

ABSTRACT
The interaction between the kidney and the coagulation system greatly affects each other because of the abundant vessel distribution and blood perfusion in the kidney. Clinically, the risks of complicated thrombosis and bleeding have become important concerns in the treatment of nephropathies, especially nephrotic syndrome, CKD, ESKD, and patients with nephropathy undergoing RRTs. Adverse effects of anticoagulant or procoagulant therapies in patients with nephropathy, especially anticoagulation-related nephropathy, heparin-induced thrombocytopenia, and bleeding, seriously worsen the prognosis of patients, which have become challenges for clinicians. Over the decades, the interaction between the kidney and the coagulation system has been widely studied. However, the effects of the kidney on the coagulation system have not been systematically investigated. Although some coagulation-related proteins and signaling pathways have been shown to improve coagulation abnormalities while avoiding additional kidney damage in certain kidney diseases, their potential as anticoagulation targets in nephropathy requires further investigation. Here, we review the progression of research on the crosstalk between the coagulation system and kidney diseases and systematically analyze the significance and shortcomings of previous studies to provide new sight into future research. In addition, we highlight the status of clinical treatment for coagulation disorder and nephropathy caused by each other, indicating guidance for the formulation of therapeutic strategies or drug development.
Keywords: AKI, CKD, dialysis, ESKD, IgA nephropathy, kidney transplantation, nephropathy, nephrotic syndrome, platelets, thrombosis
Introduction
As an important part of physiological hemostasis, coagulation is a complex process involving a series of cascade reactions between coagulation factors, which finally transform fibrinogen into insoluble fibrin, causing blood clotting (Figure 1).1 Abnormalities in coagulation factors lead to coagulation disorders, resulting in thrombosis or bleeding.1
Figure 1.
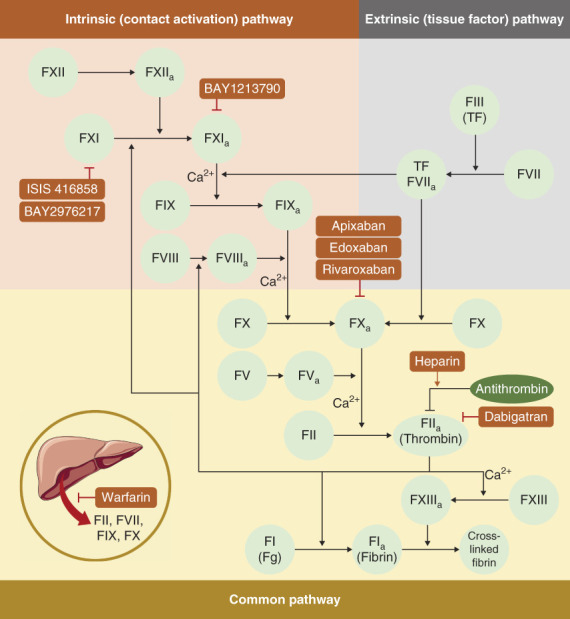
Coagulation cascade and the targets of anticoagulants. The coagulation process consists of a cascade of coagulation factors. Coagulation is initiated by both intrinsic and extrinsic pathways. In the intrinsic pathway, negatively charged surfaces, such as phospholipids and polyphosphates from activated platelets, activate FXII, triggering cascade reactions that ultimately activate FX. In the extrinsic pathway, tissue injury or inflammatory factors induce the expression of TFs that form complexes with activated FVII, thereby activating FX. Activated FX promotes the production of thrombin, which catalyzes fibrinogen (Fg) to soluble fibrin. FXIII promotes cross-linking of fibrin, resulting in the formation of blood clots. Anticoagulants inhibit thrombosis by disturbing the coagulation cascade. Warfarin inhibits the production of vitamin K–dependent coagulation factors (including FII, FVII, IX, and X) in the liver. Heparin primarily promotes the activity of AT-III. DOACs, including apixaban, edoxaban, and rivaroxaban, function as the inhibitor of FX, while dabigatran is the inhibitor of thrombin. Novel anticoagulants, including ISIS 416858 and BAY2976217, are FXI antisense oligonucleotides, while BAY1213790 is the monoclonal antibody to FXIa. AT-III, antithrombin III; DOAC, direct oral anticoagulant; FVII, coagulation factor VII; FXI, factor XI; FXIa, activated factor XI; TF, tissue factor.
As a blood filter and the most highly perfused organ, the kidney plays an important role in regulating hemodynamics and blood components, which directly affects coagulation processes.2 In fact, thrombosis is common in patients with nephrotic syndrome (NS) or CKD3–5 or patients undergoing RRTs.6 While patients with ESKD are threatened with bleeding.7 However, improper anticoagulant or procoagulant therapies may induce adverse effects, such as anticoagulation-related nephropathy (ARN), heparin-induced thrombocytopenia (HIT), or bleeding, which trouble clinicians.8,9 Over the decades, researchers have conducted extensive mechanism studies on the crosstalk between nephropathy and coagulopathy,5,10 which provide guidance for clinical treatment to some extent. However, the complex interactions between the kidney and the coagulation system are still not fully understood.
Currently, there is no review to systematically summarize and analyze the significance and shortcomings of these findings. Therefore, we review the pathogenesis and treatment of coagulation abnormalities in nephropathy, as well as the manifestations and countermeasures of adverse effects resulting from anticoagulant therapies in nephropathy patients. We emphasize the current problems in clinical practice and the corresponding deficiencies in basic research and analyze the direction of future basic research efforts on the basis of existing studies, as well as the improvement programs that could be considered in clinical treatment.
NS
NS refers to a series of kidney diseases with similar clinical and laboratory features, which are mainly characterized by proteinuria, edema, hypercholesterolemia, and hypoalbuminemia. According to different pathological features, NS can be identified as membranous nephropathy (MN), minimal change disease, focal and segmental glomerulosclerosis, and membranoproliferative glomerulonephritis.11 Thromboembolism is the most serious complication of NS, which is life-threatening. The thrombosis incidence is highest in MN (approximately 37%) and lower in other pathological types of NS (with a cumulative incidence of 24%). Venous thromboembolism (VTE) is the most common type of thrombosis, and coronary thrombosis was also detected.4,11
Mechanism of Thrombosis in NS
It was reported that complicated VTE in NS was mainly caused by the disorders of coagulation and fibrinolytic proteins, while arterial thromboembolism primarily resulted from platelet hyperreactivity.12
Disorders of the coagulation and fibrinolytic proteins were caused by the breakdown of the permselectivity barrier of the glomeruli capillary wall in the kidney of patients with NS. Low molecular weight anticoagulant proteins were lost from altered glomeruli (Figure 2A), whereas high molecular weight procoagulant proteins (HMWPPs) were retained in the plasma (Figure 2A).13,14 These processes led to a decrease in the total concentration of proteins that regulate the coagulation system, which enhanced the compensatory synthesis of both low molecular weight anticoagulant protein and HMWPP. The net effect was the accumulation of HMWPP in the blood, resulting in a hypercoagulable state that promoted thrombosis. Clinical data showed that the more severe the NS, the higher the incidence of thrombosis, which was consistent with the above mechanism.13
Figure 2.
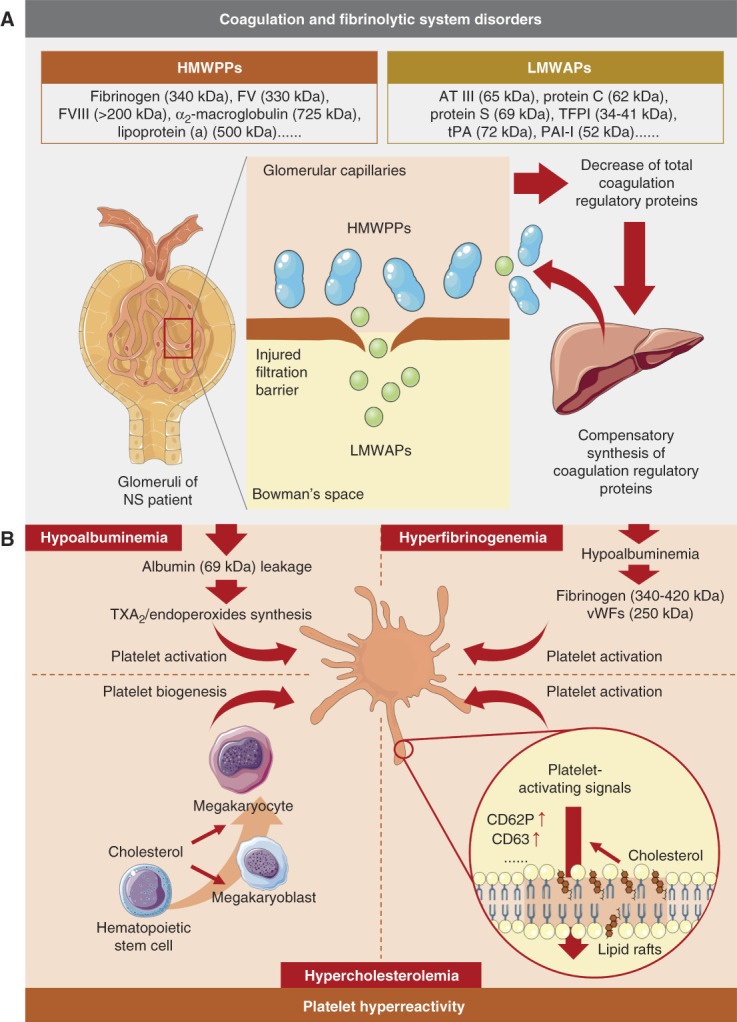
Mechanism of thrombosis in NS. (A) The injuries in the glomeruli filtration barrier led to the leakage of LMWAPs and retention of HMWPPs in plasma, resulting in a decrease in total coagulation regulatory protein levels. These processes induced compensatory synthesis of coagulation regulatory proteins in the liver, which further promoted the accumulation of HMWPPs in plasma, resulting in hypercoagulability. (B) Filtration barrier injuries also induced the leakage of albumin and hypoalbuminemia, which promoted TXA2 and endoperoxide synthesis (top left) and compensatory synthesis of fibrinogen and vWFs (top right), resulting in platelet activation. Complicated hypercholesterolemia in NS promoted platelet biogenesis by enhancing the signaling of cell surface receptors located in lipid rafts (bottom left). Hypercholesterolemia also upregulated signal transduction proteins in the lipid raft of platelets, resulting in platelet activation (bottom right). HMWPP, high molecular weight procoagulant protein; LMWAP, low molecular weight anticoagulant protein; NS, nephrotic syndrome; vWF, von Willebrand factor.
Platelet hyperreactivity in NS was mainly caused by hypoalbuminemia, hyperfibrinogenemia, and hypercholesterolemia (Figure 2B).15,16 Hypoalbuminemia resulted from the loss of albumin (69 kDa) from the altered glomeruli (Figure 2B, top left). Physiologically, albumin inhibited platelet aggregation by binding to arachidonic acid and preventing its metabolism into thromboxane A2 (TXA2) and endoperoxides. As a consequence, hypoalbuminemia resulted in the excessive production of TXA2 and endoperoxides, leading to platelet hyperreactivity.17 In addition, hypoalbuminemia also led to the compensatory synthesis of fibrinogen and von Willebrand factors (vWFs) in the liver, resulting in hyperfibrinogenemia that promoted platelet hyperreactivity (Figure 2B, top right).14 Hypercholesterolemia was one of the metabolic consequences of NS, which was confirmed as the promotor of platelet biogenesis and activation. It was reported that in various hematopoietic effector cells, membrane receptor signaling pathways located in lipid rafts were enhanced in response to membrane cholesterol accumulation. Therefore, the accumulation of cholesterol in the plasma membrane enhanced platelet biogenesis in the megakaryoblast (Figure 2B, bottom left) and procoagulant signaling transduction in the platelets (Figure 2B, bottom right), which promoted thrombosis. Indeed, numerous clinical studies detected increased platelet count and platelet hyperactivity in patients with NS, which confirmed the important role of platelets in thrombosis in patients with NS.18–23
Prophylactic and Therapeutic Anticoagulant Therapies in NS
Prophylactic and therapeutic anticoagulation is necessary for patients with NS,24,25 while thrombolytic therapy is only considered in patients with massive pulmonary embolism and severe bilateral deep vein thrombosis due to the high risk of bleeding.13
Low-molecular-weight heparin and warfarin are commonly used for prophylactic and therapeutic anticoagulation in patients with NS.24 But the advantages and disadvantages of these two drugs have not been compared. With the continuous accumulation of clinical data, arterial thromboembolism was also considered to need attention in patients with NS.24,26 Therefore, antiplatelet agents were used to prevent and treat thromboembolism in patients with NS. The Kidney Disease Improving Global Outcomes guidelines suggested aspirin may be considered for patients with MN.27 However, the risk of bleeding persists in patients with NS exposed to anticoagulants; thromboembolism risk and bleeding risk must be balanced during anticoagulation processes. As MN is the most common cause of NS, the Kidney Disease Improving Global Outcomes provides an algorithm for the formulation of prophylactic and therapeutic anticoagulant regimens for patients with MN (Figure 3), which might also be referenced for other pathological types of NS.24,27
Figure 3.
