

Abstract
Inborn errors of metabolism are a diverse group of genetic disorders including many that cause neonatal-onset epilepsy such as pyridoxine-dependent epilepsy (PDE). PDE occurs secondary to biallelic pathogenic variants in ALDH7A1 and can present with refractory neonatal seizures and status epilepticus. Neonatal seizures and encephalopathy are modifiable with pyridoxine (vitamin B6) supplementation. However, the clinical response to pyridoxine supplementation can be delayed. We present the case of a full-term neonate with PDE in which seizure cessation was seen a few hours after intravenous pyridoxine load, but the improvement in EEG background and level of clinical encephalopathy occurred 5 days later. We share this case to provide an example in which clinical improvement in PDE was gradual and required continuation of treatment for several days illustrating the necessity of continuing vitamin B6 supplementation in suspected cases until confirmatory genetic testing is obtained or an alternate cause is found.
Pearls
ALDH7A1-related pyridoxine-dependent epilepsy (PDE-ALDH7A1) is a rare cause of neonatal encephalopathy and seizures.
PDE-ALDH7A1 should be considered in the differential of neonatal epileptic encephalopathy because it is one of few treatable neonatal epilepsies.
Pyridoxine supplementation can control neonatal seizures and improve the level of neonatal encephalopathy.
Oy-sters
Because clinical and EEG improvement can be delayed after initiation of supplementation, pyridoxine should be continued until PDE-ALDH7A1 is ruled out by confirmatory testing.
Brain imaging abnormalities in neonates with PDE-ALDH7A1 can be nonspecific.
Genetic neonatal epilepsies, including PDE-ALDH7A1, should be considered if seizure burden is out of proportion to imaging findings.
Case Report
The neurology team was consulted for suspected seizures in a 1-day-old infant. Pregnancy was reported as unremarkable. The patient was born at 40 weeks' gestation through uncomplicated vaginal delivery, with normal Apgar scores (8 and 9 at 1 and 5 minutes, respectively) and no need for resuscitation. Around 5 hours of age, he was noted to have respiratory distress and episodes of rhythmic multifocal limb movements, concerning for seizures. Venous blood gas showed a pH of 6.95, pCO2 of 70.4 mm Hg, HCO3 of 16.1 mmol/L, and base deficit of 16.9. He was intubated and transferred to our center from the birth hospital. An elevated lactate (10 mmol/L) was noted on admission. Basic CSF analysis was negative for signs of infection. Plasma amino acids and CSF amino acids were unremarkable. Conventional EEG was initiated shortly after presentation and confirmed frequent multifocal electrographic seizures, arising from the bilateral centrotemporal regions and evolving to bilateral rhythmic discharges (Figure 1, A and B). Status epilepticus lasting up to 70 minutes was noted. Loading IV doses of phenobarbital, fosphenytoin, and levetiracetam were given in close succession over the first 24 hours of presentation, with improvement in seizure burden but not complete cessation. EEG background rapidly evolved to severe encephalopathy in the form of a burst-suppression pattern (Figure 1, C and D). Clinical examination also showed severe encephalopathy with absent response to stimulation, diffuse hypotonia, and ventilator dependence with high respiratory support settings. Hypoxic-ischemic encephalopathy was believed to be unlikely; therapeutic hypothermia was thus not considered.
Figure 1. Evolution of EEG Findings.

Modified neonatal double distance montage, read at 15 μV for panels A and B and at 7 μV for panels D–H, with 30 seconds per page in all panels. Electrographic seizures with right centrotemporal onset (A) and evolution to rhythmic bilateral large amplitude spike-and-wave discharges (B). Background activity before the pyridoxine load, showing a severely abnormal burst-suppression pattern for 60 consecutive seconds (C, D). Background activity 48 hours after the pyridoxine load, showing an ongoing abnormal burst-suppression pattern (E), although some activity is rarely seen within the interburst interval (F). Background activity 5 days after the pyridoxine load, showing normal activity with typical neonatal electrographic elements during wakefulness (G), and mildly excessive discontinuity during quiet sleep (H).
