

Abstract
Canonical chemokine receptor CXCR4 and atypical receptor ACKR3 both respond to CXCL12 but induce different intracellular effector responses to regulate cell migration: CXCR4 couples to G proteins and arrestins, while ACKR3 is arrestin-biased. CXCR4 also signals only in response to CXCL12, whereas ACKR3 recruits β-arrestin in response to CXCL12, CXCL12 variants, and other peptides and proteins. To investigate the role of conformational dynamics in the distinct pharmacological behaviors of CXCR4 and ACKR3, we utilized single-molecule FRET. The data revealed that apo CXCR4 preferentially populates a high-FRET inactive state while apo ACKR3 shows little conformational preference, consistent with its promiscuous ligand recognition and propensity for activation. Markedly different conformational landscapes of the receptors in response to ligands suggest that activation of ACKR3 may be achieved by a broader distribution of conformational states than CXCR4. The dynamic properties of ACKR3 may also underly its inability to couple to G proteins, making it arrestin-biased.
INTRODUCTION
CXCR4 is one of the most intensively studied chemokine receptors due to its role in driving cell migration in development and immune responses, and in cancer where it promotes tumor metastasis1, 2, 3, 4. As a class A G protein-coupled receptor (GPCR), CXCR4 activates inhibitory Gαi protein signaling pathways, is phosphorylated by GPCR kinases (GRKs), and recruits arrestins to directly control cell movement in response to the chemokine CXCL125. CXCR4 often works together with atypical chemokine receptor 3 (ACKR3) which indirectly influences cell movement by scavenging CXCL12 to regulate extracellular levels of the agonist and in turn, the responsiveness of CXCR46, 7 8. In the absence of ACKR3 scavenging, excessive CXCL12 stimulation of CXCR4 leads to downregulation of the receptor, resulting in profound effects on neuronal cell migration and development9, 10, 11.
In contrast to CXCR4, and with some exceptions noted12, 13, ACKR3 lacks G protein activity and instead is considered to be arrestin-biased14, 15. However, the molecular basis for its inability to couple to G proteins in most cells remains an unanswered question. Our recently determined structures of ACKR3-ligand complexes showed the expected hallmarks of GPCR activation, including “microswitch residues” in active state configurations, displacement of TM6 away from the helical bundle, and an open intracellular pocket, consistent with a receptor that should be able to activate G proteins16. Accordingly, we replaced the intracellular loops (ICLs) of ACKR3 with those of CXCR2, a canonical G protein-coupled chemokine receptor; however, these changes did not lead to G protein activation16, suggesting that the lack of coupling is not due to the absence of specific residue interactions. CXCL12 also adopts a distinct pose when bound to ACKR3 compared to CXCR4 and all other chemokines in chemokine-receptor complexes, but since small molecules induce similar biased effector responses, the chemokine pose cannot explain the lack of G protein coupling or arrestin-bias16. Having excluded other mechanisms we therefore surmised that the inability of ACKR3 to activate G proteins and its arrestin bias may be due to differences in receptor dynamics17. Consistent with this hypothesis, the ICLs observed in ACKR3-agonist complexes are disordered, which may preclude productive interactions with helix 5 of Gαi that are required for G protein coupling. The dynamic nature of ACKR3 is also suggested by its considerable constitutive activity in recruiting β-arrestins and possibly by its high level of constitutive internalization16, 18, 19.
In addition to their distinct effector interactions, ACKR3 and CXCR4 have dramatically different susceptibilities to activation by different ligands. CXCR4 is activated by a single chemokine agonist, CXCL12. Moreover, modifications of the CXCL12 N-terminal signaling domain (e.g. single point mutations as in CXCL12P2G or multiple mutations as in CXCL12LRHQ20, 21) transform the chemokine into an antagonist. Consistent with these observations, mutational analysis and modeling of CXCR422, 23, 24 suggest that ligand activation involves a precise network of interacting residues that stabilize the active receptor conformation. By contrast ACKR3 is activated by CXCL12P2G and CXCL12LRHQ20, 21, other chemokines (CXCL11)25, other proteins (adrenomedullin, BAM22)26, 27 as well as opioid peptides28. In fact, most ligands are agonists, which is best explained by a non-specific distortion mechanism of activation and consistent with the different binding poses of a small molecule agonist compared to the CXCL12 N-terminus in the receptor orthosteric pocket16. Thus we hypothesize that receptor conformational dynamics may also play a role in the promiscuous ligand recognition and more activation-prone nature of ACKR3 compared to CXCR4.
To investigate the role of conformational dynamics in the distinct pharmacological behaviors of ACKR3 and CXCR4, we developed a single-molecule Forster resonance energy transfer (smFRET) approach. Many ensemble methods such as EPR, NMR, and fluorescence-based methods have provided considerable insights into GPCR dynamics, conformational heterogeneity and exchange between different states29, 30, 31, 32, 33, 34, 35. However, these methods are limited in their ability to resolve the sequence of state transitions, except in rare cases where transitions can be coordinated throughout the sample36. By contrast smFRET enables detection of sparsely populated states, reveals the sequence of state transitions and provides kinetic information through analysis of state dwell times. For example, smFRET studies of the β2 adrenergic receptor (β2AR) revealed a dynamic equilibrium between inactive and active conformations that was responsive to agonist and G protein binding37. More recent smFRET studies of the glucagon receptor38 and the A2A receptor (A2AR)39, 40 have revealed the existence of stable intermediate conformations in addition to inactive and active receptor conformations. Similar to these studies, our system allows real-time observation of the conformational fluctuations of individual receptors in a native-like lipid environment, and assessment of the differences in the conformational dynamics of CXCR4 and ACKR3 in their apo states and in response to ligands.