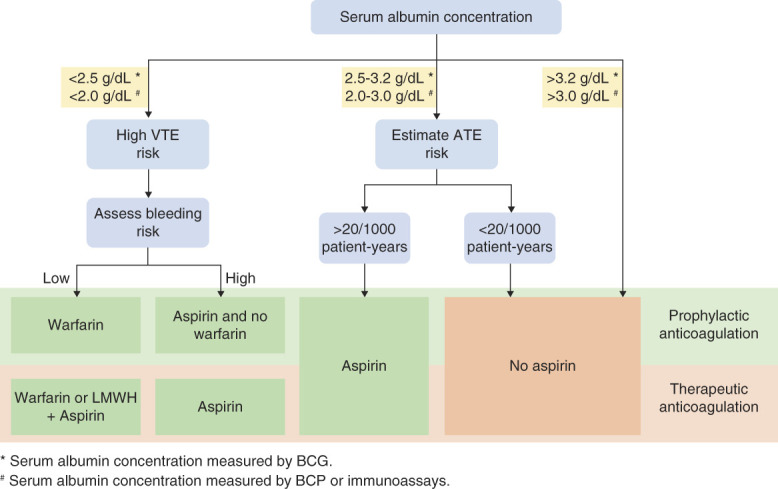
Algorithm for prophylactic and therapeutic anticoagulation in patients with MN. The formulation of an anticoagulant regimen is determined by serum albumin concentration. If the serum albumin concentration is <2.5 g/dl (by BCG) or <2.0 g/dl (by BCP), VTE risk should be considered; if the assessed bleeding risk is low, warfarin is considered for prophylactic anticoagulation, and warfarin or LMWH+aspirin are considered for therapeutic anticoagulation. If the serum albumin concentration is 2.5-3.2 g/dl (by BCG) or 2.0-3.0 g/dl (by BCP), ATE risk should be assessed; if the estimated ATE risk >20/1000 patient-years, aspirin is considered; otherwise, aspirin should not be used. If serum albumin concentration is >3.2 g/dl (by BCG) or >3.0 g/dl (by BCP), aspirin should not be used. KDIGO recommends an online tool (https://www.med.unc.edu/gntools/bleedrisk.html) to calculate the risk of bleeding in patients with MN. ATE, arterial thromboembolism; KDIGO, Kidney Disease Improving Global Outcomes; LMWH, low-molecular-weight heparin; MN, membranous nephropathy; VTE, venous thromboembolism; BCG, bromocresol green; BCP, bromocresol purple.
In addition, only a few studies reported the effectiveness of direct oral anticoagulants (DOACs) in preventing thrombosis in patients with NS (Table 1). But excretion and plasma protein binding rates varied greatly among DOACs (Table 1).33 It is still controversial whether DOACs are suitable for patients with NS. Therefore, we do not recommend DOACs as the first choice for anticoagulation in patients with NS.
Table 1.
Pharmacology of direct oral anticoagulants and their efficacy in patients with nephrotic syndrome
| DOACs | Renal Excretion Rate | Fecal Excretion Rate | Plasma Protein Binding Rate | Efficacy in NS |
|---|---|---|---|---|
| Dabigatran | 80% (iv) | — | 35% | Preventing carotid thrombosis in patients with MN28 |
| Apixaban | 27% | Approximately 50% | 87% | Preventing thromboembolism in patients with NS.29 But recurrent VTE might occur in patients with MN30 |
| Edoxaban | 50% | 50% | 55% | Inhibiting recurred renal VTE in patients with NS after warfarin treatment31 |
| Rivaroxaban | 66% | 7% | 92%–95% | Inhibiting VTE in patients with NS with low AT-III level32 |
DOACs, direct oral anticoagulants; NS, nephrotic syndrome; iv, intravenous injection; MN, membranous nephropathy; VTE, venous thromboembolism; AT-III, antithrombin III.
Faced with the current treatment dilemma, novel anticoagulants or antiplatelet drugs are urgently needed for patients with NS. Interestingly, statins were proven to not only ameliorate kidney injury but also prevent thrombosis in patients with NS.34,35 These efficacies might come from the inhibitory effect of stains on hypercholesterolemia, which reversed cholesterol-induced platelet hyperreactivity, as described above. Given the dual therapeutic effects of statins on NS and thrombosis, it may be an ideal choice for antiplatelet in patients with NS. More clinical studies are needed to clarify the benefits and shortcomings of statins in preventing thrombosis.
CKD
CKD develops from a series of underlying kidney diseases. It is characterized by decreased GFR (<60 ml/[min·1.73 m2]) and elevated renal injury markers (such as serum creatinine and urea nitrogen). CKD significantly increases the risk of thrombosis,5 especially in patients complicated with atrial fibrillation (AF), which has been recognized as an important cause of stroke.36,37
Mechanism of Thrombosis in CKD
Elevated thrombosis risk in patients with CKD mainly resulted from coagulation and fibrinolytic protein disorders, endothelial dysfunction, and platelet hyperreactivity.
Coagulation and fibrinolytic protein disorders in patients with CKD were characterized by the upregulation of procoagulant proteins, especially coagulation factor VIII, vWFs, and plasminogen activator inhibitor-I.38,39 However, patterns of change in these proteins were not the same as that in patients with NS. Current views hold that these disorders resulted not only from glomeruli injury–induced accumulation of HMWPPs as in NS (Figure 4A) but also from the release of tissue factor (TF) and coagulation activation caused by inflammation-induced vascular endothelial injury (Figure 4B).
Figure 4.
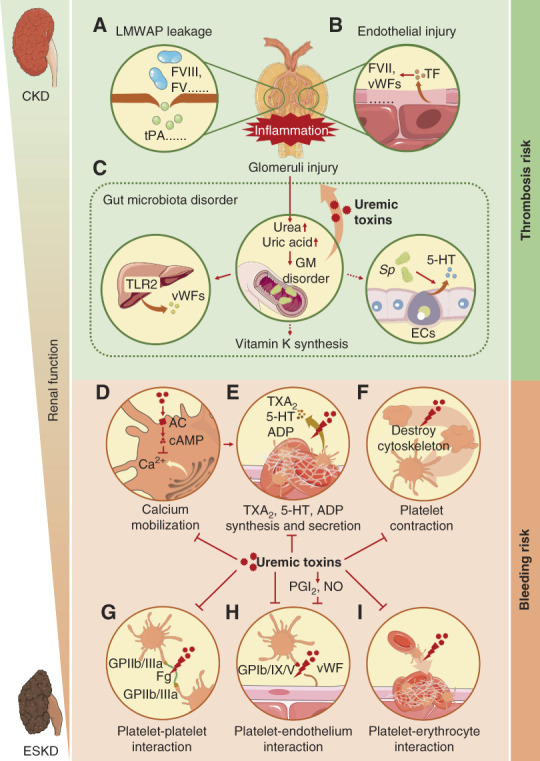
Mechanism of thrombosis in CKD and bleeding in ESKD. (A) The injuries in the glomeruli filtration barrier in CKD resulted in the leakage of LMWAPs and retention of HMWPPs, which promoted coagulation. (B) Glomeruli endothelial injuries in CKD led to the release of TF which triggered the extrinsic coagulation pathway. (C) Glomeruli injuries in CKD led to an accumulation of urea and uric acid in the gut, causing GM disorder, which on one hand enhanced the production of uremic toxins, resulting in glomeruli inflammation and injury; on the other hand, promoted vWFs, vitamin K, and 5-HT production. (D) Uremic toxins in ESKD activated AC and promoted cAMP production, which inhibited calcium mobilization, resulting in platelet dysfunction. (E) Uremic toxins and calcium mobilization abnormality inhibited the synthesis and secretion of TXA2, 5-HT, and ADP in platelets, leading to platelet dysfunction. (F) Uremic toxins destroyed the cytoskeleton of platelet, which disturbed platelet contraction, inhibiting platelet aggregation. (G) Uremic toxins inhibited platelet–platelet interaction by blocking the combination of fibrinogen (Fg) and GPIIb/IIIa, resulting in platelet aggregation dysfunction. (H) PGI2 and nitric oxide (NO) induced by uremic toxins and uremic toxins themselves inhibited platelet–endothelium interaction by blocking the combination of vWF and GPIb/IX/V, resulting in platelet aggregation dysfunction. (I) Uremic toxins inhibited platelet–erythrocyte interaction, resulting in platelet aggregation dysfunction. 5-HT, 5-hydroxytryptamine; AC, adenylate cyclase; GM, gut microbiota; HMWPP, high molecular weight procoagulant protein; LMWAP, low molecular weight anticoagulant protein; NO, nitric oxide; PGI2, prostacyclin; TF, tissue factor; TXA2, thromboxane A2; vWF, von Willebrand factor.
Platelet hyperactivity and endothelial dysfunction were caused by uremic toxins that were mainly derived from the gut microbiota (GM).40 It was reported that the decrease in GFR and impairment of renal tubular secretion in CKD resulted in increased production of nitrogen compounds (such as urea and uric acid) which were excreted partly through the intestine, but no urinary tract. In the gut, urea was converted to ammonium, leading to pH elevation and mucosal damage, resulting in GM disorder (Figure 4C, middle).40 GM disorder caused protein assimilation impairment, promoting the synthesis of uremic toxins.40 These uremic toxins were called thrombolome. Thrombolome can be divided into three groups: dietary tryptophan, phenylalanine/tyrosine, and choline/phosphatidylcholine (Table 2).40 Dietary tryptophan can combine with aryl hydrocarbon receptor (AhR) and downregulated stress-induced phosphoprotein 1 homologous and U-box containing protein 1 (STUB1). STUB1 plays an important role in TF degradation, which ubiquitinated and degraded TF in uremia environment.42 Dietary tryptophan-induced downregulation of STUB1 led to retarded TF degradation, which enhanced exogenous coagulation pathways.49 In addition, indoxyl sulfate and indole-3-acetic acid (two kinds of dietary tryptophan) could induce oxidative stress and inflammation, which also promoted platelet activation.41,43 Phenylalanine/tyrosine includes phenylacetylglutamine, p-cresol sulfate, and p-cresol glucuronide. Phenylacetylglutamine has been demonstrated to induce platelet hyperreactivity by activating α2A-, α2B-, and β2-receptors.46 But the mechanisms of platelet activation induced by p-cresol sulfate and p-cresol glucuronide remain unclear. It was suggested that they might promote platelet activation by promoting the release of phosphatidylserine (PS)-positive microparticles (MPs) from endothelial cells.47 Choline/phosphatidylcholine includes trimethylamine N-oxide which triggered platelet activation primarily by promoting 1,4,5-trisphosphate–mediated Ca2+ release.48
Table 2.
Types and functions of thrombolome in CKD
| Origin | Types | Functions |
|---|---|---|
| Dietary tryptophan | Indoxyl sulfate | a. Inhibiting endothelial cell proliferation and damaging endothelial progenitor cells by inducing oxidative stress and inflammation41 b. Increasing TF/FVII levels by upregulating the AhR-STUB1-TF axis40,42 c. Activating platelets by inducing oxidative stress-mediated p38 MAPK activation43 |
| Indole-3-acetic acid | a. Inducing COX-2 expression by activating AhR/p38 MAPK/NF-κB signaling pathways, which led to inflammation and endothelial injury44 b. Increasing TF/FVII levels by upregulating the AhR-STUB1-TF axis40,42 c. Activating platelets by inducing oxidative stress-mediated p38 MAPK activation43 |
|
| Kynurenine pathway–derived uremic toxins | a. Inducing inflammation and oxidative stress by activating AhR, which caused endothelial dysfunction and TF overexpression45 b. Causing coagulation factors disorder40,a |
|
| Phenylalanine/tyrosine | Phenylacetylglutamine | Inducing platelet hyperreactivity by activating α2A-, α2B-, β2-adrenergic receptors46 |
| P-cresol sulfate | Promoting MP release from endothelial cells47,a | |
| P-cresol glucuronide | Promoting MP release from endothelial cells47,a | |
| Choline/phosphatidylcholine | Trimethylamine N-oxide | Promoting IP3-mediated Ca2+ release, leading to platelet hyperreactivity48 |
TF, tissue factor; FVII, coagulation factor VII; AhR, aryl hydrocarbon receptor; STUB1, stress-induced phosphoprotein 1 homologous and U-box containing protein 1; p38 MAPK, p38 mitogen-activated protein kinase; NF-κB, nuclear factor-kappaB; MPs, microparticles; IP3, 1,4,5-trisphosphate.
Indicates that the underlying mechanism has not been elucidated.
In addition to uremic toxins, GM disorder could also promote hepatic vWF production by activating toll-like receptor 2 (Figure 4C, left) and might promote 5-hydroxytryptamine biosynthesis in enterochromaffin cells by upregulating tryptophan hydroxylase (Figure 4C, right).40,50–52 It was also reported that GM disorders may lead to increased vitamin K synthesis, but the evidence remains limited (Figure 4C, middle).40 These processes also contributed to thrombosis.
