

Abstract
The understanding of the mechanisms of liver fibrosis has been dominated by models in which chronic hepatocellular injury is the initiating step as is seen with viral infections. The increased prevalence of the metabolic syndrome, and the increases in liver fibrosis due to metabolic syndrome driven non-alcoholic steatohepatitis (NASH), has made it a priority to understand how this type of liver fibrosis is similar to, and different from, pure hepatocellular injury driven liver fibrosis. Both types of liver fibrosis have the transformation of the hepatic stellate cell (HSC) into a myofibroblast as a key step. In metabolic syndrome, there is little evidence that metabolite changes such as high levels of glucose and free fatty acids are directly inducing HSC transdifferentiation, however, metabolite changes may lead to reductions in immunomodulatory and hepatoprotective molecules such as lipoxins, resolvins and Interleukin (IL)-22. Cells of the innate immune system are known to be important intermediaries between hepatocellular damage and HSC transdifferentiation, primarily by producing cytokines such as transforming growth factor-β (TGF-β) and platelet derived growth factor (PDGF). Resident and infiltrating macrophages are the dominant innate immune cells, but others (dendritic cells, neutrophils, natural killer T cells and mucosal-associated invariant T cells) also have important roles in inducing and resolving liver fibrosis. CD8+ and CD4+ T cells of the adaptive immune system have been identified to have greater profibrotic roles than previously realised by inducing hepatocyte death (auto-aggressive CD8+T) cells and cytokines producing (TH17 producing CD4+T) cells. Finally, the cellular networks present in NASH fibrosis are being identified and suggest that once fibrosis has developed cell-to-cell communication is dominated by myofibroblasts autocrine signalling followed by communication with cholangiocytes and endothelial cells, with myofibroblast-hepatocyte, and myofibroblast-macrophage signalling having minor roles. Such information is essential to the development of antifibrotic strategies for different stages of fibrosis.
Keywords: fibrosis
Introduction
Chronic liver injury results in the development of liver fibrosis in approximately 25% of individuals in response to diverse insults, and several aspects of the development of liver injury appear to be common to a variety of initiating factors, while others are specific to individual types of injury.1 2 Among all of the hepatic changes due to metabolic syndrome, understanding the development of fibrosis is a priority as it is the strongest predictor of mortality.3 4 Metabolic syndrome is an umbrella term, which is characterised by obesity and insulin resistance and is the underlying driving force for non-alcoholic steatohepatitis (NASH). The increased prevalence of obesity has significantly increased the importance of liver fibrosis in the setting of metabolic syndrome and NASH. After a brief review of the common features of the liver fibrotic response we will examine the role of the metabolic syndrome in individual mechanisms of liver fibrosis.
Canonical aspects of liver fibrosis
Attempts at understanding how liver fibrosis occurs are framed around the functional properties of the myofibroblast, of which the major precursor is the hepatic stellate cell (HSC), and this has been reviewed in detail.1 2 Quiescent HSCs are recognised for their storage of vitamin A as retinyl esters and are the main source of the functional myofibroblasts in hepatocellular injury. Zone 1 distributed portal fibroblasts are a major contributor to the myofibroblast population in the setting of biliary injury and fibrosis.5,7 Fibrocytes are bone marrow derived myofibroblast precursors and may also have a minor contribution.8 Production of extracellular matrix (mostly collagens types I, III, IV) is the most characteristic feature of HSC but they take on many other functions including proliferation, release of growth factors and proinflammatory cytokines (hepatocyte growth factor (HGF), Interleukin (IL)-6, IL-8, MCP1), matrix degradation enzymes and their inhibitors.6 There are multiple functional consequences for the conversion of HSC to myofibroblasts, with the predominant ones being deposition of matrix. HSCs also provide significant support to other liver cells in part by producing HGF, which recently has been shown to have a major hepatoprotective in addition to its well-known proproliferative role.9 They also support angiogenesis during liver injury by supporting sinusoidal cell survival.10 11 Single cell analysis has allowed for regional information on portal and central vein associated HSC in healthy and damaged livers.12 13 This reveals that pericentral injury results in HSC in the pericentral region differentiating into myofibroblasts, however, HSCs further away in the periportal region undergo proliferation but do not differentiate to the same degree.14 With resolution of liver injury, there is a functional reversion of myofibroblasts into an inactivated state which looks very much like quiescent HSC, but these cells are epigenetically and functionally different from quiescent HSC, and in response to stimuli undergo differentiation much faster than quiescent HSC.15 16
Metabolite-induced HSC activation
An initial question is if any of the common changes in metabolic syndromes such as high levels of glucose and free fatty acids (FFAs) directly result in HSC activation. There are a few reports that high glucose concentration alone induced HSC proliferation and differentiation via p38-MAPK pathway and generation of free radicals.17 18 However, the concentrations of glucose used here were very high at around 25 mM (upper limit of normal fasting is 5.6 mM), and it is unlikely that this a major mechanism of HSC activation in the metabolic syndrome. Culture of the HSC cell line LX-2 with FFAs (2:1 oleate:palmitate) at physiological levels (0.5 mM) has been shown in one study to result in an approximately 2.5 fold change in α-SMA levels and anti-apoptotic molecules.19 A second study has reported that FFA induced HSC activation only occurs when HSCs are co-cultured with hepatocytes despite using a higher concentration (1.2 mM) of FFA, suggesting that FFA induce hepatotoxicity is driving HSC activation.20 As for glucose, there is little evidence for direct activation of HSC. In mice on a high cholesterol diet-free cholesterol increases, and contributes to HSC activation via increased signalling through the TLR4 pathway.21 22
Metabolite signals, however, may also be involved in the reduction of inflammation and fibrosis. A family of lipid mediators generated during inflammatory responses promote the resolution of inflammation by providing stop signals.23 These molecules include lipoxins, resolvins and protectins, which are derived from the omega-3 essential fatty acids.24 These molecules exert protective effects via dampening neutrophil and monocyte influx, and may increase phagocytosis of apoptotic neutrophils by macrophages (efferocytosis). There is very little data on proresolution lipid mediators in NASH but given the nature of chronic inflammation in NASH there may be some defects in some of these pathways. Despite the lack of evidence of a defect in these pathways as a cause of NASH, there is evidence of the potential therapeutic value of these molecules as administration of resolvin D1 to HFD fed mice was associated with reduced liver inflammation, and a synthetic lipoxin analogue reduced steatosis in mice.25 26 The proresolving lipids maresin 1 was shown to promote a prorepair M2 phenotype of liver macrophages via binding to retinoic acid-related orphan receptor and inducing a prorepair phenotype in infiltrating liver macrophages. Administration of MaR1 protected mice from high-fat diet-induced NASH in a RORα-dependent manner.27 Outside of the liver, proresolvins have been demonstrated to improve renal fibrosis.28 29 IL-22 is a cytokine that has also been shown to have broad protective effects in liver diseases including NASH, and the production of IL-22 by innate lymphoid cells is reduced in obesity.30,32 It has also been shown to reduce liver fibrosis and increased the resolution of liver fibrosis, likely by inducing HSC senescence.33 In addition to its potential as protective in NASH, it may be aetiologically important as IL-22 has prometabolic effects including improving insulin sensitivity.32