Our results indicate that ACKR3 is indeed more dynamic and conformationally heterogeneous than CXCR4, which may explain its activation prone nature and lack of G protein coupling. Four FRET states corresponding to inactive, active and two intermediate receptor conformations, all responsive to ligand were observed for apo ACKR3. The ligand-induced stabilization of not one, but two, active-like states, is consistent with a flexible mechanism of activation. By contrast and consistent with a more restricted, structurally-defined activation mechanism, CXCR4 primarily occupied an inactive high-FRET conformation with limited transitions and only a single non-responsive intermediate. Together, these data characterize the differences between ACKR3 and CXCR4 and suggest that conformational dynamics play an important role in the atypical function of ACKR3.
RESULTS
Development of smFRET experimental systems for CXCR4 and ACKR3.
In order to visualize conformational fluctuations of CXCR4 and ACKR3 by smFRET, cysteine residues were introduced into CXCR4 at positions 1504.40 in TM4 and 2336.29 in TM6, and at 1594.40 and 2456.28 in ACKR3 (numbers in superscript refer to Ballesteros-Weinstein numbering scheme for GPCRs) for covalent labeling with FRET donor (Alexa Fluor 555, A555) and acceptor (Cyanine5, Cy5). Crystal and cryo-EM structures of homologous class A GPCRs, such as β2AR, in inactive and active conformations, reveal that TM6 moves outwards from the TM helical bundle during activation, whereas the position of TM4 remains relatively fixed41. Accordingly, we anticipated that labeling at the indicated positions would be sensitive to transitions between inactive and active receptor conformations and give rise to different donor-acceptor distances and FRET efficiencies that could be resolved by smFRET measurements (Fig. 1). These positions are similar to those used effectively for β2AR37 and A2AAR39 for monitoring their conformational dynamics. Importantly, neither wild-type (WT) CXCR4 nor ACKR3 exhibited significant labeling, and the double cysteine receptor mutants retained the ability to recruit β-arrestin2 (Supplementary Fig. 1). Single receptor molecules of labeled CXCR4 or ACKR3 were then reconstituted into phospholipid nanodiscs to mimic their native membrane bilayer environment, and subsequently tethered to a quartz slide through biotinylation of the nanodisc membrane scaffolding protein (MSP) (Fig. 1). To restrict receptor incorporation to one per nanodisc, we utilized MSP1E3D1, which forms nanodiscs with a diameter of approximately 13 nm42, 43.
Figure 1:
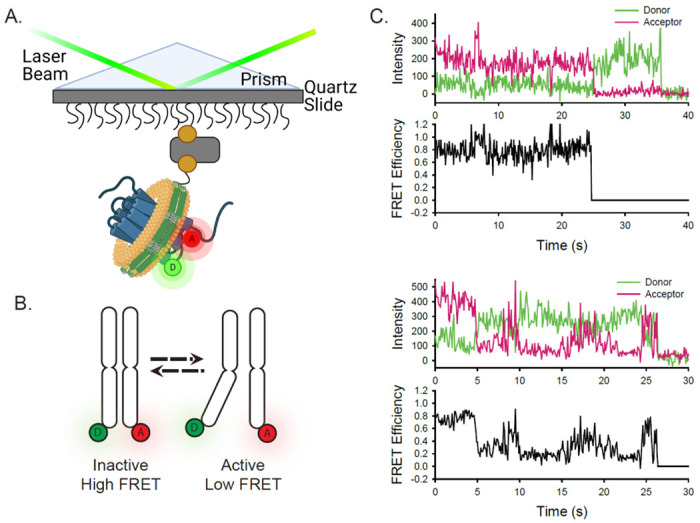
Experimental design and concept of the smFRET system. A) A single receptor molecule (blue) was labeled with donor (D) and acceptor (A) fluorophores, inserted into a phospholipid (yellow) nanodisc (green), and immobilized on a quartz slide. A prism facilitates total internal reflection of the excitation laser to excite only donor fluorophores close to the surface. B) Cartoon depicting inactive (left) and active (right) receptor conformations. C) Two representative single-molecule time traces for apo ACKR3. In each case, the donor (green) and acceptor (red) intensities are shown at the top and the corresponding FRET efficiency (black) is shown below.
ACKR3 exhibits greater conformational dynamics than CXCR4.
Receptor-nanodisc complexes were imaged on the slide surface using smFRET microscopy by exciting the A555 donor with a green (532 nm) laser and monitoring the resulting emission from both A555 and the Cy5 acceptor over time on separate segments of a CCD camera (shown schematically in Fig. 1A). Several hundred individual receptor-nanodisc complexes were typically observed in the field of view. Time traces of donor intensity, acceptor intensity, and the corresponding FRET efficiency from two representative ACKR3-nanodisc complexes are shown in Fig. 1B. These traces displayed single-step photobleaching transitions (abrupt loss of acceptor fluorescence signal or simultaneous loss of fluorescence signal in all channels), confirming that a single A555 donor and a single Cy5 acceptor were present. Anti-correlated changes in donor and acceptor emission prior to photobleaching confirmed that FRET occurred between A555 and Cy5 (Fig. 1C). Additional examples of single-molecule traces for ACKR3 and CXCR4 are shown in Supplementary Fig. 2.
