

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nucleocapsid protein (N-protein) increases early in body fluids during infection and has recently been identified as a direct inducer for lung injury. However, the signal mechanism of N-protein in the lung inflammatory response remains poorly understood. The goal of this study was to determine whether RAGE (receptor for advanced glycation endproducts) participated in N-protein–induced acute lung injury. The binding between N-protein and RAGE was examined via assays for protein–protein interaction. To determine the signaling mechanism in vitro, cells were treated with recombinant N-protein and assayed for the activation of the RAGE/MAPK (mitogen-activated protein kinase)/NF-ĸB pathway. RAGE deficiency mice and antagonist were used to study N-protein–induced acute lung injury in vivo. Binding between N-protein and RAGE was confirmed via flow cytometry–based binding assay, surface plasmon resonance, and ELISA. Pull-down and coimmunoprecipitation assays revealed that N-protein bound RAGE via both N-terminal and C-terminal domains. In vitro, N-protein activated the RAGE-ERK1/2–NF-ĸB signaling pathway and induced a proinflammatory response. RAGE deficiency subdued N-protein–induced proinflammatory signaling and response. In vivo, RAGE was upregulated in the BAL and lung tissue after recombinant N-protein insult. RAGE deficiency and small molecule antagonist partially protected mice from N-protein–induced acute lung injury. Our study demonstrated that RAGE is a receptor for N-protein. RAGE is partially responsible for N-protein–induced acute lung injury and has the potential to become a therapeutic target for treating coronavirus disease.
Keywords: SARS-CoV-2, receptor for advanced glycation endproducts, nucleocapsid protein, receptor, acute lung injury
Clinical Relevance
RAGE (receptor for advanced glycation endproducts) is a receptor for the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) N protein. RAGE is partially responsible for N protein–induced acute lung injury and has the potential to become a therapeutic target for treating coronavirus disease (COVID-19).
The coronavirus disease (COVID-19) pandemic, caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in profound public health and socioeconomic consequences around the globe (1). Acute lung injury and acute respiratory distress syndrome (ARDS) are the most serious and frequent complications of COVID-19. The mortality rate of COVID-19 ARDS is estimated to be ∼45%, whereas the incidence of ARDS among COVID-19 nonsurvivors is ∼90% (2). Both classic and COVID-19 ARDS are characterized by increased endothelial permeability, infiltration of inflammatory cells, and elevated proinflammatory cytokines, such as IL-6, IL-8, and TNF-α (3).
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA virus with a genome size of ∼30,000 nucleotides. The virion envelope is anchored with three of the four structural proteins: the spike (S), the membrane (M), and the envelope (E) proteins (4). The fourth, the nucleocapsid protein (N-protein), is located inside of the viral envelope. It packages the RNA genome into a helical ribonucleoprotein complex enclosed within the capsid, which is essential for viral replication and infection (5). The N-protein is composed of two RNA-binding domains, the N-terminal domain (NTD) and the C-terminal domain (CTD), which are connected by a Ser/Arg-rich linker region (LR) (6). Several studies have documented that SARS-CoV-2 N-protein is directly involved in lung inflammation and acute lung injury. Gao and colleagues reported that N-protein aggregated LPS-induced lung injury via binding to MASP-2, leading to complement activation (7). Pan and colleagues found that N-protein boosted NLRP3 inflammasome activation to induce lung inflammation (8). Our results showed that recombinant N-protein induced acute lung injury in mice via NF-ĸB activation (9). However, the upstream mechanism of NF-ĸB activation is unknown.
One of the upstream mediators of NF-ĸB activation is RAGE (receptor for advanced glycation endproducts). RAGE is a transmembrane glycoprotein and belongs to the immunoglobulin superfamily. In addition to binding with advanced glycation endproducts, RAGE is also a receptor for many other ligands, such as LPS, the family of S100 proteins, and HMGB1 (high mobility group box 1) (10). Compared with other tissues, RAGE is most highly expressed in the lung, particularly in the alveolar type I cells, type II cells, and macrophages (11, 12). A potential role of RAGE in the pathogenesis of COVID-19 has been proposed by several groups (13). However, the role of RAGE in N-protein–induced acute lung injury has not been reported. In the present study, we aimed to examine whether RAGE is a receptor for N-protein and determine the consequent impact on acute lung injury of N-protein–RAGE interaction.
Methods
Detailed methods are provided in the data supplement.
Plasmids and Fusion Proteins
N-protein NTD (1–174 AA), LR (175–246 AA), and CTD (247–419 AA) constructs were amplified from a full-length N-protein cDNA via PCR, confirmed via sequencing, and subcloned into the pcDNA3.1-GFP plasmid in frame for mammalian expression. The human RAGE cDNA construct was amplified from a full-length RAGE cDNA via PCR, confirmed via sequencing, and subcloned into the pcDNA3.1-Flag plasmid. The construct for full-length pET28a-His–N-protein for bacterial expression was generously provided by Dr. Feng Cong from Guangdong Laboratory Animals Monitoring Institute. Recombinant His-protein constructs for NTD, LR, and CTD were amplified from an N-protein cDNA via PCR, confirmed via sequencing, and inserted into the pET28a vector. Recombinant RAGE protein was purchased from SinoBiological.
Preparation of Recombinant N-Protein
The proteins were purified from bacterial lysate via nickel affinity chromatography (BBI Life Sciences) and dialyzed to remove extra salt and imidazole. To remove the contaminated endotoxin, the recombinant proteins were passed through a column from ToxinEraser Endotoxin Removal Kit (GenScript). The endotoxin levels were <0.1 EU/μg protein (<0.01 ng/μg) as determined by the ToxinSensor Chromogenic LAL Endotoxin Assay Kit (GenScript). To neutralize the possible protein-bound LPS, recombinant proteins were pretreated with polymyxin B (250 μg/ml, MilliporeSigma) for 1 hour at room temperature before all in vitro and in vivo experiments.
Flow Cytometry-based Binding Assay
Cells were detached by cell scrapers and rinsed with flow cytometry buffer (PBS, 0.5% BSA) to block nonspecific binding. Cells were then incubated with His-tagged full-length N-protein (5 μg/ml) at 37 °C for 1 hour. Cells were washed with flow cytometry buffer three times for 3 × 5 minutes and incubated with an Alexa Fluor 488–conjugated anti-His antibody (Biolegend) diluted 1:100 for 30 minutes. Then, cells were rinsed, detected by a BD LSRFortessa flow cytometer, and analyzed using FlowJo V10 software.
Surface Plasmon Resonance
Surface plasmon resonance was performed using a Biacore 3000 system (Cytiva). N-protein was diluted in PBS to generate a series of concentrations (6.25–400 nM). After immobilizing RAGE at the 100 resonance unit on the surface of a CM5 sensor chip (Cytiva), N-protein samples were injected for 4 minutes across the sensor surface at a flow rate of 20 μl/min. At the end of the sample injection, PBS was flowed over the sensor surface for 4 minutes to promote dissociation. The response was monitored as a function of time at room temperature. Sensorgrams were obtained by subtraction of the zero concentration curve. The binding data were fitted to a 1:1 binding model using BIAevaluation software (Cytiva).
N-Protein–induced Lung Injury Model
C57BL/6 mice were acquired from the Shanghai Laboratory Animal Center. RAGE knockout mice with C57BL/6 background were generated by the Nanjing Biomedical Research Institute of Nanjing University via CRISPR/Cas9 technology (Project number XM709828). Animal experimental protocols were approved by the Institutional Animal Care and Use Committee at Zhejiang University School of Medicine. Mice were anesthetized with an injection of phenobarbital (50 mg/kg) intraperitoneally. Acute lung injury was induced via intratracheal instillation of recombinant full-length N-protein (75 μg per mouse in 50 μl) as reported previously (9). To study the effect of RAGE inhibition on N-protein–induced lung injury, mice were subjected to intraperitoneal injection of RAGE antagonist RAP (50 or 100 μg/mouse) 1 hour before N-protein administration. Based on our experience, the optimal time point for assessing N-protein–induced lung injury was 24 hours after insult. Serum, lung, and BAL samples were harvested at 24 hours from mouse groups (48 h for Figure E1 in the data supplement).
Western Blot
Antibodies for phosphor-ERK1/2 and ERK1/2 were purchased from Abcam. Antibodies for phosphor-JNK1/2, JNK1/2, phosphor-p38, p38, phosphor–NF-ĸB p65, and NF-ĸB p65 were purchased from Cell Signaling Technology. Equal amounts of cell lysates (30 μg) were separated on 12% SDS-PAGE gels, transferred via polyvinylidenefluoride membranes, blocked in TRIS-buffered saline with Tween-20 containing 5% nonfat milk (blot buffer), and probed with the primary antibodies. After washing with blot buffer, membranes were incubated with specific secondary antibodies linked to horseradish peroxidase. Signal detection was conducted using an enzyme-linked chemiluminescence kit. Blot images were analyzed by ImageJ software.
