

Abstract
Pyridine halogenation reactions are crucial for obtaining the vast array of derivatives required for drug and agrochemical development. However, despite more than a century of synthetic endeavors, halogenation processes that selectively functionalize the carbon–hydrogen bond in the 3-position of a broad range of pyridine precursors remain largely elusive. We report a reaction sequence of pyridyl ring opening, halogenation, and ring closing whereby the acyclic Zincke imine intermediates undergo highly regioselective halogenation reactions under mild conditions. Experimental and computational mechanistic studies indicate that the nature of the halogen electrophile can modify the selectivity-determining step. Using this method, we produced a diverse set of 3-halopyridines and demonstrated late-stage halogenation of complex pharmaceuticals and agrochemicals.
Pyridines are widely found in biologically relevant molecules, and their prevalence results from an interplay of the heterocycle’s intrinsic properties and substituents (1–4). Therefore, regioselective pyridine C–H halogenation reactions are vital because installing a carbon–halogen (C–Hal) bond enables numerous subsequent bond-forming reactions (Fig. 1A). In pharmaceutical and agrochemical research, halopyridines are key intermediates to diversify candidate compounds for structure-activity relationship studies as well as target-orientated synthesis (5–10). Halopyridines are also inherently valuable in bioactive compounds such as etoricoxib. However, despite reports of pyridine halogenation reactions dating back to the late 19th century, there are still major limitations to this valuable bond construction (11–13). In particular, processes selective for the 3-position tend to be incompatible with the functionality of many substrates of interest for their bioactivity (14).
Fig. 1. Importance of pyridine halogenation reactions and a distinct strategy based on ring-opened intermediates.
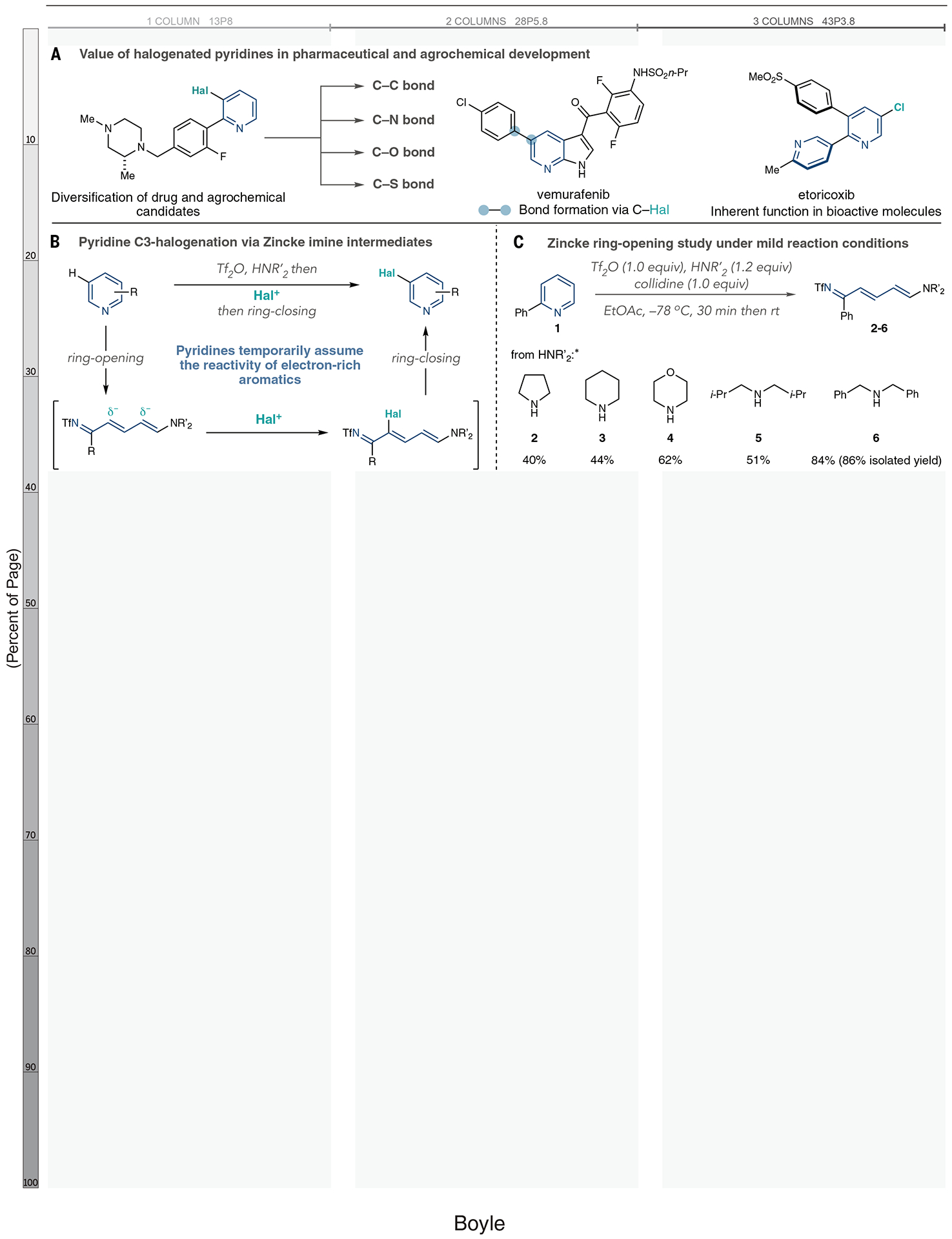
(A) Examples of pyridine halogenation products and derivatives in drug and agrochemical development. (B) A ring-opening, halogenation, ring-closing synthetic strategy. (C) Ring-opening study using amine nucleophiles. *Yields calculated from integrated 1H NMR spectra of the crude reaction mixture using triphenylmethane as an internal standard. R, general organic group; Me, methyl; Hal, halogen; Tf, trifluoromethylsulfonyl; collidine, 2,4,6-trimethylpyridine; Et, ethyl; Ac, acetate; Ph, phenyl; i-Pr, isopropyl; rt, room temperature.
