

Visual Abstract

Keywords: chronic glomerulonephritis, chronic kidney failure, diabetic glomerulosclerosis, diabetic kidney disease, podocyte
Abstract
Significance Statement
Autophagy protects podocytes from injury in diabetic kidney disease (DKD). Restoring glomerular autophagy is a promising approach to limit DKD. This study demonstrates a novel regulatory mechanism of autophagy that blocks this critical protection of the glomerular filtration barrier. We demonstrated that TRPC6 induced in podocytes in mouse models of diabetes mediates calpain activation, thereby impairing podocyte autophagy, causing injury and accelerating DKD. Furthermore, this study provides proof of principle for druggable targets for DKD because restoration of podocyte autophagy by calpain inhibitors effectively limits glomerular destruction.
Background
Diabetic kidney disease is associated with impaired podocyte autophagy and subsequent podocyte injury. The regulation of podocyte autophagy is unique because it minimally uses the mTOR and AMPK pathways. Thus, the molecular mechanisms underlying the impaired autophagy in podocytes in diabetic kidney disease remain largely elusive.
Methods
This study investigated how the calcium channel TRPC6 and the cysteine protease calpains deleteriously affect podocyte autophagy in diabetic kidney disease in mice. We demonstrated that TRPC6 knockdown in podocytes increased the autophagic flux because of decreased cysteine protease calpain activity. Diabetic kidney disease was induced in vivo using streptozotocin with unilateral nephrectomy and the BTBRob/ob mouse models.
Results
Diabetes increased TRPC6 expression in podocytes in vivo with decreased podocyte autophagic flux. Transgenic overexpression of the endogenous calpain inhibitor calpastatin, as well as pharmacologic inhibition of calpain activity, normalized podocyte autophagic flux, reduced nephrin loss, and prevented the development of albuminuria in diabetic mice. In kidney biopsies from patients with diabetes, we further confirmed that TRPC6 overexpression in podocytes correlates with decreased calpastatin expression, autophagy blockade, and podocyte injury.
Conclusions
Overall, we discovered a new mechanism that connects TRPC6 and calpain activity to impaired podocyte autophagy, increased podocyte injury, and development of proteinuria in the context of diabetic kidney disease. Therefore, targeting TRPC6 and/or calpain to restore podocyte autophagy might be a promising therapeutic strategy for diabetic kidney disease.
Introduction
Diabetic kidney disease (DKD) is one devastating microvascular complication of diabetes mellitus and among the most frequent causes of ESKD.1 Current prevention and treatment options in DKD focus on lifestyle interventions, regulation of blood glucose levels, and inhibition of the renin-angiotensin-aldosterone system (RAAS) to control blood pressure. Recently, SGLT2 inhibition was added to the therapeutic armamentarium to prevent or mitigate DKD.2–5 Of note, the prevalence of diabetes mellitus, and consequently DKD, is only anticipated to increase in the upcoming years.6–8
A key event in the pathogenesis of DKD is an injury to podocyte, leading to the development of proteinuria and eventually glomerulosclerosis.9–11 An important mechanism leading to podocyte injury in DKD is impaired autophagy.12–14 Autophagy is a crucial process of cells to generate energy and maintain cellular homeostasis during moments of cellular stress.15–17 Impaired podocyte autophagy leads to increased podocyte injury, whereas restoring podocyte autophagy prevents podocyte injury and proteinuria in experimental DKD.18–20 The molecular mechanisms underlying impaired podocyte autophagy during DKD remain elusive.
Two proteins potentially responsible for impaired podocyte autophagy during DKD are the calcium-dependent cysteine protease calpains and calcium (Ca2+)-permeable ion channel transient receptor potential channel C6 (TRPC6). TRPC6 expression and activity are increased in DKD, and TRPC6 knockout was shown to protect against the development of microvascular renal complications of diabetes mellitus in animal studies. Furthermore, TRPC6-mediated calcium influx has an inhibitory effect on podocyte autophagy.21,22 We suspected that calcium-dependent cysteine protease calpains might be activated by TRPC6 opening in podocytes. TRPC6 is a Ca2+ channel that is slit diaphragm-associated in normal podocytes. In cultured podocytes, TRPC6 directly interacts with calpain (CAPN)1/2 at the plasma membrane to regulate their activity, in a Ca2+-independent manner. This interaction is crucial to control cell motility and adhesion through regulation of Paxillin, FAK, and Talin-1.23 Along the same line, previous studies showed that the interaction between TRPC6 and calpain might contribute to podocyte injury and podocyte death by aberrant cytoskeletal remodeling in vitro.24,25 Calpains are a family of 15 cysteine proteases activated by an increase in intracellular Ca2+ concentration.26 Calpain has been recently shown as a nonconventional autophagy regulator, in part, by degrading the autophagy-initiating protein autophagy-related 5 (ATG5).27,28 Two members of the calpain superfamily, calpain-1 and calpain-2, have been extensively studied. These ubiquitously expressed calpains exist as heterodimers consisting of an isoform-specific catalytic domain encoded by Capn1 and Capn2 genes, respectively, and a common regulatory domain encoded by Capns1, a gene that is indispensable for calpain-1/2 stability and activity. The mechanisms by which calpains are activated and identify their targets are complex and poorly understood. Calpain activity is regulated by a ubiquitous endogenous inhibitor, calpastatin.29 The interplay between TRPC6 and calpain activity in the context of impaired podocyte autophagy in DKD remains unknown.
We found that TRPC6 and calpain activities are involved in impaired podocyte autophagy in DKD. In addition, we demonstrated the beneficial effect of genetic overexpression of the endogenous calpain inhibitor calpastatin and of pharmacologic calpain inhibition on the development of albuminuria in DKD in an effort to restore podocyte autophagy and integrity. These findings shed new light on the molecular mechanisms underlying this glomerular disease and may open new therapeutic avenues for the treatment of DKD by restoring podocyte autophagy.
Methods
Statistics
Data are expressed as individual dots and mean±SEM. Statistical analyses were conducted with GraphPad Prism. Comparison between two groups was performed with a two-sided unpaired t-test. A Welch correction was applied when the F test to compare variances was <0.05. Comparisons between multiple groups were performed with one-way or two-way analysis of variance (ANOVA), followed by a two-sided Tukey post hoc test or Fisher LSD post hoc test when the comparison was stand-alone. For multiple comparisons, we performed comparisons between rows and between columns only. Jamovi,30 a statistics software based on R packages,31 was used for multivariate analysis of human renal biopsies. r Correlation was computed using Pearson correlation. A P value of <0.05 was considered statistically significant. Snowcluster module32 (includes R package factoextra33) was used for principal component analysis (PCA) and PCA plot. For prediction using binomial logistic regression, R packages notably include car34 and ROCR.35 Assumptions to the model were made by calculating the collinearity index VIF.
Animals
All mice were bred and housed in a specific pathogen-free animal facility and were given free access to water and standard chow.
Type I Diabetes Model
Green fluorescent protein (GFP) light-chain 3 (LC3) calpastatin transgenic (CSTTg) mice were obtained by crossing CSTTg mice36 with GFP-LC3 mice.37 GFP-LC3 CSTTg mice and their respective control littermates (GFP-LC3) were used between 8 and 10 weeks of age. Only male mice were used in this study. GFP-LC3 and GFP-LC3 CSTTg mice were submitted for unilateral left nephrectomy. During surgical procedures, mice were anesthetized with isoflurane inhalation (2.5%). Presurgery and postsurgery analgesia was provided by buprenorphine (0.1 mg/kg) subcutaneous injection. Drugs were diluted in sterile phosphate buffer or saline. After 1 week of postsurgery recovery, mice were injected with streptozocin (STZ) at a dose of 50 mg/kg for 2 days (Merck, S0130-50MG, Darmstadt, Germany) to induce type I diabetes mellitus. Blood glycemia was monitored every week on a OneTouch Verio glucometer (Lifescan France SAS, Rueil-Malmaison, France) by tail blood collection. Urine was collected in metabolic cages once a week. Mice were killed 6 weeks after STZ injections for blood and organ collection.
Type II Diabetes Model
BTBROb/Ob mice were obtained by crossing two heterozygous BTBROb/WT mice purchased from The Jackson Laboratory (004824, Bar Harbor, ME). Male BTBROb/Ob mice and their wild-type littermates (BTBRWT/WT) were used at 6 weeks of age for experiments. They were treated with calpain pharmacologic inhibitor BDA-410, kindly provided by Mitsubishi Tanabe Pharma Corporation (Osaka, Japan), at a dose of 100 mg/kg per day administrated per os. Others received a 20 mg/kg daily dose of the TRPC6 inhibitor BI-749327 per os (MedChemTronica, Sweden). After 6 weeks of treatment, mice were killed for organ collection.
Construction and Culture of a Stable TRPC6 Knockdown Podocyte Cell Line
Conditionally immortalized mouse podocytes (MPC-5) were cultured as described previously.38 Briefly, MPC-5 were grown at permissive conditions at 33°C with 10 units of interferon gamma (IFNγ, Merck, 4777) per milliliter. To induce differentiation, IFNγ was removed from the medium and cells were grown at 37°C for 2 weeks. A podocyte cell line stably expressing a doxycycline-inducible TRPC6 silencing short hairpin RNA (shRNA) construct was created by transfecting a TRPC6 shRNA construct into undifferentiated MPC-5 using Lipofectamine 3000 (Thermofisher Scientific, Waltham, MA, L3000001). Transfected MPC-5 were maintained at 33°C in the presence of 400 µg/ml G418 (Merck, A1720) to select successfully transfected cells. To validate the TRPC6 knockdown, single clones were exposed to 5 µg/ml doxycycline (MP Biomedicals, Irvine, CA, 11420455) for 5 days to activate the knockdown construct. Because uninjured MPC-5 are known to show low TRPC6 expression levels, single clones were cultured for the last 24 hours in the absence or presence of the podocyte injury-inducing compound doxorubicin (0.25 µg/ml, Merck, D1515) to induce podocyte injury and increase TRPC6 expression. TRPC6 mRNA expression was subsequently investigated in single clones to validate the TRPC6 knockdown. Validated MPC-5 TRPC6 knockdown (KD) podocytes were thereafter cultured with a maintenance dose of 100 µg/ml G418.
In vitro Autophagic Flux Assessment
To investigate the role of TRPC6 and calpain in the context of autophagy in vitro, wild-type (WT) and TRPC6 KD podocytes were exposed for 48 hours to the calpain inhibitor calpeptin (1 µM, Merck, C8999). After 44 hours, podocytes were exposed to the autophagy inhibitor bafilomycin A (100 nM, Enzo Life Sciences, New York, NY, BML-CM110-0100) for the last 4 hours. Five days earlier and during calpeptin exposure, podocytes were exposed to 5 µg/ml doxycycline to activate the knockdown construct. In case of calcium chelation, podocytes were exposed to BAPTA-AM (5 μM, Sigma-Aldrich, A1076) for the last 2 hours.
RNA Isolation and Quantitative PCR Analysis
RNA was isolated from podocytes using Trizol (Thermofisher Scientific, 15596018). Subsequently, 1 µg RNA was reverse-transcribed into cDNA using the Transcription First Strand cDNA synthesis kit (Roche, Basel, Switzerland, 04897030001) according to the manufacturer's instructions. Quantitative TRPC6 expression levels were determined by quantitative PCR using SYBR Green (Roche, 04673484001) on a CFX 96 C1000 Thermal Cycler (Bio-rad, Hercules, CA). TRPC6 levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels using the delta–delta CT method. Three independent experiments were performed, and each experiment consisted of three samples. All samples were measured in duplicate. Sequences of gene-specific primers are GAPDH: Forward 5′-AATGCTCCCAGAAATGGCAC-3′ Reverse 5′-CCTCCACTGAACCCTGGAAA-3′ and TRPC6: Forward 5′-GACCGTTCATGAAGTTTGTAGCAC-3′ Reverse 5′-AGTATTCTTTGGGGCCTTGAGTCC-3′.