Direct hepatocyte-induced HSC activation
Hepatocyte stress and death is known to be a feature of metabolic syndrome and can result in HSC activation. Although there is modest evidence of direct activation of HSC by lipids, lipids can clearly activate proapoptotic signals in hepatocytes.34 Several studies have elucidated the apoptotic pathways activated in hepatocytes by saturated FFAs, palmitate and the phospholipid lysophosphatidylcholine35,37 (figure 1). Apoptotic pathways include TRAIL, endoplasmic stress and JNK pathways, and loss of apoptosis in experimental models such as caspase 3 knockout mice resulted in a reduction in experimental NASH.38 There is much less evidence for other modes of cell death such as necrosis and necroptosis in NASH. Apoptosis is defined as a silent death, due to degradation of cellular contents while retaining them within the plasma membrane. However, even without loss of cellular contents phagocytosis of apoptotic bodies results in activation of HSC which could occur via a number of molecules within the apoptotic body including nuclear DNA, mitochondrial DNA, miRNA.39,43 Furthermore, cell death without loss of intracellular contents certainly occurs in developmental apoptosis but in pathological apoptosis there is a mixture of stressed and partially apoptotic cells with release of intracellular damage associated molecular patterns (DAMPs) into the extracellular environment including mitochondrial and nuclear DNA.44,47 It is likely that a range of other DAMPs such as HMGB1, histones, SAP130, cytochrome c, s100 proteins and defensins have some direct HSC stimulatory activity.48,50 Such release of DAMPs is very significantly broadening the mechanistic pathways, which may be responsible for hepatocyte mediated direct HSC activation. In NASH, there is also the easily identifiable ballooning of hepatocytes which are characterised by cellular swelling and enlargement, a central nucleus, reticulated cytoplasm, loss of keratin 8 and 18, and accumulation of ubiquitinated proteins. One possible profibrotic feature of ballooning hepatocytes is secretion of sonic hedgehog, which can mediate multiple aspects of HSC biology including proliferation, activation and differentiation.51 52 Ballooning hepatocytes expressing sonic hedgehog are adjacent to HSC expression of the ligand Gli2.51 The natural loss or reduced function of hedgehog has increased steatosis and liver fibrosis demonstrating a complex relationship.53
Figure 1. Hepatocyte-mediated activation of HSC, the innate and adaptive immune activation in the metabolic syndrome. Soluble mediators are in red. DAMPs, damage-associated molecular patterns; HSC, hepatic stellate cells; MAIT, mucosal-associated invariant T cells; NK, natural killer cells.

Indirect hepatocyte-induced HSC activation
The above mechanisms of hepatocyte apoptosis, with production of apoptotic bodies, release of extracellular vesicles and release of DAMPs, which were discussed as having an important role in HSC activation, also have major impacts on many cells of the immune system.54 A dominant mechanism is the efferocytosis of apoptotic hepatocytes by macrophages resulting in the release of transforming growth factor-β (TGF-β), a major profibrogenic molecule.45 The release of TGF-β has two major effects, one is a broadly immunosuppressive and the second is a profibrotic, demonstrating the tight linkage between resolution of inflammation and repair. As such, for an acute insult such as acetaminophen, the timing of TGF-β production is at the point of resolution of inflammation and initiation of repair. For chronic insults such as NASH, injury and inflammation and repair are all occurring concurrently. Despite the importance of efferocytosis in the initiation of liver fibrosis, in a very necrotic model (thioacetamide induced hepatic toxicity) experimental data suggests that the net effect of efferocytosis is to reduce liver fibrosis, presumably by removing DAMPs which otherwise will directly activate HSC.41 The balance in non-necrotic insults such as NASH may be towards efferocytosis having a net profibrotic effect.
The macrophage population in the NASH liver is heterogeneous and complex.55 In the healthy liver most macrophages are yolk sac derived Kupffer cells (KCs), but these are significantly outnumbered in the setting of acute liver injury and in NASH by bone marrow derived infiltrating macrophages.55 56 In murine livers, KCs are clearly identified by the expression of F4/80, which does not exist in humans. Single cell analysis has, however, identified human liver macrophages with predominantly immune-tolerant functions similar to mouse KC (identified in human livers as CD68+MARCO+ ve, TIMd4+ve), and another population with genes that are predominantly proinflammatory and consistent with infiltrating cells of the mouse liver (CD68+MARCO −ve, TIMd4-ve).12 13 57 58 In human fibrotic liver another set of markers was used to identify a macrophage subset identified by CD9 and TREM-2, with vector analysis suggesting that these are derived from infiltrating macrophages.12 The direct contribution of infiltrating macrophages to liver fibrosis has been difficult to isolate as these cells are responsible for liver injury as well as promoting fibrosis. As such the dominant message from experimental studies in which infiltration of macrophages has been blocked or deleted has been one of reduced liver inflammation and fibrosis.59,62
Hepatocyte death results in activation of liver macrophages and subsequent HSC differentiation through release of soluble factors from macrophages (figure 2). TGF-β is the most potent and dominant HSC activating profibrogenic product of macrophages, however, macrophages in the liver scar tissue produce several other molecules which have cognate receptors on HSC and result in their activation. These include platelet derived growth factor (PDGF)-β, TNFSF12, SPP1 and IL-1β. These molecules show some functional biases, with, for example, SPP1 promoting HSC differentiation, TNF superfamily molecules promote proliferation, and IL-1β promotes survival, although with significant overlap.12 63 64 Macrophages also have a wide range of effects on other liver cells which indirectly effect HSC and liver fibrosis. Dominant among these is proinflammatory through macrophage production of TNF-α and IL-1β which can induce hepatocyte steatosis and injury.65 Macrophages are also very active in resolution of fibrosis. In a carbon tetrachloride-induced liver fibrosis model macrophage depletion when liver fibrosis was advanced resulted in reduced scarring and fewer myofibroblasts. In contrast, macrophage depletion during recovery led to a failure of matrix degradation.60 The identify of these fibrosis regressing or restorative macrophages was clarified as CD11Bhi F4/80int Ly-6Clo, and they were most abundant in livers during maximal fibrosis resolution and represented the principal matrix metalloproteinase (MMP)-expressing subset. Depletion of this population caused a failure of scar remodelling, and adoptive transfer and in situ labelling experiments showed that these restorative macrophages derive from recruited Ly-6Chi monocytes, demonstrating a common origin with profibrotic Ly-6Chi macrophages. This is indicative of a phenotypic switch in vivo conferring proresolution properties. Microarray profiling of the Ly-6Clo subset, compared with Ly-6Chi macrophages, showed a phenotype outside the M1/M2 classification, with increased expression of MMPs, growth factors and phagocytosis-related genes.66
Figure 2. Effects of the innate and adaptive immune system on hepatocytes and HSC in the metabolic syndrome. HSC, hepatic stellate cell; IL, interleukin; MAIT, mucosal-associated invariant T; NK-T, natural killer T.