Pyridine halogenation reactions using electrophilic aromatic substitution are electronically mismatched processes that require harsh conditions (15, 16). Specifically, elemental halides with strong Brønsted or Lewis acids are often used at elevated temperatures to compensate for the poor π nucleophilicity of pyridine rings (5, 6). Substrate scope is limited, and despite being 3-selective, reactions often result in regioisomeric mixtures. Metalation-halogenation sequences with strong bases are another approach for pyridine halogenation, but most require directing groups to access the 3-position reliably (17–19). Beyond these two approaches, there has been little progress over the past century, so the synthetic community turned to other versatile functional groups in place of halides. Iridium-catalyzed borylations and silylations are the most notable processes and are 3-selective for certain classes of substituted pyridines or through control with ligand architectures (20–26). Here, we present an alternative approach for pyridine halogenation using a ring-opening, halogenation, ring-closing strategy (Fig. 1B). This “one-pot” protocol uses a modified version of the classic Zincke ring-opening reaction that converts pyridines into azatriene intermediates, or “Zincke imines.” This synthetic maneuver temporarily transforms pyridines from electron-deficient heterocycles to a series of polarized alkenes that that undergo electrophilic substitution reactions, much like electron-rich aromatics. The halogenation process is 3-selective across a range of substituted pyridines and is functionally compatible with complex structures of pharmaceutical interest.
Current methods for Zincke ring-opening chemistry cannot enable the general process for pyridine halogenation described in Fig. 1B (27–29). The pyridine N-activation step requires forcing conditions and often fails when 2-position substituents are present. This limitation excludes many classes of substituted pyridines. To address these restrictions, we focused on ring opening of NTf-pyridinium salts. These reactive intermediates are readily formed from the pyridine and Tf2O at low temperatures and encompass most substitution patterns except 2,6-disubstitution. Toscano et al. reported a ring opening with Tf2O, but did not extend this process beyond pyridine, and observed mixtures of ring-opened products (30). With 2-phenylpyridine (1), we surveyed a series of aliphatic amines as nucleophiles (Fig. 1C). Pyrrolidine, piperidine, and morpholine gave moderate yields of ring-opened products, as did diisobutylamine (2 to 5). However, dibenzylamine proved optimal, with 6 formed in high yield. Tables S1 and S2 show further studies of pyridine ring openings using this protocol. We anticipated that the ring closure of Zincke imines such as 6 would be straightforward based on precedent in related Zincke systems (31). Examples typically use ammonium salts or Brønsted acids to obtain pyridines, and these reagents could potentially facilitate the one-pot sequence outlined in Fig. 1B (see below).
