

Abstract
Oxytocin (OXT) is a highly conserved neuropeptide that modulates social cognition, and variation in its receptor gene (Oxtr) is associated with divergent social phenotypes. The cellular mechanisms connecting Oxtr genotype to social phenotype remain obscure. We exploit an association between Oxtr polymorphisms and striatal-specific OXTR density in prairie voles to investigate how OXTR signaling influences the brain transcriptome. We discover widespread, OXTR signaling-dependent transcriptomic changes. Interestingly, OXTR signaling robustly modulates gene expression of C-type lectin-like receptors (CTLRs) in the natural killer gene complex, a genomic region associated with immune function. CTLRs are positioned to control microglial synaptic pruning; a process important for shaping neural circuits. Similar relationships between OXTR RNA and CTLR gene expression were found in human striatum. These data suggest a potential molecular mechanism by which variation in OXTR signaling due to genetic background and/or life-long social experiences, including nurturing/neglect, may affect circuit connectivity and social behavior.
Introduction
Oxytocin (OXT) is a highly conserved neuromodulator of social behaviors, and variations of this molecule have been identified in several invertebrates and most vertebrates1. This neuropeptide family has evolved from controlling gustatory plasticity and mating circuits to regulating a diverse range of complex social behaviors2. OXT signals through its associated receptor (OXTR), which enhances the saliency of social stimuli3, and facilitates various aspects of social cognition, such as social reward processing4, individual recognition5, social attachment6, and parental nurturing7. Animal studies continue to elucidate neural circuits that are modulated by OXT and shape social phenotypes8, which informs clinical studies to assess the usefulness of the OXT system as a target for the treatment of conditions of social disability, such as social anxiety or autism spectrum disorders (ASD)9. A detailed understanding of OXT signaling and its consequences on brain function will therefore provide valuable insight into the mechanisms that determine sociality in both health and disease.
The socially monogamous prairie vole (Microtus ochrogaster) shows notable similarities to human social behavior, as they console familiar conspecifics, form long-term pair bonds, and engage in biparental care10. Prairie voles produce substantial densities of OXTR in the nucleus accumbens (NAC)11, a brain region that is at the interface of sensory perception and motivated actions12. OXT signaling in the NAC facilitates the formation of enduring pair bonds, as genetically increasing or decreasing OXTR densities in this area can respectively facilitate or prevent mating-induced partner preference, a laboratory proxy for pair bond strength, which supports a direct link between NAC OXTR signaling and social attachment13–15. Interestingly, promiscuous rodent species, such as the montane vole (Microtus montanus), the house mouse (Mus musculus), and the Norway rat (Rattus norvegicus), show much lower densities of NAC OXTR11,16. A similar relationship between mating strategy and NAC OXTR levels is also observed in primates17, as the NAC of the polygamous rhesus macaque (Macaca mulatta) is devoid of OXTR18, while the socially monogamous marmoset (Callithrix jaccus) has abundant NAC OXTR19. Intriguingly, humans show abundant NAC OXTR expression as well20.
Variation in NAC OXTR abundance not only exists between species, but also within species, e.g., prairie voles exhibit considerable individual variation in NAC OXTR density. Moreover, compared to standard laboratory mouse strains, our laboratory prairie vole population contains a relatively high degree of genetic variation. This previously enabled us to identify a set of single nucleotide polymorphisms (SNPs) in the prairie vole Oxtr gene locus that strongly predicts individual OXTR density in the NAC (R2>0.7), but not in other brain regions21. Varying densities of NAC OXTR not only associate with performance in laboratory assays designed to quantify aspects of sociality such as partner preference21, alloparental care22, and resilience to neonatal neglect or paternal absence23,24, but also with divergent mating strategies in the wild25. Thus, variation in NAC OXTR signaling associates with a range of social behaviors throughout development and may promote diversity in social strategies.
In humans, SNPs in the OXTR gene predict its expression26, and not only link to indicators of sociality such as relationship quality6, resilience to childhood adversity27,28, and social recognition29, but also predict brain function in relation to social disability30–32. Understanding the mechanistic link between genetic variation in OXTR signaling and divergent sociality will provide fundamental insight into how social phenotypes are shaped by social experiences and could give clues to the complex etiologies of conditions of altered sociality, such as ASD and schizophrenia. Here, we exploited our previously reported association between genetic variation in the Oxtr gene and NAC OXTR density to investigate the molecular pathways by which variation in OXTR signaling might shape sociobehavioral phenotypes. We used ATAC and RNA sequencing to profile genotype-associated differences in chromatin accessibility and transcription. Unexpectedly, we reveal that genetic variation in the Oxtr gene associates with differences in chromatin accessibility and predicts brain gene expression beyond the NAC and Oxtr expressing cell types. Using a systemic OXTR KO prairie vole, we show that parts of the transcriptional differences associated with genotype are directly modulated by OXTR signaling. In particular, OXTR signaling controls the gene expression of C-type lectin-like receptors (CTLRs) located in the natural killer gene complex (NKC), a genomic region that is primarily known to govern innate immune responses33, although a central function has been suggested recently34. Our results give surprising clues to the cellular processes that are under control of OXT signaling and thereby provide hypotheses on the origins of diversity in social phenotypes.
Results
Non-coding genetic variation in the Oxtr gene associates with gene expression differences across brain areas
We previously demonstrated that in prairie voles, NAC OXTR density is strongly predicted by a set of non-coding SNPs in the Oxtr gene locus (R2>0.7, Figure 1A)21. This variation in OXTR abundance is brain region-specific, as genotypes show equal densities of OXTR in the insular cortex (INS). These SNPs are in perfect linkage disequilibrium (LD) with each other, and we genotyped animals at position NW_004949099:26351196 to differentiate between low (T/T) and high (C/C) NAC Oxtr expressing animals. To investigate whether region-specific associations of genotype with Oxtr expression also holds true for the expression of genes other than Oxtr and explore the molecular pathways that potentially link genetic variation to social strategy, we performed total RNA-seq on tissue from NAC, INS, and the superior colliculus (SC), where OXTR density is indistinguishable from background levels (Figure 1A–B). We quantified gene expression differences between the genotypes in each of these brain regions, while controlling for batch and sex effects (Supplementary Tables 1A–C). In line with OXTR protein distribution21, we found genotype-dependent variation in Oxtr RNA levels in the NAC (N=15, Padj=0.001), no significant differences in Oxtr expression in the INS (N=14, Padj=0.53), and low, but differentially expressed Oxtr RNA levels in the SC (N=16, Padj=0.04) (Figure 1C–D). In addition, we identified genotype-associated differentially expressed genes (DEGs, defined as Padj<0.1) in all three brain regions (30 genes in NAC, 66 genes in INS, and 147 genes in SC). Although we confirm that genotype predicts region-specific in Oxtr expression, we find, to the contrary of our expectation, that other genes show different patterns of genotype dependence across these brain regions. For example, nine genes were consistently DE across brain regions (of which two genes were consistently increased in T/T animals, and seven consistently increased in C/C animals). Notably, two of these genes (Mb4 and LOC101986986, the latter referred to as Klrb1a hereafter) locate to the same scaffold as Oxtr35. Scaffolds are computationally constructed DNA sequences, which are not necessarily as the same length as chromosomes. As such, they can be viewed as (partial) maps of chromosomes. The prairie vole genome assembly is fragmented, meaning that for many scaffolds it is not determined to which chromosome they map. Nevertheless, DEGs that locate to the Oxtr scaffold possibly indicate that differential expression is related to a genetic linkage to genotype rather than an effect of variation in NAC OXTR signaling. We previously only characterized genetic variability proximally to the Oxtr gene locus (~30 kb)21, so it is possible that additional SNPs in LD exist outside this region. So, we confirm our previous result that genotype predicts region-specific Oxtr expression, but unexpectedly find that the Oxtr genotype predicts gene expression patterns in the INS and SC as well. Thus, Oxtr genotype associations extend beyond a region-specific influence on Oxtr expression, and include widespread gene expression changes across multiple brain areas.
Figure 1. Non-coding genetic variation in the Oxtr gene associates with gene expression differences across brain areas.