Calpain Activity Assay
Calpain activity was determined by using the Calpain activity assay (Abcam, Cambridge, UK, ab65308). Cells were collected by trypsinization and resuspended in an extraction buffer, after which samples were processed according to the manufacturer's instructions. Fluorescence intensity was measured with excitation/emission at 400/505 nm using a Tecan Infinite Pro2000 plate reader. Afterward, protein content was measured for every sample by using the bicinchoninic acid assay (BCA, Merck, 71285-3) to correct for differences in cell density. The experiment was performed in sextuplicate, and samples were measured in duplicate. In case of calcium chelation, podocytes were exposed to BAPTA-AM (5 μM, Sigma-Aldrich, A1076) for the last 2 hours.
Measurements of the Intracellular Calcium Activity [Ca2+]i
Measurements of [Ca2+]i were performed in a black 96-well clear bottom plate. After differentiation, podocytes were loaded with the Ca2+-sensitive dye fura-2/AM (3 µM, Invitrogen, F1221) in an RPMI 1640 medium in the dark for 30 minutes at 37°C. The medium was removed, and the cells were washed with a CLB-HG buffer (132 mM NaCl, 5 mM KCl, 5 mM Na2HPO4, 1.2 mM NaH2PO4·H2O, 1 mM CaCl2·2H2O, 0.8 mM MgCl2·6H2O, and 10 mM glucose).39 For de-esterification, the podocytes were incubated with the CLB-HG buffer for 80 minutes at room temperature. The podocytes were imaged using the Zeiss Axio Observer 7 with Zeiss Axiocam 702 and Ibidi stage incubator at 37°C. Fluorescence intensity was recorded for 180 seconds using 340- and 380-nm excitation and 510-nm emission wavelengths. 1-Oleoyl-2-acetyl-sn-glycerol (Sigma-Aldrich, O6754) was pipetted in an end concentration of 100 µM at 10 seconds. [Ca2+]i was calculated using the ratio of 340 and 380 nm, wherein the results of five cells from a single well was pooled to obtain one measurement. In case of TRPC6 inhibition, podocytes were exposed to larixyl acetate (10 µM, Sigma-Aldrich, 02730595). In case of calcium chelation, podocytes were exposed to BAPTA-AM (5 μM, Sigma-Aldrich, A1076) for the last 2 hours. The quantitative [Ca2+]i data from three to six independent experiments were pooled.
Apoptosis Assay
Podocyte early and late apoptosis was determined by flow cytometry using the FITC annexin V apoptosis detection kit with PI (BioLegend, 640914). Cells were harvested by trypsinization and resuspended in a binding buffer. For annexin V and PI staining, cells were incubated for 15 minutes at 37°C in the dark. After addition of the binding buffer, the fluorescence was measured using the NovoCyte flow cytometer (Acea Biosciences). The cells that were annexinV+/PI− were considered early apoptotic.
Western Blot
Protein samples were isolated using a RIPA buffer (150 mM NaCl, 50 mM TrisHCl, 2 mM EDTA, 0.5% natrium deoxycholate, 0.2% sodium dodecyl sulfate [SDS], and 1% NP40 in MilliQ) supplemented with protease inhibitors (Roche, 11836170001) and phosphatase inhibitors (Roche, 4906845001). The protein content of cell extracts was determined using the BCA assay (Merck, 71285-3) to ensure equal sample loading. The experiments were performed in sextuplicate. Twenty micrograms of proteins were loaded and run on NuPAGE precast gel (4%–12% Bis-Tris, Invitrogen, WG1401BX10). Proteins were subsequently transferred to a polyvinylidene difluoride membrane using the iBlot system (Invitrogen, Waltham, MA, IB24001). Membranes were blocked in tris buffer saline (TBS) with 0.05% (v/v) Tween 20 (i.e., TBS-T) supplemented with 5% (w/v) milk overnight at 4°C. Primary antibodies were incubated for 4 hours at room temperature. The following antibodies were used: rabbit anti-LC3B (1:1000, Cell Signaling Technology, Danvers, MA, 2630), guinea pig anti-sequestosome 1 (SQSTM1) (1:10 000, Progen, Heidelberg, Germany, GP-62C), and rat anti-Tubulin (1:5000, Abcam, Ab6160). Membranes were washed in TBS-T and incubated with horseradish peroxidase-conjugated antibodies (1:2000, Cell Signaling Technology, 7074, 7076, 7077) for 2 hours. Protein bands were visualized using the ECL chemiluminescent Kit (Bio-Rad, 170-5070) on a LAS 4000 device (Fujifilm, Tokyo, Japan). Quantification was performed on ImageJ (NIH, Bethesda, MD) software (v2.3.0/1.53f).
Immunofluorescence on MPC-5
Cells were fixed with ice-cold methanol at −20°C for 20 minutes. Subsequently, cells were washed three times with phosphate-buffered saline (PBS) for 5 minutes at room temperature. Thereafter, cells were blocked for 1 hour at room temperature with a blocking solution consisting of TBS-T+3% (w/v) bovine serum albumin (BSA, Sigma-Aldrich). Subsequently, cells were incubated overnight at 4°C with primary antibodies in the blocking solution. Primary antibodies included guinea pig anti-SQSTM1 (1:400, Progen, GP62-C) and rabbit anti-LC3B (1:1000, Cell Signaling Technology, 43566S). The next day, cells were washed three times for 5 minutes with the blocking solution at room temperature. Donkey anti-rabbit Alexa 488 and goat anti–guinea pig Alexa 594 (1:500, all from Thermofisher Scientific) were subsequently incubated for 2 hours at RT in the dark. Then, cells were washed three times for 5 minutes with the blocking solution, and cell nuclei were stained with 1 µg/ml Hoechst 33342 (Thermofisher Scientific, H3570) for 10 minutes in the dark. Immediately afterward, fluorescent images were acquired using a Zeiss LSM900 inverted confocal laser scanning microscope with Airyscan 2 with a Plan-Apochromat 40×/1.2 water long-distance objective (Carl Zeiss Microscopy, Jena, Germany). The experiment was performed in sextuplicate, and at least five images were captured per experiment. The percentage of cell surface area positive for SQSTM1 and LC3B was calculated using semiautomatic macros in ImageJ (version 1.53c, National Institutes of Health). The percentage of SQSTM1 and LC3B-positive areas of the total podocyte cell surface area was calculated as a measure of the podocyte autophagic flux.
Immunofluorescence Staining of Mouse Kidney Sections
Kidneys were formalin-fixed paraffin-embedded (FFPE), sectioned (3 µm), deparaffined, and rehydrated before antigen retrieval in a heated citrate buffer (pH=6). Sections were then permeabilized with TBS-T+Triton 0.1% and blocked in TBS-T+3% BSA. Primary antibodies (Table 1) were incubated overnight at 4°C. After washing in TBS-T, secondary antibodies (Table 2) were incubated for 2 hours at RT. Nuclei were stained using Hoechst. Slides were mounted using Fluoromount-G (Aviva Systems Biology, San Diego, CA, 0100-01). Images were acquired on a Zeiss AxioPhot microscope and ZEN software. ImageJ (v2.3.0/1.53f) semiautomatic macros were used for quantifications of the percentage of NPHS1-positive and PODXL-positive areas per glomerular section on at least 30 glomeruli per mouse. Podocyte number was counted as the number of WT1+ nuclei per glomerular section on at least 30 glomeruli per mouse. SQSTM1 and GFP punctum areas within the glomerular PODXL area were measured on at least 30 glomeruli per mouse to monitor glomerular autophagic flux. Podocyte TRPC6 expression was quantified by measuring the TRPC6-positive area within the NPHS1-positive area.
Table 1.
Primary antibodies used on FFPE and FAAFPE samples
| Antigen | Host Species | Reference | Customer | Dilution | Sample |
|---|---|---|---|---|---|
| CAST | Rabbit | PAS-97541 | Invitrogen | 1:200 | Human |
| GFP | Rabbit | Ab290 | Abcam | 1:1000 | Mouse |
| NPHS1 | Guinea pig | GP-N2 | Progen | 1:100 | Mouse |
| PODXL | Goat | BAF1556 | R&D | 1:1000 | Mouse |
| SQSTM1 | Guinea pig | GP-62C | Progen | 1:400 | Human/Mouse |
| SYNPO | Mouse | #65194 | Progen | 1:20 | Human |
| TRPC6 | Rabbit | Ab233413 | Abcam | 1:500 | Mouse |
| TRPC6 | Rabbit | LS-C312900 | Lifespan Biosciences | 1:500 | Human |
| WT1 | Rabbit | Ab89901 | Abcam | 1:100 | Mouse |
Table 2.
Secondary antibodies used on FFPE and FAAFPE samples
| Antigen | Host Species | Reference | Provider | Dilution | Conjugate |
|---|---|---|---|---|---|
| Anti-goat | Donkey | A11057 | Invitrogen | 1:400 | Alexa 568 |
| Anti-guinea pig | Goat | A11073 | Invitrogen | 1:400 | Alexa 488 |
| Anti-guinea pig | Donkey | 706-605-148 | Jackson Immunol reasearch | 1:200 | Alexa 647 |
| Anti-guinea pig | Goat | A11076 | Invitrogen | 1:400 | Alexa 594 |
| Anti-mouse | Donkey | A21202 | Invitrogen | 1:400 | Alexa 488 |
| Anti-rabbit | Donkey | 711-545-158 | Jackson Immunol reasearch | 1:200 | Alexa 488 |
Analyses of Human Kidney Biopsies
The Paris Cité Université Nephropathology unit at Necker Hospital routinely received kidney biopsies for first-line histopathologic diagnosis from the nephrology unit at Georges Pompidou European Hospital. Patients referred between December 19, 2019, and December 31, 2022, and diagnosed with diabetes mellitus were identified for inclusion in this study. Control biopsies were identified as routine (month 3 or 12) post-kidney transplant biopsies. Biopsies from patients with proteinuria and minimal podocytopathy at the histology were also selected. The study was limited to patients older than 18 years. Clinical and biological data were retrospectively collected from the patients' files. The clinical data include sex, age at the time of renal biopsy, and history of diabetes mellitus. Biologic data at the time of renal biopsy included urinary albumin-to-creatinine ratio (UACR) and eGFR using the Modification of Diet in Renal Disease equation. These data are synthesized in Supplemental Tables 1 and 2.
Renal biopsies were fixed in formalin acetic acid and paraffin-embedded (FAAFPE). Three-micrometer sections were stained with Masson trichrome. Biopsies from patients with diabetes mellitus with at least two glomeruli with a class I–III glomerular diabetic lesion score40 were selected. Acellular fibrotic glomeruli were not analyzed. For control biopsies, only biopsies with no histologic glomerular abnormalities were selected. For immunofluorescence, the staining protocol was similar to the protocol described for mouse samples. Images were acquired on a Leica SP8 confocal microscope and LASX software and on a Zeiss axiophot microscope and ZEN software. Quantifications (Supplemental Table 3) were performed following the methodology previously described for mouse kidney section analysis.
Transmission Electron Microscopy Procedure
Mouse renal cortex samples were fixed in Trump fixative (Electron Microscopy Sciences, Hatfield, PE, 11750) at 4°C, processed for transmission electron microscopy as previously described,41 and were examined on a JEM1011 transmission microscope (JEOL, Tokyo, Japan) with an Orius SC1000 CCD camera (Gatan, Pleasanton, CA).