For the next stage of reaction development, we tested Zincke imine 6 with halogen electrophiles (Fig. 2A). At the outset, we did not expect that C–Hal bond formation would be selective because there was no clear electronic bias between the two δ– sites at C3 and C5 (see below) in 6 (32). Toscano et al. did observe a 3-selective reaction between an unsubstituted NTf-Zincke imine derived from pyridine and a pyridinium electrophile; however, there were no investigations into Zincke imines derived from substituted pyridines (30). When we subjected 6 to N-iodosuccinimide (NIS) at room temperature for 5 min, we observed 7-I in 92% yield and >20:1 3- versus 5-selectivity (8-I) based on analysis of the crude 1H nuclear magnetic resonance (NMR) spectrum. Using N-bromosuccinimide (NBS), we observed a 4.4:1 ratio of 7-Br and 8-Br at room temperature and >20:1 selectivity and 92% of 7-Br at −78°C in CH2Cl2. Chlorinating with N-chlorosuccinimide (NCS) was lower yielding and less selective (7-Cl and 8-Cl), and it decomposed 6. At this point, we tested conditions for ring closure and found that heating 8-I in a EtOAc/EtOH solvent mixture at 60°C with 10 equivalents of ammonium acetate smoothly formed 3-iodopyridine 9-I. Table S10 shows further studies of this ring-closing step.
Fig. 2. Reaction development and mechanistic investigation.
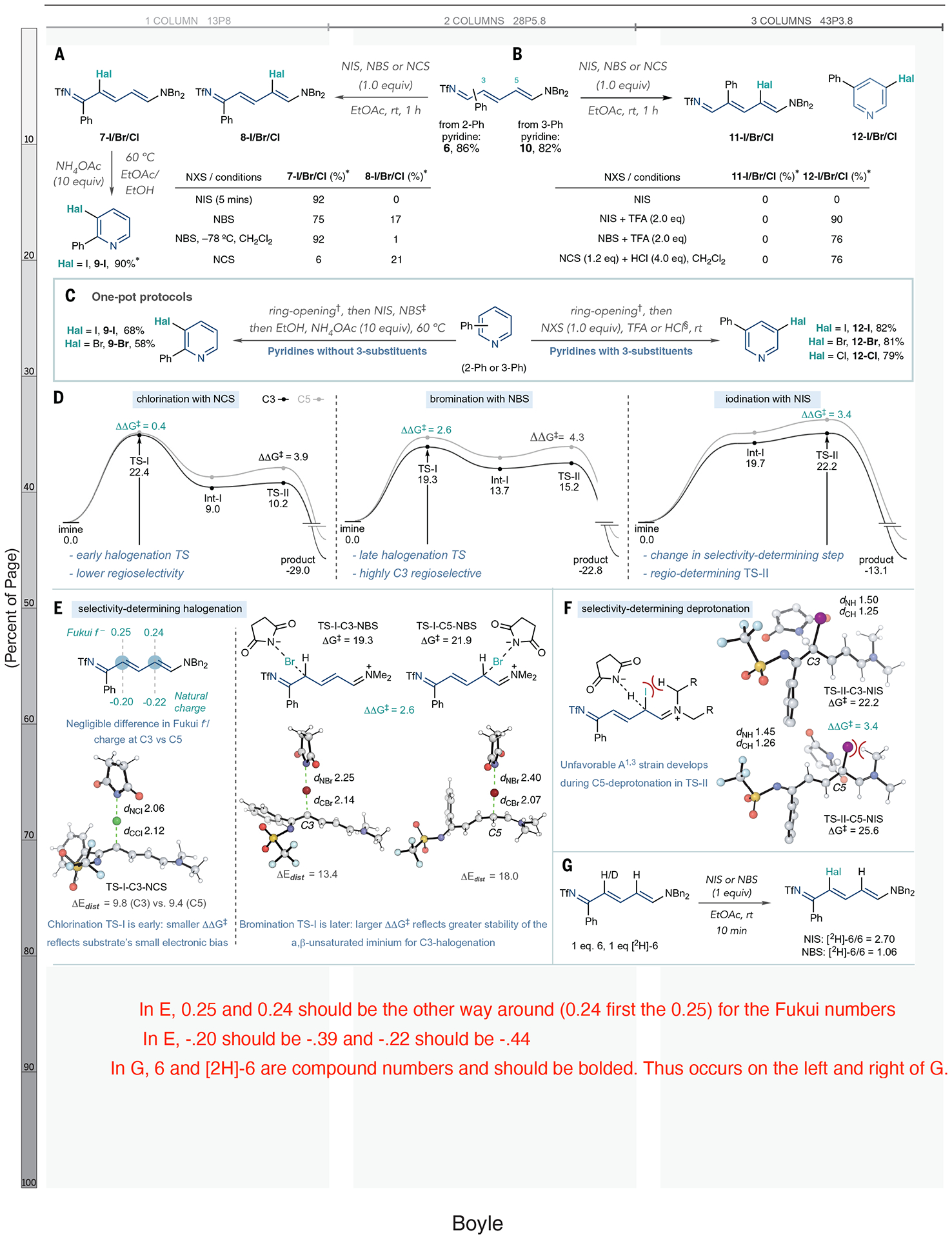
(A) Halogenation of 2-substituted Zincke imines and ring closure using ammonium salts. *Yields calculated from 1H NMR of the crude reaction mixture using triphenylmethane as an internal standard. (B) Halogenation of 3-substituted Zincke imines. (C) One-pot pyridine halogenation protocols. †Ring-opening conditions: Tf2O (1.0 equiv), HNBn2 (1.2 equiv), collidine (1.0 equiv), EtOAc, −78°C, 30 min then warm to rt. ‡Ring-opening and halogenation steps performed in CH2Cl2 instead of EtOAc when using NBS. §Halogenation–ring-closure protocol for pyridines with 3-substituents: NIS (1.0 equiv) or NBS (1.0 equiv) and TFA (2.0 equiv). NCS (1.0 equiv) and HCl (4.0 equiv) and ring opening were performed in CH2Cl2. Isolated yields are shown. (D) Computed Gibbs energy profiles (in kilocalories per mole) for imine halogenation by N-halosuccinimides. (E) Analysis of selectivity-determining chlorination and bromination transition structures TS-I. (F) Analysis of selectivity-determining deprotonation transition structure TS-II for iodination. (G). Isotope competition experiment. Bn, benzyl group; NXS, N-halosuccinimides.
