

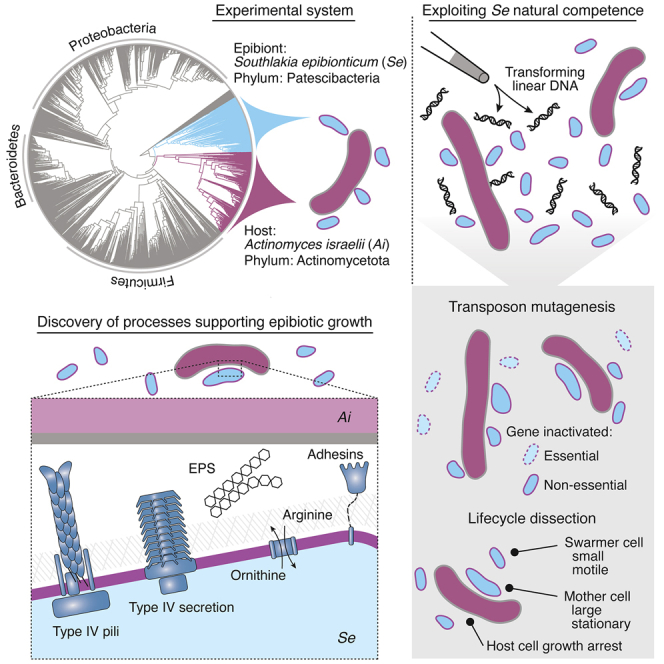
Summary
Patescibacteria, also known as the candidate phyla radiation (CPR), are a diverse group of bacteria that constitute a disproportionately large fraction of microbial dark matter. Its few cultivated members, belonging mostly to Saccharibacteria, grow as epibionts on host Actinobacteria. Due to a lack of suitable tools, the genetic basis of this lifestyle and other unique features of Patescibacteira remain unexplored. Here, we show that Saccharibacteria exhibit natural competence, and we exploit this property for their genetic manipulation. Imaging of fluorescent protein-labeled Saccharibacteria provides high spatiotemporal resolution of phenomena accompanying epibiotic growth, and a transposon-insertion sequencing (Tn-seq) genome-wide screen reveals the contribution of enigmatic Saccharibacterial genes to growth on their hosts. Finally, we leverage metagenomic data to provide cutting-edge protein structure-based bioinformatic resources that support the strain Southlakia epibionticum and its corresponding host, Actinomyces israelii, as a model system for unlocking the molecular underpinnings of the epibiotic lifestyle.
Graphical abstract
Highlights
-
•
Genetic manipulation is achieved for epibiotic Patescibacteria
-
•
Fluorescence microscopy reveals distinct cell types in the epibiotic lifecycle
-
•
Tn-seq uncovers the genetic basis for epibiont-host interactions
-
•
Epibiotic growth requires a specialized secretion system and multiple adhesins
Discovery and exploitation of natural competence in Patescibacteria enables visualization of the bacterial epibiont lifecycle and dissection of its genetic underpinnings.
Introduction
The vast majority of metagenomic DNA sequences obtained from microbial species-rich environmental sources are derived from Bacteria and Archaea that have not been cultivated. Conservative estimates suggest that these sequences, often referred to as microbial dark matter, represent organisms constituting approximately half of phylum level diversity within these domains.1,2 Microbial dark matter holds great interest as a reservoir of biosynthetic pathways and enzymes with potential for biotechnological application.3 In addition, understanding the functions of these genes is paramount to defining the molecular processes supporting a given ecosystem and for unraveling the physiology and cell biology of the organisms within.4
Patescibacteria, also known as the bacterial candidate phyla radiation (CPR), represent a large and diverse clade in the tree of bacteria with very few cultivated representatives.1,5,6,7 They share a number of unusual features that set them apart from most other bacteria.8,9 These include their small size (as little as 100–200 nm in width), reduced genomes (typically <1 Mb), and limited metabolic capability.10 This has led to the hypothesis that these organisms broadly share a requirement for host organisms to support their growth. Indeed, experiments show that most cultivated Patescibacteria attach to and proliferate on the surface of other bacteria—living as obligate epibionts.11,12,13,14,15 Genomic analyses have yielded speculation regarding the molecular functions that support the epibiotic lifestyles of Patescibacteria.9,16 However, owing to the phylogenetic distance separating Patescibacteria and well-characterized organisms, the function of much of their proteome cannot be predicted and a lack of genetic tools for these organisms has heretofore precluded the experimental investigation of genotype-phenotype relationships.
Among the Patescibacteria, members of the group Saccharibacteria, originally named TM7, were the first to be cultivated in the laboratory.13 Saccharibacteria are found in a multitude of terrestrial and marine environments; however, early interest in them stemmed from their widespread occurrence in human oral microbiomes.17,18,19,20 Archaeological findings show that this association dates to before the mesolithic period, and recent work links Saccharibacteria to human oral health.17,21,22 The growth of Saccharibacteria relies on the co-cultivation of host bacteria belonging to the class Actinomycetia within the phylum Actinomycetota, for which they exhibit strain-level specificity.23,24,25 Employing panels of Actinomycetia strains for Saccharibacterial enrichment has facilitated the isolation and sequencing dozens of strains.24,26,27 Despite this progress, the phylum remains poorly sampled, with many divergent clades uncultivated, and the extent of genetic diversity unresolved.
The genome sequences of Saccharibacteria reveal common features that provide insight into molecular functions underlying their cellular physiology and lifestyle.2,4,13,16,28,29 Akin to other Patescibacteria, Saccharibacteria generally lack a respiratory chain and pathways for the de novo generation of amino acids, nucleotides, and fatty acids.10 On the contrary, Saccharibacteria universally possess a relative wealth of specialized secretory mechanisms including the type II and IV secretion systems (T2SS and T4SS).16 These diverge significantly from related systems in bacterial pathogens that deliver toxins and effectors to eukaryotic host cells; however, it has been speculated that they function in an analogous fashion to support bacterial host co-option by Saccharibacteria.16 Saccharibacteria also possess type IV pili (T4P), which were implicated in twitching motility and host adhesion through the use of a small molecule inhibitor of pilus extrusion.25 Although such genomic analyses and experiments provide fertile ground for the formulation of hypotheses, progress toward a mechanistic understanding of the unique biology of Saccharibacteria and Patescibacteria as a whole has been stymied by a lack of genetic tools.30 Here, we discover that natural competence can be harnessed for genetic manipulation of Saccharibacteria. With this capability in hand, we go on to use fluorescent protein expression to conduct time-lapse microscopic analysis of the Saccharibacterial lifecycle, and we perform transposon mutagenesis to identify genes important for epibiotic growth. Our findings offer an initial mechanistic glimpse into the cellular functions encoded in microbial dark matter.
Results
Isolation and characterization of Saccharibacteria strains
We collected, pooled, homogenized, and filtered saliva and dental plaque from volunteers and enriched this material for Saccharibacteria using the method developed by Bor et al. (detailed in STAR Methods).26 This led to the isolation of two strains, which exhibit distinct host specificity (Figure 1A). Phylogenetic analysis using concatenated alignments of 50 core, conserved protein sequences obtained through complete genome sequencing placed the two strains within human oral subclades of the G1 clade of Saccharibacteria (Figures 1B–1D).31 Based on these assignments, we named our strains Candidatus Nanosynbacter lyticus ML1 (Nl) and Ca. Southlakia epibionticum ML1 (Se). The genome sequences of the two strains additionally indicated they bear features typical of Saccharibacteria, such as genes associated with specialized secretion systems, cell surface appendages, and competence, coupled with a lack of genes required for a multitude of biosynthetic pathways and an overall reduced genome size (Figures 1C and 1D).
Figure 1.

Phylogenetic placement and genome sequencing of newly isolated Saccharibacteria strains S. epibionticum ML1 (Se) and N. lyticus ML1 (Nl)
(A) Maximum growth (fold change) achieved by Se and Nl during co-culture with compatible host species A. israelii and Propionibacterium propionicum, respectively, and population change (growth or death) detected at equivalent time points with an incompatible host.
(B) Phylogeny constructed using 50 core, universal proteins (6,042 total amino acid positions) indicating placement of Se and Nl (blue text) within Saccharibacteria. Family names (as designated by the Genome Taxonomy Database) and groups previously designated by McLean et al. (G1, etc.) are indicated for each clade.16 HOT, human oral taxon.
(C and D) Overview of the genome sequences of S. epibionticum ML1 and N. lyticus ML1. Data in (A) represent mean ± SD.
Genetic manipulation of Saccharibacteria via natural transformation
We sought to develop methods for reverse genetic analyses within Saccharibacteria. Genes encoding the core components of the com DNA uptake system are conserved across CPR phyla, including the Saccharibacteria.9 These consist of ComEC, the central membrane DNA conduit, DprA, a catalyst of Rec-mediated recombination, and ComFC, which binds single-stranded DNA (ssDNA) and links import to recombination.32,33,34 Com proteins function in concert with T4P, which are also widely distributed in Saccharibacteria, and CPR bacteria more generally.9,33 Given the lack of nucleotide biosynthetic capability in CPR bacteria, it has been proposed that these systems facilitate nucleotide acquisition.9 Although the presence of the Com system is largely not predictive of DNA transformation in a laboratory setting,35 we sought to test whether exogenous DNA could be exploited for genetically manipulating Saccharibacteria.
As a first step toward assessing the feasibility of genetics in Saccharibacteria, we searched for antibiotics with convenient resistance determinants that potently inhibit the growth of Se without impacting that of its preferred host, Actinomyces israelii F0345 (Ai). These experiments revealed that across a wide range of concentrations, the aminoglycoside hygromycin fulfills these criteria (Figure S1A). Next, we designed and generated a linear cassette containing the hygromycin resistance gene (hph) codon optimized for Se flanked by the promoter and terminator regions of the N. lyticus TM7x tuf gene (elongation factor Tu), an open reading frame (ORF) predicted to be highly expressed (Figure 2A). For the insertion of this cassette, we selected an intergenic region located between two convergently transcribed ORFs, SEML1_0215 and SEML1_0216, hereafter referred to as neutral site 1 (NS1). To promote homologous recombination, approximately 1,000 bp on either side of the insertion site were added to the 5′ and 3′ ends of our cassette.
Figure S1.

Development and optimization of the Se transformation protocol, related to Figure 2
(A) Growth of Ai (left) and Se (right) in co-cultures containing the indicated concentrations of hygromycin. Data represent mean ± SD.
(B and C) Quantification of Se, Ai, and transformed Se populations over the course of our transformation protocol (see Figure 2B) with varying lengths (B) or concentrations (C) of transforming DNA. Transformations were performed in the presence (top panels) and absence (bottom panels) of selection for transformants with hygromycin.
Figure 2.

Harnessing natural transformation to generate mutant Saccharibacteria
(A) Schematic depicting the intergenic neutral site (blue, NS1) targeted for insertion of a hygomycin resistance cassette (yellow) in the Se genome and the linear DNA fragment employed in transformation experiments. Primer binding sites used for genotyping are indicated (sites 1–3).
(B) Overview of the Se transformation protocol. After incubation with linear DNA, Se + Ai co-cultures are enlarged concomitant with hygromycin addition and serially passage with addition of naive host at each dilution to promote Se growth (gray box). Clonal transformed Se populations were obtained by plating to isolate single colonies of Ai with accompanying Se cells, followed by growth in liquid culture, with additional Ai, to promote Se population expansion. The asterisk indicates the single insertion detected by genome sequencing.
(C) PCR-based genotyping of Se clones obtained following transformation according to the protocol shown in (B) in the presence (right) or absence (left) of selection with hygromycin during the expansion and passaging steps. Binding sites for primers targeting NS1 (1, 2) and hph (3) are shown in (A). Positive control primers (Se) target a locus distant from NS1.
(D) Se growth (red) and percent of Se transformed (black) over the course of transformation protocol depicted in (B), in the presence (squares) or absence (circles) of selection with hygromycin.
