

Abstract

In this work, we report the scalable and modular synthesis of a library of 55 monomeric and dimeric flavonoids including 14 8,8′-biflavones. The sterically demanding tetra-ortho-substituted axis of an acetophenone dimer key intermediate was constructed in a regioselective manner using Fe-mediated oxidative coupling. This step was systematically optimized and performed on up to multigram scale. The biological activities of this compound library were evaluated, including cytotoxicity against healthy and malignant human cell lines, antimicrobial activity against the apicomplexan parasite Toxoplasma gondii, and antioxidant capacity. A marked increase in activity for the 8,8′-dimeric structures compared to that of their monomeric counterparts was observed. Several biflavones were identified with high selectivity indices (low cytotoxicity and high antiprotozoal activity), showing that this class of natural products may serve as lead structures for further investigations.
Introduction
Biflavones are naturally occurring biaryl-based compounds present among a range of gymnosperms.1−4 First isolated in 1966 from Cupressus torulosa where its name is derived from,1 the naturally occurring 8,8′-biflavone cupressuflavone (CUF) was subsequently also isolated from plants of the genus Araucaria cunninghamii.5 So far, very few structural modifications with the exception of glycosylations have been observed in nature.6 While biological activities of non-C2-symmetrical biflavones are reported comprehensively in the literature (Figure 1A),7−13 8,8′-biflavones are a more elusive subclass of flavonoid natural products. CUF has thus far only been investigated in some limited capacity regarding pharmacological effects.14−19
Figure 1.

Biflavones as bioactive natural products with 8,8′-biflavones as a subclass.
In many cases, dimeric natural products show higher activities than their monomeric counterparts.20−22 Apigenin—the monomeric unit of CUF—exerts similar activities in some cases;19 in others, they are exclusively found for CUF,23 highlighting the importance of comparing the activity of dimers to monomers. Additionally, 8,8′-biflavones can also, in some cases, be thought of as structurally simplified analogues of polyketides such as gonytolid A as was proposed and successfully shown by Kikuchi et al. in their synthesis of Me,Me′-CUF (Figure 1B).24 This exemplifies a key strategy in the natural product-inspired drug design by simplifying complex natural structures to leverage the ease of synthesis but retain bioactivity.25 Further, the merging of spherical shapes of ortho-substituted-biaryl-based natural products26 with a biologically relevant flavone scaffold27 qualifies this compound class as potent drug candidates.
Total syntheses of CUF are established since the late 1960s via different methods.28−34 However, only singular examples of 8,8′-biflavones were synthesized to this date, showing the lack of a systematic and diversity-oriented approach (Figure 1B). The construction of these sterically demanding tetra-ortho-substituted biaryls poses a synthetic challenge.35,36 Syntheses of these biaryl-based natural products often suffer from a narrow substrate scope involving the coupling of highly functionalized monomers37,38 or involve multistep functionalization or coupling under harsh reaction conditions.39−42 We herein report the first modular synthesis of a dedicated library of 8,8′-biflavones (Figure 1C) and the systematic investigation of their biological activity.
Results and Discussion
To ensure scalability, which is highly desirable in natural product synthesis,43 a synthesis route was chosen that allowed for the diversification of target structures and isolation by recrystallization. We chose the protocol developed by Li et al. as the basis for our investigations.30 Starting from commercially available phenols 1a and 1b, all monomeric and dimeric target compounds should be accessible. After acylation to acetophenones 2, the key intermediates 3 and 4 should be accessible: while acetophenones 3a and 3b will be available based on the modified literature-known procedures (Scheme 1),30,44,45 the acetophenone dimers 4a and 4b were to be synthesized by the oxidative coupling without the involvement of toxicologically relevant transition metals. Finally, the Claisen–Schmidt condensation using aldehydes 5 provides chalcones 6 and bichalcones 7. Subsequent I2-catalyzed oxidation to synthesize flavones 8 and biflavones 9 was to be conducted. A variety of aldehydes were chosen to obtain the natural substitution pattern of CUF in addition to various more electron-rich and electron-deficient 8,8′-biflavones. Overall, a multigram scale synthesis route for the key intermediates 4 is outlined that avoids the use of column purification.
Scheme 1. Retrosynthesis of Flavones and Biflavones Starting from Commercially Available Phenols 1 via the Key Intermediates 4.

We initiated our studies with the synthesis of the acetophenone starting materials 3a and 3b, employed for both the oxidative coupling and the synthesis of the monomers. Acetylation and subsequent selective monomethylation of commercially available phenols 1a and 1b proceeded smoothly on decagram-scale, with yields of 74% (3a) and 70% (3b) over two steps, respectively (Scheme 2).
Scheme 2. Synthesis of Acetophenones 3a and 3b via Acetylation and Subsequent Methylation.

Starting from acetophenones 3a and 3b, the corresponding chalcone monomers 6 were synthesized via the typical Claisen–Schmidt condensations under alkaline conditions in ethanol with seven different benzaldehyde derivatives 5a–g (Scheme 2). In some cases, conversions were incomplete. This was mitigated by the addition of an additional 0.6 equiv of the corresponding aldehyde after 6 h, which led to full conversion overnight. With the synthesis of the chalcones 6 in place, we continued with the synthesis of the target flavone monomers 8. Following literature-known conditions, the oxidative cyclization using catalytic amounts of I2 in DMSO proceeded smoothly.46 All flavones were obtained in yields ranging from 36 to 93%. Thus, starting from phenols 1a and 1b, the procedure yielded 14 monomeric flavones 8aa–g and 8ba–g in four steps, requiring only a single column chromatographic purification step each (Scheme 3).
Scheme 3. Synthesis of Flavones 8.

Yields of isolated products; [a] with 1.8 equiv of aldehyde 5 and [b] at 90 °C.
With a satisfactory synthesis of the monomeric flavones 8 in place, we directed our efforts toward the synthesis of acetophenone dimers 4a and 4b to ultimately provide the target biflavones 9. The oxidative coupling of naphthol using FeCl3 to form BINOL is known since the 1870s47 and was later improved upon by solid-phase synthesis.48 The first application of FeCl3 on silica (FeCl3/SiO2) was the elimination of water from alcohols.49 Jempty et al. conducted the first oxidative coupling of arenes using FeCl3/SiO2.50 In their total synthesis, Li et al. used silica-bound FeCl3 to synthesize hexamethyl-CUF,30 in addition to the application of this method in the subsequent total syntheses of a variety of natural products.24,40,51,52 While naphthol is well-known to form radicals in the ortho position to the hydroxy-group in the presence of oxidative transition metals,48 phenols tend to form less localized radicals. The regioselectivity of Fe-based oxidative homocouplings has to the best of our knowledge hardly been investigated.53−58
We next directed our efforts toward the oxidative coupling of acetophenone 3a. A variety of recent catalytic methods were evaluated for the synthesis of 4a and 4b (Table S7). After extensive efforts, we concluded that the use of stoichiometric amounts of oxidant is crucial for the conversion of 2′-hydroxy acetophenones 3a and 3b. As such, the use of classic oxidative systems such as FeCl3/SiO2 as was shown by Li et al. seemed more promising.30 FeCl3/SiO2 was prepared using Et2O as a solvent at 40 °C. When conducting solid-phase oxidative couplings using FeCl3·6H2O on SiO2 (50% w/w), we observed the formation of small amounts of product 4a. However, we also noted a major side product that was subsequently identified as chlorinated acetophenone 10a among other minor chlorinated side products 10. This regioselective formation of the chlorinated side product has already been reported in the literature.30
To address this lack of chemoselectivity, we first conducted a full factorial screening59 regarding the %weight/weight (%w/w) composition of FeCl3/SiO2, equivalents, and reaction time (Table S5). More equivalents of FeCl3·6H2O gave a higher conversion of acetophenone 3a. A higher %w/w FeCl3·6H2O/SiO2 resulted not only in an increase in the conversion of 3a but also in a significant increase in the side product formation 10a. A longer reaction time resulted in a higher conversion of acetophenone 3a but an increase in the chlorinated side product formation 10a (Table S6).
With these assessments in place, we resumed our preparative screening efforts. The use of methanol as a cosolvent during the FeCl3/SiO2 preparation did not increase selectivity (procedure II, Table 1 entry 2). To our delight, the use of anhydrous FeCl3 instead of FeCl3·6H2O in combination with anhydrous solvents resulted in the formation of less chlorinated side product (preparation III, Table 1 entry 3). The use of anhydrous solvents and by effect the method of FeCl3/SiO2 preparation proved crucial in increasing the selectivity as the use of nonanhydrous solvents gave lower selectivity (preparation IV, Table 1 entry 4). Again, we observed an increase in the formation of side product 10a over time. FeCl3 may deteriorate over time, and thus a fast reaction may be beneficial for the selective conversion of the starting material. We also considered that the exposure to moisture might have deteriorated the FeCl3·6H2O. FeCl3/SiO2 prepared under otherwise anhydrous conditions using the newly purchased FeCl3·6H2O gave better conversion and selectivity (procedure V, Table 1 entry 4) albeit still with significant side product formation. Ultimately, 4.8 equiv of anhydrous FeCl3 with 50% w/w of SiO2 in combination with anhydrous solvents resulted in the most selective conversion to acetophenone dimer 4a in 2.5 h with a conversion of >95% (according to 1H NMR) and an isolated yield of 58%. We obtained the complementary acetophenone dimer 4b in a yield of 52% on a scale of 8 g, further underlining the utility of this reaction. We attribute the remaining mass balance to polymerization side products. This is in accordance with the previous observation of a fast reaction being more selective, whereas a long reaction time resulted in complex reaction mixtures and low yields. With the optimized protocol established, the sterically demanding products could be obtained without protecting groups and without selective functionalization of the monomeric acetophenones 3. We were able to adapt the protocol by Li et al.30 to synthesize acetophenone 4a via this route and synthesize 4b accordingly, lowering the literature-reported reaction time significantly. The X-ray structure of the acetophenone dimer 4a supports the regioselectivity of the coupling in addition to the NMR 2D data (Figure 1 and Figures S71−S73).
Table 1. Oxidative Coupling of Acetophenones 3a and 3b by Solid-Phase Synthesis, with the Main Side Product 10a Formation under Varying Conditions for the Preparation of FeCl3/SiO2.

| entry | 4 | FeCl3/SiO2 | conversiona [%] | 10a/c [%] | yieldb [%] |
|---|---|---|---|---|---|
| 1 | 4a | I | 21 | 45 | |
| 2 | 4a | II | 6 | 94 | |
| 3 | 4a | III | 76 | 18 | 39 |
| 4 | 4a | IV | 50 | 7 | 23 |
| 5 | 4a | V | 51 | 43 | |
| 6c | 4a | III | 88 | 9 | 58 |
| 7c | 4b | I | 81 | 4 | 52 |
Conversion to main product 4a/4b relative to 10 and 3 according to 1H NMR.
Isolated yield.
8 g scale, improved workup; I: FeCl3/SiO2 prepared at 40 °C in Et2O using FeCl3·6H2O; II: FeCl3/SiO2 prepared at 40 °C in Et2O/MeOH (9:1) using FeCl3·6H2O; III: FeCl3/SiO2 prepared at 40 °C, then 60 °C in anhydr. Et2O/MeOH (9:1) using FeCl3; IV: FeCl3/SiO2 prepared at 40 °C, then 60 °C in (nonanhydr.) Et2O/MeOH (9:1) using FeCl3, V: FeCl3/SiO2 prepared at 40 °C, then 60 °C in anhydr. Et2O/MeOH (9:1) using newly purchased FeCl3·6H2O (refer to Supporting Information Table S4).
With a scalable and robust synthesis of the acetophenone dimers in hand, we commenced the synthesis of the biflavone library (Scheme 4). It is noteworthy that again for some reactions, under the appropriately modified conditions of the chalcone 6 synthesis, the addition of a further 1.2 equiv of aldehyde after 6 h gave full conversion of acetophenone 4 (2.4–3.6 equiv aldehyde 5 in total). The use of an ionic liquid as a solvent to address low solubility gave chalcone dimer 7ac in comparable yield but appeared less generally applicable (Table S7). During the reaction, a complex mixture of side products and intermediates could form and as such the isolation of these highly insoluble compounds proved difficult. However, scaleup and purification by recrystallization gave chalcone dimers 7 in acceptable yield and purity. Only for 7bf and 7bg, the isolation of bichalcone in acceptable purity proved to not be possible. These compounds were obtained as a mixture of bichalcone and flavanone side products and significant amounts of an additional side product, presumably the monoaddition product.
Scheme 4. Synthesis of a Library of Bichalcones 8 and Biflavones 9.

Yields of isolated products: [a] with 3.6 equiv of aldehyde 5; [b] at 90 °C; [c] presumed mixture of chalcone and flavanones and monoaddition product; [d] 1 h reaction time; and [e] complex mixture according to 1H NMR.
With a scalable synthesis of bichalcones 7 with yields of up to 94% on a 0.2–1 g scale established, we proceeded with the synthesis of biflavones 9. Modified conditions of the monomer synthesis were successfully transferred to the chalcone dimers. Only biflavone 9bf was not obtained under the given conditions attributed to the difficulty in isolation of chalcone 7bf in sufficient purity. Overall, we were able to obtain a library of 13 8,8′-biflavones with up to 38% yield (9ae) over five steps in a scalable fashion on up to 500 mg scale in the final step, with only a single column chromatographic purification over the full synthesis route.
Biological Data
With a dedicated library at hand, we initiated the biological evaluation of flavones and biflavones (Figure 2). Bioactivity assays against Toxoplasma gondii (T. gondii) proliferation and the viability of healthy and malignant human cell lines were performed. Overall, this allowed for a systematic comparison of the natural product 8,8-biflavone analogues 9 with the corresponding monomers 8.
Figure 2.

Structure of flavones 8 and biflavones 9 and the associated library evaluated regarding their bioactivity against T. gondii proliferation and against human cell lines.
T. gondii, the causative agent of toxoplasmosis, is an obligate intracellular protozoan parasite member of the phylum Apicomplexa.60 It is referred to as one of the most successful parasites due to its ability to infect and persist in virtually all warm-blooded animals as intermediate hosts, including humans.61 According to the World Health Organization (WHO), it is estimated that up to a third of the world′s human population is infected with this parasite.62 This unmet medical need has yet to be resolved. As biflavones such as the structurally related 3′,8″-biflavone amentoflavone are known to exert activity against kinetoplastid parasites, we wished to investigate the activity of our compound library (Table 2 and Figure 3).13
Table 2. IC(50 HeLa), IC(50 T.gondii), and IC(50 Hs27) and Selectivity Index (SI) of Flavones (8) and Biflavones (9) against T. gondii Proliferationa.

SI ≥ 50 (Highlighted in Gray). [a] SI = (IC(50 Hs27))/(IC(50 T. gondii)). For Confidence Intervals of IC50, refer to Tables S1 and S2
Figure 3.
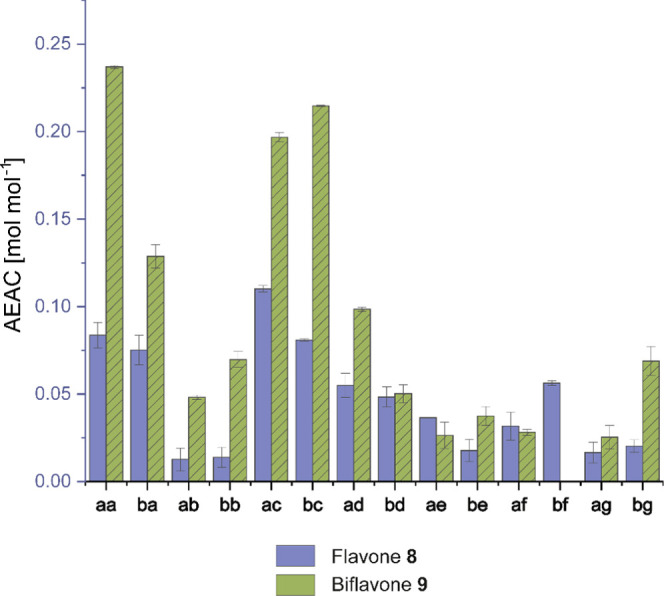
Ascorbic acid equivalent antioxidant capacity (AEAC) of flavones 8 and biflavones 9 relative to ascorbic acid (mol mol–1) determined by the ABTS decolorization assay with standard deviation.63 For absolute values, refer to Table S3.