Halogenating 3-substituted imines (Fig. 2B) required a different procedure. Ring opening of 3-phenylpyridine produced Zincke imine 10 in 82% yield, but NIS did not react with 10 at room temperature and decomposed to unidentified by-products when heated to 50°C. However, when we included two equivalents of trifluoroacetic acid (TFA), iodopyridine 12-I formed in high yield (33). We attribute this in situ rearomatization to steric interactions between the 3- and 5-substituents in 11-I; TFA can promote the alkene isomerization-recyclization sequence, as well as the subsequent elimination of dibenzylamine and N–S bond cleavage of the resulting NTf-pyridinium salt to reform the pyridine ring system. Brominating with NBS and TFA (12-Br) resulted in the same outcome, and a modified procedure using NCS with HCl in CH2Cl2 chlorinated 10 effectively to form 12-Cl. The results (Fig. 2, A and B) provide two halogenation protocols to halogenate NTf-Zincke imine intermediates depending on their substitution pattern; further details of this optimization study are described in tables S3 to S8.
Next, we combined the ring-opening, halogenation, and ring-closing steps into a one-pot process (Fig. 2C). For pyridines without 3-substituents, such as 2-phenylpyridine, ring opening using dibenzylamine and halogenation with NIS or NBS (Fig. 2A) was appropriate. Adding NH4OAc and EtOH to the reaction mixture and heating to 60°C induced ring closure to reform pyridines 9-I and 9-Br. Adding an equivalent of trimethoxybenzene after the halogenation step proved helpful in quenching any remaining N-halosuccinimide before cyclization. When 3-substituents were present, such as in 3-phenylpyridine, ring opening followed by addition of the N-halosuccinimides and acids described in Fig. 2B comprised the one-pot process (12-I, 12-Br, and 12-Cl). These protocols do not require intermediate workup or purification steps and involve adding reagents sequentially to the same reaction vessel.
We then studied the mechanism and regioselectivity of Zincke imine halogenation by N-halosuccinimides with quantum chemistry at the B3LYP-D3(BJ)/def2-TZVP//ωB97X-D/6–31+G(d,p) level of theory with a “solvation model based on density (SMD)” description of CH2Cl2. The most favorable pathway for halogenating 6 (Fig. 2D), with the NBn2 portion simplified to NMe2, consisted of electrophilic addition (TS-I) followed by deprotonation (TS-II). Calculations showed that outer-sphere electron transfer processes were inaccessible (ΔG > 34 kcal mol−1); figs. S11 to S23 provide full computational details.
Regioselectivity in electrophilic halogenation is conventionally understood in terms of differences in frontier orbital coefficients, atomic charges, or nucleophilicity parameters (34). However, the electronic environments at the C3 and C5 positions of Zincke imine 6 are very similar in terms of Fukui f− coefficients (0.24 versus 0.25), natural charges (−0.39 versus −0.44), and HOMO coefficients (both 0.26), necessitating an alternative rationale for the high levels of C3 selectivity in this transformation (Fig. 2E). Computed energy profiles (Fig. 2D) for 6 reacting with NCS, NBS, and NIS suggest an irreversible overall reaction governed by kinetically controlled regioselectivity in each case, with DDG‡ values in quantitative agreement with experiment. Breaking the extended conjugation in the Zincke imine to form positively charged s-complex Int-I is endergonic: Grel values of this high-energy intermediate increase for C–Cl, C–Br, and C–I formation, respectively (9.0, 13.7, and 19.7 kcal mol−1, respectively). These differences give rise to two distinct selectivity-determining regimes: an irreversible C–Hal bond-forming step (TS-I) determines regioselectivity for chlorination and bromination, whereas C–I bond formation is reversible, and the second deprotonation step (TS-II) dictates the regioselectivity (35).