Similar data from many individual receptor-nanodisc complexes, recorded in the presence or absence of different ligands, were globally analyzed using Hidden Markov Models (HMMs) assuming the presence of two, three, four, or five FRET states (Materials and Methods). To evaluate the appropriate level of model complexity, each distribution was fit with a Gaussian Mixture Model (GMM) and the corresponding Bayesian Information Criterion (BIC) was calculated. The BIC is a statistical measure of the likelihood that the model describes the data while penalizing addition of fitting parameters which could lead to overfitting of the noise. Theoretically, this value will be at a minimum for the model with the appropriate number of states. For ACKR3 in the apo and inverse agonist-bound distributions there is a clear minimum at 4 states (Supplementary Fig. 3). The CXCL12WT bound distribution shows only a modest decrease in BIC with the addition of a 5th state. Moreover, the resulting distributions for the intermediate FRET state (R’) in the three-state analysis exhibited substantial differences in peak position when the samples were challenged with CXCL12WT agonist or the inverse agonist VUF16840 (Supplementary Fig. 4)44. In contrast, the peak positions for the two intermediate states (R*’ and R’) in the four-state model were conserved across all conditions. We therefore conclude that ACKR3 is appropriately described by four distinct conformational states.
For CXCR4, GMM analysis indicates that three states are appropriate to describe the data, with a clear minimum in the BIC for the apo and inverse agonist-bound distributions and a modest decrease in BIC for an additional state in the CXCL12WT-bound distribution (Supplementary Fig. 5). The distributions for the CXCR4 intermediate state (R’) resulting from the three-state analysis showed similar peak positions for each sample condition (Supplementary Fig. 6). Thus, we conclude that three FRET states are present in the CXCR4 distribution.
The high-FRET states (E = 0.85) observed in both ACKR3 and CXCR4 suggest that TM4 and 6 are in close physical proximity (Figs. 2A & B), which is consistent with an inactive GPCR conformation45, 46. Accordingly, we interpret this FRET state as the inactive conformation, and designate it as R. Addition of the CXCL12WT agonist to both receptors strongly favored low-FRET states (E = 0.11 for ACKR3 and E = 0.19 for CXCR4) at the expense of high-FRET states (Figs. 2C & D). The decrease in FRET efficiency corresponds to an outward movement of TM6 away from TM4, reflecting active receptor conformations (designated R*)41, 47. The stabilization of active receptor conformations in response to agonist addition is consistent with other ACKR3 structural studies44, 48.
Figure 2:
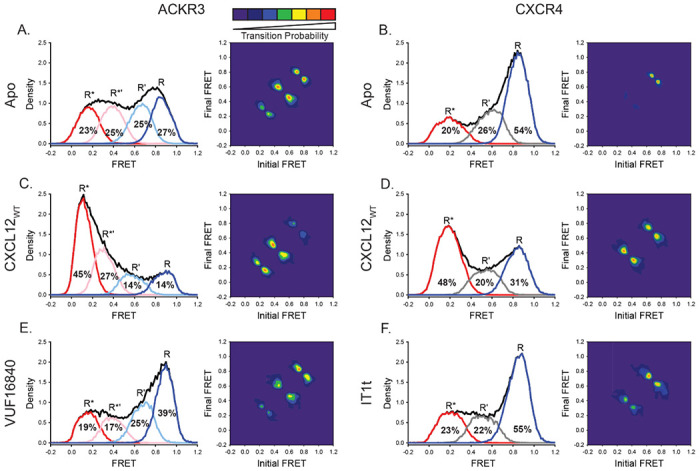
ACKR3 exhibited greater conformational flexibility compared to CXCR4. A) Apo ACKR3 showed little conformational preference across the four modeled FRET states (active R*, active-like intermediate R*’, inactive-like intermediate R’ and inactive R). Transition density probability (TDP) plot for apo ACKR3 displayed the probability of transitions from an initial FRET state (x-axis) to a final FRET state (y-axis). B) FRET populations and TDP for apo CXCR4 reveal a mostly inactive receptor population (R) with few transitions. C) Addition of CXCL12 to ACKR3 shifted the population distribution to low-FRET, R* and R*’ conformations, which was also reflected in the transition probabilities. D) CXCR4 activation by CXCL12 led to a conformational shift to low-FRET, R* state and more transitions between all three FRET states. E) Treatment of ACKR3 with VUF16480, an inverse agonist, shifted the conformational population and TDPs to high-FRET, inactive conformations. F) The CXCR4 antagonist IT1t had little impact on the FRET distribution (compared to 2B), but increased transition probabilities compared to apo receptor. Data sets represent the analysis of at least three independent experiments. The total FRET envelopes for the samples are represented by the black trace.
To gain insight into the nature of the two mid-FRET states (E = 0.39 and E = 0.66) in ACKR3 (Fig. 2A), we examined transitions between the various FRET states. Two-dimensional transition density probability (TDP) plots reveal that the four FRET states in ACKR3 were connected in a sequential fashion (Fig. 2A). Notably, non-sequential transitions that “skip over” an intervening FRET state were rarely observed. These results suggest that the mid-FRET states represent two intermediate receptor conformations that lie on the pathway between inactive and active conformations. Thus, we assigned the E = 0.66 FRET state to an inactive-like intermediate (R’) and the E = 0.39 FRET state to an active-like intermediate (R*’). The TDP indicated that all possible sequential transitions occurred in the apo receptor, with R’↔R*’ transitions being the most frequent, followed by R’↔R (Fig. 2A). However, in the presence of CXCL12WT, the TDP showed greater probability for transitions between R*’ and R* (Fig. 2C), consistent with the population shifts observed in the FRET efficiency histograms. Similar results were observed for CXCL11, the other native ACKR3 chemokine agonist, and VUF15485, a small molecule agonist49 (Supplementary Fig. 7). Treatment of ACKR3 with the small molecule inverse agonist, VUF16840, shifted the conformational distribution to the inactive R conformation with concomitant decreases of R* and R*’ (Fig. 2E), consistent with the suppression of the basal activity of ACKR316. This suppression was also evident in the TDP, which revealed higher probability transitions between R’ and R conformations and fewer transitions between R*’ and R* conformations (Fig. 2E).