Statistical Methods
Data are presented as mean ± SD. Data normality was examined using the D’Agostino-Pearson Omnibus normality test, the Shapiro-Wilk normality test, or the Kolmogorov-Smirnov test with Dallal-Wilkinson-Lilliefor–corrected P value. Student’s t test, one-way ANOVA with Tukey’s post hoc test, or two-way ANOVA with Tukey’s post hoc test was conducted for parametric values. In data sets with nonparametric values (Figure 2F, and TNF-α of Figure 3B), the Kruskal-Wallis test followed by Dunn’s post hoc test was performed for analysis. Statistical analysis was conducted using the GraphPad Prism 8.0.2. Results were deemed significant if P < 0.05.
Figure 2.
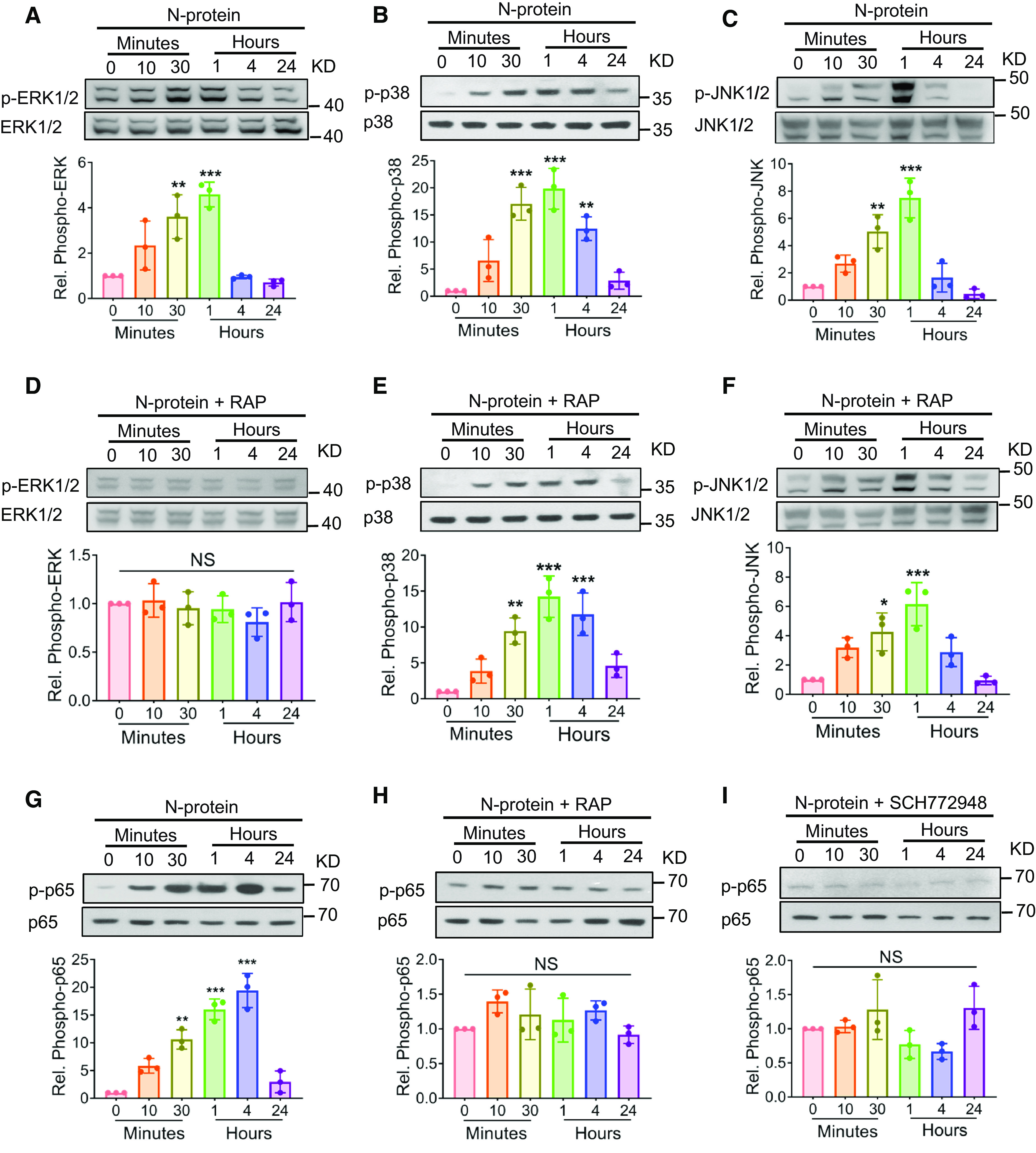
N-protein prompted ERK-mediated NF-ĸB phosphorylation via RAGE. (A–C) RAW 264.7 cells were treated with recombinant full-length N-protein (5 μg/ml) for the indicated times. Cell lysates were analyzed via Western blot using antibodies specific to phosphorylated (upper panels) or total (lower panels) ERK1/2 (A), p38 MAPK (mitogen-activated protein kinase) (B), or JNK1/2 (C). (D–F) RAW 264.7 cells were pretreated with RAGE antagonist (RAP, 20 μM) for 1 hour and then incubated with recombinant full-length N-protein (5 μg/ml) for the indicated times. Cell lysate was examined via Western blot using specific antibodies to detect phosphorylation of ERK1/2 (D), p38 MAPK (E), or JNK1/2 (F). (G and H) RAW 264.7 cells were pretreated without (G) or with (H) RAGE antagonist RAP (20 μM) for 1 hour and then stimulated with recombinant full-length N-protein (5 μg/ml) for the indicated times. Cell lysate was assessed via Western blot using antibodies specific to phospho–NF-ĸB p65 (upper panel) or total NF-ĸB p65 (lower panel). (I) RAW 264.7 cells were pretreated with ERK inhibitor (SCH772948, 500 nM) for 1 hour and then stimulated with recombinant full-length N-protein (5 μg/ml) for the indicated times. Cell lysate was examined via Western blot using antibodies specific to phosphor–NF-ĸB p65 (upper panel) or total NF-ĸB p65 (lower panel). N-protein for all groups was pretreated with polymyxin B (250 μg/ml) for 1 hour at room temperature before all experiments to neutralize endotoxin. The band density of phosphorylation was quantified with ImageJ software, normalized to that of total protein, and expressed as relative fold change compared with time 0. Data are presented as mean ± SD, n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001 versus time 0. One-way ANOVA with Tukey’s post hoc test (A–E, G–I) or Kruskal-Wallis test with Dunn’s post hoc test (F) was used for the analysis. n = number of independent experiments; NS = not significant.
Figure 3.