The higher C3 selectivity for bromination over chlorination (ΔΔG‡ of 2.5 and 0.4 kcal mol−1, respectively) can be understood on the basis of early versus late transition state arguments. Chlorination results in a more stable intermediate than bromination and, in accordance with the Hammond postulate, proceeds through an earlier TS [as shown by smaller TS distortion energies in Fig. 2E and smaller pyramidalization values at the reacting carbon atom (fig. S19)]. Given the electronic similarity between the C3 and C5 positions in the Zincke imine and the absence of appreciable intermolecular noncovalent interactions (fig. S17), there is little regioisomeric preference in the early chlorination TS. Bromination proceeds through a later TS that incurs larger substrate distortion and strongly favors addition at C3 (ΔΔEdist = 4.6 kcal mol−1). This difference in distortion energies reflects a more stable developing a,b–unsaturated iminium ion in TS-IC3-NBS versus TS-I-C5-NBS (ΔΔG‡ = 2.6 kcal mol−1). By contrast, the high regioselectivity of iodination (ΔΔG‡ = 3.4 kcal mol−1) results from a rate- and selectivity-determining deprotonation (TS-II). Restoring planarity in TS-IIC5-NIS results in increased A1,3 strain between the enamine carbon substituent and the iodide (Fig. 2F). The bond and dihedral angles in the C5-iodinated product clearly reflect this developing strain (fig. S21).
Intermolecular competition experiments validate these computational predictions (Fig. 2G). We reacted one equivalent each of 6 and [2H]-6 until this mixture consumed one equivalent of NIS. The [2H]-6/6 ratio of 2.70 measured at the end of the reaction indicates that 6 is consumed preferentially and supports the selectivity-determining deprotonation predicted by theory (Fig. 2F). The same experiment using NBS did not show a significant bias between [2H]-6 and 6 at the end of the reaction, consistent with selectivity-determining C–Br bond formation.
Next, we applied a series of azine building block-type compounds to the one-pot halogenation reaction (Fig. 3), selecting iodination for most substrates because of its convenient room temperature C–I bond-forming step. The process is compatible with pyridines with aryl, alkyl, and acetal substituents at the 2-position (13 to 17), and the acid-mediated conditions were effective for a series of pyridines (18 to 21). The one-pot iodination yield of 3-fluoropyridine (20) was low, and we reverted to a two-step process, isolating the intermediate Zincke imine. We recommend this alternative protocol to obtain higher overall yields of halogenated pyridines in these instances.
Fig. 3. One-pot pyridine halogenation: scope of building-block pyridines and dihalogenation reactions.

Yields of isolated products are shown. Yields in parentheses are those of isolated Zincke imines. *Ring-opening and halogenation steps performed in CH2Cl2 instead of EtOAc when using NBS. †Halogenation–ring-closure protocol for pyridines with 3-substituents: NIS (1.0 equiv) or NBS (1.0 equiv) and TFA (2.0 equiv). NCS (1.0 equiv) and HCl (4.0 equiv), and ring opening is performed in CH2Cl2. ‡1H NMR yields using triphenylmethane as an internal standard. Pin, pinacolato; n-Bu, normal butyl group; Ts, tosyl.
Pyridines with benzyl, cyano, and ketone substituents at the 4-position are also effective substrates (22 to 25). We then halogenated a series of 2,3-, 2,4-, and 2,5-disubstituted pyridines (26 to 34). Pyridine 27 is notable because of the sensitive 2-chloro substituent; in this case, ring opening with dibenzylamine resulted in a complex mixture of products. However, the reaction was successful using N-benzylaniline, and we attribute this outcome to its attenuated nucleophilicity. Halopyridines 32 to 34 show that the Zincke imine intermediate overrides the ortho-, para-directing benzyloxy group and maintains 3-selectivity. We formed a set of di- and trihalogenated pyridines from 3,4-disubstituted systems (35 to 37), and 38 to 40 are preliminary examples showing that this process can halogenate pyrimidines. When using 5-nitroisoquinoline as a substrate, we did not observe ring opening but instead isolated a dearomatized adduct where dibenzylamine adds to the C1 atom of the NTfisoquinolinium salt. Subjecting this intermediate to NBS then TFA resulted in 4-brominated product 41. We observed a similar intermediate in a one-pot process using 5-azaindole, resulting in bromide 42. A C6-brominated product 43 formed from 4-ethoxyquinoline; it is less clear what intermediate species operates in this case because of broadening in the 1H NMR spectra after ring opening. Figure S2 shows proposed halogenation mechanisms and spectroscopic evidence of the dearomatized adducts.