a) Illustration of the genomic location of previously identified SNPs that predict NAC OXTR levels. In red, the SNP that was genotyped to distinguish high NAC OXTR producers (C/C) from animals with low levels of NAC OXTR (T/T). Colored circles on I125-OVTA autoradiograms indicate brain areas that were sampled for sequencing experiments. b) Schematic that depicts the experimental workflow. Prairie vole picture was generated by AI c) Normalized Oxtr RNA counts for each brain area, split by genotype. d) Volcano plots depicting DEG genes between C/C and T/T animals, one for each separate brain region. e) A volcano plot showing DEGs between C/C and T/T animals across brain regions. f) Plot depicting enrichment of vole scaffolds for DEGs. g) A volcano plot showing DARs between C/C and T/T animals across brain regions. h) Plot depicting enrichment of vole scaffolds for DARs. In plot F and H, scaffolds above the red line are significantly enriched (Padj<0.1). DARs and DEGs are defined at Padj<0.1.
The Oxtr scaffold is enriched for differentially expressed genes and differentially accessible chromatin regions
To increase our power to identify genes that consistently vary between genotypes across brain regions, we determined genotype effects on gene expression after collapsing data across NAC, INS and SC, while controlling for region, sex, and batch. This analysis provided strong evidence of divergent gene expression (N=45, 1698 DEGs, Figure 1E and Supplementary Table 1D). Interestingly, 51 DEGs located to the Oxtr scaffold (NW_004949099.1), consistent with genetic linkage of these DEGs to genotype. To investigate whether more DEGs located to the Oxtr scaffold than would be expected by chance, we performed a series of Fisher’s Exact Tests, one for each scaffold that contains genes that were included in the expression analysis. We identified 5 scaffolds that are enriched for DEGs (Padj<0.1, Fisher’s Exact Test), including the Oxtr scaffold (Padj=0.029, Fisher’s Exact Test, Figure 1F and Supplementary Table 1E), indicating that a disproportionate number of genes located near Oxtr are differentially expressed between genotypes.
We next performed ATAC-seq on the same samples to determine whether Oxtr genotype was also associated with differences in chromatin accessibility. ATAC peaks preferentially located to expressed genes (P=2.47e−323, Poisson Test, Extended Data 1), supporting the notion that chromatin accessibility and gene expression are functionally linked. We quantified differential accessible regions (DARs) across brain regions, while controlling for region, sex and batch (N=45). Oxtr genotype was associated with 2686 differentially accessible regions (DARs, Padj<0.1, Figure 1G and Supplementary Table 1F), and Fisher’s Exact Tests determined that 36 scaffolds were DAR-enriched, with the Oxtr-containing scaffold showing the strongest enrichment (Padj=1.01e−18). Together, these data show strong differences between genotypes in gene expression and chromatin accessibility surrounding the Oxtr gene. This supports the idea that genotype significantly influences chromatin structure and brain transcription beyond Oxtr. The enrichment of the Oxtr scaffold for both DARs and DEGs suggest that genotype differences originate from this scaffold. However, it remains unknown how genotypes are mechanistically linked to widespread differences in gene expression.
The natural killer gene complex shows strong differential gene expression and chromatin accessibility between genotypes
To investigate biological processes that may be differentially affected by genotype, we performed a weighted gene correlation network analysis (WGCNA) that identified two divergent gene modules between genotypes (P<0.05) (Figure 2A). One of the modules was enriched for gene ontology (GO) terms related to histone modification and RNA processing, the other was linked to immune processes (Supplementary Table 2A and B). The strong GO-enrichment for immune processes corresponds with the identification of Klrb1a as the top DEG expressed from the Oxtr scaffold (NW_004949099.1) (Figure 1F). Klrb1a encodes a c-type lectin-like receptor (CTLR) that is primarily known to be involved in innate immunity36. Many CTLRs are expressed from a gene cluster that is known as the natural killer gene complex (NKC)37,38, which is located on the same chromosome as Oxtr in rats and mice39. Given the location of Klrb1a on NW_004949099.1 and the fragmented nature of the prairie vole genome assembly35, we reasoned that additional scaffolds in the prairie vole may contain parts of the NKC sequence. To identify NKC scaffolds that are likely to map to the same chromosome as NW_004949099.1, we calculated the percentage of annotated genes on a given prairie vole scaffold that are also present on mouse chromosome 6, which harbors the Oxtr gene (Figure 2B). In addition, we determined which prairie vole scaffolds contain CTLRs (Figure 2C). We mapped the combined results to the mouse NKC33, and concluded that parts of the OXTR scaffold NW_004949099.1:36120000:41749222 as well as NW_004949144.1 and NW_004949240.1 contain parts of the prairie vole NKC region, and that NW_004949097.1 flanks the NKC region (Figure 2D). Fisher’s Exact Tests determined that the constructed NKC region is enriched for DARs (P=3.36e−39) and DEGs (P=9.33e−06). These results identify the NKC as a genomic region that shows strong functional divergence between Oxtr genotypes.
Figure 2. NKC function is divergent between genotypes.

a) Schematic illustrating two gene modules that are significantly different between genotypes, with top 3 GO terms indicated. b) (Left) Plot shows the number of annotated genes that a given prairie vole scaffold shares with the mouse Oxtr chromosome. scaffolds with no matching genes are not included. Dot size represents the percentage of matching genes. (Right) Plot depicts number of CTLR genes on a given prairie vole scaffold. Scaffolds without CTLRs are not included. Dot size indicates the percentage of genes that are CTLRs. c) Upper two plots depict the fold changes of DEGs (green traces) and DARs (gold traces) across NKC scaffolds. Lower plots illustrate how NKC vole scaffolds map to the mouse Oxtr chromosome. Scaffolds are on scale, but scales of vole and mouse scaffolds differ.
Non-coding genetic variation in the Oxtr locus associates with cell-type specific differences in gene expression
Ample evidence connects NKC function to peripheral immune processes33, but much less is known about how this genomic region affects brain function. It is becoming increasingly clear that peripheral immune pathways are centrally repurposed to control neural functioning40, and that glial cells operate these often conserved signaling cascades41. To investigate whether genotype associates with differential gene expression in Oxtr-expressing cells only, or predicts expression patterns across cell types, we performed single nucleus (sn)RNA-seq (N=16) on pooled tissue from the NAC and the paraventricular nucleus (PVN) (Figure 3A). We clustered cells using the Seurat package42, resulting in 10 cell clusters (Figure 3B), which we annotated with established gene markers that strongly differentiated among major cell classes (e.g., neurons and glia) and subclasses (e.g., D1/pDyn+, D1/Mdfic+ and D2+ neurons, Supplementary Table 3A). Cluster size was not significantly different between genotypes (P=0.59, RM2W-ANOVA, Figure 3C), enabling the quantification of gene expression differences between genotypes in a pseudo-bulk analysis, while controlling for sex. In all but one cluster (Prok2+ neurons), we found evidence of differential gene expression (Figure 3C, Supplementary Table 3B). Oxtr expression was significantly different between genotypes in the Drd1/pDyn+ (Padj=5.22e−09), Drd1/Mdfic+ (Padj=9.93e−05), and Drd2+ cell clusters (Padj=1.68e−20) (Figure 3D). This is in line with our previous report, in which we show that NAC Oxtr mRNA predominantly co-localizes with mRNAs of either dopamine receptor (Drd1 or Drd2)43. Notably, the largest numbers of DEGs were observed in cell types that strongly expressed Oxtr (>100 DEGs in both Drd1/pDyn+ and Drd2+ neurons, Figure 3B–D). However, many DEGs were also observed in oligodendrocytes (>100) and Dkk+ astrocytes (>40), cells in which no to very little Oxtr expression could be detected (Figure 3C). This indicates that genotype predicts gene expression in a widespread manner, even in cell types that do not show genotype-associated variation in Oxtr RNA levels.
Figure 3. Non-coding genetic variation in the Oxtr gene associates with gene expression differences across cell types.

a) Schematic of experimental workflow. b) Annotated cell clusters. c) (Left) Size of cell clusters, split by genotype. (Right) Number of DEGs per cell cluster. OPCs are oligodendrocyte precursor cells d) Examples of three DEGs: Oxtr (Left), Klrb1a (Middle), and Clec12a (Right) that illustrate genotype effects on gene expression in a cell-type dependent manner. UMAP plots depict RNA expression with cellular resolution, violin plots show bulk expression levels. Indicated are the cell clusters in which genes were DE.
