

Abstract
Proteins and the complexes they form are central to nearly all cellular processes. Their flexibility, expressed through a continuum of states, provides a window into their biological functions. Cryogenic-electron microscopy (cryo-EM) is an ideal tool to study these dynamic states as it captures specimens in non-crystalline conditions and enables high-resolution reconstructions. However, analyzing the heterogeneous distribution of conformations from cryo-EM data is challenging. Current methods face issues such as a lack of explainability, overfitting caused by lack of regularization, and a large number of parameters to tune; problems exacerbated by the lack of proper metrics to evaluate or compare heterogeneous reconstructions. To address these challenges, we present RECOVAR, a white-box method based on principal component analysis (PCA) computed via regularized covariance estimation that can resolve intricate heterogeneity with similar expressive power to neural networks with significantly lower computational demands. We extend the ubiquitous Bayesian framework used in homogeneous reconstruction to automatically regularize principal components, overcoming overfitting concerns and removing the need for most parameters. We further exploit the conservation of density and distances endowed by the embedding in PCA space, opening the door to reliable free energy computation. We leverage the predictable uncertainty of image labels to generate high-resolution reconstructions and identify high-density trajectories in latent space. We make the code freely available at https://github.com/ma-gilles/recovar.
Proteins and their complexes play pivotal roles in cellular processes, governing essential biological functions. The function of these biological macromolecules can be elucidated through the continuum of their structural states encompassing local dynamics, large-scale rearrangement of domains, and modification of subunits.
Cryogenic electron-microscopy (cryo-EM) stands out as an ideal technique to study the dynamic conformational landscape of biomolecules and was selected as one of the “Methods to Watch” in 2022 by Nature Methods [9]. The reason lies primarily in its sample preparation: cryo-EM enables imaging of biological specimens in a frozen-hydrated condition, bypassing the need for crystallization, which can alter the structure and force all specimens to take the same conformation. Therefore, cryo-EM’s unique preservation technique offers a more faithful representation of their functional states and can capture a snapshot of their entire conformational ensemble. Furthermore, cryo-EM’s capacity to capture high-resolution images of individual particles allows the reconstruction of high-resolution structures and the identification of distinct conformational states with even tiny differences.
However, the computational and modeling challenges posed by deciphering the heterogeneous distribution of conformations from cryo-EM datasets are considerable. The workhorse of cryo-EM heterogeneity analysis is 3D-classification [34], which aims to discern discrete conformational states. While effective for a few evenly distributed states, this approach encounters difficulties with many states or uneven distributions of discrete states and cannot capture continuous changes.
A diverse array of methods has been proposed to address these challenges. These methods encompass rigid body fitting [25], linear subspace methods [42, 15, 26, 1, 29], deep-learning approaches [48, 3], manifold learning methods [12, 24, 37], strategies based on molecular dynamics [44], and methods focused on computing deformation maps [13, 30]. For comprehensive coverage of these rapidly evolving methodologies, refer to [40, 43, 10].
Though diverse in their details, most of these methods share a common framework: they employ a mapping to embed images into a “latent space”. This mapping aims to isolate image variations stemming from irrelevant factors like pose, imaging effects, and noise, focusing solely on variability due to conformational changes. A second mapping then translates this latent space into conformational states, either through a parametric map or by generating homogeneous reconstructions based on images situated in the same neighborhood of the latent space.
Significant advancements have been made in recent times, particularly with the utilization of neural networks achieving high visual resolution 1. However, present methods grapple with several issues, including a lack of explainability, the absence of regularization, and numerous parameters requiring tuning. These challenges are further exacerbated by the absence of metrics to validate or compare results from heterogeneity analysis in cryo-EM. Consequently, distinguishing between a genuinely high-resolution reconstruction and a mere overfit artifact remains difficult.
Additionally, the reconstruction phase is often only the beginning of the analysis. Estimating the energy of states or motions is often the ultimate goal. Both quantities are typically calculated based on recovering the likelihood of observing each state in the data, usually numerically estimated by density estimation in latent space. However, most methods for generating latent space do not conserve distances or densities. Thus, the energy estimates obtained this way can be entirely unrelated to the data and instead be artifacts of the method [19, 43].
To tackle these challenges, we introduce RECOVAR, a method grounded in principal component analysis (PCA) computed via REgularized COVARiance estimation. This method is white-box, automatically regularized, allows for the estimation of density, and offers results that are competitive with neural networks but with significantly reduced computational requirements.
We overcome overfitting and parameter-tuning issues using a Bayesian framework, building upon successful schemes for homogeneous reconstructions [35]. Our method belongs to the family of linear subspace methods, which have recently achieved practical success thanks to 3DVA [29], implemented in the popular software suite cryoSPARC [31]. However, linear subspace methods have often been considered to be limited to low resolution [41] or suitable only for capturing “small” motion and “simple” heterogeneity. We demonstrate that this perception is primarily based on misunderstanding their properties. Linear subspace methods do not capture the inherent heterogeneous dimension but are more akin to a change of basis. Specifically, they compute the optimal truncated basis to represent the heterogeneity and express conformational states as projections in this new coordinate system. As a result, a one-dimensional motion is typically not well-embedded in a one-dimensional subspace. However, we show that simply computing larger subspaces overcomes this limitation.
This advancement allows the exploitation of crucial properties of linear subspace methods: they approximately conserve density and distances, opening the door to rigorous energy computation. They yield predictable uncertainty, which we leverage to generate high-resolution volumes by selecting subsets of images using simple statistical tests. Finally, we leverage the density estimates to identify high-density trajectories in latent space and recover motions.
1. Results
1.1. Heterogeneity analysis by regularized covariance estimation
The method presented here uses PCA to compute an optimal linear subspace to represent the heterogeneous distribution of states. However, applying PCA to conformational states in cryo-EM data introduces a significant complexity: the observations are projection images of the conformational states, not the states themselves. Therefore, more than a straightforward application of PCA is needed. The principal components are also the eigenvectors of the covariance matrix of conformations, and remarkably, this covariance matrix can be estimated directly from projection images [15, 1]. Hence, as depicted in fig. 12, our pipeline initiates with the statistical estimation of the mean and covariance of the conformational states directly from projection images. These estimation problems are regularized by splitting the data into halfsets and extending the Fourier Shell Correlation (FSC) regularization scheme ubiquitously used in homogeneous reconstruction [35]. Once computed, the eigenbasis of the covariance matrix provides the principal components. In the subsequent stage, we employ a Bayesian framework to infer each image’s probable conformation distribution within this eigenbasis. Ultimately, the aggregated distribution in the latent space is harnessed to generate volumes and high-density motions.
Figure 1:

Illustration of the regularized covariance pipeline for heterogeneity analysis in cryo-EM. (a) The mean and covariance of the distribution of states are estimated and regularized using the FSC of two reconstructions from halfsets. The principal components are then computed. (b) Each image is assigned a probabilistic estimate, and the densities are averaged across the dataset to compute the latent density. (c) States are sampled and reconstructed. (d) Motions are generated by computing optimal continuous high-density trajectories between an initial and final state in latent space (indicated by a star and a square). Intermediate reconstructed volumes at uniform distances along the path, indicated by circles in latent space, are displayed.
The image formation process of cryo-EM, relating a 2-D image to a 3-D conformation , is typically modeled as:
| (1) |
where is a projection from 3-D to 2-D after rigid body motion parametrized by , is the contrast transfer function (CTF), and is Gaussian noise with covariance . In typical cryo-EM reconstruction, the poses are unknown and need to be inferred, often through a scheme alternating between inferring states and poses . In what follows, we assume that poses have been previously estimated, typically from a consensus reconstruction as is a common assumption for heterogeneity methods [48, 29], and fix the linear maps . This assumption is restrictive as the poses cannot be estimated from consensus reconstructions for some highly heterogeneous cryo-EM datasets. For datasets where poses can be estimated from consensus reconstructions, alignment will likely not be perfect, and the residual pose errors can result in lower-than-possible resolution or even spurious heterogeneity. While some neural methods [20, 49] have shown the ability to infer poses ab initio on some datasets, robust pose estimation for highly heterogeneous datasets remains an open problem which we come back to in the discussion.
When poses are known, the mean conformation can be straightforwardly estimated by solving a linear least-squares problem:
| (2) |
where are the Wiener filter parameters, and is the number of images. Equation (2) is also the problem usually solved during homogeneous reconstruction. In that case, the weights are often set using the FSC regularization scheme [35]; for example, it is the default method in popular cryo-EM software such as RELION [35] and cryoSPARC [31].
Analogously, the covariance of conformations - encoding the first-order deviations from the mean conformation - can be estimated using the following linear least-squares estimator [1, 15]:
| (3) |
where and and is the regularization weights. Unfortunately, the computation of this covariance estimator is a massive computational burden; e.g., representing conformations on a grid of size 1283 would result in a covariance matrix with 1286 entries—or 17 terabytes in single-precision floating-point. One solution to this problem proposed in [42] and adopted in 3DVA [28] is bypassing the covariance estimation and approximating the principal components through an iterative procedure.