Given suspicion for a genetic-metabolic epilepsy, a single IV dose of pyridoxine (100 mg) was added at 52 hours of age in addition to maintenance doses of the other antiseizure medications (ASMs). No seizures were recorded after the pyridoxine loading dose. However, the EEG background continued to show a burst-suppression pattern over the following 2 days (Figure 1, E and F), at which point the EEG was discontinued. The neurologic examination continued to show a severe encephalopathy, and ongoing mechanical ventilation was required due to a lack of respiratory drive. Brain MRI at 3 days of age showed multiple small areas of periventricular white matter injury, a small cerebellum, a thin corpus callosum, and normal spectroscopy values in the basal ganglia (Figure 2). These nonspecific MRI findings did not explain the severity of encephalopathy and extensive seizure burden. Given the questionable response to the IV load of pyridoxine, maintenance dosing of pyridoxine was initiated at 5 days of age. Because of the concern for partial response to pyridoxine, raising the possibility of pyridoxamine-5′-phosphate oxidase (PNPO) deficiency, pyridoxal-5′-phosphate (PLP) was also added a few days later as part of the local protocol for refractory neonatal seizures. Phenobarbital and fosphenytoin doses were discontinued at 6 days of age because of supratherapeutic levels; levetiracetam was continued. Seizures did not recur.
Figure 2. Brain MRI Findings.
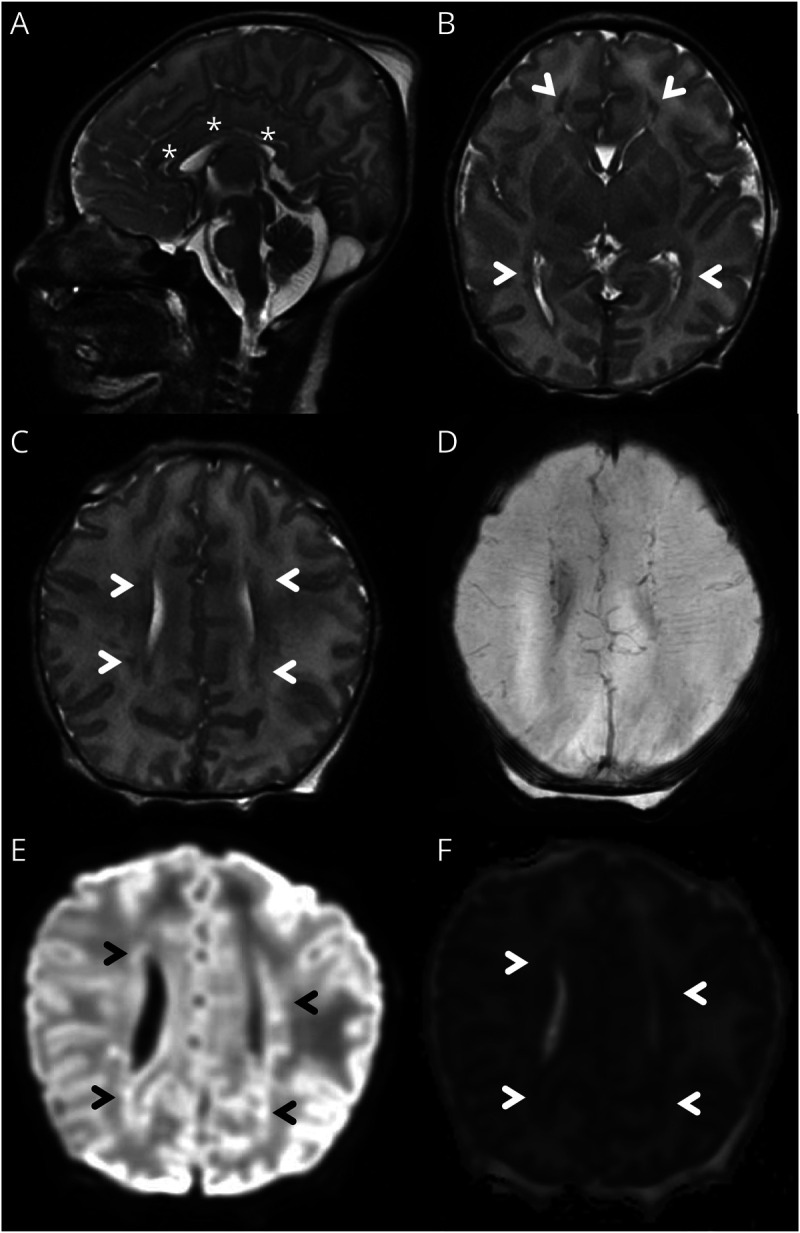
Sagittal T2 sequence showing diffusely thin corpus callosum (*, A). Axial T2 sequence showing areas of abnormal signal in the periventricular white matter anterior to the frontal horns and posterolateral to the occipital horns (arrowheads, B). Axial T2 sequence (rostral to B) showing small multifocal areas of abnormal signal in the periventricular white matter (arrowheads, C). Corresponding SWI sequence showing absence of hemorrhage (D). Corresponding DWI (E) and ADC (F) sequencing showing areas of restricted diffusion in the periventricular white matter, consistent with acute injury (arrowheads). ADC = apparent diffusion coefficient; DWI = diffusion-weighted imaging.
A repeat EEG at 7 days of age showed significant improvement in background activity (only mild excess discontinuity in sleep for age) and no seizures (Figure 1, G and H). Neurologic examination mirrored these drastic improvements, with spontaneous limb movements and normal resting tone by 9 days of age. He was extubated to room air at 10 days of age. Repeat EEG at 21 days of age remained stable, and the neurologic examination was then normal for age. Repeat brain MRI at 22 days of age showed mildly decreased white matter volume, mild thinning of the corpus callosum, mild pontine atrophy, and mega cisterna magna. He was discharged home at 30 days of age on levetiracetam, pyridoxine, and PLP maintenance; both pyridoxine and PLP were continued until the genetic diagnosis was obtained. At age 6 months, the patient remained seizure-free, although showed mild gross motor delay.