Next, we tested whether halogenating Zincke imine intermediates could outcompete neighboring electron-rich aromatic rings and if dihalogenation processes were feasible. As shown in Fig. 3, a pyridine is selectively iodinated over an anisole group (44), as evidenced by only traces of other compounds, aside from starting material, in the crude 1H NMR. As shown in fig. S10, we applied standard arene iodination conditions (NIS and TFA in MeCN at 50°C) and observed iodination on the anisole ring. Similarly, pyridines are iodinated over thiophenes, furans, and phenoxy groups (45 to 48). Furthermore, iodination selectively occurs on the pyridine in fused ring systems such as furopyridines, protected azaindoles, and thienopyridines that conventionally react at the five-membered heterocyclic portion (49 to 51) (36). C–Hal bond formation can also be applied sequentially to form 3,5-dihalogenated pyridines, a motif with two orthogonal vectors for further synthetic transformations (37–39). Although a one-pot procedure was unsuccessful, we were able to isolate Zincke imines 7-I and 7-Br and subject them to a second round of halogenation, resulting in various permutations of dihalopyridines 52 to 57. We formed dichloropyridine 58 directly from Zincke imine 6 by modifying the acid-mediated halogenation-rearomatization protocol to include 2.05 equivalents of NCS.
The final stage of this study focused on halogenating complex drug-like intermediates, pharmaceuticals, and agrochemicals (Fig. 4). These structures often contain multiple (hetero)arenes, sites of reactivity, and Lewis basic atoms, thus representing a significant synthetic challenge for the ring-opening, halogenation, ring-closing process. However, success in this endeavor would enable subsequent convergent coupling reactions, compound diversification, and late-stage functionalization efforts that are not possible with existing pyridine halogenation methods (40–42). We began our study by halogenating five polycyclic structures representative of drug-like intermediates and obtained reasonable yields of halogenated products as single regioisomers in these cases (59 to 65). A substituted quinoline containing a cyclic sultam was again brominated at C6 (66). Halopyridines 67 to 70 are notable because C–Hal bond formation occurs on one pyridine over another; we believe that this selectivity results from exclusive NTf-pyridinium salt formation involving the less hindered heterocycle in each case (43). Pyridines 71 and 72 formed atropisomers during the one-pot halogenation process, raising the possibility of controlling axial chirality in further studies.