Management of Thrombosis in CKD
Patients with CKD are prone to AF (with a prevalence of 16%-21%), which greatly increases the risk of stroke.53 Therefore, anticoagulation is necessary for patients with CKD.53–55 Currently, international guidelines permit the use of warfarin and DOACs for patients with CKD.56,57 Dabigatran, apixaban, edoxaban, and rivaroxaban have been extensively reviewed for their safety and efficacy in patients with CKD.57,58 Compared with warfarin, DOACs cause a lower incidence of severe intracranial hemorrhage, eliminating the need for drug monitoring, which simplifies the treatment of patients with CKD with VTE or AF.59 But the differences between DOACs have not been compared. With the continuous decline of renal function, bleeding risk is elevated in patients with CKD because of the accumulation of uremic toxins. Therefore, appropriate dosage adjustments according to renal function are required for warfarin or DOACs used in patients with CKD (see detail in Table 3).60
Table 3.
| eCrCl (ml/min) | Warfarin | DOACs | |||
|---|---|---|---|---|---|
| Dabigatran | Apixaban | Edoxaban | Rivaroxaban | ||
| >95 | Adjusted dose for INR 2–3 | 150 mg b.i.d. | 5 mg b.i.d.a | 60 mg QDb,c | 20 mg QD |
| 51–95 | Adjusted dose for INR 2–3 | 150 mg b.i.d. | 5 mg b.i.d.a | 60 mg QDc | 20 mg QD |
| 31–50 (25–50)d | Adjusted dose for INR 2–3 | 150 mg b.i.d. or 110 mg b.i.d.b | 5 mg b.i.d.a | 30 mg QDc | 15 mg QD |
| 15–30 | Adjusted dose for INR 2–3 (consider) | 75 mg po b.i.d.e | 2.5 mg b.i.d. (consider) | 30 mg QDc (consider) | 15 mg QD (consider) |
| <15 (not on dialysis) | Equipoise on the basis of observational data and meta-analysis | Not recommended | 2.5 mg po b.i.d.e | Not recommended | 15 mg QDe |
| <15 (on dialysis) | Equipoise on the basis of observational data and meta-analysis | Not recommended | 2.5 mg po b.i.d.e | Not recommended | 15 mg QDe |
eCrCl, estimated creatinine clearance; DOACs, direct oral anticoagulants; INR, international normalized ratio; b.i.d., twice daily; QD, once daily; po, per os.
2.5 mg twice daily if two of the three criteria are met: creatinine ≥1.5 mg/dl, body weight ≤60 kg, aged 80 years or older.
Application of this dosage in patients with CKD has not been approved by Food and Drug Administration.
The dose is halved if any of the three criteria are met: estimated creatinine clearance of 30 approximately 50 ml/min, body weight ≤60 kg, concomitant use of verapamil or quinidine.
Estimated creatinine clearance values referenced for dose adjustment of apixaban.
This application is recommended by Kidney Disease Improving Global Outcomes but lacks clinical safety or efficacy data.
Given the shorting coming of warfarin and DOACs, novel anticoagulants are urgently needed for patients with CKD. Because the AhR-STUB1-TF axis played important roles in promoting thrombosis (Table 2), blocking the AhR-STUB1-TF axis has been shown to effectively improve hypercoagulable state without increasing the risk of bleeding in patients with CKD.49 Targeting the AhR-STUB1-TF axis to develop anticoagulants may avoid the bleeding risk comes from DOACs, which is a direction worth making efforts in the future.
ESKD
Patients with CKD eventually develop ESKD (GFR <15 ml/[min·1.73m2]) with the decline of renal function.61 But unlike CKD, patients with ESKD are usually threatened by bleeding.
Mechanism of Bleeding in ESKD
Studies indicated that uremic toxin accumulation–induced platelet dysfunction was the main cause of bleeding in patients with ESKD.5,61,62 As adenylyl cyclase activators, uremic toxins could upregulate cAMP that inhibited platelet calcium mobilization, which suppressed platelet activation (Figure 4D).63 Studies found that uremic toxins could inhibit the synthesis and release of TXA2, 5-hydroxytryptamine, and ADP in platelets (Figure 4E),64,65 which may also be one of the causes of platelet dysfunction, but the underlying mechanism has not been clarified. Besides, uremic toxins have also been shown to inhibit actin, thereby disrupting the platelet cytoskeleton, resulting in platelet contraction, movement, and secretion dysfunction (Figure 4F).66 In addition, uremic toxins could inhibit the binding of GPIIb/IIIa to fibrinogen without affecting the abundance of GPIIb/IIIa on the platelet membrane, resulting in decreased platelet–platelet adhesion and signaling transduction (Figure 4G).67 But unlike GPIIb/IIIa, the glycocalicin subunits of GPIb on the platelet membrane were degraded by uremic toxins, which disturbed the binding of vWFs with GPIb/IX/V, leading to inhibition of platelet–vessel wall adhesion. Uremic toxins also induced nitric oxide and prostacyclin production in endothelial cells, causing platelet dysfunction (Figure 4H).67–70 All these effects contributed to bleeding in ESKD.
In addition, patients with ESKD were often accompanied by anemia,71 which may also be one of the causes of bleeding because erythrocytes are important in moving platelets toward the vascular wall to promote platelet aggregation (Figure 4I).61 Anemia in ESKD is mainly caused by decreased secretion of erythropoietin in the kidney, shortened lifespan of erythrocytes, and inflammation.71 Because erythropoietin is synthesized primarily in the kidney, renal failure in ESKD results in reduced erythropoietin synthesis and subsequent dyserythrogenesis.72,73 Meanwhile, patients with ESKD were usually accompanied by metabolic disorders of vitamin B12 and folic acid, which further aggravated dyserythrogenesis.74 Shortened lifespan of erythrocytes was primarily caused by uremic toxins. Uremic toxins caused oxidative and inhibited Ca2+ pump in erythrocytes which led to cytoplasm Ca2+ overload, resulting in erythrocyte apoptosis.73 Inflammation inhibited erythropoiesis by inhibiting the synthesis of erythropoietin, upregulating soluble erythropoietin receptors, and inducing erythroid progenitor apoptosis.71 Inflammation also accelerated erythrocyte destruction by activating macrophages.71 In addition, inflammation exhibited inhibition effects on the expression of transferrin and disturbed the ferric metabolism in erythrocytes, which may also be the cause of anemia.73
Management of Bleeding in ESKD
The managements of bleeding in ESKD are mainly based on its pathogenesis. For platelet dysfunction, dialysis is the first option because it removes uremic toxins from the plasma. Estrogen can also be used, which inhibits the production and release of nitric oxide from vascular endothelial cells, thus enhancing platelet–vessel wall interaction.75 For coagulation disorders, infusion of cryoprecipitate is always applied because it is rich in coagulation factor VIII, fibrinogen, fibronectin, and vWFs.76 Desmopressin is also an ideal choice because it can stimulate the release of vWFs.77 For anemia, transfusion of packed erythrocytes or application of recombinant erythropoietin can be chosen. The benefits and drawbacks of these treatments are shown in Table 4.
Table 4.
Benefits and adverse effects of different therapies for bleeding in patients with ESKD
| Therapies | Application | Benefits | Adverse Effects |
|---|---|---|---|
| Peritoneal dialysis61 | The preferred treatment for patients with ESKD who are at high risk of bleeding or have active bleeding | Anticoagulant therapy is not required | Peritonitis and loss of nutrients |
| Hemodialysis61 | The only option for patients who are not suitable for peritoneal dialysis because of insufficient clearance or intraperitoneal surgery | Mild or no anticoagulation can reduce the risk of bleeding | Thrombosis |
| Applying estrogen61 | Controlling bleeding in patients with advanced or severe kidney failure. Suitable for both male patients and female patients | Taking effect within 6 h and lasting for 2 wk by intravenous injection (0.6 mg/kg daily for 4–5 d) or taking effect within 2 d and lasting for 5 d by oral administration (25–50 mg/d). Especially suitable for patients who need continuous bleeding control Transdermal estradiol patches (50–100 μg/d, twice per week) are available, whose effects last longer, which could relieve the pain of patients |
Fluid retention, hypertension, and liver injury |
| Transfusion of cryoprecipitate61 | Only used for the emergency treatment of acute and severe bleeding | Rapidly inhibiting bleeding in patients with uremia | Therapeutic effects are short-lived, and not all patients respond Carrying the risk of infection76 |
| Applying DDAVP61 | Controlling bleeding in patients with uremia and preventing bleeding before renal biopsy | Intravenously administered at a dosage of 0.3µg/kg can inhibit bleeding within 1 h; the effect lasts for 8 h. Could also be given subcutaneously Could be given by intranasal route which is well-tolerated and safe |
Repeated administration may cause rapid depletion of vWFs, leading to hypotension, tachycardia, and thrombosis in some cases |
| Transfusion of packed erythrocytes78 | The main means to correct anemia before the use of erythropoiesis-stimulating agent | Bleeding time can be shortened by infusion of concentrated erythrocytes to a hematocrit of 30%61 | Blood volume overload, hyperkalemia, iron overload, blood-borne infection, fever, and allogenic sensitization79 |
| Application of recombinant erythropoietin78 | Maintaining the hemoglobin/hematocrit levels of patients with ESKD61 | Reducing the need for erythrocyte transfusions. Reducing fatigue and improving quality of life79 |
Increased risk of adverse cardiovascular outcomes79 Iron overload79 |
| Applying TXA80 | Should only be used in patients with severe, life-threatening bleeding that has failed to be controlled by other evidence-based treatments | A single small dose of TXA (7.5 mg/kg) intravenously could reduce the need for blood transfusion81 Could be safely used to treat severe hematuria in patients with CKD and PKD80 Could effectively treat massive hemorrhage of the upper digestive tract in hemodialysis patients82 |
Causing ureteral thrombosis and cortical necrosis, leading to acute renal failure. Contraindicated in patients with chronic kidney injuries83 Carrying the risk of systemic epilepsy because it could cross the blood–brain barrier and is neurotoxic83 |
DDAVP, desmopressin; vWFs, von Willebrand factors; TXA, tranexamic acid; PKD, polycystic kidney disease.
In addition, secondary hyperfibrinolysis resulting from the hypercoagulability of CKD is also a concern in patients with ESKD. Fibrinolytic drugs such as tranexamic acid (TXA) have been confirmed to shorten bleeding time and improve platelet function in most patients with ESKD.84 But TXA may accumulate in renal insufficiency and has not been shown to be more effective than commonly used therapies.85 Therefore, it was suggested that intravenous TXA should only be considered in acute cases where other treatments have failed to respond satisfactorily.85
RRT
RRT refers to dialysis and kidney transplantation. Patients undergoing RRT are at high risk of thrombosis.57,86 During dialysis, the formation of blood clots may block the circulation circuit, interrupting the dialysis process, and even endangering the patient's life.57 While the formation of graft thrombosis in kidney transplant recipients is one of the main causes of graft loss, which has become a concern for clinicians.86
Mechanism of Thrombosis in Dialysis
Thrombosis during hemodialysis was mainly caused by excessive activation of the contact pathway of the coagulation system and platelets.87,88
Activation of the contact pathway during hemodialysis has been demonstrated.87,88 Numerous studies have found significant increases in FXIIa levels in the blood of patients undergoing hemodialysis.87,88 FXIIa led to a surge in the production of thrombin which promoted blood clotting.87,88 Activation of FXII may be related to its contact with artificial surfaces because polyvinyl chloride, polyacrylonitrile, polymethylmethacrylate, cellulose triacetate, cuprophane, and polysulfone have been shown to induce FXII activation and thrombin production.88 However, the mechanism of contact pathway activation has not been well elucidated. It was only known that the negative charge on the artificial surface was conducive to the activation of FXII to some extent (Figure 5A).89 It was reported that FXII inhibitors, such as corn trypsin inhibitor, exhibited inhibitory effects on thrombin production induced by polyvinyl chloride membrane in whole blood or plasma.90 Therefore, elucidating the mechanism by which artificial surface activates FXII is of great significance for the development of novel anticoagulation agents or artificial surfaces that can effectively avoid thrombosis during hemodialysis.
Figure 5.
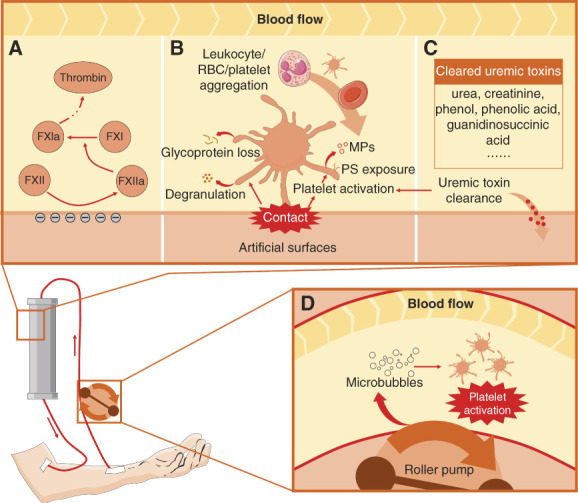
Mechanism of thrombosis in hemodialysis. (A) Contact between FXII and artificial surface activated the intrinsic pathway, which promoted thrombin production, resulting in enhanced coagulation. The negative charge on the artificial surface might contribute to the activation of FXII. (B) Contact between platelets and artificial surfaces induced platelet degranulation, PS exposure, PS-positive MP shedding, and glycoprotein loss. These effects eventually led to platelet activation and leukocyte, RBC, and platelet aggregation. (C) Hemodialysis cleared uremic toxins that induced platelet dysfunction, which promoted platelet activation. (D) Roller pump in the extracorporeal circuit induces the formation of microbubbles which promoted platelet activation. MP, microparticle; PS, phosphatidylserine; RBC, red blood cell.
The changes in platelet function during hemodialysis were complex because they were regulated by artificial surfaces, uremic toxins, and hemodynamics.57,61,91 It was reported that the contact of platelets with the artificial surface directly caused platelet activation and degranulation (Figure 5B).61,92 But artificial surface also led to the loss of platelet membrane glycoproteins at the same time (Figure 5B).92 Therefore, in the process of hemodialysis, a temporary platelet dysfunction usually occurred before platelet activation.92 Meanwhile, uremic toxins that may cause platelet dysfunction were removed during hemodialysis, which reversed platelet dysfunction (Figure 5C).61 In addition, platelets were directly exposed to the roller pump in the extracorporeal circuit; the changes in hemodynamics might induce the formation of microbubbles that promoted platelet activation (Figure 5D).91 These effects resulted in platelet hyperreactivity and thrombosis.