First, the cytotoxicity of this compound library was assessed by in vitro screening against Hs27 fibroblasts used as hosts in the T. gondii proliferation assay. Comparison of the activities against T. gondii proliferation revealed that in many cases biflavones 9 exerted higher bioactivity (lower IC50) than their respective flavone counterpart 8 (Table 2). Overall, biflavone 9ac was the most active compound regarding the inhibition of T. gondii proliferation with an IC50 of 1.6 μM. By dividing the cytotoxicity (IC50(Hs27) value against fibroblasts; Table S1) by the IC50 (T. gondii) value (T. gondii proliferation inhibition), we obtained the selectivity index (SI) for each tested compound, giving us a measure of the therapeutic potential (Table 2). Of the seven compounds with the highest SI, six are biflavones, one of which is hexa-O-methyl-CUF (9ba). Five of these most active non-natural analogues have higher SIs than hexa-O-methyl-CUF (9ba).
Next, the cytotoxicity toward malignant human cells [HeLa cells (IC50 (HeLa))] was assessed (Table 2). Broadly speaking, biflavones 9 again display higher activities compared to flavones 8, the most active compound again being biflavone 9ac. Compounds with 4-CF3 substituents 8ad, 8bd, 9ad, and 9bd appeared to be noncytotoxic against either human cell line while exhibiting moderate to good activity against T. gondii proliferation. 4-NMe2-substituted biflavone 9ac exerted the highest bioactivity (IC50 (HeLa) = 1.4 μM and IC50 (T. gondii) = 1.58 μM) as well as the highest SI (113.4) of all tested compounds (Figure 4). Additionally, biflavone 9be exerts high activity against T. gondii while exhibiting little activity against HeLa cells (IC50 (Hs27) > 200 μM, IC50 (HeLa) 96.1 μM, IC50 (T. gondii) 2.3 μM). Biflavone 9ag exerts moderate activity against HeLa cells, while no cytotoxicity and little activity against T. gondii is observed (IC50 (Hs27) > 200 μM, IC50 (HeLa) 18.5 μM, IC50 (T. gondii) 39.6 μM). Overall, all tested compounds and particularly biflavones 9 that show moderate to high cytotoxicity against malignant HeLa cells exert little to no cytotoxicity against healthy fibroblasts Hs27.
Figure 4.

Most overall active compound biflavone 9ac.
Additionally, electron-rich arenes such as flavones are known to exhibit antioxidant properties.64 Therefore, we assessed the ascorbic acid equivalent antioxidant capacity (AEAC) of our dedicated library using an ABTS decolorization assay.63 We found that electron-rich biflavone 9aa (0.25 equiv ascorbic acid [mol mol–1]) exerts the highest antioxidant activity with similarly electron-rich 9bc, 9ac, and 9ba following suit (Figure 3).
To show the usefulness of our synthesis route, we scaled-up the reaction to gram-scale. Biflavone 9ac was chosen as it exerted the lowest IC50 regarding HeLa cells while also having one of the highest selectivity indices (SI) toward T. gondii proliferation inhibition. We found that slightly lower yields were obtained, which we attribute to the necessity for an intermediate workup as no further conversion was observed after adding additional 1.2 equiv of aldehyde 5c. Following the procedure of the chalcone synthesis, we performed the flavone synthesis on a 575 mg scale. The pure biflavone was isolated in a yield of 21%. This scaleup allowed for additional modification of the biflavone by cleavage of the methoxy groups using BCl3 (Scheme 5). This was to showcase the potential for further functionalization. The obtained biflavone 11 was isolated in a 44% yield and showed no biological activity (IC50 (HeLa) > 100 μM, IC50 (T. gondii) > 50 μM, IC50 (Hs27) > 200 μM) and less antioxidant capacity (0.037 equiv (11) vs 0.197 equiv (9ac) ascorbic acid (mol mol–1)) compared to the protected biflavones 9ac.
Scheme 5. Selective Deprotection of the Methoxy Groups of Biflavone 9ac for the Synthesis of Biflavone 11. Yield of Isolated Product.

Conclusions
In summary, we were able to establish a scalable synthesis of a library of non-natural 8,8′-biflavones. The lack of a diversity-oriented approach for the synthesis of 8,8′-biflavones was addressed, and synthesis protocols were set in place that allow for further investigations. To provide the necessary acetophenone dimer key intermediates, we investigated a one-step oxidative coupling procedure. By doing so, we were able to overcome chemoselectivity issues by investigating the chlorinated side product formation. The reaction was then performed on a scale of up to 8 g without the need for column chromatographic isolation. Using our established protocols, we managed to synthesize a library of 14 flavones and 13 biflavones. Further, we were able to show the antiproliferative biological activity of the said library against T. gondii as well as selective cytotoxicity against malignant human cell lines (HeLa) and antioxidant capacity. Most biflavones 9 were more active than their respective monomeric flavone counterparts 8. The most potent biflavone 9ac showed the strongest activity against both T. gondii proliferation and HeLa viability while exerting low cytotoxicity against healthy fibroblasts (SI = 113.4, IC50 (T. gondii) = 1.6 μM, IC50 (HeLa) = 1.4 μM), making it a lead structure in further studies. The synthesis of this compound was scaled up to 500 mg scale to show the viability of our approach. While this study highlights the potential use of flavones and biflavones as novel anti-Toxoplasma agents, further research is needed to assess their efficacy and safety in humans. The modes of action are undergoing further investigations.
Experimental Section
Experimental Synthesis Procedures
General Information
All chemicals not synthesized or present in the group were purchased from Sigma-Aldrich Co., Alfa Aesar GmbH & Co. KG, Merck KGaA, or Fluorochem Ltd. All reactants were used without any further purification unless stated otherwise. Methanol was dried using the activated molecular sieve (3 Å). DMSO was degassed via freeze–pump–thaw. Anhyd. diethyl ether and dichloromethane were taken from the solvent purifier MB SPS-800 by MBraun. Silica gel 60 (0.040–0.063 mm, 230–400 mesh) by Merck used for synthesis was dried in an oven at 110 °C overnight. Anhyd. solid reagents were stored in a desiccator under an atmosphere of N2. All glassware and stirring bars used for reactions under anhyd. or inert conditions were put in an oven at 110 °C for at least 12 h. When removing glassware from the oven, it was sealed airtight using septa and stopcocks. It was then attached to a Schlenk line and left to cool under nitrogen gas, which was itself dried over SICAPENT, for several minutes. Glassware was then dried using the Schlenk technique by heating the glassware under vacuum for several minutes and then letting it cool to room temperature under a N2 flow. This process was repeated three times. Septa were only opened briefly during the addition of reactants under a N2 countercurrent. Liquid reactants, solvents, and solutions of reactants were transferred using syringes flushed three times with N2. Solvents were removed using rotary evaporators at a bath temperature of 40 °C under reduced pressure. Analytical balance AE 163 by Mettler Toledo was used to determine and weigh yields and reactants. Sonication of reactions was conducted using an ultrasonic cleaning bath T310 by Elma Schmidbauer GmbH. Distillations of liquid aldehydes were conducted using Kugelrohrofen Glass Oven B-580 and Glass Oven B-585 by Büchi under reduced pressure. For thin-layer chromatography (TLC), silica gel plates (Polygram SIL G/UV 254) by Machery-Nagel with a fluorescent indicator were used. Spots were made visible under UV light, using aqueous potassium permanganate solution, CAM, Molydip, anisaldehyde. Column chromatographic purification was conducted using the appropriate solvent mixture and silica gel 60 (0.040–0.063 mm, 230–400 mesh) by Merck in cylindric glass columns by applying pressure with compressed air. IR spectra were recorded using a SpectrumTwo FT-IR by PerkinElmer with attenuated total reflection (ATR). The absorption bands were given in units of wave numbers (cm–1). 1H-, 13C-, 135DEPT-, COSY-, HSQC-, and HMBC-NMR spectra were measured on the spectrometer Bruker Avance/DRX 600 at a frequency of 600 MHz (1H) and 151 MHz (13C). 19F- and select 1H- and 13C NMR spectra were measured on the spectrometer Bruker Avance/DRX 300 at a frequency of 282 MHz (19F), 300 MHz (1H), and 75 MHz (13C). Deuterated chloroform with 0.03 vol % of TMS (CDCl3) or deuterated d6-DMSO was used as a solvent. The 1H- and 13C-spectra were referenced to the solvent peak (CDCl3 δ = 7.26 ppm (1H), δ = 77.16 ppm (13C); DMSO-d6 δ = 2.50 ppm (1H), δ = 39.52 ppm (13C)). Data was evaluated using the software MNova (MestReNova) version 14.1 by Mestrelab Research. Coupling constants J were given in Hz, and chemical shifts δ were given in ppm (parts per million). Multiplicities are abbreviated as the following: singlet (s), broad singlet (brs), doublet (d), triplet (t), quartet (q), and multiplet (m).
General Procedures
General Procedure A Chalcone Monomer (6)
A vial with a stir bar was charged with acetophenone (3) (250 mg, 1.0 equiv), ethanol (2.50 mL), and aq. KOH solution (3 m, 6.0 equiv, 2.78 mL). The mixture was stirred for 5 min until all solids were dissolved. Aldehyde (5) (1.2 equiv) was added at once. The reaction was stirred at room temperature unless stated otherwise. If stated, after 6 h, another portion of aldehyde was added (0.6 equiv). The reaction mixture was left to stir overnight. After the completion of the reaction, aq. HCl (1 M, 10 mL) was added and solids precipitated. The suspension was filtered, and the filtrate was washed with some methanol. The filtrate was then dissolved in CH2Cl2 (10 mL), MeOH (10% v/v) was added, and the solvent was removed in vacuo. The crude product was washed with MeOH (4 mL) at 70 °C and left to cool to room temperature, and the solids were filtered off to isolate the title compound.
General Procedure B Chalcone Dimer (7)
A vial with a stir bar was charged with acetophenone (4) (200 mg, 1.0 equiv), ethanol (2.50 mL), and aq. KOH solution (3 M, 12.0 equiv). The mixture was stirred for 5 min until all solids were dissolved. Aldehyde (5) (2.4 equiv) was added at once. The reaction was stirred at room temperature unless stated otherwise. If stated, after 6 h, another portion of aldehyde was added (1.2 equiv). The reaction mixture was left to stir overnight unless stated otherwise. After the completion of the reaction, aq. HCl (1 M, 10 mL) was added unless stated otherwise, and solids precipitated. The suspension was filtered, and the filtrate was washed with some methanol. The filtrate was then dissolved in CH2Cl2, MeOH (10% v/v) was added, and the solvent was removed in vacuo. The crude product was washed with MeOH (10 mL) at 22 °C and filtered off to isolate the title compound.
General Procedure C Flavone Monomer (8)
A 10 mL microwave vial with a stir bar was charged with chalcone (6) (100 mg, 1.00 equiv) and capped with a septum. The degassed DMSO (1.70 mL) and a solution of I2 in degassed DMSO (78.8 mM, 4 mol %) were added. The reaction solution was then heated to 150 °C and stirred for 3 h. After complete conversion, sat. aq. Na2SO3 solution (4 mL) was added. The aq. phase was extracted with ethyl acetate (3 × 20 mL). The combined org. layers were washed with cold water (3 × 20 mL) and sat. NaCl solution (2 × 20 mL), and the solvent was removed in vacuo. The product was isolated by filtration over a plug of silica (CH2Cl2/MeOH, 9:1 v/v).
General Procedure D Flavone Dimer (9)
A 10 mL microwave vial with a stir bar was charged with bichalcone (7) (30 mg, 1.00 equiv) and capped with a septum. Degassed DMSO (0.24 mL) and a solution of I2 in degassed DMSO (78.8 mM, 10 mol %) were added. The reaction solution was then heated to 150 °C and stirred for 3 h unless stated otherwise. After complete conversion, sat. aq. Na2SO3 solution (4 mL) was added. The aq. phase was extracted with ethyl acetate (3 × 20 mL). The combined org. layers were washed with cold water (3 × 20 mL) and sat. NaCl solution (2 × 20 mL), and the solvent was removed in vacuo. The product was isolated by column chromatography unless otherwise stated.
(E)-1-(2-Hydroxy-6-methoxy-4-methylphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (6aa)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 4-methoxybenzaldehyde (5a) (0.30 mL, 2.50 mmol, 1.2 + 0.6 equiv). The product was isolated as orange solids (286 mg, 0.96 mmol, 69%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 3.86 (s, 3H), 3.94 (s, 3H), 6.24 (d, J = 1.5 Hz, 1H), 6.44 (dd, J = 1.5, 0.8 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.8 Hz, 2H), 7.80 (s, 2H), 13.49 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.53, 55.56, 56.01, 103.00, 110.01, 111.52, 114.56, 125.51, 128.43, 130.34, 142.87, 147.51, 161.02, 161.64, 165.33, 193.94. IR (ATR film): 3003, 2838, 1627, 1605, 1561, 1511, 1483, 1457, 1422, 1410, 1305, 1289, 1255, 1223, 1207, 1172, 1113, 1035, 982, 828, 814, 768, 742, 722, 662, 557, 539 HR-MS (ESI):m/z calcd for [C18H19O4]+ ([M + H+]): 299.1278, found: 299.1284. Melting point: 113–114 °C.
(E)-1-(2-Hydroxy-6-methoxy-4-methylphenyl)-3-phenylprop-2-en-1-one (6ab)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and benzaldehyde (5b) (0.26 mL, 2.50 mmol, 1.2 + 0.6 equiv). The product was isolated as orange solids (209 mg, 0.78 mmol, 56%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 3.94 (s, 3H), 6.24 (s, 1H), 6.45 (s, 1H), 7.40–7.61 (m, 5H), 7.80 (d, J = 15.6 Hz, 1H), 7.89 (d, J = 15.6 Hz, 1H), 13.37 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.59, 56.01, 102.97, 109.89, 111.46, 127.83, 128.56, 129.04, 130.31, 135.61, 142.72, 147.89, 161.04, 165.31, 194.02. IR (ATR film): 3091, 3036, 2980, 2951, 1641, 1576, 1495, 1459, 1418, 1378, 1344, 1284, 1232, 1216, 1168, 1124, 1082, 985, 954, 878, 853, 824. HR-MS (ESI):m/z calcd for [C17H17O3]+ ([M + H+]): 269.1172, found: 269.1173. Melting point: 169.6–170.2 °C.
(E)-3-(4-(Dimethylamino)phenyl)-1-(2-hydroxy-6-methoxy-4-methylphenyl)prop-2-en-1-one (6ac)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 4-(dimethylamino)benzaldehyde (5c) (373 mg, 2.50 mmol, 1.2 + 0.6 equiv) at 90 °C. The product was isolated as red solids (264 mg, 0.85 mmol, 61%). 1H NMR (600 MHz, CDCl3): δ 2.31 (s, 3H), 3.04 (s, 6H), 3.93 (s, 3H), 6.22 (d, J = 1.6 Hz, 1H), 6.43 (d, J = 1.6 Hz, 1H), 6.70 (d, J = 8.6 Hz, 2H), 7.50–7.55 (m, 2H), 7.74 (d, J = 15.4 Hz, 1H), 7.84 (d, J = 15.4 Hz, 1H), 13.72 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.47, 40.28, 55.93, 102.90, 110.06, 111.40, 112.00, 122.40, 123.41, 130.59, 144.39, 146.89, 152.02, 160.88, 165.24, 193.67. IR (ATR film): 3121, 3009, 2968, 2808, 2645, 2169, 2052, 1628, 1593, 1525, 1476, 1462, 1435, 1413, 1375, 1336, 1296, 1257, 1223, 1198, 1168, 1113, 1069, 1031, 984, 948, 883, 838, 819, 807, 768, 732, 711, 680, 665, 638, 600, 546, 529, 514, 501, 470. HR-MS (ESI):m/z calcd for [C19H22O3N]+ ([M + H+]): 312.1594, found: 312.1596. Melting point: 182–183 °C.