Next, we set out to confirm our earlier finding that the NKC region is enriched for DEGs. We determined DEGs across cell clusters (Supplementary Table 3C), and consistent with our previous analysis, the NKC region was enriched for DEGs (P = 0.024). In total, 19 out of 116 DE genes were located in the NKC region (NKCDEGs). Two CTLRs were top three NKCDEGs: Clec12a and Klrb1a. CLEC and KLR proteins are known to form ligand-receptor pairs that modulate natural killer (NK) cell function44, a peripheral cell type that bears functional similarities to microglia. We found specific expression of Clec12a in microglia (Figure 3D), and preferential expression of Klrb1a on Oxtr expressing cells (P=9.24e−16, Fisher’s Exact Test), raising the intriguing possibility of functional interactions between Clec12a+ microglia and Klrb1a/Oxtr+ neurons in the NAC. In addition, these results independently support functional divergence in the NKC region associated with genetic variation in the Oxtr gene locus. However, it remains unclear how variation in OXTR signaling is involved in this association. Are effects on gene expression solely driven by genotype, or are they (partly) mediated by OXTR signaling?
Genetic deletion of OXTR reveals that OXTR signaling mediates genotype effects on gene expression
To determine whether variation in OXTR abundance was causally linked to NKC gene expression, we performed bulk RNA-seq on NAC tissue from C/C, T/T and systemic OXTR knock-out voles with a C/C genetic background (CΔ/CΔ, Figure 4A)45. Interestingly, CΔ/CΔ animals produced similar levels of NAC Oxtr RNA as C/C animals, indicating that genotype effects on Oxtr expression are not disturbed by genetic deletion of OXTR (Figure 4B). However, as expected, we observed 60 base pair deletions in the first exon of all CΔ/CΔ Oxtr reads (Extended Data 2). Moreover, we have previously shown that this mutation leads to a complete absence of receptor binding and downstream signaling45. So, although Oxtr is expressed in CΔ/CΔ animals, the resulting RNA does not translate into functional OXTR protein. We next determined expression differences between genotypes (N=24), while controlling for sex. Oxtr showed the strongest association out of 77 DEGs (Padj=3.34e−13) (Figure 4B and Supplementary Table 4A). In addition, we confirmed for the third time that the NKC region is enriched for DEGs (P=5.012e−08, Fisher’s Exact Test).
Figure 4. Genetic deletion of OXTR reveals that OXTR signaling mediates genotype effects.

a) Schematic that depicts experimental workflow. b) Normalized expression of genotype driven genes located on the OXTR scaffold: (Left) A2m, (Middle) Oxtr and (Right) Rpusd3 RNA. Gray lines above plots indicate significant genotype driven effects (Padj<0.1). c) Normalized counts of OXTR mediated RNAs located in the NKC region: (Left) Clec12a, (Middle) CUNH12orf57 and (Right) Klrb1a RNA. Purple lines indicate significant OXTR mediated effects (Padj<0.1). d) Schematic of identified cis effects of genotype on gene expression, separated in genotype driven (gray lines), or OXTR-mediated effects (purple lines). The question mark indicates an unknown genetic module that is causal to Oxtr gene expression differences between genotypes.
To directly investigate DEGs driven by OXTR signaling (OXTRDEGs), we first used Fisher’s method to combine the p-values of genes that showed directionally consistent fold changes and were P<0.05 across all three independent C/C vs T/T comparisons (Figures 1, 3 and 4). After correction for multiple comparisons, we identified 71 high-confidence genes that were consistently and significantly DE (HCDEG) between C/Cs and T/Ts (Supplementary Table 4B). We then identified OXTRDEGs by determining which HCDEGs were DE between C/C and CΔ/CΔ animals. As animals of these genotypes were produced by the same breeder pairs, gene expression differences between these animals can only be caused by a difference in the presence of functional OXTR protein. We identified 24 OXTRDEGs genes, 3 of which are located to the NKC region: Klrb1a, Clec12a and CUNH12orf57 (Figure 4C and Supplementary Table 4C). All HCDEGs in the NKC region were OXTRDEGs. We also identified 8 OXTR-independent DEGs (GTDEGs: HCDEG DE in T/T versus CΔ/CΔ animals), which included 3 genes that locate to the Oxtr scaffold, but not to the NKC region: Oxtr, Rpusd3 and A2m (Figure 4B and Supplementary Table 4D). These genes may span the region that harbor the genetic variation causal to individual variation in NAC OXTR densities. More surprising however, is the finding that OXTR signaling modulates gene expression in the NKC region, as it provides compelling evidence that NKC transcriptional activity is not genetically linked to non-coding genetic variation in the Oxtr gene. Instead, NKC gene expression appears to be modulated by OXTR signaling. In addition, the identification of OXTRDEGs does support the notion that OXTR-signaling has widespread effects on brain cell transcription, i.e., across brain regions and cell types. As demonstrated by the expression patterns of OXTRDEGs in our previous bulk RNA-seq and snRNA-seq experiments, altered expression of OXTRDEGs is not limited to the NAC or Oxtr-expression cells only, but is also affected in brain regions and cell types that do not show genotype-associated differences in Oxtr expression (Extended Data Figure 3). Interestingly, one OXTRDEG - Plgrkt - is involved in lactation46, a well-known function of OXTR signaling. Other OXTRDEGs, Hrh2, Kynu and Recq1, have been associated with social disability47–49, supporting the notion that OXTR signaling influences social phenotypes in both health and disease.
Variation in human NAC OXTR expression associates with altered NKC transcription
In humans, OXTR and the NKC region are not syntenic, and located on chromosome 3 and 12 respectively39. However, given our findings in the prairie vole, it is conceivable that variation in OXTR abundance has non-genetic effects on NKC transcriptional activity in humans as well. To explore this possibility, we investigated which gene expression levels significantly correlate with OXTR RNA abundance in the human NAC. We used GTEx bulk RNA-seq data stemming from human NAC tissue (N=246) for a gene expression analysis with OXTR Transcript Per Kilobase Million (TPM) values as the independent variable26. Genes that were significantly associated with OXTR abundance (N=1385 genes, Padj<0.1 and log2 fold change > 0.2, Supplementary Table 5) were disproportionately located within the human NKC region (Chr12:9000000:10600000, containing 24 genes) (N=7 genes, P=3.36e−3, Fisher’s Exact Test). These genes included six CTLRs (CLEC2C, CLEC8A, KLRC2, KLRC3, KLRF1 and KLRK1) and PZP, an alpha-2-macroglobulin (A2M)-like gene (Figure 5A).
Figure 5. The human NKC expresses several neural CTLRs that correlate with OXTR abundance.

a) Schematic that depicts the NKC genes that significantly correlate with OXTR abundance in the human NAC. b) Cell cluster specific expression levels of OXTR correlated genes that reside in the NKC as well as OXTR in the human NAC.
CTLRs are selectively expressed in dopaminoceptive neurons and microglia in the human NAC
To determine whether NKC gene expression patterns in the human NAC mirrored patterns in prairie vole, we used a publicly available human snRNA-seq data to determine which cell types express these CTLRs50. Although these data were enriched for neuronal cells, we observed evidence for divergent CLEC and KLR expression patterns across cell clusters, with predominant CLEC expression in microglia and KLR expression in dopaminoceptive neurons (Figure 5B). Together, these findings raise the possibility of a conserved relationship between OXTR signaling and central NKC function in prairie voles and humans.