In contrast to ACKR3, CXCR4 showed a preference for the inactive R state in the absence of ligands and only a single intermediate (R’, E = 0.59) conformation (Fig. 2C). Transitions among states R’ and R* were less probable compared to ACKR3 in the absence of ligands (Figs. 2A & B), suggesting that CXCR4 is intrinsically less dynamic than ACKR3. However, transitions among states R’ and R* were more probable when CXCL12WT was present (Fig. 2D), suggesting that agonist binding enhanced CXCR4 dynamics towards the active state. Surprisingly, a small molecule CXCR4 inhibitor (IT1t) had little effect on the conformational distribution of the receptor, despite previous reports of the molecule acting as an inverse agonist (Fig. 2F)50, 51, 52. However, IT1t decreased the probabilities of the R’ and R* state-to-state transitions relative to CXCL12WT perhaps indicating it is more of a neutral antagonist than an inverse agonist.
In summary, the smFRET analyses revealed that ACKR3 and CXCR4 are both conformationally heterogeneous. ACKR3 showed more conformational heterogeneity than CXCR4, populating inactive, active and two intermediate conformations, with little preference for any particular conformation in the absence of ligands. The conformational plasticity is consistent with previous reports of constitutive activity16, 19 (Fig. 2A). In contrast, CXCR4 showed a preference for an inactive conformation in the absence of ligands and exhibited just a single intermediate conformation. Nevertheless, despite these differences, both receptors respond to their shared chemokine agonist CXCL12WT, revealing substantial population shifts towards active or active-like conformations.
CXCL12 N-terminal mutants promote open receptor conformations despite their contrasting pharmacological effects on ACKR3 and CXCR4.
CXCR4 is sensitive to N-terminal mutations of CXCL12 while ACKR3 is relatively insensitive. For example, the variant CXCL12P2G, containing a proline to glycine mutation in the second position, and CXCL12LRHQ, where the first three residues of CXCL12WT were replaced with the four residue motif LRHQ starting with L0, are antagonists of CXCR4 but agonists of ACKR320, 21. To gain further insight into the ligand-dependent responses of these receptors, we examined how these mutant chemokines influence the conformational states and dynamics of CXCR4 and ACKR3.
CXCL12P2G increased the population of the active R* conformation of ACKR3 relative to the apo receptor by 5% and also promoted formation of the intermediate R*’ conformations (Figs. 3A & B, Supplementary Fig. 8), rather than the predominantly stabilizing the R* state as observed for CXCL12WT (Fig. 2C). This suggests that activation of ACKR3 can be achieved by populating intermediate states, not just the R* state, which is consistent with a flexible, distortion mechanism. CXCL12LRHQ also promoted the active R* conformation of ACKR3, although to a lesser extent than CXCL12WT (Figs. 2C & 3C). Additionally, transitions to the active R*’and R* conformations of ACKR3 appeared to be more likely in the presence of CXCL12LRHQ compared to CXCL12P2G (Figs. 3B & C), which could be a consequence of its longer residence time on the receptor53.
Figure 3:

CXCL12 variants containing mutations to the N-terminus promoted low FRET active states in ACKR3 and CXCR4. A) Apo receptor FRET distributions and TDPs for ACKR3 repeated from Fig. 2A for reference. B) Treatment of ACKR3 with CXCL12P2G displayed a shift to low-FRET, R*’ and R* states while suppressing the transitional probabilities relative to the apo receptor. C) CXCL12LRHQ treatment of ACKR3 shifted the FRET distribution to low-FRET states and transitions. D) FRET distribution and TDP for Apo CXCR4 repeated from Fig. 2B. E) Addition of CXCL12P2G to CXCR4 promoted a shift to the low-FRET (R*) conformation and increase in transition probabilities. F) CXCL12LRHQ led to a subtle shift to more R* CXCR4 FRET states without affecting the transition probabilities. Data sets represent the analysis of at least three independent experiments. The total FRET envelopes for the samples are represented by the black trace.
Surprisingly, despite acting as a CXCR4 antagonist21, CXCL12P2G also promoted a shift to the active R* conformation of CXCR4 compared to the apo receptor (R* increased by 16%, Supplementary Fig. 8); in fact, the shift was more pronounced than for ACKR3, whereas CXCL12P2G is pharmacologically activating but only produces a 5% increase in R* (11% in R* + R*’) (Fig. 3). The shift is also evident in the TDPs where the state-to-state transitions, especially involving the R* active state, were more probable for the CXCL12P2G complex compared with apo CXCR4 as well as for ACKR3 bound to CXCL12P2G (Fig. 3). CXCL12LRHQ also promoted the R* active conformation of CXCR4 (R* increased by 9%), although to a lesser extent than CXCL12P2G (16%) and far less than CXCL12WT (28%) (Figs. 3E & F, 2D, Supplementary Fig. 8). T he probabilities of state-to-state transitions were also unchanged relative to the apo receptor (Figs. 3D & F). However in the presence of either mutant chemokine, the overall CXCR4 conformational state remained predominantly inactive, consistent with the pharmacological antagonism observed for these variants. Together the data suggest that while both ligands produce an apparent opening of the intracellular cleft but do not promote CXCR4 coupling to G proteins or other effectors, the intracellular cleft is not sufficient to drive G protein activation of CXCR4. Instead a precise network of interacting residues is required to stabilize a productive conformation capable of signaling24.