Public Databases
Single-cell RNAseq and single-nuclei RNAseq data were extracted from public databases. For KPMP, the results are based on data generated by KPMP: DK114886, DK114861, DK114866, DK114870, DK114908, DK114915, DK114926, DK114920, DK114923, DK114933, and DK114937. https://www.kpmp.org. Data were downloaded on April 19, 2022. Other single-cell RNAseq data were extracted from Kidney Interactive Transcriptomics (https://humphreyslab.com/SingleCell/). Bulk transcriptomics data were downloaded from the Nephroseq public database: https://www.nephroseq.org/.
Study Approval
Animal experiments were conducted according to the French veterinary guidelines and those formulated by the European Community for experimental animal use (L358-86/609EEC) and were approved by the Institut National de la Santé et de la Recherche Médicale, local University Research Ethics Committee, and French Ministry of Research (APAFIS-#22373).
The part of the research involving human samples was approved by the Ethics Evaluation Committee of Inserm (CEEI) (IRB00003888, FWA00005831). Archived samples were only used after obtaining informed consent during routine clinical follow-up consultation in the HEGP hospital if not previously done at the time of the initial consultation. No sample was obtained for the project itself; thus, the project did not modify medical care of the patients. Codes were used to ensure strict anonymity of patients. The study complies with the 2000 Declaration of Helsinki.
Results
Podocyte-Specific TRPC6 Overexpression is Associated with Autophagy Blockade and Podocyte Dedifferentiation in Murine Diabetes
First, we investigated the link between impaired podocyte autophagy and TRPC6 activation in DKD. We analyzed the expression of TRPC6, LC3B, and the autophagic cargo protein sequestosome 1 (SQSTM1, also called P62) in podocytes in two murine diabetic models: (1) a type 1 diabetes mellitus model of accelerated DKD combining streptozotocin (STZ)-induced diabetes with unilateral nephrectomy42 and (2) a type 2 diabetes model of DKD with the use of BTBRob/ob mice.43,44 GFP-LC3 reporter mice were used in the type 1 diabetes mellitus model to monitor the autophagic flux in vivo.37 In both models, we demonstrated increased podocyte-specific TRPC6 expression (Figure 1, A–C).
Figure 1.
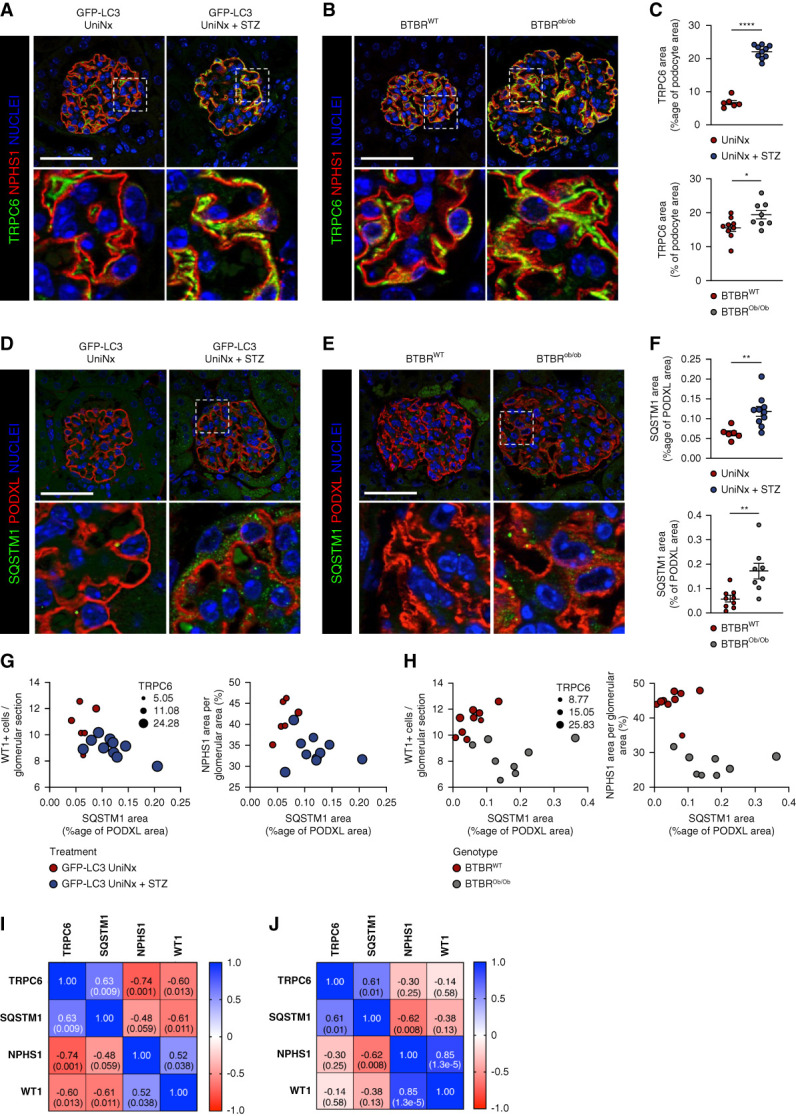
Increased TRPC6 expression and podocyte autophagy flux blockade in diabetic kidney disease. (A and B) Representative immunofluorescence images of TRPC6 (green) and NPHS1 (red) expression in the glomerulus from GFP-LC3 UniNx mice and GFP-LC3 UniNx+STZ mice (A) and from BTBRWT and BTBROb/Ob mice at 12 weeks of age (B). Nuclei are stained in blue with Hoechst. Scale bar: 50 μm. (C) Quantification of the TRPC6-positive area within the NPHS1 area in type I and type II diabetes models showing increased TRPC6 expression in DKD. n=6 GFP-LC3 UniNx, n=10 GFP-LC3 UniNx+STZ, n=9 BTBRWT, n=8 BTBROb/Ob mice. Values are presented as individual plots and mean±SEM. Unpaired t test: ****P < 0.0001 for GFP-LC3 UniNx versus GFP-LC3 UniNx+STZ and *P = 0.047 for BTBRWT versus BTBROb/Ob. (D and E) Representative immunofluorescence images of SQSTM1 (green) and podocalyxin/PODXL (red) expression in the glomerulus from GFP-LC3 UniNx mice and GFP-LC3 Unix+STZ mice (D) and from BTBR WT and BTBROb/Ob mice at 12 weeks of age (E). Nuclei are stained in blue with Hoechst. Scale bar: 50 μm. (F) Quantification of the SQSTM1 area within the PODXL area in type I and type II diabetes models showing increased SQSTM1 expression in DKD. n=6 GFP-LC3 UniNx, n=10 GFP-LC3 UniNx+STZ, n=9 BTBRWT, n=8 BTBROb/Ob mice. Values are presented as individual plots and mean±SEM. Unpaired t test: **P = 0.006 for GFP-LC3 UniNx versus GFP-LC3 UniNx+STZ and **P = 0.009 for BTBRWT versus BTBROb/Ob. (G–H) Multivariate representation of glomerular SQSTM1 area, NPHS1 area, WT1+ cells, and TRPC6 area in GFP-LC3 UniNx and GFP-LC3 UniNx+STZ mice (G) and in BTBR WT and BTBROb/Ob mice at 12 weeks of age (H). (I and J) Correlation matrix of the glomerular areas of SQSTM1, TRPC6, and NPHS1 in GFP-LC3 UniNx and GFP-LC3 UniNx+STZ mice (I) and in BTBR WT and BTBROb/Ob mice at 12 weeks of age (J). Numbers in squares represent Pearson r (P value). *P < 0.05, **P < 0.01, and ****P < 0.0001. Podocyte dedifferentiation and loss in mouse models of DKD are closely associated with the raises in podocyte TRPC6 and SQSTM1 expressions. Podocyte accumulation of SQSTM1 also correlates with podocyte TRPC6 raise in DKD models.
Next, we measured the GFP-LC3 punctum number, representing the number of autophagosomes and SQSTM1 expression. The number of GFP-LC3 puncta in podocytes was identical in STZ-treated and non–STZ-treated uninephrectomized mice (Supplemental Figure 1, A and B). In association with SQSTM1 expression, the GFP-LC3 punctum number per cell may point toward a blockade or an increase of the autophagic flux. SQSTM1 expression was increased in podocytes in both models (Figure 1, D and E). These data suggested decreased podocyte autophagic flux in the pathogenesis of DKD.
In the type 1 diabetes model, we confirmed that TRPC6 expression correlated positively with SQSTM1 expression and negatively with both NPHS1 expression and the number of podocytes (quantified as Wilms tumor 1 (WT1)-positive [+] cells per glomerulus) (Figure 1, G and I). Furthermore, SQSTM1 expression correlated negatively with the number of WT1+ cells per glomerulus and NPHS1 expression (Figure 1, G and I). In the type 2 diabetes model, we also confirmed that TRPC6 expression correlated positively with SQSTM1 expression, whereas SQSTM1 correlated negatively with NPHS1 expression (Figure 1, H and J). Therefore, our data indicate that podocyte TRPC6 overexpression in experimental DKD correlates with podocyte autophagic flux blockade and podocyte injury.
Regulation of Autophagic Flux in Podocytes Depends on TRPC6 Activity
We generated a doxycycline-inducible TRPC6 knockdown (KD) podocyte cell line. TRPC6 expression decreased by approximately 50% when TRPC6 KD podocytes were induced with doxycycline (Supplemental Figure 2A). Because uninjured podocytes show low TRPC6 expression levels, podocytes were exposed to doxorubicin to increase TRPC6 expression. Doxorubicin caused a five-fold increase in TRPC6 expression in podocytes in the absence of doxycycline. However, in the presence of doxycycline-induced TPRC6 gene knockdown, doxorubicin treatment no longer resulted in increased TRPC6 expression compared with uninjured podocytes, thereby validating the TRPC6 knockdown.
TRPC6 KD podocytes displayed decreased calpain activity in comparison with WT podocytes (Figure 2A). Moreover, treatment with the calpain inhibitor calpeptin reduced calpain activity in WT podocytes. No effect of calpeptin treatment was observed on calpain activity in TRPC6 KD (Figure 2A). Of note, calpain activity in WT podocytes treated with calpeptin was similarly low as the one in TRPC6 KD podocytes without calpeptin treatment, suggesting that calpeptin treatment and TRPC6 KD led to a similar order-of-magnitude effect on calpain activity.
Figure 2.