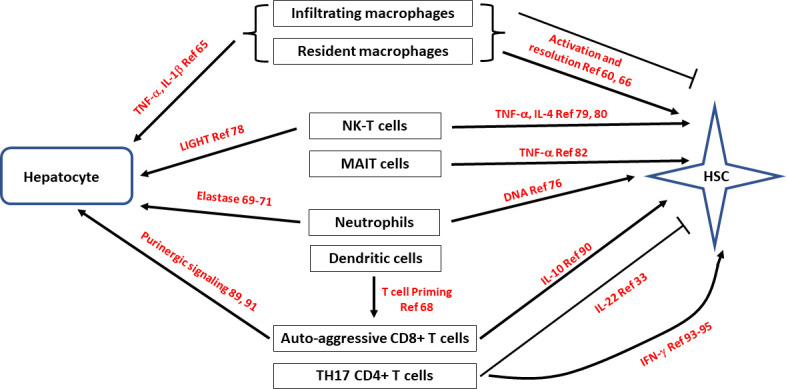
Dendritic cells (DCs) are another innate immune cells and have some similarities to macrophages in that they can be activated by DAMPs, phagocytose apoptotic cells and produce proinflammatory cytokines.67 They are, however, much more efficient at antigen presentation and several types have been identified. Conventional DCs consist of the subtypes plasmacytoid DCs, inflammatory DCs and Langerhans cells. Conventional DCs (cDCs) are the main type in the liver and have recently been found to have a major role in NASH using single-cell transcriptomic analysis which identified an increase in hepatic cDCs and further defined their source as NASH-induced boost in cycling of cDC progenitors in the bone marrow.68 Analysis of blood and liver from patients on the non-alcoholic fatty liver disease (NAFLD)/NASH spectrum showed that these type 1 cDCs (cDC1) were more abundant and activated in disease. Sequencing of physically interacting cDC-T cell pairs from liver-draining lymph nodes revealed that cDCs in NASH promote inflammatory T cell reprogramming, previously associated with NASH worsening, and depletion or blocking of cDC1 attenuated liver pathology in NASH mouse models.68 This demonstrates that DCs have an important role in programming T cells in NASH (see The adaptive immune system). In contrast to these data supporting a role of DCs in NASH earlier studies using coculture and in vivo ablation experiments revealed only a minor contribution to NF-κB activation in HSCs by DCs, and no contribution of DCs to liver fibrosis development, respectively.64
Although macrophages are by far the dominant cell type in NASH, other immune cells also have important roles. Neutrophils are the earliest responders in tissue injury, and this appears to be the case in NASH as well. Conceptually they are given dual roles of removal of cellular debris and as such reducing inflammation, but also able to release proteases and DAMPs and potentially increasing inflammation. They are frequently overlooked as NASH is usually examined in its chronic stage. Neutrophils are certainly increased in NASH liver tissue and the levels of plasma elastase correlate with the severity of NASH.69,71 In mouse models of NASH removal of neutrophils, in mice lacking myeloperoxidase or neutrophil elastase, there was reduced liver damage, but at least in one model of toxic injury no effect of fibrosis.72,75 In mouse models of NASH neutrophils release extracellular traps could certainly provide proinflammatory and differentiation signals via TLR9 on immune cells and HSC.76
Natural killer T (NKT) cells are a subset of CD1d-restricted T cells at the interface between the innate and adaptive immune system. NKT cells can be subdivided into functional subsets that respond rapidly to a wide variety of glycolipids and stress-related proteins using T-cell or NK cell-like effector mechanisms.77 NK-T cells are activated in mouse models of NASH, and NK-T cells primarily cause steatosis via secretion of the cytokine LIGHT (TNFSF14).78 Activation of NK-T cells in murine models of NASH was shown to be dependent on a hedgehog dependent pathway, and adoptive transfer of NK-T cells has been shown to induce liver fibrosis.79 80 Collectively, therefore, there are strong data for NK-T cells having a role in steatosis and hepatocyte injury in NASH, and data for NK-T cells having a role in liver fibrosis in other mechanism, suggesting that will also have a role in NASH mediated fibrosis.
Mucosal-associated invariant T (MAIT) cells are T cells with a semi-variant αβ T cell receptor which recognises biosynthetic derivatives of riboflavin synthesis presented on MR1.81 These derivatives of riboflavin are produced by some bacterial and fungal species, which presumably provide the activating signals. As such MAIT cells are activated by metabolites, however, there is no evidence that these metabolites are increased in the metabolic syndrome. Circulating MAIT cells were shown to be reduced in the blood of patients with alcoholic or non-alcoholic fatty liver disease-related cirrhosis, and in contrast accumulate in liver fibrotic septa. In two models of liver fibrosis (CCl4 and bile duct ligation), MAIT cell-enriched mice show increased liver fibrosis and accumulation of hepatic fibrogenic cells, whereas MAIT cell-deficient mice are resistant. Coculture experiments indicate that MAIT cells enhance the proinflammatory properties of monocyte-derived macrophages, and promote mitogenic and proinflammatory functions of fibrogenic cells, via distinct mechanisms.82 Supportive data were generated from the peripheral blood of patients with autoimmune hepatitis where there was also a reduction in MAIT cells and there was a correlation between decreased frequency of MAIT cells and the degree of liver fibrosis.83 So although these data are not from metabolic syndrome induced fibrosis they are consistent with MAIT cells having a profibrogenic effect via liver macrophage stimulation and directly via HSC activation.
The above discission has been about hepatocyte death-related and injury-related activation of the adaptive immune system, but it is relevant to ask if there is evidence of direct activation of the immune system by metabolites, and in general there is lack of such evidence for this except for macrophages. Saturated FFA cholesterol has been demonstrated to activate macrophages in NASH.55 84 In addition, in the early stages of murine NASH resident KC take up lipid to a degree that it triggers their death with replacement by the more proinflammatory bone marrow-derived macrophages.85 86
The adaptive immune system
Metabolic syndrome-associated liver injury is dominated by the innate immune system, and epitope independent liver injury. There is, however, a very significant degree of activation of the adaptive immune system. Simple histology reveals infiltration of T and B cells, with CD8+T cells being a dominant cell type.87,89 Total loss of cells of the adaptive immune system (T, cells, B cells and NK T cells) results in less steatosis and inflammation and injury in a choline deficient high fat diet (CDHFD) model.78 In addition to injury CD8+T cells appear to have an even more proximal effect of driving the insulin resistance that occurs in diet induced NASH.89 Mice lacking CD8+T cells had better metabolic parameters and adoptive transfer of CD8+T cells reversed this.89 Specific to fibrosis is evidence that CD8+T cells can activate HSC in vitro and in vivo via possibly an IL-10 mechanism.90 It is likely that human NASH is a heterogeneous disease, and this study noted a difference in the role of CD8+T cells in models of lean and obese NASH. In a lean NASH model (CDHFD diet), intrahepatic CD8+T cells were elevated but depletion did not influence liver injury or HSC activation, which was in contrast from the situation in obese NASH where both were reduced. A detailed study of CD8+T cells in a mouse model of CDHFD, and in human NASH, identified a population of CD8+T cells with markers of tissue residency (CXCR6), effector function (granzyme positive) and exhaustion (PD1 positive).91 The authors presented information that this phenotype was due to a combination of IL-15 upregulation and metabolic signals (acetate and extracellular ATP). Furthermore, these CXCR6+CD8+ T cells can kill hepatocytes in purinergic receptor dependent but MHCI independent manner, which was termed autoaggression. The net effect of such autoaggression may, however, not be increased liver fibrosis as CD8+T cells have also shown to induce HSC death via a classical Fas/Fas-L pathway.92 Much of the role of CD8+T cells may, therefore, depend on the stage of disease, in early disease having a NASH driving role by increasing insulin resistance and hepatocyte damage, and later in fibrosis having a fibrosis resolution effect by killing HSC.