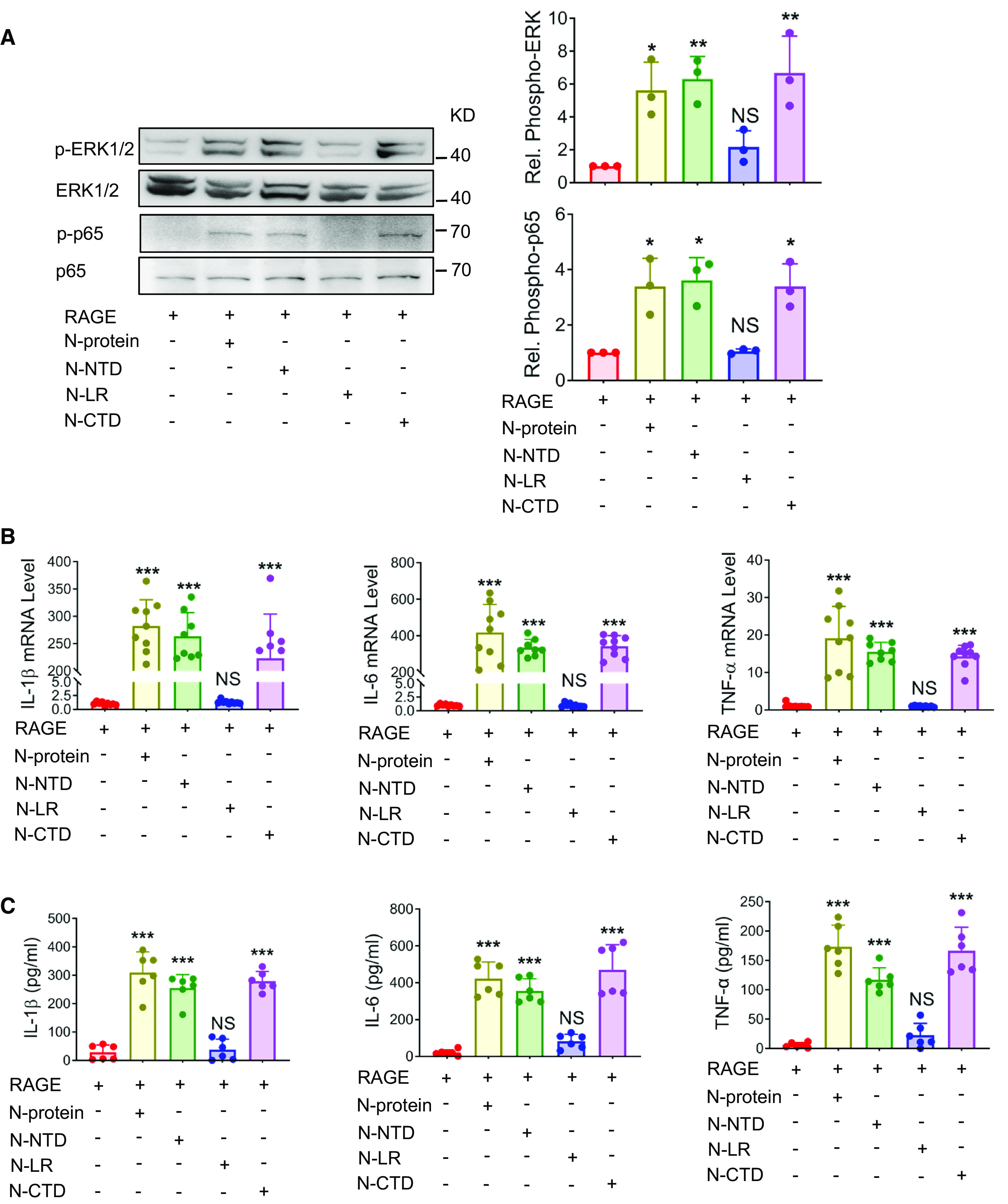
N-NTD and N-CTD induced proinflammatory signaling and immune response. (A) HEK293 cells were transiently transfected with RAGE for 24 hours and treated with recombinant N-proteins expressing full-length, N-NTD, N-CTD, or N-LR (5 μg/ml) for 30 minutes. Cell lysates were analyzed via Western blot using antibodies specific to phosphorylated (upper panels) or total (lower panels) ERK1/2 or NF-ĸB p65. The levels of phosphorylation were quantified with ImageJ software (n = 3). (B and C) HEK293 cells were transiently transfected with RAGE for 24 hours and treated with recombinant proteins expressing full-length, NTD, CTD, or LR of N-protein (5 μg/ml) for 24 hours. (B) The mRNA expression of proinflammatory cytokines IL-1β, IL-6, and TNF-α in HEK293 cells was quantitated by qRT-PCR (n = 8–9). (C) Culture supernatants were analyzed by ELISA for IL-1β, IL-6, and TNF-α (n = 6). Data are presented as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 versus no N-protein treatment. One-way ANOVA with Tukey’s post hoc test (A, B, and C, except TNF-α in B) or Kruskal-Wallis test with Dunn’s post hoc test (TNF-α in B) was used for the analysis.
Results
RAGE Is a Receptor for N-Protein
RAGE is constitutively expressed in A549 human alveolar epithelial cells (14). The first experiment was to test whether recombinant full-length N-protein could bind to the surface of A549 cells using a flow cytometry–based binding assay. Detached cells were incubated with or without His-tagged N-proteins and then analyzed by flow cytometry after incubation with an anti-His antibody. The results suggested that N-protein bound with surface molecules of A549 cells (Figure 1A). To determine whether N-protein could specifically bind with RAGE on the cell surface, bone marrow–derived macrophages (BMDMs) were isolated from RAGE knockout mice with C57BL/6 background (RAGE−/−) and wild-type (WT) C57BL/6 mice. We observed that full-length N-protein bound BMDMs from WT mice, whereas the binding was substantially reduced in BMDMs from RAGE−/− mice (Figure 1B). Surface plasmon resonance analysis demonstrated that N-protein had a strong binding affinity with RAGE, with an equilibrium dissociation constant (Kd) of 34.5 ± 10.6 nM (Figure 1C). It has been documented that the binding between the receptor binding domain of the SARS-CoV-2 S-protein and ACE2 (angiotensin-converting enzyme 2) has a Kd in the range of 6–133 nM (15). To demonstrate that RAGE possesses potent binding with N-protein, N-protein was coated on plates, and the binding of various concentrations of RAGE was assayed via ELISA. The half-maximal effective concentration (EC50) was 3.87 ± 0.57 μg/ml for N-protein (Figure 1D). Lopez and colleagues reported that ACE2 bound with the receptor binding domain of SARS-CoV2 S-protein with a half-maximal effective concentration value of 13.5 μg/ml (16).
Figure 1.
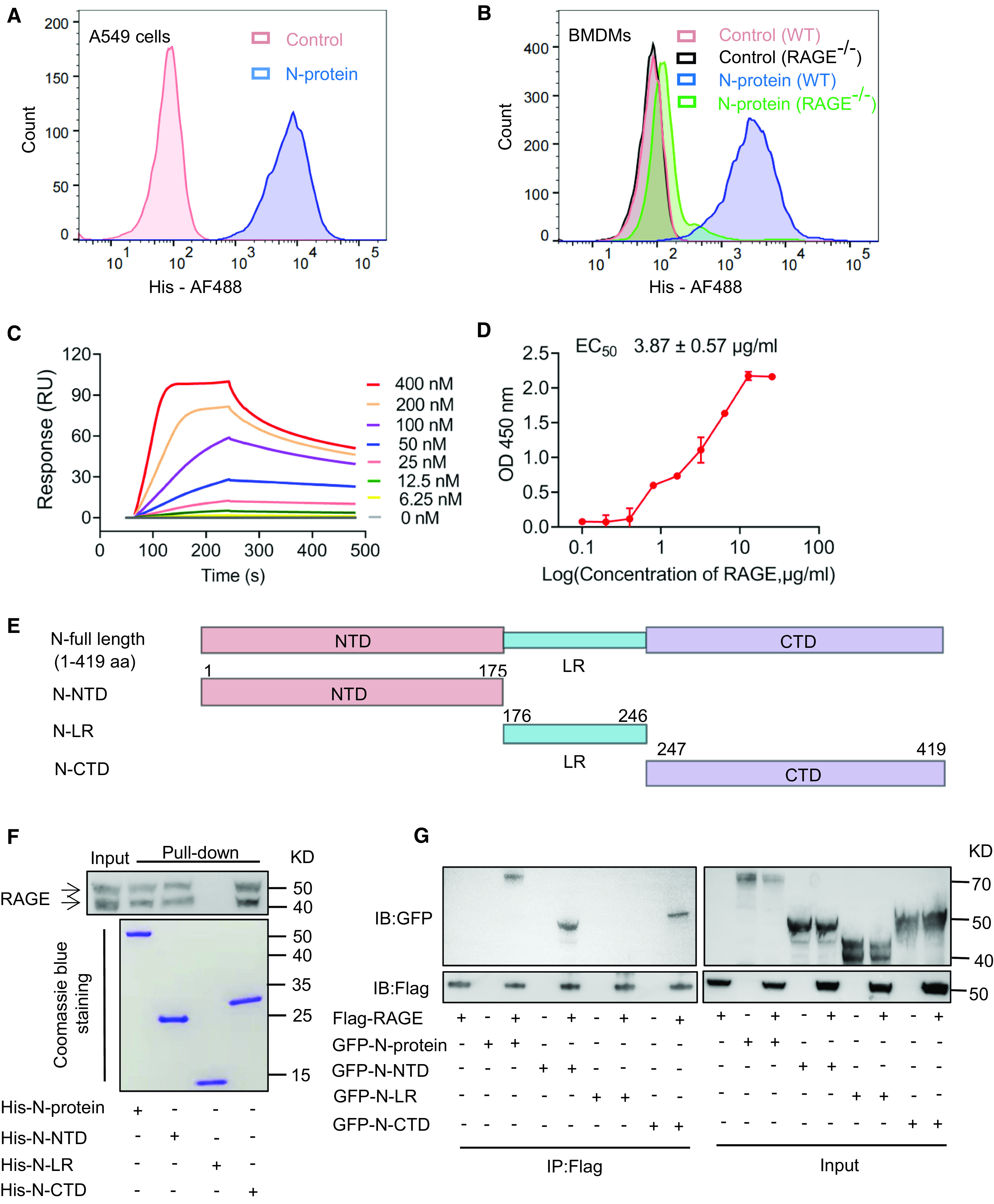
N-protein bound to RAGE (receptor for advanced glycation endproducts). (A) Detached A549 cells were incubated with or without His-tagged full-length N-protein under unpermeablized conditions for 1 hour and washed with PBS + 0.5% BSA three times. Binding of N-protein to A549 cells was measured by flow cytometry using an Alexa Fluor 488–conjugated anti-His antibody. (B) Detached bone marrow–derived macrophages (BMDMs) from RAGE−/− and wild-type (WT) C57BL/6 mice were incubated with or without His-tagged full-length N-protein under unpermeablized conditions for 1 hour and washed with PBS + 0.5% BSA three times. Binding of N-protein to BMDMs was measured by flow cytometry using an Alexa Fluor 488–conjugated anti-His antibody. (C) Surface plasmon resonance sensorgrams show the binding of immobilized RAGE to N-protein at concentrations of 400, 200, 100, 50, 25, 12.5, and 6.25 nM. The data were fitted to the one-to-one kinetic model. (D) Recombinant RAGE of different concentrations (0.1–25.6 μg/ml) was applied to ELISA plates coated with N-protein. Bound RAGE was detected using a rabbit anti-RAGE antibody followed by a goat anti-rabbit secondary antibody conjugated to horseradish peroxidase. After adding substrate, OD was measured at 450 nm and normalized against the negative control. Data are presented as mean ± SD. (E) A schematic diagram shows full-length and truncated N-proteins. (F) Lung lysates were prepared from C57BL/6 mice and pulled down with His-tagged full-length N-protein, N–N-terminal domain (N-NTD), N–C-terminal domain (N-CTD), or N-linker region (N-LR). The lysates and pull-down products were examined via Western blot using an anti-RAGE antibody. The two bands detected in the pull-down assay represent the RAGE protein before and after glycosylation (upper panel). Bait proteins were visualized with Coomassie staining (lower panel). (G) Cell lysates from HEK 293 cells expressing FLAG-RAGE and GFP–N-proteins of full-length, N-NTD, N-CTD, or N-LR were subjected to immunoprecipitation using an anti-FLAG antibody. The cell lysates and immunoprecipitation fractions were examined by Western blot using an anti-GFP or anti-Flag antibody. Data shown are representative of three independent experiments. EC50 = half-maximal effective concentration; OD = optical density; RU = resonance unit.