(E)-1-(2-Hydroxy-6-methoxy-4-methylphenyl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (6ad)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 4-(trifluoromethyl)benzaldehyde (5d) (0.34 mL, 2.50 mmol, 1.2 + 0.6 equiv). The product was isolated as orange solids (299 mg, 0.89 mmol, 64%). 1H NMR (600 MHz, CDCl3): δ 2.36 (s, 3H), 3.97 (s, 3H), 6.27 (s, 1H), 6.48 (s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.78 (d, J = 15.7 Hz, 1H), 7.94 (d, J = 15.7 Hz, 1H), 13.26 (d, J = 2.3 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 22.63, 56.05, 103.00, 109.77, 111.53, 124.05 (q, J = 272.1 Hz), 125.98 (q, J = 3.8 Hz), 128.53, 130.21, 131.62 (q, J = 32.6 Hz), 139.06, 140.37, 148.42, 161.03, 165.40, 193.63. 19F NMR (282 MHz, CDCl3): δ −62.77. IR (ATR film): 3746, 3123, 3051, 2974, 1635, 1614, 1565, 1487, 1456, 1410, 1368, 1317, 1287, 1266, 1223, 1169, 1066, 1031, 1015, 983, 909, 843, 813, 768, 747, 732, 683, 666, 626, 593, 558, 530, 506, 492 HR-MS (ESI):m/z calcd for [C18H16O3F3]+ ([M + H+]): 337.1046, found: 337.1050. Melting point: 124–126 °C.
(E)-3-(4-Bromophenyl)-1-(2-hydroxy-6-methoxy-4-methylphenyl)prop-2-en-1-one (6ae)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 4-bromobenzaldehyde (5e) (309 mg 1.66 mmol, 1.2 equiv). The product was isolated as yellow solids (261 mg, 0.75 mmol, 54%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 3.93 (s, 3H), 6.23 (d, J = 1.6 Hz, 1H), 6.45 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 15.6 Hz, 1H), 7.85 (d, J = 15.6 Hz, 1H), 13.30 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.60, 56.04, 102.98, 109.82, 111.51, 124.48, 128.43, 129.88, 132.28, 134.56, 141.20, 148.13, 161.00, 165.36, 193.74. IR (ATR film): 2970, 2850, 1632, 1571, 1486, 1454, 1402, 1369, 1331, 1270, 1222, 1206, 1159, 1114, 1072, 1031, 1009, 979, 945, 874, 820, 790, 764, 712, 595, 572, 557, 529. HR-MS (ESI):m/z calcd for [C17H16O3Br]+ ([M + H+]): 347.0277, found: 347.0279. Melting point: 122–124 °C.
(E)-3-(3-Bromophenyl)-1-(2-hydroxy-6-methoxy-4-methylphenyl)prop-2-en-1-one (6af)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 3-bromobenzaldehyde (5f) (0.20 mL, 1.66 mmol, 1.2 equiv). The product was isolated as yellow solids (279 mg, 0.81 mmol, 58%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 3.94 (s, 3H), 6.24 (d, J = 1.5 Hz, 1H), 6.43–6.46 (m, 1H), 7.28 (t, J = 7.8 Hz, 1H), 7.51 (dd, J = 7.9, 1.8 Hz, 2H), 7.67 (d, J = 15.6 Hz, 1H), 7.74 (t, J = 1.8 Hz, 1H), 7.84 (d, J = 15.6 Hz, 1H), 13.27 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.61, 56.09, 102.99, 109.79, 111.49, 123.15, 127.21, 129.18, 130.53, 131.03, 132.97, 137.80, 140.70, 148.23, 161.03, 165.35, 193.65. IR (ATR film): 2918, 2850, 1633, 1571, 1454, 1410, 1368, 1329, 1307, 1276, 1222, 1204, 1159, 1114, 1072, 1031, 978, 945, 898, 863, 815, 787, 760, 699, 667, 626, 606, 583, 557, 529, 485. HR-MS (ESI):m/z calcd for [C17H16O3Br]+ ([M + H+]): 347.0277, found: 347.0278. Melting point: 123–124 °C.
(E)-3-(2-Bromophenyl)-1-(2-hydroxy-6-methoxy-4-methylphenyl)prop-2-en-1-one (6ag)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3a (250 mg, 1.39 mmol, 1.0 equiv) and 2-bromobenzaldehyde (5g) (0.19 mL, 1.66 mmol, 1.2 equiv). The product was isolated as yellow solids (347 mg, 1.00 mmol, 72%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 3.92 (s, 3H), 6.23 (s, 1H), 6.45 (s, 1H), 7.23 (td, J = 7.6, 1.6 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.63 (dd, J = 8.0, 1.2 Hz, 1H), 7.69 (dd, J = 7.9, 1.6 Hz, 1H), 7.80 (d, J = 15.4 Hz, 1H), 8.11 (d, J = 15.5 Hz, 1H), 13.29 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 22.61, 56.03, 102.95, 109.80, 111.50, 126.07, 127.77, 128.03, 130.45, 131.10, 133.69, 135.66, 140.78, 148.17, 161.02, 165.38, 193.65. IR (ATR film): 3065, 1629, 1577, 1487, 1466, 1451, 1438, 1367, 1334, 1278, 1225, 1208, 1191, 1159, 1113, 1049, 1025, 996, 966, 944, 893, 858, 839, 813, 768, 745, 713, 656, 625, 606, 561, 530, 511, 489. HR-MS (ESI):m/z calcd for [C17H16O3Br]+ ([M + H+]): 347.0277, found: 347.0281. Melting point: 162–164 °C.
(E)-1-(2-Hydroxy-4,6-dimethoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one or Flavokavain A (6ba)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 4-methoxybenzaldehyde (5a) (0.28 mL, 2.29 mmol, 1.2 + 0.6 equiv). The product was isolated as yellow solids (292 mg, 0.93 mmol, 73%). The analytical data was in accordance with the literature.651H NMR (600 MHz, CDCl3): δ 3.84 (s, 3H), 3.86 (s, 3H), 3.92 (s, 3H), 5.97 (dd, J = 5.1, 2.4 Hz, 1H), 6.11 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 7.80 (m, 2H), 14.43 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.39, 55.56, 55.84, 91.26, 93.86, 106.41, 114.38, 125.19, 128.37, 130.10, 142.45, 161.38, 162.48, 166.03, 168.38, 192.61. IR (ATR film): 3005, 2838, 1622, 1580, 1559, 1511, 1488, 1455, 1440, 1421, 1391, 1344, 1304, 1289, 1255, 1216, 1172, 1158, 1112, 984, 939, 870, 828, 766, 721, 697, 674, 615, 559, 539, 520. HR-MS (ESI):m/z calcd for [C18H19O5]+ ([M + H+]): 315.1227, found: 315.1230. Melting point: 113–114 °C (110 °C).66
(E)-1-(2-Hydroxy-4,6-dimethoxyphenyl)-3-phenylprop-2-en-1-one (6bb)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and benzaldehyde (5b) (0.23 mL, 2.29 mmol, 1.2 + 0.6 equiv) at 50 °C. The product was isolated as yellow solids (130 mg, 0.46 mmol, 36%). The analytical data was in accordance with the literature.651H NMR (600 MHz, CDCl3): δ 3.84 (s, 3H), 3.92 (s, 3H), 5.97 (d, J = 2.4 Hz, 1H), 6.11 (d, J = 2.4 Hz, 1H), 7.36–7.44 (m, 3H), 7.58–7.63 (m, 2H), 7.79 (d, J = 15.6 Hz, 1H), 7.91 (d, J = 15.6 Hz, 1H), 14.31 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.59, 55.87, 91.31, 93.86, 106.41, 127.60, 128.36, 128.88, 130.04, 135.64, 142.32, 162.55, 166.27, 168.43, 192.67. IR (ATR film): 3082, 3059, 3025, 3004, 2969, 2940, 2849, 1615, 1558, 1494, 1415, 1340, 1284, 1211, 1155, 1112, 1072, 1055, 1033, 984, 939, 888, 869, 818, 788, 742, 691, 673, 647, 623, 577, 562, 534, 495, 459 HR-MS (ESI):m/z calcd for [C17H17O4]+ ([M + H+]): 285.1121, found: 285.1122. Melting point: 83–84 °C (85–86 °C).65
(E)-3-(4-(Dimethylamino)phenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (6bc)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 4-(dimethylamino)benzaldehyde (5c) (228 mg, 1.53 mmol, 1.2 equiv) at 90 °C. The product was isolated as red solids (296 mg, 0.91 mmol, 71%). The analytical data was in accordance with the literature.671H NMR (600 MHz, CDCl3): δ 3.03 (s, 6H), 3.82 (s, 3H), 3.91 (s, 3H), 5.95 (d, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 15.3 Hz, 1H), 7.83 (d, J = 15.4 Hz, 1H), 14.66 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 40.16, 55.52, 55.78, 91.12, 93.83, 106.45, 111.90, 122.09, 123.40, 130.37, 143.99, 151.84, 162.39, 165.63, 168.31, 192.47. IR (ATR film): 3004, 2962, 2850, 1621, 1587, 1528, 1481, 1436, 1415, 1379, 1346, 1296, 1209, 1172, 1154, 1111, 1031, 1001, 986, 941, 920, 864, 810, 765, 711, 677, 638, 616, 547, 516, 503, 461. HR-MS (ESI):m/z calcd for [C19H22O4]+ ([M + H+]): 328.1543, found: 328.1546. Melting point: 203–204 °C (153 °C).66
(E)-1-(2-Hydroxy-4,6-dimethoxyphenyl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (6bd)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 4-(trifluoromethyl)benzaldehyde (5d) (0.31 mL, 2.29 mmol, 1.2 + 0.6 equiv). The product was isolated as yellow solids (293 mg, 0.83 mmol, 65%). 1H NMR (600 MHz, CDCl3): δ 3.84 (s, 3H), 3.92 (s, 3H), 5.97 (d, J = 2.4 Hz, 1H), 6.11 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.68 (d, J = 8.3 Hz, 2H), 7.74 (d, J = 15.6 Hz, 1H), 7.93 (d, J = 15.6 Hz, 1H), 14.14 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.78, 56.05, 91.53, 94.00, 106.43, 124.07 (q, J = 272.1 Hz), 125.96 (q, J = 3.8 Hz), 128.47, 130.12, 131.51 (q, J = 32.6 Hz), 139.19, 140.13, 162.65, 166.72, 168.66, 192.32. 19F NMR (282 MHz, CDCl3): δ −62.76. IR (ATR film): 3022, 2980, 2948, 1633, 1613, 1579, 1488, 1456, 1438, 1414, 1342, 1324, 1287, 1216, 1101, 1067, 1030, 1016, 955, 936, 906, 872, 838, 827, 815, 768, 750, 732, 693, 676, 605, 590, 535, 504, 461. HR-MS (ESI):m/z calcd for [C18H16O4F3]+ ([M + H+]): 353.0995, found: 353.0997. Melting point: 148–149 °C.
(E)-3-(4-Bromophenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (6be)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 4-bromobenzaldehyde (5e) (423 mg, 2.29 mmol, 1.2 + 0.6 equiv). The product was isolated as a yellow solid (356 mg, 0.98 mmol, 77%). The analytical data was in accordance with the literature.651H NMR (600 MHz, CDCl3): δ 3.82 (s, 3H), 3.90 (s, 3H), 5.94 (d, J = 2.4 Hz, 1H), 6.09 (d, J = 2.4 Hz, 1H), 7.44 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 15.6 Hz, 1H), 7.85 (d, J = 15.6 Hz, 1H), 14.23 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.72, 55.99, 91.42, 93.95, 106.39, 124.30, 128.26, 129.78, 132.20, 134.63, 140.88, 162.58, 166.49, 168.57, 192.40. IR (ATR film): 3016, 2996, 2978, 1627, 1586, 1564, 1484, 1440, 1420, 1336, 1303, 1289, 1272, 1215, 1158, 1114, 1069, 1029, 1007, 986, 973, 936, 891, 818, 792, 758, 709, 666, 604, 564, 534, 505, 483, 460. HR-MS (ESI):m/z calcd for [C17H16O4Br]+ ([M + H+]): 363.0226, found: 363.0231. Melting point: 169–170 °C (166 °C)68 (150–151 °C).65
(E)-3-(3-Bromophenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (6bf)
Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 3-bromobenzaldehyde (5f) (0.27 mL, 2.29 mmol, 1.2 + 0.6 equiv). The product was isolated as a yellow solid (310 mg, 0.85 mmol, 67%). The analytical data was in accordance with the literature.691H NMR (600 MHz, CDCl3): δ 3.83 (s, 3H), 3.92 (s, 3H), 5.96 (d, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 1H), 7.27 (t, J = 7.9 Hz, 1H), 7.49 (dd, J = 7.9, 1.8 Hz, 2H), 7.66 (d, J = 15.6 Hz, 1H), 7.72 (t, J = 1.8 Hz, 1H), 7.85 (d, J = 15.6 Hz, 1H), 14.18 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.75, 56.07, 91.47, 93.96, 106.42, 123.12, 127.12, 129.05, 130.49, 130.99, 132.84, 137.90, 140.43, 162.63, 166.59, 168.59, 192.35. IR (ATR film): 2942, 2852, 1619, 1578, 1469, 1454, 1440, 1416, 1392, 1340, 1319, 1303, 1262, 1216, 1158, 1114, 1072, 1055, 1030, 983, 939, 911, 863, 819, 787, 758, 688, 670, 647, 624, 582, 564, 533, 492. HR-MS (ESI):m/z calcd for [C17H16O4Br]+ ([M + H+]): 363.0226, found: 363.0230. Melting point: 115–116 °C.
(E)-3-(2-Bromophenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (6bg)
Synthesized in accordance with General Procedure A. Starting from acetophenone 3b (250 mg, 1.27 mmol, 1.0 equiv) and 2-bromobenzaldehyde (5g) (0.18 mL, 1.53 mmol, 1.2 equiv). The product was isolated as yellow solids (385 mg, 1.06 mmol, 83%). The analytical data was in accordance with the literature.701H NMR (600 MHz, CDCl3): δ 3.83 (s, 3H), 3.89 (s, 3H), 5.95 (d, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 1H), 7.21 (td, J = 7.6, 1.6 Hz, 1H), 7.34 (td, J = 7.6, 1.3 Hz, 1H), 7.62 (dd, J = 8.1, 1.2 Hz, 1H), 7.67 (dd, J = 7.8, 1.7 Hz, 1H), 7.81 (d, J = 15.5 Hz, 1H), 8.09 (d, J = 15.5 Hz, 1H), 14.21 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 55.73, 56.01, 91.41, 93.96, 106.41, 125.96, 127.75, 127.99, 130.32, 130.98, 133.62, 135.73, 140.51, 162.62, 166.55, 168.60, 192.34. IR (ATR film): 3005, 2976, 1627, 1579, 1565, 1490, 1464, 1437, 1416, 1343, 1321, 1307, 1269, 1216, 1160, 1113, 1048, 1027, 971, 939, 874, 814, 797, 770, 744, 695, 663, 646, 622, 586, 535, 507. HR-MS (ESI):m/z calcd for [C17H16O4Br]+ ([M + H+]): 363.0226, found: 363.0233. Melting point: 147–148 °C (146–147 °C).70
5-Methoxy-2-(4-methoxyphenyl)-7-methyl-4H-chromen-4-one (8aa)
Synthesized in accordance with General Procedure C. Starting from chalcone 6aa (100 mg, 0.34 mmol, 1.0 equiv) and I2 (78.8 mM, 173 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (81.0 mg, 0.28 mmol, 81%). 1H NMR (600 MHz, CDCl3): δ 2.46 (s, 3H), 3.88 (s, 3H), 3.98 (s, 3H), 6.62 (d, J = 1.4 Hz, 1H), 6.63 (s, 1H), 6.94 (dd, J = 1.6, 0.8 Hz, 1H), 7.00 (d, J = 8.9 Hz, 2H), 7.83 (d, J = 8.9 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 22.42, 55.63, 56.59, 107.88, 110.39, 112.60, 114.54, 124.13, 127.86, 145.01, 158.44, 159.67, 161.04, 162.28, 178.34. IR (ATR film): 2840, 1639, 1606, 1575, 1512, 1483, 1423, 1377, 1340, 1300, 1220, 1180, 1115, 1050, 955, 903, 833, 611, 586. HR-MS (ESI):m/z calcd for [C18H17O4]+ ([M + H+]): 297.1121, found: 297.1126. Melting point: 173–174 °C.