An epilepsy gene panel identified a homozygous missense variant in ALDH7A1 (c.1559C>T; p.Ser520Phe), inherited from both parents. This was reported by the laboratory as a variant of uncertain significance. However, given his clinical phenotype, allelic homozygosity, and the pathogenic features of the variant, including multiple streams of in silico testing (SIFT, PROVEAN, Mutation assessor, LIST-S2, PolyPhen-2) that predicted a deleterious effect, we concluded the variant was disease-causing and consistent with PDE. Once the diagnosis was established, we discontinued PLP, and the infant was continued on pyridoxine and levetiracetam.
Discussion
Genetic disorders are rare but important causes of neonatal seizures that often lead to a complex clinical picture that may be difficult to diagnose and treat.1 In the Neonatal Seizure Registry, 13% of neonates (79/611) with seizures were diagnosed with neonatal epilepsy, 30% of which (24/79) were found to have a genetic cause.2 PDE-ALDH7A1 is a rare genetic cause of refractory neonatal seizures that is well-described in the literature, characterized by a remarkable response to pyridoxine supplementation.3–5 This disorder is caused by biallelic pathogenic variants in ALDH7A1, a gene that encodes for alpha-aminoadipic semialdehyde (α-AASA) dehydrogenase—also known as antiquitin.4–10 Antiquitin deficiency leads to an abnormality in lysine catabolism and accumulation of intermediate metabolites, including delta-1-piperideine-6-carboxylate, which binds to and deactivates PLP.4,5 PLP is an active B6 vitamer that acts as a cofactor for multiple essential enzymes; downstream dysregulation of these enzymatic pathways through PLP deficiency causes neonatal epileptic encephalopathy which is salvaged by pyridoxine supplementation.3–5,10
Although atypical cases are described,4,8,10 PDE-ALDH7A1 classically presents in the neonatal period with prolonged and drug-refractory seizures, status epilepticus, and encephalopathy.6,7,9 Various seizure types are described, including clonic, tonic, myoclonic, autonomic, sequential, and electrographic only.11 Poor feeding, respiratory distress, apneas, lactic acidosis, and electrolyte disturbances have been described.10 Typical EEG patterns include high-voltage delta activity, multifocal epileptiform discharges, and a burst-suppression pattern.4,7,9,12 Seizures are usually unresponsive to common ASM, although reports of partial or temporary response to initial therapy are described. Refractory seizures, EEG background, and clinical encephalopathy usually respond rapidly to pyridoxine loading and supplementation.9,10,13 Delayed response of a few days to weeks have been reported, but this is believed to be infrequent.5 Thus, in the appropriate clinical setting, we recommend initiating a pyridoxine trial even with a partial response to ASM and continuing pyridoxine maintenance therapy even if the clinical impact is not immediately striking.5,13 Repeat EEG monitoring and serial neurologic examinations are indicated to assess for this possible delayed response.