To provide more evidence for N-protein and RAGE association, His-tagged proteins expressing full-length, NTD (N-NTD), LR (N-LR), and CTD (N-CTD) (Figure 1E) were used in a pull-down assay of mouse lung lysate. Full-length N-protein, N-NTD, and N-CTD were able to pull down a considerable amount of RAGE, as detected by Western blot, whereas N-LR did not show binding activity (Figure 1F). To confirm the physical association of N-protein and RAGE in a cellular context, constructs of GFP–N-protein and Flag-RAGE were expressed in HEK-293 cells either alone or in combination via transient transfection. When cell lysates were immunoprecipitated with an anti-FLAG antibody, the cotransfected full-length N-protein, N-NTD, and N-CTD, but not N-LR, were robustly coimmunoprecipitated (Figure 1G). These results indicate that N-protein has two binding sites for RAGE, one at the CTD and the other at the NTD. Similarly, it has been reported that HMGB1 has two RAGE-binding domains (17).
N-Protein Activates the RAGE-ERK1/2–NF-ĸB Pathway
Previously, our group reported that recombinant N-protein induced lung injury and NF-ĸB activation, independent of endotoxin (9). To investigate the upstream signaling mechanism of NF-ĸB activation, RAW 264.7 macrophages were treated with recombinant full-length N-protein for a series of time points. Cell lysates were examined for the phosphorylation of MAPK (mitogen-activated protein kinase; ERK1/2, p38 MAPK, and JNK1/2) via Western blot analysis. The results showed that phosphorylation of ERK1/2, p38 MAPK, and JNK1/2 was elevated in a time-dependent manner, reaching a peak between 30 minutes and 1 hour (Figures 2A–2C). Pretreatment of cells with RAGE antagonist (RAP, 20 μM) blocked N-protein–induced ERK1/2 activation, indicating that RAGE is upstream of ERK1/2 (Figure 2D). The phosphorylation of p38 MAPK and JNK1/2 evoked by N-protein was unaltered by RAGE antagonist, suggesting that the activation of p38 MAPK and JNK1/2 is independent of RAGE (Figures 2E and 2F). This phenomenon is similar to a previous report that S100B increased the phosphorylation of p38 MAPK and JNK1/2 independent of RAGE (18). Compared with the phosphorylation pattern of MAPK induced by N-protein (Figures 2A–2C), NF-ĸB p65 phosphorylation lasted longer, reaching a peak between 1 and 4 hours (Figure 2G). Similar to that of ERK1/2, N-protein–induced NF-ĸB p65 phosphorylation was blocked by RAGE antagonist (Figure 2H). Furthermore, ERK1/2 inhibition (SCH772948, 500 nM) abolished the effect of N-protein on NF-ĸB activation, suggesting ERK1/2 is upstream of NF-ĸB (Figure 2I). These findings support that N-protein induces signaling through RAGE-dependent activation of the ERK1/2–NF-ĸB pathway.
N-NTD and N-CTD Mimic Full-Length N-Protein in Signaling and Inflammatory Response
It has been reported that HEK 293 cells do not express RAGE or express low levels of RAGE (19). To examine whether N-NTD and N-CTD could promote RAGE-mediated signaling, HEK 293 cells were transfected with Flag-tagged RAGE and stimulated without or with recombinant proteins expressing full-length N-protein, N-NTD, N-CTD, or N-LR (5 μg/ml) for 30 minutes. Without stimulation, the phosphorylation of ERK1/2 and NF-ĸB p65, as detected by Western blot, was almost undetectable. Treatment with full-length N-protein, N-NTD, and N-CTD, but not N-LR, elevated the phosphorylation of ERK1/2 and NF-ĸB p65 (Figure 3A). To study whether N-NTD and N-CTD could enhance the RAGE-mediated inflammatory response, transfected HEK293 cells were stimulated without or with recombinant proteins expressing full-length, N-NTD, N-CTD, or N-LR (5 μg/ml) for 24 hours. Full-length N-protein, N-NTD, and N-CTD significantly elevated both mRNA expression (Figure 3B) and cellular release (Figure 3C) of proinflammatory cytokines IL-1β, IL-6, and TNF-α, as determined by qRT-PCR and ELISA, respectively.
RAGE Deficiency Reduces N-Protein–induced ERK1/2–NF-ĸB Signaling and Proinflammatory Response
To further explore the role of RAGE in the N-protein–induced proinflammatory pathway, BMDMs were isolated from RAGE−/− and WT C57BL/6 mice and treated with recombinant full-length N-protein for different time points. Compared with WT BMDMs, phosphorylation of ERK1/2 and NF-ĸB p65 was abolished in RAGE−/− BMDMs in response to N-protein (Figure 4A). Compared with WT, RAGE deficiency significantly decreased the effect of N-protein on mRNA expression and cellular release of IL-1β, IL-6, and TNF-α at 24 hours (Figure 4B). However, RAGE deficiency did not totally wipe out the proinflammatory effect of N-protein. To further support the role of ERK1/2 in regulation of the N-protein–induced proinflammatory response, WT BMDMs were treated with ERK1/2 inhibitor (SCH772948, 500 nM) before N-protein insult. ERK1/2 inhibition significantly inhibited the mRNA expression and cellular release of proinflammatory cytokines IL-1β, IL-6, and TNF-α at 24 hours (Figure 4C). Taken together, these findings indicate that RAGE-ERK1/2–NF-ĸB pathway is essential in the N-protein–induced proinflammatory response.
Figure 4.