ACKR3 constitutive activity is linked to receptor conformational heterogeneity
Apo ACKR3 displays little conformational selectivity, with similar occupancies observed for all four conformational states (Fig. 2A). We hypothesized that this might be related to the constitutive activity of the receptor13, 16 and tied to the presence of Tyr at position 2576.40, which is a hydrophobic residue in all other chemokine receptors (V, I, or L). In many other class A GPCRs, mutating the residue at 6.40 results in constitutive activity54, 55, 56 and previous analysis of rhodopsin suggests that this is a consequence of lowering the energy barrier between different receptor conformations57. In our previous work, we showed that Y2576.40L reduces basal arrestin recruitment to ACKR3 and acts as a constitutively inactivating mutation (CIM), while preserving the ability of the receptor to be activated by CXCL12 (Supplementary Fig. 9)16. Adding this single mutation to our ACKR3 smFRET construct converted the broad conformational distribution of the wild-type apo receptor (Fig. 4B) to a narrower distribution concentrated in the high-FRET states (Fig. 4C), similar to what we observe for apo CXCR4 (Fig. 2B). State-to-state transitions were also suppressed relative to WT ACKR3 (Fig. 4B & C). This suggests that Y2576.40 contributes to the ability of ACKR3 to be constitutively active.
Figure 4:

Mutating Y2576.40L in ACKR3 led to a loss of constitutive activity and reduces conformational heterogeneity. A) Structure of ACKR3 bound with CXCL12WT (PDBID: 7SK3) highlighting the location of Y2576.40.16 B, C) FRET distributions and TPDs of apo and CXLC12 treated WT ACKR3 repeated from Fig. 2A and C. D) The mutation Y2576.40L shifted the conformational landscape of the apo receptor to mostly high-FRET, inactive-like conformations and lowered the probability of state-to-state transitions. E) Treatment of Y2576.40L ACKR3 with 500 nM CXCL12 promoted greater low-FRET active states. Data sets represent the analysis of at least three independent experiments. The total FRET envelopes for the samples are represented by the black trace.
The effectiveness of CXCL12WT in promoting an active (R*) receptor conformation was substantially less for Y2576.40L compared to the WT receptor (Figs. 4C & E); however, the mutant receptor is reported to still recruit β-arrestin in response to CXCL12WT, with an Emax value that was only slightly reduced relative to the WT receptor (Supplementary Fig. 9)16, suggesting again that the population of intermediate conformational states may contribute to receptor activation.
Point mutations in intracellular loop 3 of ACKR3 decouple CXCL12-induced receptor activation from outward movement of helix 6
While searching for appropriate labeling sites, we observed two positions with unusual properties in intracellular loop 3 (ICL3) of ACKR3. Mutation of the Asp at position 2446.27 to an Ala (D2446.27A) greatly increased basal arrestin association (Fig. 5B), while preserving the robust arrestin response with CXCL12WT treatment (Fig. 5C). A second mutation, which swapped the charge at the so-called ionic lock residue at position 6.3058 (K2476.30E), acted as a CIM and suppressed both basal arrestin recruitment as well as the response to CXCL12WT (Figs. 5B & C). To gain insight into the mechanisms of these mutations, the conformational landscape of ACKR3 was probed by smFRET. For D2446.27A, the overall FRET envelope was shifted to low-FRET R* and R*’ populations (67% D2446.27A, Fig. 5D, vs 48% for WT, Fig. 2A), as might be expected given its greater constitutive activity (Fig. 5D). In the case of K2476.30E, the overall FRET envelope was generally similar to that of WT ACKR3, with little conformational preference (Fig. 5E). However, the R* state appeared more frequently than one might expect for an inactivating mutation when compared to WT ACKR3 (28% for K2476.30E vs 23% for WT ACKR3). Unexpectedly, treatment of either ACKR3 mutant with CXCL12WT lead to only minor changes to the FRET distributions (Figs. 5F & G), even though the mutants are pharmacologically responsive and recruit β-arrestin. These data suggest that receptor activation may result from conformational changes such as helical movements or side chain rearrangements undetectable using our smFRET constructs, consistent with a more non-specific activation mechanism.
Figure 5:
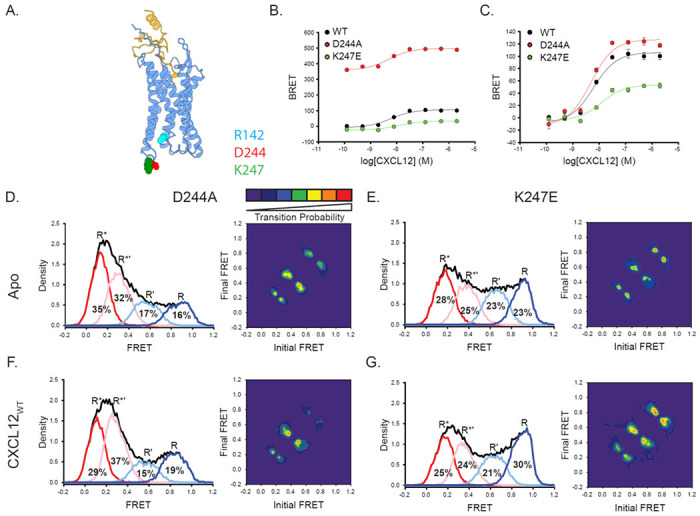
Mutations in ICL3 of ACKR3 affect the conformational state of the receptor leading to constitutive activating or inactivating effects. A) Structure of ACKR3 with CXCL12 (PDBID 7SK3) highlighting the mutation positions of D2446.27 and K2476.30 as well as the counterion in the “ionic lock” motif, R1423.50.16 B, C) β-arrestin2 recruitment to WT, D2446.27A, and K2476.30E ACKR3 constructs reported as a percentage of WT ACKR3 BRET (B) or baseline corrected (C). D) Apo, D2446.27A ACKR3 showed a shifted FRET distribution and TDPs to predominantly low-FRET states. D) K2476.30E ACKR3 showed little difference FRET distribution compared to WT, although the transition probabilities were reduced. F, G) CXCL12 treatment did not alter the FRET distributions of D2446.27A (F) nor K2476.30E (G) compared to the apo receptors; however, the probability of transitions increased. The WT ACKR3 data from B and C was adapted in part from Gustavsson et al. 201953. Data sets represent the analysis of at least three independent experiments. The total FRET envelopes for the samples are represented by the black trace.