Discussion
Here, we performed three independent sequencing experiments to investigate the causes and consequences of natural variation in NAC Oxtr expression. These experiments provide converging evidence that genetic variation in the prairie vole Oxtr gene locus is associated with widespread differences in the brain transcriptome, even in regions and cell types that do not show genotype-dependent differences in Oxtr expression. Use of systemic OXTR knock-out voles demonstrates that a portion of the genotype effects on neural transcription are caused independently of OXTR signaling and highlight regions outside the Oxtr locus as regions of interest that may be part of the genetic mechanism that drives individual variation in NAC Oxtr expression. Most remarkable however, is our finding that OXTR signaling modulates transcription in the NKC, for which we found strong evidence in three independent experiments. Moreover, we report an association between NAC OXTR expression and NKC transcriptional activity in human NAC as well. Thereby, our results reveal unexpected transcriptional programs that are under control of OXTR signaling and may be conserved across species. The fact that OXTR signaling-dependent transcription was altered in cells where Oxtr expression is not dependent on genotype or in which Oxtr expression could not be detected is surprising. We speculate that altered activity of Oxtr expressing cells during development in response to social environments, e.g., parental nurturing and social play, may alter associated microglial activity and thereby affect downstream neural circuits.
The NKC region has traditionally been linked to immune function, because it harbors a multitude of CTLR genes that collectively enable natural killer (NK) cells to discriminate self from other33,38. CTLRs recognize a wide variety of protein ligands and are pivotal in the shaping of innate immune responses by regulating the cytotoxic processes that occur in defense to pathogens51. We show that the expression levels of two CTLR genes, Klrb1a and Clec12a, diverge between genotypes and are regulated by OXTR signaling. We find that Klrb1a is preferentially expressed in Oxtr-expressing dopaminoceptive cells, while Clec12a expression is restricted to microglia in the prairie vole brain. Intriguingly, our analyses of publicly available human snRNA sequencing data suggest functional similarities in NAC OXTR RNA and NKC transcriptional activity between humans and prairie vole. In humans, we found that NAC OXTR RNA abundance correlates with the expression of several CTLR genes, and that the NKC region is enriched for OXTR correlated genes, even though the NKC and OXTR are not syntenic in humans. Moreover, CTLR genes are expressed across cell types in the human NAC, with CLEC expression predominantly found in microglia and KLR expression observed in dopaminoceptive cells, mirroring prairie vole NAC CTLR expression patterns.
Microglia, like NK cells, use lectins to interact with their environment52, and microglial CTLR expression has been shown to play role in synaptic pruning53. In the periphery, CLEC and KLR proteins can act as ligand-receptor pairs44, which guide NK cells to initiate immune responses by endowing them with the ability to distinguish self from other51. As we show that OXTR signaling regulates the expression of neural CTLR genes, central CLEC:KLR interactions are a candidate mechanism via which OXTR signaling can shape the development of neural circuits. Through region and cell-type specific expression patterns, central CTLRs are positioned to instruct microglia to prune only targeted synapses, while leaving other neural connections intact54. It remains to be determined if OXTR signaling indeed controls microglial activity, and if so, what the exact consequences on circuit structure and function are. Nevertheless, our results indicate a molecular pathway by which variation in OXTR signaling, either by social experience dependent regulation of OXT release or by varying OXTR densities, may potently modulate the neurodevelopmental trajectories of social phenotypes.
These results represent an important advancement in our understanding of how OXTR signaling affects neural circuit functioning. We know from decades of rodent, primate, and human studies that OXTR signaling has the capacity to modulate emotionality and social behaviors in adult individuals16, but the influence of OXTR signaling on neurodevelopment has been much less explored. Previous work in the prairie vole shows that adult social behaviors are susceptible to variation in OXTR signaling interacting with social contexts during critical early life periods23. Early-life neonatal separations (3 hours/day for the first 2 weeks of life) lead to impairments in social bonding in adult prairie voles with low NAC OXTR, while high NAC OXTR voles are resilient to the same neglect model. Presumably, they are rescued by the heightened OXTR signaling upon reunion with parents afforded by the high OXTR density. Additionally, OXTR genotype and the resulting variation in NAC OXTR density interacts with parental composition (uniparental versus biparental care) to influence adult social attachment. Similar gene*environment interactions have been suggested to exist in humans as well. A multitude of human studies have suggested a link between genetic variation in the OXTR gene and diversity in social behavior, psychiatric outcome and brain functional connectivity, including but not limited to resilience to social neglect55, facial recognition29, parental behavior28, and conditions of altered sociality27, such as ASD31,32,56. These data underscore the impact that individual variation in OXTR signaling, whether mediated by variation in OXTR density or social experience, can have on adult sociality and/or emotionality. However, none of these studies provide mechanistic clues as to how OXTR signaling, in interaction with the social environment, shapes social phenotypes. Our results provide a first step in this direction, by showing that OXTR signaling modulates the expression of CTLR genes, which we hypothesize to be involved in shaping neural circuit development.
Concluding, our findings demonstrate that NAC OXTR signaling modulates NKC transcription and provide mechanistic clues into how OXTR signaling might influence neural circuits development and social behaviors. Therefore, we hope that this work will guide future studies to investigate whether OXTR signaling influences microglial pruning of synapses by modulation of CTLR expression, and whether this affects neurodevelopmental trajectories into adult sociality.
Material and methods
Animal husbandry
All male (N=45) and female (N=45) prairie voles (Microtus ochrogaster) used for experimental were virgins between 60–120 days old (early adulthood) and taken from our colony, which consists of breeders derived from field captured voles in Illinois, USA. Prairie voles were housed in same-sex groups (2–3 animals) from post-natal day (PND) 21–23 in ventilated plexiglass cages (36×18×19 cm) filled with bedding and nesting material under a 14:10 h light/dark cycle at 20–22°C and 40–60% humidity with ad libitum access to water and food (Lab Rabbit Diet HF #5236, LabDiet). All experimental procedures were approved by the Emory University Institutional Animal Care and Use Committee.
Genotyping and breeding of C/C, T/T and CΔ/CΔ
We created breeder pairs producing low- or high nucleus accumbens (NAC) OXTR offspring by randomly genotyping prairie voles from our colony at position NW_004949099.1:26351196. A single-nucleotide polymorphism (SNP; C or T) at this locus strongly predicts NAC Oxtr expression. Genomic DNA was isolated from ear clips with the DNAeasy Blood and TIssue kit (Qiagen, Germany). Allele-specific PCR amplification of the SNP locus was achieved using HiDi® DNA Polymerase (myPols Biotec., Germany), allele-specific forward primers (FW_CC: 5’- GTG GAA TCA TCC CAC CGT GC −3’, FW_TT: 5’ - GTG GAA TCA TCC CAC CGT GT −3’) and a reverse primer (RV:5’- TCC CAA TGT GCT AGA GCA GG −3’). Cycling conditions consisted of initial denaturation: 95 °C for 2 min, 35 amplification cycles: 95 °C for 15 sec, 53 °C for 15 sec, 72 °C for 30 sec, and a final elongation phase: 72 °C for 5 min. Heterozygous (CT) animals were selected for further breeding. Their homozygous offspring (either CC or TT) were subsequently crossed to create breeder pairs to solely produce low Oxtr (TT)- or high Oxtr (CC)-expressing animals. For genotyping of OXTR KO animals, we PCR-amplified the region around the 60 bp deletion in the Oxtr gene using these primers (FW: AGATCAGTGCCCGGGTGCCC, RV: TCGAGCGACATAAGCAGCAG). Cycling conditions were: 95 °C for 2 min, 35 amplification cycles: 95 °C for 15 sec, 53 °C for 15 sec, 72 °C for 30 sec, and a final elongation phase: 72 °C for 5 min. Heterozygous OXTR KO animals were first bred with either CC or TT animals. Subsequently, their heterozygous OXTR KO offspring were crossed (C/CΔxC/CΔ and T/CΔxT/CΔ breeder pairs) such that their combined offspring would include TT, CC and CΔ/CΔ animals. Care was taken to prevent inbreeding; all mates had a kinship coefficient of <12.5%.
I125-OVTA autoradiography
Fresh frozen brains were sectioned on a cryostat at 20 µM and mounted on Fisher Superfrost plus slides and stored at −80 °C. The slides were thawed and fixed for two min with 0.1% paraformaldehyde in PBS at RT and then incubated in 50 pM I125-OVTA (2200 Ci/mmol; PerkinElmer; Boston, MA), a selective, radiolabeled OXTR ligand, for one hour. Unbound I125-OVTA was then removed with Tris-MgCl2 buffer and sections were allowed to dry. Sections were exposed to BioMax MR film for 7 days, after which films were developed and images were taken using an MCID camera set-up (InterFocus Ltd, UK).