We take a different route that does not require iteration—an expensive computation for large cryo-EM datasets—and instead computes the principal component using modern linear algebra techniques. The algorithm presented relies on two main computational tricks; the first exploits the hidden structure in eq. (3): it can be reduced to a Hadamard linear system, whose entries can be solved efficiently and independently, see appendix A.1. The second trick is based on the observation that for low-rank covariance matrices , only a subset of the columns is required to estimate the entire matrix and its leading eigenvectors, which are the principal components we seek. This celebrated fact in numerical linear algebra is the basis of numerical schemes such as the Nyström extension [46], see appendix A.2.
Entries of are estimated with a high dynamic range of signal-to-noise ratio (SNR). This is analogous to the homogeneous reconstruction problem: low-frequency coefficients are easier to estimate as they are more often observed in images due to the Fourier slice theorem. The covariance estimation problem presents an even wider range of SNR across pairs of frequencies [15]; making careful regularization even more crucial. To that end, we generalize the FSC regularization used in homogeneous reconstruction to the covariance estimation problem by carefully accounting for the highly nonuniform sampling of entries of the covariance matrix. Briefly, this regularization proceeds by splitting the dataset into two halves and computing two independent copies of the same object. The correlation scores between the two copies provide estimates of the SNR, which can be used to set the regularization weights; see appendix A.3 for details. Optionally, we use a real-space mask to focus the analysis on the part of the molecule or to boost the SNR of the covariance estimation. Finally, after the regularized covariance is estimated, principal components are extracted with a singular value decomposition.
These computational advances, coupled with new regularization strategies for the covariance matrix, allow us to robustly and efficiently compute many principal components at high resolution that can encode a rich distribution of heterogeneous conformations. This helps alleviate a main limitation of linear subspace methods: their sometimes limited representation power for low dimensional subspace.
1.2. Estimation of random conformations
We next turn to the problem of estimating the conformation present in each image. Due to noise and projection ambiguity, confidently matching an image to a single conformation is often impossible. Instead, we use a Bayesian framework to find a distribution of likely states for each image; Under the likelihood model in eq. (1), the posterior probability that an image comes from a particular state can be computed using Bayes’ law:
| (4) |
We approximate the distribution of states as a normal distribution , plug the truncated eigenvalue expansion of the estimated covariance matrix where the columns of are the estimated principal components and the diagonal entries of are the estimated eigenvalues, and reparametrize by its coordinates in the low-dimensional basis , where is the number of chosen principal components. We refer to the coordinate as living in latent space and refer to states as living in volume space to make the distinction. We can now estimate explicitly a distribution of possible states present in a given image :
| (5) |
where and are the mean and covariance estimate of the conformation in image , with explicit formulas given in appendix A.5. The covariance conformation estimate reflects the uncertainty in the latent variable assigned to image , and is leveraged to generate volumes and trajectories in subsequent steps. We further allow for contrast variation in each image by computing a per-image optimal scaling parameter similar to [29, 39], and describe a method to estimate this parameter at virtually no extra cost; see appendices A.4 and A.5 for details.
We define the latent density as the average of densities across all images:
| (6) |
The true density of conformational states is of great biological interest as it is a measure of the free energy of a particular state through the Boltzmann statistics. The latent density in eq. (6) is a kernel density estimate of the volume density, and may be used as a proxy of this true density for some purposes. It should be noted that it differs from it in important ways: is defined over a different space (volumes instead of atomic positions), and it encodes not only states but also uncertainty around states due to noise and projection ambiguities. Most importantly, the dimension of the linear subspace required to represent a certain heterogeneity is often higher than the intrinsic dimension of that heterogeneity; thus, only a small subset of positions in latent space encodes a valid state. For this reason, it is essential to sample conformational states only at meaningful places in the latent space, typically those of high density.
1.3. Volume generation
Once a latent space coordinate is selected, the corresponding volume can be reconstructed by different methods. We highlight two here and dicuss others in section 2. One approach, coined reprojection, uses the explicit inverse map , which entails taking a linear combination of principal components. This reprojection scheme works well when the SNR is sufficiently high to compute all the relevant principal components. This is illustrated in fig. A.9, for instance.
In practice, limited SNR often allows access to only a subset of necessary principal components, and it is preferable to generate volumes from a different method. A simple alternative is to use a reweighting scheme, which generates homogeneous reconstructions from particle subsets. It is a common strategy in heterogeneity methods [29, 5], with the choice of subsets heavily dependent on the employed embedding.
These two schemes for volume generation highlight two different interpretations of linear subspace methods. While the reprojection scheme might be interpreted as explicitly modeling heterogeneous distributions as linear combinations of volumes, the reweighting scheme underscores an alternative and sometimes more appealing interpretation: it merely computes the projection of conformational states onto a low-dimensional subspace. PCA identifies the optimal subspace, and endows the resulting embedding with desirable properties: distances, density, and uncertainty are preserved between volume and latent space up to truncation error in the subspace.
We propose a reweighting scheme that leverages these benefits: given a state represented by a latent vector , we compute the likelihood that each image belongs to that state. Images with a high likelihood of originating from state are selected according to the likelihood model in eq. (5). Specifically, we identify the set of images as defined by:
| (7) |
where is the quantile function for probability of the chi-squared distribution with degrees of freedom (equal to the dimension of ). This is the high-dimensional generalization of the familiar interval estimation, and unless stated otherwise, we set corresponding to a two-standard deviation interval when . That is, under this model, we expect that of images in the dataset truly coming from state are included in the set , though subspace truncation may introduce some covariance estimation inaccuracy. Increasing increases the number of images coming from included in (i.e., it decreases false negatives), but also includes images coming from increasingly different states (it increases false positives).
To resolve the high-resolution frequencies, homogeneous reconstructions carefully average individual frequencies across a large number of images, thereby averaging out the noise. Ideally, one would like to do the same in heterogeneous reconstruction, but that averaging comes with a tradeoff as each image may come from a slightly different conformation. Here lies the delicate tradeoff at the heart of every heterogeneous reconstruction algorithm: aggregating images is necessary to overcome the noise but degrades the amount of heterogeneity captured. When the mapping from latent space to volume space is parametric, this choice is made implicitly by the parametrization, often by imposing some degree of smoothness. Here, the parameter makes this tradeoff explicit.
After the subset of images is identified, a homogeneous reconstruction, including FSC regularization, is employed to reconstruct a volume from that subset of images. The different strategies for volume reconstruction are illustrated in fig. 2.
Figure 2:

Illustration of recovered volumes and trajectories from dataset described in section 1.5 (a). Visualization of the true trajectory (black dashed) and recovered trajectory (blue dashed) plotted over the estimated latent density. The trajectories and latent densities are four-dimensional, and each plot is a projection onto a pair of dimensions. (b) Visualization of the true volumes and reconstructed volumes along the estimated trajectory by reweighting and reprojection at the end states (star and square) and intermediate states sampled at uniform intervals along the trajectory (circles). (c) Scatter plot of log-likelihoods at the decoded along the recovered trajectory. Each of the 50,000 particles is represented at its MAP-estimate . The threshold used for reweighting is .
1.4. Motion recovery
Traditionally, linear subspace methods attempt to recover motions by moving along straight lines in the latent space, corresponding to linear interpolation in volume space. For instance, in 3DVA [29], individual principal components are traversed to generate axes of motion. However, this straightforward linear approach falls short because even a simple linear motion of atoms translates to a highly nonlinear trajectory when observed in volume space. Consequently, a linear path in volume or latent space cannot adequately capture rigid motion, let alone general atomic movements. Fortunately, linear functions locally approximate smooth functions well, allowing small motions to occasionally be captured by traversing a single principal component. In most cases, multiple principal components are needed to accurately represent a single motion, which can lead to significant artifacts when using only one, as illustrated in fig. A.8. Furthermore, multiple axes of motion are typically embedded in the same principal component, as we illustrate below. This lack of separation is clear from the theory: independent variables are not encoded in different principal components by PCA. Alternative decompositions such as Independent Component Analysis (ICA) [7] have that property but require estimating higher-order statistics.
We adopt an alternate strategy based on physical considerations: molecules display stochastic motions, randomly walking from one state to neighboring ones with probability depending on the rate of change of free energy, generally preferring to move towards low free energy states. A physically more meaningful way to estimate trajectory would thus be to find the most likely trajectory between two states. This trajectory could, in theory, be computed using linear subspace methods; as both distances and density, which can be used to infer energy, are approximately conserved by the embedding. However, high noise levels and the motion’s stochastic nature make this task difficult.