Given that PNPO deficiency is on the differential diagnosis of refractory neonatal epileptic encephalopathy,3,4,6,9 the addition of enteral PLP should be considered when there is an incomplete response to pyridoxine, as discussed in published protocols.5 Caused by biallelic pathogenic variants in the PNPO gene, PNPO deficiency also ultimately leads to a decrease in PLP, causing downstream effects on PLP-dependent enzymes.5 Some newborns with PNPO deficiency will respond to pyridoxine therapy, whereas many have a better response to specific PLP supplementation.5,9
Recommendations for investigation of suspected PDE-ALDH7A1 include urine α-AASA, plasma pipecolic acid, and sequencing of ALDH7A1, as well as other genes of interest.3,12 Brain MRI is performed to assess for structural causes of neonatal seizures,12 although nonspecific imaging findings have been described in PDE-ALDH7A1: corpus callosum abnormalities, white matter lesions, parenchymal atrophy, small cerebellum, and mega cisterna magna.4,8,14 PDE-ALDH7A1 should be considered even when the brain MRI is abnormal, especially if seizures are out of proportion to the imaging findings. These neonates may also have a history of low tone, poor Apgar scores, and abnormal blood gas, which may be misdiagnosed as hypoxic-ischemic encephalopathy. Lack of a clear perinatal event, drug-resistant seizures, and discordance between the degree of MRI abnormalities and the level of encephalopathy should raise suspicion for PDE-ALDH7A1.
In confirmed cases of PDE-ALDH7A1, enteral supplementation with pyridoxine should continue lifelong; the specifics of therapy and monitoring are well described.3,4,6,9 Neurologic outcomes in treated patients are variable. Most patients have developmental delays and intellectual disability, although some are reported to be cognitively normal.9,15 There may be a correlation between early onset of seizures or delayed initiation of pyridoxine and poorer outcomes, although this finding has not been consistent in the literature.4,9
Appendix. Authors
| Name | Location | Contribution |
| Olivier Fortin, MD | Prenatal Pediatrics Institute, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Kelsey Christoffel, MD | Prenatal Pediatrics Institute, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Youssef Kousa, DO, PhD | Prenatal Pediatrics Institute, Children's National Hospital; Department of Neurology, and Department of Pediatrics, The George Washington University School of Medicine and Health Sciences; Division of Neurology, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content |
| Ilana Miller, MS, CGC | Division of Medical Genetics, and Rare Disease Institute, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Eyby Leon, MD | Division of Medical Genetics, and Rare Disease Institute, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content |
| Kelsey Donoho, MD | Department of Pediatrics, The George Washington University School of Medicine and Health Sciences; Division of Neonatology, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content |
| Sarah B. Mulkey, MD, PhD | Prenatal Pediatrics Institute, Children's National Hospital; Department of Neurology, and Department of Pediatrics, The George Washington University School of Medicine and Health Sciences, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Tayyba Anwar, MD | Department of Neurology, and Department of Pediatrics, The George Washington University School of Medicine and Health Sciences; Division of Neurology, Children's National Hospital, Washington, DC | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
Study Funding
Extramural funding for this work was provided by the NIH (K08NS119882 and T32HD098066).
