell JW., Blat IC., Wrobel MJ., Lerner J., Brunet J-P., Subramanian A., Ross KN., et al. (2006). The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- 35.Perry JM, Tao F, Roy A, Lin T, He XC, Chen S, Lu X, Nemechek J, Ruan L, Yu X, et al. (2020). Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol. 22, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu J-M, Xia W, Hsu Y-H, Chan L-C, Yu W-H, Cha J-H, Chen C-T, Liao H-W, Kuo C-W, Khoo K-H, et al. (2018). STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao C, Wang Y, Broaddus R, Sun L, Xue F, and Zhang W. (2018). Exon 3 mutations of drive tumorigenesis: a review. Oncotarget 9, 5492–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, and Akiyama T. (2004). DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene 23, 8520–8526. [DOI] [PubMed] [Google Scholar]
- 39.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, and He X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847. [DOI] [PubMed] [Google Scholar]
- 40.Kurnit KC, Kim GN, Fellman BM, Urbauer DL, Mills GB, Zhang W, and Broaddus RR (2017). CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol. 30, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myers A, Barry WT, Hirsch MS, Matulonis U, and Lee L. (2014). β-Catenin mutations in recurrent FIGO IA grade I endometrioid endometrial cancers. Gynecol. Oncol. 134, 426–427. [DOI] [PubMed] [Google Scholar]
- 42.Phillips KA, Deverka PA, Trosman JR, Douglas MP, Chambers JD, Weldon CB, and Dervan AP (2017). Payer coverage policies for multigene tests. Nat. Biotechnol. 35, 614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trosman JR, Weldon CB, Gradishar WJ, Benson AB 3rd, Cristofanilli M, Kurian AW, Ford JM, Balch A, Watkins J, and Phillips KA (2018). From the Past to the Present: Insurer Coverage Frameworks for Next-Generation Tumor Sequencing. Value Health 21, 1062–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong R, Liu W, DeLair D, Razavian N, and Fenyö D. (2021). Predicting endometrial cancer subtypes and molecular features from histopathology images using multi-resolution deep learning models. Cell Rep Med 2, 100400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davoli T, Uno H, Wooten EC, and Elledge SJ (2017). Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355. 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. (2018). Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray Chaudhuri A, and Nussenzweig A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinho SS, and Reis CA (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555. [DOI] [PubMed] [Google Scholar]
- 49.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oaknin A., Gilbert L., Tinker AV., Brown J., Mathews C., Press J., Sabatier R., O’Malley DM., Samouelian V., Boni V., et al. (2022). Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: interim results from GARNET-a phase I, single-arm study. J Immunother Cancer 10. 10.1136/jitc-2021-003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soliman PT, Westin SN, Iglesias DA, Fellman BM, Yuan Y, Zhang Q, Yates MS, Broaddus RR, Slomovitz BM, Lu KH, et al. (2020). Everolimus, Letrozole, and Metformin in Women with Advanced or Recurrent Endometrioid Endometrial Cancer: A Multi-Center, Single Arm, Phase II Study. Clin. Cancer Res. 26, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hall C, Stone RL, Gehlot A, Zorn KK, and Burnett AF (2016). Use of Metformin in Obese Women With Type I Endometrial Cancer Is Associated With a Reduced Incidence of Cancer Recurrence. Int. J. Gynecol. Cancer 26, 313–317. [DOI] [PubMed] [Google Scholar]
- 53.Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L, and Bae-Jump VL (2015). Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 4, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beausoleil SA, Villén J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 55.Blumenberg L, Kawaler E, Cornwell M, Smith S, Ruggles K, and Fenyö D. (2019). BlackSheep: A Bioconductor and Bioconda package for differential extreme value analysis. bioRxiv, 825067. 10.1101/825067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, and Durbin R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao Y, Wang J, and Zhao F. (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindgren CM, Adams DW, Kimball B, Boekweg H, Tayler S, Pugh SL, and Payne SH (2021). Simplified and Unified Access to Cancer Proteogenomic Data. J. Proteome Res. 20, 1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benelli M, Pescucci C, Marseglia G, Severgnini M, Torricelli F, and Magi A. (2012). Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 28, 3232–3239. [DOI] [PubMed] [Google Scholar]