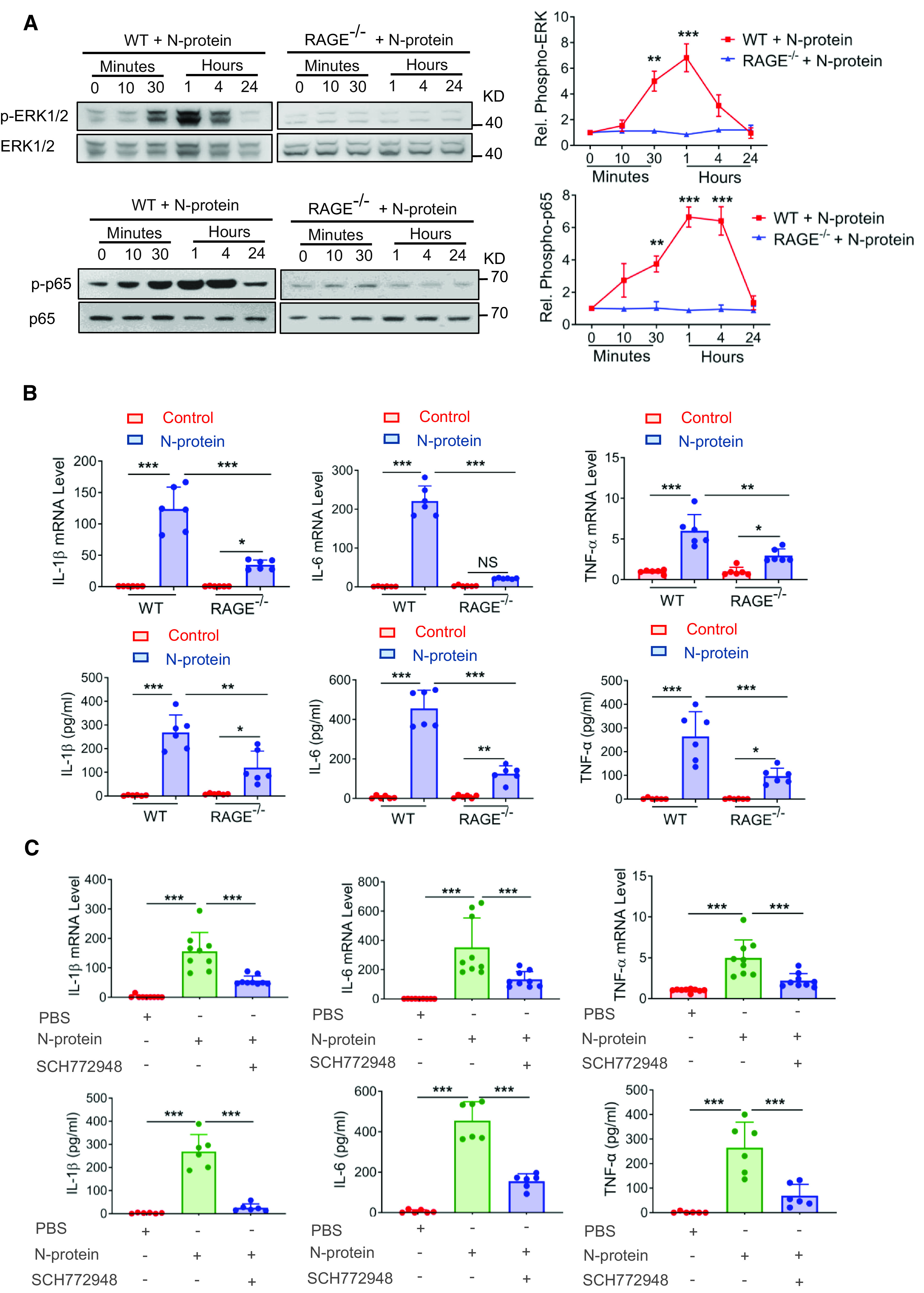
RAGE knockout reduced inflammatory response induced by N-protein in BMDMs. (A) BMDMs from WT and RAGE knockout mice were treated with recombinant full-length N-protein (5 μg/ml) for the indicated times. Cell lysate was analyzed via Western blot using antibodies specific to phosphorylated (upper panels) or total (lower panels) of ERK1/2 or NF-ĸB p65. The levels of phosphorylation were quantified with ImageJ software (n = 3). (B) BMDMs from WT and RAGE knockout mice were treated with N-protein (5 μg/ml) for 24 hours. The mRNA expression of proinflammatory cytokines IL-1β, IL-6, and TNF-α in BMDMs was quantitated by qRT-PCR (upper panel, n = 6). Culture supernatants were analyzed by ELISA for IL-1β, IL-6, and TNF-α (lower panel, n = 6). (C) BMDMs from WT mice were pretreated with ERK1/2 inhibitor (SCH772948, 500 nM) for 1 hour and then stimulated with recombinant N-protein (5 μg/ml) for 24 hours. The mRNA expression of proinflammatory cytokines IL-1β, IL-6, and TNF-α was quantitated by qRT-PCR (upper panel, n = 9). Culture supernatants were analyzed by ELISA for IL-1β, IL-6, and TNF-α (lower panel, n = 6). Data are presented as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. One-way ANOVA with Tukey’s post hoc test versus time 0 (A). Two-way ANOVA with Tukey’s post hoc test (B). One-way ANOVA with Tukey’s post hoc test (C).
RAGE Deficiency and Inhibition Alleviate N-Protein–induced Acute Lung Injury
Previously, our group reported that recombinant N-protein–induced lung injury was dose dependent. Intratracheal administration of a low dose of N-protein (3 μg/mouse) failed to trigger an acute lung injury at 24 hours, whereas a high dose (75 μg/mouse) had a more pronounced effect than a medium dose (15 μg/mouse) (9). To study whether N-protein induces RAGE expression in vivo, WT C57BL/6 mice were treated intratracheally with recombinant full-length N-protein (75 μg/mouse) or PBS. At 24 hours, N-protein insult significantly increased RAGE levels in the BAL and serum relative to the control in the ELISA assay (Figure 5A). The treatment also elevated the total RAGE mRNA levels in BAL cells (Figure 5B). In addition, lungs with N-protein insult showed extensive RAGE reactivity in immunohistochemistry (Figure 5C), indicating that N-protein upregulates both membrane and soluble RAGE.
Figure 5.
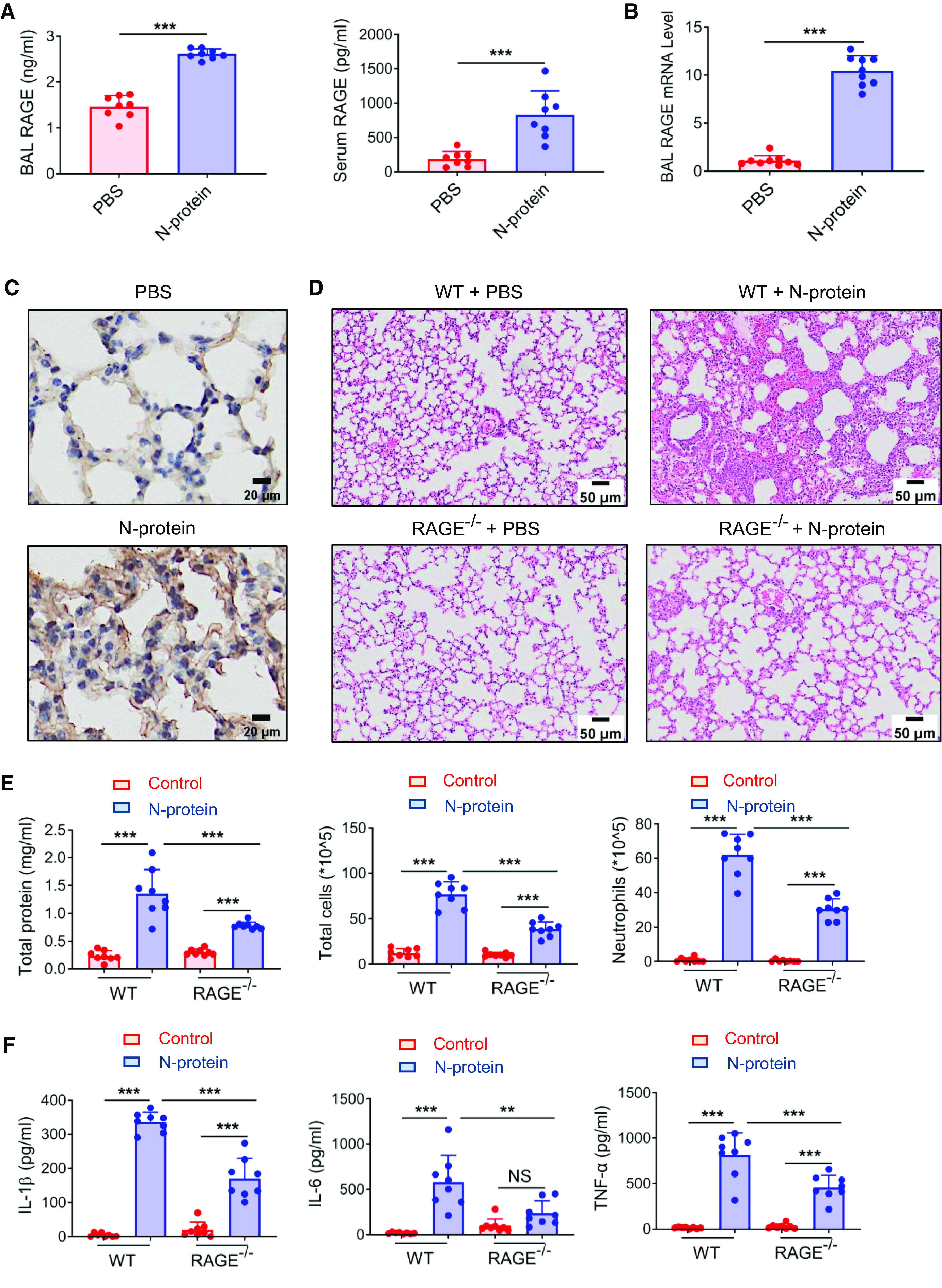
RAGE mediated N-protein–induced acute lung injury. (A–C) WT C57BL/6 mice were administered intratracheally with His-tagged full-length N-protein (75 μg/mouse in 50 μl) or PBS. (A) BAL and serum samples were harvested at 24 hours. RAGE levels in the BAL and serum were determined by ELISA (n = 8). (B) Total RAGE mRNA expression in BAL cells was examined via qRT-PCR (n = 9). (C) RAGE expression in lung tissue was detected via immunohistochemistry (n = 3). Scale bars, 20 μm. (D–F) RAGE−/− mice with C57BL/6 background and WT C57BL/6 mice were treated with PBS or N-protein (75 μg/mouse in 50 μl) intratracheally. Lung tissue and BAL samples were collected at 24 hours after N-protein insult. (D) Tissue sections were stained with hematoxylin and eosin (n = 3). Scale bars, 50 μm. (E) Total protein, total cells, and neutrophils in the BAL were assayed to evaluate lung injury (n = 8). (F) Levels of IL-1β, IL-6, and TNF-α in the BAL were determined via ELISA (n = 8). Data are presented as mean ± SD. **P < 0.01 and ***P < 0.001. Student’s two-tailed t test (A and B). Two-way ANOVA with Tukey’s post hoc test (E and F).