Differently, thrombosis is rare in patients undergoing peritoneal dialysis because of the lack of artificial surfaces.93
Mechanism of Thrombosis in Kidney Transplantation
The underlying diseases and perioperative treatments may be the causes of thrombosis in kidney transplant recipients.94
Before kidney transplantation, underlying diseases (such as NS) and preoperative treatments (such as dialysis) always resulted in a hypercoagulable state. Patients with ESKD usually required invasive treatments or experienced arteriosclerosis that led to endothelial injury.95 Besides, the use of diuretics increased blood viscosity and slowed blood flow.3 These disturbances led to coagulation and fibrinolytic system disorders, resulting in thrombosis.96
During kidney transplantation, physiological factors such as shorter and thinner renal veins,97 younger ages, and different blood types between recipients and donors may promote graft thrombosis.98 Prolonged ischemia during transplantation may induce acute tubular necrosis with graft edema, leading to thrombosis.99 Operation-induced vascular torsion, anastomotic stenosis, and endothelial injury can also lead to thrombosis.100
After kidney transplantation, patients usually received immunosuppressive therapies. Many immunosuppressants, such as cyclosporine, tacrolimusand, high doses of pulsed methylprednisolone, anti-CD3 antibody, thymoglobulin, mycophenolate mofetil, and azathioprine have been confirmed to disturb coagulation and fibrinolytic systems or cause vascular dysfunction, which were also an important cause of thrombosis.101 The mechanisms by which these immunosuppressants promoted thrombosis are described in detail in Table 5.
Table 5.
Immunosuppressants that induced thrombosis after kidney transplantation
| Immunosuppressant | Function | Prothrombotic Effects |
|---|---|---|
| Cyclosporine (calcineurin inhibitors) | Inhibiting Il-2 transcription and T-cell activation | Inducing renal vascular endothelial injury through its nephrotoxicity, which promoted thromboplastin generation, FVIII activation, TXA2 release, platelet activation, and suppressed thrombomodulin activity, hence downregulating the protein C anticoagulant pathway102 Elevating PAI and LDL levels and inducing hypofibrinolysis, which increased thrombogenicity and accelerated atherosclerosis39,103 |
| Tacrolimus (calcineurin inhibitors) | Inhibiting IL-2 release and T-cell activation | Inducing vascular dysfunction by causing increases in vasoconstrictive factors (such as endothelin and thromboxane), activation of the renin-angiotensin system, and decreases in vasodilative factors (such as PGE2, PGI2, and NO)104 |
| High doses of pulsed methylprednisolone (corticosteroid) | Inhibiting the phagocytosis of neutrophils and inducing T-cell apoptosis105 | Inducing overproduction of procoagulant factors106 |
| OKT3 antibody (antithymocyte/antilymphocyte globulin) | Directly targeting the TCR-CD3 complex, which is presently the most potent agent to treat acute renal allograft rejection107 | Inducing TF expression at the surface of both monocytes and endothelial cells, which activated the extrinsic coagulation pathway107 |
| Thymoglobulin (antithymocyte/antilymphocyte globulin) | Inducing lymphocyte depletion in the peripheral blood by activating complement-dependent cell lysis and inducing T-cell apoptosis | Thymoglobulin was not specific for T cells; it contained antibodies directed against platelets, which could activate platelets, inducing thrombosis108 |
| Mycophenolate mofetil (antimetabolite) | Noncompetitive, reversible inhibiting inosine monophosphate dehydrogenase, which is required for purine synthesis during lymphocyte activation109 | Although mycophenolate mofetil did not affect platelet function, it may act as a cofactor in the coagulation cascade, which promoted thrombosis by facilitating the production of Leiden-mutated FV (which cannot be inactivated by protein C)109,110 |
| Azathioprine (antimetabolite) | Azathioprine is converted into six-mercaptopurine which inhibits DNA synthesis, thereby inhibiting lymphocyte proliferation111 | May induce warfarin resistance by enhancing the synthesis of vitamin K–dependent coagulation factors111 |
FVIII, coagulation factor VIII; TXA2, thromboxane A2; PAI, plasminogen activator inhibitor; PGE2, prostaglandin E2; PGI2, prostacyclin; NO, nitric oxide; OKT3, anti-CD3 antibody; TCR, T-cell antigen receptor; FV, coagulation factor V; TF, tissue factor.
Anticoagulation in Dialysis
Anticoagulation is necessary for patients undergoing continuous RRT (CRRT). At present, guidelines recommend heparin or thrombin inhibitors for anticoagulant treatment in dialysis patients (Table 6).6,112,115 Warfarin is not recommended for dialysis patients with AF because it may induce stroke and bleeding.116,117 However, the application of heparin might cause HIT (the underlying mechanism is shown in Figure 6),9,118 which is a severe immune response characterized by severe thrombosis with a high mortality rate. Therefore, timely HIT detection is necessary for dialysis patients receiving heparin anticoagulation. But the diagnosis of HIT is complex because it relies on clinical suspicion followed by stepwise testing, which delays the treatment of HIT.119 Inspiring, our team has developed the first HIT antibody assay kit in China that could be used for fast bedside HIT diagnosis, which brought benefits to patients undergoing CRRT. An effective method to avoid HIT in CRRT is to use thrombin inhibitors, but the irreversibility and narrow therapeutic window limit its application.
Table 6.
| Anticoagulants | Dosages | Advantages | Disadvantages |
|---|---|---|---|
| Unfractionated heparin | 5–15 IU/kg (initial) 5–10 IU/kg per hour (continuous) |
Ease to use and could be reversed by protamine with a short half-life and lower cost | May induce bleeding, hypertriglyceridemia, HIT, osteoporosis, and hyperkalemia |
| Regional heparin with protamine | Heparin prefilter: 1000–1500 U/h Protamine postfilter: 10–12 mg/h |
Lower risk of bleeding | Complexity in administration and high cost |
| Enoxaparin | 0.15 mg/kg (initial) 0.05 mg/kg per hour (continuous) |
Lower risk of HIT | Lacking reversal agent, resulting in bioaccumulation risk. Higher cost |
| Dabigatran | 15–25 IU/kg (initial) 5 IU/kg per hour (continuous) |
||
| Argatroban | 0.1 mg/kg (initial) 0.05–0.2 mg/kg per minute (continuous) |
Can be used in patients with HIT | With a narrow therapeutic window and bleeding and anaphylaxis risk.113 Lacking reversal agent114 |
| Bivalirudin | 2 mg/h | ||
| RCA | Infused to achieve a citrate blood concentration of 3–4 mmol/L | Lower risk of bleeding and may improve inflammatory profile | Might cause metabolic acidosis because of citrate accumulation. Needing calcium monitoring |
HIT, heparin-induced thrombocytopenia; RCA, regional citrate anticoagulation.
Figure 6.
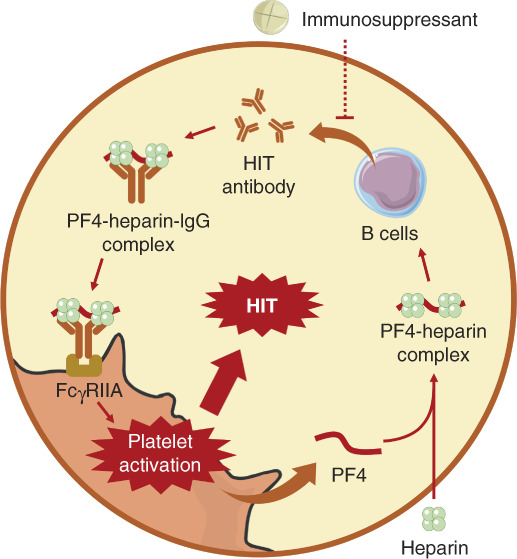
Mechanism of HIT. Heparin bound to PF4 to form the PF4-heparin complex that induced HIT antibody production from B cells. HIT antibodies bound to the PF4-heparin complex to form a PF4-heparin-IgG complex. PF4-heparin-IgG complex interacted with FcγRIIA, which induced platelet activation and secondary PF4 release. This circulation promoted the activation of a large number of platelets, resulting in HIT. Immunosuppressants might inhibit B cells from producing HIT antibodies, which inhibited HIT. HIT, heparin-induced thrombocytopenia; PF4, platelet factor 4.
Given the defects of heparin and thrombin inhibitors, regional citrate anticoagulation (RCA) has emerged in recent years, which is reversible and more effective in anticoagulation with less bleeding and HIT risk.120 However, excessive RCA treatment might cause metabolic acidosis because of citrate accumulation. As a consequence, calcium monitoring is necessary for patients receiving RCA.121
In addition, DOACs were also used in dialysis patients, but apixaban was the only drug that has been proven to be safely used for acute VTE treatment in dialysis patients. Dabigatran and rivaroxaban are not recommended for dialysis patients because of the lack of insufficient evidence.
In recent years, the benefits of factor XI (FXI) inhibitors in dialysis patients have attracted extensive attention. Different from FX, FXI is only involved in the internal coagulation pathway (as shown in Figure 1). Because the hypercoagulable state during dialysis is mainly affected by internal pathways, targeting FXI may gain more advantages than targeting FX in the anticoagulation treatment.122 Several clinical trials have been conducted on the anticoagulant effects of FXI inhibitors in dialysis patients (Table 7); these inhibitors may be the dawn of dialysis patients in the future.
Table 7.
Clinical trials on the anticoagulation effects of factor XI inhibitors in dialysis patients
| Identifier | Drugs | Types | Phases | Status |
|---|---|---|---|---|
| NCT03358030 | ISIS 416858 | FXI antisense oligonucleotide | Phase 2 | Completed |
| NCT04534114 | BAY2976217 | FXI antisense oligonucleotide | Phase 2 | Completed |
| NCT04523220 | BAY1213790 (osocimab) | Monoclonal antibody to FXIa | Phase 2 | Completed |
FXI, factor XI; FXIa, activated factor XI.
Anticoagulation in Kidney Transplantation
Guidelines for anticoagulant/antiplatelet treatment in kidney transplant recipients are still lacking, but continuous anticoagulation is necessary because the risk of thrombosis persists.123 Heparin, low-molecular-weight heparin, and warfarin have been shown to be effective in kidney transplant recipients. Interestingly, unlike dialysis patients, kidney transplant recipients undergoing heparin treatment rarely experienced HIT even if they have a history of HIT. This phenomenon might result from the inhibitory effect of immunosuppressants on the synthesis of HIT antibodies (Figure 6),124 which indicated that heparin may be safe for kidney transplant recipients. Antiplatelet drugs, including clopidogrel and aspirin, could also be used but is not the first choice because available evidences showed that antiplatelet drugs did not improve graft loss in kidney transplant recipients.125
DOACs may not be suitable for kidney transplant recipients because it is cleared by the kidney and has drug interactions with certain immunosuppressants, especially calcineurin inhibitors.33
IgA Nephropathy
IgA nephropathy is an autoimmune nephropathy that is manifested by the deposition of IgA immune complexes in the mesangial region of glomeruli.126,127 Although the incidence of thrombosis is low, many studies have also found hypercoagulable states in patients with IgA nephropathy.128,129
Generation of Hypercoagulable States in IgA Nephropathy
The hypercoagulable state of patients with IgA nephropathy may be associated with excessive accumulation of coagulation factors in injured glomeruli, especially coagulation factor V, coagulation factor VII, FVIII, and FIX.130,131 The deposition of coagulation factors may be secondary to IgA deposition. It was reported that the glomeruli capillary wall offered the first contact with circulating macromolecular IgA1, which triggered the secondary complement deposition and alternative pathway activation. Activation of complement destroyed vascular endothelium, which induced collagen exposure and TF release, resulting in coagulation activation (Figure 7B).
Figure 7.
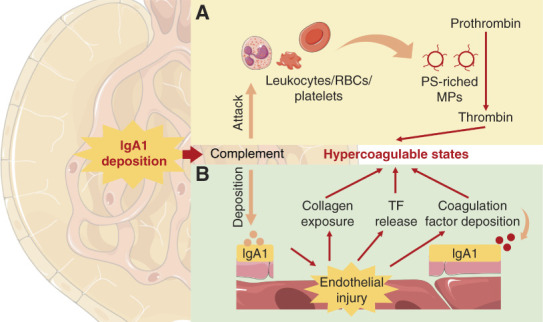
Mechanism of the generation of hypercoagulable state in IgA nephropathy. (A) The deposited complement attacked PS-positive MP-originating cells, leading to the shedding of PS-riched MPs, which promoted the production of thrombin, resulting in hypercoagulable states. (B) The deposited complement induced endothelial injury, which promoted collagen exposure, TF release, and coagulation factor deposition, resulting in hypercoagulable states. MP, microparticle; PS, phosphatidylserine; TF, tissue factor.
MPs originated from leukocytes, red blood cells, and platelets might also contribute to the hypercoagulable states in patients with IgA nephropathy. It was reported that terminal complement complex (C5b–C9) may attack MP-originating cells, pouncing PS to the outer leaflet of the plasma membrane accompanied by the shedding of MPs; MPs promoted thrombin production by providing binding sites for the assembly of the prothrombinase and tenase complexes (Figure 7A).132 Indeed, patients with IgA nephropathy usually showed a high level of PS-positive MPs and increased PS exposure in MP-originating cells.133 It was reported that the addition of MPs derived from patients with IgA nephropathy or normal ECs cultured with serum from patients with IgA nephropathy to MP-depleted plasma containing erythrocytes and leukocytes significantly reduced the median coagulation time of MP-depleted plasma, which could be restored by lactadherin (a PS blocker),133 confirming the procoagulant roles of MPs in patients with IgA nephropathy.