5-Methoxy-7-methyl-2-phenyl-4H-chromen-4-one (8ab)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ab (100 mg, 0.38 mmol, 1.0 equiv) and I2 (78.8 mM, 192 μL, 0.02 mmol, 4 mol %). The product was isolated as white solids (90.3 mg, 0.34 mmol, 90%). 1H NMR (600 MHz, CDCl3): δ 2.45 (s, 3H), 3.98 (s, 3H), 6.62 (s, 1H), 6.70 (s, 1H), 6.95 (s, 1H), 7.49 (m, 3H), 7.87 (dd, J = 7.5, 2.3 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 22.40, 56.55, 107.93, 109.17, 110.39, 112.62, 126.14, 129.05, 131.34, 131.77, 145.26, 158.45, 159.65, 160.97, 178.28. IR (ATR film): 3068, 2914, 2851, 2236, 1637, 1610, 1577, 1566, 1482, 1463, 1449, 1410, 1375, 1337, 1298, 1263, 1219, 1188, 1163, 1118, 1079, 1049, 1014, 1001, 973, 956, 919, 901, 848, 825, 769, 729, 691, 676, 610, 566, 545, 528, 488. HR-MS (ESI):m/z calcd for [C17H15O3]+ ([M + H+]): 267.1016, found: 267.1020. Melting point: 162–163 °C.
2-(4-(Dimethylamino)phenyl)-5-methoxy-7-methyl-4H-chromen-4-one (8ac)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ac (100 mg, 0.32 mmol, 1.0 equiv) and I2 (78.8 mM, 162 μL, 0.01 mmol, 4 mol %). The product was isolated as orange solids (50.0 mg, 0.16 mmol, 50%). 1H NMR (600 MHz, CDCl3): δ 2.44 (s, 3H), 3.05 (s, 6H), 3.97 (s, 3H), 6.57 (s, 1H), 6.60 (s, 1H), 6.73 (d, J = 8.9 Hz, 2H), 6.92 (s, 1H), 7.76 (d, J = 8.9 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 22.38, 40.24, 56.54, 106.13, 107.60, 110.36, 111.76, 112.55, 118.36, 127.49, 144.54, 152.37, 158.38, 159.53, 161.89, 178.42. IR (ATR film): 2924, 2853, 2235, 1629, 1597, 1523, 1481, 1463, 1445, 1412, 1366, 1339, 1300, 1253, 1223, 1197, 1170, 1118, 1092, 1049, 1015, 972, 947, 914, 776, 758, 728, 677, 643, 611, 577, 541, 528, 510, 480 HR-MS (ESI):m/z calcd for [C19H20NO3]+ ([M + H+]): 310.1438, found: 310.1441. Melting point: 198 °C (decomposition).
5-Methoxy-7-methyl-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (8ad)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ad (100 mg, 0.30 mmol, 1.0 equiv) and I2 (78.8 mM, 152 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (65.7 mg, 0.20 mmol, 66%). 1H NMR (600 MHz, CDCl3): δ 2.46 (s, 3H), 3.98 (s, 3H), 6.64 (s, 1H), 6.74 (s, 1H), 6.96 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.98 (d, J = 8.1 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 22.44, 56.59, 108.19, 110.34, 112.63, 122.91 (q, J = 271.2 Hz), 126.07 (q, J = 3.8 Hz), 126.48, 132.97 (q, J = 32.8 Hz), 135.23, 145.73, 158.35, 159.23, 159.73, 177.89. 19F NMR (282 MHz, CDCl3): δ −62.95. IR (ATR film): 3074, 2920, 1645, 1612, 1567, 1518, 1486, 1466, 1417, 1379, 1319, 1295, 1257, 1219, 1202, 1159, 1114, 1071, 1049, 1016, 958, 902, 889, 849, 823, 776, 731, 702, 677, 649, 635, 612, 585, 566, 551, 530, 497. HR-MS (ESI):m/z calcd for [C18H14O3F3]+ ([M + H+]): 335.0890, found: 335.0895. Melting point: 226–228 °C.
2-(4-Bromophenyl)-5-methoxy-7-methyl-4H-chromen-4-one (8ae)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ae (100 mg, 0.29 mmol, 1.0 equiv) and I2 (78.8 mM, 147 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (90.5 mg, 0.26 mmol, 91%). 1H NMR (600 MHz, CDCl3): δ 2.46 (s, 3H), 3.98 (s, 3H), 6.64 (s, 1H), 6.69 (s, 1H), 6.94 (s, 1H), 7.63 (d, J = 8.6 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 22.48, 56.60, 108.05, 109.29, 110.35, 112.54, 126.02, 127.61, 130.71, 132.39, 145.53, 158.34, 159.67, 159.95, 178.13. IR (ATR film): 3072, 2236, 1612, 1588, 1567, 1483, 1463, 1404, 1375, 1334, 1300, 1265, 1218, 1188, 1164, 1120, 1074, 1048, 1008, 956, 828, 776, 731, 677, 644, 566, 550, 529, 495, 475. HR-MS (ESI):m/z calcd for [C17H14O3Br]+ ([M + H+]): 345.0121, found: 345.0121. Melting point: 203–206 °C.
2-(3-Bromophenyl)-5-methoxy-7-methyl-4H-chromen-4-one (8af)
Synthesized in accordance with General Procedure C. Starting from chalcone 6af (100 mg, 0.29 mmol, 1.0 equiv) and I2 (78.8 mM, 147 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (92.1 mg, 0.27 mmol, 92%). 1H NMR (600 MHz, CDCl3): δ 2.39 (s, 3H), 3.91 (s, 3H), 6.57 (d, J = 8.3 Hz, 2H), 6.86 (t, J = 1.1 Hz, 1H), 7.29 (t, J = 7.9 Hz, 1H), 7.54 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.68 (dt, J = 7.9, 1.4 Hz, 1H), 7.92 (t, J = 1.9 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 22.28, 56.37, 107.88, 109.44, 110.17, 112.29, 123.08, 124.44, 128.84, 130.40, 133.49, 133.99, 145.44, 158.03, 158.97, 159.40, 177.71. IR (ATR film): 2921, 1644, 1613, 1560, 1483, 1465, 1417, 1375, 1301, 1266, 1217, 1165, 1121, 1098, 1077, 1050, 998, 977, 956, 846, 825, 791, 744, 721, 693, 612, 566, 529, 487. HR-MS (ESI):m/z calcd for [C17H14O3Br]+ ([M + H+]): 345.0121, found: 345.0125. Melting point: 165–166 °C.
2-(2-Bromophenyl)-5-methoxy-7-methyl-4H-chromen-4-one (8ag)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ag (100 mg, 0.29 mmol, 1.0 equiv) and I2 (78.8 mM, 147 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (76.4 mg, 0.22 mmol, 76%). 1H NMR (600 MHz, CDCl3): δ 2.45 (s, 3H), 3.99 (s, 3H), 6.45 (s, 1H), 6.64 (s, 1H), 6.88 (s, 1H), 7.35 (td, J = 7.7, 1.7 Hz, 1H), 7.43 (td, J = 7.5, 1.2 Hz, 1H), 7.55 (dd, J = 7.7, 1.7 Hz, 1H), 7.70 (dd, J = 8.0, 1.1 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 22.43, 56.58, 108.06, 110.45, 112.62, 114.33, 122.05, 127.70, 130.98, 131.81, 134.02, 134.05, 145.50, 158.71, 159.75, 161.65, 177.90. IR (ATR film): 2931, 1650, 1616, 1483, 1466, 1436, 1411, 1332, 1298, 1267, 1217, 1164, 1117, 1061, 1040, 1026, 854, 764, 727, 683, 567, 545, 500. HR-MS (ESI):m/z calcd for [C17H14O3Br]+ ([M + H+]): 345.0121, found 345.0125. Melting point: 146–148 °C.
5,7-Dimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one or Apigenin Trimethyl Ether (8ba)
Synthesized in accordance with General Procedure C. Starting from chalcone 6ba (100 mg, 0.32 mmol, 1.0 equiv) and I2 (78.8 mM, 162 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (35.7 mg, 0.12 mmol, 36%). The analytical data was in accordance with the literature.711H NMR (600 MHz, CDCl3): δ 3.88 (s, 3H), 3.91 (s, 3H), 3.96 (s, 3H), 6.37 (d, J = 2.3 Hz, 1H), 6.56 (d, J = 2.3 Hz, 1H), 6.60 (s, 1H), 7.00 (d, J = 8.9 Hz, 2H), 7.82 (d, J = 8.9 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 55.62, 55.87, 56.59, 93.02, 96.25, 107.92, 109.46, 114.53, 124.09, 127.76, 160.03, 160.83, 161.12, 162.22, 164.07, 177.75. IR (ATR film): 2941, 1640, 1605, 1513, 1491, 1460, 1422, 1347, 1301, 1259, 1218, 1202, 1180, 1161, 1114, 1057, 1032, 908, 833, 773, 620, 599, 559. HR-MS (ESI):m/z calcd for [C18H17O5]+ ([M + H+]): 313.1071, found: 313.1075. Melting point: 155–157 °C (155–157 °C).71
5,7-Dimethoxy-2-phenyl-4H-chromen-4-one or Dimethyl-chrysin (8bb)
Synthesized in accordance with General Procedure C. Starting from chalcone 6bb (100 mg, 0.35 mmol, 1.0 equiv) and I2 (78.8 mM, 178 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (80.1 mg, 0.28 mmol, 80%). The analytical data was in accordance with the literature.721H NMR (600 MHz, CDCl3): δ 3.91 (s, 3H), 3.95 (s, 3H), 6.37 (d, J = 2.3 Hz, 1H), 6.57 (d, J = 2.3 Hz, 1H), 6.68 (s, 1H), 7.47–7.52 (m, 3H), 7.84–7.90 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 55.89, 56.57, 93.02, 96.35, 109.26, 109.52, 126.09, 129.06, 131.29, 131.75, 160.08, 160.78, 161.12, 164.22, 177.69. IR (ATR film): 3017, 2948, 2922, 2844, 2326, 2226, 2015, 1646, 1605, 1491, 1465, 1451, 1422, 1392, 1348, 1302, 1268, 1215, 1204, 1189, 1161, 1120, 1104, 1079, 1058, 1035, 1022, 1000, 962, 949, 915, 851, 819, 803, 766, 723, 689, 642, 614, 556, 530, 483. HR-MS (ESI):m/z calcd for [C17H15O4]+ ([M + H+]): 283.0965, found: 283.0969. Melting point: 80.0 °C (brown discoloration), 141–142 °C (145–146 °C).72
2-(4-(Dimethylamino)phenyl)-5,7-dimethoxy-4H-chromen-4-one (8bc)
Synthesized in accordance with General Procedure C. Starting from chalcone 6bc (100 mg, 0.31 mmol, 1.0 equiv) and I2 (78.8 mM, 157 μL, 0.01 mmol, 4 mol %). The product was isolated as orange solids (37.1 mg, 0.11 mmol, 37%). 1H NMR (600 MHz, CDCl3): δ 3.06 (s, 6H), 3.91 (s, 3H), 3.95 (s, 3H), 6.36 (d, J = 2.3 Hz, 1H), 6.55 (s, 2H), 6.74 (d, J = 9.0 Hz, 2H), 7.75 (d, J = 9.0 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 40.25, 55.83, 56.57, 93.03, 96.03, 106.23, 111.83, 112.16, 118.48, 127.44, 152.39, 160.00, 161.04, 161.72, 163.79, 177.87. IR (ATR film): 2943, 1635, 1601, 1525, 1489, 1458, 1369, 1347, 1303, 1253, 1217, 1200, 1161, 1117, 1057, 1029, 1002, 908, 819, 729, 674, 642, 618, 592, 511, 467. HR-MS (ESI):m/z calcd for [C19H20NO4]+ ([M + H+]): 326.1387, found: 326.1388. Melting point: 211–214 °C.
5,7-Dimethoxy-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (8bd)
Synthesized in accordance with General Procedure C. Starting from chalcone 6bd (100 mg, 0.29 mmol, 1.0 equiv) and I2 (78.8 mM, 147 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (26.8 mg, 0.08 mmol 27%). The analytical data was in accordance with the literature.731H NMR (600 MHz, CDCl3): δ 3.91 (s, 3H), 3.95 (s, 3H), 6.38 (d, J = 2.3 Hz, 1H), 6.57 (d, J = 2.3 Hz, 1H), 6.70 (s, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.97 (d, J = 8.1 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 55.80, 56.45, 92.88, 96.41, 109.36, 110.30, 123.68 (q, J = 272.2 Hz), 125.91 (q, J = 3.8 Hz), 126.26, 132.77 (q, J = 32.8 Hz), 135.02, 158.87, 159.84, 161.04, 164.34, 177.11. 19F NMR (282 MHz, CDCl3): δ −62.93. IR (ATR film): 2945, 2240, 1645, 1574, 1491, 1459, 1416, 1386, 1322, 1295, 1264, 1218, 1203, 1161, 1070, 1057, 1027, 1016, 908, 841, 807, 731, 675, 635, 616, 586, 564, 528, 514, 493, 468. HR-MS (ESI):m/z calcd for [C18H14F3O4]+ ([M + H+]): 351.0839, found: 351.0843. Melting point: 185–186 °C.
2-(4-Bromophenyl)-5,7-dimethoxy-4H-chromen-4-one (8be)
Synthesized in accordance with General Procedure C. Starting from chalcone 6be (100 mg, 0.28 mmol, 1.0 equiv) and I2 (78.8 mM, 142 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (92.6 mg, 0.26 mmol, 93%). The analytical data was in accordance with the literature.741H NMR (600 MHz, CDCl3): δ 3.90 (s, 3H), 3.94 (s, 3H), 6.36 (d, J = 2.3 Hz, 1H), 6.54 (d, J = 2.3 Hz, 1H), 6.63 (s, 1H), 7.61 (d, J = 8.6 Hz, 2H), 7.71 (d, J = 8.6 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 55.91, 56.57, 92.93, 96.40, 109.33, 109.38, 125.88, 127.47, 130.60, 132.33, 159.64, 159.91, 161.06, 164.28, 177.44. IR (ATR film): 2842, 1644, 1607, 1572, 1488, 1459, 1422, 1404, 1380, 1341, 1277, 1217, 1202, 1161, 1118, 1105, 1073, 1057, 1008, 905, 827, 773, 738, 718, 674, 635, 616, 529, 491, 475. HR-MS (ESI):m/z calcd for [C17H14O4Br]+ ([M + H+]): 361.0070, found: 361.0073. Melting point: 194–195 °C (197–198 °C).74
2-(3-Bromophenyl)-5,7-dimethoxy-4H-chromen-4-one (8bf)
Synthesized in accordance with General Procedure C. Starting from chalcone 6bf (100 mg, 0.28 mmol, 1.0 equiv) and I2 (78.8 mM, 142 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (84.9 mg, 0.24 mmol, 85%). The analytical data was in accordance with the literature.751H NMR (600 MHz, CDCl3): δ 3.92 (s, 3H), 3.96 (s, 3H), 6.39 (d, J = 2.3 Hz, 1H), 6.58 (d, J = 2.3 Hz, 1H), 6.65 (s, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.63 (dd, J = 8.0, 1.5 Hz, 1H), 7.77 (dt, J = 7.9, 1.4 Hz, 1H), 8.03 (d, J = 1.9 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 55.82, 56.46, 92.85, 96.41, 109.35, 109.75, 123.16, 124.50, 128.93, 130.45, 133.66, 134.00, 158.95, 159.84, 161.00, 164.26, 177.21. IR (ATR film): 2949, 1649, 1609, 1571, 1489, 1421, 1384, 1268, 1201, 1160, 1115, 1099, 1063, 1024, 910, 854, 825, 764, 728, 636, 611, 566, 554, 531, 504, 480. HR-MS (ESI):m/z calcd for [C17H14O4Br]+ ([M + H+]): 361.0070, found: 361.0077. Melting point: 134–136 °C (278–280 °C)75 The melting point deviated strongly from the literature-reported value.