DISCUSSION
Since structures of ACKR3 showed evidence of intracellular loop disorder and progressive structural substitutions failed to promote G protein coupling16, we recently proposed that the atypical behavior of ACKR3 may be related to receptor conformational dynamics16, 17. Consistent with this hypothesis, in the present study we found that the conformational landscapes of ACKR3 and the canonical GPCR CXCR4 are markedly different. Our smFRET studies revealed four distinct conformations of apo ACKR3 with approximately equal populations corresponding to inactive R, active R* and two intermediate receptor conformations, R*’ and R’. The state-to-state transition density probability plots further reinforced that ACKR3 is a flexible receptor that readily exchanges between different conformational states, consistent with our structural studies where a flexible intracellular interface in the absence of interaction partners was observed16. By contrast, the smFRET results reveal that apo CXCR4 is more conformationally static and primarily populates an inactive high-FRET conformation with limited transitions and only a single intermediate. Thus, ACKR3 must have a relatively flat energy landscape, with similar local minima and low energy barriers between states (Fig. 6), which is consistent with a dynamic receptor that is constitutively active and readily activated. Moreover, the flat energy landscape of ACKR3 may contribute to its ligand promiscuity and lack of G protein activation. In contrast, CXCR4 has a more rugged landscape with deeper valleys and higher barriers (Fig. 6).
Figure 6:
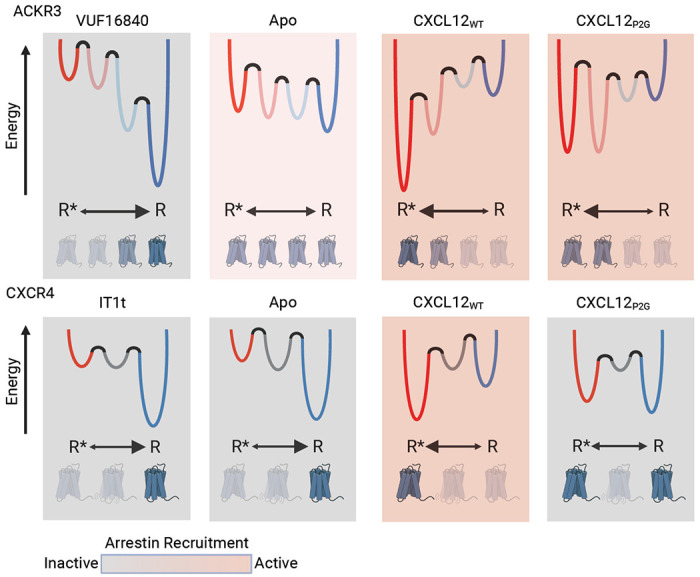
Modeled conformational energy landscape for ACKR3 and CXCR4 highlights the differences in the responsiveness of the two receptors to ligands. ACKR3 is shows little conformational preference in the apo form. While inverse agonist, VUF16840, treatment shifts the population to the inactive R conformation, the agonists CXCL12WT and CXCL12P2G promote the R* and R*’ populations. Despite stabilizing different levels of the active-intermediates, both CXCL12WT variants are pharmacologically active for ACKR3. The flexibility of ACKR3 might explain the promiscuity of this atypical receptor. In contrast, CXCR4 is predominantly inactive, R state, in the apo form and less efficiently converted to R* with CXCL12WT treatment, whereas IT1t had little impact. Though CXCL12P2G is pharmacologically inactive, the ligand promoted a detectable shift to the R* state for the canonical receptor, suggesting TM6 movement is not sufficient for CXCR4 activation.
Two other class A GPCRs have been studied by smFRET, β2AR37 and the A2A adenosine receptor39. These receptors were labeled with donor and acceptor probes within TM4 and TM6, as we have done here for ACKR3 and CXCR4. Inactive and active conformations of β2AR and A2A were resolved as separate FRET states, although in the case of β2AR, G proteins were required to substantially stabilize the active conformation. In addition, a stable intermediate conformation was resolved in the A2A receptor39. Recent smFRET studies of the class C glucagon receptor have also revealed inactive and active conformations, as well as a single intermediate conformation38. These observations for A2A and the glucagon receptor echo the behavior we have observed for CXCR4. In contrast, we resolved two intermediate conformational states (R’ and R*’) in ACKR3 that were both responsive to ligand treatments. The two intermediates appear to be unique to ACKR3 and may be integral to its atypical behavior.