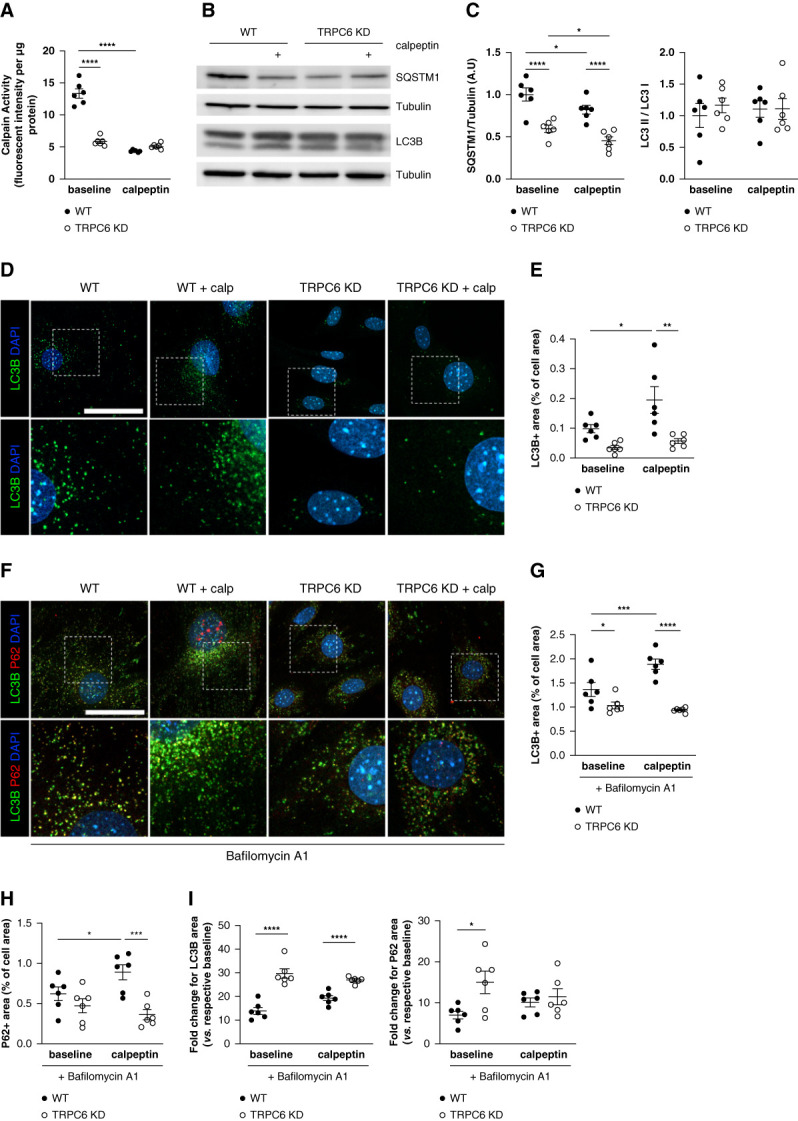
Calpain inhibition and TRPC6 KD enhance autophagic flux in vitro through decreased calpain activation. (A) Calpain activity measured in WT and TRPC6 KD MPC-5 podocytes in the absence or presence of the calpain inhibitor calpeptin (48 hours at 1 μM) normalized to protein content per sample. Two-way analysis of variance: treatment, ****P < 0.0001; phenotype, ****P < 0.0001. Tukey multiple comparison test: ****P < 0.0001 for baseline WT versus baseline TRPC6 KD, ****P < 0.0001 for baseline WT versus calpeptin WT. TRPC6 KD podocytes exhibit lower calpain activity than WT podocytes, within a similar range to podocytes treated with a calpain inhibitor. (B–C) Western blot analysis of the expression of SQSTM1/P62 and LC3B in MPC-5 WT and TRPC6 KD podocytes in the absence or presence of calpeptin (1 μM) for 48 hours and associated quantification. TRPC6 KD cells and calpeptin-treated cells have less SQSTM1/P62 abundance than WT podocytes, suggesting increased autophagy flux in these cells. Representative of n=6 replicates per condition. Protein expression was normalized to the tubulin level. Two-way analysis of variance for SQSTM1: treatment, **P = 0.008; phenotype, ****P < 0.0001. Tukey multiple comparison test: ****P < 0.0001 for baseline WT versus baseline TRPC6 KD, ****P < 0.0001 for calpeptin WT versus calpeptin TRPC6 KD, *P = 0.038 for baseline WT versus calpeptin WT, *P = 0.038 for baseline TRPC6 KD versus calpeptin TRPC6 KD. Two-way analysis of variance for LC3 II/LC3 I: treatment, P = 0.89; phenotype, P = 0.59. (D–E) Representative immunofluorescence images of LC3B (green) in WT and TRPC6 KD MPC-5 podocytes treated or not with calpeptin for 48 hours and associated quantification. TRPC6 KD and calpeptin-treated cells have a lower number of LC3+ dots per cell. Nuclei were stained with Hoechst (blue). Scale bar: 50 μm. Images are representative of n=6 replicates per condition. Two-way analysis of variance: treatment, *P = 0.02; phenotype, ***P = 0.0005. Tukey multiple comparison test: P = 0.28 for baseline WT versus baseline TRPC6 KD, **P = 0.003 for calpeptin WT versus calpeptin TRPC6 KD, *P = 0.047 for baseline WT versus calpeptin WT, P = 0.62 for baseline TRPC6 KD versus calpeptin TRPC6 KD. (F–H) LC3B (green) and SQSTM1 (red) immunofluorescences in WT and TRPC6 KD MPC-5 podocytes treated or not with calpeptin for 48 hours and associated quantification. Cells were treated with bafilomycin A1 for 4 hours to block lysosomal acidification and autophagosome–lysosome fusion. Nuclei were stained with Hoechst (blue). Scale bar: 50 μm. Images are representative of n=6 replicates per condition. Two-way analysis of variance for LC3B: treatment: *P = 0.0378; phenotype: ****P < 0.0001. Tukey; interaction: **P = 0.004 multiple comparison test: *P = 0.03 for baseline WT versus baseline TRPC6 KD, ****P < 0.0001 for calpeptin WT versus calpeptin TRPC6 KD, ***P = 0.001 for baseline WT versus calpeptin WT, P = 0.49 for baseline TRPC6 KD versus calpeptin TRPC6 KD. Two-way analysis of variance for SQSTM1: treatment: P = 0.35; phenotype: ***P < 0.0006; interaction: *P = 0.03. Tukey multiple comparison test: P = 0.22 for baseline WT versus baseline TRPC6 KD, ***P = 0.0002 for calpeptin WT versus calpeptin TRPC6 KD, *P = 0.03 for baseline WT versus calpeptin WT, P = 0.36 for baseline TRPC6 KD versus calpeptin TRPC6 KD. Calpeptin treatment induces SQSTM1+ and LC3+ dot accumulation in WT podocytes, showing that calpain activation inhibition increases the autophagy flux in podocytes. (I) Fold change expression for LC3B and SQSTM1 in bafilomycin A1 condition compared with non–bafilomycin A1 condition. Multiple unpaired t test Holm-Sidak method. For LC3B: WT bafilomycin A1 versus TRPC6 KD bafilomycin A1 ****P < 0.0001; WT bafilomycin A1 calpeptin versus TRPC6 KD bafilomycin A1 calpeptin ****P < 0.0001. For SQSTM1: WT bafilomycin A1 versus TRPC6 KD bafilomycin A1 *P = 0.02; WT bafilomycin A1 calpeptin versus TRPC6 KD bafilomycin A1 calpeptin P = 0.55. Whereas TRPC6 KD cells showed less LC3+ dots at baseline, bafilomycin A1 treatment unmask the higher autophagy flux in these cells with a higher fold change in LC3+ dots and SQSTM1+ dots number when compared with WT podocytes.
Because calpain activity depends on intracellular Ca2+, we assessed [Ca2+]i in WT and TRPC6 KD podocytes after stimulation of TRPC6 channels using its agonist 1-Oleoyl-2-acetyl-sn-glycerol (OAG). As expected, OAG induced a fast Ca2+ response in podocytes and this response was decreased in TRPC6 KD cells. Larixyl acetate, a TRPC6 inhibitor, completely blocked the [Ca2+]i response in podocytes (Supplemental Figure 2B). Similarly, the Ca2+ chelator BAPTA abolished the OAG-mediated Ca2+ response in podocytes (Supplemental Figure 2C). BAPTA-treated podocytes had decreased calpain activity when compared with nontreated podocytes, confirming the important role of Ca2+ to activate calpain (Supplemental Figure 2D). Of note, calpeptin or BAPTA treatment did not induce cell apoptosis (Supplemental Figure 2E).
We next investigated the effect of TRPC6 knockdown and calpain inhibition on the autophagic flux. LC3B exists in a cytosolic form LC3B-I and autophagosomal form LC3B-II. Quantification of the LC3B-II/LC3B-I ratio in Western blot is used to assess the autophagic flux. The LC3B-II/LC3B-I ratio did not differ between WT and TRPC6 KD podocytes (Figure 2C). In addition, calpeptin treatment did not result in an altered LC3B-II/LC3B-I ratio for both WT and TRPC6 KD podocytes. By contrast, calpeptin decreased SQSTM1 expression in WT podocytes. In addition, TRPC6 KD podocytes showed lower SQSTM1 expression compared with WT podocytes, and calpeptin treatment further enhanced this drop in SQSTM1 level (Figure 2, B and C). Immunofluorescence analysis confirmed that Western blot analysis as quantification of LC3B punctiform expression (i.e., autophagosomes) demonstrated a significant increase in autophagosomes in podocytes treated with calpeptin. We observed fewer LC3B puncta in TRPC6 KD cells than in WT podocytes (Figure 2, D and E). Ca2+ chelation using BAPTA also promoted LC3B punctum accumulation in WT podocytes but not in TRPC6 KD podocytes, thus suggesting autophagy induction by Ca2+ chelation (Supplemental Figure 2F). These findings indicated that TRPC6 knockdown, inhibition of Ca2+ signaling, and calpain inhibition stimulate the autophagic flux in podocytes.
Because autophagy is a highly dynamic process, we subsequently investigated the effect of calpain inhibition and TRPC6 KD on autophagy in the presence of bafilomycin A1, which prevents autolysosome degradation. Calpeptin treatment increased LC3B and SQTM1 accumulation in WT podocytes after bafilomycin A1 treatment (Figure 2, F–H). By contrast, calpeptin treatment had no effect on LC3B and SQTM1 expression in TRPC6 KD podocytes (Figure 2, F–H). We demonstrated a greater increase in LC3B and SQSTM1 accumulation in TRPC6 KD podocytes than WT podocytes on bafilomycin A1 treatment, indicating increased autophagic flux in the absence of full TRPC6 activity. No potentiator effect was measured in TRPC6 KD podocytes treated with calpeptin (Figure 2I).
Overall, these results showed that calpain inhibition by calpeptin enhances the autophagic flux in WT podocytes. Moreover, the inhibitory effect of calpain on autophagy is TRPC6-dependent.
Calpastatin Overexpression Prevents Podocyte Injury and Restores Podocyte Autophagic Flux in the Type I Diabetes Model
Next, we studied whether the diabetes-induced glomerular injury could be prevented by modulating calpain activity and thereby autophagy in vivo. We used mice that constitutively overexpressed the endogenous calpain inhibitor calpastatin and induced type I diabetes mellitus with accelerated DKD in these mice (GFP-LC3 CSTTg UniNx+STZ) and compared them with wild-type diabetic mice (GFP-LC3 UniNx+STZ). Transgenic overexpression of calpastatin did not alter glycemia levels or body weight loss after inducing type I diabetes mellitus (Supplemental Figure 3, A–B). The baseline urinary albumin-to-creatinine ratio was identical between the two genotypes (Supplemental Figure 3C). However, transgenic overexpression of calpastatin prevented the development of microalbuminuria 6 weeks after inducing type I diabetes (Figure 3A). Six weeks after the STZ injections, podocyte injury and podocyte loss were observed in GFP-LC3 UniNx+STZ mice as evidenced by reduced NPHS1 expression and decreased number of WT1+ cells, respectively. By contrast, calpastatin overexpression prevented NPHS1 loss but not podocyte loss (Figure 3, B and C). Consistently, ultrastructure analysis with transmission electron microscopy showed a more prominent reduced foot process effacement in GFP-LC3 CSTTg UniNx+STZ mice than in GFP-LC3 UniNx+STZ mice (Figure 3E).
Figure 3.