We instead propose a simpler heuristic with some of the properties of this trajectory that overcomes the clear pitfalls of linear trajectories: we generate non-linear motions by computing short and high-density continuous trajectories of volumes between two predetermined conformational states. Since high-density states correspond to low energy, we expect this trajectory to behave like the most likely one. Furthermore, exploiting density allows the trajectory to stay in meaningful regions of latent space.
Given that distances and density are approximately preserved between volume and latent space due to the PCA embedding, we can directly approximate these trajectories in the latent space. Specifically, we compute a trajectory between two states that minimizes the cumulative inverse density. That is, we find the trajectory that minimizes the expression
| (8) |
subject to , , with and is the latent density defined in eq. (6). Minimizing eq. (8) falls under the category of classical optimal control problems, see e.g. [2, 38], and appendix A.6 for our implementation details. Next, we demonstrate on real and simulated datasets that this approach can recover large and intricate motions.
1.5. Reconstruction of large synthetic motion
We commence our evaluation by applying our method to a synthetic dataset introduced in [48]. This dataset depicts an artificial rotational motion of a protein complex comprising 50,000 images, each sized at 128 × 128. Despite being characterized by relatively high SNR and having known uniform poses, this motion is large, presenting a scenario where linear subspace methods are traditionally expected to perform poorly.
First, we obtain a mask using real-space correlation based on mean estimates obtained from two half-sets through the EMDA toolbox [45]. Next, we retrieve principal components estimated with varying levels of accuracy, see fig. A.6. We use the top four for analysis. We then project the two ground-truth extreme states into the latent space through the mapping , recover a trajectory between these states by solving eq. (8), and generate volumes by reweighting. As observed in fig. 2, we recover the correct motion with high visual resolution. The computation requires 8 minutes on one GPU, with detailed timings in table 1.
Table 1:
Timing and complexity of each step on simulated dataset of images of size , computing columns, using the first principal components, with contrast interval and latent space discretization parameters defined in appendices A.5 and A.6 respectively. Since all , , are fixed in practice, the run time is typically for large datasets (), and for small datasets. Timings were performed on an NVIDIA A100 80GB GPU.
| Step | Complexity | Timing |
|---|---|---|
| Covariance and principal component estimation | 5m58s | |
| Per-image means and covariance estimations | 12s | |
| Trajectory computation | 1m55s | |
| Volume generation (per conformation, amortized) | 2s | |
| Total | 8m21s |
Despite the simplicity of the rigid motion, its representation forms a highly nonlinear curve in latent and volume space. This example illustrates the issues of attempting to decode motion solely by traversing a principal component: a single motion is present in the dataset, yet its accurate recovery requires multiple principal components. Thus, generating multiple motions from different principal components would yield multiple erroneous trajectories. Furthermore, the motion is not monotonous within any of the principal components. Therefore, traversing a single principal component coupled with a reweighted strategy results in averaging images from very different states, as illustrated in the output of 3DVA in fig. A.8.
In appendix A.8, we also show results of our method on the same trajectory but with images sampled nonuniformly across states, and a comparison with the neural network method cryoDRGN on that dataset. Our method performs similarly to the uniform case, only with lower-resolution reconstructions of states less frequently observed in images. In contrast, while cryoDRGN embeds the distribution effectively, its embedding is disordered in this case. This uninformative structure results from the lack of conservation of distances in latent space. Consequently, the trajectories generated in this embedding show either no motion or discontinuous jumps. Furthermore, the recovered trajectory visits low-occupancy parts of the latent space, resulting in hallucinated states not present in the data but still displaying high-resolution features. This difference between the trajectories recovered by the two methods highlights the importance of density, distance, and uncertainty preservation in the embedding.
1.6. Reconstruction of synthetic multi-dimensional motion
Next, we evaluate our method using a more complex synthetic dataset that better resembles real-world conditions with realistic factors such as SNR, pose distribution, image contrast, and motion patterns, see fig. 3. This dataset comprises approximately 300,000 images and is designed to mimic the heterogeneity observed in the precatalytic spliceosome structure, as reported in previous studies [27, 25]. The dataset involves three bodies that move independently, creating multidimensional heterogeneity. We construct two types of motions: one two-dimensional landscape of top-down motions where two bodies move independently (80% of images) and one trajectory of left-right motions of both bodies (20% of images). One trajectory is significantly more frequent in the images among the top-down motions.
Figure 3:

Pipeline applied to a realistic synthetic dataset. (a) Simulated dataset of the precatalytic spliceosome with three bodies moving independently. A landscape of motions consisting of 80% of images where the orange and pink bodies move independently in top-down motions, including a high-density trajectory where both bodies first move down together, and the orange body then locks back into place. A second motion, consisting of 20% of images, shows the two bodies’ left-right motion. (b) Depiction of the first five estimated principal components displaying a decaying SNR, as expected in PCA. (c) Scatter plot of each image’s inferred versus ground truth amplitude contrast. (d-e) The two ground truth and two recovered trajectories in volume and latent space. The star and the square represent the endpoints, and the circles are intermediary states at equidistant points along the trajectory. The bottom row shows the recovered trajectory over pairs of dimensions overlayed on the latent density integrated over the remaining dimensions.
We again form a mask from the halfset mean estimates, compute the first four principal components, and project the ground truth extremal states into latent space. We then generate trajectories by solving eq. (8), and generate volumes along the recovered trajectories by the reweighting scheme.
We recover the correct motion in both cases, highlighting the capability of linear subspace methods to recover large, intricate motions in realistic conditions. The reconstructed volumes closely match the ground truth volumes, even though trajectories do not exactly match in latent space. This discrepancy is due to the regularization of latent coordinates in eq. (19), which improves the robustness of the trajectory at the cost of biasing the estimated latent coordinates towards the origin. This further regularization makes the method robust to the number of and noise in principal components; e.g., even though the fourth central component displays some noise, the ultimate reconstructions do not suffer from it. The resolution of the reconstructed states in these trajectories is not constant, a variation that stems from the uneven distribution of images along each trajectory. This observation underscores the data-hungry nature of reweighting schemes [43] and points to the possible advantages of parametric mapping, which can trade off bias for improved resolution.
The results also highlight that interpreting principal components as motions as in 3DVA is misleading since two motions in orthogonal directions are partially embedded into the same principal component direction .
1.7. Motions of the spliceoseaume
We apply the method on the precatalytic spliceosome dataset [27] (EMPIAR-10180), which is a well-characterized dataset commonly employed for benchmarking continuous cryo-EM heterogeneity methods. We downsample the stack of approximately 300,000 images to a size of 256 × 256.
Our analysis focuses on a specific region, the head of the structure, for which we create a loose focusing mask to highlight the capability and boost SNR. We compute principal components with contrast correction (see fig. 4(a)) and use the first ten to generate 40 different states using the centers of k-means clustering on the collection of . Six of these states, superimposed on a UMAP [23] visualization of the set of are shown in fig. 4(b).
Figure 4:

Pipeline applied to the precatalytic spliceosome dataset (EMPIAR-10180). (a) Computed mean, with focusing mask overlayed and first six principal components computed. (b) Six conformational states which were obtained by clustering the distribution of . (c) A top-down (blue-dotted) and left-right (black-dotted) trajectories were reconstructed by using identified k-means centers as endpoints and solving eq. (8). The two trajectories are also plotted in latent space over pairs dimensions. All reconstructions are displayed with a B-factor correction of 170Å2.
Similar to the findings in [48], our observations reveal a combination of discrete and continuous heterogeneity. The discrete heterogeneity is due to a denatured state characterized by a partially absent head region, and the continuous heterogeneity is due to a large range of top-down displacements and a smaller range of left-right displacements of the head from a central base state (visualized here as the position). We generate a top-down motion and a left-right motion by picking states generated from the k-means centers as endpoints (see fig. 4(c)). Notably, the latent space exhibits less structured behavior than the simulated dataset in fig. 3. This lack of structure could be due to a single central state being stable, with all other states representing perturbations of it. However, unmodeled error sources such as miss-alignments or junk particles could contribute to this less structured latent space configuration. Our method computes this embedding in 1 hour and 42 minutes on 1 GPU, 12× faster than cryoDRGN on the same hardware, see table A.2 for details.
1.8. Assembly states of the ribosome
We next analyze the ribosomal subunit dataset (EMPIAR-10076) [6], known for its significant compositional heterogeneity. This dataset has been extensively studied, revealing 14 distinct assembly states through repeated 3D classification [6] in the original research and one additional state identified by cryoDRGN [48].