Fig. 4. One-pot halogenation of complex azines.
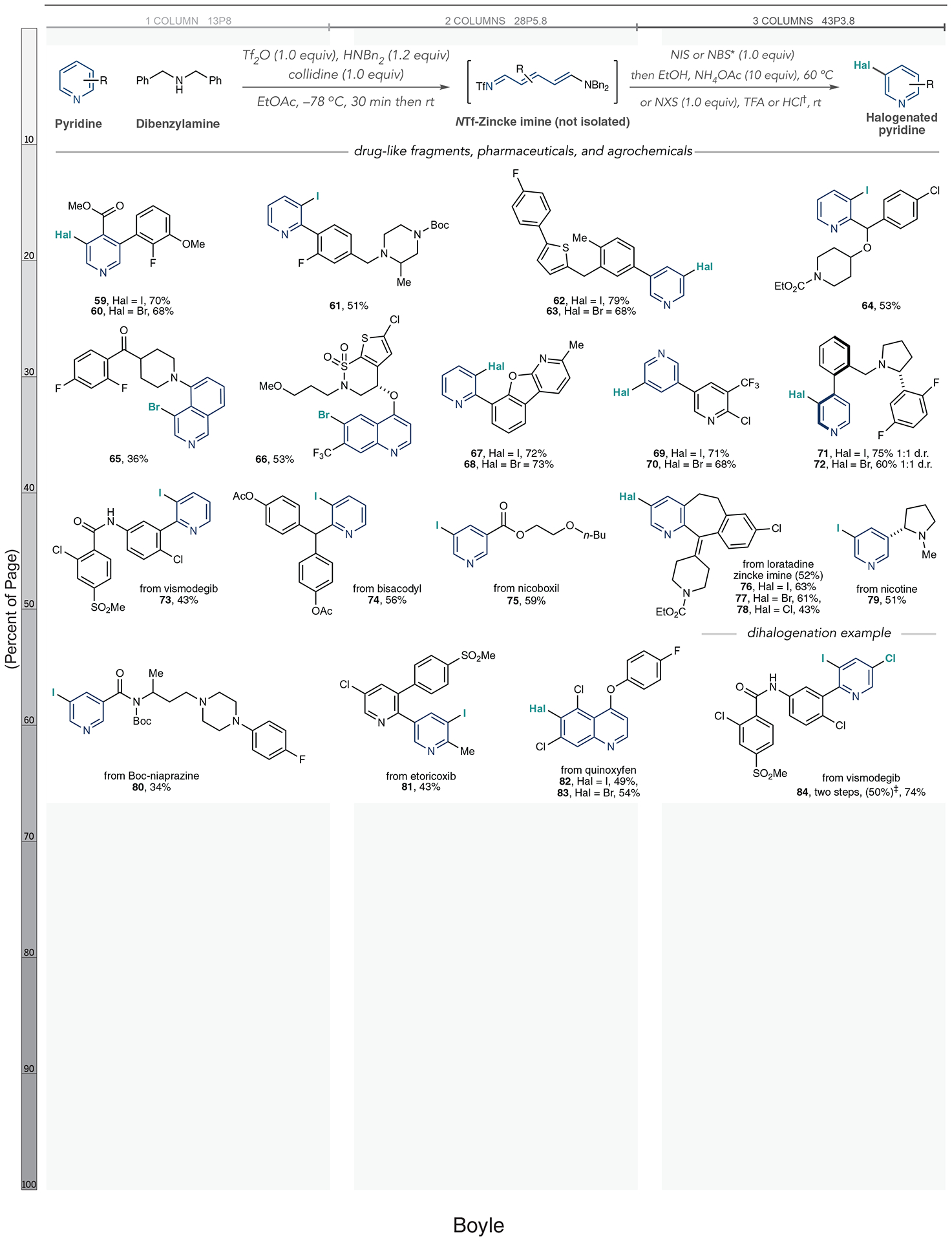
Yields of isolated products are shown. Yields in parentheses are those of isolated Zincke imines. *Ring-opening and halogenation steps performed in CH2Cl2 instead of EtOAc when using NBS. †Halogenation–ring-closure protocol for pyridines with 3-substituents: NIS (1.0 equiv) or NBS (1.0 equiv) and TFA (2.0 equiv). NCS (1.0 equiv) and HCl (4.0 equiv), and ring opening is performed in CH2Cl2. ‡Isolated yield of iodinated Zincke imine. Boc, tert-butoxycarbonyl group.
This protocol successfully halogenated a diverse collection of pharmaceuticals and agrochemicals, demonstrating its utility for late-stage functionalization. We obtained moderate yields of iodinated vismodegib, bisacodyl, and nicoboxil (73 to 75). Loratadine proved challenging in the one-pot process, but we could synthesize iodo-, bromo-, and chloro-analogs when we isolated the intermediate Zincke imine (76 to 78). Nicotine, Boc-protected niaprazine, etoricoxib, and quinoxyfen were competent substrates (79 to 83). The dihalogenation sequence is also amenable to complex pyridines. Using the basal cell carcinoma drug vismodegib, we obtained iodo-chloro analog 84 in reasonable yield over two steps.
Given the lack of alternative methods for this transformation and the high value of the halopyridine products, we anticipate that this strategy will be useful for developing medicines and agrochemicals. This approach for pyridine functionalization is likely amenable to numerous other bond constructions as well.
Supplementary Material
ACKNOWLEDGMENTS
Funding:
This work was supported by Colorado State University with funds from the National Institutes of Health (NIH grant R01 GM144591-01). R.S.P. acknowledges support from the National Science Foundation (NSF grant CHE-1955876); use of computational resources from the RMACC Summit Supercomputer, which is funded by NSF grants ACI-1532235 and ACI-1532236, the University of Colorado Boulder, and Colorado State University; and support from XSEDE through allocation TG-CHE180056.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials. Cartesian coordinates and absolute thermochemical data for all stationary points in computational studies are available in data S1.