Analogous to CD8+T cells, CD4+T cells are also increased in numbers in NASH and show differentiation features of Th1, and Th17 differentiation.93 As can be expected they also demonstrated IFN-γ production, and loss of IFN-γ resulted in a lower inflammatory infiltrate.94 95 Evidence for the human relevance of these data was significantly increased by the demonstration in chimeric mice with a human immune system that on a high fat and high cholesterol diet Human CD4+ central and effector memory T cells increased within the liver and in the peripheral blood, accompanied by marked upregulation of proinflammatory cytokines (IL-17A and IFNγ). Furthermore, in vivo depletion of human CD4+ T cells in the chimeric mice reduced liver inflammation and fibrosis, but not steatosis. These results highlighted CD4+ T cell subsets as important drivers of NAFLD progression from steatosis to fibrosis.96 In non-chimeric mice lacking IL-17, multiple diet-induced NASH models also resulted in reduced hepatocellular damage and liver inflammation.97,99 In view of the increase in gut permeability in NASH, it is very relevant that a microbial signature known to drive TH-17 cells was able to accelerate liver damage.98 100 101
Cell network interactions of HSC in metabolic syndrome liver fibrosis
The above data have mostly been generated from the perspective of individual molecules (eg, PDGF), by using in vitro addition, and in vivo deletion studies. The development of single-cell and single-nucleus data and vector analysis allows modelling of HSC cell-to-cell interaction, and the development of an understanding of importance of HSC network interactions in liver fibrosis. This has recently been performed for established NASH fibrosis in humans and mice.102 HSC from NASH fibrotic livers and control livers differed in a relatively small number of genes (169 upregulated and 291 downregulated in human HSC, with roughly double that number in murine HSC). Using CellphoneDB software to predict interactions, it was found that the top three interactions were autocrine HSC interactions, HSC–cholangiocyte interactions and HSC–endothelial interactions (figure 3). Notably hepatocyte–HSC interactions did not dominate. Liver fibrosis in NASH is typically perisinusoidal with much of the fibrotic septa concentrated around sinusoids, which may explain the dominance of HSC–endothelial interactions.2 103 Biliary fibrosis is different from NASH fibrosis and results from proliferation of myofibroblasts and bile ductules, and is dominated by septa joining portal tracts. Viral hepatitis-driven fibrosis is the third major pattern, with fibrous septa ranging from portal tract to central vein. It is of great interest if the HSC–cell interactions will be significantly different in these varied types of fibrosis.
Figure 3. Single cell data from human NASH and mouse models reveals dominant autocrine signalling of hepatic stellate cells as well as paracrine signalling with cholangiocytes and endothelial cells.102 NASH, non-alcoholic steatohepatitis.
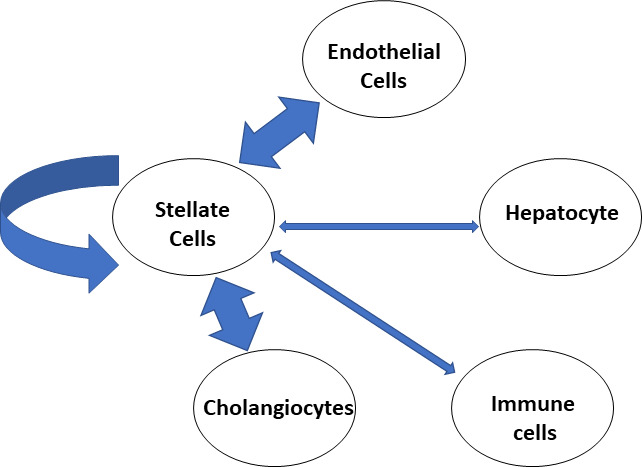
A variety of data suggest that the nature of a significant number of these interactions is short distances, with the majority possibly requiring direct contact. This is based on approximately half of the interactions were between ligand pairs involved in short-range interactions (defined as either non-secreted or collagen/integrin interactions). This conclusion was supported by imaging data which showed that with the development of fibrosis HSC projections were elongated and HSC made greater direct contact with other HSC, increasing from approximately 10% HSC having HSC–HSC interactions in healthy mouse livers, to over 80% in NASH fibrotic livers. These data provide valuable new information which highlights the large gaps in knowledge generated mostly by a hepatocyte centric view of the development of liver fibrosis. It also raises interesting questions about how these HSC cell–cell interactions progress from early initiation to the full development of liver fibrosis. It is still possible that at the early stages of NASH fibrosis hepatocyte–HSC interactions are dominant. Finally, it may also begin to provide an understanding of the troubling, yet underdiscussed finding, that although many HSC activators have been identified, suggesting a highly redundant system, loss of many individual molecules seems to halt the development of liver fibrosis.
Effects of common minor alleles in the development of liver fibrosis in NASH
In common with other complex diseases such as hypertension and dementia the development of the disease phenotype in NASH is due to a combination of genetics and environmental factor. Based on comparison between siblings, monogenic twins and other family members, there is a very wide range in the estimation of the genetic contribution to NASH, ranging between 20% and 70%.104,106 This can be understood in part due to different frequency of the minor alleles in the different study population, differences in the definition of NASH and because the effect of the genetic contribution of the currently identified alleles seems to increase with increasing obesity. The assessment of the genetic contribution to fibrosis in NASH is further limited by the available studies which have focused more on the genetic contribution to the development of NAFLD or NASH as these are easier to identify in a large population, as compared with fibrosis. Also, the bulk of the data is from European populations.
Studies on the genetic susceptibility to NASH have identified some clear and well-known minor alleles which are relatively common (allele frequency greater than 5%). The five best characterised are minor alleles of PNPLA3, TM6SF2, HSD17B13, MBOAT7 and GCKR, and all except HSD17B13 result in a greater probability of a disease phenotype.107 The function of these alleles is overwhelmingly related to lipid biology in hepatocytes with greater steatosis and hepatocyte stress. The role of hepatocyte stress stimulating activation of the innate immune system and possibly HSC directly suggests that the genetic susceptibility to NASH would also extend to greater risk of liver fibrosis and this appears to be the case, at least for the PNPLA3 gene.108 In addition to having an in-direct effect on HSC activation via hepatocyte stress, it is possible that some of these proteins, and their variant forms may have a direct effect on HSC activation as they are expressed in HSC, PNPLA 3, for example, is highly expressed in human HSC, and this is increased on HSC activation. Increased PNPLA3 levels results in reduced HSC lipid droplet content, and the common mutant 148M has reduced enzymatic activity.109 Knockdown of PNPLA3 in HSC significantly reduces the profibrogenic protein α-SMA, and HSC with the common mutant PNPLA3 had significantly higher release of proinflammatory cytokines.110 Overall, the data support PNPLA3 as being required in HSC activation, with the common mutant 148M potentiating the profibrogenic phenotype of HSC. An interesting question is if there exist minor alleles which predispose to liver fibrosis directly, or at least independent of negative effects on hepatocytes. This does not seem to be the case for the best characterised five genes but may be the case for the tyrosine kinase MerTK which transduces signals from a variety of extracellular stimuli into the cytoplasm, and mutations in which have been shown to provide protection against liver fibrosis.111 For fibrosis this is relevant because MerTK activation in macrophages promotes liver fibrosis by the production of TGF-β which activates HSC. All transretinoic acid cleavage of MerTK blocks this fibrogenic signal, and there is evidence that this downregulatory pathway is defective in NASH.112 Accounting for all of the major and minor genetic associations, it is estimated that the basis for less than 50% of the genetic susceptibility for NASH has been identified.113
Diabetes mellitus type II and NASH induce liver fibrosis
The development of diabetes mellitus (DM) and NASH shares many aetiological factors such as overnutrition and a high carbohydrate intake, and also share the key pathophysiological feature of total body insulin resistance, and it is therefore not surprising that there is a greater prevalence of NASH in the DM population. An important question is if these are the expected statistical overlap, or if there are specific features of DM which drive liver fibrosis. For the years 2014–2017, the global presence of NAFLD in a DM population was 55%, for NASH was 37% and the estimated prevalence of advance fibrosis among the NAFLD population was 17%.114 Other studies have also identified a greater than twofold increase in cirrhosis and complications of cirrhosis in patients with DM.115 These factors suggest features of DM may be driving liver fibrosis. The connection between DM and fibrosis also extends beyond NASH and the liver as DM is well known to result in cardiac, renal and pulmonary fibrosis, all organs which lack the parenchymal pathology of NASH such as steatosis.