Starting with a filtered particle stack containing around 87,000 particles from the cryoDRGN study, we perform an initial run of our algorithm by downsampling images to a box size of 128 and using a loose spherical mask. We then compute the real-space variance map3 allowing us to pinpoint regions with high variance used to generate a mask for subsequent analysis (as seen in fig. 5(a)). Using the mask, we run the algorithm once more on the stack with a box size of 256. We find that this strategy of generating a focusing mask around regions of high variance helps boost the SNR of highly heterogeneous datasets. We use a 20-dimensional PCA to embed the particles in latent space and display UMAP visualization of the . We then produce volumes by reweighting to highlight the wealth of states captured by the linear subspace in fig. 5. Reprojected states, stability under different numbers of principal components, and the embedding obtained by cryoDRGN are also shown in fig. A.9 for comparison. Interestingly, the UMAP representation of cryoDRGN and our method are strikingly similar despite the two approaches using vastly different embedding strategies. This result illustrates the high representation capacity of linear subspace methods with a large subspace size comparable to a highly non-linear neural method. We compute the embedding with box size 128 in 7 minutes, 11× faster than cryoDRGN, and the embedding with box size 256 in 48 minutes, 6× faster than cryoDRGN, see table A.2 for details.
Figure 5:

Pipeline applied to a ribosome dataset (EMPIAR-10076). (a) Initial run using a spherical mask. We use the computed mean and real-space variance map to identify a region of high variability to form a mask. (b) Estimated principal components with a box size of 256 and the generated mask. (c) UMAP visualization of the 20-dimensional embedding color-coded with published labels. Circles denote the median embedding of images with each published label, and the states reconstructed at those positions with in eq. (7) chosen corresponding to a 8 standard deviation in one dimension, selected so that > 90% of images are in at least one of the sets . All states are displayed with a B-factor correction of 170Å2.
2. Discussion
2.1. Related work
We introduced a heterogeneity analysis pipeline based on PCA with a novel automatic regularization technique and improved efficiency, enabling the fast and stable computation of large numbers of principal components at high resolution. Additionally, we designed customized volume generation and motion recovery schemes that leverage the favorable properties of linear subspace methods.
Several linear subspace methods have been proposed for cryo-EM heterogeneity, including bootstrapping from 3D reconstructions [26], covariance estimation methods [15, 1], probabilistic PCA (PPCA) methods [42, 29], and methods employing molecular dynamics [44]. Our approach shares similarities with the PPCA method 3DVA [29] implemented in the popular software suite cryoSPARC, but there are key differences. Our method requires a small number of passes through the data to estimate the principal components, while 3DVA relies on an iterative expectation-maximization procedure. Moreover, our method allows for automatic regularization so that we can stably compute hundreds of principal components with no parameter tuning. In contrast, 3DVA requires the user to pick at least two coupled parameters to compute the principal components: a number of principal components, and the resolution of a low-pass filter applied to images before computation. Under poor choice of either parameter, 3DVA is unstable, leading to poor results or program errors. While our method also requires choosing a number of principal components to embed images, it does not require an a priori choice to compute the principal components. This foregoes an expensive parameter tuning step to identify the correct number of principal components. Furthermore, embedding images is much cheaper than computing the principal components, e.g., 30 times faster in table 1, allowing the evaluation of different embedding sizes with little additional cost.
PPCA methods, such as 3DVA, have their advantages. Fixing the number of components a priori can be an effective regularizer for the reconstructed principal components. Indeed, in fig. A.8, we show that 3DVA outperforms our method at estimating accurately some of the principal components for some parameter choices, likely due to this rank constraint. Furthermore, the flexible iterative framework of PPCA methods offers the potential for future incorporation of more sophisticated priors, imposing sparsity or non-uniform resolution, as demonstrated in [32] for homogeneous reconstruction.
A further critical distinction between 3DVA and our method is in interpreting latent space. 3DVA traverses individual principal components to generate motion and uses a reweighting scheme considering a single principal component at a time. We showed that this interpretation is often erroneous, particularly for large motions.
Another popular class of method for heterogeneity analysis is deep-learning methods, which have received a lot of research interest following the success of cryoDRGN [48] (see [10] for a recent review). Deep learning methods seem to have a high expressive power and often achieve higher visual resolution than other methods. However, quantifying this improvement can be challenging due to the misleading nature of the typical FSC metric in heterogeneous reconstruction (see fig. A.7). The superior resolution of deep-learning methods is likely due to the sophisticated implicit priors learned by the neural networks. However, an even better resolution may be achievable by incorporating explicit deformation modeling, as demonstrated in 3DFlex [30].
The drawback of deep-learning methods for heterogeneity analysis is their black-box nature, making interpretation, validation, and hyperparameter tuning (network structure, number of epochs, weight decay, mini-batch size, etc.) particularly difficult. While some default hyperparameters perform well on many datasets, the same hyperparameters applied to other datasets can result in overfitting artifacts in the reconstructed volumes [16]. Furthermore, the lack of explicit regularization and metrics for heterogeneity can make identifying suitable parameters difficult, and the user must resort to repeated, expensive computations and subjective visual inspection. Finally, deep-learning methods lack key properties: they do not preserve distances or distributions, uncertainty estimates are unavailable, and embeddings exhibit unexpected behavior [11]. As a result, interpreting their latent space is particularly difficult, and there is no principled way to estimate motion. This, coupled with the lack of explainability, may lead to false conclusions about the heterogeneity in cryo-EM datasets as demonstrated in fig. A.10.
2.2. Validation of heterogeneity analysis methods
Emerging methods for heterogeneity analysis in cryo-EM encompass a wide range of techniques, from deep learning, manifold learning, and PCA to deformation fields and molecular dynamics. Each approach has its strengths, but a notable challenge arises when validating and comparing the outcomes of these diverse techniques. While all heterogeneity methods yield probability distributions of conformational states as output, the challenge stems from their differing representations and coordinate systems. These differences make direct comparisons a complex task.
Even comparing individual reconstructions is not straightforward when generated by heterogeneous algorithms. As highlighted in appendix A.7, the omnipresent FSC score, typically employed to evaluate homogeneous reconstructions, is misleading when applied to volumes obtained by heterogeneity analysis. One reason is that different states within a heterogeneous dataset are often highly correlated. Thus, using a correlation-based score to evaluate the reconstruction of a specific state can inadvertently reflect the homogeneous component of the reconstruction.
Covariance estimation and linear subspace methods offer promising avenues to address the challenge of quantifying the consistency of heterogeneous reconstructions across different techniques. First, sampling from each method’s distributions makes it possible to estimate the covariance of each method’s output and compare it with the covariance computed directly from minimally processed images as in appendix A.1. Furthermore, the covariance estimates can be compared using the covariance FSC proposed in appendix A.3. Second, the mapping from volume space to latent space provided by linear subspace methods can embed volumes obtained by different algorithms into the same coordinate system. This mapping could allow for comparisons and alignment of different distributions of volumes obtained through other heterogeneity methods.
2.3. Limitations and opportunities
One current limitation of linear subspace methods is their relatively simple priors compared to intricate implicit priors learned by deep neural networks. This distinction suggests that achieving higher volume resolution could be possible by incorporating more sophisticated priors. One potential avenue for achieving enhanced resolution involves improving either the resolution or the number of computed principal components. One route for such advancement may include using sparse-PCA [50] in a wavelet basis or utilizing non-uniform priors, similar to [32].
Another strategy is to introduce priors on the mapping from the latent space to the volume space. For example, the method in [24] utilizes manifold learning techniques built on the PCA embedding. Another possibility for continuous heterogeneity could be fitting nonlinear mappings, such as deformation fields [30] or polynomial maps [13] from PCA embedding to volume space. This fusion could retain the desirable properties of linear subspace embedding while harnessing the data efficiency offered by these methods.
A shortcoming of the method presented here is the lack of an automatic choice of the optimal number of principal components for downstream tasks. While our approach can compute hundreds of principal components stably and efficiently, typically, only up to a few dozen exhibit sufficient SNR, and picking the number can substantially impact the quality of the result. We recommend heuristic choices based on the decay of eigenvalues (e.g., the Scree test [14]) and visualizing principal components and their embeddings. Fortunately, our method computes embeddings of different sizes at virtually no extra time, and thus, we calculate the embeddings with , 10, 20 in our implementation by default. Furthermore, our method demonstrates resilience against overestimation due to the regularization steps as shown in fig. A.9, and thus, we suggest slightly overestimating the number of principal components. We note that related theory predicts the optimal choice based on observation count and noise levels [8] but does not cover prediction for the method presented here. This area presents an opportunity for future theoretical exploration.
The introduction of the Bayesian framework was central to the resolution revolution in cryo-EM [17] and has dramatically contributed to its democratization thanks to its robust parameter estimation framework that requires relatively little user input. In contrast, the current state of heterogeneity analysis typically involves ad-hoc filtering and hand-tuning of various parameters. The framework presented here generalizes the Bayesian framework used for homogeneous reconstruction and thus offers similar potential in robustly and confidently reconstructing wide ranges of heterogeneous distributions from cryo-EM datasets with little need for tuning.