(E) Luminescence production from Se-Ai co-cultures (left) or co-culture filtrates (right) in which Se contains a nanoluciferase expression cassette inserted at NS1 (shown at bottom).
(F) Fluorescence and phase contrast micrographs of Se-Ai co-cultures in which Se carries an mCherry (top) or sfgfp (bottom) expression cassette inserted at NS1. Scale bar, 1 μm. Data in (D) and (E) represent mean ± SD. Asterisks indicate statistically significant differences (unpaired two-tailed Student’s t test; ∗p < 0.05).
See also Figures S1 and S2.
To transform Se, we incubated Se-Ai co-cultures with 2.0 ng/μL of our linear cassette for 6 h before initiating selection with hygromycin (Figure 2B). Naive host was added concomitantly to permit the outgrowth of successfully transformed Se cells. Reasoning that transformation may be inefficient, we passaged these co-cultures twice, at 48-h intervals, with continued hygromycin selection and the addition of naive host. Cultures were then diluted and plated to obtain colonies, which were selected and propagated with naive host without selection before genotyping (Figure 2B). The latter step was included to bottleneck the Se population and facilitate the isolation of clonal populations. Remarkably, each Se-infected culture we tested—accounting for the majority of colonies selected—contained our synthetic cassette inserted at the expected location (Figure 2C). Whole genome sequencing confirmed these PCR results, and it further showed that cassette integration occurred without introducing off-target mutations.
Quantitative PCR (qPCR) analysis of total Se, transformed Se, and Ai at regular intervals during our transformation procedure demonstrated that approximately 0.2% of Se contains the integrated cassette by the conclusion of the initial incubation period (Figure 2D). Although Se levels remain low through the second passage under selection with hygromycin, all surviving Se cells bear the cassette at this time point. In the final passage, the population of Se continues to maintain the cassette and expands markedly, far surpassing levels of the host (Figure S1B). In the absence of hygromycin, similar quantities of initially transformed Se are observed; however, this small proportion fails to expand despite overall robust growth of Se. We observed similar transformation behavior using lengths of DNA with homology to the insertion site flanking regions as short as 221 bp (Figure S1B) and with as little as 0.02 ng/μL (Figure S1C). Finally, to probe the generality of our methods, we identified predicted NSs within Nl and the previously published Saccharibacterial strain N. lyticus TM7x.13,36 We then subjected these strains to an analogous transformation protocol. Genotyping of transformed populations by both PCR and whole genome sequencing of N. lyticus TM7x purified clonal populations indicated cassette insertion at the desired locations also occurred within these strains (Figure S2).
Figure S2.

Transformation of two strains of Nanosynbacter lyticus, related to Figure 2
(A, C, and E) Schematics depicting the intergenic neutral sites (blue, NS1 and NS2) targeted for insertion of a hygomycin resistance cassette (yellow) in the N. lyticus ML1 (Nl ML1) (A) or N. lyticus TM7x (Nl TM7x) (C and E) genomes and the linear DNA fragment employed in transformation experiments with these strains. Primer binding sites used for genotyping are indicated (sites 1–3).
(B) Genotyping of Nl ML1-P. propionicum co-cultures transformed with the linear DNA fragment depicted in (A) (right) or parallel negative control co-cultures with no DNA added (left) at the end of passage 4 (see STAR Methods) with primers targeting NS1 (1,2) or hph (3). Positive control Nl ML1 primers target a genomic locus distant from NS1.
(D and F) Genotyping of Nl TM7x-Schaalia odontolytica co-cultures transformed with the linear DNA fragment depicting in (C) or (E), respectively, at the end of passage 4. Cultures were grown in the presence (right) or absence (left) of continuous selection with hygromycin. Primers targeted NS1 (D) or NS2 (F) (1,2) or hph (3). Positive control Nl TM7x primers target a genomic locus distant from NS1 and NS2.
The ability to introduce heterologous DNA into Se has numerous foreseeable applications, one of which is the expression of reporter genes that allows Se to be distinguished and studied within the context of co-culture with their hosts. To explore this possibility, we designed and generated NS1 insertion cassettes containing genes encoding nanoluciferase, mCherry, and green fluorescent protein (GFP) under the control of the tuf promoter and upstream of hph driven by a second predicted strong promoter of N. lyticus TM7x, that of rpsJ (Figures 2E and 2F). Using our transformation protocol, we obtained clonal populations of hygromycin-resistant Se containing each of these genes. Luminescence assays and fluorescence microscopy demonstrated robust activity of each reporter gene (Figures 2E and 2F). We did not detect their activity in host cells, indicating the feasibility of achieving specific manipulation of Se in the context of a co-culture.
The expression of fluorescent proteins within Se provided the opportunity to visualize CPR bacterial growth with extended, time-lapse fluorescence imaging. Over the course of 20 h, both Se and Ai populations—deposited from co-cultures at a low Se:Ai ratio onto an agar substrate containing growth media—showed clear evidence of expansion (representative time-lapse fluorescence microscopy videos are open for access via https://mougouslab.org/data). Apparent T4P-mediated motility of Se was also observed, as reported by Xie et al.25 Our time-lapse imaging further captured features of the Patescibacteria lifecycle at unprecedented spatiotemporal resolution. For instance, we could distinguish productive (Se growth supporting) versus non-productive (Se adhered without concomitant growth) interactions and directly measure their respective impact on individual host cells (Figure 3). Additionally, we observed productively adhered mother cells producing small swarmer cell progeny via repeated polar budding and the differentiation of a subset of these progeny into mother cells. Altogether, these findings show that natural transformation can be exploited to genetically manipulate Patescibacteria in a directed manner and open a window into the distinctive biology of this largely unexplored group of organisms.
Figure 3.

Fluorescent protein expression and quantitative microscopy enable tracking of the S. epibionticum lifecycle
(A and D) Snapshots captured at the indicated time points from time-lapse fluorescence and phase contrast microscopy of GFP-expressing Se grown in co-culture with Ai. Arrows indicate example Se cells exhibiting productive (pink, purple) and non-productive (blue) interactions with Ai cells. White outlines in the fluorescent channel depict an Ai cell affected by Se infection (Ai 1). Scale bar, 1 μm.
(B and E) Omnipose-generated segmentation of Se and Ai cells depicted in (A) and (D), at the start (left) and end (right) of the 20- or 22-h growth period.
(C and F) Growth of individual Ai cells as impacted by productive (light gray) or non-productive Se cells (black, dark gray). Colors correspond to cell masks shown in (B) and (E). Time-lapse fluorescence microscopy videos from which (A) and (D) derive, as well as additional raw and annotated videos of Se-Ai co-cultures, are open for access via https://mougouslab.org/data.
Tn-seq identifies genes essential for epibiotic growth
The ability to genetically manipulate Saccharibacteria enables myriad avenues of investigation. As a first step toward genetic dissection of the Se epibiotic relationship with Ai, we conducted transposon-insertion sequencing (Tn-seq) within Se during growth on Ai. To this end, we performed in vitro Tn5-based transposition on purified Se genomic DNA, repaired gaps as described by Manoil and colleagues, and used sequencing to confirm high-frequency, homogeneous insertion across the genome.37 This DNA was then used to transform Se, with slight modifications from our basic transformation protocol (see STAR Methods). Most notably, we elected to increase the scale of the experiment to account for our previously measured transformation efficiency and thereby avoid population bottlenecking following the onset of selection (Figure S3A). We collected three samples for Tn-seq analysis, an initial sample following the transformation and recovery period (T0), and two additional samples representing the population after 48 h serial passages with the addition of naive host (T1 and T2). Measurements of Se and Ai levels at each of these time points revealed a drop in Se levels prior to T1 that is not observed at similar time points in transformations targeting NS1, suggesting that a majority of Se cells received lethal mutations (Figure S3B). Our measurements also showed that, as expected, Ai levels drop relative to those of Se at later passages, such that by T2, Se outnumbers Ai by approximately 10-fold.
Figure S3.

Population dynamics of Se and Ai during transposon mutagenesis, related to Figure 4
(A) Schematic depicting the protocol employed for transposon mutagenesis of Se.
(B) Quantitative PCR-based measurements of Se and Ai levels during the course of the transposon mutagenesis experiment. Data in (B) represent mean ± SD.
Insertion site sequencing of our Tn-seq samples indicated a high density of insertions across the genome at each of the time points collected (40,382 unique sites among T0-T2; Figure 4A). These data were then applied to the Hiden Markov Model (HMM)-based essential gene identification algorithm within the TRANSIT software package.38 This method compares the depth of sequencing reads obtained at each insertion site and calls essential genes based on the deviation of this value across a given ORF from the genome average. In total, we identified 295 essential genes in Se, which fell into two classes (1) genes called as essential at T0 (61 genes, class I) and (2) genes called essential only after T0 (234 genes, class II) (Figure 4A; Table S1). KEGG analysis showed that class I genes are enriched in genetic information processing (53%). Many class II genes also belong to this category (34%); however, a higher proportion of genes in this group fall within metabolism and signaling categories. It is worth noting that one-third and one-half of class I and II genes, respectively, could not be classified by this method.
Figure 4.

Identification of genes important for fitness of S. epibionticum during co-culture with Ai identified by Tn-seq
(A) Overview of normalized transposon-insertion frequency across the Se genome detected in input DNA used for mutagenesis (dark gray), and from samples collected 48 h after the onset of selection (T0, dark blue) and subsequent outgrowth time points (T1-T2, shades of blue). Genes encoding proteins belonging to Patescibacteria-enriched protein families (Pb enriched) and class I and II essential genes (ES I and II) are indicated in the outer circles (shades of green). The locations of the essential arginine deiminase system (ADS) genes, T4SS genes, and two loci containing T4P genes (T4P1 and T4P2) are indicated outside of the circle.
(B) Se population levels detected in Se-Ai co-cultures following transformation with constructs designed to replace the indicated genes with hph.
(C and D) Total Se population (C) and proportion transformed (D) following transformation with an unmarked cassette targeted to NS1 in the indicated strains of Se. See also Figures S4 and S5 and Table S1. Data in (B)–(D) represent mean ± SD. Asterisks indicate statistically significant differences (B, one-way ANOVA followed by Dunnett’s compared to no DNA control; D, unpaired two-tailed Student’s t test; ∗p < 0.05, ns, not significant).
See also Figures S3 and S4 and Table S1.
To validate our Tn-seq results, we selected two class I and two class II essential genes, along with four non-essential control genes, and designed constructs to replace each with an hph expression cassette via a double-crossover recombination event. The constructs shared equivalent length flanking sequences to enable the direct comparison of their behavior in our transformation protocol. We assessed the fitness impact of inactivating each gene by quantifying Se populations following transformation and outgrowth under selection with hygromycin. For the control genes not predicted to contribute to fitness, we achieved robust levels of Se growth by this time point, comparable with that achieved when introducing the hph expression cassette at NS1 (Figure 4B). In contrast, in transformations targeting each of the four predicted essential genes examined, Se failed to proliferate, consistent with the inactivation of these genes strongly impacting fitness. Together, these findings provide confirmation that our Tn-seq analysis successfully identified the relative fitness contributions of Se genes during co-culture with Ai.
We noted that genes encoding homologs of com system components ComEC, ComF, and DprA were not among those genes defined as contributing significantly to Se fitness in our Tn-seq study. This finding suggests that this DNA competence machinery is not required for acquiring nucleotides to support Se growth, as had been previously suggested.9 To determine whether the com system of Se instead functions to mediate natural transformation, we generated a Se strain in which the comEC ORF is replaced by the hph expression cassette (Se ΔcomEC::hph). To assess whether this mutation affects Se transformation, we measured the efficiency of inserting a second, unmarked cassette at NS1. At the conclusion of the transformation protocol, insertion at the NS1 site was only detectable in the wild-type background, supporting the hypothesis that the com system mediates natural transformation in this species (Figures 4C and 4D).