Glomerular filtration function and glomerular injury in diabetic mice with calpastatin overexpression. (A) Evolution of the urinary albumin-to-creatinine ratio (UACR) in GFP-LC3 UniNx+STZ and GFP-LC3 CSTTg UniNx+STZ mice. Values are presented as individual plots and mean±SEM. Two-way analysis of variance: Weeks, P = 0.003; Genotype, P = 0.13. Tukey multiple comparison test: ***P = 0.0004 for GFP-LC3 UniNx+STZ versus GFP-LC3 CSTTg UniNx+STZ at week 6. Calpastatin overexpression partially prevents diabetes-induced microalbuminuria. (B) Representative immunofluorescence of WT1 (Green) and NPHS1 (Red) expression in glomeruli from GFP-LC3 UniNx and GFP-LC3 CSTTg UniNx mice 6 weeks after being injected or not with STZ. Nuclei are stained in blue with Hoechst. Scale bar: 50 μm (C) Quantification of NPHS1 area and WT1+ nuclei per glomerular area from GFP-LC3 UniNx+STZ and GFP-LC3 CSTTg UniNx+STZ compared with control UniNx mice. n=6 GFP-LC3 UniNx, n=9 GFP-LC3 UniNx+STZ, n=7 GFP-LC3 CSTTg UniNx, n=8 GFP-LC3 UniNx+STZ mice. Values are presented as individual plots and mean±SEM. Two-way analysis of variance for NPHS1: genotype, *P = 0.02; treatment, ****P < 0.0001. Tukey multiple comparison test: ***P = 0.0002 for GFP-LC3 UniNx versus GFP-LC3 UniNx+STZ, *P = 0.03 for GFP-LC3 UniNx+STZ versus GFP-LC3 CSTTg UniNx+STZ. Two-way analysis of variance for WT1+: genotype, P = 0.20; treatment, ***P = 0.0002. Tukey multiple comparisons test: *P = 0.03 for GFP-LC3 UniNx versus GFP-LC3 UniNx+STZ and *P = 0.02 for GFP-LC3 CSTTg UniNx versus GFP-LC3 CSTTg UniNx+STZ. (D) Transmission electron microscopy images of the podocyte ultrastructure in GFP-LC3 UniNx+STZ and GFP-LC3 CSTTg UniNx+STZ mice. Scale bar: 1 μm. Calpastatin overexpression partially protects podocytes from diabetes-induced podocyte dedifferentiation.
Finally, we showed that glomerular SQSTM1 expression was decreased in GFP-LC3 CSTTg UniNx+STZ mice compared with GFP-LC3 UniNx+STZ mice (Figure 4, A and B) and that glomerular LC3B expression was increased (Figure 4, C and D). These results indicate that calpastatin overexpression prevented DKD-induced glomerular autophagic flux blockade.
Figure 4.

Calpastatin overexpression restores glomerular autophagic flux in the type I diabetes model. (A) Representative immunofluorescence of SQSTM1 (green) and PODXL (red) in the glomerulus from GFP-LC3 UniNx and GFP-LC3 CSTTg UniNx mice 6 weeks after being injected or not with STZ. Nuclei are stained in blue with Hoechst. Scale bar: 50 μm (B) Quantification of the SQSTM1 area in the PODXL area from GFP-LC3 UniNx and GFP-LC3 CSTTg UniNx mice 6 weeks after being injected or not with STZ. For ethical reasons, we did not duplicate GFP-LC3 UniNx and GFP-LC3 CSTTg uniNx groups, and quantifications of these groups are already presented in Figure 1H and Supplemental Figure 1B. n=7 GFP-LC3 UniNx, n=10 GFP-LC3 UniNx+STZ, n=7 GFP-LC3 CSTTg uniNx, n=8 GFP-LC3 CSTTg UniNx+STZ mice. Two-way analysis of variance: genotype, P = 0.13; treatment, *P = 0.0223. Tukey multiple comparison test: **P = 0.002 for GFP-LC3 UniNx versus GFP-LC3 UniNx+STZ and **P = 0.007 for GFP-LC3 UniNx versus GFP-LC3 CSTTg UniNx+STZ. SQSTM1 accumulates in WT diabetic mice but not in mice with calpastatin overexpression. (C) Representative immunofluorescence of GFP-LC3 (green) and PODXL (red) in glomeruli from GFP-LC3 UniNx and GFP-LC3 CSTTg UniNx mice 6 weeks after being injected or not with STZ. Nuclei are stained in blue with Hoechst. (D) Quantification of the GFP-LC3 punctum area in the PODXL area from GFP-LC3 UniNx and GFP-LC3 CSTTg UniNx mice 6 weeks after being injected or not with STZ. n=6 GFP-LC3 UniNx, n=10 GFP-LC3 UniNx+STZ, n=7 GFP-LC3 CSTTg uniNx, n=8 GFP-LC3 CSTTg UniNx+STZ mice. Two-way analysis of variance: genotype, P = 0.09; treatment,**P = 0.008. Tukey multiple comparison test: **P = 0.02 GFP-LC3 UniNx+STZ versus GFP-LC3 CSTTg UniNx+STZ and **P = 0.02 for GFP-LC3 CSTTg UniNx versus GFP-LC3 CSTTg UniNx+STZ. We observe an increased number of LC3+ dots in podocytes from diabetic mice with calpastatin overexpression when compared with WT diabetic mice.
Pharmacologic Calpain and TRPC6 Inhibition Restore Podocyte Autophagic Flux and Prevent Podocyte Injury in the Type II Diabetes Model
BD4-410, a calpain inhibitor,45–47 and BI-749327, a TRPC6 antagonist, were used to treat diabetic BTBRob/ob mice. Six weeks of daily administration of BDA-410 or BI-749327 significantly prevented the development of microalbuminuria in BTBRob/ob mice (Figure 5A). Calpain and TRPC6 inhibition partially prevented podocyte dedifferentiation in BTBRob/ob mice, as evidenced by WT1 and NPHS1 immunostaining, but not podocyte loss (Figure 5, B and C). COL4 deposition was also significantly reduced in BTBRob/ob mice treated with BDA-410 (Supplemental Figure 4, A and B). TEM ultrastructure analysis confirmed the mesangial expansion with collagen structure in BTBRob/ob mice (Supplemental Figure 4C). The capillary loops almost completely disappeared in BTBRob/ob mice with focal complete podocyte foot process effacement. Strikingly, BDA-410–treated mice displayed expanded capillary loops that maintained properly flattened endothelial cells and preserved podocyte foot processes (Figure 5D).
Figure 5.
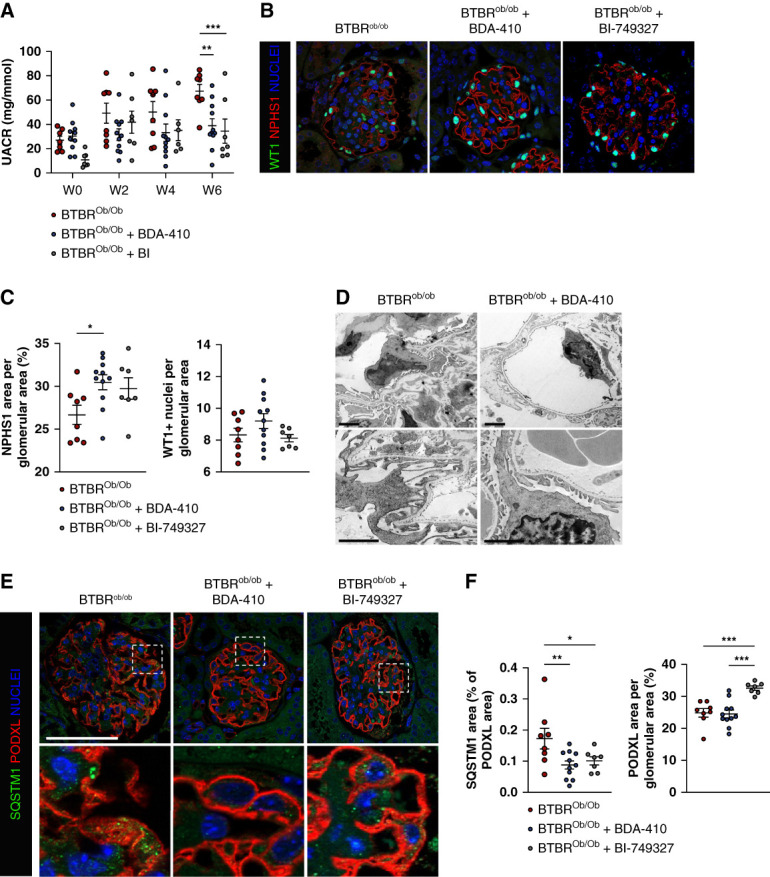
Pharmacologic calpain-1 and TRPC6 inhibition restores glomerular autophagic flux and preserve the glomerular structure in the type II diabetes model. (A) Evolution of the albumin-to-creatinine ratio (UACR) in BTBRob/ob mice treated or not with BDA-410 or BI-749327 for 6 weeks. Values are presented as individual plots and mean±SEM. Two-way analysis of variance: weeks, P = 0.001; treatment, P = 0.0007. Fisher LSD multiple comparison test: P = 0.001 for BTBRob/ob and BTBRob/ob+BDA-410 mice at week 6, P = 0.0006 for BTBRob/ob and BTBRob/ob+BI-749327 mice at week 6. Pharmacologic calpain-1 and TRPC6 inhibition with BDA-410 and BI-749327 partially protects mice from diabetes-induced microalbuminuria. (B) Representative immunofluorescence of WT1 (green) and NPHS1 (red) expression in glomeruli from BTBRob/ob mice treated or not with BDA-410 or BI-749327 for 6 weeks. Nuclei are stained in blue with Hoechst. Scale bar: 50 μm. (C) Quantification of the NPHS1 area and the WT1+ nuclei per glomerular area. n=8 BTBRob/ob, n=11 BTBRob/ob+BDA-410, n=7 BTBRob/ob+BI-749327. Values are presented as individual plots and mean±SEM. WT1: one-way analysis of variance, P = 0.17; Fisher LSD test: P = 0.15 for BTBRob/ob versus BTBRob/ob+BDA-410 and P = 0.77 for BTBRob/ob versus BTBRob/ob+BI-749327. NPHS1: one-way analysis of variance, *P = 0.039; Fisher LSD test: *P = 0.013 for BTBRob/ob versus BTBRob/ob+BDA-410 and P = 0.066 for BTBRob/ob versus BTBRob/ob BI-749327. (D) Transmission electron microscopy images of the podocyte ultrastructure in BTBRob/ob and BTBRob/ob+BDA-410 mice. Scale bar: 2 μm. BDA-410 protects podocytes from diabetes-induced dedifferentiation. (E) Representative immunofluorescence of SQSTM1 (green) and PODXL (red) in the glomerulus from BTBRob/ob mice treated or not with BDA-410 or BI-749327 for 6 weeks. Nuclei are stained in blue with Hoechst. Scale bar: 50 μm. (F) Quantification of the PODXL area and SQSTM1 area in the PODXL area from BTBRob/ob mice treated or not with BDA-410 or BI-749327 for 6 weeks. n=8 BTBRob/ob, n=11 BTBRob/ob+BDA-410, n=7 BTBRob/ob+BI-749327. Values are presented as individual plots and mean±SEM. SQSTM1: one-way analysis of variance, P = 0.018; Fisher LSD test: **P = 0.006 for BTBRob/ob versus BTBRob/ob+BDA-410 and *P = 0.03 for BTBRob/ob versus BTBRob/ob+BI-749327. PODXL: one-way analysis of variance, ***P = 0.0001; Fisher's LSD Test: P = 0.83 for BTBRob/ob versus BTBRob/ob+BDA-410 and ***P = 0.0003 ofr BTBRob/ob versus BTBRob/ob+BI-749327. BDA-410 and BI-749327 prevent diabetes-induced SQSTM1 accumulation in podocytes.