The Bayesian framework was particularly critical in developing robust homogeneous methods that infer poses and volumes. These algorithms typically alternate between predicting poses and predicting volumes. However, poor alignment of poses produces inaccurate volumes, and inaccurate volumes, particularly from overfitting, also cause poor alignment. The significant advance of the FSC regularization introduced in [35] was to resolve this issue: thanks to the adaptive regularization recomputed at each step, resolution and alignment can progress slowly together. Our regularization strategy generalizes this scheme, and our method’s high efficiency allows it to be run repeatedly within an alternating framework. Therefore, it is a promising method to address the problem of jointly inferring conformations and poses. This framework may result in more accurate pose estimation for highly heterogeneous datasets, thereby improving the input to all heterogeneity reconstruction algorithms.
Acknowledgments
The authors thank Eric Verbeke, Joakim Anden, Roy Lederman, Edgar Dobriban, William Leeb, Bogdan Toader, and Willem Diepeveen for helpful discussions. The authors thank Ellen Zhong and other developers of cryoDRGN for making the software and code accessible, which our implementation relied on. The authors also thank the developers of RELION, cryoSPARC, EMDA, and ChimeraX for making their software freely available, all of which were used to generate the results presented here.
The authors are supported in part by AFOSR under Grant FA9550-20-1-0266, in part by Simons Foundation Math+X Investigator Award, in part by NSF under Grant DMS-2009753, and in part by NIH/NIGMS under Grant R01GM136780-01. Part of this research was performed while the first author was visiting the Institute for Pure and Applied Mathematics (IPAM), which is supported by the National Science Foundation (Grant No. DMS- 1925919).
A. Methods
A.1. Solutions to least-squares problems
The solution to the mean estimation problem in eq. (2) is obtained through the following matrix equation:
Here, denotes a diagonal matrix with entries from vector . When the projection operator is discretized using nearest-neighbors and noise covariance is modeled as diagonal, as is often the case in cryo-EM, the matrices become diagonal in the Fourier domain. This matrix equation can then be efficiently solved in operations, analogous to the E-step of the E-M algorithm used in iterative refinement, as seen in works like [34].
The solution to the covariance estimator in eq. (3) can be expressed similarly:
| (9) |
| (10) |
| (11) |
where · denotes the Hadamard (pointwise) matrix product. Solving matrix equations of this form is computationally intensive, with a complexity of for a general system. The technique presented in [1] addresses this by utilizing the convolution structure to achieve a solution in operations where . Nevertheless, storage and computational requirements remain excessive for moderate volume sizes ().
Taking advantage of the diagonal structure of in eq. (10), obtained through a nearest-neighbor discretization, we rewrite the system of equations in eq. (9) as a Hadamard system:
| (12) |
| (13) |
Remarkably, this formulation allows for efficient and independent computation of entries in by constructing entries of matrices , , and , followed by pointwise division. In this way, columns of may be computed in in a single pass through the data. This computational technique heavily relies on the diagonal structure of in eq. (10), which stems from a nearest-neighbor discretization of the projection operator. In contrast, the projection of the mean in eq. (11) does not necessitate a nearest-neighbor discretization, so we utilize linear interpolation. Although discretization errors are typically not the dominant source of error, numerical experiments have shown that this choice can significantly reduce discretization errors with minimal additional computational cost.
Additionally, experimental images often contain multiple particles within each image. To mitigate this, we use a real space mask to filter out these unwanted particles while enhancing the dataset’s SNR. To generate per-image masks, we dilate a three-dimensional loose mask and project it using inferred poses for each image. We then threshold and smooth by a cosine-softening kernel to generate mask , which we then apply to the mean-subtracted images. Specifically, the right-hand side is computed as follows:
| (14) |
where is used to emphasize projection operators computed using linear interpolation. While this scheme enhances the efficiency of evaluating individual covariance matrix elements, computing all of them still demands operations and memory, which is impractical. We next describe numerical algebra techniques to approximate leading eigenvectors of using a subset of entries to address this issue.
A.2. Approximate SVD from subsets of columns
We estimate the principal components from a subset of the entries of through an approach inspired by the randomized SVD algorithm [22]. To motivate the approach, suppose we observe the volumes directly; in that case, we could approximate the principal components by performing a randomized SVD on the mean-subtracted volume matrix , as presented in algorithm 1 (replicated from [22]).
| Algorithm 1 Randomized SVD of the matrix [22] | |
|---|---|
|
|
Line 1 of algorithm 1 is the rangefinder step, typically implemented as the subspace spanned by the product for a small random matrix , and it aims to find a subspace containing the top singular vectors. Line 2 computes the coordinate of each column in that subspace. However, we cannot perform lines 1 and 2 directly since we do not directly observe volumes but rather images. Instead, we use the subspace spanned by a subset of the columns of and estimate the coordinates of the volumes through their MAP estimate in eq. (5). Steps 3-5 of algorithm 1 remain unchanged. We summarize the proposed method in algorithm 2.
| Algorithm 2 Proposed approximate SVD | |
|---|---|
|
|
The strategy of approximating a matrix from a subset of its columns is a celebrated trick in numerical linear algebra and the basis for schemes such as the Nyström extension [46] and the CUR decomposition [21]. The approximation accuracy depends on the matrix’s properties and the chosen columns. While near-optimal choices exist in theory (see, e.g. [18, 4]), our case differs due to our goal of computing eigenvectors of the true covariance using noisy estimates through the estimation problem. Unfortunately, this case has limited existing theory, so we rely on heuristic parameter choices.
Our approach prioritizes selecting columns of with a high SNR. In this context, we choose columns corresponding to lower frequencies, motivated by the fact that low-frequency entries of the covariance matrix are sampled more frequently. For example, the sampling of the frequency pair (, ) is inversely proportional to their cross product in an idealized case [15]. We compute columns associated with frequencies that satisfy and , where is the Fourier space grid spacing and is a specified radius. We set in all experiments, resulting in 298 computed columns. We also take advantage of the Hermitian symmetry property of the covariance matrix, allowing us to generate all columns satisfying , without significant computational overhead. We denote the block of columns of as , where is a matrix that subsamples these columns.
In the noiseless case, and under the assumption that exactly spans the columns of and the matrices are full rank, we recover the true . It follows that the estimated SVD of computed from algorithm 2 is exact.4 In other cases, we only recover an approximate SVD of , and the noise and possible ill-conditioning of necessitates a regularization in solving eq. (5). This regularization is set using eigenvalues estimates of in the MAP framework, see appendix A.5. The eigenvalues of are the square of the singular values of , but initializing them is necessary to compute eq. (5) in line 2 of algorithm 2.
We initialize them to , where denotes the -th singular value. This estimate is chosen so that it is exact when is rank-one. That is, if for some and , then and so that .
We compute the initial singular value decomposition in the spatial domain by applying standard a randomized SVD to where is the 3D Fourier transform matrix of size and applies the 3D softened mask.
A.3. A generalized FSC regularization scheme
We introduce a scheme that generalizes the Fourier Shell Correlation (FSC) regularization initially proposed in [34] for homogeneous reconstruction. The foundation of this regularization scheme is rooted in the simplifying assumption that both the signal and the noise are independently and identically distributed within one frequency shell, though they follow different distributions in different shells. This simplification allows us to decouple computations over these shells.
Thus, we consider the signal and noise in a specific frequency shell where the signal follows a Gaussian distribution with zero mean and variance , while the noise follows a Gaussian distribution with zero mean and variance . This leads to the simple scalar-valued forward model:
where represents frequency within the shell, and is the —th observation of frequency corrupted by noise . We then estimate the underlying signal by solving the following least-squares problem:
| (15) |
Here, serves as a regularization weight. The solution to eq. (15) is:
Figure A.6:

Estimation of principal components and eigenvalues. (a) Sine of principal angles between the subspaces spanned by the estimated and exact top eigenvectors for different estimators. The approximate SVD improves on the accuracy across all principal components compared to the inital one. (b) Eigenvalue estimates for the initial and approximate SVD estimator. The first estimator is on the correct order of magnitude but underestimates eigenvalues. A lower eigenvalue estimate is expected due to regularization. The second estimator is more accurate, especially for eigenvalues corresponding to well-estimated eigenvectors. (c) Visualization of the first six exact and estimated eigenvectors, showing increasing frequency and noise. Note that eigenvectors are unique up to sign, which accounts for the sign flip between exact and estimated.
where . The optimal value of , which minimizes the mean-squared error (MMSE) is known to be . However, estimating is our goal. To do this, we begin with an initial value and calculate two estimates and by randomly splitting the dataset into two halves. We then estimate their correlation using the concept from [33]:
Solving for , we obtain the estimate:
| (16) |
For comparison, under our assumptions and notation, the estimate proposed in [35] is equivalent to:
| (17) |
Notably, if the pose distribution is uniform (more precisely if are constant within a frequency shell), then the two estimates in eq. (16) and eq. (17) agree, but eq. (16) also accounts for the non-uniform case. This new estimator can be applied to homogeneous reconstruction, and we expect it to perform better for datasets with highly non-uniform pose distributions. However, the effect is most pronounced for covariance estimation since sampling of the entries of the covariance matrix in a given column and frequency shell is highly non-uniform even when the pose distribution is uniform.