One general driver of fibrosis in DM may be exacerbation of hepatocyte steatosis due to de novo lipogenesis which is known to be essential for NAFLD and NASH. Two key features of DM are hyperglycaemia and high insulin levels, and both can drive de novo lipogenesis. Hyperglycaemia induces de novo lipogenesis via the transcription factor ChREBP, and hyperinsulinaemia via the transcription factor SREBP1c. A further key feature is high levels of FFAs which are converted to triglycerides in hepatocytes in patients with DM, in response to insulin resistance of adipose tissue with loss of insulin mediated inhibition of lipolysis. These features of DM which feed into the pathways of hepatic steatosis are likely supplemented by other direct fibrogenic mechanism, which may be common to the increased fibrosis of multiple organs seen in DM.
Although acceleration of de novo lipogenesis is likely the key mechanism of increased NASH and NASH-induced fibrosis, there are data on a range of metabolite abnormalities found in DM which may directly effect cell types and pathways and promote fibrosis other than de novo lipogenesis.116 Most of this data are of limited scope coming from in vitro studies or single in vivo animal studies, but collectively it suggests that metabolic derangement may be important. High blood glucose has been shown to result in glycated collagen which can result in upregulation of actin, and e-cadherin in cardiac fibroblasts as well as inducing chemotaxis and contraction, and this is consistent with clinical data that shows correlation of hyperglycaemia with increased fibrosis in multiple organs.117,119 Macrophages may also be activated by hyperglycaemia via upregulation of intercellular adhesion molecule (ICAM) and release of chemokines CCL2, as has been demonstrated in renal fibroblasts.120 121 Mast cells are not considered to be major contributors to organ fibrosis but mast cell deficiency has been shown to attenuate cardiac fibrosis and hyperglycaemia results in release of mast cell granules.122,124 The role of hyperglycaemia is highlighted by the increased cardiac mast cell content even in models of type 1 DM which have hyperglycaemia but lack insulin resistance.125 Hyperglycaemia can also activate the renin–angiotensin aldosterone system which increases fibrosis in organs with a major haemodynamic role such as the heart and the kidneys.126,128 Hyperglycaemia may also directly activate the profibrogenic TGF-β pathway by increase TGF-β expression, activating latent TGF-β and upregulating TGF-β receptors.129,131
In addition to hyperglycaemia, there are limited data that fatty acids can promote fibrosis by modifying T cell biology with a reduction in PD1+CD4+ T-cell frequency, and by in vitro treatment with fatty acids, but the in vivo relevance of this is unclear.87 132 There are limited data of direct lipotoxic injury with hepatocyte lipotoxicity resulting in release of inflammatory mediators including BMP-23, a member of the TGF-β superfamily, which can activate HSC via conventional Smad pathways.133 134 Elevations in adipokines such as leptin have also been shown to have profibrogenic effects by promoting fibroblast to myofibroblast conversion, inducing collagen synthesis and induction of profibrogenic factors such as TGF-β on macrophages.135,137 Insulin itself has direct proliferative effects on HSC.138 139
Adipose tissue fibrosis
Finally, it is important to point out that the liver is not the only organ which develops fibrosis in the setting of obesity and metabolic syndrome, as this is also well established in white adipose tissue (WAT). As expected with overnutrition WAT undergoes massive expansion, and in addition, there is accumulation of inflammatory cells which directly contribute to insulin resistance.140 Obesity results in both hypertrophy and hyperplasia of WAT, and the factors which determine the relative amounts of each of these are not well understood. Adipocyte hypertrophy is associated with abnormal morphology including dilated endoplasmic reticulum, ruptured plasma membrane and debris in the extracellular space.141 142 These morphological abnormalities are associated with increased secretion of proinflammatory cytokines by macrophages which worsen insulin resistance, including IL-6, IL-8 and TNF-α.143 The increased mass of adipose tissue requires increased vasculature and angiogenesis which is stimulated by the production of angiogenic factors from the adipocytes (epidermal growth factor-2 (EGF-2), vascular epithelial growth factor (VEGF), hepatocyte growth factor (HGF), insulin like growth factor (IGF) and others).144 Surprisingly, and in contrast to the liver, one of the main drivers of adipocyte death and adipose tissue pathology appears to be lack of adequate angiogenesis resulting in hypoxic death of adipocytes. This is supported by the ability of increased angiogenesis to result in less adipocyte death, and less adipose tissue inflammation and fibrosis.145 146 The current paradigm is that relative under-vascularisation results in local hypoxia, resulting in infiltration of proinflammatory macrophages with insulin resistance and adipose tissue fibrosis.147 One interesting feature of adipose tissue fibrosis is the ability of fibrosis to physically constrain adipose tissue expansion, as demonstrated by increased adipose tissue mass in collagen VI null mice.148 A final difference between adipose and liver fibrosis is that PDGFRα+ cells appear to have the capacity to differentiate into adipocytes or myofibroblasts, although there is some suggestion that PDGFRa+CD9+ progenitors are driven towards myofibroblasts and PDGFRa+CD9- progenitors are driven towards adipocytes.149 In summary, adipose tissue shares with the liver the development of fibrosis in the setting of obesity and the metabolic syndrome with some important differences that fibrosis appears to be driven in part by hypovascularisation. It is unclear if there are any direct consequences of adipose tissue fibrosis for liver fibrosis, but there are clear negative implication for hepatic steatosis. This is due to increased adipose inflammation resulting in insulin resistance and release of FFAs into the blood stream, and the adipose fibrosis limiting the growth of the WAT pool, thus directing nutrients to other organs including the liver.
Conclusions
Although liver fibrosis is a common feature of all types of chronic liver injury, there appear to be unique aspects to metabolic syndrome driven liver fibrosis. Fibrosis due to chronic hepatocellular injury appears dominated by hepatocyte driven signals resulting in activation of resident Kupffer cells and recruitment of infiltrating macrophages which produce profibrogenic cytokines. In contrast metabolic syndrome induced liver fibrosis appears to have many points of initiation and progression. Hepatocyte damage due to insulin resistance and lipotoxicity is still a major driver, in addition, there are several innate immune cells which are activated by hepatocyte signals as well as cells of adaptive immune system demonstrating autotoxicity (CD8+T cells) and profibrotic signals (TH17, CD4+T cells). Finally, the nature of the cell-to-cell interactions in established liver fibrosis in NASH demonstrate predominantly autocrine loops, with a minimal role for hepatocyte–HSC interactions. These sets of relationships may, however, be very different at stage of early fibrosis or fibrosis resolution. Understanding of such cellular interactions is key to developing therapies which are effective at a variety of stages of liver fibrosis.