Bulk total RNA and ATAC sequencing sample preparation
Animals were sacrificed using CO2 asphyxiation, after which brains were dissected out and sliced on a ice-cold mouse brain matrix (Zivic Instruments, USA ). For combined RNA and ATAC sequencing, slices were then transferred to ice-cold HBSS and nucleus accumbens (NAC), insular cortex (INS) and superior colliculus (SC) were isolated using micro punches (1.2 and 2 mm diameter). Punches were digested with papain dissociation enzyme (Thermo-Scientific, USA) in 250 ul HBSS containing 500 units of DNAse (Sigma-Aldrich, Germany) at 37 °C for 30 minutes. Cells were mechanically dissociated and filtrated into single-cell solution using FlowMi filters (40 and 70 um, BelArt, USA). Viability and yield were checked by tryptophan staining after which cells were cryopreserved in HBSS supplemented with 10% DMSO and stored at −80 °C before sending for sequecning (Genewiz, LLC., NJ, USA). Each sample was both ATAC- and RNA-sequenced, except for samples CMI and TMI, that were only ATAC-sequenced. For bulk RNA sequencing of the OXTR KO animals, NAC punches were made from flash-frozen brain slides, stored at −80 °C and sent for sequencing (Genewiz. LLC).
Bulk total RNA sequencing library preparation and processing
Total RNA was extracted from samples using a standard TRIzol extraction method. RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies, Palo Alto, CA, USA). rRNA depletion sequencing library was prepared by using QIAGEN FastSelect rRNA HMRKit (Qiagen, Hilden, Germany), per the manufacturer’s instructions. RNA sequencing library preparation uses NEB Next Ultra II RNA Library Preparation Kit for Illumina by following the manufacturer’s recommendations (NEB, Ipswich, MA, USA). Briefly, enriched RNAs are fragmented for 15 minutes at 94 °C. First strand and second strand cDNA are subsequently synthesized. cDNA fragments are end repaired and adenylated at 3’ends, and universal adapters are ligated to cDNA fragments, followed by index addition and library enrichment with limited cycle PCR. Sequencing libraries were validated using the Agilent Tapestation 4200 (Agilent Technologies, CA, USA), and quantified using Qubit 2.0 Fluorometer (ThermoFisher Scientific, MA, USA) as well as by quantitative PCR (KAPA Biosystems, MA, USA). The sequencing libraries were clustered on 1 lane of a flowcell. After clustering, the flowcell was loaded on an Illumina HiSeq instrument, according to manufacturer’s instructions. The samples were sequenced using a 2×150bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Bulk total RNA-seq processing and DEG identification
Read quality was checked with FastQC and adapter sequences were trimmed with Cutadapt57. Reads were aligned to the prairie vole genome (GCA_000317375.1) with STAR 2.7.10a in default mode58. FeatureCounts was used count reads within genes (exonic and intronic), and genes that had >5 counts in >22 samples were input to Deseq2 to identify differentially expressed genes (DEGs)59,60. The model used to look at region-specific effects of genotype was ~group+sex+batch, where group had six levels (NAC_CC, NAC_TT, INS_CC, INS_TT, SC_CC and SC_TT), while for differential expression analysis between genotypes the model ~genotype+area+sex+batch was used. For the analysis of KO animals, genes with >5 counts in >11 individuals were included and the model ~genotype+sex was used. Genes were differentially expressed if Padj< 0.1.
ATAC sequencing library preparation and processing
Cryopreserved cell samples were thawed, washed, and treated with DNAse I (Life Tech, CA, USA) to remove genomic DNA contamination. Cell samples were quantified and assessed for viability using a Countess Automated Cell Counter (ThermoFisher Scientific, Waltham, MA, USA). After cell lysis and cytosol removal, nuclei were treated with Tn5 enzyme (Illumina, CA, USA) for 30 minutes at 37°C and purified with Minelute PCR Purification Kit (Qiagen) to produce tagmented DNA samples. Tagmented DNA was barcoded with Nextera Index Kit v2 (Illumina) and amplified via PCR prior to a SPRI Bead cleanup to yield purified DNA libraries. The sequencing libraries were clustered on a single lane of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq according to manufacturer’s instructions. The samples were sequenced using a 2×150bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
ATAC sequencing data processing and DAR identification
Read quality was checked with FastQC and adapter sequences were trimmed with Cutadapt57. Reads were aligned to the prairie vole genome (GCA_000317375.1) with Bowtie2 with the --very-sensitive option enabled61. Next, regions of open chromatin were determined with Genrich using -j -y -r -v options and a cut off value of P < 0.05. Peak sets were constructed by merging samples from each genotype with a significance cutoff Padj=0.1. These peak sets served as input to Diffbind 3.8.4, which was used to calculate differences in chromatin accessibility between genotypes, using the model - genotype+area+sex+batch62. Regions were differentially accessible if Padj < 0.1.
Weighted gene correlation network analysis
Modules of co-expressed DEGs were analyzed using weighted gene correlation network analysis (WGCNA) using the “WGCNA” package in R63. The normalized gene expression data for DEGs was used as the input gene expression matrix and the function pickSoftThreshold was used to determine the optimal soft-thresholding power. We determined the optimal soft-thresholding power to be 10 because it was the lowest power for which the scale-free topology fit index reached 0.8. Then an adjacency matrix was created from the input gene expression matrix using the adjacency function with power = 10, and otherwise default parameters. The adjacency matrix was transformed into a topological overlap matrix, and modules were detected by hierarchical average linkage clustering analysis for the gene dendrogram. After the modules were identified, the module eigengene (ME) was summarized by the first principal component of the module expression levels. Module–trait relationships were estimated using the correlation between MEs and genotype. To evaluate the correlation strength, we calculated the module significance (MS), which was defined as the average absolute gene significance (GS) of all the genes involved in the module. Modules were considered significantly associated with genotyoe if P<0.05 and visualized with the iGraph package. Additionally, we extracted the corresponding gene information for each module which was used as in input to ShinyGO64.
Single nuclei isolation
Animals were sacrificed using CO2 asphyxiation, after which brains were dissected out and sliced on a ice-cold mouse brain matrix (Zivic Instruments). NAC and paraventricular hypothalamus (PVN) punches were taken from flash-frozen brain slides and stored at −80 °C. Immediately prior to nuclei isolation, frozen tissue samples were pooled by Oxtr genotype and sex, so PVN and NAC tissue were combined. Pooled samples were then incubated in 150 μl chilled lysis buffer containing 10 mM Tris-HCL (Sigma), 10 mM NACl (Sigma), 3 mM MgCl2 (Sigma), 0.1% Nonidet P40 Substitute (Sigma), and Nuclease-free H2O for 30 minutes with gentle rotation. Following lysis, 150 μl Hibernate AB Complete nutrient medium (HAB; with 2% B27 and 0.5 mM Glutamax; BrainBits), and samples were rapidly triturated using silanized glass Pasteur pipettes (500 μm internal diameter, BrainBits) until complete tissue dissociation. Samples were then centrifuged (400 x g, 5 minutes, 4°C) and resuspended in 1–1.5 ml chilled wash and resuspension buffer containing 2% Bovine Serum Albumin (Sigma) and 0.2 U/μl RNase Inhibitor (Sigma) in 1X PBS (Thermo Fisher). The nuclei suspensions were filtered through 40 μm Flowmi® cell strainers (Sigma) and 30 μm MACS® SmartStrainers (Milltenyi) to remove large debris and aggregations of nuclei prior to fluorescence activated cell sorting (FACS). Singlet nuclei were then further purified using FACS (BD FACSAria™ Fusion Cell Sorter, BD Biosciences) and FACSDiva software (v8.0.1, BD Biosciences). Sizing beads (6 μm; BD Biosciences) and 1.0 μg/ml DAPI (Sigma) were used to set gating parameters, enabling selection of singlet nuclei based on size (forward scatter), shape (side scatter), and DNA content (DAPI fluorescence). Approximately 250,000 nuclei/pool (ranging from 242,421–252,159) were collected into 1 mL wash and resuspension buffer for downstream library preparation and sequencing.