To examine the role of RAGE in N-protein–induced acute lung injury, RAGE−/− and WT C57BL/6 mice were treated intratracheally with recombinant full-length N-protein (75 μg/mouse) or PBS. Lung samples and BAL were harvested at 24 hours. Treatment of N-protein elicited edema and higher cellularity in WT mice compared with control mice. However, inflammatory cell infiltration and septal thickening were remarkably less prominent in treated RAGE−/− mice compared with WT mice (Figure 5D). Total protein levels, the number of total cells, and the number of neutrophils in the BAL were significantly reduced (by 42.4%, 49.5%, and 50.8%, respectively) in treated RAGE−/− versus WT mice (Figure 5E). Proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, in the BAL were substantially decreased (by 49.1%, 59.1%, and 43.7%, respectively) in treated RAGE−/− mice versus WT mice (Figure 5F). However, lung injury was not totally abolished in RAGE−/− mice compared with control mice (Figures 5E and 5F). Reduced lung injury in treated RAGE−/− versus WT mice was still observed at 48 hours after N-protein insult. However, increased protein leakage in BAL was no longer present in treated RAGE−/− mice versus their control mice (Figure E1). These results show that N-protein–induced acute lung injury is partially protected by RAGE deficiency. Using a lentivirus-mediated N-protein expression system, Pan and colleagues reported that N-protein mediated NLRP3 inflammasome activation and prompted hyperinflammation in the lung (8). Intraabdominal administration of the NLRP3 inhibitor MCC950 (10 mg/kg) alleviated lung injury induced by recombinant N-protein (Figure E2), indicating NLRP3 activation is also partially responsible for N-protein–induced acute lung injury.
To determine the therapeutic potential of RAGE inhibition in N-protein–induced lung injury, C57BL/6 mice were treated with recombinant full-length N-protein intratracheally (75 μg/mouse), N-protein + RAGE antagonist RAP (100 μg/mouse intraperitoneally 1 h before N-protein insult), or PBS. RAGE inhibition alleviated the effect of N-protein on lung pathology (Figure 6A). It also significantly reduced total protein concentration (by 51.1%), total cell count (by 49.1%), and neutrophil infiltration (by 51.2%) in the BAL (Figure 6B). Proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, in the BAL were significantly decreased (by 53.1%, 62.6%, and 45.2%, respectively) by RAGE inhibition (Figure 6C). RAGE inhibition with a lower dose of RAP (50 μg/mouse) had a similar effect as the 100-μg dose in alleviating protein leakage in the BAL, while failing to reduce neutrophil infiltration (Figure E3). Several studies have reported that RAGE ligands, such as S100A8 and S100A9, were upregulated in patients with COVID-19 (20). S100A8 and S100A9 were shown to exacerbate lung inflammation via neutrophil accumulation in tuberculosis (21). Our results revealed that N-protein had an effect similar to S100A8 and A9 in inducing acute lung injury (Figure E4). These findings suggest RAGE inhibition might be beneficial in alleviating lung injury from both N-protein and other RAGE agonists in COVID-19.
Figure 6.
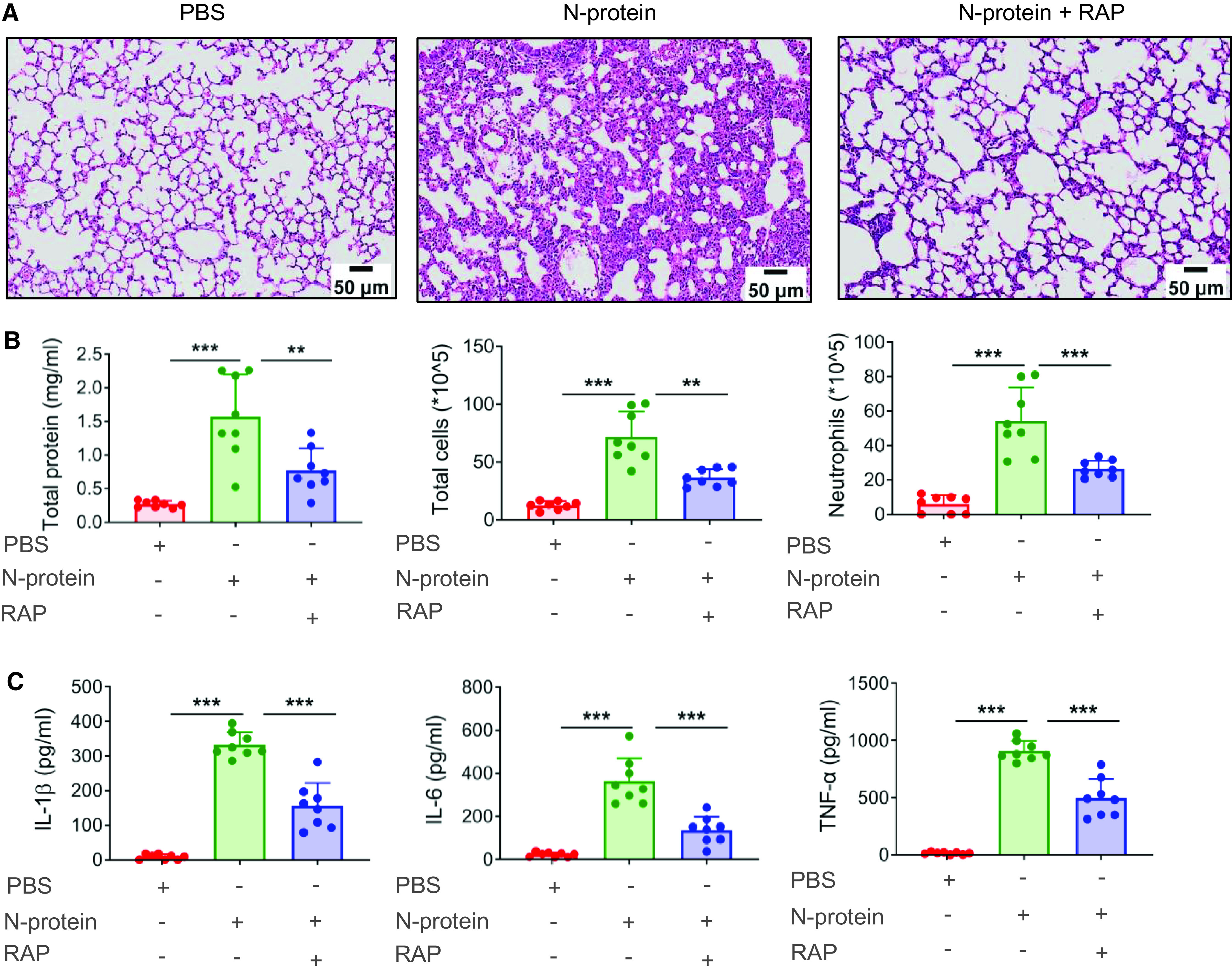
RAGE antagonist reduced N-protein–induced acute lung injury. (A–C) WT C57BL/6 mice were treated with PBS, full-length N-protein (75 μg/mouse), or N-protein + RAGE antagonist RAP (100 μg/mouse, intraperitoneal injection 1 h before N-protein administration). Lung tissue and BAL samples were collected at 24 hours after N-protein insult. (A) Tissue sections were stained with hematoxylin and eosin (n = 3). Scale bars, 50 μm. (B) Total protein, total cells, and neutrophils in the BAL were determined to evaluate lung injury (n = 8). (C) Levels of IL-1β, IL-6, and TNF-α in the BAL were analyzed via ELISA (n = 8). Data are presented as mean ± SD. **P < 0.01 and ***P < 0.001. One-way ANOVA with Tukey’s post hoc test (B and C).