REFERENCES AND NOTES
- 1.Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 57, 10257–10274 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Baumann M, Baxendale IR, Beilstein J. Org. Chem 9, 2265–2319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zakharychev VV, Kuzenkov AV, Martsynkevich AM, Chem. Heterocycl. Compd 56, 1491–1516 (2020). [Google Scholar]
- 4.Stolar M, Baumgartner T, Chem. Commun 54, 3311–3322 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Joule JA, Mills K, Heterocyclic Chemistry (Wiley, ed. 5, 2009). [Google Scholar]
- 6.Grimmett MR, Adv. Heterocycl. Chem 58, 271–345 (1993). [Google Scholar]
- 7.Murakami K, Yamada S, Kaneda T, Itami K, Chem. Rev 117, 9302–9332 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Crawley ML, Trost BM, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective (Wiley, 2012). [Google Scholar]
- 9.Bunnett JF, Zahler RE, Chem. Rev 49, 273–412 (1951). [Google Scholar]
- 10.Bailey WF, Patricia JJ, J. Organomet. Chem 352, 1–46 (1988). [Google Scholar]
- 11.Hoffmann AW, Ber. Dtsch. Chem. Ges 12, 984–990 (1879). [Google Scholar]
- 12.Sell WJ, Dootson FW, J. Chem. Soc. Trans 73, 442–445 (1898). [Google Scholar]
- 13.Sell WJ, Dootson FW, J. Chem. Soc. Trans 73, 432–441 (1898). [Google Scholar]
- 14.Fier PS, Hartwig JF, Science 342, 956–960 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Olah GA, Acc. Chem. Res 4, 240–248 (1971). [Google Scholar]
- 16.Galabov B, Nalbantova D, Schleyer Pv., Schaefer HF 3rd, Acc. Chem. Res 49, 1191–1199 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Manolikakes SM, Barl NM, Samann C, Knochel P, Naturforsch Z. B. J. Chem. Sci 68, 411–422 (2013). [Google Scholar]
- 18.El-Hiti GA, Smith K, Hegazy AS, Alshammari MB, Masmali AM, ChemInform 46, 1 (2015). [Google Scholar]
- 19.El-Hiti GA, Smith K, Hegazy AS, Heterocycles 91, 479–504 (2015). [Google Scholar]
- 20.Larsen MA, Hartwig JF, J. Am. Chem. Soc 136, 4287–4299 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Sadler SA et al. , Org. Biomol. Chem 12, 7318–7327 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Wright JS, Scott PJH, Steel PG, Angew. Chem. Int. Ed 60, 2796–2821 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Uemura N, Nakao Y, J. Am. Chem. Soc 141, 7972–7979 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Trouvé J, Zardi P, Al-Shehimy S, Roisnel T, Gramage-Doria R, Angew. Chem. Int. Ed 60, 18006–18013 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Cheng C, Hartwig JF, J. Am. Chem. Soc 137, 592–595 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Zhou XY, Zhang M, Liu Z, He JH, Wang XC, J. Am. Chem. Soc 144, 14463–14470 (2022). [DOI] [PubMed] [Google Scholar]
- 27.Vanderwal CD, J. Org. Chem 76, 9555–9567 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Cheng WC, Kurth MJ, Org. Prep. Proced. Int 34, 585–608 (2002). [Google Scholar]
- 29.Sowmiah S, Esperanca JMSS, Rebelo LPN, Afonso CAM, Org. Chem. Front 5, 453–493 (2018). [Google Scholar]
- 30.Toscano RA et al. , Chem. Pharm. Bull. (Tokyo) 45, 957–961 (1997). [Google Scholar]
- 31.Becher J, Synthesis 1980, 589–612 (1980). [Google Scholar]
- 32.Stämpfli U, Neuenschwander M, Helv. Chim. Acta 66, 1427–1435 (1983). [Google Scholar]
- 33.Nguyen TM, del Rayo Sanchez-Salvatori M, Wypych JC, Marazano C, J. Org. Chem 72, 5916–5919 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Tomberg A, Johansson MJ, Norrby PO, J. Org. Chem 84, 4695–4703 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Liljenberg M, Stenlid JH, Brinck T, J. Phys. Chem. A 122, 3270–3279 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Klemm LH, Louris JN, J. Heterocycl. Chem 21, 785–789 (1984). [Google Scholar]
- 37.Reeves EK, Entz ED, Neufeldt SR, Chemistry 27, 6161–6177 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Keylor MH, Niemeyer ZL, Sigman MS, Tan KL, J. Am. Chem. Soc 139, 10613–10616 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Ji J, Li T, Bunnelle WH, Org. Lett 5, 4611–4614 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, Ritter T, J. Am. Chem. Soc 144, 2399–2414 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blakemore DC et al. , Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Dolewski RD, Fricke PJ, McNally A, J. Am. Chem. Soc 140, 8020–8026 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Cartesian coordinates and absolute thermochemical data for all stationary points in computational studies are available in data S1.