We use this regularization scheme to set entries of the regularization matrix in eq. (3) by applying it independently in each column. We replace the EFSC with the empirical FSC computed between halfsets, initializing from a multiple of the regularization weights of the mean in eq. (2) and iterating this process three times, which has been observed to converge in practice.5 That is, we compute the entries of the weight matrix as follows:
| (18) |
Here, is the frequency shell of radius , is the FSC between column of the two covariance matrices and computed from the two half-datasets, and is the average of the two values of calculated from eq. (13). We perform these computations only for the columns chosen in appendix A.2, indicated by the subscript col. Additionally, we enforce symmetry on matrix to ensure the symmetry of matrix . The outlined algorithmic steps are summarized in algorithm 3.
| Algorithm 3 Computation of the regularization parameters | |
|---|---|
|
|
A.4. Contrast correction in covariance estimation
Cryo-EM images sometimes display variations in contrast. This effect is captured in the image formation model through an additional scaling factor :
If not accounted for, this contrast variation can manifest as a spurious source of heterogeneity, often displayed in recovered trajectories with appearing or disappearing parts of the volume. While some algorithms for consensus reconstruction attempt to infer the scale parameters as part of the algorithm [36], the estimation is poor when significant heterogeneity is present, so they should be estimated as part of a heterogeneous reconstruction. As the contrast is not factored into the covariance estimation in eq. (3), our covariance estimate reflects the covariance of the contrasted distribution of states, which we denote by . Assuming that contrast factors and states are independent, can be related to the unconstrained covariance of the distribution as follows [39]:
That is, the contrasted covariance differs by an unknown scaling factor close to 1 (e.g., in fig. 4) and is corrupted by a rank-one component . We assume that is low-rank and , and we recover the original subspace by projecting out the mean component:
where . Furthermore, the low-rank decomposition of can be efficiently computed from the low-rank decomposition of as showed in algorithm 4:
A.5. Estimation of per-image latent distribution and contrast
We estimate the mean of the conformation distribution and the contrast parameter associated with the image using a maximum a posteriori estimate. Assuming follows a Gaussian distribution and using a flat prior on , the MAP estimate is written as the solution of the following optimization problem:
| Algorithm 4 Update of low-rank decomposition by projecting out mean component | |
|---|---|
|
|
| (19) |
To solve this minimization, we perform a grid search over the contrast variable and explicitly minimize over the latent variables . Solving eq. (19) for a fixed contrast involves the normal equations:
| (20) |
The computational complexity of constructing and solving eq. (20) is . Therefore, the naïve computational complexity of solving this problem for all contrast values of is operations. However, if the matrix , and vectors , are precomputed and stored, the complexity drops to . In our experiments, we set , and the additional cost of optimizing over contrast is small compared to the cost of optimizing over alone since the term dominates.
The covariance of the state, denoted as , is directly computed from eq. (4) as: in operations per image.
A.6. Traversing latent space
We generate a motion between two latent space coordinates and by solving the minimization problem described in eq. (8). This problem can be approached using dynamic programming by first computing the value function , defined as:
where is a continuous trajectory satisfying , with . This value function is the viscosity solution of the Eikonal equation [2]:
| (21) |
where is the domain of interest. Thus, can be computed by solving this partial differential equation. Once eq. (21) is solved, the optimal trajectory can be obtained by finding the path orthogonal to the level curves of , which can be computed numerically using the steepest descent method on starting from .
The Eikonal equation can be discretized and solved using variants of Dijkstra’s algorithm for finding shortest paths on graphs [38]6. We choose the domain to be a —dimensional rectangle with lower and upper bounds in dimension equal to the 1st and 99th percentile of the distribution of . We then discretize the problem by evaluating on a —dimensional uniform grid. Adding a small constant ensures the existence of a solution and the stability of the numerical method. We set .
The computation of the trajectory is dominated by the evaluation of on the grid, resulting in a computational cost of , where is the number of grid points in one dimension which we set to in all experiments. This limits the applicability of this method to low values of , and therefore, we use this method only in dimensions up to 4. To compute a higher dimensional trajectory, we iteratively increase the dimension of the trajectory by one while keeping the previous dimensions fixed, starting from the 4-dimensional trajectory. This heuristic is not guaranteed to solve the high-dimensional minimization problem, but it performs well in practice.
A.7. FSC scores for heterogeneity
We illustrate the deceptive nature of FSC scores in determining the resolution of volumes generated by heterogeneous algorithms in fig. A.7. The intriguing observation is that while the reprojected state appears visually as low resolution, the conventional interpretation of the FSC score would lead us to conclude that it is a Nyquist resolution estimate of the true state. On the contrary, despite being visually a much better estimate, the FSC score indicates that the reweighted reconstruction’s resolution is around half the reprojected resolution. This puzzling phenomenon can be explained by how the reprojected state is computed: . That is, it is a linear combination of the mean and the principal components. The principal components are estimated at a lower resolution than the mean and are regularized adequately so they do not contribute much noise. Consequently, the state essentially equals the mean in high-frequency shells. Furthermore, the mean is highly correlated to the true state (as the blue curve depicts). Thus, the reprojected state appears highly correlated to the ground truth. While the impact is not as pronounced in the reweighted reconstruction, a similar phenomenon occurs on a more localized scale since any reweighted reconstruction necessarily incurs some averaging over different states.
Other methods for heterogeneity reconstruction might not offer the same level of interpretability as the reprojection scheme. However, we anticipate that they would exhibit similar behavior since the static parts of the molecule are more easily resolved to a higher resolution. Adjustments to the FSC calculation, such as the mean-subtracted FSC proposed in [24], or using a local FSC, might provide some relief but will likewise be misleading.
Figure A.7:
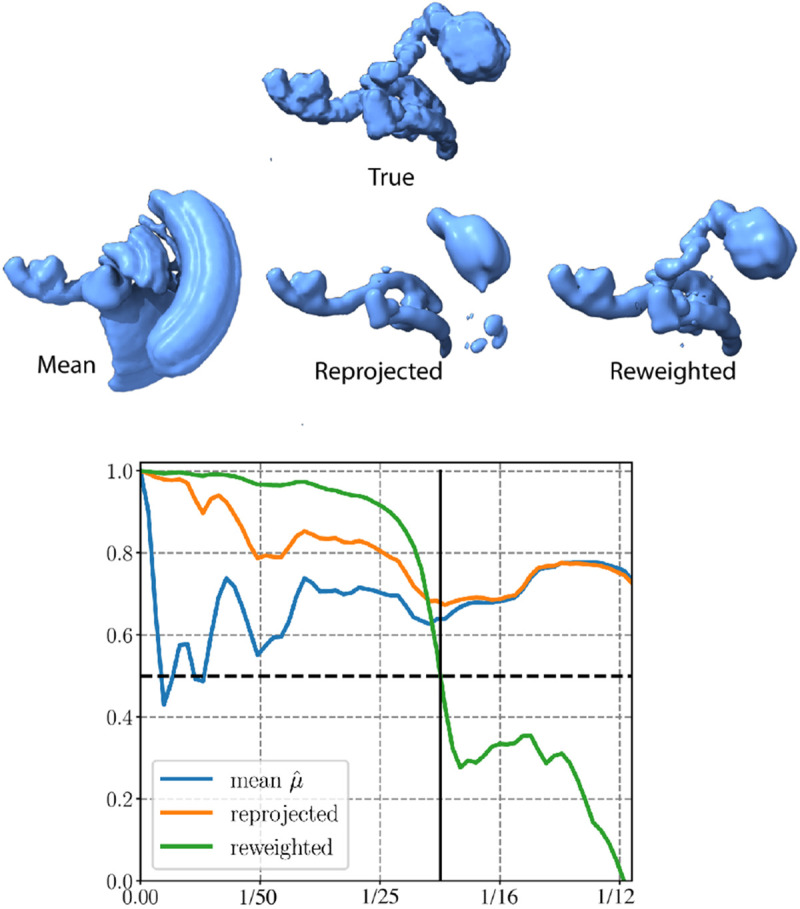
Illustration of the deceptive nature of FSC scores in heterogeneity analysis. Top: the true state, the estimated mean conformation, and reconstructed states using reprojection and reweighting from the dataset detailed in fig. 2. Bottom: Fourier Shell Correlation curves, computed with a loose mask, between the true state and the reprojected reconstruction (orange), the reweighted reconstruction (green), and the estimated mean conformational state (blue).