2-(2-Bromophenyl)-5,7-dimethoxy-4H-chromen-4-one (8bg)
Synthesized in accordance with General Procedure C. Starting from chalcone 6bg (100 mg, 0.28 mmol, 1.0 equiv) and I2 (78.8 mM, 142 μL, 0.01 mmol, 4 mol %). The product was isolated as white solids (92.3 mg, 0.26 mmol, 92%). 1H NMR (600 MHz, CDCl3): δ 3.88 (s, 3H), 3.96 (s, 3H), 6.39 (d, J = 2.3 Hz, 1H), 6.43 (s, 1H), 6.50 (d, J = 2.3 Hz, 1H), 7.35 (td, J = 7.7, 1.7 Hz, 1H), 7.43 (td, J = 7.5, 1.2 Hz, 1H), 7.54 (dd, J = 7.6, 1.7 Hz, 1H), 7.70 (dd, J = 8.1, 1.2 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 55.89, 56.59, 92.99, 96.51, 109.48, 114.41, 122.05, 127.71, 130.98, 131.80, 133.97, 134.02, 160.35, 161.20, 161.34, 164.35, 177.28. IR (ATR film): 2843, 1644, 1607, 1489, 1459, 1421, 1383, 1334, 1306, 1269, 1217, 1202, 1161, 1119, 1101, 1079, 1057, 1028, 997, 965, 953, 916, 876, 846, 824, 790, 772, 754, 726, 692, 674, 643, 616, 566, 529, 483 HR-MS (ESI):m/z calcd for [C17H14O4Br]+ ([M + H+]): 361.0070, found: 361.0074. Melting point: 162–163 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-methoxyphenyl)prop-2-en-1-one) (7aa)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 4-methoxybenzaldehyde (5a) (0.24 mL, 2.01 mmol, 2.4 equiv +1.2 equiv). The product was isolated as orange solids (278 mg, 0.46 mmol, 83%). 1H NMR (600 MHz, CDCl3): δ 2.11 (s, 6H), 3.86 (s, 6H), 3.99 (s, 6H), 6.43 (s, 2H), 6.94 (d, J = 8.7 Hz, 4H), 7.58 (d, J = 8.7 Hz, 4H), 7.79 (d, J = 15.6 Hz, 2H), 7.84 (d, J = 15.6 Hz, 2H), 13.80 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 20.87, 55.40, 55.77, 103.30, 109.96, 114.38, 117.73, 125.66, 128.37, 130.16, 142.50, 147.05, 160.12, 161.41, 162.70, 194.03. IR (ATR film): 2970, 2839, 1623, 1603, 1558, 1510, 1464, 1422, 1363, 1327, 1304, 1291, 1256, 1216, 1170, 1114, 1037, 908, 871, 828, 770, 647, 619, 559, 536, 521, 487. HR-MS (ESI):m/z calcd for [C36H35O8]+ ([M + H+]): 595.2327, found: 595.2335. Melting point: 212–214 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-phenylprop-2-en-1-one) (7ab)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and benzaldehyde (5b) (0.20 mL, 2.01 mmol, 2.4 + 1.2 equiv). The product was isolated as orange solids (263 mg, 0.49 mmol, 88%). 1H NMR (600 MHz, CDCl3): δ 2.12 (s, 6H), 4.00 (s, 6H), 6.44 (s, 2H), 7.37–7.45 (m, 6H), 7.59–7.65 (m, 4H), 7.80 (d, J = 15.6 Hz, 2H), 7.94 (d, J = 15.6 Hz, 2H), 13.71 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 21.06, 55.96, 103.50, 110.07, 117.82, 128.16, 128.56, 129.04, 130.24, 135.76, 142.55, 147.57, 160.38, 162.89, 194.30. IR (ATR film): 3104, 3026, 2970, 2942, 2250, 1628, 1609, 1564, 1448, 1388, 1361, 1329, 1272, 1214, 1179, 1115, 1073, 1038, 976, 948, 907, 869, 817, 789, 758, 725, 688, 647, 565, 534, 494. HR-MS (ESI):m/z calcd for [C34H31O6]+ ([M + H+]): 535.2115, found: 535.2122. Melting point: 213–215 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-(dimethylamino)phenyl)prop-2-en-1-one) (7ac)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 4-(dimethylamino)benzaldehyde (5c) (200 mg, 1.34 mmol, 2.4 equiv) at 90 °C. The product was isolated as red solids (175 mg, 0.28 mmol, 50%).
Scaleup: in a 50 mL round-bottom flask, acetophenone 4a (1.00 g, 2.80 mmol, 1.0 equiv) and 4-(dimethylamino)benzaldehyde (5c) (1.00 g, 6.70 mmol, 2.4 equiv) were given in EtOH (10 mL). Aq. KOH solution was added (3 M, 11 mL, 33.5 mmol, 12.0 equiv). The reaction mixture was stirred at 90 °C. After 24 h, 4-(dimethylamino)benzaldehyde (5c) (500 mg, 3.40 mmol, 1.2 equiv) was added. After an additional 24 h heating was stopped, KPi-buffer (1 M, pH 7, 20 mL) was added. The organic phases were extracted using CH2Cl2 (3x 100 mL). The combined organic phases were washed with sat. aq. NaCl solution (50 mL) and dried over MgSO4. The solvent was removed in vacuo. The mixture was then resuspended in MeOH (10 mL). Aq. KOH solution was added (3 M, 11.0 mL, 33.5 mmol, 12.0 equiv). Then, 4-(dimethylamino)benzaldehyde (5c) (500 mg, 3.40 mmol, 1.2 equiv) was added and the mixture was stirred at 90 °C for 16 h. The reaction was stopped, and KPi-buffer (1 M, pH 7, 20 mL) was added. The organic phases were extracted using CH2Cl2 (3 × 100 mL). The combined organic phases were washed with sat. aq. NaCl solution (50 mL) and dried over MgSO4. MeOH (15 mL) was added to the solution, and the solvent was carefully removed in vacuo. The resulting mixture was macerated with MeOH (15 mL). The solids were filtered off and washed with copious amounts of MeOH. The product was obtained as red solids (607 mg, 0.98 mmol, 35%). 1H NMR (600 MHz, CDCl3): δ 2.10 (s, 6H), 3.04 (s, 12H), 3.98 (s, 6H), 6.41 (s, 2H), 6.70 (d, J = 8.6 Hz, 4H), 7.53 (d, J = 8.5 Hz, 4H), 7.78 (d, J = 15.4 Hz, 2H), 7.84 (d, J = 15.5 Hz, 2H), 13.96 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 20.98, 40.30, 55.87, 103.40, 110.29, 112.07, 118.01, 122.95, 123.69, 129.01, 130.54, 144.05, 146.58, 152.03, 160.15, 162.83, 193.97. IR (ATR film): 1602, 1543, 1525, 1472, 1445, 1414, 1364, 1316, 1298, 1228, 1211, 1167, 1113, 1067, 979, 947, 866, 815, 732, 703, 651, 613, 569, 541, 469. HR-MS (ESI):m/z calcd for [C38H41N2O6]+ ([M + H+]): 621.2959, found: 621.2962. Melting point: 265 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one) (7ad)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 4-(trifluoromethyl)benzaldehyde (5d) (0.25 mL, 2.01 mmol, 2.4 + 1.2 equiv). The product was isolated as orange solids (232 mg, 0.35 mmol, 62%). 1H NMR (600 MHz, CDCl3): δ 2.12 (s, 6H), 4.00 (s, 6H), 6.45 (s, 2H), 7.67 (d, J = 8.2 Hz, 4H), 7.71 (d, J = 8.1 Hz, 4H), 7.76 (d, J = 15.6 Hz, 2H), 7.97 (d, J = 15.7 Hz, 2H), 13.59 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 21.10, 56.03, 103.55, 109.97, 117.77, 124.09 (q, J = 271.5), 126.00 (q, J = 3.8 Hz), 128.54, 130.49, 131.65 (q, J = 33.4 Hz), 139.17, 140.30, 148.08, 160.41, 162.94, 193.93. 19F NMR (282 MHz, CDCl3): δ −62.76. IR (ATR film): 2946, 2852, 1609, 1570, 1481, 1452, 1414, 1389, 1362, 1321, 1288, 1273, 1216, 1180, 1116, 1068, 1034, 1017, 979, 954, 834, 734, 716, 673, 652, 593, 571, 535. HR-MS (ESI):m/z calcd for [C36H29F6O6]+ ([M + H+]): 671.1863, found: 671.1866. Melting point: 208–209 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-bromophenyl)prop-2-en-1-one) (7ae)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 4-bromobenzaldehyde (5e) (248 mg 1.34 mmol, 2.4 equiv). The product was isolated as orange solids (364 mg, 0.53 mmol, 94%). 1H NMR (600 MHz, CDCl3): δ 2.11 (s, 6H), 3.99 (s, 6H), 6.43 (s, 2H), 7.47 (d, J = 8.5 Hz, 4H), 7.54 (d, J = 8.4 Hz, 3H), 7.71 (d, J = 15.6 Hz, 2H), 7.90 (d, J = 15.6 Hz, 2H), 13.65 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 21.06, 56.00, 103.51, 110.00, 117.80, 124.43, 128.73, 129.88, 132.29, 134.68, 141.07, 147.79, 160.35, 162.90, 194.02. IR (ATR film): 2970, 2941, 2848, 2251, 1627, 1605, 1559, 1485, 1389, 1359, 1323, 1213, 1178, 1141, 1114, 1072, 1035, 1009, 979, 946, 908, 875, 819, 801, 786, 731, 648, 632, 604, 571, 535, 507, 491. HR-MS (ESI):m/z calcd for [C34H29O6Br2]+ ([M + H+]): 691.0325, found: 691.0324. Melting point: 230–236 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(3-bromophenyl)prop-2-en-1-one) (7af)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 3-bromobenzaldehyde (5f) (0.16 mL, 1.34 mmol, 2.4 equiv). The product was isolated as orange solids (335 mg, 0.48 mmol, 86%). 1H NMR (600 MHz, CDCl3): δ 2.11 (s, 6H), 4.00 (s, 6H), 6.44 (s, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.51 (dt, J = 8.1, 1.5 Hz, 4H), 7.68 (d, J = 15.6 Hz, 2H), 7.74 (t, J = 1.8 Hz, 2H), 7.89 (d, J = 15.6 Hz, 2H), 13.63 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 21.09, 56.04, 103.51, 109.92, 117.71, 123.15, 127.20, 129.43, 130.53, 131.06, 132.94, 137.88, 140.62, 147.92, 160.37, 162.90, 193.93. IR (ATR film): 2922, 1630, 1469, 1389, 1323, 1274, 1180, 1116, 1035, 907, 864, 795, 778, 730, 648. HR-MS (ESI):m/z calcd for [C34H29O6Br2]+ ([M + H+]): 691.0325, found: 691.0324. Melting point: 110 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′-dimethoxy-6,6′-dimethyl-[1,1′-biphenyl]-3,3′-diyl)bis(3-(2-bromophenyl)prop-2-en-1-one) (7ag)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4a (200 mg, 0.56 mmol, 1.0 equiv) and 2-bromobenzaldehyde (5g) (0.16 mL, 1.34 mmol, 2.4 equiv). The product was isolated as orange solids (253 mg, 0.36 mmol, 65%). 1H NMR (600 MHz, CDCl3): δ 2.11 (s, 6H), 3.98 (s, 6H), 6.43 (s, 2H), 7.24 (td, J = 7.7, 1.6 Hz, 2H), 7.36 (td, J = 7.4, 1.0 Hz, 2H), 7.64 (dd, J = 8.0, 1.3 Hz, 2H), 7.70 (dd, J = 7.8, 1.6 Hz, 2H), 7.85 (d, J = 15.5 Hz, 2H), 8.11 (d, J = 15.6 Hz, 2H), 13.61 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 20.93, 55.83, 103.33, 109.85, 117.63, 125.88, 127.62, 127.96, 130.69, 130.86, 133.54, 135.70, 140.43, 147.70, 160.21, 162.79, 193.76. IR (ATR film): 2851, 1610, 1573, 1465, 1361, 1214, 1180, 1117, 1027, 907, 730, 535. HR-MS (ESI):m/z calcd for [C34H29O6Br2]+ ([M + H+]): 691.0325, found: 691.0323. TLC (petroleum ether/EtOAc, 6:4 v/v): Rf = 0.44 Melting point: 220–221 °C.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-methoxyphenyl)prop-2-en-1-one) (7ba)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.56 mmol, 1.0 equiv) and 4-methoxybenzaldehyde (5a) (0.15 mL, 1.23 mmol, 2.4 equiv). The product was isolated as yellow solids (145 mg, 0.25 mmol, 45%). The analytical data is in accordance with the literature.291H NMR (600 MHz, CDCl3): δ 3.86 (s, 12H), 4.01 (s, 6H), 6.14 (s, 2H), 6.93 (d, J = 8.6 Hz, 4H), 7.56 (d, J = 8.5 Hz, 4H), 7.76 (d, J = 15.5 Hz, 2H), 7.82 (d, J = 15.6 Hz, 2H), 14.20 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 55.55, 55.98, 56.08, 87.20, 103.27, 106.93, 114.50, 125.90, 128.69, 130.18, 142.07, 161.40, 162.93, 164.02, 164.78, 193.18. IR (ATR film): 1739, 1622, 1604, 1510, 1466, 1407, 1371, 1290, 1255, 1215, 1171, 1122, 829, 801, 776, 603, 559, 539, 520. HR-MS (ESI):m/z calcd for [C36H35O10]+ ([M + H+]): 627.2225, found: 627.2227. Melting point: 284–285 °C (282–285 °C).29
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-phenylprop-2-en-1-one) (7bb)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.51 mmol, 1.0 equiv) and benzaldehyde (5b) (0.19 mL, 1.85 mmol, 2.4 + 1.2 equiv). The product was isolated as yellow solids (65.8 mg, 0.13 mmol, 23%). 1H NMR (600 MHz, CDCl3): δ 3.86 (s, 6H), 4.02 (s, 6H), 6.15 (s, 2H), 7.37–7.44 (m, 6H), 7.61 (d, J = 7.1 Hz, 4H), 7.77 (d, J = 15.6 Hz, 2H), 7.91 (d, J = 15.6 Hz, 2H), 14.11 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 56.00, 56.10, 87.20, 103.20, 106.90, 128.25, 128.47, 129.00, 130.04, 135.94, 141.97, 163.06, 164.25, 164.80, 193.25. IR (ATR film): 2918, 2850, 1737, 1617, 1560, 1470, 1450, 1435, 1373, 1331, 1287, 1217, 1179, 1155, 1122, 1037, 870, 804, 762, 726, 703, 685, 662, 633, 576, 532, 477 HR-MS (ESI):m/z calcd for [C34H31O8]+ ([M + H+]): 567.2013, found: 567.2020. Melting point: 272 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-(dimethylamino)phenyl)prop-2-en-1-one) (7bc)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.51 mmol, 1.0 equiv) and 4-(dimethylamino)benzaldehyde (5c) (183.4 mg, 1.23 mmol, 2.4 equiv) at 90 °C. The product was isolated as red solids (80.2 mg, 0.13 mmol, 24%). (Due to poor solubility in CDCl3, and d6-DMSO, only a 1H NMR spectrum could be obtained.) 1H NMR (600 MHz, CDCl3): δ 3.04 (s, 12H), 3.84 (s, 6H), 4.00 (s, 6H), 6.13 (s, 2H), 6.70 (d, J = 8.6 Hz, 4H), 7.52 (d, J = 8.5 Hz, 4H), 7.76 (d, J = 15.4 Hz, 2H), 7.81 (d, J = 15.4 Hz, 2H), 14.37 (s, 2H). IR (ATR film): 2850, 1598, 1542, 1527, 1467, 1434, 1411, 1370, 1334, 1295, 1214, 1168, 1114, 1038, 996, 982, 970, 863, 818, 776, 722, 702, 662, 605, 543. HR-MS (ESI):m/z calcd for [C38H41N2O8]+ ([M + H+]): 653.2857, found: 653.2851. Melting point: 302 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one) (7bd)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.51 mmol, 1.0 equiv) and 4-(trifluoromethyl)benzaldehyde (5d) (0.17 mL, 1.23 mmol, 2.4 equiv). The product was isolated as yellow solids (95.0 mg, 0.15 mmol, 27%). 1H NMR (600 MHz, CDCl3): δ 3.87 (s, 6H), 4.02 (s, 6H), 6.15 (s, 2H), 7.66 (d, J = 8.2 Hz, 4H), 7.69 (d, J = 8.2 Hz, 4H), 7.73 (d, J = 15.7 Hz, 2H), 7.94 (d, J = 15.7 Hz, 2H), 13.99 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 56.05, 56.12, 87.21, 103.14, 106.80, 124.12 (q, J = 271.3 Hz), 125.96 (q, J = 3.8 Hz), 128.45, 130.61, 131.46 (q, J = 32.7 Hz), 139.36, 139.75, 163.13, 164.57, 164.83, 192.79. 19F NMR (282 MHz, CDCl3): δ −62.74. IR (ATR film): 2921, 2852, 1731, 1611, 1566, 1467, 1405, 1321, 1286, 1215, 1122, 1067, 1032, 1016, 954, 907, 834, 775, 732, 649, 597, 531. HR-MS (ESI):m/z calcd for [C36H29F6O8]+ ([M + H+]): 703.1761, found: 703.1767. Melting point: 251 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(4-bromophenyl)prop-2-en-1-one) (7be)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.56 mmol, 1.0 equiv) and 4-bromobenzaldehyde (5e) (227 mg, 1.23 mmol, 2.4 equiv). The product was isolated as yellow solids (202 mg, 0.31 mmol, 55%). 1H NMR (600 MHz, CDCl3): δ 3.86 (s, 6H), 4.01 (s, 6H), 6.14 (s, 2H), 7.45 (d, J = 8.4 Hz, 4H), 7.53 (d, J = 8.3 Hz, 4H), 7.67 (d, J = 15.6 Hz, 2H), 7.88 (d, J = 15.6 Hz, 2H), 14.05 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 56.03, 56.10, 87.20, 103.17, 106.83, 124.19, 128.83, 129.79, 132.24, 134.86, 140.51, 163.05, 164.38, 164.80, 192.92. IR (ATR film): 2918, 2850, 1738, 1628, 1556, 1486, 1471, 1436, 1399, 1372, 1328, 1291, 1214, 1180, 1154, 1090, 1073, 1033, 1009, 974, 820, 648, 631. HR-MS (ESI):m/z calcd for [C34H29Br2O8]+ ([M + H+]): 723.0220, found: 723.0224. Melting point: 274 °C (decomposition).