Whereas BTBRob/ob mice presented glomerular accumulation of SQSTM1, BDA-410 or BI-749327 treatment prevented such SQSTM1 glomerular accumulation in BTBRob/ob mice, showing that the glomerular autophagic flux is maintained in BDA-410–treated and BI-749327–treated mice. Interestingly, BTBRob/ob mice treated with a TRPC6 antagonist displayed preserved glycocalyx compared with BTBRob/ob, whereas BTBRob/ob BDA-410–treated mice did not (Figure 5, E and F). Both BTBRob/ob-treated mice showed a reduction in the podocyte-specific TRPC6 overexpression previously described in BTBRob/ob mice, whereas GFP-LC3 CSTTg mice had similar TRPC6 expression to GFP-LC3 mice (Supplemental Figure 5). BI-749327 also prevented podocyte injury in the type 1 diabetes model (UniNx+STZ mice) together with podocyte autophagic flux maintenance (Supplemental Figure 6).
Together, these results demonstrated that pharmacologic calpain or TRPC6 inhibition using BDA-410 or BI-749327 promotes glomerular autophagic flux maintenance and partially prevents glomerular injury by preserving the glomerular filtration barrier ultrastructure and function in BTBRob/ob diabetic mice.
TRPC6 Overexpression, Calpastatin Decline, and SQSTM1 Accumulation Correlate with Glomerular Injury in Human Kidneys from Patients with Diabetes
Next, we analyzed the expression of TRPC6, calpastatin/CAST, and SQSTM1 in renal biopsies from patients with DKD. Whereas TRPC6 abundance was very low in the podocytes of control renal biopsies, it was significantly increased in podocytes from patients with DKD (Figure 6, A and B). Furthermore, SQSTM1 accumulated in podocytes from patients with DKD compared with the ones from control biopsies (Figure 6, A and B). A glomerulus-by-glomerulus Pearson correlation analysis showed a negative correlation between podocyte-specific TRPC6 abundance and expression of the podocyte maturity marker synaptopodin/SYNPO. TRPC6 expression was also positively correlated with glomerular diabetic lesion score. Moreover, SQSTM1 expression correlated negatively with SYNPO expression, correlated positively with TRPC6 expression, and correlated positively with glomerular diabetic lesion score (Figure 6C). CAST was predominantly expressed in podocytes in glomeruli from controls and its expression tended to decrease in glomeruli from patients with DKD (Figure 6D and Supplemental Figure 7). Pearson correlation analysis showed a positive correlation between CAST expression and SYNPO expression. CAST expression also correlated negatively with glomerular diabetic lesion score. SQSTM1 did not correlate with CAST expression (Supplemental Figure 7). Podocyte TRPC6 overexpression, SYNPO decrease, and autophagy deficiency were also correlated with renal function, as evaluated by eGFR and UACR (Figure 6E). Taken together, we showed that when compared with control kidneys, DKD is associated with podocyte TRPC6 overexpression, podocyte autophagic flux blockade, and glomerular and podocyte injury.
Figure 6.

TRPC6 expression, autophagic flux assessment, and podocyte injury in patients with diabetic kidney disease. (A) Representative immunofluorescence of SYNPO (white), TRPC6 (green), and SQSTM1 (red) expression in the glomerulus from controls and patients with diabetes. Nuclei are stained in blue with Hoechst. Scale bar: 75 μm. (B) Quantifications of SYNPO, TRPC6, SQSTM1, and CAST expressions. Values are presented as individual plots of each patient and mean±SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001. SYNPO and CAST expressions are low in patients with diabetes while TRPC6 expression increases and SQSTM1 accumulates in podocytes. (C) Correlation matrix of the glomerular areas of SQSTM1, TRPC6, and SYNPO and the glomerular diabetic lesion score (GDLS) in patients by individual glomerulus. Numbers in squares represent Pearson r (P value). (D) Quantifications of CAST expression in glomeruli. Values are presented as individual plots of each patient and mean±SEM. (E) Correlation between the mean glomerular expression of SQSTM1, TRPC6, and SYNPO and biologic parameters of patients (eGFR and UACR). Numbers in squares represent: Pearson r (P value); glomerular diabetic lesion score; UACR, urinary albumin-to-creatinine ratio.
Finally, to determine whether changes in TRPC6, SQSTM1, SYNPO, and CAST expressions were specific to DKD, we also compared their expression in renal biopsies from patients with DKD and patients with proteinuria but nondiabetic podocytopathy (minimal change disease and membranous nephropathy) (Figure 7A). Analysis of variances demonstrated significant differential glomerular expression of SQSTM1, SYNPO, and CAST, but not TRPC6, between DKD and other proteinuric nondiabetic patients (Supplemental Table 4). Principal component analysis using SQSTM1, SYNPO, CAST, and TRPC6 expressions showed clustering of DKD patients with proteinuria on one hand and other proteinuric nondiabetic patients on the other hand (Figure 7B). Finally, to decipher whether a combination of the four markers was sufficient to discriminate patients with DKD from patients with proteinuria, we performed a binomial logistic regression using SQSTM1, TRPC6, SYNPO, and CAST as covariables. The best models were the one combining SYNPO, CAST, and SQSTM1 and the one with the 4 markers (Supplemental Tables 5 and 6), and the last (model 4) predicted patients with DKD and proteinuria with 82.9% accuracy, 85% specificity, and 81% sensitivity for a cutoff value of 0.6 (Supplemental Table 7). The ROC curve had an AUC of 0.9 (Figure 7C). Taken together, we demonstrated that changes in SQSTM1, CAST, TRPC6, and SYNPO expressions in podocytes are closely linked to diabetes-associated podocytopathy.
Figure 7.
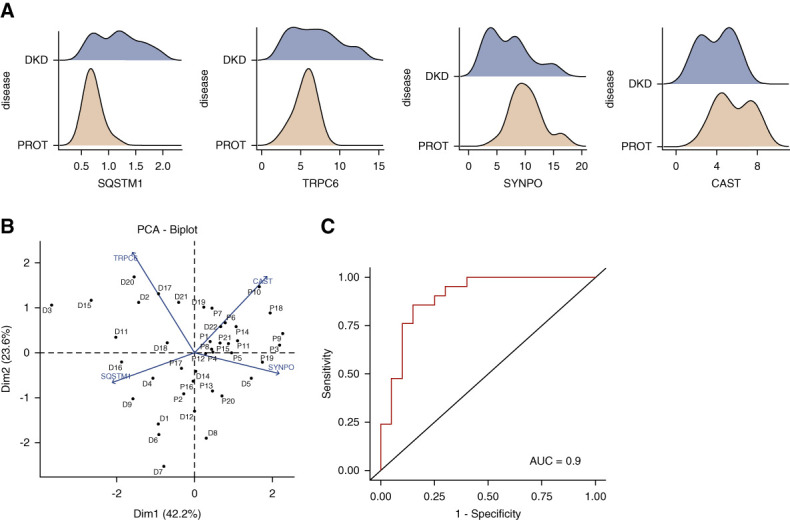
SQSTM1, TRPC6, SYNPO, and CAST glomerular area discriminate patients with DKD from those with proteinuria. (A) Dispersion of SQSTM1, TRPC6, SYNPO, and CAST glomerular area in glomeruli in patients with DKD (blue) or patients with proteinuria (yellow, PROT). (B) Principal component analysis of patients with DKD and PROT using SQSTM1, TRPC6, SYNPO, and CAST glomerular areas as covariables. (C) We generated a binomial logistic regression model with SQSTM1, TRPC6, SYNPO, and CAST glomerular areas as covariables, and a ROC curve illustrating prediction was plotted. AUC=0.9.
Discussion
This study uncovers the involvement of the Ca2+ channel TRPC6 and cysteine protease calpain as a crucial pathway impairing the cytoprotective podocyte autophagy and promoting diabetic glomerular damage. Our data suggest that TRPC6-mediated Ca2+ influx results in elevated calpain activity, impaired podocyte autophagy, podocyte injury, and development of proteinuria in DKD (Figure 8). Transgenic overexpression of calpastatin and pharmacologic inhibition of TRPC6 and calpain normalized podocyte autophagic flux and prevented podocyte injury and proteinuria in two models of DKD.
Figure 8.
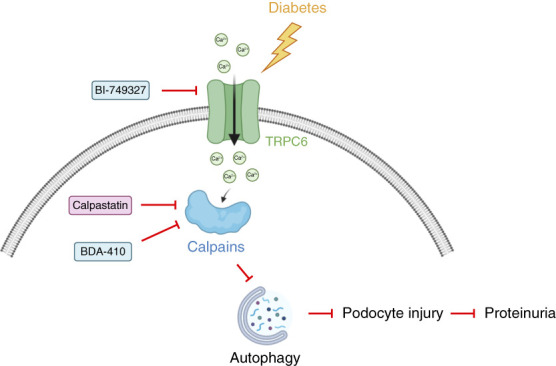
Interplay between calpain and TRPC6 results in impaired podocyte autophagy and podocyte injury in diabetic kidney disease (DKD). DKD results in enhanced transient receptor potential channel 6 (TRPC6) expression and subsequently elevated TRPC6-mediated Ca2+ influx in podocytes. Increased TRPC6-mediated Ca2+ influx leads to enhanced calpain activation and autophagy inhibition. Impaired autophagy will lead to podocyte injury and proteinuria formation. Inhibition of calpain (by calpastatin or pharmacologic inhibition) or TRPC6 inhibition restores podocyte autophagy flux and prevents proteinuria development in DKD.
Normal calpain activity plays a role in key signaling processes: They regulate cell behavior, actin cytoskeletal dynamics, cell adhesion and motility, endoplasmic reticulum stress, apoptosis, and inflammation on interaction with their multiple substrates. Conversely, abnormal calpain activation is responsible for the degradation of most of the cellular protein pool.
Expression of CAPN1 is not detected in renal cells in single-cell RNAseq data from Kidney Interactive Transcriptomics and the Kidney Tissue Atlas public database, whereas CAPN2 and CAST are highly enriched in podocytes (Supplemental Figure 8). We confirmed enrichment of CAST expression in podocytes in mouse and human kidneys. Using in silico analysis, we previously found that calpains may cleave some constituents of the podocyte,41 besides targeting cytoskeleton proteins48,49 and proteins of the autophagy machinery. High coexpression of CAPN2 and its inhibitor CAST within the same cells may, therefore, suggest that tight regulation of calpain activation/inhibition is required to maintain podocyte homeostasis.
We observed a substantial decrease in CAST expression in podocytes in human kidney biopsies from patients with diabetes. The Nephroseq database showed that glomerular changes in CAPN1 and CAST mRNA expression are associated with NPHS1 mRNA expression and GFR in patients with DKD (Supplemental Figure 9). Interestingly, CAPN1 expression increased in glomeruli from patients with DKD concomitantly to CAST, and CAPN2 decreased. These data support the notion that modulation of calpain activity may affect podocyte homeostasis in DKD. Future studies should elucidate the molecular mechanisms responsible for changes in calpains and calpastatin expression and activity in DKD.