A distinctive set of essential cellular features characterize the Patescibacteria lifestyle
The number of essential genes we identified in Se (295) falls within the typical range of essential genes in free-living bacteria. This is despite Se lacking the genes that typically comprise a significant proportion of essential gene sets, such as those participating in the biosynthesis of fatty acids, nucleotides, and amino acids. We hypothesize that in lieu of these biosynthetic pathways, the Se-essential gene set includes Patescibacteria-specific genes that specifically enable its unique host-cell-associated lifestyle. Banfield and colleagues previously leveraged the large number of Patescibacteria genomes available from metagenomic and traditional genome sequencing datasets (n = 2,321) to define Patescibacteria-enriched protein families.9 To gain insight into which of these are essential for Se growth on Ai, we identified Se proteins belonging to these families and cross-referenced these against the essential gene list obtained from our Tn-seq experiment (Figures 4A and S4). This revealed that 73 of the 116 Patescibacteria-enriched genes within Se are essential for growth, a dramatic enrichment (∼5-fold) relative to their proportion in the genome.
Figure S4.

Distribution of 921 core protein families across Patescibacteria and other bacterial genomes, including Se and N. lyticus ML1, related to Figure 4
Columns represent core families (derived from Meheust et al.9) and rows represent individual genomes from the indicated bacterial groups. Patescibacteria-enriched protein families indicated at top (blue), and dendrogram at left represents clustering of bacterial strains based on protein family content.
The phylogenetic distance of Se from well-studied bacterial systems poses a challenge for standard genome annotation methods and thus limited our capacity to leverage our Tn-seq data to define Patescibacteria-specific processes important for epibiotic growth. To improve functional predictions associated with ORFs in the Se genome, we applied a battery of sequence and structure-based computational tools. ProtNLM is a natural language model trained on UniProt to predict protein names given a sequence.39 Applying ProtNLM to the Se proteome yielded functional annotations for 337 of the 855 proteins (ProtNLM score ≥ 0.5, Table S2). This sequence-based annotation was further improved by mapping each protein to Pfam domains using hmmscan, detecting Pfam domains in an additional 270 proteins, 245 of which could be assigned functions (Table S2).40,41
To complement our sequence-based annotations, we took advantage of recent advances in protein structure prediction to conduct genome-wide structure-based homology analyses of the Se proteome.42,43 Generation of structural models using AlphaFold (AF) relies on evolutionary information extracted from multiple sequence alignments (MSAs).43 For nearly 25% of the Se proteome (220 proteins), our initial MSAs based on HHblits searches of UniRef and BFD (the default databases used by AF) were too shallow for high confidence structure prediction (<500 sequences post-filtering; Figure 5A).44,45,46 To improve the MSA depth for these proteins, we implemented a hidden Markov model (HMM)-based approach to identify and align additional homologs for each protein from multiple metagenomic datasets. This resulted in considerably deeper MSAs (>500 sequences) for an additional 9% of the proteome (Figure 5B). Using the highest depth MSA obtained for each protein, we obtained AF models for >99% of the predicted Se proteome. For comparison, we also computed AF models using the original 220 MSAs that contained <500 sequences and compared the model confidence metric obtained using these and the improved depth MSAs. Particularly, for low-confidence models obtained using MSAs generated with the default databases (average pLDDT < 50), we found that the use of the deeper MSAs for structural prediction resulted in substantial model confidence improvement (Figure 5C). The structure model improvements enabled by using extensive metagenomic databases for MSA generation were further underscored by structural homology search results obtained using Foldseek (FS).47 For some proteins, the improved structural models led to the identification of structural homologs for proteins that initially had no FS matches passing our cutoffs, whereas for others, the changes in the overall predicted structure led to the identification of different and more closely aligning top matches (Figures 5D, 5E, and S5A–S5C). In total, using FS, we were able to identify similar structures for 89% of modeled proteins (761/852, Table S2).
Figure 5.

Inclusion of extensive metagenomic data in MSAs enables proteome-wide AF modeling of S. epibionticum protein structures
(A) Histograms depicting MSA depths obtained for Se proteins using HHblits.
(B) Maximum depths obtained for Se protein MSAs that initially contained <500 sequences. Additional sequences were sourced from metagenomic sequence databases and incorporated into MSAs using Jackhmmer or Phmmer (see STAR Methods).
(C) Comparison of the AF confidence metric (pLDDT) determined using Hhblits or Jackhmmer/Phmmer (metagenome)-generated MSAs for Se proteins with initially shallow MSAs (<500). Se proteins shown in (D) and (E) are highlighted in blue.
(D and E) Example Se protein structure models and associated predicted alignment matrices obtained using shallow (right) or metagenomic sequence-improved (left) MSAs. Se protein models (blue) are aligned to models from top Foldseek (FS) hits (light gray, AF database50 numbers A0A1F6S045, D and A0A7W4ES58, E), when available. The annotation in (D) and (E) derives from the best FS hit. Some structures are trimmed to highlight the alignment.
See also Figures S5A–S5C and Table S2.
Figure S5.

Structural models for the Se proteome generated using metagenomic sequence enriched MSAs and the Ai genome, related to Figures 5 and 6
(A–C) Example Se protein structure models and associated predicted alignment matrices obtained using shallow (right) or metagenomic sequence-improved (left) MSAs. Se proteins models (blue) are aligned to models for top FS hits (light gray, AF database50 numbers A0A8B1YQG7, A; A0A660M2Z7, metagenome and R7KEI0, shallow, B; A0A563D6X1, metagenome and A0A563CX08, shallow, C), when available. Some structures are trimmed to highlight the alignment.
(D) Overview of the genome sequence of A. israelii F0345.
The incorporation of structural information resulted in functional predictions for 70 Se proteins that had none assigned by sequence-based approaches (Table S2). However, many of the AF models with similar structures identified for Se proteins are uncharacterized (12%), providing little information regarding function. Moreover, unrelated proteins may share similar structures due to convergence, and additional evidence is usually needed to assess the homologous relationships. Domain Parser for AF Models (DPAM) is a recently developed tool that parses structural domains from AF-modeled structures and integrates both structure and sequence searches to map protein domains to evolutionary classification of protein domains (ECOD).48,49 Using DPAM, we detected homologous ECOD domains for 80% of the predicted Se proteins (Table S2). This included 31 proteins for which no function was assigned by other methods. In supplementary material accompanying this report, we provide alignments for each mapped Se protein domain, along with links to corresponding Protein Data Bank identifiers (PDB IDs) and ECOD classifications (https://conglab.swmed.edu/ECOD_Se/Se_ECOD.html).
Aided by our improved annotation of the Se genome, we identified cellular functions that are uniquely essential to the epibiotic lifestyle (Figures 6A and 6B). Consistent with its adhesion to and reliance on a host cell for sustenance, many of these are localized outside of the Se cytoplasm. This includes T4P, which typically mediate surface attachment and twitching motility. A prior study utilizing quercetin, a small molecule inhibitor of pilus retraction, reported that T4P are important for host attachment in the Saccharibacterial species Leucosynbacter cicadicola.25 We find that the majority of genes associated with T4P function (SEML1_0104-0116, SEML1_0689-0693, and SEML1_0758-0768) are class I essential genes, indicating that disrupting this appendage is immediately detrimental to Se survival and further suggesting that T4P remain critical even after a productive interaction is established (Figures 4 and 6A; Table S1). Of note, in naturally competent bacteria, T4P work in concert with the com system to bind and mediate the uptake of extracellular DNA.50,51 Our observation that Se cells lacking comEC are viable suggests that the essentiality of T4P is unrelated to its role in competence.
Figure 6.

Unusual essential genes of S. epibionticum encode numerous envelope-associated functions predicted to mediate host-cell interaction
(A) Schematic of genes encoding the core T4P components (T4P1 locus, top), and locations of transposon insertions detected in input mutagenized DNA (gray) and Tn-seq samples T0-T2 (shades of blue). Gene essentiality is indicated by shading (class I essential genes [ES I], dark gray; class II essential genes [ES II], light gray).
(B) Model of the Se/Ai interface during a productive infection, depicting selected envelope-associated functions found to be essential in our Tn-seq screen. These include macromolecular structures predicted to mediate adhesion (T4P) and DNA or protein delivery (T4SS), protein adhesins and the ADS system for arginine catabolism. Mother and swarmer cell designations derive from the analysis shown in Figure 3. See also Figure S5D. EPS, extracellular polysaccharide; Orn, ornithine; Arg, arginine.
A second essential cell envelope-associated machinery we identified is an apparent T4SS (SEML1_0006-0019; Figure 6B). This specialized secretion system can participate in myriad functions but is most often implicated in the direct transfer of DNA (conjugation) or effector proteins to neighboring prokaryotic or eukaryotic cells. Our structure-based annotation shows that the subunit composition of the Se T4SS is unusual, but most closely resembles that found in conjugative T4SSs of Gram-positive bacteria (Table S2). A notable distinction is the presence of a series of predicted pilin subunits in Se, which are essential and encoded within the T4SS gene cluster; Gram-positive conjugative T4SSs generally lack a pilus. Other differences include the lack of recognizable coupling and relaxase proteins in the Se T4SS. These proteins directly participate in and are required for DNA transport, suggesting that the T4SS of Se may not function in this manner and may instead export proteins or serve in an alternative capacity.
In addition to macromolecular machines, we found an assortment of other essential cell envelope-associated proteins whose predicted function is not typically associated with critical cellular processes in bacteria (Figure 6B). Several of these proteins contain domains implicated in polysaccharide binding or degradation, including the lectin-like VCBS domain (SEML1_0891), the cellosome-associated dockerin domain (SEML1_0315), and the glycan-binding fibronectin III domain (SEML1_0890). We speculate that the proteins containing these domains target host-cell-associated glycans, either for promoting adhesion or for degradative purposes. An additional essential protein and two adjacent genes classified as conferring a growth deficit if inactivated are predicted components of extracellular polysaccharide (EPS) biosynthesis machinery (SEML1_0081-0083). Therefore, an EPS synthesized by Se may also contribute to productive host-cell interaction.
The arginine deiminase system (ADS) is an adenosine triphosphate (ATP)-generating catabolic pathway prevalent in mammalian-adapted Saccharibacteria species.36 A prior study demonstrated that arginine supplementation to N. lyticus TM7x-Schaalia odontolytica co-cultures permits acid neutralization via ammonia production, supporting the viability of both strains during acid stress.36 The authors of this study put forth a model in which hosts lacking ADS benefit from and are specifically permissive to colonization by Saccharibacteria containing the pathway. In Se, the ADS consists of ArcA, a fusion of arginine deiminase and ornithine carbamoyltransferase, ArcC, a carbamate kinase, and ArcDE, an arginine/ornithine antiporter (SEML1_0801-0803); all were classified as essential in our Tn-seq study (Table S1). Interestingly, in contrast to the proposed model for ADS function in Saccharibacterial-host cell interactions, sequencing of the Ai genome revealed intact homologs of each ADS gene (Figure S5D). Taken together with our Tn-seq data, this observation suggests that in some co-culture pairs, the organisms may compete for arginine. In summary, the uniqueness of the epibiotic lifestyle of Se is borne out by the unusual collection of essential functions revealed by our Tn-seq data.
Discussion
Scientists have been aware of Patescibacteria in environmental samples for many years; however, our understanding of this group of organisms has lagged.11 One major challenge is their apparent strict requirement for host bacteria, the identity of which cannot currently be determined a priori, thus adding significant complexity to Patescibacteria isolation.12,26,52,53 It is foreseeable that methods bringing to bear both experimental and computational approaches could provide predictions of host-epibiont associations in the future. Here, we addressed a second impediment to studying Patescibacteria—a lack of genetic tools for their manipulation. Indeed, despite streamlined methods for isolating Saccharibacteria on host Actinobacteria that have been employed by several laboratories, this latter challenge has, to date, prohibited a molecular dissection of their epibiotic lifestyle.24,25,53 Our discovery that natural transformation can facilitate targeted mutagenesis in Saccharibacteria opens the door to direct interrogation of genotype-phenotype relationships within these Patescibacteria; however, challenges remain. The most fundamental hurdle is that factors required for the Saccharibacteria-host cell interaction are also required for viability. Indeed, our Tn-seq analysis led to the identification of numerous essential genes not typically required for bacterial growth that encode predicted functions consistent with their involvement in mediating the interaction between Se and Ai. In analogous situations, researchers overcome this challenge using assorted conditional inactivation strategies (e.g., modulated expression, temperature sensitivity, and inducible degradation). Although it is conceivable that such methods could be implemented in Saccharibacteria, a powerful approach in this system may involve exploiting host genetics to identify gene interactions that suppress otherwise lethal mutations in the epibiont.