Benefits of Antithrombotic Agents for Patients with IgA Nephropathy
At present, there are no guidelines indicating the need for anticoagulation in patients with IgA nephropathy. But antithrombotic agents have been widely applied to patients with IgA nephropathy in China and Japan (Table 8).150 Several multicenter randomized controlled trials found that anticoagulants/antiplatelet drugs significantly ameliorated renal injury caused by IgA immune complex deposition (Table 8).138,150 These effects may be related to the downregulation of MPs by anticoagulants/antiplatelet drugs because MPs could also bind to complement C3 and induce proinflammatory cytokines in IgA nephropathy.151
Table 8.
Antithrombotic agents that showed therapeutic effects on IgA nephropathy
| Agents | Known Efficacy |
|---|---|
| Anticoagulation | |
| Danaparoid | One-week treatment with danaparoid (400 U/kg, twice daily, ip) significantly improved kidney injury in the IgA nephropathy mouse model134 |
| Heparin | Continuous intravenous heparin infusion (10,000-25,000 U/d) combined with glucocorticoid (40 mg/d) improved kidney injuries by downregulating caldesmon and inhibiting mesangial cell activation in patients with IgA nephropathy135 |
| Antiplatelet | |
| Dipyridamole | a. Dipyridamole (300 mg/d, po) significantly reduced proteinuria in patients with IgA nephropathy139,140 |
| b. Dipyridamole alone (5 mg/kg·days, po) slightly improved albuminuria in childhood IgA nephropathy141 | |
| Aspirin | Aspirin (100-200 mg/d) combined with eicosapentaenoic acid (1800-2700 mg/d) effectively inhibited IgA and C3 deposition in the glomeruli and improved renal function in patients with progressive IgA nephropathy142 |
| Dilazep | a. Dilazep (300 mg/kg·d, oral administration at age 12-60 wk or 20-60 wk) effectively improved renal dysfunction in the IgA nephropathy mouse model143 |
| b. Dilazep significantly inhibited proteinuria and improved renal injury in patients with IgA nephropathy144 | |
| Ferulate | Erazine ferulate tablets combined with eucalyptol limonene pinene enteric soft capsules effectively improved kidney injury in childhood IgA nephropathy145 |
| Anticoagulation+ antiplatelet | |
| Warfarin+ dipyridamole | Combining warfarin (a single morning dose maintains the thrombotest at 20%-50%) and dipyridamole (6 mg/kg·days, up to 300 mg/d, po) with prednisolone and mizoribine for 2 yr effectively inhibited albuminuria in severe childhood IgA nephropathy but showed a tendency to promote glomerulosclerosis138 |
| Heparin+ warfarin+ dipyridamole | a. Pretreatment of continuous intravenous heparin infusion (10,000-25,000 U/d) for 8 wk followed by warfarin (1-2 mg/d) effectively enhanced the therapeutic effects of dipyridamole or ACEi/ARB combined with dipyridamole on patients with progressive IgA nephropathy136 |
| b. Combining heparin-warfarin (a single morning dose to maintain the thrombotest at 30%-50%) and dipyridamole (5 mg/kg·days, up to 400 mg/d, po) with prednisolone and azathioprine for 2 yr effectively improved glomerulosclerosis in childhood IgA nephropathy137 | |
| Thrombolysis | |
| Urokinase | a. A single injection of urokinase induced significant fibrinolytic activity, effectively improving albuminuria and/or hematuria in patients with IgA nephropathy146 |
| b. Urokinase significantly reduced deposition of C3 in patients with IgA nephropathy, but did not affect IgA and fibrin deposition147 | |
| c. In patients with severe IgA nephropathy, therapeutic effects of urokinase (100,000 IU/d) combined with benazepril (10 mg/d) were superior to benazepril alone148 | |
| d. Continuous urokinase therapy gained better prognoses than antiplatelet therapies in patients with moderate-to-advanced degrees of IgA nephropathy149 |
ip, intraperitoneal injection; ACEi, angiotensin-converting enzyme inhibitors; ARB, angiotensin II receptor blockers; po, per os.
However, IgA nephropathy is considered as the most common underlying disease of ARN.152 Warfarin,153,154 dabigatran,155,156 and aspirin157 have been confirmed to induce ARN in patients with IgA nephropathy. Therefore, unnecessary anticoagulation in patients with IgA nephropathy may carry the risk of aggravating kidney injuries. The use of warfarin and DOACs should be minimized if not necessary.
ARN
ARN is defined as AKI caused by oral anticoagulants such as warfarin and DOACs.152,158 It is mainly manifested by impairment of the glomeruli filtration barrier with Bowman's space and tubular hemorrhage, resulting in tubular obstruction and occlusion.159
Pathogenesis of ARN
The pathogenesis of ARN has not been fully elucidated. It was found that dabigatran-treated CKD rats developed ARN phenotypes, which could be enhanced by protease-activated receptor (PAR)–1 inhibitor SCH79797.160 These findings suggested that DOACs may be the inhibitor of PARs by inhibiting thrombin production. Because PARs provide nutritional support for endothelial cells, the glomeruli destruction hemorrhage might be caused by the inhibitory effect of DOACs on PARs.161 However, it was also found that application of anticoagulant therapies alone cannot cause ARN, and knockout of PAR or application of PAR-1 inhibitor vorapaxar did not induce ARN in experimental models or human patients.161,162 Clinical data also suggested that DOACs induced ARN only when there was significant nephron reduction, hyperperfusion, and hypertension163 or acute glomeruli injury.164 Therefore, the current views hold that ARN is the result of a second hit of anticoagulants on fragile glomeruli destroyed by the primary disease.158
Treatment of ARN
The diagnosis of ARN is shown in Figure 8. However, treatment options for ARN are limited. One of the most effective methods is to closely monitor the patient's coagulation and renal function and adopt effective coping strategies during warfarin or DOAC treatment.161 For patients with ARN, treatment is supportive; control of BP and reduction of anticoagulation were commonly used.161 If the patient is anticoagulated with warfarin, the anticoagulation strategy should be switched to DOACs. If the patient is anticoagulated with DOACs, the dose should be reduced.158 In patients with primary or secondary glomerulopathy without ARN, anticoagulation should be combined with glomerular anti-inflammatory therapy to effectively prevent ARN.158
Figure 8.
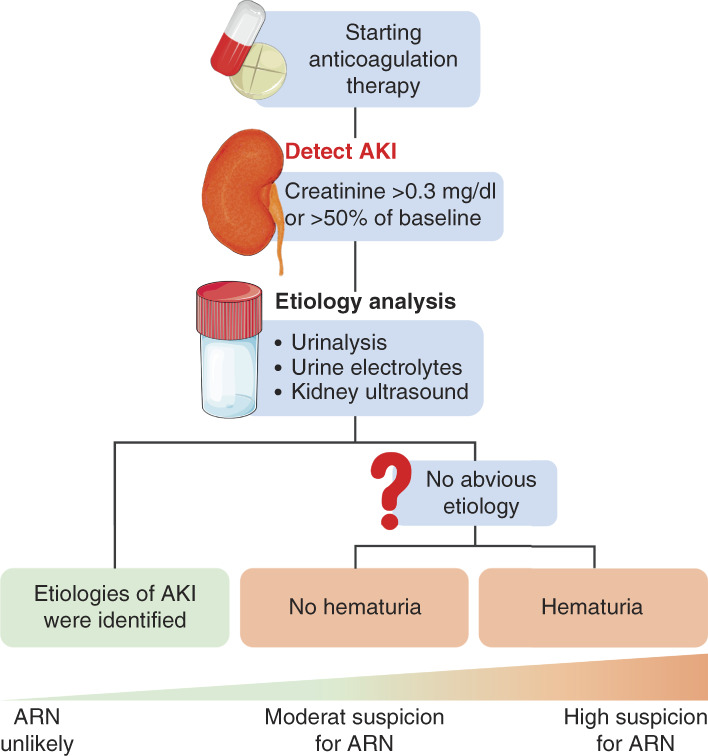
Diagnosis of ARN. When AKI occurs in patients undergoing anticoagulant therapy with an INR >3.0, ARN should be considered. The first diagnosis is to identify the cause of AKI by urinalysis, urine electrolytes, and kidney ultrasound. If the cause of AKI is clear, ARN could be excluded. If the cause of AKI is unclear, further diagnosis is needed to determine whether the patient develops hematuria. ARN is diagnosed if the patient develops hematuria. If the patient does not develop hematuria, the ARN is not fully determined, but the patient is always treated as ARN. ARN, anticoagulation-related nephropathy; INR, international normalized ratio.
Conclusion
Although the crosstalk between nephropathy and coagulation disorder has attracted the attention of clinicians and researchers for decades, practices and studies in this field are still at a relatively preliminary stage. For thromboembolism, heparin, warfarin, DOACs, and antiplatelet drugs are widely used, but adverse effects such as bleeding, HIT, and ARN have become lingering nightmares for clinicians. Owing to the lack of novel drugs, clinicians only have the option of adjusting the dosage (such as adjusting the dosage of DOACs in patients with CKD), which reduces the therapeutic effects at the same time. For bleeding in patients with ESKD, all treatment options have major defects, but there is no better alternative method at present. Encouragingly, some novel ideals point the direction for future drug development, such as inhibiting hypercholesterolemia in NS, targeting the AhR-STUB1-TF axis in CKD, and targeting FXI in dialysis. These methods bypass the limitations of classical drugs, which might bring benefits to patients with nephropathy. But more novel targets and drugs are urgently needed, which requires the joint efforts of researchers and clinicians.
Acknowledgments
We would like to thank Qiufen Xie for the guidance of clinical anticoagulant medication for patients with kidney disease and thank for the funding of National Key R&D Program of China, National High Level Hospital Clinical Research Funding, and National Natural Science Foundation of China for this work.
Disclosures
All authors have nothing to disclose.
Funding
This work was funded by National High Level Hospital Clinical Research Funding (Scientific Research Seed Fund of Peking University First Hospital, No. 2023SF51. Science and Technology Achievement Transformation and Incubation Guidance Fund of Peking University First Hospital, No. 2022CX04, No. 2022CX14, No. 2022CX11, and No. 2022RT01). National Key R&D Program of China (No. 2020YFC2008304); National Natural Science Foundation of China (No. 82274024, No. 81973395, No. 82073935, and No. 82274015).
Author Contributions
Conceptualization: Yimin Cui.
Funding acquisition: Yimin Cui, Xiaocong Pang, Qian Xiang, Zhiwei Qiu.
Investigation: Zhiwei Qiu.
Project administration: Yimin Cui.
Resources: Yimin Cui, Xiaocong Pang.
Supervision: Yimin Cui, Qian Xiang.
Visualization: Zhiwei Qiu.
Writing – original draft: Zhiwei Qiu.
Writing – review & editing: Yimin Cui, Xiaocong Pang, Zhiwei Qiu, Qian Xiang.