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(3-bromophenyl)prop-2-en-1-one) (7bf)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.56 mmol, 1.0 equiv) and 3-bromobenzaldehyde (5f) (0.14 mL, 1.23 mmol, 2.4 equiv). The product was obtained as yellow solids (124 mg). A 3:1 mix of chalcone:flavanone with monoaddition product present (according to 1H NMR). 1H NMR reported for the major chalcone product. 1H NMR (600 MHz, CDCl3): δ 3.86 (d, 6H), 4.03 (s, 6H), 6.15 (s, 2H), 7.29 (t, J = 7.9 Hz, 2H), 7.50 (d, J = 7.8 Hz, 4H), 7.65 (d, J = 15.5 Hz, 2H), 7.73 (s, 2H), 7.87 (d, J = 15.6 Hz, 2H), 14.03 (s, 2H). HR-MS (ESI):m/z calcd for [C34H29Br2O8]+ ([M + H+]): 723.0221, found: 723.0224.
rac-(2E,2′E)-1,1′-(2,2′-Dihydroxy-4,4′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(3-(2-bromophenyl)prop-2-en-1-one) (7bg)
Synthesized in accordance with General Procedure B. Starting from acetophenone 4b (200 mg, 0.56 mmol, 1.0 equiv) and 2-bromobenzaldehyde (5g) (0.14 mL, 1.23 mmol, 2.4 equiv), 1.5 h reaction time. The product was obtained as yellow solids (84.7 mg). A 5:1 mix of chalcone:flavanone with monoaddition product present (according to 1H NMR). 1H NMR reported for the major chalcone product. 1H NMR (600 MHz, CDCl3): δ 3.86 (s, 6H), 4.00 (s, 6H), 6.13 (s, 2H), 7.22 (t, J = 7.7 Hz, 2H), 7.35 (t, J = 7.8 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 7.6 Hz, 3H), 7.82 (d, J = 15.6 Hz, 2H), 8.07 (d, J = 15.6 Hz, 2H), 13.96 (s, 1H). HR-MS (ESI):m/z calcd for [C34H29Br2O8]+ ([M + H+]): 723.0216, found: 723.0224.
rac-5,5′-Dimethoxy-2,2′-bis(4-methoxyphenyl)-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9aa)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ab (30.0 mg, 50.7 μmol, 1.00 equiv) and I2 (78.8 mM, 63 μL, 5.1 μmol, 10 mol %) in 1 h. The product was isolated by column chromatography (EtOAc/MeOH, 95:5 v/v) and was obtained as white solids (15.8 mg, 30.2 μmol, 53%). The analytical data was in accordance with the literature.241H NMR (600 MHz, CDCl3): δ 2.18 (s, 6H), 3.79 (s, 6H), 4.11 (s, 6H), 6.64 (s, 2H), 6.78 (d, J = 8.9 Hz, 4H), 6.88 (s, 2H), 7.23 (d, J = 8.9 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 20.76, 55.63, 56.72, 107.23, 108.34, 112.96, 114.66, 116.56, 123.48, 127.32, 144.67, 155.77, 159.28, 160.94, 162.32, 178.62. IR (ATR film): 2844, 2238, 1605, 1576, 1512, 1496, 1478, 1464, 1442, 1423, 1371, 1335, 1301, 1207, 1116, 1062, 1031, 976, 956, 909, 730, 644, 590. TLC (EtOAc/MeOH, 95:5 v/v): Rf = 0.26 HR-MS (ESI):m/z calcd for [C36H31O8]+ ([M + H+]): 591.2013, found: 591.2020. Melting point: 295–296 °C.
rac-5,5′-Dimethoxy-7,7′-dimethyl-2,2′-diphenyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9ab)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ab (30.0 mg, 56.5 μmol, 1.00 equiv) and I2 (78.8 mM, 72 μL, 5.7 μmol, 10 mol %). The product was isolated by column chromatography (EtOAc/MeOH, 98:2 v/v) and was obtained as white solids (6.2 mg, 12 μmol, 21%). 1H NMR (600 MHz, CDCl3): δ 2.19 (s, 6H), 4.11 (s, 6H), 6.73 (s, 2H), 6.89 (s, 2H), 7.26–7.33 (m, 8H), 7.34–7.40 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 20.65, 56.57, 108.28, 108.50, 112.92, 116.35, 125.44, 129.01, 131.09, 131.29, 144.81, 155.66, 159.20, 160.64, 178.44. IR (ATR film): 3005, 2848, 1640, 1597, 1578, 1495, 1479, 1464, 1450, 1370, 1333, 1307, 1281, 1258, 1207, 1189, 1122, 1062, 976, 955, 908, 850, 771, 730, 689, 665, 645, 612, 551, 531. TLC (EtOAc/MeOH, 98:2 v/v): Rf = 0.28 HR-MS (ESI):m/z calcd for [C34H27O6]+ ([M + H+]): 531.1802, found: 531.1805. Melting point: 296 °C.
rac-2,2′-Bis(4-(dimethylamino)phenyl)-5,5′-dimethoxy-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9ac)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ac (30.0 mg, 48.6 μmol, 1.00 equiv) and I2 (78.8 mM, 62 μL, 4.9 μmol, 10 mol %). The product was isolated by column chromatography and was obtained as orange solids (6.3 mg, 10 μmol, 21%). The reaction was repeatedly scaled up with bichalcone 7ac (575 mg, 0.93 mmol, 1.00 equiv). The product was isolated as orange solids (123 mg, 0.20 μmol, 21%). The purity of biflavone 9ac was assessed by reverse-phase HPLC (97.3%) and was in accordance with purity determined by 1H NMR (96.9%). Stability experiments revealed that biflavone 9ac was stable in DMSO at up to 40 °C over 24 h. 1H NMR (600 MHz, CDCl3): δ 2.17 (s, 3H), 2.97 (s, 6H), 4.10 (s, 3H), 6.51 (d, J = 9.1 Hz, 4H), 6.58 (s, 1H), 6.86 (s, 1H), 7.16 (d, J = 9.0 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 20.71, 40.17, 56.64, 105.38, 108.02, 111.79, 112.83, 116.66, 117.70, 127.05, 144.20, 152.28, 155.70, 159.00, 161.92, 178.80. IR (ATR film): 2923, 2237, 1731, 1604, 1591, 1524, 1495, 1364, 1335, 1302, 1284, 1251, 1197, 1171, 1120, 1063, 908, 819, 761, 728, 662, 642, 582, 570, 544, 531, 512. TLC (EtOAc/CH2Cl2/MeOH, 7:2.5:0.5 v/v): Rf = 0.16 HR-MS (ESI):m/z calcd for [C38H37N2O6]+ ([M + H+]): 617.2646, found: 617.2653. Melting point: 196–197 °C.
rac-5,5′-Dimethoxy-7,7′-dimethyl-2,2′-bis(4-(trifluoromethyl)phenyl)-4H,4′H-[8,8′-bichromene]-4,4′-dione (9ad)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ad (30.0 mg, 45.0 μmol, 1.00 equiv) and I2 (78.8 mM, 57 μL, 4.5 μmol, 10 mol %) for 2 h. The product was isolated by column chromatography (petroleum ether/EtOAc/isopropanol, 6:3:1 v/v) and was obtained as a white viscous semisolid (8.5 mg, 13 μmol, 28%). 1H NMR (600 MHz, CDCl3): δ 2.21 (s, 6H), 4.12 (s, 6H), 6.78 (s, 2H), 6.93 (s, 2H), 7.40 (d, J = 8.2 Hz, 4H), 7.56 (d, J = 8.3 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 20.83, 56.78, 108.68, 109.89, 113.06, 116.21, 123.61 (q, J = 272.1 Hz), 125.84, 126.22 (q, J = 3.7 Hz), 133.12 (q, J = 33.1 Hz), 134.59, 145.37, 155.68, 159.03, 159.52, 178.08. 19F NMR (282 MHz, CDCl3): δ −63.13. IR (ATR film): 2854, 1817, 1646, 1600, 1579, 1497, 1480, 1466, 1445, 1417, 1323, 1295, 1207, 1125, 1063, 1015, 977, 957, 909, 844, 777, 731, 625, 557, 532, 518. TLC (petroleum ether/EtOAc/isopropanol, 6:3:1 v/v): Rf = 0.21 HR-MS (ESI):m/z calcd for [C36H25F6O6]+ ([M + H+]): 667.1555, found: 667.1550. Melting point: 263–265 °C.
rac-2,2′-Bis(4-bromophenyl)-5,5′-dimethoxy-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9ae)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ae (30.0 mg, 43.6 μmol, 1.00 equiv) and I2 (78.8 mM, 55 μL, 4.4 μmol, 10 mol %). The product was obtained as white solids (27.7 mg, 40.1 μmol, 94%). 1H NMR (600 MHz, CDCl3): δ 2.18 (s, 6H), 4.10 (s, 6H), 6.69 (s, 2H), 6.89 (s, 2H), 7.14 (d, J = 8.7 Hz, 4H), 7.42 (d, J = 8.7 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 20.82, 56.74, 108.49, 108.77, 112.94, 116.23, 126.19, 126.93, 130.08, 132.49, 145.14, 155.62, 159.37, 159.72, 178.26. IR (ATR film): 3005, 2931, 2851, 2240, 1773, 1638, 1597, 1562, 1479, 1464, 1403, 1368, 1329, 1303, 1275, 1260, 1207, 1187, 1167, 1122, 1061, 1030, 1008, 977, 955, 907, 830, 794, 681, 645, 626, 573, 557, 532, 499, 478. HR-MS (ESI):m/z calcd for [C34H25O4Br2]+ ([M + H+]): 687.0012, found: 687.0017. Melting point: 245 °C (decomposition).
rac-2,2′-Bis(3-bromophenyl)-5,5′-dimethoxy-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9af)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7af (30.0 mg, 43.6 μmol, 1.00 equiv) and I2 (78.8 mM, 55 μL, 4.4 μmol, 10 mol %) for 1 h. The product was isolated by column chromatography (EtOAc 100%) and was obtained as white solids (25.4 mg, 37.5 μmol, 86%). 1H NMR (600 MHz, CDCl3): δ 2.24 (s, 6H), 4.11 (s, 6H), 6.70 (s, 2H), 6.94 (s, 2H), 7.18 (t, J = 7.9 Hz, 2H), 7.31 (dt, J = 8.0, 1.4 Hz, 2H), 7.34 (t, J = 1.9 Hz, 2H), 7.49 (ddd, J = 8.0, 2.0, 1.0 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 20.84, 56.73, 108.62, 109.16, 113.03, 116.16, 123.38, 123.91, 128.77, 130.58, 133.21, 134.18, 145.20, 155.49, 158.77, 159.50, 178.11. IR (ATR film): 2851, 2241, 1642, 1598, 1561, 1496, 1478, 1442, 1417, 1367, 1299, 1266, 1206, 1124, 1062, 998, 956, 917, 847, 790, 730, 692, 646, 619, 571, 551, 532. TLC (EtOAc 100%): Rf = 0.26 HR-MS (ESI):m/z calcd for [C34H25O4Br2]+ ([M + H+]): 687.0012, found: 687.0005. Melting point: 276–278 °C.
rac-2,2′-Bis(2-bromophenyl)-5,5′-dimethoxy-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9ag)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ag (30.0 mg, 43.6 μmol, 1.00 equiv) and I2 (78.8 mM, 55 μL, 4.4 μmol, 10 mol %) for 2 h. The product was obtained as white solids (16.5 mg, 24.0 μmol, 55%). 1H NMR (600 MHz, CDCl3): δ 2.12 (s, 6H), 4.02 (s, 6H), 6.46 (s, 2H), 6.78 (s, 2H), 7.20 (td, J = 7.4, 6.9, 2.0 Hz, 2H), 7.21–7.29 (m, 4H), 7.51 (dd, J = 7.8, 1.5 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 20.93, 56.54, 108.54, 113.00, 114.33, 116.56, 121.57, 127.67, 130.73, 131.78, 133.49, 134.22, 144.91, 156.39, 159.23, 161.62, 178.19. IR (ATR film): 3300, 2924, 2869, 2852, 1733, 1653, 1600, 1562, 1496, 1464, 1437, 1370, 1327, 1279, 1263, 1206, 1115, 1065, 976, 954, 912, 855, 763, 733, 703, 680, 645, 612. HR-MS (ESI):m/z calcd for [C34H25O4Br2]+ ([M + H+]): 687.0012, found: 687.0011. Melting point: 250 °C (decomposition).