Discussion
It has been well established that the association between SARS-CoV-2 S-protein and ACE2 mediates the viral entry to cells. To our knowledge, this is the first study to report the binding between N-protein and RAGE, linking another SARS-CoV-2 structural protein with a membrane receptor. This conclusion is substantiated by the following findings: 1) there was a direct high-affinity physical association between N-protein and RAGE (Figures 1A–1D); 2) N-protein bound RAGE via both the NTD and the CTD (Figures 1F and 1G); 3) N-protein activated the ERK1/2–NF-ĸB pathway through RAGE (Figure 2); 4) N-NTD and N-CTD mimicked full-length N-protein to induce an inflammatory response (Figure 3); 5) genetic deletion of RAGE significantly decreased N-protein–induced signaling and proinflammatory response in vitro (Figures 4A and 4B); 6) in mice, N-protein treatment elevated RAGE levels in the BAL and the lung (Figures 5A–5C); 7) RAGE deficiency protected against N-protein–induced acute lung injury in mice (Figures 5D–5F); and 8) inhibition of RAGE signaling via antagonist significantly alleviated N-protein–induced acute lung injury (Figure 6).
Previously, our group reported that N-protein induced acute lung injury in mice via activation of NF-ĸB (9). We speculated that a membrane receptor may mediate the binding with N-protein and activation of NF-ĸB. In the preliminary experiments using pull-down and coimmunoprecipitation assays, we found that N-protein was unable to physically associate with Toll-like receptor 4, the receptor for LPS (data not shown). Then, we turned our attention to exploring the binding between N-protein and RAGE. Our results demonstrated that the NTD and CTD of N-protein, but not the LR, bound with RAGE. Similarly, HMGB1 has two RAGE-binding domains, one in the box A domain (23–50 AA) (22) and the other in the box B domain (150–183 AA) (23). It has been documented that both the NTD and the CTD of N-protein possess the ability of RNA binding (24). The presence of two domains with similar functions may help to reinforce the activation of the RAGE-mediated signaling pathway. However, we did not examine the ligand-binding domains for N-protein in RAGE. The mature human RAGE is a glycoprotein with 404 amino acids. It consists of an extracellular domain (1–342 AA), a single transmembrane domain (amino acids 343–363), and a cytoplasmic tail (amino acids 364–404). The extracellular part consists of one variable domain (V) and two constant domains (C1 and C2). The majority of binding studies showed that ligands bind with the V domain of RAGE; however, the C1 domain also participates in the recognition of ligands (25). The molecular mechanism underlying the binding between N-protein and RAGE warrants further investigation.
The association between RAGE and COVID-19 has been reported in the literature. Lim and colleagues showed that serum sRAGE is a biomarker for predicting disease severity of COVID-19 and the need for mechanical ventilation (26). mRNA and protein levels of RAGE ligands, such as S100A8, S100A9, S100A11, S100A12, and S100 P, were significantly higher in the lungs of COVID-19 fatal cases compared with healthy control subjects (27). Serum S100B levels were elevated in patients with COVID-19 and correlated with disease severity (28). In addition, serum levels of S100A8/A9 and HMGB1 at admission had prognostic value for ICU admission and hospital death in patients with COVID-19 (20). Furthermore, Jessop and colleagues found that RAGE antagonist improved the survival of SARS-CoV-2–infected mice and alleviated lung inflammation and perivascular pathology (29). The present study brings new light to the role of RAGE in COVID-19.
Using immunohistochemistry, Massoth and colleagues found that N-protein localized predominantly extracellularly and within the hyaline membrane in a large percentage of patients who died of COVID-19 (3). During SARS-CoV-2 infection, N-protein can be released into extracellular space and blood via several mechanisms. First, N-protein can be naturally secreted from SARS-COV-2–infected cells. When transfected alone in HEK 293 cells, N-protein and S-protein, but not E- and M-proteins, were readily detected in the cell lysate (30). Second, several studies showed that SARS-CoV-2 infection induced cell necrosis, leading to the passive release of virions, viral proteins, and cytoplasmic components into the extracellular space and blood (31). Third, one report showed that extracellular vesicles carrying S-protein were generated by host cells and budded from the plasma membrane, indicating that N-protein might also be released via the same machinery (32). Clinical studies showed that serum N-protein concentration was closely correlated with disease severity and had a peak value from 1 ng/ml to 2.7 μg/ml in patients with COVID-19 (33, 34). The present study revealed that N-protein activated the membrane receptor RAGE on target cells and mediated the inflammatory response, providing the mechanism of N-protein in SARS-CoV-2–induced lung injury.
In the present study, inhibition or knockout of RAGE did not completely abolish the effects of N-protein on acute lung injury, which suggests that N-protein may have other binding partners. It has been reported that N-protein interacted directly with NLRP3 and promoted NLRP3 inflammasome activation to induce an inflammatory response in the lung (8). Here, we showed that N-protein–induced lung injury was partially blocked by the NLRP3 inhibitor MCC950. Another study showed that N-protein bound with Smad3 and enhanced TGF-β/Smad3 signaling to induce acute kidney injury via the G1 cell cycle arrest mechanism (35). Moreover, the interaction between N-protein and α-synuclein accelerated amyloid formation and cell death, which might explain the reported correlation between COVID-19 and Parkinsonism (36).
There are several other findings worth mentioning. In the SDS-PAGE analysis, we noticed that N-NTD migrated slightly more slowly than the predicted 20 kD, whereas the N-CTD (also expected 20 kD) moved even more slowly than NTD (Figure 1D). The same phenomenon was also seen in the report from Nakayama and colleagues (37). This finding suggests that N-NTD and N-CTD have differences in post-translation modification. Second, our study demonstrated that N-protein induced activation of ERK1/2, p38 MAPK, and JNK1/2. However, the activation of p38 MAPK and JNK1/2 was not affected by RAGE inhibition, suggesting that this effect is independent of RAGE. Ishihara and colleagues found that RAGE bound specifically with ERK but not with JNK or p38 MAPK (38). Somensi and colleagues documented that heat shock protein 70 induced activation of ERK1/2, p38 MAPK, and JNK. However, only ERK1/2 activation was blocked by RAGE knockdown (39).
In conclusion, the present study reveals that RAGE is a cell surface receptor for N-protein. N-protein activates the RAGE-ERK1/2–NF-ĸB pathway and promotes the production of proinflammatory cytokines. N-protein–induced acute lung injury is partially mediated by RAGE. Therapeutic interventions targeting RAGE may alleviate cytokine storm and ARDS in COVID-19.
Acknowledgments
Acknowledgment
The authors thank Dr. Feng Cong, Guangdong Laboratory Animals Monitoring Institute, China for the kind gift of the construct of His-tagged N-protein of SARS-CoV-2.
Footnotes
Supported by Natural Science Foundation of Zhejiang Province grant LQY19H190001, Health Commission of Zhejiang Province grant 2021KY188, and National Natural Science Foundation of China grants 82370080, 82070074, and 82272191.