The small genome of Saccharibacteria stands in contrast to the apparent complexity of their lifecycle. Xie et al. proposed a model for the epibiotic growth of Saccharibacteria that consists of four stages: T4P-mediated infection, growth, bud formation, and asymmetric division.25 Our time-lapse microscopy generally supports this model; however, it further revealed facets of the Saccharibacteria-host interaction, which were not previously captured. Although the aforementioned model implies uniform progression, our data suggest the lifecycle is more complex. We find that only a small subset of Se achieve productive infections and that after an extended enlargement period, these cells rapidly bud a large number of highly motile swarmer cell progeny (>10 in a 20-h period observed in many instances). Quantitative analyses of our data additionally permitted us to link productive and non-productive Se infections with corresponding host cell outcomes. We observed a striking inverse relationship between Se growth and that of Ai; cells productively infected with as few as one Se not only failed to divide but diminished in size, whereas those infected unproductively by multiple Se readily proliferated.
Our microscopy observations suggest a potential division of labor within Se populations, wherein one subpopulation is devoted to reproduction, and a second, motile subpopulation searches for new compatible host cells. Operating under the assumption that the Saccharibacteria-Actinobacteria relationship is chiefly one of parasitism, which we maintain is not fully resolved in the literature, then the relevant prevailing evolutionary theory predicts that Se fitness is dependent on both the rate at which it causes new infections and the duration of those infections.54 The reproductive strategy of Se appears consistent with this framework; release of progeny cells destined to infect other host cells tempers host cell burden and prolongs the infection, while concurrently maximizing Se reproduction. A similar outcome could be achieved if Se employed active mechanisms to block superinfection, reminiscent of those utilized by phage; however, swarmer cell production has the added advantage of increasing Se dispersal in the face of low host cell densities.55 Interestingly, most nascent swarmer cells we observed do not themselves establish a productive infection within our 20-h observation period, despite close proximity and apparent adherence to host cells. It may be that the time required to establish a productive interaction varies, and many of these cells would go on to become productive. It is also conceivable that infected host cells defend against secondary infections, perhaps even inducing a defensive state in neighboring kin cells. Future studies coupling quantitative microscopy with genetics to further dissect the Se-Ai interaction will no doubt address these questions and shed light on this fascinating interphyla relationship.
The set of essential genes we identified in Se differs markedly from those described in other bacteria. One striking omission from the list is its predicted F-type ATP synthase, all the components of which are dispensable for growth on Ai. This raises the question of why Se maintains the capacity to assemble this large macromolecular machine. Like most Patescibacteria, Se lacks a respiratory chain and likely grows via anaerobic fermentation.10,11 It likely generates ATP by substrate-level phosphorylation, including via the arginine deiminase pathway, which we found is essential.56 During fermentative growth, ATP synthase can operate in reverse, hydrolyzing ATP and extruding protons, which is thought to contribute to maintaining the proton motive force (PMF).57 However, the lack of essentiality of ATP synthase in Se suggests that it is not the primary driver of the PMF. It has been hypothesized that the close physical proximity maintained between Saccharibacteria and their host could allow the epibionts to harness the host-generated proton gradient, either to facilitate solute transport or to fuel ATP synthesis.10 However, we find that growth of Ai is arrested rapidly upon productive association with Se; it is unclear how long such non-growing host cells maintain their proton gradient. Interestingly, several oral Streptoccocus species rely on proton extrusion by an ATP synthase acting in reverse to resist the acid stress generated by cariogenic oral bacteria.58 Given that Saccharibacteria are common oral cavity inhabitants, we speculate that they may employ the complex in a similar fashion.
We anticipate that the methods and genetic tools presented here will facilitate molecular-level characterization of Saccharibacteria and Patescibacteria more broadly. In this regard, one key question is the extent to which natural competence is active and able to be similarly exploited across Patescibacteria. The ability to manipulate Patescibacteria outside of Saccharibacteria, particularly those with phylogenetically distinct hosts and inhabiting diverse niches, should aid in elucidating the core requirements of the epibiotic lifestyle. Regardless of the precise methods utilized, genetic manipulation of Patescibacteria will open the door to phenotypic studies of the rich reserves of microbial dark matter these organisms contain, potentially revealing unprecedented biological mechanisms.
Limitations of the study
Although the methods we developed for genetic manipulation of Saccharibacteria unlock many previously inaccessible routes of investigation, they are nevertheless subject to limitations. Chief among these is the reliance on a single antibiotic-resistance cassette, which precludes facile construction of strains with multiple mutations and may limit the breadth of organisms that can be targeted. Additionally, we have yet to develop an approach for generating unmarked mutants and have not established whether genetic manipulation via natural transformation can be applied to Patescibacteria outside of Saccharibacteria. Beyond the technical limitations of our approach, applying the methods we describe toward genotype-phenotype investigations of Se is limited by (1) the difficulty of distinguishing genes required for basic physiology from those encoding host interaction determinants and (2) screening under in vitro growth conditions that differ from those encountered in nature (e.g., the oral cavity for Se).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Southlakia epibionticum ML1 | This study | N/A |
| Nanosynbacter lyticus ML1 | This study | N/A |
| N. lyticus TM7x | He et al.13 | N/A |
| Actinomyces israelii F0345 | Dewhirst et al.59 | N/A |
| A. odontolyticus F0309 | Batty60 | N/A |
| Schaalia odontolytica XH001 | He et al.13 | N/A |
| A. urogenitalis S6-C4 | Nikolaitchouk et al.61 | N/A |
| Actinomyces sp. F0386 | Dewhirst et al.59 | N/A |
| Propionibacterium propionicum F0230 | Dewhirst et al.59 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Instagene matrix | Bio-Rad | Cat#732-6030 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#1725272 |
| EZ-Tn5 Transposase | Biosearch Technologies | Cat#TNP92110 |
| Benzonase Nuclease | Sigma | Cat#E1014 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69506 |
| Nano-Glo Luciferase Assay System | Promega | Cat#N1110 |
| Deposited data | ||
| The complete genome sequences of S. epibionticum ML1, N. lyticus ML1 and A. israelii F0345 | This study | GenBank: PRJNA957798 |
| Transposon insertion sequencing data | This study | NCBI Sequence Read Archive: PRJNA957798 |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S3. | N/A | N/A |
| Recombinant DNA | ||
| DNA fragments used in this study are listed in Table S4. | N/A | N/A |
| Software and algorithms | ||
| Geneious Prime 2023.1.2 | Geneious, Software, Newark, New Jersey, USA | https://www.geneious.com; RRID:SCR_010519 |
| Prism 9 for macOS | GraphPad, Software, La Jolla, California, USA | https://www.graphpad.com; RRID:SCR_022798 |
| Adobe Illustrator 27.3.1 | Adobe Systems Incorporated, San Jose, California, USA | https://www.adobe.com/products/illustrator; RRID:SCR_010279 |
| UCSF ChimeraX version 1.6.1 | UCSF, Software, San Francisco, California, USA | https://www.cgl.ucsf.edu/chimerax/; RRID:SCR_015872 |
| Trycycler v0.5.2 | Wick et al.62 | N/A |
| Filtlong v0.2.1 | https://github.com/rrwick/Filtlong | N/A |
| Flye v2.9 | https://github.com/fenderglass/Flye/releases/tag/2.9. Accessed 4 October 2021 | N/A |
| Raven v1.8.1 | Vaser and Šikić63 | N/A |
| Miniasm v0.3r179 | Li64 | N/A |
| Minipolish v0.1.3 | https://github.com/rrwick/Minipolish/blob/main/miniasm_and_minipolish.sh | N/A |
| Polypolish v0.5.0 | Wick et al.62 | N/A |
| MaSuRCA v4.0.9 | Zimin et al.65 | N/A |
| PROKKA v1.14.5 | Seemann66 | N/A |
| BGME | Criscuolo and Gribaldo67 | N/A |
| IQtree | Trifinopoulos et al.68 | N/A |
| Omnipose | Cutler et al.69 | N/A |
| TRANSIT suite | DeJesus et al.38 | N/A |
| ProtNLM | Gane et al.39 | N/A |
| hmmscan | Eddy et al.40 | N/A |
| KofamKOALA version 2023-04-01 | Aramaki et al.70 | N/A |
| HHblits | Remmert et al.44 | N/A |
| phmmer and jackhmmer | Eddy40 | N/A |
| AlphaFold | Jumper et al.43 | N/A |
| FoldSeek v6.0 | van Kempen et al.47 | N/A |
| Domain Parser for AlphaFold Models (DPAM) | Zhang et al.49 | N/A |
| Other | ||
| 5 μm mixed cellulose esters (MCE) membrane | Millipore | SMWP04700 |
| 5 μm syringe filters, Surfactant-Free Cellulose Acetate and Cellulose Acetate | Thermo Scientific | 723-2545 |
| 0.1mm zirconia/silica beads | BioSpec | Catalog#11079101z |
| Mini-BeadBeater-16 | Biospec | Model 607 |
| Breath-Easy sealing membrane | Sigma | Z380059 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joseph Mougous (mougous@uw.edu).
Materials availability
Plasmids and bacterial strains generated in this study are available upon request from the lead contact.
Experimental model and study participant details
Strains, media and growth conditions
Saccharibacteria strains employed in this work include Southlakia epibionticum ML1 and Nanosynbacter lyticus ML1, both isolated in this study, and Nanosynbacter lyticus TM7x.13 Host bacterial strains used include Actinomyces israelii F0345, A. odontolyticus F0309, Schaalia odontolytica XH001 (XH001),13 A. urogenitalis S6-C4, Actinomyces sp. F0386 and Propionibacterium propionicum F0230 (Pp).59,60,61 Ai and Pp host bacteria mono-cultures, Se-Ai co-cultures and Nl ML1-Pp co-cultures were routinely grown statically under an atmosphere of ambient air supplemented with 5% CO2 at 37°C or anaerobically with shaking at 37°C using the GasPak EZ Anaerobe Container System with Indicator (BD 260626 and 260001) in TSY media (30 g/L tryptic soy and 5 g/L yeast extract) or TSYR media (TSY media supplemented with 10 mM arginine), or anaerobically on TSBY agar plates (30 g/L tryptic soy, 5 g/L yeast extract, 15 g/L agar, supplemented with 5% (v/v) horse blood). XH001 host bacteria mono-cultures and Nl TM7x-XH001 co-cultures were grown at 37°C anaerobically in Brain Heart Infusion (BHI) media or on BHI agar plates. For selection of hygromycin-resistant Saccharibacteria, hygromycin was used at 150 μg/mL. Se-Ai and Nl ML1-Pp co-cultures were stored at -80°C in TSY or TSYR supplemented with 10% (v/v) Dimethylsulfoxide (DMSO). Nl TM7x-XH001 co-cultures were stored at -80°C in BHI supplemented with 25% (v/v) glycerol.