The ACKR3 agonists CXCL12P2G and VUF15485 populated the R*’ as well as the R* state of ACKR3, reflecting a broad distribution of conformations that are permissive for arrestin recruitment (Fig. 6). By contrast, the single intermediate R’ state of CXCR4 was unaffected by ligand treatment, unlike the responsive intermediate observed with A2A and glucagon receptors38, 39. Moreover, CXCL12P2G shifted CXCR4 to the R* state, yet this variant is an antagonist of CXCR4 and an agonist of ACKR3. These data are consistent with a non-specific distortion mechanism where multiple conformations lead to activation of ACKR3 versus a more conformationally specific mechanism for CXCR4. Cryo-EM structures of ACKR3 with CXCL12 and with the small molecule agonist CCX662 support a distortion mechanism for this receptor16. Although their binding poses overlap, CCX662 reaches deeper into the binding pocket and makes different interactions than the N-terminus of CXCL12 (Supplementary Fig. 10A); it also induces a 1-2 Å outward shift of TM5 (residues 212-219) relative to the CXCL12 complex16. The loose structural requirements for activation of ACKR3 contrast with the strict requirements for CXCR4 activation by CXCL12 suggested by experimentally enhanced models and extensive mutagenesis (Supplementary Fig. 10B & C)22, 23, 24.The less specific nature of the distortion mechanism may also explain why antagonizing ACKR3 through targeting the orthosteric pocket has proven to be challenging and suggests allosteric strategies may be required.
Structural features that make ACKR3 conformationally flexible include Y2576.40, which we previously showed contributes to the constitutive activity of ACKR316. As for a constitutively active M2576.40Y mutant of rhodopsin, Y6.40 in ACKR3 stacks against Y5.58, and Y7.53 and helps coordinate the active-like conformation32,59. Additionally, the broad conformational distribution and high probability of state-to-state transitions of ACKR3 parallel the dramatically lower energy barrier of the M2576.40Y rhodopsin mutant relative to WT rhodopsin57. By contrast, mutation of Y2576.40 to Leu, the corresponding amino acid in CXCR4, promoted the dominance of inactive high-FRET receptor conformations similar to CXCR4; it also reduced the transition probabilities and the ability of ACKR3 to constitutively recruit arrestin16.
In addition to the contribution of Y2576.40 to the dynamic behavior of ACKR3, a disulfide bond between Cys34 in the receptor N-terminus and Cys287 in extracellular loop 3 (ECL3), observed in all other reported chemokine receptor:chemokine structures60, 61, 62 is conspicuously missing in cryo-EM structures of ACKR3 with chemokine and small molecule agonists16. The disulfide might constrain the relative positions of TM1 and TM6/7 and the opening of the orthosteric pocket. Therefore, its absence may confer ACKR3 with greater conformational flexibility than other chemokine receptors, consistent with our observations, and may allow it to recognize and be activated by diverse ligands.
Why ACKR3 does not couple to G proteins, at least in most cells, is not entirely clear. But it does not appear to be a consequence of any obvious sequence differences since insertion of a DRY box motif and substitution of all ICLs from a canonical GPCR failed to confer G protein activity16. Based on docking of G proteins to static structures of ACKR3, it is possible that a steric clash between the G protein helix 5 and ICL2 and/or ICL3 of the receptor prevents coupling as previously proposed16. Alternatively, the flexible dynamics and conformational transitions revealed here could prevent formation of productive contacts between ACKR3 and G protein that lead to coupling, even though G proteins appear to constitutively associate with the receptor13, 16, 63. Lack of a well-organized intracellular pocket due to frequent conformational transitions may also be the underlying reason that the fingerloop of arrestin fails to bind to the pocket, as observed for other GPCRs64, 65, but instead inserts into membranes/micelles adjacent to the receptor17. Nevertheless, arrestins are still recruited to ACKR3 because GRKs phosphorylate the receptor C-terminal tail in response to CXCL12; in turn, the phosphorylated C-tail provides docking sites for direct interaction of arrestins that are reinforced by arrestin fingerloop interactions with the membrane. Since GRKs interact with the cytoplasmic pocket to facilitate phosphorylation, it is yet unclear how dynamics might decouple G protein activation and arrestin binding to the receptor cytoplasmic pocket17, but support pocket-mediated GRK activity. However, given the fleeting interaction between GRKs and GPCRs66, rapid state sampling by ACKR3 may not necessarily be a detriment to GRK engagement and phosphorylation. Additional studies will be required to further assess whether and how dynamics, sterics, or a combination of both prevent canonical G protein and arrestin interactions with the ACKR3 intracellular pocket while being permissive to GRK interactions. To this end, recent rhodopsin complex structures reveal a larger TM4-TM6 distance for GRK (35.9 Å) engagement than for G protein (32.6 Å) or arrestin (33.4 Å) (distances measured between Ca atoms of N151 4.40 and K2456.28)67, 68, 69.
In summary, the conformational energy landscape of ACKR3 is flatter and has more local minima reflecting its greater conformational heterogeneity than CXCR4 or other previously studied GPCRs37, 38, 40. The structural heterogeneity is consistent with the ability of ACKR3 to bind and be activated by different ligands via a distortion mechanism, and the fact that most ligands are activating rather than inhibiting. ACKR3 resembles the human cytomegalovirus chemokine receptor US28, which also recognizes diverse chemokines and constitutively internalizes. It has been proposed to respond to distortion of the orthosteric binding pocket rather than by specific side chain contacts between receptor and ligand70, 71, similar to that we propose here for ACKR3. On the other hand, US28 not only activates Gai and Gaq in response to diverse chemokines but it constitutively activates G proteins72. Whether it has a relatively flat energy landscape and/or more conformational states than canonical GPCRs remains to be seen. These conformational features may also be operative in other G protein-coupled chemokine receptors that respond to multiple ligands and have considerable constitutive activity such as CCR1, CCR2, and CCR360, 73.