References
- 1.Gui M, Zhao B, Huang J, Chen E, Qu H, Mao E. Pathogenesis and therapy of coagulation disorders in severe acute pancreatitis. J Inflamm Res. 2023;2023(16):57–67. doi: 10.2147/JIR.S388216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans RG, Smith DW, Lee CJ, Ngo JP, Gardiner BS. What makes the kidney susceptible to hypoxia? Anat Rec (Hoboken). 2020;303(10):2544–2552. doi: 10.1002/ar.24260 [DOI] [PubMed] [Google Scholar]
- 3.Sharp W, Olivero JJ. Venous thrombosis in nephrotic syndrome. Methodist Debakey Cardiovasc J. 2018;14(3):237–238. doi: 10.14797/mdcj-14-3-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu T, Tang LV, Hu Y. Venous thromboembolism in kidney diseases and genetic predisposition. Kidney Dis (Basel). 2022;8(3):181–189. doi: 10.1159/000523777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlou EG Georgatzakou HT Fortis SP, et al. Coagulation abnormalities in renal pathology of chronic kidney disease: the interplay between blood cells and soluble factors. Biomolecules. 2021;11(9):1309. doi: 10.3390/biom11091309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Legrand M, Tolwani A. Anticoagulation strategies in continuous renal replacement therapy. Semin Dial. 2021;34(6):416–422. doi: 10.1111/sdi.12959 [DOI] [PubMed] [Google Scholar]
- 7.Garlapati P, Gajjar B, Then EO, Gayam V. End-stage renal disease and lower gastrointestinal bleeding-A propensity-matched analysis of nationwide inpatient sample. Int J Clin Pract. 2021;75(2):e13633. doi: 10.1111/ijcp.13633 [DOI] [PubMed] [Google Scholar]
- 8.Mezue K, Ram P, Egbuche O, Menezes RG, Lerma E, Rangaswami J. Anticoagulation-related nephropathy for the internist: a concise review. Am J Cardiovasc Dis. 2020;10(4):301–305. PMID: 33224577. [PMC free article] [PubMed] [Google Scholar]
- 9.Hvas AM, Favaloro EJ, Hellfritzsch M. Heparin-induced thrombocytopenia: pathophysiology, diagnosis and treatment. Expert Rev Hematol. 2021;14(4):335–346. doi: 10.1080/17474086.2021.1905512 [DOI] [PubMed] [Google Scholar]
- 10.Gigante A Di Mario F Pierucci A, et al. Kidney disease and venous thromboembolism: does being woman make the difference? Eur J Intern Med. 2017;39:18–23. doi: 10.1016/j.ejim.2017.02.012 [DOI] [PubMed] [Google Scholar]
- 11.Chen F, Peng Y, Chen M. Spontaneous coronary thrombosis in a young patient with nephrotic syndrome. Am J Med Sci. 2020;359(6):378–381. doi: 10.1016/j.amjms.2020.02.006 [DOI] [PubMed] [Google Scholar]
- 12.Freedman JE. Molecular regulation of platelet-dependent thrombosis. Circulation. 2005;112(17):2725–2734. doi: 10.1161/CIRCULATIONAHA.104.494468 [DOI] [PubMed] [Google Scholar]
- 13.Kerlin BA, Ayoob R, Smoyer WE. Epidemiology and pathophysiology of nephrotic syndrome-associated thromboembolic disease. Clin J Am Soc Nephrol. 2012;7(3):513–520. doi: 10.2215/CJN.10131011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gigante A Barbano B Sardo L, et al. Hypercoagulability and nephrotic syndrome. Curr Vasc Pharmacol. 2014;12(3):512–517. doi: 10.2174/157016111203140518172048 [DOI] [PubMed] [Google Scholar]
- 15.Bang NU, Trygstad W, Schroeder JE, Heidenreich RO, Csiscko BM. Enhanced platelet function in glomerular renal disease. J Lab Clin Med. 1973;81(5):651–660. doi: 10.5555/uri:pii:0022214373902461 [DOI] [PubMed] [Google Scholar]
- 16.Shattil SJ, Cooper RA. Role of membrane lipid composition, organization, and fluidity in human platelet function. Prog Hemost Thromb. 1978;4:59–86. PMID: 362482. [PubMed] [Google Scholar]
- 17.Jackson CA, Greaves M, Patterson AD, Brown CB, Preston FE. Relationship between platelet aggregation, thromboxane synthesis and albumin concentration in nephrotic syndrome. Br J Haematol. 1982;52(1):69–77. doi: 10.1111/j.1365-2141.1982.tb03862.x [DOI] [PubMed] [Google Scholar]
- 18.Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362(9384):629–639. doi: 10.1016/S0140-6736(03)14184-0 [DOI] [PubMed] [Google Scholar]
- 19.Eneman B, Levtchenko E, van den Heuvel B, Van Geet C, Freson K. Platelet abnormalities in nephrotic syndrome. Pediatr Nephrol. 2016;31(8):1267–1279. doi: 10.1007/s00467-015-3173-8 [DOI] [PubMed] [Google Scholar]
- 20.Wasilewska AM, Zoch-Zwierz WM, Tomaszewska B, Biernacka A. Platelet-derived growth factor and platelet profiles in childhood nephrotic syndrome. Pediatr Nephrol. 2005;20(1):36–41. doi: 10.1007/s00467-004-1620-z [DOI] [PubMed] [Google Scholar]
- 21.Tkaczyk M, Baj Z. Surface markers of platelet function in idiopathic nephrotic syndrome in children. Pediatr Nephrol. 2002;17(8):673–677. doi: 10.1007/s00467-002-0865-7 [DOI] [PubMed] [Google Scholar]
- 22.Sirolli V Ballone E Garofalo D, et al. Platelet activation markers in patients with nephrotic syndrome. Nephron. 2002;91(3):424–430. doi: 10.1159/000064282 [DOI] [PubMed] [Google Scholar]
- 23.Gao C Xie R Yu C, et al. Procoagulant activity of erythrocytes and platelets through phosphatidylserine exposure and microparticles release in patients with nephrotic syndrome. Thromb Haemost. 2012;107(04):681–689. doi: 10.1160/TH11-09-0673 [DOI] [PubMed] [Google Scholar]
- 24.Kelddal S, Nykjaer KM, Gregersen JW, Birn H. Prophylactic anticoagulation in nephrotic syndrome prevents thromboembolic complications. BMC Nephrol. 2019;20(1):139. doi: 10.1186/s12882-019-1336-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glassock RJ. Prophylactic anticoagulation in nephrotic syndrome: a clinical conundrum. J Am Soc Nephrol. 2007;18(8):2221–2225. doi: 10.1681/ASN.2006111300 [DOI] [PubMed] [Google Scholar]
- 26.Mahmoodi BK ten Kate MK Waanders F, et al. High absolute risks and predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome: results from a large retrospective cohort study. Circulation. 2008;117(2):224–230. doi: 10.1161/CIRCULATIONAHA.107.716951 [DOI] [PubMed] [Google Scholar]
- 27.Li X, Xie X, Zhao Y, Wang G, Shao H, Zhang X. Some points for the KDIGO 2021 guideline for prophylactic anticoagulation in membranous nephropathy: is it clear enough for us to follow?. Nephron. 2023;147(3-4):193–198. doi: 10.1159/000525913 [DOI] [PubMed] [Google Scholar]
- 28.Sasaki Y, Raita Y, Uehara G, Higa Y, Miyasato H. Carotid thromboembolism associated with nephrotic syndrome treated with dabigatran. Case Rep Nephrol Dial. 2014;4(1):42–52. doi: 10.1159/000362162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sexton DJ de Freitas DG Little MA, et al. Direct-acting oral anticoagulants as prophylaxis against thromboembolism in the nephrotic syndrome. Kidney Int Rep. 2018;3(4):784–793. doi: 10.1016/j.ekir.2018.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds ML, Nachman PH, Mooberry MJ, Crona DJ, Derebail VK. Recurrent venous thromboembolism in primary membranous nephropathy despite direct Xa inhibitor therapy. J Nephrol. 2019;32(4):669–672. doi: 10.1007/s40620-018-0552-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada Y Nagaba Y Nagaba H, et al. Edoxaban was effective for treating renal vein thrombosis in a patient with nephrotic syndrome. Intern Med. 2017;56(17):2307–2310. doi: 10.2169/internalmedicine.8742-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L, Zhang H, Zhang J, Tian H, Liang J, Liu Z. Rivaroxaban for the treatment of venous thromboembolism in patients with nephrotic syndrome and low AT-III: a pilot study. Exp Ther Med. 2018;15(1):739–744. doi: 10.3892/etm.2017.5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derebail VK, Rheault MN, Kerlin BA. Role of direct oral anticoagulants in patients with kidney disease. Kidney Int. 2020;97(4):664–675. doi: 10.1016/j.kint.2019.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resh M, Mahmoodi BK, Navis GJ, Veeger NJ, Lijfering WM. Statin use in patients with nephrotic syndrome is associated with a lower risk of venous thromboembolism. Thromb Res. 2011;127(5):395–399. doi: 10.1016/j.thromres.2010.12.020 [DOI] [PubMed] [Google Scholar]
- 35.Yashiro M, Muso E, Shio H, Sasayama S. Amelioration of hypercholesterolaemia by HMG-CoA reductase inhibitor (Pravastatin) improved platelet hyperaggregability in nephrotic patients. Nephrol Dial Transpl. 1994;9(12):1842–1843. doi: 10.1093/ndt/9.12.1842 [DOI] [PubMed] [Google Scholar]
- 36.Wattanakit K, Cushman M, Stehman-Breen C, Heckbert SR, Folsom AR. Chronic kidney disease increases risk for venous thromboembolism. J Am Soc Nephrol. 2008;19(1):135–140. doi: 10.1681/ASN.2007030308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32(5):S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470 [DOI] [PubMed] [Google Scholar]
- 38.Huang MJ Wei RB Wang Y, et al. Blood coagulation system in patients with chronic kidney disease: a prospective observational study. BMJ Open. 2017;7(5):e014294. doi: 10.1136/bmjopen-2016-014294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Opatrny K, Jr., Zemanova P, Opatrna S, Vit L. Fibrinolysis in chronic renal failure, dialysis and renal transplantation. Ann Transplant. 2002;7(1):34–43. PMID: 12221902 [PubMed] [Google Scholar]
- 40.Fryc J, Naumnik B. Thrombolome and its emerging role in chronic kidney diseases. Toxins (Basel). 2021;13(3):223. doi: 10.3390/toxins13030223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu M, Kim YJ, Kang DH. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin J Am Soc Nephrol. 2011;6(1):30–39. doi: 10.2215/CJN.05340610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gondouin B Cerini C Dou L, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013;84(4):733–744. doi: 10.1038/ki.2013.133 [DOI] [PubMed] [Google Scholar]
- 43.Dias GF Bonan NB Steiner TM, et al. Indoxyl sulfate, a uremic toxin, stimulates reactive oxygen species production and erythrocyte cell death supposedly by an organic anion transporter 2 (OAT2) and NADPH oxidase activity-dependent pathways. Toxins (Basel). 2018;10(7):280. doi: 10.3390/toxins10070280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dou L Sallee M Cerini C, et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J Am Soc Nephrol. 2015;26(4):876–887. doi: 10.1681/ASN.2013121283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolachalama VB Shashar M Alousi F, et al. Uremic solute-aryl hydrocarbon receptor-tissue factor Axis associates with thrombosis after vascular injury in humans. J Am Soc Nephrol. 2018;29(3):1063–1072. doi: 10.1681/ASN.2017080929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nemet I Saha PP Gupta N, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180(5):862–877.e22. doi: 10.1016/j.cell.2020.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meijers BK Van Kerckhoven S Verbeke K, et al. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54(5):891–901. doi: 10.1053/j.ajkd.2009.04.022 [DOI] [PubMed] [Google Scholar]
- 48.Zhu W Gregory JC Org E, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111–124. doi: 10.1016/j.cell.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shashar M Belghasem ME Matsuura S, et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci Transl Med. 2017;9(417):eaam8475. doi: 10.1126/scitranslmed.aam8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackel S Kiouptsi K Lillich M, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood. 2017;130(4):542–553. doi: 10.1182/blood-2016-11-754416 [DOI] [PubMed] [Google Scholar]
- 51.Gao Y, Zhang J, Chen H, Wang Z, Hou J, Wang L. Dimethylamine enhances platelet hyperactivity in chronic kidney disease model. J Bioenerg Biomembr. 2021;53(5):585–595. doi: 10.1007/s10863-021-09913-4 [DOI] [PubMed] [Google Scholar]
- 52.Yano JM Yu K Donaldson GP, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161(2):264–276. doi: 10.1016/j.cell.2015.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turakhia MP Blankestijn PJ Carrero JJ, et al. Chronic kidney disease and arrhythmias: conclusions from a kidney disease: improving global Outcomes (KDIGO) controversies conference. Eur Heart J. 2018;39(24):2314–2325. doi: 10.1093/eurheartj/ehy060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhaese SAM, De Vriese AS. Oral anticoagulation in patients with advanced chronic kidney disease and atrial fibrillation: beyond anticoagulation. Mayo Clinic Proc. 2023;98(5):750–770. doi: 10.1016/j.mayocp.2023.01.007 [DOI] [PubMed] [Google Scholar]
- 55.Park H Yu HT Kim TH, et al. Oral anticoagulation therapy in atrial fibrillation patients with advanced chronic kidney disease: CODE-AF registry. Yonsei Med J. 2023;64(1):18–24. doi: 10.3349/ymj.2022.0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahaffey KW, Turakhia MP. Initiating anticoagulation with the intention of cardioverting: does drug choice matter? Eur Heart J. 2018;39(32):2972–2974. doi: 10.1093/eurheartj/ehy303 [DOI] [PubMed] [Google Scholar]