Method details
Isolation of Saccharibacteria strains Se ML1 and Nl ML1
Isolation of Saccharibacteria strains in co-culture with host bacteria was carried out as essentially as previously described.53,71 Anonymous volunteers aged over 18 years provided oral samples for Saccharibacteria isolation. Supragingival plaque samples were collected with toothpicks and dispersed in 1 mL of Maximum Recovery Diluent (MRD; Peptone 1.0 g/L, Sodium Chloride 8.5 g/L, pH 7.0) buffer. 5 mL of saliva was collected by voluntary expectoration into sterile 50 mL conical tubes. All saliva samples and plaque samples were then pooled into 20 mL MRD. Pooled samples were then vigorously resuspended by vortexing and filtered with a 0.22 μm filter, and the flowthrough was collected. Residual bacteria in the sample tube and filter were collected by washing once with 10 mL MRD, filtered again, and combined with the previous flowthrough. Saccharibacteria present in filtrates were pelleted by centrifuging at 60,000 rcf for 1 hr at 4°C. Supernatant was removed, and pellets were resuspended in 1 mL MRD. The presence of Saccharibacteria in these samples were confirmed by PCR, using phylum specific primers71 (Table S3). The collected Saccharibacteria were then added to mono-cultures of a panel of five potential host species (A. odontolyticus, A. urogenitalis, Actinomyces sp. F0386, Ai, and Pp) and the cultures were passaged every 24 or 48 hr. The presence of Saccharibacteria cells in the final cultures was confirmed by qPCR with universal Saccharibacteria primers (Table S3) and microscopic imaging. The cultures were then streaked on TSY or TSBY agar, isolated colonies of host bacteria were tested for Saccharibacteria by PCR, and positive colonies were re-cultured in liquid medium and stored as clonal co-cultures.
To create purified suspensions of Saccharibacteria from co-cultures, the co-culture was centrifuged at 3,000 rcf for 5 min at room temperature, to pellet host cells. Supernatant containing suspended Saccharibacteria was then collected and filtered twice using a pre-sterilized 5 μm mixed cellulose esters (MCE) filter. Filtrate was centrifuged at 15,000 rcf for 30 min at room temperature to pellet Saccharibacteria. The resulting pellet was resuspended in a small volume of TSYR supplemented with 10% (v/v) DMSO and stored at -80°C.
Saccharibacteria host compatibility testing
Purified populations of Se and Nl ML1 for host compatibility testing were generated by growing 100 mL co-cultures of each with their respective host strains according to the standard protocol described above. Co-cultures were passed through a 0.45 μm SFCA filter to remove host cells then spun at 80,000 rcf for 20 min to pellet Saccharibacteria. Supernatant was removed and cell pellets were resuspended in 1 mL fresh medium. Purified Saccharibacteria cells were then added to OD600 = 0.2 cultures of Ai and Pp (compatible host for Nl) at an MOI of 0.2. Cultures were then incubated statically at 37°C under ambient air enriched with 5% CO2 for up to 72 hr, and populations of Se and Nl were monitored over time using qPCR.
Whole genome sequencing
Genomic DNA of Se, Nl ML1and Ai F0345 was isolated using the Wizard HMW DNA Extraction kit (Promega). Sequencing was performed on Illumina iSeq and MiSeq and Oxford Nanopore (ONT) MinION instruments after standard sequencing library preparation protocols (Illumina and Oxford Nanopore). De-novo assemblies were generated using the Trycyler pipeline.62 Specifically, ONT long reads were filtered using Filtlong v0.2.1 (https://github.com/rrwick/Filtlong) with Illumina re–rence reads, using --keep_percent 95 to retain approximately 95% of the reads. Long reads were then subsampled into 12 bins and assembled into 12 assemblies using the Flye v2.9 (https://github.com/fenderglass/Flye/releases/tag/2.9. Accessed 4 October 2021), Raven v1.8.163 Miniasm v0.3r17964 and Minipolish v0.1.3 (https://github.com/rrwick/Minipolish/blob/main/miniasm_and_minipolish.sh) assemblers. Assembly contigs were manually curated and then reconciled using Trycycler v0.5.2.62 A consensus assembly for each bacterium was generated and then polished with Illumina short reads using Polypolish v0.5.062 and POLCA from MaSuRCA v4.0.9.65 Initial annotations were generated using PROKKA v1.14.5.66
To sequence clonal transformed Se and Nl TM7x (see Results and below), genomic DNA was isolated from frozen pellets of the purified Saccharibacteria by re-suspending in Buffer PB (Qiagen) to a total volume of 500 μL, adding 250 μL of 0.1mm zirconia/silica beads (BioSpec Catalog # 11079101z), 250 μL of 20% SDS, and 550 μL of phenol:chloroform:IAA (25:24:1) (Invitrogen Catalog #15593-031), and bead-beating in a Mini-BeadBeater-16 (Biospec Model 607) with settings 3450 RPM, 115V, 10A, and ½ HP, for four 30-second intervals, each followed by cooling on ice for 1 minute. Purification of the DNA was performed by applying the aqueous phase directly to a DNeasy Blood & Tissue Prep Kit (Qiagen) purification column and following the recommended protocol for washing and elution. Sequencing was performed on an Illumina iSeq using standard library preparation protocols (Illumina). Reads were mapped to the assembled Se ML1 genome or the Nl TM7x genome (GenBank: NZ_CP007496) using minimap2 and variants were called using LoFreq v2.
Phylogenetic analysis
The Se and Nl ML1 genomes were phylogenetically placed using whole genome information. A genome tree was generated from these newly isolated strain genomes with a manually curated set of high-quality Patescibacteria genomes (complete and partial) and metagenome assembled genomes (MAGS) to remove any contaminants deposited with the original assemblies as described previously.16 This species tree was constructed using a set of 50 core, universal proteins defined by COG (Clusters of Orthologous Groups) gene families with KBase.72 Sequences of each of the 50 selected proteins were individually aligned using MUSCLE, then concatenated into a curated single curated multiple sequence alignment (Data S2). The alignments were trimmed using BGME67 (default settings with gap rate cut-off 0.1) to remove poorly aligned sections resulting in 6042 columns, 3949 distinct patterns, 2686 parsimony-informative, 1525 singleton sites and 1831 constant sites (Data S3). A phylogenetic tree was reconstructed from this concatenated and trimmed alignment using IQtree (-st AA -m TEST -bb 1000 -alrt 1000).68 Genomes were annotated with the latest GTDB taxonomy (Release 214).7
Measuring hygromycin sensitivities of Ai and Se
Hygromycin sensitivity of Ai and Se was measured in liquid co-cultures. Duplicate co-cultures were initiated by mixing purified Se with Ai at an OD600 of 0.2 in TSY and at an approximate cellular ratio of 1:2 (Se:Ai). The co-cultures were divided into multiple aliquots in 96-well culture plates and hygromycin was added to the final concentrations shown in Figure S1. The plates were covered with Breath-Easy sealing membrane (Sigma Z380059) and incubated without lids at 37°C with 5% CO2. At 1 day and 3 days, the cells within individual wells were pelleted (>15 min at >15,000 rcf) and stored at -20°C for later genomic DNA isolation and qPCR-based quantification of Se and Ai (see below).
Design and generation of cassettes for heterologous gene expression in Se and Nl
Cassettes for heterologous gene expression in Se and Nl ML1were designed by appending promoter and terminator sequences from the Nl TM7x genome13 to the 5’ and 3’ ends, respectively, of ORFs codon optimized for Se. The promoter and terminator elements were sourced from Nl TM7x rather than Se to reduce the likelihood of off-target integration at corresponding Se loci. The Nl TM7x promoters were chosen from genes expected to be highly and constitutively expressed, tuf and rpsJ. Se codon usage was calculated using the Dynamic Codon Biaser (DCB).73 Heterologous ORFs were optimized to match relative codon usage frequencies found in Se, but omitting codons with less than 10% usage. Heterologous genes utilized included hph (hygromycin B phosphotransferase) from Streptomyces hygroscopicus, superfolder GFP (www.fpbase.org/protein/superfolder-gfp/), mCherry2 (www.fpbase.org/protein/mcherry2/) and NanoLuc Luciferase.74,75 The cassette used for transformation of Nl TM7x used the same codon-optimized hph ORF described above, but the promoter and terminator elements were sourced from the Se tuf gene. Table S4 reports the composition and complete sequences of the designed cassettes. Cassettes were obtained as gBlocks from Integrated DNA Technologies, Inc (IDT) or gene fragments from Twist Biosciences.
Linear fragments used for transformations were generated by adding Se, or Nl genomic sequences corresponding to the targeted insertion or allelic replacement sites to the left and right sides of a heterologous gene expression cassette or of two cassettes joined together. Overlap extension PCR was used to join gBlock cassettes and genomic fragments, the latter of which were individually amplified from Se or Nl genomic DNA. In some cases, complete fragments including genomic sequences were obtained as gBlocks from IDT or gene fragments from Twist Biosciences. Fragments used for flank-length tests were generated by amplification using larger fragments as templates followed by gel-purification. Table S4 reports the composition and complete sequences of the fragments utilized and primers used are listed in Table S3.
Genetic transformation of Se and Nl with targeted insertion and allelic replacement constructs
To prepare Se-Ai or Nl ML1-Pp co-cultures for transformation, 1-mL aliquots of a previously frozen co-culture (see methods on co-culturing) were thawed on ice and added to 9 mL of TSY supplemented with freshly cultured Ai to a final OD600 of 0.2, incubated statically for 2 d at 37°C in ambient air enriched with 5% CO2, then enlarged by the addition of 10 volumes of TSY and incubated anaerobically using the GasPak EZ Anaerobe Container System with Indicator (BD 260626 and 260001) at 37°C for 2 days with shaking at 160 rpm.
For transformation of Se or Nl ML1 with linear targeted insertion constructs, 0.22 - 0.3 mL aliquots of prepared co-culture were incubated statically with transforming DNA for 6 h at 37°C in ambient air enriched with 5% CO2 in culture tubes. The mixtures were subsequently enlarged by the addition of TSYR to approximately 5 mL and supplemented with recently passaged Ai (for Se) or Pp (for Nl ML1) to a final OD600 of approximately 0.06. This time point was designated “TF” (time zero after transformation). After removal of a sample for later qPCR analysis, the cultures were divided and one half was supplemented with hygromycin to 150 μg/mL. The cultures were then incubated as above for an additional 2-3 days. This time point was designated “P1” (end of initial passage). Cultures were subsequently serially passaged as many as four times by five-fold dilution into fresh TSYR supplemented with Ai (for Se) or Pp (for Nl) to a final OD600 of 0.06 and, for the hygromycin containing cultures, with additional hygromycin to 150 μg/mL. These passages (designated “P2”, “P3”, etc.) were each incubated for 2-3 days at 37°C in ambient air enriched with 5% CO2 and without agitation. Cultures were sampled at each passage by removing and pelleting (>15 min at >15,000 rcf) of 0.1 to 1 mL, and the pellets were stored at -20°C for later qPCR analysis.
For transformation of Nl TM7x with linear targeted insertion constructs, frozen Nl TM7x-S. odontolytica XH001 co-culture glycerol stock was inoculated into BHI media. Co-cultures were grown anaerobically and were passaged every 24 h until the end of passage 2 (P2). 0.9 mL co-cultures were incubated with 300 ng transforming DNA for 6 h at 37°C anaerobically. The rest of the transformation procedure is similar to that of Se and Nl ML1 described above, with the following differences: host supplemented during each passage was S. odontolytica XH001, media used was BHI, cultures were passaged every 24 h, and cultures were passaged to P4.
qPCR assays
We employed qPCR for quantification of Saccharibacteria and host populations in co-cultures, as well as to monitor Se transformation. For these assays, genomic DNA was isolated from frozen co-culture pellets using Instagene matrix (Bio-Rad) and quantitative PCR (qPCR) was performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad). To quantify Se and Ai, amplification employed primers targeting Se uvrB or the 16S rRNA gene of Ai and was performed in 20 μL reactions with 1X SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), 300 nM each primer and 4 μL template DNA (primer sequences provided in Table S3). Thermocycling conditions were 95°C for 5 min followed by 35-40 cycles of 95°C for 20 s, 60°C for 30 s and a read of fluorescence. To quantify transformed Se (insertions at NS1), amplification employed primer pairs targeting the insertion sequence and sequence adjacent to NS1 but outside of the linear transformation construct arms and was performed in 20 μL reactions with 1X Phusion HF Buffer, 0.2 mM dNTPs, 250 nM each primer, 0.5X SYBR Green I (ThermoFisher), 4 μL template DNA and 0.4 units of Phusion High-Fidelity DNA Polymerase (NEB) (primer sequences provided in Table S3). Thermocycling conditions were 98°C for 90 s followed by 35-40 cycles of 98°C for 15 s, 70°C for 20 s, 72°C for 90 s and a read of fluorescence. Melt curves were performed following each amplification to verify product homogeneity. Absolute target sequence abundance was determined by comparison to duplicate standard curve reactions performed in parallel with each assay. The standard curve templates were generated by serial dilution of previously amplified and gel-purified products quantified by Qubit (ThermoFisher).