Author Contributions: J. Xia, J.W., Q.S., and J. Xu conceived and designed the experiments. J. Xia, J.W., L.Y., R.H., K.Z., R. Zhang, W.T., Q.X., D.L., Y.Z., Y.H., X.Z., R. Zang, and J.F. performed the experiments. J. Xia, J.W., L.Y., R.H., K.Z., R.Z., W.T., Q.X., D.L., Y.Z., Y.H., X.Z., R.Z., J.F., Q.S., and J. Xu analyzed and interpreted the data. All authors were involved in drafting the manuscript and approved the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0351OC on July 21, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Miller IF, Becker AD, Grenfell BT, Metcalf CJE. Disease and healthcare burden of COVID-19 in the United States. Nat Med . 2020;26:1212–1217. doi: 10.1038/s41591-020-0952-y. [DOI] [PubMed] [Google Scholar]
- 2. Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit Care . 2020;24:516. doi: 10.1186/s13054-020-03240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Massoth LR, Desai N, Szabolcs A, Harris CK, Neyaz A, Crotty R, et al. Comparison of RNA in situ hybridization and immunohistochemistry techniques for the detection and localization of SARS-CoV-2 in human tissues. Am J Surg Pathol . 2021;45:14–24. doi: 10.1097/PAS.0000000000001563. [DOI] [PubMed] [Google Scholar]
- 4. V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol . 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alsaadi EAJ, Jones IM. Membrane binding proteins of coronaviruses. Future Virol . 2019;14:275–286. doi: 10.2217/fvl-2018-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ribeiro-Filho HV, Jara GE, Batista FAH, Schleder GR, Costa Tonoli CC, Soprano AS, et al. Structural dynamics of SARS-CoV-2 nucleocapsid protein induced by RNA binding. PLOS Comput Biol . 2022;18:e1010121. doi: 10.1371/journal.pcbi.1010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao T, Zhu L, Liu H, Zhang X, Wang T, Fu Y, et al. Highly pathogenic coronavirus N protein aggravates inflammation by MASP-2-mediated lectin complement pathway overactivation. Signal Transduct Target Ther . 2022;7:318. doi: 10.1038/s41392-022-01133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun . 2021;12:4664. doi: 10.1038/s41467-021-25015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia J, Tang W, Wang J, Lai D, Xu Q, Huang R, et al. SARS-CoV-2 N protein induces acute lung injury in mice via NF-ĸB activation. Front Immunol . 2021;12:791753. doi: 10.3389/fimmu.2021.791753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pickering RJ, Tikellis C, Rosado CJ, Tsorotes D, Dimitropoulos A, Smith M, et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J Clin Invest . 2019;129:406–421. doi: 10.1172/JCI99987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fehrenbach H, Kasper M, Tschernig T, Shearman MS, Schuh D, Müller M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol . 1998;44:1147–1157. [PubMed] [Google Scholar]
- 12. Katsuoka F, Kawakami Y, Arai T, Imuta H, Fujiwara M, Kanma H, et al. Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem Biophys Res Commun . 1997;238:512–516. doi: 10.1006/bbrc.1997.7263. [DOI] [PubMed] [Google Scholar]
- 13. Chiappalupi S, Salvadori L, Vukasinovic A, Donato R, Sorci G, Riuzzi F. Targeting RAGE to prevent SARS-CoV-2-mediated multiple organ failure: hypotheses and perspectives. Life Sci . 2021;272:119251. doi: 10.1016/j.lfs.2021.119251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pouwels SD, Hesse L, Wu X, Allam VSRR, van Oldeniel D, Bhiekharie LJ, et al. LL-37 and HMGB1 induce alveolar damage and reduce lung tissue regeneration via RAGE. Am J Physiol Lung Cell Mol Physiol . 2021;321:L641–L652. doi: 10.1152/ajplung.00138.2021. [DOI] [PubMed] [Google Scholar]
- 15. Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of common mutations in the SARS-CoV-2 spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife . 2021;10:e70658. doi: 10.7554/eLife.70658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopez E, Haycroft ER, Adair A, Mordant FL, O’Neill MT, Pymm P, et al. Simultaneous evaluation of antibodies that inhibit SARS-CoV-2 variants via multiplex assay. JCI Insight . 2021;6:e150012. doi: 10.1172/jci.insight.150012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H, Liu H, Zeng Q, Imperato GH, Addorisio ME, Li J, et al. Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol Med . 2019;25:13. doi: 10.1186/s10020-019-0081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsoporis JN, Izhar S, Leong-Poi H, Desjardins JF, Huttunen HJ, Parker TG. S100B interaction with the receptor for advanced glycation end products (RAGE): a novel receptor-mediated mechanism for myocyte apoptosis postinfarction. Circ Res . 2010;106:93–101. doi: 10.1161/CIRCRESAHA.109.195834. [DOI] [PubMed] [Google Scholar]
- 19. Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J . 2008;22:3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 20. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol Immunol . 2020;17:992–994. doi: 10.1038/s41423-020-0492-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med . 2013;188:1137–1146. doi: 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. LeBlanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, et al. An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem . 2014;289:7777–7786. doi: 10.1074/jbc.M113.541474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res . 2002;62:4805–4811. [PubMed] [Google Scholar]
- 24. Chauhan A, Avti P, Shekhar N, Prajapat M, Sarma P, Bhattacharyya A, et al. Structural and conformational analysis of SARS CoV 2 N-CTD revealing monomeric and dimeric active sites during the RNA-binding and stabilization: insights towards potential inhibitors for N-CTD. Comput Biol Med . 2021;134:104495. doi: 10.1016/j.compbiomed.2021.104495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci . 2011;36:625–632. doi: 10.1016/j.tibs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 26. Lim A, Radujkovic A, Weigand MA, Merle U. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COVID-19 disease severity and indicator of the need for mechanical ventilation, ARDS and mortality. Ann Intensive Care . 2021;11:50. doi: 10.1186/s13613-021-00836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu M, Chen Y, Xia H, Wang C, Tan CY, Cai X, et al. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc Natl Acad Sci USA . 2020;117:28336–28343. doi: 10.1073/pnas.2018030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aceti A, Margarucci LM, Scaramucci E, Orsini M, Salerno G, Di Sante G, et al. Serum S100B protein as a marker of severity in Covid-19 patients. Sci Rep . 2020;10:18665. doi: 10.1038/s41598-020-75618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jessop F, Schwarz B, Scott D, Roberts LM, Bohrnsen E, Hoidal JR, et al. Impairing RAGE signaling promotes survival and limits disease pathogenesis following SARS-CoV-2 infection in mice. JCI Insight . 2022;7:e155896. doi: 10.1172/jci.insight.155896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Plescia CB, David EA, Patra D, Sengupta R, Amiar S, Su Y, et al. SARS-CoV-2 viral budding and entry can be modeled using BSL-2 level virus-like particles. J Biol Chem . 2021;296:100103. doi: 10.1074/jbc.RA120.016148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morais da Silva M, Lira de Lucena AS, Paiva Júnior SSL, Florêncio De Carvalho VM, Santana de Oliveira PS, da Rosa MM, et al. Cell death mechanisms involved in cell injury caused by SARS-CoV-2. Rev Med Virol . 2022;32:e2292. doi: 10.1002/rmv.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verta R, Grange C, Skovronova R, Tanzi A, Peruzzi L, Deregibus MC, et al. Generation of spike-extracellular vesicles (S-EVs) as a tool to mimic SARS-CoV-2 interaction with host cells. Cells. 2022;11:146. doi: 10.3390/cells11010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Ong CM, Yun C, Mo W, Whitman JD, Lynch KL, et al. Diagnostic value of nucleocapsid protein in blood for SARS-CoV-2 infection. Clin Chem . 2021;68:240–248. doi: 10.1093/clinchem/hvab148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollock NR, Savage TJ, Wardell H, Lee RA, Mathew A, Stengelin M, et al. Correlation of SARS-CoV-2 nucleocapsid antigen and RNA concentrations in nasopharyngeal samples from children and adults using an ultrasensitive and quantitative antigen assay. J Clin Microbiol . 2021;59:e03077-20. doi: 10.1128/JCM.03077-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang W, Chen J, Hu D, Pan P, Liang L, Wu W, et al. SARS-CoV-2 N protein induces acute kidney injury via Smad3-dependent G1 cell cycle arrest mechanism. Adv Sci (Weinh) . 2022;9:e2103248. doi: 10.1002/advs.202103248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Semerdzhiev SA, Fakhree MAA, Segers-Nolten I, Blum C, Claessens MMAE. Interactions between SARS-CoV-2 N-protein and α-synuclein accelerate amyloid formation. ACS Chem Neurosci . 2022;13:143–150. doi: 10.1021/acschemneuro.1c00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakayama EE, Kubota-Koketsu R, Sasaki T, Suzuki K, Uno K, Shimizu J, et al. Anti-nucleocapsid antibodies enhance the production of IL-6 induced by SARS-CoV-2 N protein. Sci Rep . 2022;12:8108. doi: 10.1038/s41598-022-12252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003;550:107–113. doi: 10.1016/s0014-5793(03)00846-9. [DOI] [PubMed] [Google Scholar]
- 39. Somensi N, Brum PO, de Miranda Ramos V, Gasparotto J, Zanotto-Filho A, Rostirolla DC, et al. Extracellular HSP70 activates ERK1/2, NF-kB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell Physiol Biochem . 2017;42:2507–2522. doi: 10.1159/000480213. [DOI] [PubMed] [Google Scholar]