- 57.Cheung CYS, Parikh J, Farrell A, Lefebvre M, Summa-Sorgini C, Battistella M. Direct oral anticoagulant use in chronic kidney disease and dialysis patients with venous thromboembolism: a systematic review of thrombosis and bleeding outcomes. Ann Pharmacother. 2021;55(6):711–722. doi: 10.1177/1060028020967635 [DOI] [PubMed] [Google Scholar]
- 58.Wang Z Xiang Q Hu K, et al. Comparison of the safety and efficacy of direct oral anticoagulants and warfarin in atrial fibrillation or venous thromboembolism in patients with renal impairment: systematic review, meta-analysis and network meta-analysis. Am J Cardiovasc Drugs. 2021;21(6):643–657. doi: 10.1007/s40256-021-00469-7 [DOI] [PubMed] [Google Scholar]
- 59.Karcioglu O Zengin S Ozkaya B, et al. Direct (new) oral anticoagulants (DOACs): drawbacks, bleeding and reversal. Cardiovasc Hematol Agents Med Chem. 2022;20(2):103–113. doi: 10.2174/1871525719666210914110750 [DOI] [PubMed] [Google Scholar]
- 60.Mavrakanas TA, Charytan DM, Winkelmayer WC. Direct oral anticoagulants in chronic kidney disease: an update. Curr Opin Nephrol Hypertens. 2020;29(5):489–496. doi: 10.1097/MNH.0000000000000634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaw D, Malhotra D. HEMATOLOGY: issues in the dialysis patient: platelet dysfunction and end-stage renal disease: platelet dysfunction and ESRD. Semin Dial. 2006;19(4):317–322. doi: 10.1111/j.1525-139X.2006.00179.x [DOI] [PubMed] [Google Scholar]
- 62.Evans EP, Branch RA, Bloom AL. A clinical and experimental study of platelet function in chronic renal failure. J Clin Pathol. 1972;25(9):745–753. doi: 10.1136/jcp.25.9.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vlachoyannis J, Schoeppe W. Adenylate cyclase activity and cAMP content of human platelets in uraemia. Eur J Clin Invest. 1982;12(5):379–381. doi: 10.1111/j.1365-2362.1982.tb00683.x [DOI] [PubMed] [Google Scholar]
- 64.Di Minno G, Martinez J, McKean ML, De La Rosa J, Burke JF, Murphy S. Platelet dysfunction in uremia. Multifaceted defect partially corrected by dialysis. Am J Med. 1985;79(5):552–559. doi: 10.1016/0002-9343(85)90051-8 [DOI] [PubMed] [Google Scholar]
- 65.Macconi D Vigano G Bisogno G, et al. Defective platelet aggregation in response to platelet-activating factor in uremia associated with low platelet thromboxane A2 generation. Am J Kidney Dis. 1992;19(4):318–325. doi: 10.1016/s0272-6386(12)80447-1 [DOI] [PubMed] [Google Scholar]
- 66.Escolar G, Diaz-Ricart M, Cases A, Castillo R, Ordinas A, White JG. Abnormal cytoskeletal assembly in platelets from uremic patients. Am J Pathol. 1993;143(3):823–831. PMID: 8362980. [PMC free article] [PubMed] [Google Scholar]
- 67.Benigni A Boccardo P Galbusera M, et al. Reversible activation defect of the platelet glycoprotein IIb-IIIa complex in patients with uremia. Am J Kidney Dis. 1993;22(5):668–676. doi: 10.1016/s0272-6386(12)80429-x [DOI] [PubMed] [Google Scholar]
- 68.Sloand EM Sloand JA Prodouz K, et al. Reduction of platelet glycoprotein Ib in uraemia. Br J Haematol. 1991;77(3):375–381. doi: 10.1111/j.1365-2141.1991.tb08587.x [DOI] [PubMed] [Google Scholar]
- 69.Remuzzi G, Mecca G, Cavenaghi A, Donati MB, de Gaetano G. Prostacyclin-like activity and bleeding in renal failure. Lancet. 1977;310(8050):1195–1197. doi: 10.1016/s0140-6736(77)90437-8 [DOI] [PubMed] [Google Scholar]
- 70.Noris M Benigni A Boccardo P, et al. Enhanced nitric oxide synthesis in uremia: implications for platelet dysfunction and dialysis hypotension. Kidney Int. 1993;44(2):445–450. doi: 10.1038/ki.1993.264 [DOI] [PubMed] [Google Scholar]
- 71.Georgatzakou HT, Antonelou MH, Papassideri IS, Kriebardis AG. Red blood cell abnormalities and the pathogenesis of anemia in end-stage renal disease. Proteomics Clin Appl. 2016;10(8):778–790. doi: 10.1002/prca.201500127 [DOI] [PubMed] [Google Scholar]
- 72.Provenzano R Shutov E Eremeeva L, et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrol Dial Transplant. 2021;36(9):1717–1730. doi: 10.1093/ndt/gfab051 [DOI] [PubMed] [Google Scholar]
- 73.Bieber E. Erythropoietin, the biology of erythropoiesis and epoetin alfa. An overview. J Reprod Med. 2001;46(5 suppl l):521–530. PMID: 11396386. [PubMed] [Google Scholar]
- 74.Capelli I Cianciolo G Gasperoni L, et al. Folic acid and vitamin B12 administration in CKD, why not? Nutrients. 2019;11(2):383. doi: 10.3390/nu11020383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heistinger M Stockenhuber F Schneider B, et al. Effect of conjugated estrogens on platelet function and prostacyclin generation in CRF. Kidney Int. 1990;38(6):1181–1186. doi: 10.1038/ki.1990.331 [DOI] [PubMed] [Google Scholar]
- 76.Janson PA, Jubelirer SJ, Weinstein MJ, Deykin D. Treatment of the bleeding tendency in uremia with cryoprecipitate. N Engl J Med. 1980;303(23):1318–1322. doi: 10.1056/NEJM198012043032302 [DOI] [PubMed] [Google Scholar]
- 77.Kaufmann JE, Oksche A, Wollheim CB, Gunther G, Rosenthal W, Vischer UM. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J Clin Invest. 2000;106(1):107–116. doi: 10.1172/JCI9516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Livio M, Marchesi D, Remuzzi G, Gotti E, Mecca G, de Gaetano G. Uraemic bleeding: role of anaemia and beneficial effect of red cell transfusions. Lancet. 1982;320(8306):1013–1015. doi: 10.1016/s0140-6736(82)90050-2 [DOI] [PubMed] [Google Scholar]
- 79.Hanna RM, Streja E, Kalantar-Zadeh K. Burden of anemia in chronic kidney disease: beyond erythropoietin. Adv Ther. 2021;38(1):52–75. doi: 10.1007/s12325-020-01524-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alameel T, West M. Tranexamic acid treatment of life-threatening hematuria in polycystic kidney disease. Int J Nephrol. 2011;2011:203579. doi: 10.4061/2011/203579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma TK, Chow KM, Kwan BC, Leung CB, Szeto CC, Li PK. Manifestation of tranexamic acid toxicity in chronic kidney disease and kidney transplant patients: a report of four cases and review of literature. Nephrology (Carlton). 2017;22(4):316–321. doi: 10.1111/nep.12762 [DOI] [PubMed] [Google Scholar]
- 82.Sabovic M, Lavre J, Vujkovac B. Tranexamic acid is beneficial as adjunctive therapy in treating major upper gastrointestinal bleeding in dialysis patients. Nephrol Dial Transplant. 2003;18(7):1388–1391. doi: 10.1093/ndt/gfg117 [DOI] [PubMed] [Google Scholar]
- 83.Bhowmik D, Vibha D, Dogra M, Agarwal S, Bhat A. Tranexamic acid overdosage-induced generalized seizure in renal failure. Saudi J Kidney Dis Transpl. 2014;25(1):130–132. doi: 10.4103/1319-2442.124529 [DOI] [PubMed] [Google Scholar]
- 84.Panes O Muñoz B Pais E, et al. Tranexamic acid inhibits fibrinolysis, shortens the bleeding time and improves platelet function in patients with chronic renal failure. Thromb Haemost. 1999;82(10):1250–1254. doi: 10.1055/s-0037-1614370 [DOI] [PubMed] [Google Scholar]
- 85.Galbusera M, Remuzzi G, Boccardo P. Treatment of bleeding in dialysis patients. Semin Dial. 2009;22(3):279–286. doi: 10.1111/j.1525-139X.2008.00556.x [DOI] [PubMed] [Google Scholar]
- 86.Jeong R Quinn RR Ravani P, et al. Graft function, albuminuria, and the risk of hemorrhage and thrombosis after kidney transplantation. Can J Kidney Health Dis. 2020;7:205435812095219. doi: 10.1177/2054358120952198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Francois K Orlando C Jochmans K, et al. Hemodialysis does not induce detectable activation of the contact system of coagulation. Kidney Int Rep. 2020;5(6):831–838. doi: 10.1016/j.ekir.2020.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skinner SC, Derebail VK, Poulton CJ, Bunch DO, Roy-Chaudhury P, Key NS. Hemodialysis-related complement and contact pathway activation and cardiovascular risk: a narrative review. Kidney Med. 2021;3(4):607–618. doi: 10.1016/j.xkme.2021.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Renaux JL, Thomas M, Crost T, Loughraieb N, Vantard G. Activation of the kallikrein-kinin system in hemodialysis: role of membrane electronegativity, blood dilution, and pH. Kidney Int. 1999;55(3):1097–1103. doi: 10.1046/j.1523-1755.1999.0550031097.x [DOI] [PubMed] [Google Scholar]
- 90.Hong J, Larsson A, Ekdahl KN, Elgue G, Larsson R, Nilsson B. Contact between a polymer and whole blood: sequence of events leading to thrombin generation. J Lab Clin Med. 2001;138(2):139–145. doi: 10.1067/mlc.2001.116486 [DOI] [PubMed] [Google Scholar]
- 91.Daugirdas JT, Bernardo AA. Hemodialysis effect on platelet count and function and hemodialysis-associated thrombocytopenia. Kidney Int. 2012;82(2):147–157. doi: 10.1038/ki.2012.130 [DOI] [PubMed] [Google Scholar]
- 92.Sloand JA, Sloand EM. Studies on platelet membrane glycoproteins and platelet function during hemodialysis. J Am Soc Nephrol. 1997;8(5):799–803. doi: 10.1681/ASN.V85799 [DOI] [PubMed] [Google Scholar]
- 93.Lindsay RM, Friesen M, Koens F, Linton AL, Oreopoulos D, de Veber G. Platelet function in patients on long term peritoneal dialysis. Clin Nephrol. 1976;6(2):335–339. PMID: 954241. [PubMed] [Google Scholar]
- 94.Stallone G, Pontrelli P, Rascio F, Castellano G, Gesualdo L, Grandaliano G. Coagulation and fibrinolysis in kidney graft rejection. Front Immunol. 2020;11:1807. doi: 10.3389/fimmu.2020.01807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tran L Pannier B Lacolley P, et al. A case-control study indicates that coagulation imbalance is associated with arteriosclerosis and markers of endothelial dysfunction in kidney failure. Kidney Int. 2021;99(5):1162–1172. doi: 10.1016/j.kint.2020.12.011 [DOI] [PubMed] [Google Scholar]
- 96.Li Y, Chen Y, Qi X, Hu B, Du Q, Qian Y. Poor response to rivaroxaban in nephrotic syndrome with acute deep vein thrombosis: a case report. Medicine (Baltimore). 2019;98(31):e16585. doi: 10.1097/MD.0000000000016585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amezquita Y Mendez C Fernandez A, et al. Risk factors for early renal graft thrombosis: a case-controlled study in grafts from the same donor. Transplant Proc. 2008;40(9):2891–2893. doi: 10.1016/j.transproceed.2008.09.014 [DOI] [PubMed] [Google Scholar]
- 98.Keller AK, Jorgensen TM, Jespersen B. Identification of risk factors for vascular thrombosis may reduce early renal graft loss: a review of recent literature. J Transplant. 2012;2012:793461–793469. doi: 10.1155/2012/793461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gander R Asensio M Royo GF, et al. Vascular thrombosis in pediatric kidney transplantation: graft survival is possible with adequate management. J Pediatr Urol. 2018;14(3):222–230. doi: 10.1016/j.jpurol.2018.01.027 [DOI] [PubMed] [Google Scholar]
- 100.Cardinal H, Dieude M, Hebert MJ. Endothelial dysfunction in kidney transplantation. Front Immunol. 2018;9:1130. doi: 10.3389/fimmu.2018.01130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.El Zorkany K, Bridson JM, Sharma A, Halawa A. Transplant renal vein thrombosis. Exp Clin Transpl. 2017;15(2):123–129. doi: 10.6002/ect.2016.0060 [DOI] [PubMed] [Google Scholar]
- 102.Song SY, Wang ZA, Ding YC, Ji XM, Meng R. Cyclosporine-A-induced intracranial thrombotic complications: systematic review and cases report. Front Neurol. 2020;11:563037. doi: 10.3389/fneur.2020.563037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Verpooten GA Cools FJ Van der Planken MG, et al. Elevated plasminogen activator inhibitor levels in cyclosporin-treated renal allograft recipients. Nephrol Dial Transplant. 1996;11(2):347–351. doi: 10.1093/oxfordjournals.ndt.a027265 [DOI] [PubMed] [Google Scholar]
- 104.Bobadilla NA, Gamba G. New insights into the pathophysiology of cyclosporine nephrotoxicity: a role of aldosterone. Am J Physiol Renal Physiol. 2007;293(1):F2–F9. doi: 10.1152/ajprenal.00072.2007 [DOI] [PubMed] [Google Scholar]
- 105.Scholz GA, Fux M, Christ L, Iype J, Banz Y, Villiger PM. Divergent regulatory T cell responses to high-dose methylprednisolone and tocilizumab in giant cell arteritis. J Autoimmun. 2022;133:102909. doi: 10.1016/j.jaut.2022.102909 [DOI] [PubMed] [Google Scholar]
- 106.Go RC Nyirenda T Bojarian M, et al. Methylprednisolone, venous thromboembolism, and association with heparin to 30 days in hospital survival in severe Covid-19 pneumonia. BMC Pulm Med. 2022;22(1):6. doi: 10.1186/s12890-021-01810-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shankar R, Bastani B, Salinas-Madrigal L, Sudarshan B. Acute thrombosis of the renal transplant artery after a single dose of OKT3. Am J Nephrol. 2001;21(2):141–144. doi: 10.1159/000046237 [DOI] [PubMed] [Google Scholar]
- 108.Thiyagarajan UM, Ponnuswamy A, Bagul A. Thymoglobulin and its use in renal transplantation: a review. Am J Nephrol. 2013;37(6):586–601. doi: 10.1159/000351643 [DOI] [PubMed] [Google Scholar]
- 109.Cherney DZ, Zaltzman JS. Mycophenolate mofetil causing deep venous thrombosis in a renal transplant patient with factor V Leiden. Nephrol Dial Transplant. 2001;16(8):1702–1704. doi: 10.1093/ndt/16.8.1702 [DOI] [PubMed] [Google Scholar]