Table A.2:
Embedding time across datasets and box size. All timings were performed on an NVIDIA A100 80GB GPU using version 2.3 of cryoDRGN and default parameters.
| Dataset name | dataset size | box size | our method | cryoDRGN |
|---|---|---|---|---|
| EMPIAR-10180 | 327490 | 256 | 1h14m | 20h22m |
| EMPIAR-10076 | 87327 | 128 | 7m1s | 1h21m |
| EMPIAR-10076 | 87327 | 256 | 48m38s | 4h49m |
A.8. Illustration of favorable latent space properties
We illustrate the importance of preserving distances and density between latent and volume space by comparing our method to cryoDRGN on a synthetic dataset from the introduced in the original cryoDRGN paper [48]. This synthetic dataset uses the same trajectory as in section 1.5, but the distribution of states among images is different. Instead, some states are more likely than others, and a few states along the trajectory are not observed in the data entirely. We note that cryoDRGN and our method recover the correct motion in the uniform dataset.
Our method uses the same parameter as in section 1.5 (), and we train a cryoDRGN network with default parameters (version 2.3, , 25 epochs).
Figure A.8:

Result of 3DVA on a synthetic dataset with large motion with the same mask as used in section 1.5. (a) Accuracy of the computed principal components for different choices of dimension (3DVA-4,8,12,16,20) and the method presented here (covariance). At 20 dimensions, the lack of regularization of 3DVA causes instability in the algorithm, and the program returns an error (depicted as 100% error). (b) The estimated latent coordinates using dimension 4 are very similar across methods (see fig. 2 for comparison). (c) True trajectory and reconstruction along the first principal component using a reweighting strategy (option “intermediate” in cryoSPARC) and reprojected (option “linear” in cryoSPARC). (d) Latent depiction of the recovered motions: in blue are the particles used in the reweighting scheme, and in red is the latent coordinate used to reproject the first principal component. In either case, large artifacts are present due to the averaging of particles from distinct states in the case of reweighting and the early truncation of the principal component in the reprojection case.
We first wish to visualize how each method embeds the trajectory in latent space (see fig. A.10(a)). Our method uses the map to embed the trajectory. CryoDRGN does not provide a straightforward way to embed volumes into latent space, as only images used to train the network can be embedded. We follow the suggestion in the original paper to embed volumes given ground truth labels: we average the latent coordinates of the collection of images for a particular state, and then assign the volume to the nearest latent vector. We embed the ground truth trajectory using this method (for cryoDRGN, we skip over states with less than 5 images).
We observe that the ground-truth trajectory displays large discontinuities across the unobserved states. This observation is expected as the VAE does not conserve distances between latent and volume space. We next recover a trajectory using cryoDRGN’s graph algorithm, which seeks the shortest path on a pruned nearest-neighbor graph in latent space. No path is found with the default parameter, so we increase neighborhood size (average and maximum) in increments of 5, starting from 5 until we find a path. In this case, we found a trajectory for a neighborhood size of 20.
The results in fig. A.10(a) show that the graph algorithm fails to recover the correct trajectory; an expected outcome as nearby states are embedded at opposite ends of the latent space by cryoDRGN. In contrast, our method recovers the correct trajectory thanks to the conservation of distance and density between latent and volume space.
In fig. A.10(b), we show ten volumes reconstructed at approximately equidistant points, as measured by latent distances, along the recovered trajectories for each method. We overlay the ground truth trajectory in transparent yellow. Our method recovers the correct motion with a highly variable resolution. It recovers high-resolution volumes for states with many observed images in the dataset (volumes 2 and 10), a lower resolution volume for states with fewer images (volumes 3 and 8); and the FSC regularization forces the volume to be small (dropping below the visual threshold) for states with no images at all (volumes 4 and 6).
In contrast, all volumes produced by cryoDRGN display high-resolution features, but even as the trajectory moves in latent space, no motion is observed in the volumes (volumes 1 to 4 and 8 to 10). In parts of the trajectory that are sparsely populated (states 5 and 7), the network seems to hallucinate states by interpolating two states.
This difference illustrates the importance of uncertainty preservation in heterogeneity analysis. Our method returns low-resolution volumes or even 0 when there is too little information to form high-resolution reconstructions. In contrast, when the network has no information; it hallucinates a volume with high-resolution features. While this phenomenon is easy to spot in this simple, high SNR dataset, it may not be as evident in real datasets with high noise levels and outliers.
Figure A.9:

(a) UMAP visualization of latent space under different principal component dimensions (d=4,12,20,50). Also shown is the embedding computed by the deep-learning method cryoDRGN [48]. Each dot corresponds to an image in both cases, and its color reflects its label published in the original analysis [6]. (b) Comparison of reconstructions by reweighting and reprojection. Some parts of the reprojected scheme appear in higher resolution, but they also show artifacts for some minor states; see also appendix A.7.
Figure A.10:

Comparison of embeddings and trajectories between cryoDRGN and our method on a dataset with noncontiguous states. (a) Embeddings for both methods overlayed with embedded ground truth and recovered trajectory. For the cryoDRGN plot, we show scatter plots of the embedded dataset over pairs of PCs of the latent space (see appendix A.8 for details). For our method, we show the estimated latent density over pairs of axes, where the density was integrated among the remaining axes. (b) Reconstructed volumes with the two methods at approximately equidistant points along the trajectory, overlayed in transparent yellow by the ground truth trajectory. The star and square are the two chosen endpoints and intermediary states, denoted in latent space with triangles for cryoDRGN and circles for our method. All volumes are visualized at the isosurface level 0.025 (ground truth outline 0.02).
Footnotes
Competing interests
The authors declare no competing interests.
We use the term “visual resolution” to refer to volume reconstructions which display visually high-resolution features, although quantifying that resolution is typically challenging; see appendix A.7 for clarification
The spatial domain variance map is displayed in fig. 1 only for illustration, the regularization is computed between columns of the covariance matrix in the Fourier domain, see appendix A.3.
We compute the real-space variance map from the low-rank form of in the Fourier domain by , that is, the squared-norm of the rows of where is the 3D inverse discrete Fourier transform matrix.
Under these assumptions, the Nyström extension would also be exact. However, we have observed empirically that it performs much worse in the presence of noise.
Notably, this iteration does not necessitate iterating through the data.
Our implementation uses scikit-fmm, see https://github.com/scikit-fmm.
Code availability
The software is available at https://github.com/ma-gilles/recovar, and will be incorporated in the ASPIRE software system [47].
Data availability
Data for synthetic experiments in section 1.5 and appendix A.8 are deposited in https://zenodo.org/records/4355284. Data for synthetic experiment in section 1.6 are deposted in https://zenodo.org/records/10030109. Experimental datasets are deposited in EMPIAR with entry ID 10076 and 10180.