rac-5,5′,7,7′-Tetramethoxy-2,2′-bis(4-methoxyphenyl)-4H,4′H-[8,8′-bichromene]-4,4′-dione or Hexa-O-methylcupressuflavone (9ba)
Synthesized in accordance with General Procedure D. Starting from bichalcone 7ba (30.0 mg, 48.2 μmol, 1.00 equiv) and I2 (78.8 mM, 61 μL, 4.8 μmol, 10 mol %). The product was isolated by column chromatography (EtOAc/MeOH, 9:1 v/v) and was obtained as white solids (23.1 mg, 37.1 μmol, 77%). The analytical data was in accordance with the literature.291H NMR (600 MHz, CDCl3): δ 3.77 (s, 6H), 3.86 (s, 6H), 4.12 (s, 6H), 6.58 (s, 2H), 6.59 (s, 2H), 6.77 (d, J = 8.9 Hz, 4H), 7.29 (d, J = 8.9 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 55.47, 56.13, 56.60, 91.78, 102.13, 106.80, 109.05, 114.39, 123.54, 127.15, 156.57, 160.66, 161.29, 161.74, 162.03, 178.14. IR (ATR film): 1637, 1593, 1465, 1424, 1388, 1338, 1304, 1180, 1111, 1033, 919, 834, 588, 567. TLC (EtOAc/MeOH, 9:1 v/v): Rf = 0.22 HR-MS (ESI):m/z calcd for [C36H31O10]+ ([M + H+]): 623.1912, found: 623.1912. Melting point: 240.0 °C (brown discoloration) 285–286 °C (294–295 °C).29
rac-5,5′,7,7′-Tetramethoxy-2,2′-diphenyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (9bb)
Synthesized in accordance with General Procedure D. Starting from 7bb (30.0 mg, 53.3 μmol, 1.00 equiv) and I2 (78.8 mM, 68 μL, 5.3 μmol, 10 mol %) for 2 h. The product was isolated by column chromatography (EtOAc/MeOH, 98:2 v/v) and was obtained as white solids (17.6 mg, 31.4 μmol, 59%). The analytical data was in accordance with the literature.341H NMR (600 MHz, CDCl3): δ 3.85 (s, 6H), 4.12 (s, 6H), 6.59 (s, 2H), 6.68 (s, 2H), 7.28 (d, J = 7.4 Hz, 4H), 7.33–7.40 (m, 6H). 13C NMR (151 MHz, CDCl3): δ 56.25, 56.72, 91.92, 102.19, 108.36, 109.29, 125.56, 129.01, 131.23, 131.42, 156.77, 160.62, 161.51, 161.99, 178.19. IR (ATR film): 3067, 2844, 2238, 1635, 1590, 1508, 1491, 1465, 1450, 1435, 1388, 1333, 1299, 1265, 1214, 1190, 1172, 1125, 1109, 1087, 1036, 1022, 957, 917, 849, 812, 771, 728, 689, 672, 644, 618, 570, 546, 514. TLC (EtOAc/MeOH, 98:2 v/v): Rf = 0.12 HR-MS (ESI):m/z calcd for [C34H27O8]+ ([M + H+]): 563.1700, found: 563.1707. Melting point: 312–313 °C (318–319 °C).34
rac-2,2′-Bis(4-(dimethylamino)phenyl)-5,5′,7,7′-tetramethoxy-4H,4′H-[8,8′-bichromene]-4,4′-dione (9bc)
Synthesized in accordance with General Procedure D. Starting from 7bc (30.0. mg, 46.3 μmol, 1.00 equiv) and I2 (78.8 mM, 59 μL, 4.6 μmol, 10 mol %). The product was isolated by column chromatography (CH2Cl2/EtOAc/MeOH, 5:4.5:0.5 v/v) and obtained as orange solids (7.7 mg, 12 μmol, 26%). 1H NMR (600 MHz, CDCl3): δ 2.95 (s, 12H), 3.86 (s, 6H), 4.11 (s, 6H), 6.50 (d, J = 8.9 Hz, 4H), 6.53 (s, 2H), 6.56 (s, 2H), 7.21 (d, J = 9.0 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 40.19, 56.24, 56.71, 91.82, 102.44, 105.21, 109.21, 111.78, 118.11, 127.09, 152.25, 156.70, 161.21, 161.61, 161.82, 178.44. IR (ATR film): 2942, 2234, 1630, 1601, 1586, 1524, 1487, 1465, 1434, 1387, 1366, 1336, 1305, 1253, 1214, 1198, 1171, 1124, 1064, 1030, 1001, 947, 917, 840, 819, 783, 766, 729, 664, 643, 582, 563, 513, 486. TLC (CH2Cl2/EtOAc/MeOH, 5:4.5:0.5 v/v): Rf = 0.14 HR-MS (ESI):m/z calcd for [C38H37N2O8]+ ([M + H+]): 649.2544, found: 649.2546.
rac-5,5′,7,7′-Tetramethoxy-2,2′-bis(4-(trifluoromethyl)phenyl)-4H,4′H-[8,8′-bichromene]-4,4′-dione (9bd)
Synthesized in accordance with General Procedure D. Starting from 7bd (30.0 mg, 42.9 μmol, 1.00 equiv) and I2 (78.8 mM, 54 μL, 4.3 μmol, 10 mol %) for 2 h. The product was isolated by column chromatography (petroleum ether/EtOAc/iPrOH, 2:7:1 v/v) and obtained as white solids (17.7 mg, 25.3 μmol, 59%). 1H NMR (600 MHz, CDCl3): δ 3.89 (s, 6H), 4.15 (s, 6H), 6.62 (s, 2H), 6.73 (s, 2H), 7.46 (d, J = 8.2 Hz, 4H), 7.56 (d, J = 8.3 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 56.32, 56.77, 92.04, 101.94, 109.24, 109.63, 123.64 (q, J = 271.8 Hz), 125.80, 126.09 (q, J = 3.6 Hz), 132.92 (q, J = 32.9 Hz), 134.79, 156.67, 158.84, 161.72, 162.24, 177.67. 19F NMR (282 MHz, CDCl3): δ −63.08. IR (ATR film): 1622, 1594, 1416, 1388, 1324, 1216, 1170, 1128, 1070, 1027, 958, 919, 843, 812. TLC (petroleum ether/EtOAc/iPrOH, 2:7:1 v/v): Rf = 0.35 HR-MS (ESI):m/z calcd for [C36H25N2O8]+ ([M + H+]): 699.1448, found: 699.1450. Melting point: 261–262 °C.
rac-2,2′-Bis(4-bromophenyl)-5,5′,7,7′-tetramethoxy-4H,4′H-[8,8′-bichromene]-4,4′-dione (9be)
Synthesized in accordance with General Procedure D. Starting from 7be (30.0 mg, 41.6 μmol, 1.00 equiv) and I2 (78.8 mM, 53 μL, 4.2 μmol, 10 mol %) for 2 h. The product was isolated by column chromatography (petroleum ether/EtOAc/iPrOH, 2:7:1 v/v) and obtained as white solids (20.9 mg, 29.1 μmol, 70%). 1H NMR (600 MHz, CDCl3): δ 3.86 (s, 6H), 4.12 (s, 6H), 6.58 (s, 2H), 6.64 (s, 2H), 7.20 (d, J = 8.4 Hz, 4H), 7.42 (d, J = 8.5 Hz, 4H). 13C NMR (151 MHz, CDCl3): δ 56.28, 56.74, 91.93, 101.96, 108.51, 109.17, 125.95, 126.92, 130.30, 132.35, 156.60, 159.53, 161.60, 162.07, 177.85. IR (ATR film): 2885, 2239, 1638, 1590, 1508, 1487, 1465, 1435, 1403, 1388, 1331, 1302, 1274, 1215, 1190, 1171, 1127, 1107, 1073, 1027, 1009, 958, 828, 782, 730, 687, 644, 626, 572, 545, 517, 477. TLC (petroleum ether/EtOAc/iPrOH, 2:7:1 v/v): Rf = 0.21 HR-MS (ESI):m/z calcd for [C34H25O8Br2]+ ([M + H+]): 718.9911, found: 718.9915. Melting point: 251–255 °C.
rac-2,2′-Bis(2-bromophenyl)-5,5′,7,7′-tetramethoxy-4H,4′H-[8,8′-bichromene]-4,4′-dione (9bg)
Synthesized in accordance with General Procedure D. Starting from 7bg (30.0 mg, 41.6 μmol, 1.00 equiv) and I2 (78.8 mM, 53 μL, 4.2 μmol, 10 mol %) for 1 h. The product was isolated by column chromatography (EtOAc/MeOH, 9:1 v/v) and obtained as white solids (16.7 mg, 23.3 μmol, 56%). 1H NMR (600 MHz, CDCl3): δ 3.84 (s, 6H), 4.06 (s, 6H), 6.47 (s, 2H), 6.50 (s, 2H), 7.23 (ddd, J = 7.8, 4.0, 2.4 Hz, 4H), 7.27–7.29 (m, 2H), 7.53–7.57 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 56.20, 56.58, 91.98, 102.18, 109.12, 114.04, 121.57, 127.62, 130.63, 131.65, 133.50, 134.24, 157.33, 161.14, 161.41, 162.13, 177.82. IR (ATR film): 3493, 2940, 2842, 2244, 1646, 1593, 1472, 1434, 1389, 1336, 1214, 1109, 1025, 730. TLC (EtOAc/MeOH, 9:1 v/v): Rf = 0.32 HR-MS (ESI):m/z calcd for [C34H25O8Br2]+ ([M + H+]): 718.9911, found: 718.9916 Melting point: 260 °C (decomposition).
rac-2,2′-Bis(4-(dimethylamino)phenyl)-5,5′-dihydroxy-7,7′-dimethyl-4H,4′H-[8,8′-bichromene]-4,4′-dione (11)
A dry 25 mL Schlenk round-bottom flask was charged with biflavone 9ac (40.0 mg, 0.06 mmol, 1.0 equiv) and anhydr. CH2Cl2 (2.5 mL). The solution was cooled to −78 °C, and BCl3 (1 M in CH2Cl2, 0.14 mL, 0.14 mmol, 2.2 equiv) was added dropwise. The cooling bath was left to warm up overnight. After 16 h of reaction time, KPi-buffer (K2HPO4/KH2PO4, 1 M, pH 7, 10 mL) was added. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL), and the combined organic phases were washed with sat. aq. NaCl solution (20 mL). The crude product was isolated by column chromatography (EtOAc/MeOH, 9:1 v/v) and obtained as orange crystals (16.9 mg, 0.03 μmol, 44%). 1H NMR (600 MHz, CDCl3): δ 2.15 (s, 6H), 3.01 (s, 12H), 6.56 (s, 2H), 6.58 (d, J = 9.0 Hz, 4H), 6.87 (s, 2H), 7.28 (d, J = 9.0 Hz, 4H), 12.99 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 20.78, 40.17, 102.04, 109.12, 111.85, 112.58, 113.33, 117.29, 127.63, 146.42, 152.81, 153.79, 160.16, 165.32, 183.41. IR (ATR film): 2923, 1649, 1588, 1520, 1482, 1360, 1246, 1201, 1163, 1110, 1034, 814. TLC (petroleum ether/EtOAc, 55:45 v/v): Rf = 0.3 HR-MS (ESI):m/z calcd for [C36H33N2O6]+ ([M + H+]): 589.2333, found: 589.2340 Melting point: 280–283 °C.
Acknowledgments
The authors gratefully acknowledge the DFG (GRK2158), the Forschungszentrum Jülich GmbH, and the Heinrich Heine University (HHU) for their generous support. The authors thank Ruth Ganardi, Sebastian Myllek, Karin Buchholz, Daniel Degrandi, and Ursula Sorg for their scientific consultation, Birgit Henßen for HPLC support, and Vera Ophoven for synthesis support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c06503
Coordinates of the X-ray structure of compound 4a; bioactivity data; additional synthesis procedures; computational details including all coordinates; copies of all 1H and 13C spectral data; HPLC chromatograms; and material and methods for biological assays as well as biological activity graphs (PDF)
Compound 4a piet13 (CIF)
Author Present Address
Institute of Bioorganic Chemistry, Heinrich Heine University Düsseldorf, Forschungszentrum Jülich, Stetternicher Forst, Geb.15.8, 52426 Jülich (Germany)
Author Contributions
# F.M., L.B. and J.G. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Murti V.; Raman P.; Seshadri T. Cupressuflavone, a new biflavonyl pigment. Tetrahedron 1967, 23, 397–404. 10.1016/S0040-4020(01)83325-1. [DOI] [Google Scholar]
- Molyneux R.; Waiss A. Jr; Haddon W. Oxidative coupling of apigenin. Tetrahedron 1970, 26, 1409–1416. 10.1016/S0040-4020(01)92970-9. [DOI] [Google Scholar]
- Rahman W.; Bhatnagar S. A new biflavonyl AC3 from Araucaria cunninghamii. Tetrahedron Lett. 1968, 9, 675–678. 10.1016/S0040-4039(00)75610-3. [DOI] [Google Scholar]
- Murti V.; Raman P.; Seshadri T. Cupressuflavone, a new member of the biflavonyl group. Tetrahedron Lett. 1964, 5, 2995–2997. 10.1016/0040-4039(64)83077-X. [DOI] [Google Scholar]
- Natarajan S.; Murti V.; Seshadri T. Biflavones of some Cupressaceae plants. Phytochemistry 1970, 9, 575–579. 10.1016/S0031-9422(00)85693-9. [DOI] [Google Scholar]
- Inatomi Y.; Iida N.; Murata H.; Inada A.; Murata J.; Lang F. A.; Iinuma M.; Tanaka T.; Nakanishi T. A pair of new atropisomeric cupressuflavone glucosides isolated from Juniperus communis var. depressa. Tetrahedron Lett. 2005, 46, 6533–6535. 10.1016/j.tetlet.2005.07.091. [DOI] [Google Scholar]
- Yu S.; Yan H.; Zhang L.; Shan M.; Chen P.; Ding A.; Li S. F. Y. A review on the phytochemistry, pharmacology, and pharmacokinetics of amentoflavone, a naturally-occurring biflavonoid. Molecules 2017, 22, 299 10.3390/molecules22020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. P.; Park H.; Son K. H.; Chang H. W.; Kang S. S. Biochemical pharmacology of biflavonoids: implications for anti-inflammatory action. Arch. Pharmacal. Res. 2008, 31, 265–273. 10.1007/s12272-001-1151-3. [DOI] [PubMed] [Google Scholar]
- Lin Y.-M.; Flavin M. T.; Schure R.; Chen F.-C.; Sidwell R.; Barnard D. I.; Huffmann J. H.; Kern E. R. Antiviral activities of biflavonoids. Planta Med. 1999, 65, 120–125. 10.1055/s-1999-13971. [DOI] [PubMed] [Google Scholar]
- Adnan M.; Rasul A.; Hussain G.; Shah M. A.; Zahoor M. K.; Anwar H.; Sarfraz I.; Riaz A.; Manzoor M.; Adem Ş.; Selamoglu Z. Ginkgetin: A natural biflavone with versatile pharmacological activities. Food Chem. Toxicol. 2020, 145, 111642 10.1016/j.fct.2020.111642. [DOI] [PubMed] [Google Scholar]
- Yamaguchi L. F.; Kato M. J.; Di Mascio P. Biflavonoids from Araucaria angustifolia protect against DNA UV-induced damage. Phytochemistry 2009, 70, 615–620. 10.1016/j.phytochem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Gontijo V. S.; dos Santos M. H.; Viegas C. Jr Biological and chemical aspects of natural biflavonoids from plants: a brief review. Mini Rev. Med. Chem. 2017, 17, 834–862. 10.2174/1389557517666161104130026. [DOI] [PubMed] [Google Scholar]
- Tasdemir D.; Kaiser M.; Brun R.; Yardley V.; Schmidt T. J.; Tosun F.; Rüedi P. Antitrypanosomal and antileishmanial activities of flavonoids and their analogues: in vitro, in vivo, structure-activity relationship, and quantitative structure-activity relationship studies. Antimicrob. Agents Chemother. 2006, 50, 1352–1364. 10.1128/AAC.50.4.1352-1364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sayed E.; Gad H. A.; El-Shazly M.; Abdel-Daim M. M.; Singab A. N. Anti-inflammatory and analgesic activities of cupressuflavone from Cupressus macrocarpa: Impact on pro-inflammatory mediators. Drug Dev. Res. 2018, 79, 22–28. 10.1002/ddr.21417. [DOI] [PubMed] [Google Scholar]
- Freitas A.; Almeida M.; Andrighetti-Fröhner C.; Cardozo F.; Barardi C.; Farias M.; Simões C. Antiviral activity-guided fractionation from Araucaria angustifolia leaves extract. J. Ethnopharmacol. 2009, 126, 512–517. 10.1016/j.jep.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Siddiqui J. A.; Swarnkar G.; Sharan K.; Chakravarti B.; Sharma G.; Rawat P.; Kumar M.; Khan F. M.; Pierroz D.; Maurya R. 8, 8 ″-Biapigeninyl stimulates osteoblast functions and inhibits osteoclast and adipocyte functions: Osteoprotective action of 8, 8 ″-biapigeninyl in ovariectomized mice. Mol. Cell. Endocrinol. 2010, 323, 256–267. 10.1016/j.mce.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Al-Sayed E.; Abdel-Daim M. M. Protective role of Cupressuflavone from Cupressus macrocarpa against carbon tetrachloride-induced hepato-and nephrotoxicity in mice. Planta Med. 2014, 80, 1665–1671. 10.1055/s-0034-1383211. [DOI] [PubMed] [Google Scholar]
- Sasaki H.; Kitoh Y.; Tsukada M.; Miki K.; Koyama K.; Juliawaty L. D.; Hakim E. H.; Takahashi K.; Kinoshita K. Inhibitory activities of biflavonoids against amyloid-β peptide 42 cytotoxicity in PC-12 cells. Bioorg. Med. Chem. Lett. 2015, 25, 2831–2833. 10.1016/j.bmcl.2015.04.106. [DOI] [PubMed] [Google Scholar]
- DeForest J. C.; Du L.; Joyner P. M. 4′, 4‴, 7, 7 ″-Tetra-O-methylcupressuflavone Inhibits Seed Germination of Lactuca sativa. J. Nat. Prod. 2014, 77, 1093–1096. 10.1021/np4010739. [DOI] [PubMed] [Google Scholar]