Footnotes
Funding: NIH award: 5U01AA026962-05.
prepub: Prepublication history for this paper is available online. To view these files, please visit the journal online (http://dx.doi.org/10.1136/egastro-2023-100015).
Patient consent for publication: Not applicable.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
References
- 1.Trivedi P, Wang S, Friedman SL. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33:242–57. doi: 10.1016/j.cmet.2020.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75:473–88. doi: 10.1002/hep.32285. [DOI] [PubMed] [Google Scholar]
- 3.Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149:389–97. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced Nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology. 2018;155:443–57. doi: 10.1053/j.gastro.2018.04.034. [DOI] [PubMed] [Google Scholar]
- 5.Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42. doi: 10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 7.Lemoinne S, Thabut D, Housset C. Portal myofibroblasts connect angiogenesis and fibrosis in liver. Cell Tissue Res. 2016;365:583–9. doi: 10.1007/s00441-016-2443-5. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Kisseleva T. Bone marrow-derived fibrocytes contribute to liver fibrosis. Exp Biol Med (Maywood) 2015;240:691–700. doi: 10.1177/1535370215584933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filliol A, Saito Y, Nair A, et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610:356–65. doi: 10.1038/s41586-022-05289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desroches-Castan A, Tillet E, Ricard N, et al. Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology. 2019;70:1392–408. doi: 10.1002/hep.30655. [DOI] [PubMed] [Google Scholar]
- 11.DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61:1740–6. doi: 10.1002/hep.27376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–8. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong X, Kuang H, Ansari S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. 2019;75:644–60. doi: 10.1016/j.molcel.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobie R, Wilson-Kanamori JR, Henderson BEP, et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–47. doi: 10.1016/j.celrep.2019.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–53. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Xu J, Rosenthal S, et al. Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology. 2020;158:1728–44. doi: 10.1053/j.gastro.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu LK, Liu YC, Ma G, et al. High levels of glucose promote the activation of hepatic Stellate cells via the P38-mitogen-activated protein kinase signal pathway. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15038419. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto R, Enjoji M, Kohjima M, et al. High glucose stimulates hepatic Stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int. 2005;25:1018–26. doi: 10.1111/j.1478-3231.2005.01130.x. [DOI] [PubMed] [Google Scholar]
- 19.Bechmann LP, Zahn D, Gieseler RK, et al. Resveratrol amplifies profibrogenic effects of free fatty acids on human hepatic stellate cells. Hepatol Res. 2009;39:601–8. doi: 10.1111/j.1872-034X.2008.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbero-Becerra VJ, Giraudi PJ, Chávez-Tapia NC, et al. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicol In Vitro. 2015;29:1753–8. doi: 10.1016/j.tiv.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 21.Teratani T, Tomita K, Suzuki T, et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic Stellate cells. Gastroenterology. 2012;142:152–64. doi: 10.1053/j.gastro.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 22.Teratani T, Tomita K, Furuhashi H, et al. Lipoprotein lipase up-regulation in hepatic stellate cells exacerbates liver fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun. 2019;3:1098–112. doi: 10.1002/hep4.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128:2657–69. doi: 10.1172/JCI97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rius B, Titos E, Morán-Salvador E, et al. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB J. 2014;28:836–48. doi: 10.1096/fj.13-235614. [DOI] [PubMed] [Google Scholar]
- 26.Börgeson E, Johnson AMF, Lee YS, et al. Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. 2015;22:125–37. doi: 10.1016/j.cmet.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han Y-H, Shin K-O, Kim J-Y, et al. A Maresin 1/RORα/12-Lipoxygenase Autoregulatory circuit prevents inflammation and progression of Nonalcoholic Steatohepatitis. J Clin Invest. 2019;129:1684–98. doi: 10.1172/JCI124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennan EP, Nolan KA, Börgeson E, et al. Lipoxins attenuate renal fibrosis by inducing Let-7C and suppressing TGFbetaR1. J Am Soc Nephrol. 2013;24:627–37. doi: 10.1681/ASN.2012060550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qu X, Zhang X, Yao J, et al. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J Pathol. 2012;228:506–19. doi: 10.1002/path.4050. [DOI] [PubMed] [Google Scholar]
- 30.Hwang S, He Y, Xiang X, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology. 2020;72:412–29. doi: 10.1002/hep.31031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Gao B. From basic liver immunology to therapeutic opportunities for liver diseases. Cell Mol Immunol. 2021;18:1–3. doi: 10.1038/s41423-020-00607-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Ota N, Manzanillo P, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–41. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- 33.Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–9. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsova P, Ibrabim SH, Gores GJ, et al. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57:1758–70. doi: 10.1194/jlr.R066357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malhi H, Barreyro FJ, Isomoto H, et al. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–31. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malhi H, Bronk SF, Werneburg NW, et al. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 37.Kakisaka K, Cazanave SC, Fingas CD, et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G77–84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thapaliya S, Wree A, Povero D, et al. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig Dis Sci. 2014;59:1197–206. doi: 10.1007/s10620-014-3167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canbay A, Taimr P, Torok N, et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–63. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 40.Eguchi A, Yan R, Pan SQ, et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1Beta and IL-17 upregulation in alcoholic hepatitis mice. J Mol Med (Berl) 2020;98:1021–34. doi: 10.1007/s00109-020-01926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An P, Wei L-L, Zhao S, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020;11:2362. doi: 10.1038/s41467-020-16092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh R, Wang Y, Xiang Y, et al. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan S-S, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–43. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 44.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765–83. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15:738–52. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Martinez I, Santoro N, Chen Y, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of Tlr9. J Clin Invest. 2016;126:859–64. doi: 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1Beta in mice. Gastroenterology. 2010;139:323–34. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847. [DOI] [PubMed] [Google Scholar]
- 49.Arriazu E, Ge X, Leung T-M, et al. Signalling via the osteopontin and high mobility group Box-1 axis drives the fibrogenic response to liver injury. Gut. 2017;66:1123–37. doi: 10.1136/gutjnl-2015-310752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Zeng C, Zheng B, et al. Hmgb1-induced autophagy facilitates hepatic stellate cells activation: a new pathway in liver fibrosis. Clin Sci (Lond) 2018;132:1645–67. doi: 10.1042/CS20180177. [DOI] [PubMed] [Google Scholar]
- 51.Rangwala F, Guy CD, Lu J, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224:401–10. doi: 10.1002/path.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang JJ, Tao H, Li J. Hedgehog signaling pathway as key player in liver fibrosis: new insights and perspectives. Expert Opin Ther Targets. 2014;18:1011–21. doi: 10.1517/14728222.2014.927443. [DOI] [PubMed] [Google Scholar]