Isolation of isogenic mutant co-cultures
To obtain co-cultures with isogenic mutant (transformed) Se and Nl TM7x, transformation mixtures grown for three passages (Se) or four passages (Nl TM7x) under hygromycin selection were serially diluted, plated on TSBY agar (Se) or BHI agar (Nl TM7x) without antibiotic and incubated for 5-7 days anaerobically. For Se, isolated colonies (representing Ai potentially colonized with Se) were picked into 0.2 mL TSYR supplemented with Ai at a final OD600 of 0.2 and incubated statically at 37°C in ambient air enriched with 5% CO2 for 2-4 days, then screened for the presence of Se and for the mutant (genomic insertion) and wild-type (no insertion) alleles by PCR (e.g., Figure 2A; primer sequences in Table S3). For Nl TM7x, isolated colonies (representing XH001 potentially colonized with Nl TM7x) were picked into 50 μL BHI media and 1 μL from which was used as template for PCR screening (Figures S2C and S2E). Cultures were incubated statically at 37°C anaerobically for 3 days with passaging every 24 h. Cultures containing pure mutant Se or Nl TM7x were frozen and/or further propagated for purification of the Se or Nl TM7x for WGS (see above).
Nanoluciferase assay
Nanoluciferase assays were performed using isogenic wild-type Se-Ai co-cultures and Se NS1::nluc-hph2-Ai co-cultures grown statically in ambient air supplemented with 5% CO2 at 37°C in TSY media for three passages as described above. To separate Se from host cells, 20 mL of each co-culture was passed through a 0.45 μM SCFM filter and the resulting filtrate was spun at 80,000 rcf for 30 min to pellet Se, supernatant was removed, and the pelleted Se was resuspended in 750 μL TSY. 100 μL of these purified Se cells or 100 μL of the corresponding Se-Ai co-cultures were mixed with 100 μL Nano-Glo Luciferase assay reagent (50:1 mixture of substrate:buffer, Promega N1110) in a 96-well plate. Luminescence signal indicative of nanoluciferase activity was detected using a Cytation 2 plate reader. Luminescence signal was later normalized by Se abundance in co-cultures and filtrates as measured by qPCR using Se-specific primers as described above.
Microscopy
Imaging was performed on a Nikon Eclipse Ti-E wide-field epi-fluorescence microscope, equipped with a sCMOS camera (Hamamatsu) and X-cite LED for fluorescence imaging. We imaged through a Nikon Plan Apo λ 60X 1.4 NA oil-immersion Ph3 objective. The microscope was controlled by NIS-Elements 3.30.02. Isogenic Se NS1::mcherry-hph2-Ai co-cultures and Se NS1::sfgfp-hph2-Ai co-cultures were grown statically in ambient air supplemented with 5% CO2 at 37°C in TSYR media as described above. Cell samples were spotted on a 3% (w/v) agarose pad made with TSYR media supplemented with 0.4% glucose placed on a microscope slide. The microscope chamber was heated to 37°C for time-lapse experiments. Time-lapse images were aligned and segmented with Omnipose using the phase contrast channel and the published bact_phase_omni model.69 Masks were manually linked and corrected for segmentation errors in regions of Ai cell overlap using Napari. Regions corresponding to Se were also removed to accurately track host Ai growth alone. For visualization purposes, fluorescence intensity was gamma-corrected to normalize background levels on a frame-by-frame basis while not distorting the Se signal. No bleaching correction was implemented. Figures 3A–3F and associated videos were generated using Python and annotated in Adobe Premiere.
Mutagenesis of Sac1a genomic DNA and transposon mutant library generation
The transposon used for in vitro transposition (here named T36) was generated by amplification of the hph1 insertion cassette using the 5’-phosphorylated primers T36-ampF and T36-ampR, which add required 19-bp Tn5 mosaic end sequences to each end of the amplicon, followed by purification using the Qiagen PCR Purification Kit with elution in TE (primer sequences provided in Table S3). In vitro transposition was performed in multiple 50-μl reactions, each containing 1.0 μg Se genomic DNA, 82 ng purified T36, 1X EZ-Tn5 Reaction Buffer and 3.4 U EZ-Tn5 Transposase (Biosearch Technologies) with incubation for 2 hr at 37 °C. The reactions were stopped, DNA was ethanol precipitated, and gaps were repaired as described.37 Following gap repair, the DNA was purified using Qiagen PCR Purification Kit with elution in water after the columns were washed twice with Buffer PE.
Transformation of Se to generate transposon mutant pools was performed similarly to the procedure described above for transformation with targeted insertion fragments, but in quadruplicate, at larger scale and with modifications. Specifically, for each replicate the initial 6 hr incubation contained 3.3 μg of in vitro transposon-mutagenized genomic DNA and 0.75 mL of Ai-Se co-culture that had been concentrated 42-fold by centrifugation for 35 min at 15,000 rcf followed by re-suspension in a small volume of TSYR. Each replicate was then enlarged by adding 330 mL of TSYR supplemented with fresh Ai at a final OD600 of 0.12, then incubated for two additional hours before hygromycin addition. Concomitantly with hygromycin addition, MgCl2 (to 1 mM) and benzonase (Sigma) were added to 25 U/mL to degrade extracellular DNA, and the cultures were similarly supplemented with benzonase approximately every 24 h during passaging. After the initial 2-day growth passage (ending at time point T0), the culture was serially passaged three additional times by dilution of 160 mL culture into 640 mL in TSYR supplemented with Ai at OD600 of 0.15, hygromycin at 150 μg/mL and MgCl2 at 1 mM. These passages were each incubated statically for 2 days at 37°C in ambient air enriched with 5% CO2. Samples for Tn-seq analysis were taken at the end of the initial passage (170 mL per replicate, T0) and at the end passages 1, 2 and 3 (250 mL per replicate, T1-T3). These samples were treated with additional benzonase (50 U/mL) for 30 min, then EDTA was added to 15 mM and pellets were collected by centrifugation for 30 min at 15,000 rcf in multiple tubes per replicate and stored at -80°C.
Tn-seq library preparation and sequencing
Genomic DNA was isolated from frozen co-culture pellets using the bead-beating method described above (section on whole genome sequencing). Tn-seq libraries were prepared by the C-Tailing method as described.76 The transposon-specific primers utilized are listed in Table S3. Sequencing was performed in multiplex as 50-bp single-end reads on an Illumina MiniSeq with 25-40% PhiX spike-in.
Custom scripts (https://github.com/lg9/Tn-seq37,76) were used to process the Illumina reads. In brief, reads were first filtered for those displaying transposon end sequence as their initial bases (the sequencing primer was designed to anneal six bases from the end of the transposon). These reads were then mapped to the Se genome after removing the transposon end sequences. Reads per unique mapping position and orientation were tallied. For downstream analysis, the mapped read counts per site from all four replicates were combined for each time point, after normalizing the counts to 500,000 total reads per replicate. Read counts per gene were calculated by summing reads from all unique sites within a given gene.
Se gene essentiality analysis
Gene essentiality was determined at each time point (T0-T2) using the Hidden Markov Model method, HMM, part of the TRANSIT suite.38 Default parameters were used for the HMM analysis. HMM takes into account the read counts per insertion(s) in each gene to identify those with over- or under-represented insertion read counts. HMM classifies genes among four potential categories: essential, not essential, growth defect, or growth advantage. We focused on genes classified as essential. Although HMM was originally developed for analyzing Himar1 data, we were able to use HMM to analyze our Tn5 data by sequencing the in vitro transposon-mutagenized genomic DNA we originally used to transform Se. A total of 35,792 insertions were detected in that samples (with one or more reads). We expanded the universe of potential insertions by including insertions detected at any sampled time point (T0-T2). Thus, in contrast to the general idea of each nucleotide being a potential insertion site in Tn5 data (that would result in low saturation of the transposon mutant libraries and low performance of HMM),77 we estimated a total of 65,890 potential insertion sites in our Tn-seq experiments. The set of potential insertions was specified as part of the input for HMM runs (i.e.,.WIG file).
Transformations to validate gene essentiality predictions
To validate Tn-seq essentiality predictions, we designed constructs to replace target genes with hph. Each construct contained 335-bp sequences targeting the desired site flanking the hph expression cassette described above. 100 ng synthesize linear DNA or water were added to 300 μL wild-type Se-Ai co-cultures (4 biological replicates per condition) prepared according to our standard protocol, and transformation and subsequent passaging were performed using our general transformation protocol described above. At the end of passage 3, 1 mL of each transformed co-culture was treated with 1 mM MgCl2 and 25 U/mL benzonase for 30 min at 37°C to remove extracellular DNA. 15 mM EDTA was then added to stop the benzonase reaction, samples were centrifuged at 21,000 rcf for 1 hr and the resulting pellets were stored at -80°C. Se abundance was then measured by qPCR as described above.
Generation and transformation efficiency of Se ΔcomEC::hph
A clonal population of Se ΔcomEC::hph was generated by replacing comEC with our hygromycin resistance cassette following the transformation and clonal mutant isolation protocols described above. To assess Se ΔcomEC::hph transformability, 50 ng of synthesized linear gene product for inserting unmarked sfGFP at NS1 (with 545 bp flanking regions) was used to transform 150 μL isogenic wild-type or ΔcomEC Se-Ai co-cultures (4 biological replicates per condition). After 6.5 h of incubation between DNA and co-cultures at 37°C, the entire transformation culture was treated with 1 mM MgCl2 and 25 U/mL benzonase for 30 min at 37°C to degrade extracellular DNA. 15 mM EDTA was added to stop the benzonase reaction and the samples were centrifuged at 21,000 rcf for 1 hr and the resulting pellets were stored at -80°C. We then used qPCR to measure the abundance of transformed and total Se using NS1-sfGFP or uvrB- specific primers as described above.
Patescibacteria-enriched protein families analysis
To assess the protein family distribution of the Se and Nl genomes, we collected amino acid sequences for 22,977 protein clusters (protein families) from Meheust et al.9 These protein families were built through their occurrences across at least 5 distinct non-redundant or draft Patescibacteria, bacteria outside of Patescibacteria and Archaeal genomes. The presence or absence of any given family in Se or Nl were determined through all-vs-all protein sequence search of every protein in the genome against the database of protein family sequences. Protein sequence search was performed by using the MMseqs2 (version: 14-7e284) algorithm with the following parameters: module: easy-search; sensitivity (-s): 7.5; alignment coverage (-c): 0.5; greedy-best-hits: 1. The resulting presence or absence matrix was combined with the core family matrix of 921 protein families across 2890 genomes (collected from Meheust et al.9) for clustering analysis. A Jaccard distance based on the presence or absence of core families across Se, Nl and 2890 other genomes was calculated in R by using proxyC package (version: 0.3.3). Agglomerative hierarchical clustering was performed by using cluster package (version: 2.1.4) in R with the “wand” method. A hierarchical clustering heatmap was built using complexHeatmap package in R (version: 2.14.0) by plotting presence (black) or absence (white) of protein families (columns) in a given genome (rows). Enrichment or depletion of core protein family clusters in Se and Nl was performed using Fisher’s exact test in R with input table of presence or absence of protein families in the Se and Nl genomes. A threshold of Benjamini-Hochberg corrected p-values of < 5e-05 was used to determine the enrichment or depletion.