- 110.Knudsen GH, Nielsen C, Nielsen CB, Frederiksen H, Vinholt PJ. The effect of mycophenolate mofetil on platelet function. Blood Coagul Fibrinolysis. 2020;31(2):132–139. doi: 10.1097/MBC.0000000000000886 [DOI] [PubMed] [Google Scholar]
- 111.Vazquez SR, Rondina MT, Pendleton RC. Azathioprine-induced warfarin resistance. Ann Pharmacother. 2008;42(7):1118–1123. doi: 10.1345/aph.1L077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Claudel SE, Miles LA, Murea M. Anticoagulation in hemodialysis: a narrative review. Semin Dial. 2021;34(2):103–115. doi: 10.1111/sdi.12932 [DOI] [PubMed] [Google Scholar]
- 113.Kong Y, Chen H, Wang YQ, Meng L, Wei JF. Direct thrombin inhibitors: patents 2002-2012 (review). Mol Med Rep. 2014;9(5):1506–1514. doi: 10.3892/mmr.2014.2025 [DOI] [PubMed] [Google Scholar]
- 114.Kam PC, Kaur N, Thong CL. Direct thrombin inhibitors: pharmacology and clinical relevance. Anaesthesia. 2005;60(6):565–574. doi: 10.1111/j.1365-2044.2005.04192.x [DOI] [PubMed] [Google Scholar]
- 115.Holbrook A Schulman S Witt DM, et al. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141(2):e152S–e184S. doi: 10.1378/chest.11-2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chan KE, Lazarus JM, Thadhani R, Hakim RM. Warfarin use associates with increased risk for stroke in hemodialysis patients with atrial fibrillation. J Am Soc Nephrol. 2009;20(10):2223–2233. doi: 10.1681/ASN.2009030319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shah M Avgil Tsadok M Jackevicius CA, et al. Warfarin use and the risk for stroke and bleeding in patients with atrial fibrillation undergoing dialysis. Circulation. 2014;129(11):1196–1203. doi: 10.1161/CIRCULATIONAHA.113.004777 [DOI] [PubMed] [Google Scholar]
- 118.Lee HW An JN Lee HS, et al. Neutrophil extracellular traps and heparin-induced antibodies contribute to vascular access thrombosis in hemodialysis patients. Kidney Res Clin Pract. 2021;40(4):712–723. doi: 10.23876/j.krcp.21.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hogan M, Berger JS. Heparin-induced thrombocytopenia (HIT): review of incidence, diagnosis, and management. Vasc Med. 2020;25(2):160–173. doi: 10.1177/1358863X19898253 [DOI] [PubMed] [Google Scholar]
- 120.Zarbock A Kullmar M Kindgen-Milles D, et al. Effect of regional citrate anticoagulation vs systemic heparin anticoagulation during continuous kidney replacement therapy on dialysis filter life span and mortality among critically ill patients with acute kidney injury: a randomized clinical trial. JAMA. 2020;324(16):1629–1639. doi: 10.1001/jama.2020.18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fiaccadori E Regolisti G Cademartiri C, et al. Efficacy and safety of a citrate-based protocol for sustained low-efficiency dialysis in AKI using standard dialysis equipment. Clin J Am Soc Nephrol. 2013;8(10):1670–1678. doi: 10.2215/CJN.00510113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eikelboom J, Floege J, Thadhani R, Weitz JI, Winkelmayer WC. Anticoagulation in patients with kidney failure on dialysis: factor XI as a therapeutic target. Kidney Int. 2021;100(6):1199–1207. doi: 10.1016/j.kint.2021.08.028 [DOI] [PubMed] [Google Scholar]
- 123.Alonso-Escalante JC, Machado L, Tabar KR, Tindall R, Thai N, Uemura T. Is continuing anticoagulation or antiplatelet therapy safe prior to kidney transplantation? Ann Transplant. 2021;26:e931648. doi: 10.12659/AOT.931648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bradbury CA Pell J Hill Q, et al. Mycophenolate mofetil for first-line treatment of immune thrombocytopenia. N Engl J Med. 2021;385(10):885–895. doi: 10.1056/NEJMoa2100596 [DOI] [PubMed] [Google Scholar]
- 125.Natale P Palmer SC Saglimbene VM, et al. Antiplatelet agents for chronic kidney disease. Cochrane Database Syst Rev. 2022;2(2):CD008834. doi: 10.1002/14651858.CD008834.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rajasekaran A, Julian BA, Rizk DV. IgA nephropathy: an interesting autoimmune kidney disease. Am J Med Sci. 2021;361(2):176–194. doi: 10.1016/j.amjms.2020.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402–2414. doi: 10.1056/NEJMra1206793 [DOI] [PubMed] [Google Scholar]
- 128.Omae T, Ishikawa T, Nakajima Y, Nogami K. Coagulation potential in pediatric patients with immunoglobulin A nephropathy. Pediatr Int. 2022;64(1):e15042. doi: 10.1111/ped.15042 [DOI] [PubMed] [Google Scholar]
- 129.Xia M Liu D Peng L, et al. Coagulation parameters are associated with the prognosis of immunoglobulin a nephropathy: a retrospective study. BMC Nephrol. 2020;21(1):447. doi: 10.1186/s12882-020-02111-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Matsubara M, Akiu N, Ootaka T, Saito T, Yoshinaga K. Glomerular deposition of coagulation factors VII, VIII, and IX in IgA nephropathy: possible coagulation system involvement in IgA nephropathy. Nephron. 1989;53(4):381–383. doi: 10.1159/000185788 [DOI] [PubMed] [Google Scholar]
- 131.Liu N Ono T Suyama K, et al. Mesangial factor V expression colocalized with fibrin deposition in IgA nephropathy. Kidney Int. 2000;58(2):598–606. doi: 10.1046/j.1523-1755.2000.00206.x [DOI] [PubMed] [Google Scholar]
- 132.Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31(1):15–26. doi: 10.1161/ATVBAHA.109.200956 [DOI] [PubMed] [Google Scholar]
- 133.He Z Zhang Y Cao M, et al. Increased phosphatidylserine-exposing microparticles and their originating cells are associated with the coagulation process in patients with IgA nephropathy. Nephrol Dial Transplant. 2016;31(5):747–759. doi: 10.1093/ndt/gfv403 [DOI] [PubMed] [Google Scholar]
- 134.Shimosawa M Sakamoto K Tomari Y, et al. Lipopolysaccharide-triggered acute aggravation of mesangioproliferative glomerulonephritis through activation of coagulation in a high IgA strain of ddY mice. Nephron Exp Nephrol. 2009;112(4):e81–e91. doi: 10.1159/000224798 [DOI] [PubMed] [Google Scholar]
- 135.Ando Y Moriyama T Miyazaki M, et al. Enhanced glomerular expression of caldesmon in IgA nephropathy and its suppression by glucocorticoid-heparin therapy. Nephrol Dial Transplant. 1998;13(5):1168–1175. doi: 10.1093/ndt/13.5.1168 [DOI] [PubMed] [Google Scholar]
- 136.Ishii T, Kawamura T, Tsuboi N, Ogura M, Utsunomiya Y, Hosoya T. Prospective trial of combined therapy with heparin/warfarin and renin-angiotensin system inhibitors in progressive IgA nephropathy. Contrib Nephrol. 2007;157:114–119. doi: 10.1159/000102314 [DOI] [PubMed] [Google Scholar]
- 137.Yoshikawa N Ito H Sakai T, et al. A controlled trial of combined therapy for newly diagnosed severe childhood IgA nephropathy. J Am Soc Nephrol. 1999;10(1):101–109. doi: 10.1681/ASN.V101101 [DOI] [PubMed] [Google Scholar]
- 138.Shima Y Nakanishi K Kaku Y, et al. Combination therapy with or without warfarin and dipyridamole for severe childhood IgA nephropathy: an RCT. Pediatr Nephrol. 2018;33(11):2103–2112. doi: 10.1007/s00467-018-4011-6 [DOI] [PubMed] [Google Scholar]
- 139.Yagame M Tomino Y Miura M, et al. Clinical effect of dipyridamole in patients with IgA nephropathy. Tokai J Exp Clin Med. 1986;11(5):329–333. PMID: 2960035. [PubMed] [Google Scholar]
- 140.Camara S de la Cruz JP Frutos MA, et al. Effects of dipyridamole on the short-term evolution of glomerulonephritis. Nephron. 1991;58(1):13–16. doi: 10.1159/000186370 [DOI] [PubMed] [Google Scholar]
- 141.Kano K, Nishikura K, Yamada Y, Arisaka O. Effect of fluvastatin and dipyridamole on proteinuria and renal function in childhood IgA nephropathy with mild histological findings and moderate proteinuria. Clin Nephrol. 2003;60(8):85–89. doi: 10.5414/cnp60085 [DOI] [PubMed] [Google Scholar]
- 142.Hirahashi J Hanafusa N Wada T, et al. Aspirin and eicosapentaenoic acid may arrest progressive IgA nephropathy: a potential alternative to immunosuppression. Intern Med. 2015;54(18):2377–2382. doi: 10.2169/internalmedicine.54.4623 [DOI] [PubMed] [Google Scholar]
- 143.Hayashi T, Kaneko S, Thang NT, Shou I, Shirato I, Tomino Y. Effect of dilazep hydrochlorideon the Im munohistopathology of IgA nephropathy in ddY mice. Nephron. 2000;86(3):327–332. doi: 10.1159/000045788 [DOI] [PubMed] [Google Scholar]
- 144.Yoshida H Kanatsu K Muso E, et al. Effects of an anti-platelet drug (dilazep) in IgA nephropathy: comparison of clinical effects with renal biopsy findings. Nihon Jinzo Gakkai Shi. 1994;36(4):339–344. PMID: 8022106. [PubMed] [Google Scholar]
- 145.Liu Z, Pan J, Sun C, Zhou J, Li NA. Clinical effects of perazine ferulate tablets combined with eucalyptol limonene pinene enteric soft capsules for treatment of children with IgA nephropathy. Exp Ther Med. 2016;12(1):169–172. doi: 10.3892/etm.2016.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tomino Y Miura M Suga T, et al. Effects of a "single shot" of urokinase on fibrinolytic activities in patients with IgA nephropathy. Tokai J Exp Clin Med. 1984;9(1):43–47. [PubMed] [Google Scholar]
- 147.Watanabe T, Takahashi S, Nakajo S, Hamasaki M. Pathological improvement of IgA nephropathy and Henoch-Schonlein purpura nephritis with urokinase therapy. Pediatr Int. 1996;38(6):622–628. doi: 10.1111/j.1442-200x.1996.tb03720.x [DOI] [PubMed] [Google Scholar]
- 148.Chen X Qiu Q Tang L, et al. Effects of co-administration of urokinase and benazepril on severe IgA nephropathy. Nephrol Dial Transplant. 2004;19(4):852–857. doi: 10.1093/ndt/gfh069 [DOI] [PubMed] [Google Scholar]
- 149.Miura M, Endoh M, Nomoto Y, Sakai H. Long-term effect of urokinase therapy in IgA nephropathy. Clin Nephrol. 1989;32(5):209–216. PMID: 2510959. [PubMed] [Google Scholar]
- 150.Liu XJ Geng YQ Xin SN, et al. Antithrombotic drug therapy for IgA nephropathy: a meta analysis of randomized controlled trials. Intern Med. 2011;50(21):2503–2510. doi: 10.2169/internalmedicine.50.5971 [DOI] [PubMed] [Google Scholar]
- 151.Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc Biol. 2014;34(2):313–320. doi: 10.1161/ATVBAHA.113.302378 [DOI] [PubMed] [Google Scholar]
- 152.Trujillo H Sandino J Cavero T, et al. IgA nephropathy is the most common underlying disease in patients with anticoagulant-related nephropathy. Kidney Int Rep. 2022;7(4):831–840. doi: 10.1016/j.ekir.2022.01.1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ishii H Hirai K Yanai K, et al. Warfarin-related nephropathy with acute kidney injury in a patient with immunoglobulin A nephropathy. CEN Case Rep. 2018;7(2):198–203. doi: 10.1007/s13730-018-0325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sevillano AM Diaz M Caravaca-Fontan F, et al. IgA nephropathy in elderly patients. Clin J Am Soc Nephrol. 2019;14(8):1183–1192. doi: 10.2215/CJN.13251118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ikeda M Tanaka M Shimoda S, et al. Dabigatran-induced anticoagulant-related nephropathy with undiagnosed IgA nephropathy in a patient with normal baseline renal function. CEN Case Rep. 2019;8(4):292–296. doi: 10.1007/s13730-019-00410-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sharfuddin N, Nourbakhsh M, Box A, Benediktsson H, Muruve DA. Anticoagulant related nephropathy induced by dabigatran. Case Rep Nephrol. 2018;2018:1–7. doi: 10.1155/2018/7381505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Moeckel GW, Luciano RL, Brewster UC. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin Kidney J. 2013;6(5):507–509. doi: 10.1093/ckj/sft076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brodsky S, Eikelboom J, Hebert LA. Anticoagulant-related nephropathy. J Am Soc Nephrol. 2018;29(12):2787–2793. doi: 10.1681/ASN.2018070741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Golbin L Vigneau C Touchard G, et al. Warfarin-related nephropathy induced by three different vitamin K antagonists: analysis of 13 biopsy-proven cases. Clin Kidney J. 2017;10(3):381–388. doi: 10.1093/ckj/sfw133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ryan M Ware K Qamri Z, et al. Warfarin-related nephropathy is the tip of the iceberg: direct thrombin inhibitor dabigatran induces glomerular hemorrhage with acute kidney injury in rats. Nephrol Dial Transplant. 2014;29(12):2228–2234. doi: 10.1093/ndt/gft380 [DOI] [PubMed] [Google Scholar]
- 161.Wheeler DS, Giugliano RP, Rangaswami J. Anticoagulation-related nephropathy. J Thromb Haemost. 2016;14(3):461–467. doi: 10.1111/jth.13229 [DOI] [PubMed] [Google Scholar]
- 162.Morrow DA Braunwald E Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–1413. doi: 10.1056/NEJMoa1200933 [DOI] [PubMed] [Google Scholar]
- 163.Brodsky SV Mhaskar NS Thiruveedi S, et al. Acute kidney injury aggravated by treatment initiation with apixaban: another twist of anticoagulant-related nephropathy. Kidney Res Clin Pract. 2017;36(4):387–392. doi: 10.23876/j.krcp.2017.36.4.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Naimark DM. Warfarin-related nephropathy. Kidney Int. 2012;81(3):322. doi: 10.1038/ki.2011.383 [DOI] [PubMed] [Google Scholar]