- 53.Guillen-Sacoto MJ, Martinez AF, Abe Y, et al. Human germline hedgehog pathway mutations predispose to fatty liver. J Hepatol. 2017;67:809–17. doi: 10.1016/j.jhep.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peiseler M, Schwabe R, Hampe J, et al. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77:1136–60. doi: 10.1016/j.jhep.2022.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Kazankov K, Jørgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16:145–59. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 56.Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-SAC-derived erythro-myeloid progenitors. Nature. 2015;518:547–51. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacParland SA, Liu JC, Ma X-Z, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9:4383. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao J, Zhang S, Liu Y, et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020;6:22. doi: 10.1038/s41421-020-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fan X, Zhang Q, Li S, et al. Attenuation of CCL4-induced hepatic fibrosis in mice by vaccinating against TGF-Beta1. PLoS One. 2013;8:e82190. doi: 10.1371/journal.pone.0082190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miura K, Yang L, van Rooijen N, et al. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310–21. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270–83. doi: 10.1002/hep.29544. [DOI] [PubMed] [Google Scholar]
- 63.Syn W-K, Choi SS, Liaskou E, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–15. doi: 10.1002/hep.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pradere J-P, Kluwe J, De Minicis S, et al. Hepatic Macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stienstra R, Saudale F, Duval C, et al. Kupffer cells promote hepatic steatosis via interleukin-1Beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–22. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 66.Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186–95. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cabeza-Cabrerizo M, Cardoso A, Minutti CM, et al. Dendritic cells revisited. Annu Rev Immunol. 2021;39:131–66. doi: 10.1146/annurev-immunol-061020-053707. [DOI] [PubMed] [Google Scholar]
- 68.Deczkowska A, David E, Ramadori P, et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. 2021;27:1043–54. doi: 10.1038/s41591-021-01344-3. [DOI] [PubMed] [Google Scholar]
- 69.Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–405. doi: 10.1002/hep.26937. [DOI] [PubMed] [Google Scholar]
- 70.Rensen SS, Slaats Y, Nijhuis J, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175:1473–82. doi: 10.2353/ajpath.2009.080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mirea A-M, Toonen EJM, van den Munckhof I, et al. Increased proteinase 3 and neutrophil elastase plasma concentrations are associated with non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. Mol Med. 2019;25:16. doi: 10.1186/s10020-019-0084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zang S, Wang L, Ma X, et al. Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem Biophys. 2015;73:479–87. doi: 10.1007/s12013-015-0682-9. [DOI] [PubMed] [Google Scholar]
- 73.Pulli B, Ali M, Iwamoto Y, et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal. 2015;23:1255–69. doi: 10.1089/ars.2014.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen J, Liang B, Bian D, et al. Knockout of neutrophil elastase protects against Western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun. 2019;518:691–7. doi: 10.1016/j.bbrc.2019.08.111. [DOI] [PubMed] [Google Scholar]
- 75.Moles A, Murphy L, Wilson CL, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60:782–91. doi: 10.1016/j.jhep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao X, Yang L, Chang N, et al. Neutrophils undergo switch of apoptosis to netosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. 2020;11:379. doi: 10.1038/s41419-020-2582-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krijgsman D, Hokland M, Kuppen PJK. The role of natural killer T cells in cancer-A phenotypical and functional approach. Front Immunol. 2018;9:367. doi: 10.3389/fimmu.2018.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of Intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–64. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 79.Syn W-K, Oo YH, Pereira TA, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wehr A, Baeck C, Heymann F, et al. Chemokine receptor CXCR6-dependent hepatic NK T cell accumulation promotes inflammation and liver fibrosis. J Immunol. 2013;190:5226–36. doi: 10.4049/jimmunol.1202909. [DOI] [PubMed] [Google Scholar]
- 81.Hinks TSC, Zhang XW. MAIT cell activation and functions. Front Immunol. 2020;11:1014. doi: 10.3389/fimmu.2020.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hegde P, Weiss E, Paradis V, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. 2018;9:2146. doi: 10.1038/s41467-018-04450-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Böttcher K, Rombouts K, Saffioti F, et al. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic Stellate cell activation. Hepatology. 2018;68:172–86. doi: 10.1002/hep.29782. [DOI] [PubMed] [Google Scholar]
- 84.Ioannou GN, Haigh WG, Thorning D, et al. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res. 2013;54:1326–34. doi: 10.1194/jlr.M034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Daemen S, Gainullina A, Kalugotla G, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. 2021;34:108626. doi: 10.1016/j.celrep.2020.108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Remmerie A, Martens L, Thoné T, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity. 2020;53:641–57. doi: 10.1016/j.immuni.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol. 2020;17:81–92. doi: 10.1038/s41575-019-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhattacharjee J, Kirby M, Softic S, et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun. 2017;1:299–310. doi: 10.1002/hep4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghazarian M, Revelo XS, Nøhr MK, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol. 2017;2:eaai7616. doi: 10.1126/sciimmunol.aai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Breuer DA, Pacheco MC, Washington MK, et al. Cd8(+) T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2020;318:G211–24. doi: 10.1152/ajpgi.00040.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dudek M, Pfister D, Donakonda S, et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444–9. doi: 10.1038/s41586-021-03233-8. [DOI] [PubMed] [Google Scholar]
- 92.Koda Y, Teratani T, Chu P-S, et al. Cd8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. 2021;12:4474. doi: 10.1038/s41467-021-24734-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirsova P, Bamidele AO, Wang H, et al. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (Lausanne) 2021;12:760860. doi: 10.3389/fendo.2021.760860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo X-Y, Takahara T, Kawai K, et al. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol. 2013;305:G891–9. doi: 10.1152/ajpgi.00193.2013. [DOI] [PubMed] [Google Scholar]
- 95.Rau M, Schilling A-K, Meertens J, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of TH17 cells in the liver and an increased TH17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. 2016;196:97–105. doi: 10.4049/jimmunol.1501175. [DOI] [PubMed] [Google Scholar]
- 96.Her Z, Tan JHL, Lim Y-S, et al. CD4(+) T cells mediate the development of liver fibrosis in high fat diet-induced NAFLD in humanized mice. Front Immunol. 2020;11:580968. doi: 10.3389/fimmu.2020.580968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Giles DA, Moreno-Fernandez ME, Stankiewicz TE, et al. Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PLoS One. 2016;11:e0149783. doi: 10.1371/journal.pone.0149783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harley ITW, Stankiewicz TE, Giles DA, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830–9. doi: 10.1002/hep.26746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tang Y, Bian Z, Zhao L, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. 2011;166:281–90. doi: 10.1111/j.1365-2249.2011.04471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luther J, Garber JJ, Khalili H, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol. 2015;1:222–32. doi: 10.1016/j.jcmgh.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang W, Wu N, Wang X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep. 2015;5:8096. doi: 10.1038/srep08096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang S, Li K, Pickholz E, et al. An autocrine signaling circuit in hepatic stellate cells underlies advanced fibrosis in nonalcoholic steatohepatitis. Sci Transl Med. 2023;15:eadd3949. doi: 10.1126/scitranslmed.add3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Dig Liver Dis. 2004;36:231–42. doi: 10.1016/j.dld.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 104.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide Association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palmer ND, Musani SK, Yerges-Armstrong LM, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology. 2013;58:966–75. doi: 10.1002/hep.26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol. 2020;17:40–52. doi: 10.1038/s41575-019-0212-0. [DOI] [PubMed] [Google Scholar]