References
- [1].Andén Joakim and Singer Amit. Structural variability from noisy tomographic projections. SIAM Journal on Imaging Sciences, 11(2):1441–1492, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bardi Martino, Dolcetta Italo Capuzzo, et al. Optimal control and viscosity solutions of Hamilton-Jacobi-Bellman equations, volume 12. Springer, 1997. [Google Scholar]
- [3].Chen Muyuan and Ludtke Steven J. Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Nature methods, 18(8):930–936, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen Yifan, Epperly Ethan N, Tropp Joel A, and Webber Robert J. Randomly pivoted Cholesky: Practical approximation of a kernel matrix with few entry evaluations. arXiv preprint arXiv:2207.06503, 2022. [Google Scholar]
- [5].Dashti Ali, Mashayekhi Ghoncheh, Shekhar Mrinal, Danya Ben Hail Salah Salah, Schwander Peter, Georges Amedee des, Singharoy Abhishek, Frank Joachim, and Ourmazd Abbas. Retrieving functional pathways of biomolecules from single-particle snapshots. Nature communications, 11(1):4734, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davis Joseph H, Tan Yong Zi, Carragher Bridget, Potter Clinton S, Lyumkis Dmitry, and Williamson James R. Modular assembly of the bacterial large ribosomal subunit. Cell, 167(6):1610–1622, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lieven De Lathauwer Bart De Moor, and Vandewalle Joos. A multilinear singular value decomposition. SIAM journal on Matrix Analysis and Applications, 21(4):1253–1278, 2000. [Google Scholar]
- [8].Dobriban Edgar, Leeb William, and Singer Amit. Optimal prediction in the linearly transformed spiked model. The Annals of Statistics, 48(1):491 – 513, 2020. [Google Scholar]
- [9].Doerr Allison. A dynamic direction for cryo-EM. Nature Methods, 19(1):29–29, 2022. [DOI] [PubMed] [Google Scholar]
- [10].Donnat Claire, Levy Axel, Poitevin Frederic, Zhong Ellen D, and Miolane Nina. Deep generative modeling for volume reconstruction in cryo-electron microscopy. Journal of Structural Biology, page 107920, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Edelberg Daniel G and Lederman Roy R. Using VAEs to learn latent variables: Observations on applications in cryo-EM. arXiv preprint arXiv:2303.07487, 2023. [Google Scholar]
- [12].Frank Joachim and Ourmazd Abbas. Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM. Methods, 100:61–67, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Herreros David, Lederman Roy R, Krieger James M, Jiménez-Moreno Amaya, Martínez Marta, Myška David, Strelak D, Filipovic J, Sorzano Carlos OS, and Carazo José M. Estimating conformational landscapes from cryo-EM particles by 3d Zernike polynomials. Nature Communications, 14(1):154, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jolliffe Ian T. Choosing a subset of principal components or variables. Principal component analysis, pages 111–149, 2002. [Google Scholar]
- [15].Katsevich Eugene, Katsevich Alexander, and Singer Amit. Covariance matrix estimation for the cryo-EM heterogeneity problem. SIAM journal on imaging sciences, 8(1):126–185, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kinman Laurel F, Powell Barrett M, Zhong Ellen D, Berger Bonnie, and Davis Joseph H. Uncovering structural ensembles from single-particle cryo-EM data using cryoDRGN. Nature Protocols, 18(2):319–339, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kühlbrandt Werner. The resolution revolution. Science, 343(6178):1443–1444, 2014. [DOI] [PubMed] [Google Scholar]
- [18].Kumar Sanjiv, Mohri Mehryar, and Talwalkar Ameet. Sampling methods for the Nyström method. The Journal of Machine Learning Research, 13(1):981–1006, 2012. [Google Scholar]
- [19].Lederman Roy R and Toader Bogdan. On manifold learning in Plato’s cave: Remarks on manifold learning and physical phenomena. arXiv preprint arXiv:2304.14248, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Levy Axel, Wetzstein Gordon, Julien NP Martel Frederic Poitevin, and Zhong Ellen. Amortized inference for heterogeneous reconstruction in cryo-em. Advances in Neural Information Processing Systems, 35:13038–13049, 2022. [PMC free article] [PubMed] [Google Scholar]
- [21].Mahoney Michael W and Drineas Petros. Cur matrix decompositions for improved data analysis. Proceedings of the National Academy of Sciences, 106(3):697–702, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Martinsson Per-Gunnar and Tropp Joel A. Randomized numerical linear algebra: Foundations and algorithms. Acta Numerica, 29:403–572, 2020. [Google Scholar]
- [23].Leland McInnes John Healy, and Melville James. UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426, 2018. [Google Scholar]
- [24].Moscovich Amit, Halevi Amit, Andén Joakim, and Singer Amit. Cryo-EM reconstruction of continuous heterogeneity by Laplacian spectral volumes. Inverse Problems, 36(2):024003, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakane Takanori, Kimanius Dari, Lindahl Erik, and Scheres Sjors HW. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. elife, 7:e36861, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Penczek Pawel A, Kimmel Marek, and Spahn Christian MT. Identifying conformational states of macromolecules by eigen-analysis of resampled cryo-EM images. Structure, 19(11):1582–1590, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Plaschka Clemens, Lin Pei-Chun, and Nagai Kiyoshi. Structure of a pre-catalytic spliceosome. Nature, 546(7660):617–621, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Punjani Ali, Brubaker Marcus A, and Fleet David J. Building proteins in a day: Efficient 3D molecular structure estimation with electron cryomicroscopy. IEEE Transactions on Pattern Analysis and Machine Intelligence, 39(4):706–718, 2016. [DOI] [PubMed] [Google Scholar]
- [29].Punjani Ali and Fleet David J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. Journal of structural biology, 213(2):107702, 2021. [DOI] [PubMed] [Google Scholar]
- [30].Punjani Ali and Fleet David J. 3DFlex: determining structure and motion of flexible proteins from cryo-EM. Nature Methods, pages 1–11, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Punjani Ali, Rubinstein John L, Fleet David J, and Brubaker Marcus A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature methods, 14(3):290–296, 2017. [DOI] [PubMed] [Google Scholar]
- [32].Punjani Ali, Zhang Haowei, and Fleet David J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nature methods, 17(12):1214–1221, 2020. [DOI] [PubMed] [Google Scholar]
- [33].Rosenthal Peter B and Henderson Richard. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. Journal of molecular biology, 333(4):721–745, 2003. [DOI] [PubMed] [Google Scholar]
- [34].Scheres Sjors HW. A Bayesian view on cryo-EM structure determination. Journal of molecular biology, 415(2):406–418, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Scheres Sjors HW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of structural biology, 180(3):519–530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Scheres Sjors HW, Valle Mikel, Grob Patricia, Nogales Eva, and Carazo José-María. Maximum likelihood refinement of electron microscopy data with normalization errors. Journal of structural biology, 166(2):234–240, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Seitz Evan, Acosta-Reyes Francisco, Maji Suvrajit, Schwander Peter, and Frank Joachim. Geometric machine learning informed by ground truth: Recovery of conformational continuum from single-particle cryo-EM data of biomolecules. BioRxiv, pages 2021–06, 2021. [Google Scholar]
- [38].Sethian James A et al. Level set methods and fast marching methods, volume 98. Cambridge Cambridge UP, 1999. [Google Scholar]
- [39].Shi Yunpeng and Singer Amit. Ab-initio contrast estimation and denoising of cryo-EM images. Computer Methods and Programs in Biomedicine, 224:107018, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sorzano C. O. S., Jiménez A., Mota J., Vilas J. L., Maluenda D., Martínez M., Ramírez-Aportela E., Majtner T., Segura J., Sánchez-García R., Rancel Y., del Caño L., Conesa P., Melero R., Jonic S., Vargas J., Cazals F., Freyberg Z., Krieger J., Bahar I., Marabini R., and Carazo J. M.. Survey of the analysis of continuous conformational variability of biological macromolecules by electron microscopy. Acta Crystallographica Section F, 75(1):19–32, Jan 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Oscar Carlos Sorzano S and Carazo Jose Maria. Principal component analysis is limited to low-resolution analysis in cryoEM. Acta Crystallographica Section D: Structural Biology, 77(6):835–839, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tagare Hemant D, Kucukelbir Alp, Sigworth Fred J, Wang Hongwei, and Rao Murali. Directly reconstructing principal components of heterogeneous particles from cryo-EM images. Journal of structural biology, 191(2):245–262, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Toader Bogdan, Sigworth Fred J, and Lederman Roy R. Methods for cryo-EM single particle reconstruction of macromolecules having continuous heterogeneity. Journal of Molecular Biology, 435(9):168020, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vuillemot Rémi, Miyashita Osamu, Tama Florence, Rouiller Isabelle, and Jonic Slavica. NMMD: Efficient cryo-EM flexible fitting based on simultaneous normal mode and molecular dynamics atomic displacements. Journal of Molecular Biology, 434(7):167483, 2022. [DOI] [PubMed] [Google Scholar]
- [45].Warshamanage Rangana, Yamashita Keitaro, and Murshudov Garib N. EMDA: A Python package for electron microscopy data analysis. Journal of Structural Biology, page 107826, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Williams Christopher and Seeger Matthias. Using the Nyström method to speed up kernel machines. Advances in neural information processing systems, 13, 2000. [Google Scholar]
- [47].Wright Garrett, Andén Joakim, Bansal Vineet, Xia Junchao, Langfield Chris, Carmichael Josh, Sowattanangkul Kris, Brook Robbie, Shi Yunpeng, Heimowitz Ayelet, Pragier Gabi, Sason Itay, Moscovich Amit, Shkolnisky Yoel, and Singer Amit. Computationalcryoem/aspire-python: v0.12.0 10.5281/zenodo.5657281, September 2023. [DOI] [Google Scholar]
- [48].Ellen D Zhong Tristan Bepler, Berger Bonnie, and Davis Joseph H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nature methods, 18(2):176–185, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhong Ellen D, Lerer Adam, Davis Joseph H, and Berger Bonnie. CryoDRGN2: Ab initio neural reconstruction of 3d protein structures from real cryo-EM images. In Proceedings of the IEEE/CVF International Conference on Computer Vision, pages 4066–4075, 2021. [Google Scholar]
- [50].Zou Hui, Hastie Trevor, and Tibshirani Robert. Sparse principal component analysis. Journal of computational and graphical statistics, 15(2):265–286, 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The software is available at https://github.com/ma-gilles/recovar, and will be incorporated in the ASPIRE software system [47].
Data for synthetic experiments in section 1.5 and appendix A.8 are deposited in https://zenodo.org/records/4355284. Data for synthetic experiment in section 1.6 are deposted in https://zenodo.org/records/10030109. Experimental datasets are deposited in EMPIAR with entry ID 10076 and 10180.