Single nucleus RNA library preparation and sequencing
Suspensions of purified singlet nuclei were loaded onto the 10x Genomics Chromium Controller (10x Genomics) at concentrations ranging from 240–250 nuclei/ul with a target range of 4,000 nuclei per sample. Downstream cDNA synthesis and library preparation using Single Cell 3’ GEM, Library and Gel Bead Kit v3.1 and Dual Index Kit TT, Set A (PN-1000215/PN-3000431) were performed according to manufacturer instructions (Chromium Single Cell 3’ Reagent Kits User Guide v3.1 Chemistry, 10X Genomics). Sample quality was assessed using high sensitivity DNA analysis on the Bioanalyzer 2100 system (Agilent) and libraries were quantified using a Qubit 2.0 (Invitrogen). Barcoded cDNA libraries were pooled and sequenced on the NovaSeq 6000 platform (Illumina) on a single flow cell (2×150 bp paired end reads; Illumina).
Single nucleus RNA pre-processing and quality control
FASTQ files were processed with Cell Ranger version 3.1.0 (10X Genomics). Reads were aligned to the Microtus ochrogaster genome assembly using a splice-aware alignment algorithm (STAR) within Cell Ranger, and gene annotations were obtained from the same assembly (NCBI RefSeq assembly accession: GCA_000317375.1, MicOch1.0). Because nuclear RNA contains intronic sequence, they were included in the Cell Ranger count step. Cell Ranger filtered out UMIs that were homopolymers, contained N, or contained any base with a quality score less than 10. Following these steps, Cell Ranger generated four filtered feature-barcode matrices (one per pool) containing expression data for a total of 26,397 features (corresponding to annotated genes) and a total of 21,838 barcodes (corresponding to droplets and putative nuclei) that were used passed through additional quality control steps in the Seurat package in R42. Total transcripts and total genes varied slightly across pools, and therefore slightly different criteria were used to remove potentially dead or dying nuclei from each pool. Mitochondrial genes were represented at low levels in every pool and were not used as a filtering criterion. To reduce risk of analyzing dead or dying nuclei, barcodes associated with fewer than 400–500 (depending on pool) total genes were excluded from downstream analysis. To reduce risk of doublets or multiplets, barcodes associated with more than 4,000–5,000 total genes and 10,400–18,000 total transcripts were also excluded. This step filtered out a total of 819 barcodes (3.7%).
Prediction of nuclei from distinct individual animals
To deconvolute nuclei derived from distinct individuals within pools, we used Souporcell65, a tool that clusters nuclei based on genetic variants detected in RNA reads. Reads were first filtered for base quality scores greater than 10, high confidence mapping to the genome (-F 3844), and exceeding Cell Ranger criteria for UMI counting (tag xf:i:25). Next, high-quality reads were used to call variants in each nucleus using the GATK v4.1.8.1 HaplotypeCaller function with default parameters, but without the MappingQualityAvailableReadFilter to retain reads that were confidently mapped to the genome by Cell Ranger (MAPQ score of 255). Vartrix v1.1.22 was then used to count alleles in each nucleus (using recommended parameters). Lastly, Souporcell v2.0 was used to cluster cells by genotype, using default parameters but with the number of clusters set to 4 based on the known number of individuals in each pool. Thus, four clusters were identified for each pool, with each cluster comprising nuclei that were predicted to have originated from the same individual animal. Souporcell also uses clustering information to predict multiplets (based on individual barcodes that are associated with reads assigned to multiple individuals). We filtered out an additional 435 (2%) barcodes that were predicted to be multiplets by Souporcell.
Dimensionality reduction and clustering
To normalize the data, we used the SCTransform function in Seurat. To examine dimensionality, we first performed a linear dimensional reduction using the RunPCA command with the maximum possible number of dimensions (“dim” set to 50). All 50 PCs were used as input for non-linear dimensional reduction (Uniform Manifold Approximation and Projection, UMAP) using the RunUMAP function in Seurat. For RunUMAP, “min.dist” was set to 0.5, “n.neighbors” was set to 50, “spread” was set to 0.2, “n.epochs” was set to 1000, and “metric” was set to “euclidean”. Prior to clustering, nuclei were embedded into a K nearest-neighbor (KNN) graph based on euclidean distance in UMAP space, with edge weights based on local Jaccard similarity, using the FindNeighbors function in Seurat (reduction=”umap”,k.param=50, dims=1:2, n.trees=500, prune.SNN=0). Global clustering was then performed using the Seurat FindClusters function using the Louvain algorithm with multilevel refinement (algorithm=2) with resolution set to 0.2. Clusters that were not present in all individuals (N=1) were excluded from further analysis.
Cluster marker gene analysis
Cluster identities were assigned using a combination of unbiased analysis of cluster-specific marker genes as well as a supervised examination of previously established marker genes. Cluster-specific marker genes were identified using the FindAllMarkers function in Seurat. Briefly, this function compares gene expression within each cluster to gene expression across all other clusters and calculates Bonferroni-adjusted p values using a Wilcoxon rank-sum test. Clusters were annotated based on well-known marker genes that differentiate between major cell-types (neurons, astrocytes, oligodendrocytes, oligodendrocyte precursor cells and microglia) and further subdivided using genes that further separated cell clusters (e.g., D1-neurons and D2-neurons).
Identification of DEGs in single nucleus RNA sequencing experiment
We used a pseudo-bulk analysis based on DeSeq2 to identify differentially expressed genes (DEGs) between genotypes in each cluster. Genes that had >1 counts in >7 individual were included in the analysis. For each independent cell cluster, the model ~genotype+sex was used, and P-values were adjusted for multiple comparisons across cell clusters. For comparison with bulk RNA sequencing experiments, a standard DeSeq analysis was performed across all cells, not taking cluster into account, with the model ~genotype+sex. Gene were significant if Padj < 0.1.
Identification of OXTR-correlated genes and CTLR expression in human NAC
To identity genes of which the expression correlates with OXTR in the human NAC, we used human GTEx bulk RNA sequencing data containing raw gene read counts from NAC tissue, which was aligned to GRCh38/hg38 using STAR v2.5.3a, based on the GENCODE v26 annotation. Genes that showed >10 counts in >132 individual were included. OXTR TPM values were used as the independent variable in a DeSeq2 analysis. Genes were considered significantly correlated with OXTR if Padj < 0.1 and log2 fold change > 0.2. To determine cell-type specific NAC expression of OXTR-correlated genes localized in the NKC we used the publicly available Seurat object from Tran et al., with existing cluster annotation. We used the plotExpression function to plot raw counts in each NAC cell type.
Extended Data
Extended Data 1.

Number of ATAC peaks present in the transcription start site (TSS), exonic, or intronic regions of expressed versus low/not expressed genes, as well as intergenic regions. ATAC peaks preferentially localize to expressed genes rather than low/not expressed genes.
Extended Data 2.

Read plots of a part of the Oxtr gene of representative individuals of each genotype. Red box indicates 60bp region in the Oxtr coding sequence that is deleted in the systemic OXTR KO but not in wildtype animals.
Extended Data 3. OXTR signaling affects neural transcription across brain regions and cell types.