- 61.Toghi Eshghi S, Shah P, Yang W, Li X, and Zhang H. (2015). GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem. 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hänzelmann S, Castelo R, and Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J., White NM., Schmidt HK., Fulton RS., Tomlinson C., Warren WC., Wilson RK., and Maher CA. (2016). INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasaikar SV, Straub P, Wang J, and Zhang B. (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Monroe ME, Shaw JL, Daly DS, Adkins JN, and Smith RD (2008). MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wen B, Mei Z, Zeng C, and Liu S. (2017). metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinformatics 18, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim S, and Pevzner PA (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, and Ding L. (2014). MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30, 1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen L, Zhang B, Schnaubelt M, Shah P, Aiyetan P, Chan D, Zhang H, and Zhang Z. (2018). MS-PyCloud: An open-source, cloud computing-based pipeline for LC-MS/MS data analysis. bioRxiv, 320887. 10.1101/320887. [DOI] [Google Scholar]
- 70.Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis ER, et al. (2012). MuSiC: identifying mutational significance in cancer genomes. Genome Res. 22, 1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gibbons BC, Chambers MC, Monroe ME, Tabb DL, and Payne SH (2015). Correcting systematic bias and instrument measurement drift with mzRefinery. Bioinformatics 31, 3838–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, and Kohlbacher O. (2014). OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wen B, Wang X, and Zhang B. (2019). PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 29, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayakonda A, Lin D-C, Assenov Y, Plass C, and Koeffler HP (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye K., Schulz MH., Long Q., Apweiler R., and Ning Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N, and Saez-Rodriguez J. (2018). Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruggles KV, Tang Z, Wang X, Grover H, Askenazi M, Teubl J, Cao S, McLellan MD, Clauser KR, Tabb DL, et al. (2016). An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol. Cell. Proteomics 15, 1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tan VYF, and Févotte C. (2013). Automatic relevance determination in nonnegative matrix factorization with the β-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 35, 1592–1605. [DOI] [PubMed] [Google Scholar]
- 81.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haas BJ, Dobin A, Stransky N, Li B, Yang X, Tickle T, Bankapur A, Ganote C, Doak TG, Pochet N, et al. (2017). STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv, 120295. 10.1101/120295. [DOI] [Google Scholar]
- 83.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- 84.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen T, and Guestrin C. (2016). XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 785–794. [Google Scholar]
- 86.Kuhn M. (2008). Building Predictive Models inRUsing thecaretPackage. Journal of Statistical Software 28. 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- 87.Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 88.Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, Madhavan S, and Ketchum KA (2015). The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 14, 2707–2713. [DOI] [PubMed] [Google Scholar]
- 89.Chen YT., Scanlan MJ., Sahin U., Türeci O., Gure AO., Tsang S., Williamson B., Stockert E., Pfreundschuh M., and Old LJ. (1997). A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. U. S. A. 94, 1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, et al. (2014). NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 42, D975–D979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, and Staudt LM (2016). Toward a Shared Vision for Cancer Genomic Data. New England Journal of Medicine 375, 1109–1112. 10.1056/nejmp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Consortium UniProt (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xi R, Lee S, Xia Y, Kim T-M, and Park PJ (2016). Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 44, 6274–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- 96.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li M, Xie X, Zhou J, Sheng M, Yin X, Ko E-A, Zhou T, and Gu W. (2017). Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 33, 2131–2139. [DOI] [PubMed] [Google Scholar]
- 98.Kim S, Gupta N, and Pevzner PA (2008). Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lange V, Picotti P, Domon B, and Aebersold R. (2008). Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, Xie F, Wu S, Kim J-S, Jones N, et al. (2012). Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Natl. Acad. Sci. U. S. A. 109, 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sharma V, Eckels J, Schilling B, Ludwig C, Jaffe JD, MacCoss MJ, and MacLean B. (2018). Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteomics 17, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma Z-Q., Dasari S., Chambers MC., Litton MD., Sobecki SM., Zimmerman LJ., Halvey PJ., Schilling B., Drake PM., Gibson BW., et al. (2009). IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patro R, and Kingsford C. (2013). Predicting protein interactions via parsimonious network history inference. Bioinformatics 29, i237–i246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang B, Chambers MC, and Tabb DL (2007). Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih T-SM, Chang H-Y, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hu Y, Pan J, Shah P, Ao M, Thomas SN, Liu Y, Chen L, Schnaubelt M, Clark DJ, Rodriguez H, et al. (2020). Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 33, 108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ku AT, Young AIJ, Ibrahim AA, Bu W, Jiang W, Lin M, Williams LC, McCue BL, Miles G, Nagi C, et al. (2023). Short-term PI3K Inhibition Prevents Breast Cancer in Preclinical Models. Cancer Prev. Res. 16, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, and Trajanoski Z. (2017). Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 18, 248–262. [DOI] [PubMed] [Google Scholar]
- 110.Mani DR, Maynard M, Kothadia R, Krug K, Christianson KE, Heiman D, Clauser KR, Birger C, Getz G, and Carr SA (2021). PANOPLY: a cloud-based platform for automated and reproducible proteogenomic data analysis. Nat. Methods 18, 580–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kather JN, Heij LR, Grabsch HI, Loeffler C, Echle A, Muti HS, Krause J, Niehues JM, Sommer KAJ, Bankhead P, et al. (2020). Pan-cancer image-based detection of clinically actionable genetic alterations. Nat Cancer 1, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, et al. (2018). HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frolkis A, Knox C, Lim E, Jewison T, Law V, Hau DD, Liu P, Gautam B, Ly S, Guo AC, et al. (2010). SMPDB: The Small Molecule Pathway Database. Nucleic Acids Res. 38, D480–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, A Miller R, Digles D, Lopes EN, Ehrhart F, et al. (2021). WikiPathways: connecting communities. Nucleic Acids Res. 49, D613–D621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang R, Grishagin I, Wang Y, Zhao T, Greene J, Obenauer JC, Ngan D, Nguyen D-T, Guha R, Jadhav A, et al. (2019). The NCATS BioPlanet - An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 10, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, and Ma’ayan A. (2016). The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016. 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tabula Sapiens Consortium*, Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR, Salzman J, Yosef N, Bulthaup B, Brown P, et al. (2022). The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Poole W, Gibbs DL, Shmulevich I, Bernard B, and Knijnenburg TA (2016). Combining dependent P-values with an empirical adaptation of Brown’s method. Bioinformatics 32, i430–i436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rykunov D, Beckmann ND, Li H, Uzilov A, Schadt EE, and Reva B. (2016). A new molecular signature method for prediction of driver cancer pathways from transcriptional data. Nucleic Acids Res. 44, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genotype, phenotype, immune scores, clinical, and pathological characteristics of tumor and normal samples. Related to Figure 1.
Table S2. Mutation signatures, CNV drivers, CNV arm immune score correlations, and PARP-i drug sensitivity data. Related to Figures 1 and 6.
Table S3. PIK3R1 mutation status, AKT inhibitor response of EC cell lines, and sequencing of CRISPR-Cas9 edited cells. Related to Figure 2.
Table S4. Immune subgroup, immune scores and SRM peptide levels for ICI treated samples. Related to Figure 3.
Table S5. Inferred MYC activity in CMAP, TCGA, Exploratory and Independent cohorts, total MYC IHC scores. Related to Figure 4.
Table S6. Transporters scores and classes, CTNNB1 mutation protein fold change, and top classifying genes for hotspot mutation. Related to Figure 5.
Table S7. Deep learning models, multi-omic NMF clustering, and glycopeptide NMF clustering. Related to Figures 6 and 7.
Data Availability Statement
Clinical and proteomic (raw MS files and processed data files) data reported in this paper, including both exploratory and independent CPTAC datasets, can be accessed via the Proteomic Data Commons (PDC) at: https://pdc.cancer.gov/ (PDC000125, PDC000126, PDC000226). Genomic and transcriptomic data files for both CPTAC cohorts (phs001287) and TCGA cohort (phs000178) can be accessed via Genomic Data Commons (GDC) at: https://portal.gdc.cancer.gov/projects/CPTAC-3. The DepMap datasets can be accessed via the DepMap portal: https://depmap.org/portal/. Processed CPTAC data for both cohorts used in this publication can also be found in the PDC, the Python package called ‘cptac’ (https://pypi.org/project/cptac/, install via pip) to allow programmatic access and LinkedOmics via http://www.linkedomics.org/data_download/CPTAC-UCEC-independent/ 64. Histological and radiological images for both CPTAC cohorts (10.7937/k9/tcia.2018.3r3juisw) and TCGA cohort (10.7937/k9/tcia.2016.gkj0zwac) can be accessed via Imaging Data Commons (IDC) at https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ucec, and The Cancer Imaging Archive at https://wiki.cancerimagingarchive.net/pages/viewpage.action?pageId=33948263. Deidentified digitized H&E slides from NYU reported in this paper will be shared by the lead contact upon request.