BDA-410 is an orally active calpain inhibitor with relatively selective inhibition of calpain-1 (Ki value of 130 nM) rather than calpain-2 (Ki value of 630 nM).47,50 BDA-410 has proven promising effects in neurologic disorders and aging in rodents. It improved the memory and synaptic transmission in Alzheimer disease.50 It also alleviated neurodegeneration in inherited cerebellar ataxia.51 In aging-related disease models, BDA-410 ameliorated the aging phenotype by increased lipogenesis, body weight reduction,52 and enhancement of muscle force.53 It also allowed recovery from the age-related phenotype in α-klotho deficient mice, a model of accelerated aging.45 No significant side effects were reported in any of these studies, suggesting good tolerance of systemic administration of this calpain 1 inhibitor in mice, at least at the doses used and for the duration of treatment. We showed that BDA-410 ameliorated diabetic-induced glomerular injury in a mouse model of type 2 diabetes mellitus, with preservation of the autophagic flux in glomeruli. Interestingly, BDA-410 treatment not only prevented podocyte injury but also preserved the glomerular structure overall. Although protection of podocytes is pivotal for the glomerular architecture, we cannot rule out that calpain-1 inhibition may also be beneficial for other cells than podocytes in kidneys. Because physiologic roles of calpains are generally auxiliary, their inhibition is usually well-tolerated and supports the rationale for using conventional calpain inhibitors to treat diseases aggravated by increased calpain activity. BDA-410 is not currently implicated in a clinical trial, but other calpain inhibitors are.54,55 Therefore, targeting calpains might be a promising therapeutic strategy for the treatment of DKD by restoring podocyte autophagy. Nevertheless, the safety of chronic calpain inhibition in DKD would require careful attention and clinical trials with long-term follow-up.
In addition to calpain inhibitors, the results of this study also support the use of TRPC6 inhibitors for preventing podocyte injury in DKD by restoring autophagy.56,57 Our data linking the deleterious TRPC6/calpain axis to autophagy inhibition and podocyte damage might be generalizable to other podocytopathies beyond DKD. Likewise, increased podocyte-specific expression of TRPC6 is observed in other glomerulopathies than DKD, for example, focal segmental glomerulosclerosis (FSGS)58 and impaired podocyte autophagy is known to contribute to further disease progression of FSGS.59,60 Interestingly, the TRPC6 inhibitor BI-764198 is being tested in a phase 2 clinical trial for treatment of primary FSGS (NCT05213624). Therefore, it would be interesting to investigate in future studies whether TRPC6 also contributes to impaired podocyte autophagy and further disease progression in the context of FSGS through aberrant calpain activation.
The control of podocyte autophagy by the TRPC6–calpain cascade in two models of type I and type II diabetes with strong correlative expression data in human tissues is a salient novelty of this study. We and others have previously demonstrated that maintenance of the autophagic flux is crucial to maintain podocyte homeostasis in DKD.19,20 However, the tuning of podocyte autophagy in DKD remains a challenge. Calpains have been found to participate in autophagic flux regulation through different actions.27,28,61 The mechanisms linking calpain-mediated inhibition of podocyte autophagy during DKD progression may involve, at least in part, the activation of the endogenous RAAS. Accordingly, we previously demonstrated that calpastatin constitutive overexpression in mice could stimulate the autophagic flux in podocytes and prevented podocyte injury in a model of angiotensin II–induced hypertensive glomerular injury, limiting glomerular oxidative and ER stress.41 The regulation of calpains in DKD had not been explored, nor its links with the TRPC6 channel. It may well be that blunting the TRPC6–calpain cascade exerts cytoprotective action beyond the promotion of autophagy. For example, in a genetic model of podocyte injury induced by deletion of the cyclin G-associated kinase GAK, pharmacologic calpain inhibition or Capns1 deletion mitigated the severity of glomerulosclerosis and proteinuria through the regulation of the NF-κB/GADD45B pathway.62 Our study complements experimental data generated in specific inflammatory or genetic models of kidney disease and in vitro. In a model of antiglomerular basement membrane nephritis, calpain activation promoted Talin-1 cleavage and actin cytoskeleton disorganization in podocytes, participating in podocyte injury.49 Thus, our current study raises the hypothesis of an interplay between podocyte cytoskeletal dynamics and crucial maintenance of an intact autophagy.
Importantly, we could support the clinical relevance of these findings in patients with diabetes. The expression of both SQSTM1, physiologically degraded by autophagy, and TRPC6 were enhanced in podocytes from patients with diabetes. Interestingly, SQSTM1 expression showed a negative correlation with the expression of the podocyte marker synaptopodin, suggesting that altered autophagy is somehow related to podocyte dedifferentiation. The additional significant association of SQSTM1 expression with albuminuria is also consistent with a key role of podocyte autophagy for the maintenance of the glomerular filtration barrier in humans as shown in mice. Furthermore, such a pattern of high SQSTM1 and TRPC6 podocyte abundance was significantly correlated with eGFR, a crucial functional measure of DKD progression.
In conclusion, the data of this study shed new light on the underlying pathogenic mechanisms of diabetic glomerular injury and highlights the therapeutic potential of sustaining the podocyte autophagy and integrity through inhibition of TRPC6-mediated signaling and/or calpain as treatment options for DKD.
Supplementary Material
Acknowledgments
We thank Nicolas Perez, Corina Suldac, and the ERI U970 team for assistance in animal care and handling; Nicolas Sorhaindo for biochemical measurements (ICB-IFR2, laboratoire de Biochimie, Hôpital Bichat, Paris, France); Corinne Lesaffre for histological staining (PH2I, histology platform PARCC, Paris, France); and Alain Schmitt and Jean-Marc Masse for transmission electron microscopy (Institut Cochin, Paris, France). We thank the Radboudumc Technology Center Microscopy for use of their microscopy facilities. We acknowledge administrative support from Véronique Oberweis, Bruno Pillard, and Cyrille Mahieux (PARCC, Paris, France).
Footnotes
Y.S. and D.Y. contributed equally to this work.
Disclosures
A. Karras reports Consultancy: Alnylam, GSK, Novartis, Otsuka, Vifor; Honoraria: ASTRAZENECA, Bohringer-Ingelheim, GSK, NOVARTIS, OTSUKA, PFIZER, VIFOR; Advisory or Leadership Role: Novartis, Otsuka, Vifor; and Speakers Bureau: AstraZeneca, Boehringer-Ingelheim, Otsuka, Pfizer, Vifor. E. Letavernier reports Consultancy: Biocodez, IKI, Withings; Research Funding: Advicenne, Coloplast; Honoraria: Biocodex, IKI, Withings; and Patents or Royalties: Insert transfert. T. Nijenhuis reports Research Funding: Dutch Kidney Foundation, Radboudumc; and Advisory or Leadership Role: Scientific board Dutch Society for Nephrology, Scientific advisory board Dutch Kidney Foundation, Speaker and chair Tubulopathies Expert Working Group and Executive Committee member, European Rare Kidney Disorders Network (ERKNet), and Board member ERA Working Group Genes & Kidney. M. Rabant reports Honoraria: Pfizer. R. Saito reports Employer: Mitsubishi Tanabe Pharma Corporation; and Other Interests or Relationships: IQ consortium; Life intelligence consortium (LINC). D.C. 't Hart reports Employer: Genome Diagnostics; Radboud university medical center. P.-L. Tharaux reports Consultancy: Alentis Therapeutics; Research Funding: Alentis Therapeutics; Honoraria: Travere Therapeutics; and Advisory or Leadership Role: French National Institute for Medical Research (INSERM), French Society of Cardiology (SFC), French Society of Hypertension (SFHTA), Associate Editor: Kidney International; Advisory Board Member: Nature Review Nephrology. J. Van der Vlag reports Other Interests or Relationships: Secretary Science committee Dutch federation of Nephrology. D. Yildiz reports Other Interests or Relationships: Nierstichting (The Dtuch Kidney Foundation). All remaining authors have nothing to disclose.
Funding
This work was supported by the French institute for health and medical research (Inserm). Y.S. was supported by a graduate fellowship from the “Fondation de France Recherche Cardiovasculaire” and “Fondation pour la Recherche Médicale (FRM).” The project was supported by the “Société francophone du diabète (SFD)” and “Laboratoire Servier” as part of a SFD-Laboratoire Servier research grant 2020 and supported by the EFSD/Novo Nordisk Programme for Diabetes Research in Europe (O.L.). D.C.H. and D.Y. were supported by a Kolff Senior Postdoc Career Stimulation Grant 13OKS023 (to T.N.) and a PhD Student Grant 19OP023 (to J.V. and T.N.) from the Dutch Kidney Foundation.
Author Contributions
Conceptualization: Olivia Lenoir, Tom Nijenhuis, Yann Salemkour, Pierre-Louis Tharaux, Johan van der Vlag, Dilemin Yildiz.
Data curation: Léa Dionet, Olivia Lenoir, Nassim Mahtal, Yann Salemkour, Ryuta Saito, Daan C. ‘t Hart, Kim A.T. Verheijden, Dilemin Yildiz.
Formal analysis: Léa Dionet, Olivia Lenoir, Nassim Mahtal, Tom Nijenhuis, Yann Salemkour, Ryuta Saito, Daan C. ‘t Hart, Pierre-Louis Tharaux, Johan van der Vlag, Kim A.T. Verheijden, Dilemin Yildiz.
Funding acquisition: Olivia Lenoir, Yann Salemkour, Tom Nijenhuis.
Investigation: Jean-Daniel Delbet, Alexandre Karras, Olivia Lenoir, Emmanuel Letavernier, Marion Rabant.
Methodology: Olivia Lenoir, Yann Salemkour, Tom Nijenhuis, Dilemin Yildiz, Johan van der Vlag.
Project administration: Olivia Lenoir.
Resources: Jean-Daniel Delbet, Alexandre Karras, Olivia Lenoir, Emmanuel Letavernier, Marion Rabant, Ryuta Saito.
Supervision: Olivia Lenoir, Tom Nijenhuis, Pierre-Louis Tharaux, Johan van der Vlag.
Validation: Jean-Daniel Delbet, Alexandre Karras, Olivia Lenoir, Tom Nijenhuis, Marion Rabant, Ryuta Saito, Pierre-Louis Tharaux, Johan van der Vlag.
Visualization: Léa Dionet, Olivia Lenoir, Tom Nijenhuis, Ryuta Saito, Daan C. ‘t Hart, Pierre-Louis Tharaux, Kim A.T. Verheijden, Dilemin Yildiz, Yann Salemkour.
Writing – original draft: Jean-Daniel Delbet, Alexandre Karras, Olivia Lenoir, Emmanuel Letavernier, Nassim Mahtal, Tom Nijenhuis, Marion Rabant, Yann Salemkour, Daan C. ‘t Hart, Pierre-Louis Tharaux, Johan van der Vlag, Dilemin Yildiz.
Writing – review & editing: Olivia Lenoir, Tom Nijenhuis, Yann Salemkour, Daan C. ‘t Hart, Pierre-Louis Tharaux, Johan van der Vlag, Dilemin Yildiz.
Data Sharing Statement
All data are available in the main text or the Supplemental materials.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/E527.
Supplemental Figure 1. Autophagic flux assessment in type diabetes mellitus in mice.
Supplemental Figure 2. Validation of TRPC6 KD construction in MPC-5.
Supplemental Figure 3. Biologic parameters in mice with calpastatin overexpression.
Supplemental Figure 4. Pharmacologic calpain-1 inhibition with BDA-410 or TRPC6 inhibition with BI-749327 prevents glomerulosclerosis in the type II diabetes model.
Supplemental Figure 5. TRPC6 expression in diabetes models.
Supplemental Figure 6. Pharmacologic TRPC6 inhibition with BI-749327 prevents podocyte injury and restores glomerular autophagic flux in the type I diabetes model.
Supplemental Figure 7. CAST expression, autophagic flux assessment, and podocyte injury in patients with diabetic kidney disease.
Supplemental Figure 8. Single-cell RNAseq database.
Supplemental Figure 9. Nephroseq transcriptomic database.
Supplemental Table 1. Patient characteristics.
Supplemental Table 2. Patients' descriptive statistics.
Supplemental Table 3. Staining descriptive statistics.
Supplemental Table 4. Analyses of variances for patients with DKD and proteinuria.