Sequence-based gene annotation of the Se genome
We used ProtNLM (https://github.com/google-research/google-research/tree/master/protnlm) to predict the functions of proteins in the Se proteome from sequence.39 A prediction of score ≥0.5 corresponds to >70% accuracy for proteins without close UniRef50 matches and ∼95% accuracy for proteins with close matches. We thus considered the 337 proteins with score ≥0.5 to have confident annotations (Table S2). Additionally, we mapped each protein to Pfam domains (Nov, 2021 release v35.046) using hmmscan.40 We filtered the hit Pfam domains by full sequence E-value ≤ 1e-3 and domain-based CE-value ≤ 1e-5 or CI-value ≤ 1e-5, domain coverage ≥ 50% (coverage of aligned portion of domain to full length domain) and selected the top hit for each aligned region. Finally, we used KofamKOALA70 (version 2023-04-01) to conduct an HMM search against the KEGG Orthology (KO) database78 (release 106.0). We assigned broad functional categories based on KO to the 353 proteins which mapped to KOs above the adaptive significance threshold (E-value ≤ 0.001) which are computed for each KO family as described in Aramaki et al.70
Generation of multiple sequence alignments
To construct MSAs for each protein in the Se proteome, we initially used HHblits44 to search against UniRef (2022 release46) and BFD (2019 release45). For those MSAs that contained <500 sequences after this approach, we conducted extensive homology searches against metagenome databases similar to Anishchenko et al.79 We first converted the initial HHblits (E-value ≤ 1e-3) MSAs for each protein into a hidden Markov model (HMM) which we used as a seed sequence profile to search against metagenomic and metatranscriptomic sequences from JGI,80 MGnify,81 and UniRef with hmmsearch,40 respectively. Hits from JGI, MGnify, and UniRef were gathered and aligned to the query protein by phmmer and jackhmmer,40 to retain sequences with E-value ≤ 1e-5 at each stage. We removed columns containing gaps in the query sequence from the MSAs, which were then subjected to redundancy filtering at 95% sequence identity and 50% sequence coverage. We selected the deepest MSA for each protein (from HHblits against UniRef+BFD or phmmer/jackhmmer against metagenome) for downstream analysis.
Structure-based annotations
Using MSAs described above, we computed AlphaFold (AF) models (model_1 without structural template search, 10 recycles, and version 1.0 weights) for 852 of the 855 proteins (3 were excluded because of MSA depth or protein size).43 We then applied FoldSeek47 (v6.0, sensitivity 9.5, and alignment type 1) against AFdatabase50 (AF Protein Structure Database v4 clustered at 50% sequence identity and 80% coverage, accessed May 2023). We report the top 3 hits in Table S2 based on normalized TM-alignment score ≥ 0.5 using alignment type 1 (TM-score; which correlates well to fold-family level similarity52), and query coverage and target coverage ≥ 50%. To classify proteins into evolutionary contexts based on structure and sequence similarity, we used Domain Parser for AlphaFold Models (DPAM),49 and the full results are available at https://conglab.swmed.edu/ECOD_Se/Se_ECOD.html.
Quantification and statistical analysis
Statistical significance in Figures 2E and 4D was assessed by unpaired two-tailed student’s t-tests between relevant samples. Statistical significance in Figure 4B was assessed by one-way ANOVA with Dunnett’s multiple comparison test compared to a no DNA control. Statistical details for each experiment can be found in the figure legends.
Acknowledgments
The authors wish to thank Simon Dove and Joshua Woodward for insightful comments on the manuscript, Yekaterina Shulgina for assistance with ribosomal gene annotation, and members of the Mougous laboratory for helpful suggestions. This work was supported by grants from the NIH (R01AI128215 and R01AI141953 to N.S.B. and R01DE023810 to J.S.M.), the National Science Foundation (MCB-2105570 to N.S.B. and S.T., and IIBR-2042948 and IOS-2050550 to N.S.B.), the Department of Defense, Defense Threat Reduction Agency (HDTRA1-21-1-0007 [DTRA J5B] to D.B.), the Bill and Melinda Gates Foundation (OPP1156262 to D.B.), and the Welch Foundation (I-2095-20220331 to Q.C.). Q.C. is a Southwestern Medical Foundation endowed scholar. J.D.M. and D.B. are HHMI Investigators, and J.D.M. is supported by the Lynn M. and Michael D. Garvey Endowed Chair at the University of Washington.
Author contributions
L.A.G., A.L., S.B.P., and J.D.M. conceived the study. Y.W., L.A.G., A.L., I.R.H., S.T., K.J.C., Q.C., N.S.B., S.B.P., and J.D.M. designed the study. Y.W., L.A.G., P.A.A., A.L., and K.J.C. performed experiments. Y.W., L.A.G., I.R.H., S.T., K.J.C., M.L.A.-O., Y.L., M.C.R., J.S.M., and Q.C. processed, analyzed, and visualized the data. Y.W., L.A.G., I.R.H., S.B.P., and J.D.M. wrote the manuscript. D.B., N.S.B., S.B.P., and J.D.M. provided supervision. Q.C., D.B., N.S.B., and J.D.M. funded the study. All authors contributed to manuscript editing and support the conclusions.
Declaration of interests
The authors declare no competing interests.
Published: September 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.08.017.
Supplemental information
Light gray shaded rows indicate class I essential genes, and dark gray shading indicates class II essential genes. Blue text indicates genes discussed in the text.
Data and code availability
-
•
The complete genome sequences of Se ML1, Nl ML1and Ai F0345 have been deposited in GenBank under BioProject PRJNA957798 with accession numbers SAMN34266291, SAMN34266292 and SAMN34266293, respectively. Tn-seq data generated in this study have been deposited in the Sequence Reads Archive (SRA, BioProject PRJNA957798). These datasets are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hug L.A., Baker B.J., Anantharaman K., Brown C.T., Probst A.J., Castelle C.J., Butterfield C.N., Hernsdorf A.W., Amano Y., Ise K., et al. A new view of the tree of life. Nat. Microbiol. 2016;1 doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 2.Marcy Y., Ouverney C., Bik E.M., Lösekann T., Ivanova N., Martin H.G., Szeto E., Platt D., Hugenholtz P., Relman D.A., Quake S.R. Dissecting biological "dark matter" with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. USA. 2007;104:11889–11894. doi: 10.1073/pnas.0704662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katz M., Hover B.M., Brady S.F. Culture-independent discovery of natural products from soil metagenomes. J. Ind. Microbiol. Biotechnol. 2016;43:129–141. doi: 10.1007/s10295-015-1706-6. [DOI] [PubMed] [Google Scholar]
- 4.Rinke C., Schwientek P., Sczyrba A., Ivanova N.N., Anderson I.J., Cheng J.F., Darling A., Malfatti S., Swan B.K., Gies E.A., et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 5.Coleman G.A., Davín A.A., Mahendrarajah T.A., Szánthó L.L., Spang A., Hugenholtz P., Szöllősi G.J., Williams T.A. A rooted phylogeny resolves early bacterial evolution. Science. 2021;372 doi: 10.1126/science.abe0511. [DOI] [PubMed] [Google Scholar]
- 6.Megrian D., Taib N., Jaffe A.L., Banfield J.F., Gribaldo S. Ancient origin and constrained evolution of the division and cell wall gene cluster in Bacteria. Nat. Microbiol. 2022;7:2114–2127. doi: 10.1038/s41564-022-01257-y. [DOI] [PubMed] [Google Scholar]
- 7.Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 8.Brown C.T., Hug L.A., Thomas B.C., Sharon I., Castelle C.J., Singh A., Wilkins M.J., Wrighton K.C., Williams K.H., Banfield J.F. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–211. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 9.Méheust R., Burstein D., Castelle C.J., Banfield J.F. The distinction of CPR bacteria from other bacteria based on protein family content. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-12171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castelle C.J., Brown C.T., Anantharaman K., Probst A.J., Huang R.H., Banfield J.F. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 2018;16:629–645. doi: 10.1038/s41579-018-0076-2. [DOI] [PubMed] [Google Scholar]
- 11.Ji Y., Zhang P., Zhou S., Gao P., Wang B., Jiang J. Widespread but poorly understood bacteria: candidate phyla radiation. Microorganisms. 2022;10 doi: 10.3390/microorganisms10112232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batinovic S., Rose J.J.A., Ratcliffe J., Seviour R.J., Petrovski S. Cocultivation of an ultrasmall environmental parasitic bacterium with lytic ability against bacteria associated with wastewater foams. Nat. Microbiol. 2021;6:703–711. doi: 10.1038/s41564-021-00892-1. [DOI] [PubMed] [Google Scholar]
- 13.He X., McLean J.S., Edlund A., Yooseph S., Hall A.P., Liu S.Y., Dorrestein P.C., Esquenazi E., Hunter R.C., Cheng G., et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. USA. 2015;112:244–249. doi: 10.1073/pnas.1419038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuroda K., Yamamoto K., Nakai R., Hirakata Y., Kubota K., Nobu M.K., Narihiro T. Symbiosis between Candidatus Patescibacteria and Archaea discovered in wastewater-treating bioreactors. mBio. 2022;13 doi: 10.1128/mbio.01711-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yakimov M.M., Merkel A.Y., Gaisin V.A., Pilhofer M., Messina E., Hallsworth J.E., Klyukina A.A., Tikhonova E.N., Gorlenko V.M. Cultivation of a vampire: 'Candidatus Absconditicoccus praedator'. Environ. Microbiol. 2022;24:30–49. doi: 10.1111/1462-2920.15823. [DOI] [PubMed] [Google Scholar]
- 16.McLean J.S., Bor B., Kerns K.A., Liu Q., To T.T., Solden L., Hendrickson E.L., Wrighton K., Shi W., He X. Acquisition and adaptation of ultra-small parasitic reduced genome bacteria to mammalian Hosts. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.107939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bor B., Bedree J.K., Shi W., McLean J.S., He X. Saccharibacteria (TM7) in the human oral microbiome. J. Dent. Res. 2019;98:500–509. doi: 10.1177/0022034519831671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brinig M.M., Lepp P.W., Ouverney C.C., Armitage G.C., Relman D.A. Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl. Environ. Microbiol. 2003;69:1687–1694. doi: 10.1128/AEM.69.3.1687-1694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ouverney C.C., Armitage G.C., Relman D.A. Single-cell enumeration of an uncultivated TM7 subgroup in the human subgingival crevice. Appl. Environ. Microbiol. 2003;69:6294–6298. doi: 10.1128/AEM.69.10.6294-6298.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paster B.J., Boches S.K., Galvin J.L., Ericson R.E., Lau C.N., Levanos V.A., Sahasrabudhe A., Dewhirst F.E. Bacterial diversity in human subgingival plaque. J. Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chipashvili O., Utter D.R., Bedree J.K., Ma Y., Schulte F., Mascarin G., Alayyoubi Y., Chouhan D., Hardt M., Bidlack F., et al. Episymbiotic Saccharibacteria suppresses gingival inflammation and bone loss in mice through host bacterial modulation. Cell Host Microbe. 2021;29:1649–1662.e7. doi: 10.1016/j.chom.2021.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adler C.J., Dobney K., Weyrich L.S., Kaidonis J., Walker A.W., Haak W., Bradshaw C.J., Townsend G., Sołtysiak A., Alt K.W., et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 2013;45:450–455. doi: 10.1038/ng.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bor B., McLean J.S., Foster K.R., Cen L., To T.T., Serrato-Guillen A., Dewhirst F.E., Shi W., He X. Rapid evolution of decreased host susceptibility drives a stable relationship between ultrasmall parasite TM7x and its bacterial host. Proc. Natl. Acad. Sci. USA. 2018;115:12277–12282. doi: 10.1073/pnas.1810625115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie J., Utter D.R., Kerns K.A., Lamont E.I., Hendrickson E.L., Liu J., Wu T., He X., McLean J., Bor B. Strain-level variation and diverse Host Bacterial Responses in Episymbiotic Saccharibacteria. mSystems. 2022;7 doi: 10.1128/msystems.01488-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie B., Wang J., Nie Y., Tian J., Wang Z., Chen D., Hu B., Wu X.L., Du W. Type IV pili trigger episymbiotic association of Saccharibacteria with its bacterial host. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2215990119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bor B., Collins A.J., Murugkar P.P., Balasubramanian S., To T.T., Hendrickson E.L., Bedree J.K., Bidlack F.B., Johnston C.D., Shi W., et al. Insights obtained by culturing saccharibacteria with their bacterial Hosts. J. Dent. Res. 2020;99:685–694. doi: 10.1177/0022034520905792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cross K.L., Campbell J.H., Balachandran M., Campbell A.G., Cooper C.J., Griffen A., Heaton M., Joshi S., Klingeman D., Leys E., et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol. 2019;37:1314–1321. doi: 10.1038/s41587-019-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albertsen M., Hugenholtz P., Skarshewski A., Nielsen K.L., Tyson G.W., Nielsen P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013;31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 29.Podar M., Abulencia C.B., Walcher M., Hutchison D., Zengler K., Garcia J.A., Holland T., Cotton D., Hauser L., Keller M. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl. Environ. Microbiol. 2007;73:3205–3214. doi: 10.1128/AEM.02985-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He X. Culture-based approaches to studying "microbial dark matter". Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2219691120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stackebrandt E., Goebel B.M. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 1994;44:846–849. [Google Scholar]