Disclosure
O. Fortin reports no disclosures relevant to the manuscript. K. Christoffel reports extramural funding from the NIH for this work (T32HD098066). Y. Kousa reports extramural funding from the NIH for this work (K08NS119882). I. Miller, E. Leon, K. Donoho, S.B. Mulkey, and T. Anwar report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Yamamoto H, Okumura A, Fukuda M. Epilepsies and epileptic syndromes starting in the neonatal period. Brain Dev. 2011;33(3):213-220. doi: 10.1016/j.braindev.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 2.Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al. Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology. 2017;89(9):893-899. doi: 10.1212/wnl.0000000000004284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coughlin CR II, Tseng LA, Abdenur JE, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2021;44(1):178-192. doi: 10.1002/jimd.12332 [DOI] [PubMed] [Google Scholar]
- 4.Coughlin CR II, Gospe SM Jr. Pyridoxine-dependent epilepsy: current perspectives and questions for future research. Ann Child Neurol Soc. 2023;1:24-37. doi: 10.1002/cns3.20016 [DOI] [Google Scholar]
- 5.Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B6 metabolism. J Inherit Metab Dis. 2019;42(4):629-646. doi: 10.1002/jimd.12060 [DOI] [PubMed] [Google Scholar]
- 6.Gospe SM. Pyridoxine-dependent epilepsy: ALDH7A1. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews [online]. University of Washington, Seattle; 1993. Accessed January 16, 2023. ncbi.nlm.nih.gov/books/NBK1486/. [Google Scholar]
- 7.Kaminiów K, Pająk M, Pająk R, Paprocka J. Pyridoxine-dependent epilepsy and antiquitin deficiency resulting in neonatal-onset refractory seizures. Brain Sci. 2021;12(1):65. doi: 10.3390/brainsci12010065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain. 2010;133(7):2148-2159. doi: 10.1093/brain/awq143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockler S, Plecko B, Gospe SM, et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104(1-2):48-60. doi: 10.1016/j.ymgme.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 10.van Karnebeek CDM, Tiebout SA, Niermeijer J, et al. Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatr Neurol. 2016;59:6-12. doi: 10.1016/j.pediatrneurol.2015.12.013 [DOI] [PubMed] [Google Scholar]
- 11.Pressler RM, Cilio MR, Mizrahi EM, et al. The ILAE classification of seizures and the epilepsies: modification for seizures in the neonate. Position paper by the ILAE Task Force on Neonatal Seizures. Epilepsia. 2021;62(3):615-628. doi: 10.1111/epi.16815 [DOI] [PubMed] [Google Scholar]
- 12.Mastrangelo M, Celato A, Leuzzi V. A diagnostic algorithm for the evaluation of early onset genetic-metabolic epileptic encephalopathies. Eur J Paediatr Neurol. 2012;16(2):179-191. doi: 10.1016/j.ejpn.2011.07.015 [DOI] [PubMed] [Google Scholar]
- 13.Bok LA, Maurits NM, Willemsen MA, et al. The EEG response to pyridoxine-IV neither identifies nor excludes pyridoxine-dependent epilepsy. Epilepsia. 2010;51(12):2406-2411. doi: 10.1111/j.1528-1167.2010.02747.x [DOI] [PubMed] [Google Scholar]
- 14.Friedman SD, Ishak GE, Poliachik SL, et al. Callosal alterations in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2014;56(11):1106-1110. doi: 10.1111/dmcn.12511 [DOI] [PubMed] [Google Scholar]
- 15.Basura GJ, Hagland SP, Wiltse AM, Gospe SM. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. 2009;168(6):697-704. doi: 10.1007/s00431-008-0823-x [DOI] [PubMed] [Google Scholar]