- 107.Sharma D, Mandal P. NAFLD: Genetics and its clinical implications. Clin Res Hepatol Gastroenterol. 2022;46:102003. doi: 10.1016/j.clinre.2022.102003. [DOI] [PubMed] [Google Scholar]
- 108.Bruschi FV, Tardelli M, Herac M, et al. Metabolic regulation of hepatic PNPLA3 expression and severity of liver fibrosis in patients with NASH. Liver Int. 2020;40:1098–110. doi: 10.1111/liv.14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pirazzi C, Valenti L, Motta BM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23:4077–85. doi: 10.1093/hmg/ddu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bruschi FV, Claudel T, Tardelli M, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. 2017;65:1875–90. doi: 10.1002/hep.29041. [DOI] [PubMed] [Google Scholar]
- 111.Petta S, Valenti L, Marra F, et al. MERTK Rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2016;64:682–90. doi: 10.1016/j.jhep.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 112.Cai B, Dongiovanni P, Corey KE, et al. Macrophage mertk promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020;31:406–21. doi: 10.1016/j.cmet.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283:356–70. doi: 10.1111/joim.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. 2019;71:793–801. doi: 10.1016/j.jhep.2019.06.021. [DOI] [PubMed] [Google Scholar]
- 115.Lomonaco R, Godinez Leiva E, Bril F, et al. Advanced liver fibrosis is common in patients with type 2 diabetes followed in the outpatient setting: the need for systematic screening. Diabetes Care. 2021;44:399–406. doi: 10.2337/dc20-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tuleta I, Frangogiannis NG. Diabetic fibrosis. Biochim Biophys Acta Mol Basis Dis. 2021;1867:166044. doi: 10.1016/j.bbadis.2020.166044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yuen A, Laschinger C, Talior I, et al. Methylglyoxal-modified collagen promotes myofibroblast differentiation. Matrix Biol. 2010;29:537–48. doi: 10.1016/j.matbio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 118.Wang Q, Wang J, Wang P, et al. Glycemic control is associated with atrial structural remodeling in patients with type 2 diabetes. BMC Cardiovasc Disord. 2019;19:278. doi: 10.1186/s12872-019-1249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cao Y, Zeng W, Cui Y, et al. Increased myocardial extracellular volume assessed by cardiovascular magnetic resonance T1 mapping and its determinants in type 2 diabetes mellitus patients with normal myocardial systolic strain. Cardiovasc Diabetol. 2018;17:7. doi: 10.1186/s12933-017-0651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chow FY, Nikolic-Paterson DJ, Ozols E, et al. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–22. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 121.Chow FY, Nikolic-Paterson DJ, Ma FY, et al. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–80. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 122.Nagai K, Fukushima T, Oike H, et al. High glucose increases the expression of proinflammatory cytokines and secretion of TNFalpha and beta-hexosaminidase in human mast cells. Eur J Pharmacol. 2012;687:39–45. doi: 10.1016/j.ejphar.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 123.He A, Fang W, Zhao K, et al. Mast cell-deficiency protects mice from streptozotocin-induced diabetic cardiomyopathy. Transl Res. 2019;208:1–14. doi: 10.1016/j.trsl.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Uemura K, Kondo H, Ishii Y, et al. Mast cells play an important role in the pathogenesis of hyperglycemia-induced atrial fibrillation. J Cardiovasc Electrophysiol. 2016;27:981–9. doi: 10.1111/jce.12995. [DOI] [PubMed] [Google Scholar]
- 125.Huang ZG, Jin Q, Fan M, et al. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS One. 2013;8:e60827. doi: 10.1371/journal.pone.0060827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Koszegi S, Molnar A, Lenart L, et al. RAAS inhibitors directly reduce diabetes-induced renal fibrosis via growth factor inhibition. J Physiol. 2019;597:193–209. doi: 10.1113/JP277002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Asbun J, Manso AM, Villarreal FJ. Profibrotic influence of high glucose concentration on cardiac fibroblast functions: effects of losartan and vitamin E. Am J Physiol Heart Circ Physiol. 2005;288:H227–34. doi: 10.1152/ajpheart.00340.2004. [DOI] [PubMed] [Google Scholar]
- 128.Zhou Y, Poczatek MH, Berecek KH, et al. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun. 2006;339:633–41. doi: 10.1016/j.bbrc.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 129.Zhang D, Jin W, Wu R, et al. High glucose intake exacerbates autoimmunity through reactive-oxygen-species-mediated TGF-beta cytokine activation. Immunity. 2019;51:671–81. doi: 10.1016/j.immuni.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ko S-H, Hong O-K, Kim J-W, et al. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin-angiotensin system. J Cell Biochem. 2006;98:343–55. doi: 10.1002/jcb.20797. [DOI] [PubMed] [Google Scholar]
- 131.Wu L, Derynck R. Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell. 2009;17:35–48. doi: 10.1016/j.devcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Seike T, Mizukoshi E, Yamada K, et al. Fatty acid-driven modifications in T-cell profiles in non-alcoholic fatty liver disease patients. J Gastroenterol. 2020;55:701–11. doi: 10.1007/s00535-020-01679-7. [DOI] [PubMed] [Google Scholar]
- 133.Haczeyni F, Wang H, Barn V, et al. The selective peroxisome proliferator-activated receptor-Delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. 2017;1:663–74. doi: 10.1002/hep4.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vacca M, Leslie J, Virtue S, et al. Bone morphogenetic protein 8B promotes the progression of non-alcoholic steatohepatitis. Nat Metab. 2020;2:514–31. doi: 10.1038/s42255-020-0214-9. [DOI] [PubMed] [Google Scholar]
- 135.Choi SS, Syn W-K, Karaca GF, et al. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J Biol Chem. 2010;285:36551–60. doi: 10.1074/jbc.M110.168542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zibadi S, Cordova F, Slack EH, et al. Leptin’s regulation of obesity-induced cardiac extracellular matrix remodeling. Cardiovasc Toxicol. 2011;11:325–33. doi: 10.1007/s12012-011-9124-0. [DOI] [PubMed] [Google Scholar]
- 137.Wang J, Leclercq I, Brymora JM, et al. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology. 2009;137:713–23. doi: 10.1053/j.gastro.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Svegliati-Baroni G, Ridolfi F, Di Sario A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology. 1999;29:1743–51. doi: 10.1002/hep.510290632. [DOI] [PubMed] [Google Scholar]
- 139.Adachi M, Osawa Y, Uchinami H, et al. The forkhead transcription factor Foxo1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology. 2007;132:1434–46. doi: 10.1053/j.gastro.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 140.Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism. Cell. 2018;172:22–40. doi: 10.1016/j.cell.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 142.Giordano A, Murano I, Mondini E, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. 2013;54:2423–36. doi: 10.1194/jlr.M038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jernås M, Palming J, Sjöholm K, et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. 2006;20:1540–2. doi: 10.1096/fj.05-5678fje. [DOI] [PubMed] [Google Scholar]
- 144.Herold J, Kalucka J. Angiogenesis in adipose tissue: the interplay between adipose and endothelial cells. Front Physiol. 2020;11:624903. doi: 10.3389/fphys.2020.624903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Michailidou Z, Turban S, Miller E, et al. Increased angiogenesis protects against adipose hypoxia and fibrosis in metabolic disease-resistant 11Beta-hydroxysteroid dehydrogenase type 1 (HSD1)-deficient mice. J Biol Chem. 2012;287:4188–97. doi: 10.1074/jbc.M111.259325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.An YA, Sun K, Joffin N, et al. Angiopoietin-2 in white adipose tissue improves metabolic homeostasis through enhanced angiogenesis. Elife. 2017;6:e24071. doi: 10.7554/eLife.24071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Khan T, Muise ES, Iyengar P, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–91. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Marcelin G, Ferreira A, Liu Y, et al. A Pdgfralpha-mediated switch toward CD9(High) adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 2017;25:673–85. doi: 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]