- Frank M.; Niemann H.; Böhler P.; Stork B.; Wesselborg S.; Lin W.; Proksch P. Phomoxanthone A-from mangrove forests to anticancer therapy. Curr. Med. Chem. 2015, 22, 3523–3532. 10.2174/0929867322666150716115300. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Krohn K.; Flörke U.; Pescitelli G.; Di Bari L.; Antus S.; Kurtán T.; Rheinheimer J.; Draeger S.; Schulz B. New Mono-and Dimeric Members of the Secalonic Acid Family: Blennolides A–G Isolated from the Fungus Blennoria sp. Chem. - Eur. J. 2008, 14, 4913–4923. 10.1002/chem.200800035. [DOI] [PubMed] [Google Scholar]
- Kikuchi H.; Isobe M.; Sekiya M.; Abe Y.; Hoshikawa T.; Ueda K.; Kurata S.; Katou Y.; Oshima Y. Structures of the dimeric and monomeric chromanones, gonytolides A–C, isolated from the fungus Gonytrichum sp. and their promoting activities of innate immune responses. Org. Lett. 2011, 13, 4624–4627. 10.1021/ol2018449. [DOI] [PubMed] [Google Scholar]
- Sirimangkalakitti N.; Juliawaty L. D.; Hakim E. H.; Waliana I.; Saito N.; Koyama K.; Kinoshita K. Naturally occurring biflavonoids with amyloid β aggregation inhibitory activity for development of anti-Alzheimer agents. Bioorg. Med. Chem. Lett. 2019, 29, 1994–1997. 10.1016/j.bmcl.2019.05.020. [DOI] [PubMed] [Google Scholar]
- Kikuchi H.; Hoshikawa T.; Kurata S.; Katou Y.; Oshima Y. Design and synthesis of Structure-Simplified derivatives of Gonytolide for the promotion of innate immune responses. J. Nat. Prod. 2016, 79, 1259–1266. 10.1021/acs.jnatprod.5b00829. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49. 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- Brown D. G.; Bostrom J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? Miniperspective. J. Med. Chem. 2016, 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kaur M.; Silakari O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. 10.1016/j.ejmech.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Razaq S. A new approach to the synthesis of symmetrical biflavones. Tetrahedron Lett. 1971, 12, 4633–4636. 10.1016/S0040-4039(01)97549-5. [DOI] [Google Scholar]
- Ahmad S.; Razaq S. New synthesis of biflavones of cupressuflavone series. Tetrahedron 1976, 32, 503–506. 10.1016/0040-4020(76)80071-3. [DOI] [Google Scholar]
- Li H.-Y.; Nehira T.; Hagiwara M.; Harada N. Total Synthesis and Absolute Stereochemistry of the Natural Atropisomer of the Biflavone 4′, 4″′, 7, 7″-Tetra-O-methylcupressuflavone. J. Org. Chem. 1997, 62, 7222–7227. 10.1021/jo970670w. [DOI] [PubMed] [Google Scholar]
- Lin G.-Q.; Zhong M. The first enantioselective synthesis of optically pure (R)-and (S)-5, 5″-dihydroxy-4′, 4″′, 7, 7″-tetramethoxy-8, 8″-biflavone and the reconfirmation of their absolute configuration. Tetrahedron Lett. 1997, 38, 1087–1090. 10.1016/S0040-4039(96)02475-6. [DOI] [Google Scholar]
- Parthasarathy M.; Gupta S. Oxidative coupling of phloroacetophenone dimethyl ether, resacetophenone and resacetophenone monomethyl ether using silica-bound FeCl3. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1984, 23, 227–230. [Google Scholar]
- Lee C. Y.; Cheon C. H. Diastereomeric Resolution of a Racemic Biarylboronic Acid and Its Application to Divergent Asymmetric Total Syntheses of Some Axially Chiral Natural Products. Adv. Synth. Catal. 2016, 358, 549–554. 10.1002/adsc.201500798. [DOI] [Google Scholar]
- Zhang F. J.; Lin G. Q. Synthesis of the optically pure 5, 5″-dihydroxy-7, 7″-dimethoxy-8, 8″-biflavone and its derivatives. Chin. J. Chem. 1997, 15, 464–471. 10.1002/cjoc.19970150513. [DOI] [Google Scholar]
- Bringmann G.; Mortimer A. J. P.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atropselektive Synthese axial-chiraler Biaryle. Angew. Chem. 2005, 117, 5518–5563. 10.1002/ange.200462661. [DOI] [Google Scholar]
- Bringmann G.; Günther C.; Ochse M.; Schupp O.; Tasler S.. Biaryls in nature: a multi-facetted class of stereochemically, biosynthetically, and pharmacologically intriguing secondary metabolites. In Progress in the Chemistry of Organic Natural Products; Springer, 2001; Vol. 82, pp 1–249. [DOI] [PubMed] [Google Scholar]
- Wu X.; Iwata T.; Scharf A.; Qin T.; Reichl K. D.; Porco J. A. Jr Asymmetric synthesis of gonytolide A: strategic use of an aryl halide blocking group for oxidative coupling. J. Am. Chem. Soc. 2018, 140, 5969–5975. 10.1021/jacs.8b02535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T.; Skraba-Joiner S. L.; Khalil Z. G.; Johnson R. P.; Capon R. J.; Porco J. A. Jr Atropselective syntheses of (−) and (+) rugulotrosin A utilizing point-to-axial chirality transfer. Nat. Chem. 2015, 7, 234–240. 10.1038/nchem.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles R. G.; Sargent M. Naturally-Occurring Dibenzofurans. X. A New Synthesis of Di-O-Methylstrepsilin. Aust. J. Chem. 1986, 39, 2177–2181. 10.1071/CH9862177. [DOI] [Google Scholar]
- Drochner D.; Hüttel W.; Bode S. E.; Müller M.; Karl U.; Nieger M.; Steglich W. Dimeric orsellinic acid derivatives: Valuable intermediates for natural product synthesis. Eur. J. Org. Chem. 2007, 2007, 1749–1758. 10.1002/ejoc.200600899. [DOI] [Google Scholar]
- Hauser F. M.; Gauuan P. J. F. Total synthesis of (±)-Biphyscion. Org. Lett. 1999, 1, 671–672. 10.1021/ol990758r. [DOI] [PubMed] [Google Scholar]
- Rahman M.; Riaz M.; Desai U. R. Synthesis of biologically relevant biflavanoids–a review. Chem. Biodiversity 2007, 4, 2495–2527. 10.1002/cbdv.200790205. [DOI] [PubMed] [Google Scholar]
- Kuttruff C. A.; Eastgate M. D.; Baran P. S. Natural product synthesis in the age of scalability. Nat. Prod. Rep. 2014, 31, 419–432. 10.1039/C3NP70090A. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhou R.-G.; Wu T.; Yang T.; Qin Q.-X.; Li I.; Yang B.; Yang J. Total synthesis of apigenin. J. Chem. Res. 2012, 36, 121–122. 10.3184/174751912X13285269293913. [DOI] [Google Scholar]
- Königs P.; Rinker B.; Maus L.; Nieger M.; Rheinheimer J.; Waldvogel S. Structural revision and synthesis of altechromone A. J. Nat. Prod. 2010, 73, 2064–2066. 10.1021/np1005604. [DOI] [PubMed] [Google Scholar]
- Freitas M.; Ribeiro D.; Tome S. M.; Silva A. M.; Fernandes E. Synthesis of chlorinated flavonoids with anti-inflammatory and pro-apoptotic activities in human neutrophils. Eur. J. Med. Chem. 2014, 86, 153–164. 10.1016/j.ejmech.2014.08.035. [DOI] [PubMed] [Google Scholar]
- Dianin A. About Products from the Oxidation of Naphthols with Ferric Chloride. Zh. Russ. Fiz.-Khim. O-va. 1874, 6, 183–193. [Google Scholar]
- Tanaka K.; Toda F. Oxidative coupling reactions of phenols with FeCl3 in the solid state. Mol. Cryst. Liq. Cryst. Incorporating Nonlinear Opt. 1990, 187, 49–52. 10.1080/00268949008036026. [DOI] [Google Scholar]
- Keinan E.; Mazur Y. Reactions in dry media. Ferric chloride adsorbed on silica gel. A multipurpose, easily controllable reagent. J. Org. Chem. 1978, 43, 1020–1022. 10.1021/jo00399a057. [DOI] [Google Scholar]
- Jempty T. C.; Miller L. L.; Mazur Y. Oxidative coupling reactions using silica-bound ferric chloride. J. Org. Chem. 1980, 45, 749–751. 10.1021/jo01292a051. [DOI] [Google Scholar]
- Drochner D.; Hüttel W.; Nieger M.; Müller M. Unselective Phenolic Coupling of Methyl 2-Hydroxy-4-methoxy-6-methylbenzoate—A Valuable Tool for the Total Synthesis of Natural Product Families. Angew. Chem. 2003, 115, 961–963. 10.1002/ange.200390216. [DOI] [PubMed] [Google Scholar]
- Hüttel W.; Nieger M.; Müller M. A Short and Efficient TotalSynthesis of the Naturally Occurring Coumarins Siderin, Kotanin, Isokotanin A and Desertorin C. Synthesis 2003, 1803–1808. 10.1055/s-2003-41027. [DOI] [Google Scholar]
- Nieves-Quinones Y.; Paniak T. J.; Lee Y. E.; Kim S. M.; Tcyrulnikov S.; Kozlowski M. C. Chromium-salen catalyzed cross-coupling of phenols: mechanism and origin of the selectivity. J. Am. Chem. Soc. 2019, 141, 10016–10032. 10.1021/jacs.9b03890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libman A.; Shalit H.; Vainer Y.; Narute S.; Kozuch S.; Pappo D. Synthetic and predictive approach to unsymmetrical biphenols by iron-catalyzed chelated radical–anion oxidative coupling. J. Am. Chem. Soc. 2015, 137, 11453–11460. 10.1021/jacs.5b06494. [DOI] [PubMed] [Google Scholar]
- Kang H.; Herling M. R.; Niederer K. A.; Lee Y. E.; Vasu Govardhana Reddy P.; Dey S.; Allen S. E.; Sung P.; Hewitt K.; Torruellas C.; et al. Enantioselective vanadium-catalyzed oxidative coupling: development and mechanistic insights. J. Org. Chem. 2018, 83, 14362–14384. 10.1021/acs.joc.8b02083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. E.; Cao T.; Torruellas C.; Kozlowski M. C. Selective oxidative homo-and cross-coupling of phenols with aerobic catalysts. J. Am. Chem. Soc. 2014, 136, 6782–6785. 10.1021/ja500183z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalit H.; Libman A.; Pappo D. meso-Tetraphenylporphyrin iron chloride catalyzed selective oxidative cross-coupling of phenols. J. Am. Chem. Soc. 2017, 139, 13404–13413. 10.1021/jacs.7b05898. [DOI] [PubMed] [Google Scholar]
- Shalit H.; Dyadyuk A.; Pappo D. Selective oxidative phenol coupling by iron catalysis. J. Org. Chem. 2019, 84, 1677–1686. 10.1021/acs.joc.8b03084. [DOI] [PubMed] [Google Scholar]
- Montgomery D. C.Design and Analysis of Experiments; John Wiley & Sons, 2017. [Google Scholar]
- Dardé M.; Ajzenberg D.; Smith J.. Population structure and epidemiology of Toxoplasma gondii. In Toxoplasma Gondii; Elsevier, 2007; pp 49–80. [Google Scholar]
- Flegr J.; Prandota J.; Sovičková M.; Israili Z. H. Toxoplasmosis–a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS One 2014, 9, e90203 10.1371/journal.pone.0090203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA, E. 2019; p e05926. [DOI] [PMC free article] [PubMed]
- Re R.; Pellegrini N.; Proteggente A.; Pannala A.; Yang M.; Rice-Evans C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Biol. Med. 1999, 26, 1231–1237. 10.1016/S0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
- Catarino M. D.; Alves-Silva J. M.; Pereira O. R.; Cardoso S. M. Antioxidant capacities of flavones and benefits in oxidative-stress related diseases. Curr. Top. Med. Chem. 2015, 15, 105–119. 10.2174/1568026615666141209144506. [DOI] [PubMed] [Google Scholar]
- Boeck P.; Falcão C. A. B.; Leal P. C.; Yunes R. A.; Cechinel Filho V.; Torres-Santos E. C.; Rossi-Bergmann B. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg. Med. Chem. 2006, 14, 1538–1545. 10.1016/j.bmc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Thieury C.; Lebouvier N.; Le Guével R.; Barguil Y.; Herbette G.; Antheaume C.; Hnawia E.; Asakawa Y.; Nour M.; Guillaudeux T. Mechanisms of action and structure-activity relationships of cytotoxic flavokawain derivatives. Bioorg. Med. Chem. 2017, 25, 1817–1829. 10.1016/j.bmc.2017.01.049. [DOI] [PubMed] [Google Scholar]
- Mai C. W.; Yaeghoobi M.; Abd-Rahman N.; Kang Y. B.; Pichika M. R. Chalcones with electron-withdrawing and electron-donating substituents: anticancer activity against TRAIL resistant cancer cells, structure–activity relationship analysis and regulation of apoptotic proteins. Eur. J. Med. Chem. 2014, 77, 378–387. 10.1016/j.ejmech.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Sinyeue C.; Matsui M.; Oelgemöller M.; Bregier F.; Chaleix V.; Sol V.; Lebouvier N. Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents. Molecules 2022, 27, 1781 10.3390/molecules27061781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akçok İ.; Çağır A. Synthesis of stilbene-fused 2′-hydroxychalcones and flavanones. Bioorg. Chem. 2010, 38, 139–143. 10.1016/j.bioorg.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Cabrera M.; Simoens M.; Falchi G.; Lavaggi M. L.; Piro O. E.; Castellano E. E.; Vidal A.; Azqueta A.; Monge A.; de Cerain A. L. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure–activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. 10.1016/j.bmc.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Schwarz M.; Eno R. F.; Freitag-Pohl S.; Coxon C. R.; Straker H. E.; Wortley D. J.; Hughes D. J.; Mitchell G.; Moore J.; Cummins I.; et al. Flavonoid-based inhibitors of the Phi-class glutathione transferase from black-grass to combat multiple herbicide resistance. Org. Biomol. Chem. 2021, 19, 9211–9222. 10.1039/D1OB01802G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basílio N.; Lima J. C.; Cruz L.; de Freitas V.; Pina F.; Ando H.; Kimura Y.; Oyama K. I.; Yoshida K. Unveiling the 6, 8-rearrangement in 8-phenyl-5, 7-dihydroxyflavylium and 8-methyl-5, 7-dihydroxyflavylium through host–guest complexation. Eur. J. Org. Chem. 2017, 2017, 5617–5626. 10.1002/ejoc.201701009. [DOI] [Google Scholar]
- Zheng X.; Cao J.-G.; Meng W.-D.; Qing F.-L. Synthesis and anticancer effect of B-ring trifluoromethylated flavonoids. Bioorg. Med. Chem. Lett. 2003, 13, 3423–3427. 10.1016/S0960-894X(03)00752-2. [DOI] [PubMed] [Google Scholar]
- Shih T.-L.; Chou C.-E.; Liao W.-Y.; Hsiao C.-A. Copper-mediated trimethylsilyl azide in amination of bromoflavonoids to synthesize unique aminoflavonoids. Tetrahedron 2014, 70, 3657–3664. 10.1016/j.tet.2014.04.022. [DOI] [Google Scholar]
- Wu C.; Dunaway-Mariano D.; Mariano P. S. Design, synthesis, and evaluation of inhibitors of pyruvate phosphate dikinase. J. Org. Chem. 2013, 78, 1910–1922. 10.1021/jo3018473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.