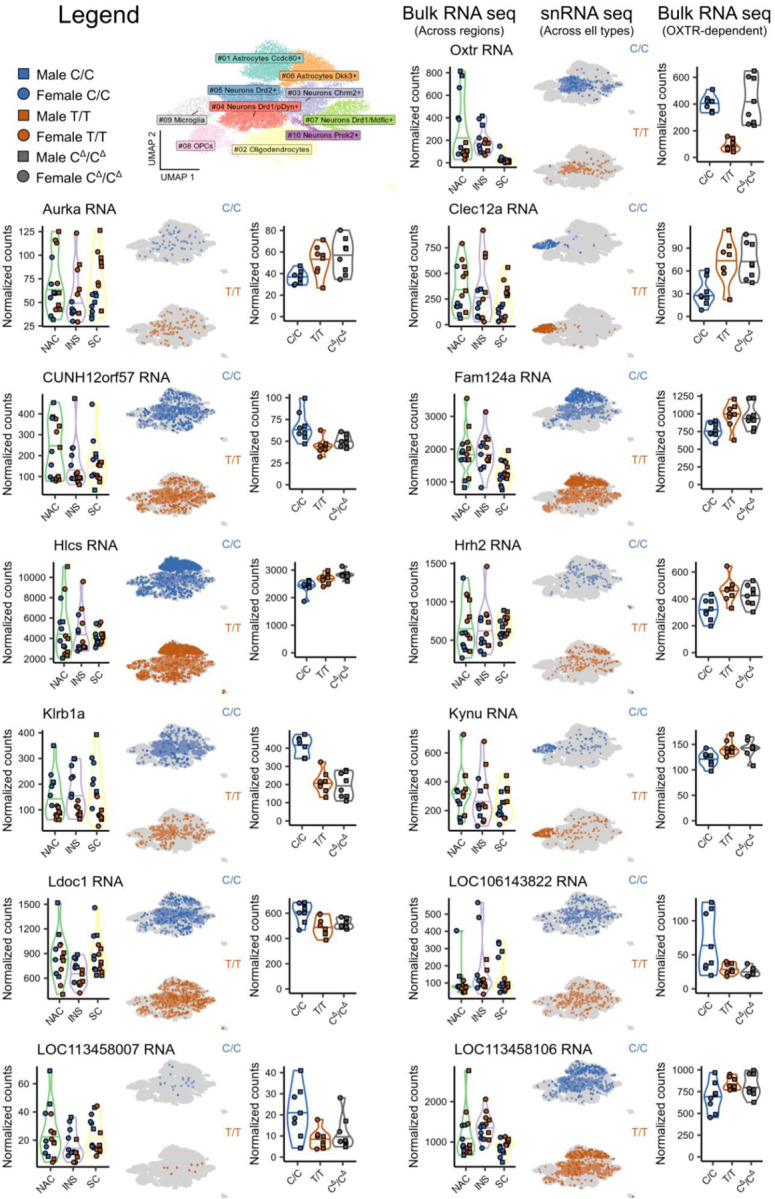

Illustrated are expression levels for each OXTRDEG genes across the separate RNA-seq experiments.
Supplementary Material
Acknowledgements
We thank Lorra Julian and the Emory Primate Center veterinary staff for their dedicated animal care. We thank Zakia Sathi for help with genotyping.
Funding Statement
This project was supported by NIH grants P50MH100023 and R01MH112788 to LJY, R01GM144560 to JTS for support to BEH, GWG, ZVJ and JTS, and OD P51OD011132 to Emory National Primate Research Center.
Footnotes
Competing interests
The authors declare that to have no competing interests.
Data availability
Sequencing data will be made available on the NIH GEO repository, and associated scripts and materials will be uploaded to the Dryad repositor after peer review.
References
- 1.Donaldson Z. R. & Young L. J. Oxytocin, vasopressin, and the neurogenetics of sociality. Science 322, 900–904 (2008). https://doi.org: 10.1126/science.1158668 [DOI] [PubMed] [Google Scholar]
- 2.Beets I., Temmerman L., Janssen T. & Schoofs L. Ancient neuromodulation by vasopressin/oxytocin-related peptides. Worm 2, e24246 (2013). https://doi.org: 10.4161/worm.24246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shamay-Tsoory S. G. & Abu-Akel A. The Social Salience Hypothesis of Oxytocin. Biol Psychiatry 79, 194–202 (2016). https://doi.org: 10.1016/j.biopsych.2015.07.020 [DOI] [PubMed] [Google Scholar]
- 4.Hung L. W. et al. Gating of social reward by oxytocin in the ventral tegmental area. Science 357, 1406–1411 (2017). https://doi.org: 10.1126/science.aan4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferguson J. N. et al. Social amnesia in mice lacking the oxytocin gene. Nat Genet 25, 284–288 (2000). https://doi.org: 10.1038/77040 [DOI] [PubMed] [Google Scholar]
- 6.Walum H. et al. Variation in the oxytocin receptor gene is associated with pair-bonding and social behavior. Biol Psychiatry 71, 419–426 (2012). https://doi.org: 10.1016/j.biopsych.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marlin B. J., Mitre M., D’Amour J A., Chao M. V. & Froemke R. C. Oxytocin enables maternal behaviour by balancing cortical inhibition. Nature 520, 499–504 (2015). https://doi.org: 10.1038/nature14402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valtcheva S. et al. Neural circuitry for maternal oxytocin release induced by infant cries. Nature 621, 788–795 (2023). https://doi.org: 10.1038/s41586-023-06540-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ford C. L. & Young L. J. Translational opportunities for circuit-based social neuroscience: advancing 21st century psychiatry. Curr Opin Neurobiol 68, 1–8 (2021). https://doi.org: 10.1016/j.conb.2020.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walum H. & Young L. J. The neural mechanisms and circuitry of the pair bond. Nat Rev Neurosci 19, 643–654 (2018). https://doi.org: 10.1038/s41583-018-0072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young L. J. & Wang Z. The neurobiology of pair bonding. Nat Neurosci 7, 1048–1054 (2004). https://doi.org: 10.1038/nn1327 [DOI] [PubMed] [Google Scholar]
- 12.Castro D. C. & Bruchas M. R. A Motivational and Neuropeptidergic Hub: Anatomical and Functional Diversity within the Nucleus Accumbens Shell. Neuron 102, 529–552 (2019). https://doi.org: 10.1016/j.neuron.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keebaugh A. C., Barrett C. E., Laprairie J. L., Jenkins J. J. & Young L. J. RNAi knockdown of oxytocin receptor in the nucleus accumbens inhibits social attachment and parental care in monogamous female prairie voles. Soc Neurosci 10, 561–570 (2015). https://doi.org: 10.1080/17470919.2015.1040893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keebaugh A. C. & Young L. J. Increasing oxytocin receptor expression in the nucleus accumbens of pre-pubertal female prairie voles enhances alloparental responsiveness and partner preference formation as adults. Horm Behav 60, 498–504 (2011). https://doi.org: 10.1016/j.yhbeh.2011.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross H. E. et al. Variation in oxytocin receptor density in the nucleus accumbens has differential effects on affiliative behaviors in monogamous and polygamous voles. J Neurosci 29, 1312–1318 (2009). https://doi.org: 10.1523/JNEUROSCI.5039-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Froemke R. C. & Young L. J. Oxytocin, Neural Plasticity, and Social Behavior. Annu Rev Neurosci 44, 359–381 (2021). https://doi.org: 10.1146/annurev-neuro-102320-102847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freeman S. M. & Young L. J. Comparative Perspectives on Oxytocin and Vasopressin Receptor Research in Rodents and Primates: Translational Implications. J Neuroendocrinol 28 (2016). https://doi.org: 10.1111/jne.12382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman S. M., Inoue K., Smith A. L., Goodman M. M. & Young L. J. The neuroanatomical distribution of oxytocin receptor binding and mRNA in the male rhesus macaque (Macaca mulatta). Psychoneuroendocrinology 45, 128–141 (2014). https://doi.org: 10.1016/j.psyneuen.2014.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schorscher-Petcu A., Dupré A. & Tribollet E. Distribution of vasopressin and oxytocin binding sites in the brain and upper spinal cord of the common marmoset. Neurosci Lett 461, 217–222 (2009). https://doi.org: 10.1016/j.neulet.2009.06.016 [DOI] [PubMed] [Google Scholar]
- 20.Quintana D. S. et al. Oxytocin pathway gene networks in the human brain. Nat Commun 10, 668 (2019). https://doi.org: 10.1038/s41467-019-08503-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King L. B., Walum H., Inoue K., Eyrich N. W. & Young L. J. Variation in the Oxytocin Receptor Gene Predicts Brain Region-Specific Expression and Social Attachment. Biol Psychiatry 80, 160–169 (2016). https://doi.org: 10.1016/j.biopsych.2015.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olazabal D. E. & Young L. J. Oxytocin receptors in the nucleus accumbens facilitate “spontaneous” maternal behavior in adult female prairie voles. Neuroscience 141, 559–568 (2006). https://doi.org: 10.1016/j.neuroscience.2006.04.017 [DOI] [PubMed] [Google Scholar]
- 23.Barrett C. E., Arambula S. E. & Young L. J. The oxytocin system promotes resilience to the effects of neonatal isolation on adult social attachment in female prairie voles. Transl Psychiatry 5, e606 (2015). https://doi.org: 10.1038/tp.2015.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahern T. H., Olsen S., Tudino R. & Beery A. K. Natural variation in the oxytocin receptor gene and rearing interact to influence reproductive and nonreproductive social behavior and receptor binding. Psychoneuroendocrinology 128, 105209 (2021). https://doi.org: 10.1016/j.psyneuen.2021.105209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ophir A. G., Gessel A., Zheng D. J. & Phelps S. M. Oxytocin receptor density is associated with male mating tactics and social monogamy. Horm Behav 61, 445–453 (2012). https://doi.org: 10.1016/j.yhbeh.2012.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium G. T. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). https://doi.org: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smearman E. L. et al. Oxytocin Receptor Genetic and Epigenetic Variations: Association With Child Abuse and Adult Psychiatric Symptoms. Child Dev 87, 122–134 (2016). https://doi.org: 10.1111/cdev.12493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley B., Davis T. A., Wingo A. P., Mercer K. B. & Ressler K. J. Family environment and adult resilience: contributions of positive parenting and the oxytocin receptor gene. Eur J Psychotraumatol 4 (2013). https://doi.org: 10.3402/ejpt.v4i0.21659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skuse D. H. et al. Common polymorphism in the oxytocin receptor gene (OXTR) is associated with human social recognition skills. Proc Natl Acad Sci U S A 111, 1987–1992 (2014). https://doi.org: 10.1073/pnas.1302985111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujisawa T. X. et al. Oxytocin receptor DNA methylation and alterations of brain volumes in maltreated children. Neuropsychopharmacology 44, 2045–2053 (2019). https://doi.org: 10.1038/s41386-019-0414-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez L. M. et al. Imaging-genetics of sex differences in ASD: distinct effects of OXTR variants on brain connectivity. Transl Psychiatry 10, 82 (2020). https://doi.org: 10.1038/s41398-020-0750-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez L. M. et al. Additive effects of oxytocin receptor gene polymorphisms on reward circuitry in youth with autism. Mol Psychiatry 22, 1134–1139 (2017). https://doi.org: 10.1038/mp.2016.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokoyama W. M. & Plougastel B. F. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol 3, 304–316 (2003). https://doi.org: 10.1038/nri1055 [DOI] [PubMed] [Google Scholar]
- 34.Yang X. et al. Characterization of large-scale genomic differences in the first complete human genome. Genome Biol 24, 157 (2023). https://doi.org: 10.1186/s13059-023-02995-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGraw L. A., Davis J. K., Young L. J. & Thomas J. W. A genetic linkage map and comparative mapping of the prairie vole (Microtus ochrogaster) genome. BMC Genet 12, 60 (2011). https://doi.org: 10.1186/1471-2156-12-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giorda R. et al. NKR-P1, a signal transduction molecule on natural killer cells. Science 249, 1298–1300 (1990). https://doi.org: 10.1126/science.2399464 [DOI] [PubMed] [Google Scholar]
- 37.Yokoyama W. M. et al. cDNA cloning of mouse NKR-P1 and genetic linkage with LY-49. Identification of a natural killer cell gene complex on mouse chromosome 6. J Immunol 147, 3229–3236 (1991). [PubMed] [Google Scholar]
- 38.Brown M. G., Scalzo A. A., Matsumoto K. & Yokoyama W. M. The natural killer gene complex: a genetic basis for understanding natural killer cell function and innate immunity. Immunol Rev 155, 53–65 (1997). https://doi.org: 10.1111/j.1600-065x.1997.tb00939.x [DOI] [PubMed] [Google Scholar]
- 39.Renedo M. et al. The human natural killer gene complex is located on chromosome 12p12-p13. Immunogenetics 46, 307–311 (1997). https://doi.org: 10.1007/s002510050276 [DOI] [PubMed] [Google Scholar]
- 40.Stevens B. & Johnson M. B. The complement cascade repurposed in the brain. Nat Rev Immunol 21, 624–625 (2021). https://doi.org: 10.1038/s41577-021-00621-z [DOI] [PubMed] [Google Scholar]
- 41.Ferro A., Auguste Y. S. S. & Cheadle L. Microglia, Cytokines, and Neural Activity: Unexpected Interactions in Brain Development and Function. Front Immunol 12, 703527 (2021). https://doi.org: 10.3389/fimmu.2021.703527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021). https://doi.org: 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue K., Ford C. L., Horie K. & Young L. J. Oxytocin receptors are widely distributed in the prairie vole (Microtus ochrogaster) brain: Relation to social behavior, genetic polymorphisms, and the dopamine system. J Comp Neurol 530, 2881–2900 (2022). https://doi.org: 10.1002/cne.25382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogler I. & Steinle A. Vis-à-vis in the NKC: genetically linked natural killer cell receptor/ligand pairs in the natural killer gene complex (NKC). J Innate Immun 3, 227–235 (2011). https://doi.org: 10.1159/000324112 [DOI] [PubMed] [Google Scholar]
- 45.Horie K. et al. Oxytocin receptor knockout prairie voles generated by CRISPR/Cas9 editing show reduced preference for social novelty and exaggerated repetitive behaviors. Horm Behav 111, 60–69 (2019). https://doi.org: 10.1016/j.yhbeh.2018.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miles L. A. et al. The plasminogen receptor(,) Plg-R(KT)(,) is essential for mammary lobuloalveolar development and lactation. J Thromb Haemost 16, 919–932 (2018). https://doi.org: 10.1111/jth.13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Q. et al. Histamine H(2) receptor deficit in glutamatergic neurons contributes to the pathogenesis of schizophrenia. Proc Natl Acad Sci U S A 120, e2207003120 (2023). https://doi.org: 10.1073/pnas.2207003120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang S. et al. Frontostriatal circuitry and the tryptophan kynurenine pathway in major psychiatric disorders. Psychopharmacology (Berl) (2023). https://doi.org: 10.1007/s00213-023-06466-9 [DOI] [PubMed] [Google Scholar]
- 49.Hou Y. et al. RecQ dysfunction contributes to social and depressive-like behavior and affects aldolase activity in mice. Neurobiol Dis 180, 106092 (2023). https://doi.org: 10.1016/j.nbd.2023.106092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran M. N. et al. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 109, 3088–3103.e3085 (2021). https://doi.org: 10.1016/j.neuron.2021.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown G. D., Willment J. A. & Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol 18, 374–389 (2018). https://doi.org: 10.1038/s41577-018-0004-8 [DOI] [PubMed] [Google Scholar]
- 52.Siew J. J. & Chern Y. Microglial Lectins in Health and Neurological Diseases. Front Mol Neurosci 11, 158 (2018). https://doi.org: 10.3389/fnmol.2018.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marin I. A. et al. The nonclassical MHC class I Qa-1 expressed in layer 6 neurons regulates activity-dependent plasticity via microglial CD94/NKG2 in the cortex. Proc Natl Acad Sci U S A 119, e2203965119 (2022). https://doi.org: 10.1073/pnas.2203965119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevens B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). https://doi.org: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- 55.Olazabal D. E. et al. Oxytocin system polymorphisms rs237887 and rs2740210 variants increase the risk of depression in pregnant women with early abuse. Dev Psychobiol 65, e22400 (2023). https://doi.org: 10.1002/dev.22400 [DOI] [PubMed] [Google Scholar]
- 56.Andari E. et al. Epigenetic modification of the oxytocin receptor gene: implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacology 45, 1150–1158 (2020). https://doi.org: 10.1038/s41386-020-0610-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3 (2011). https://doi.org: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 58.Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). https://doi.org: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao Y., Smyth G. K. & Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). https://doi.org: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 60.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). https://doi.org: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). https://doi.org: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross-Innes C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). https://doi.org: 10.1038/nature10730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langfelder P. & Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). https://doi.org: 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ge S. X., Jung D. & Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020). https://doi.org: 10.1093/bioinformatics/btz931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heaton H. et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods 17, 615–620 (2020). https://doi.org: 10.1038/s41592-020-0820-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.