Supplemental Table 5. Binomial logistic regression.
Supplemental Table 6. Model comparison.
Supplemental Table 7. Model coefficients.
Supplemental Table 8. Prediction with model 4.
References
- 1.Yuan CM, Nee R, Ceckowski KA, Knight KR, Abbott KC. Diabetic nephropathy as the cause of end-stage kidney disease reported on the medical evidence form CMS2728 at a single center. Clin Kidney J. 2017;10(2):257–262. doi: 10.1093/ckj/sfw112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onyenwenyi C, Ricardo AC. Impact of lifestyle modification on diabetic kidney disease. Curr Diab Rep. 2015;15(9):60. doi: 10.1007/s11892-015-0632-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6:319–330. doi: 10.1038/nrneph.2010.58 [DOI] [PubMed] [Google Scholar]
- 4.Neuen BL, Young T, Heerspink HJL, Neal B, Perkovic V, Billot L. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019;7(11):845–854. doi: 10.1016/S2213-8587(19)30256-6 [DOI] [PubMed] [Google Scholar]
- 5.Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–1446. doi: 10.1056/NEJMoa2024816 [DOI] [PubMed] [Google Scholar]
- 6.Jiang G, Luk AOY, Tam CHT, Xie F, Carstensen B, Lau ESH. Progression of diabetic kidney disease and trajectory of kidney function decline in Chinese patients with Type 2 diabetes. Kidney Int. 2019;95(1):178–187. doi: 10.1016/j.kint.2018.08.026 [DOI] [PubMed] [Google Scholar]
- 7.Molitch ME, Gao X, Bebu I, de Boer IH, Lachin J, Paterson A. Early glomerular hyperfiltration and long-term kidney outcomes in type 1 diabetes: the DCCT/EDIC experience. Clin J Am Soc Nephrol. 2019;14(6):854–861. doi: 10.2215/CJN.14831218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pichaiwong W, Homsuwan W, Leelahavanichkul A. The prevalence of normoalbuminuria and renal impairment in type 2 diabetes mellitus. Clin Nephrol. 2019;92:73–80. doi: 10.5414/CN109606 [DOI] [PubMed] [Google Scholar]
- 9.Fogo AB. The targeted podocyte. J Clin Invest. 2011;121(6):2142–2145. doi: 10.1172/JCI57935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagtalunan ME Miller PL Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54(6):1626–1634. doi: 10.2337/diabetes.54.6.1626 [DOI] [PubMed] [Google Scholar]
- 12.Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. 2015;224(1):R15–R30. doi: 10.1530/JOE-14-0437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez CD, Carro Negueruela MP, Nicora Santamarina C, Resnik R, Vaccaro MI. Autophagy dysregulation in diabetic kidney disease: from pathophysiology to pharmacological interventions. Cells. 2021;10(9):2497. doi: 10.3390/cells10092497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120(4):1084–1096. doi: 10.1172/jci39492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chun Y, Kim J. Autophagy: an essential degradation program for cellular homeostasis and Life. Cells. 2018;7(12):278. doi: 10.3390/cells7120278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. doi: 10.1038/nrm2239 [DOI] [PubMed] [Google Scholar]
- 18.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181–2196. doi: 10.1172/jci44771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. 2015;11(7):1130–1145. doi: 10.1080/15548627.2015.1049799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65(3):755–767. doi: 10.2337/db15-0473 [DOI] [PubMed] [Google Scholar]
- 21.Staruschenko A, Spires D, Palygin O. Role of TRPC6 in progression of diabetic kidney disease. Curr Hypertens Rep. 2019;21(7):48. doi: 10.1007/s11906-019-0960-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou X, Xiao H, Zhang Y, Zeng X, Huang M, Chen X. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018;9(10):1015. doi: 10.1038/s41419-018-1052-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farmer LK, Rollason R, Whitcomb DJ, Ni L, Goodliff A, Lay AC. TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J Am Soc Nephrol. 2019;30(10):1910–1924. doi: 10.1681/ASN.2018070729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verheijden KAT, Sonneveld R, Bakker-van Bebber M, Wetzels JFM, van der Vlag J, Nijenhuis T. The calcium-dependent protease calpain-1 links TRPC6 activity to podocyte injury. J Am Soc Nephrol. 2018;29(8):2099–2109. doi: 10.1681/ASN.2016111248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y, Tang X, Wang Y, Yu D, Zhu C, Yu J. Tetrandrine alleviates podocyte injury via calcium-dependent calpain-1 signaling blockade. BMC Complement Med Ther. 2021;21(1):296. doi: 10.1186/s12906-021-03469-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352(23):2413–2423. doi: 10.1056/NEJMra043361 [DOI] [PubMed] [Google Scholar]
- 27.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124–1132. doi: 10.1038/ncb1482 [DOI] [PubMed] [Google Scholar]
- 28.Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6(1):61–66. doi: 10.4161/auto.6.1.10326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wendt A, Thompson VF, Goll DE. Interaction of calpastatin with calpain: a review. Biol Chem. 2004;385(6):465–472. doi: 10.1515/bc.2004.054 [DOI] [PubMed] [Google Scholar]
- 30.Project Tj. Jamovi Version 2.3; 2022. [Google Scholar]
- 31.Team RC. R: A Language and Environment for Statistical Computing. Version 4.1. R Core Team; 2021. [Google Scholar]
- 32.Seol H. snowCluster: Multivariate Anaysis. Version 7.1.3. 2023. [Google Scholar]
- 33.Kassambara AM. F: Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R package; 2020. [Google Scholar]
- 34.Fox J, Weisberg S. car: Companion to Applied Regression. R package; 2020. [Google Scholar]
- 35.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: Visualizing the Performance of Scoring Classifiers; 2015. [DOI] [PubMed] [Google Scholar]
- 36.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res. 2008;102(6):720–728. doi: 10.1161/circresaha.107.160077 [DOI] [PubMed] [Google Scholar]
- 37.Mizushima N, Kuma A. Autophagosomes in GFP-LC3 transgenic mice. Methods Mol Biol. 2008;445:119–124. doi: 10.1007/978-1-59745-157-4_7 [DOI] [PubMed] [Google Scholar]
- 38.Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8(5):697–705. doi: 10.1681/ASN.v85697 [DOI] [PubMed] [Google Scholar]
- 39.Kumar SS, Ward ML, Mountjoy KG. Quantitative high-throughput assay to measure MC4R-induced intracellular calcium. J Mol Endocrinol. 2021;66(4):285–297. doi: 10.1530/JME-20-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21(4):556–563. doi: 10.1681/ASN.2010010010 [DOI] [PubMed] [Google Scholar]
- 41.Bensaada I, Robin B, Perez J, Salemkour Y, Chipont A, Camus M. Calpastatin prevents Angiotensin II-mediated podocyte injury through maintenance of autophagy. Kidney Int. 2021;100(1):90–106. doi: 10.1016/j.kint.2021.02.024 [DOI] [PubMed] [Google Scholar]
- 42.Uil M, Scantlebery AML, Butter LM, Larsen PWB, de Boer OJ, Leemans JC. Combining streptozotocin and unilateral nephrectomy is an effective method for inducing experimental diabetic nephropathy in the 'resistant' C57Bl/6J mouse strain. Sci Rep. 2018;8(1):5542. doi: 10.1038/s41598-018-23839-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21(9):1533–1542. doi: 10.1681/ASN.2009121290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finch N, Fawaz S, Neal C, Butler M, Lee V, Salmon A. Reduced glomerular filtration in diabetes is attributable to loss of density and increased resistance of glomerular endothelial cell fenestrations. J Am Soc Nephrol. 2022;33(6):1120–1136. doi: 10.1681/ASN.2021030294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nabeshima Y, Washida M, Tamura M, Maeno A, Ohnishi M, Shiroishi T. Calpain 1 inhibitor BDA-410 ameliorates alpha-klotho-deficiency phenotypes resembling human aging-related syndromes. Sci Rep. 2014;4:5847. doi: 10.1038/srep05847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Franceschi L, Franco RS, Bertoldi M, Brugnara C, Matte A, Siciliano A. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease. FASEB J. 2013;27(2):750–759. doi: 10.1096/fj.12-217836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Chen H, Jeong JJ, Chishti AH. BDA-410: a novel synthetic calpain inhibitor active against blood stage malaria. Mol Biochem Parasitol. 2007;155(1):26–32. doi: 10.1016/j.molbiopara.2007.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebart MC, Benyamin Y. Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 2006;273(15):3415–3426. doi: 10.1111/j.1742-4658.2006.05350.x [DOI] [PubMed] [Google Scholar]
- 49.Tian X, Kim JJ, Monkley SM, Gotoh N, Nandez R, Soda K. Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J Clin Invest. 2014;124(3):1098–1113. doi: 10.1172/JCI69778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118(8):2796–2807. doi: 10.1172/JCI34254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simoes AT, Goncalves N, Nobre RJ, Duarte CB, Pereira de Almeida L. Calpain inhibition reduces ataxin-3 cleavage alleviating neuropathology and motor impairments in mouse models of Machado-Joseph disease. Hum Mol Genet. 2014;23(18):4932–4944. doi: 10.1093/hmg/ddu209 [DOI] [PubMed] [Google Scholar]
- 52.Pereyra AS, Wang ZM, Messi ML, Zhang T, Wu H, Register TC. BDA-410 treatment reduces body weight and fat content by enhancing lipolysis in sedentary senescent mice. J Gerontol A Biol Sci Med Sci. 2017;72(8):1045–1053. doi: 10.1093/gerona/glw192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang T, Pereyra AS, Wang ZM, Birbrair A, Reisz JA, Files DC. Calpain inhibition rescues troponin T3 fragmentation, increases Cav1.1, and enhances skeletal muscle force in aging sedentary mice. Aging Cell. 2016;15(3):488–498. doi: 10.1111/acel.12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dokus LE, Yousef M, Banoczi Z. Modulators of calpain activity: inhibitors and activators as potential drugs. Expert Opin Drug Discov. 2020;15(4):471–486. doi: 10.1080/17460441.2020.1722638 [DOI] [PubMed] [Google Scholar]
- 55.Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. 2016;15(12):854–876. doi: 10.1038/nrd.2016.212 [DOI] [PubMed] [Google Scholar]
- 56.Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci U S A. 2019;116(20):10156–10161. doi: 10.1073/pnas.1815354116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Urban N, Wang L, Kwiek S, Rademann J, Kuebler WM, Schaefer M. Identification and validation of Larixyl acetate as a potent TRPC6 inhibitor. Mol Pharmacol. 2016;89(1):197–213. doi: 10.1124/mol.115.100792 [DOI] [PubMed] [Google Scholar]
- 58.Sonneveld R, Hoenderop JG, Isidori AM, Henique C, Dijkman HB, Berden JH. Sildenafil prevents podocyte injury via PPAR-gamma-mediated TRPC6 inhibition. J Am Soc Nephrol. 2017;28(5):1491–1505. doi: 10.1681/ASN.2015080885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin Q, Banu K, Ni Z, Leventhal JS, Menon MC. Podocyte autophagy in homeostasis and disease. J Clin Med. 2021;10(6):1184. doi: 10.3390/jcm10061184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol. 2014;234(2):203–213. doi: 10.1002/path.4382 [DOI] [PubMed] [Google Scholar]
- 61.Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2(4):e144. doi: 10.1038/cddis.2011.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian X, Inoue K, Zhang Y, Wang Y, Sperati CJ, Pedigo CE. Inhibiting calpain 1 and 2 in cyclin G associated kinase-knockout mice mitigates podocyte injury. JCI Insight. 2020;5(22):e142740. doi: 10.1172/jci.insight.142740 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in the main text or the Supplemental materials.