- 32.Damke P.P., Celma L., Kondekar S.M., Di Guilmi A.M., Marsin S., Dépagne J., Veaute X., Legrand P., Walbott H., Vercruyssen J., et al. ComFC mediates transport and handling of single-stranded DNA during natural transformation. Nat. Commun. 2022;13 doi: 10.1038/s41467-022-29494-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubnau D., Blokesch M. Mechanisms of DNA uptake by naturally competent bacteria. Annu. Rev. Genet. 2019;53:217–237. doi: 10.1146/annurev-genet-112618-043641. [DOI] [PubMed] [Google Scholar]
- 34.Sharma D.K., Misra H.S., Bihani S.C., Rajpurohit Y.S. Biochemical properties and roles of DprA protein in bacterial natural transformation, virulence, and Pilin variation. J. Bacteriol. 2023;205 doi: 10.1128/jb.00465-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston C., Martin B., Fichant G., Polard P., Claverys J.P. Bacterial transformation: distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014;12:181–196. doi: 10.1038/nrmicro3199. [DOI] [PubMed] [Google Scholar]
- 36.Tian J., Utter D.R., Cen L., Dong P.T., Shi W., Bor B., Qin M., McLean J.S., He X. Acquisition of the arginine deiminase system benefits epiparasitic Saccharibacteria and their host bacteria in a mammalian niche environment. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2114909119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallagher L.A., Bailey J., Manoil C. Ranking essential bacterial processes by speed of mutant death. Proc. Natl. Acad. Sci. USA. 2020;117:18010–18017. doi: 10.1073/pnas.2001507117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeJesus M.A., Ambadipudi C., Baker R., Sassetti C., Ioerger T.R. Transit--A software tool for Himar1 TnSeq analysis. PLOS Comp. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gane A., Bileshi M.L., Dohan D., Speretta E., Heliou A., Meng-Papaxanthos L., Zellner H., Brevdo E., Parikh A., Martin M.J., et al. ProtNLM: model-based natural language protein annotation. Preprint. 2022 https://storage.googleapis.com/brain-genomics-public/research/proteins/protnlm/uniprot_2022_04/protnlm_preprint_draft.pdf. [Google Scholar]
- 40.Eddy S.R. Accelerated profile HMM Searches. PLoS Comp. Biol. 2011;7 doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J., et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D., et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Remmert M., Biegert A., Hauser A., Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2011;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 45.Steinegger M., Mirdita M., Söding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods. 2019;16:603–606. doi: 10.1038/s41592-019-0437-4. [DOI] [PubMed] [Google Scholar]
- 46.Suzek B.E., Huang H., McGarvey P., Mazumder R., Wu C.H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 47.van Kempen M., Kim S.S., Tumescheit C., Mirdita M., Lee J., Gilchrist C.L.M., Söding J., Steinegger M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2023 doi: 10.1038/s41587-023-01773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H., Schaeffer R.D., Liao Y., Kinch L.N., Pei J., Shi S., Kim B.H., Grishin N.V. ECOD: an evolutionary classification of protein domains. PLoS Comp. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J., Schaeffer R.D., Durham J., Cong Q., Grishin N.V. DPAM: A domain parser for AlphaFold models. Protein Sci. 2023;32 doi: 10.1002/pro.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellison C.K., Dalia T.N., Vidal Ceballos A., Wang J.C., Biais N., Brun Y.V., Dalia A.B. Retraction of DNA-bound type IV competence pili initiates DNA uptake during natural transformation in Vibrio cholerae. Nat. Microbiol. 2018;3:773–780. doi: 10.1038/s41564-018-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hepp C., Maier B. Kinetics of DNA uptake during transformation provide evidence for a translocation ratchet mechanism. Proc. Natl. Acad. Sci. USA. 2016;113:12467–12472. doi: 10.1073/pnas.1608110113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira D., Zivanovic Y., López-Archilla A.I., Iniesto M., López-García P. Reductive evolution and unique predatory mode in the CPR bacterium Vampirococcus lugosii. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-22762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murugkar P.P., Collins A.J., Chen T., Dewhirst F.E. Isolation and cultivation of candidate phyla radiation Saccharibacteria (TM7) bacteria in coculture with bacterial hosts. J. Oral Microbiol. 2020;12 doi: 10.1080/20002297.2020.1814666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cressler C.E., McLEOD D.V., Rozins C., VAN DEN Hoogen J., Day T. The adaptive evolution of virulence: a review of theoretical predictions and empirical tests. Parasitology. 2016;143:915–930. doi: 10.1017/S003118201500092X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egido J.E., Costa A.R., Aparicio-Maldonado C., Haas P.J., Brouns S.J.J. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2022;46 doi: 10.1093/femsre/fuab048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zúñiga M., Pérez G., González-Candelas F. Evolution of arginine deiminase (ADI) pathway genes. Mol. Phylogenet. Evol. 2002;25:429–444. doi: 10.1016/s1055-7903(02)00277-4. [DOI] [PubMed] [Google Scholar]
- 57.Trchounian A., Trchounian K. Fermentation revisited: how do microorganisms survive under energy-limited conditions? Trends Biochem. Sci. 2019;44:391–400. doi: 10.1016/j.tibs.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 58.Sekiya M. Proton pumping ATPases: rotational catalysis, physiological roles in oral pathogenic bacteria, and inhibitors. Biol. Pharm. Bull. 2022;45:1404–1411. doi: 10.1248/bpb.b22-00396. [DOI] [PubMed] [Google Scholar]
- 59.Dewhirst F.E., Chen T., Izard J., Paster B.J., Tanner A.C., Yu W.H., Lakshmanan A., Wade W.G. The human oral microbiome. J. Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Batty I. Actinomyces odontolyticus, a new species of actinomycete regularly isolated from deep carious dentine. J. Pathol. Bacteriol. 1958;75:455–459. doi: 10.1002/path.1700750225. [DOI] [PubMed] [Google Scholar]
- 61.Nikolaitchouk N., Hoyles L., Falsen E., Grainger J.M., Collins M.D. Characterization of Actinomyces isolates from samples from the human urogenital tract: description of Actinomyces urogenitalis sp. nov. Int. J. Syst. Evol. Microbiol. 2000;50:1649–1654. doi: 10.1099/00207713-50-4-1649. [DOI] [PubMed] [Google Scholar]
- 62.Wick R.R., Judd L.M., Cerdeira L.T., Hawkey J., Méric G., Vezina B., Wyres K.L., Holt K.E. Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02483-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vaser R., Šikić M. Time- and memory-efficient genome assembly with Raven. Nat Comput. Sci. 2021;1:332–336. doi: 10.1038/s43588-021-00073-4. [DOI] [PubMed] [Google Scholar]
- 64.Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics. 2016;32:2103–2110. doi: 10.1093/bioinformatics/btw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimin A.V., Puiu D., Luo M.C., Zhu T., Koren S., Marçais G., Yorke J.A., Dvořák J., Salzberg S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017;27:787–792. doi: 10.1101/gr.213405.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 67.Criscuolo A., Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010;10 doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trifinopoulos J., Nguyen L.T., von Haeseler A., Minh B.Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–W235. doi: 10.1093/nar/gkw256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cutler K.J., Stringer C., Lo T.W., Rappez L., Stroustrup N., Brook Peterson S., Wiggins P.A., Mougous J.D. Omnipose: a high-precision morphology-independent solution for bacterial cell segmentation. Nat. Methods. 2022;19:1438–1448. doi: 10.1038/s41592-022-01639-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., Ogata H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2252. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Collins A.J., Murugkar P.P., Dewhirst F.E. Establishing stable binary cultures of symbiotic saccharibacteria from the oral cavity. J. Vis. Exp. 2021 doi: 10.3791/62484. [DOI] [PubMed] [Google Scholar]
- 72.Arkin A.P., Cottingham R.W., Henry C.S., Harris N.L., Stevens R.L., Maslov S., Dehal P., Ware D., Perez F., Canon S., et al. KBase: the United States Department of Energy systems biology KnowledgeBase. Nat. Biotechnol. 2018;36:566–569. doi: 10.1038/nbt.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dehlinger B., Jurss J., Lychuk K., Putonti C. The Dynamic Codon Biaser: calculating prokaryotic codon usage biases. Microb. Genom. 2021;7 doi: 10.1099/mgen.0.000663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall M.P., Unch J., Binkowski B.F., Valley M.P., Butler B.L., Wood M.G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zalacain M., González A., Guerrero M.C., Mattaliano R.J., Malpartida F., Jiménez A. Nucleotide sequence of the hygromycin B phosphotransferase gene from Streptomyces hygroscopicus. Nucleic Acids Res. 1986;14:1565–1581. doi: 10.1093/nar/14.4.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallagher L.A. Methods for TN-seq analysis in Acinetobacter baumannii. Methods Mol. Biol. 2019;1946:115–134. doi: 10.1007/978-1-4939-9118-1_12. [DOI] [PubMed] [Google Scholar]
- 77.Ioerger T.R. Analysis of gene essentiality from TnSeq data using transit. Methods Mol. Biol. 2022;2377:391–421. doi: 10.1007/978-1-0716-1720-5_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anishchenko I., Baek M., Park H., Hiranuma N., Kim D.E., Dauparas J., Mansoor S., Humphreys I.R., Baker D. Protein tertiary structure prediction and refinement using deep learning and Rosetta in CASP14. Proteins. 2021;89:1722–1733. doi: 10.1002/prot.26194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen I.A., Chu K., Palaniappan K., Ratner A., Huang J., Huntemann M., Hajek P., Ritter S., Varghese N., Seshadri R., et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 2021;49:D751–D763. doi: 10.1093/nar/gkaa939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mitchell A.L., Almeida A., Beracochea M., Boland M., Burgin J., Cochrane G., Crusoe M.R., Kale V., Potter S.C., Richardson L.J., et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 2020;48:D570–D578. doi: 10.1093/nar/gkz1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Light gray shaded rows indicate class I essential genes, and dark gray shading indicates class II essential genes. Blue text indicates genes discussed in the text.
Data Availability Statement
-
•
The complete genome sequences of Se ML1, Nl ML1and Ai F0345 have been deposited in GenBank under BioProject PRJNA957798 with accession numbers SAMN34266291, SAMN34266292 and SAMN34266293, respectively. Tn-seq data generated in this study have been deposited in the Sequence Reads Archive (SRA, BioProject PRJNA957798). These datasets are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.