MATERIALS AND METHODS
Unless otherwise stated all chemicals and reagents were purchased from SigmaAldrich or Fisher Scientific. Methoxy e-Coelenterazine (Prolume Purple) was purchased from Nanolight Technologies (Prolume LTD).
Cloning
ACKR3 (residues 2-362) preceded by an N-terminal HA signal sequence and followed by C-terminal 10His and FLAG tags was cloned into pFasBac vector. For cell-based assays, ACKR3 (residues 2-362) with or without an N-terminal FLAG tag was inserted into pcDNA3.1 expression vector followed C-terminally by Renilla luciferase II to form FLAG_ACKR3_rlucII and ACKR3_rlucII respectively. CXCR4 with an N-terminal FLAG tag and C-terminal 10His was inserted into pFasBac. Site-directed mutagenesis was performed by overlap extension and confirmed by Sanger sequencing.
Arrestin recruitment by BRET
Arrestin recruitment to ACKR3 and CXCR4 was detected using a BRET2 assay as previously described15, 22, 53. Briefly, HEK293T cells (ATCC) were plated at 750k/well in a 6-well dish in Dulbecco’s modified eagle media (DMEM) with 10% fetal bovine serum (FBS) and transfected 24 hrs later with 50 ng receptor DNA, 1 μg GFP10_β-arrestin2 (a kind gift from N. Heveker, Université de Montréal, Canada), and 1.4 μg empty pcDNA3.1 vector using TransIT-LT1 transfection system (MirusBio) and expressed for 40 hrs. The cells were then washed with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and mechanically lifted in Tyrode’s buffer (25 mM HEPES, 140 mM NaCl, 1 mM CaCl2, 2.7 mM KCl, 12 mM NaHCO3, 5.6 mM Glucose, 0.5 mM MgCl2, 0.37 mM NaH2PO4, pH 7.5). 100k cells were plated per 96-well white BRET plate (BD Fisher) and reattached for 45 min at 37 °C. GFP expression was checked using a SpectraMax M5 plate fluorometer (Molecular Devices) with 485 nm excitation, 538 nm emission, and 530 nm cutoff. 5 μM Prolume Purple substrate was subsequently added and total luminescence detected using a TECAN Spark Luminometer (TECAN Life Sciences) at 37 °C. CXCL12 was then added to each well at the indicated final concentrations and BRET was read using default BRET2 settings (blue emission 360-440 nm, red emission 505-575 nm) and an integration time of 0.5 sec. Experiments were baseline matched and normalized to the Emax of WT receptor. The reported data is the average of three independent experiments performed in duplicate. Points were fit to a sigmoidal dose-response model using SigmalPlot 11.0 (Systat Software, Inc).
Receptor purification, labeling, and nanodisc reconstitution
M1594.40C/Q2456.28C ACKR3 and L1504.40C/Q2336.29C CXCR4 were purified from Sf9 cells as previously described16. Briefly, Sf9 cells were infected with baculovirus (prepared using Bac-to-Bac Baculovirus Expression System, Invitrogen) containing either the mutant ACKR3 or CXCR4. Cells were harvested after 48 hrs and membranes dounce homogenized in hypotonic buffer (10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl) followed three more times with hypotonic buffer with 1 M NaCl. The membranes were spun down at 50k x g for 30 min between each round of douncing. After the final round, membranes were incubated with 100 μM CCX662 (Chemocentryx Inc.) for ACKR3 or 100 μM IT1t for CXCR4 and solubilized in 50 mM HEPES pH 7.5, 400 mM NaCl, 0.75/0.15% dodecyl maltoside/cholesteryl hemisuccinate (DDM/CHS) with a protease inhibitor tablet (Roche) for 4 hrs. Insoluble material was then removed by centrifugation at 50k x g for 30 min and Talon resin (Clontech) with 20 mM imidazole was added to bind the expressed receptors overnight at 4 °C. The next morning, the resin was transferred to a column and washed with WB1 (50 mM HEPES pH 7.5, 400 mM NaCl, 0.1/0.02% DDM/CHS, 10% glycerol, 20 mM imidazole) followed by WB2 (WB1 with 0.025/0.005% DDM/CHS) and finally eluted with WB2 with 250 mM imidazole. The imidazole was removed by desalting column (PD MiniTrap G-25, GE Healthcare). Final protein concentration was determined by A280 using an extinction coefficient of 75000 M−1cm−1 (ACKR3) and 58850 M−1cm−1 (CXCR4), snap frozen in liquid nitrogen, and stored at −80 °C until use.
When ready to prepare samples for smFRET measurements, two nanomoles of receptor was thawed and incubated with fourteen nanomoles of Alexa Fluor 555 (A555) and Cy5 overnight at 4 °C with rotation. The next morning, free label was removed using a 100k Da cut-off spin concentrator (Amicon) and the sample concentrated to ~100 μl. Label incorporation was evaluated by measuring the absorbance at A280 (ε280 = 75000 M−1cm−1 for ACKR3 and 58850 M−1cm−1 for CXCR4), A555 (ε555 = 150000 M−1cm−1), and A645 (ε645 = 250000 M−1cm−1) to detect the concentration of the labeled receptor, A555, and Cy5, respectively. The contribution of the fluorophores to A280 was removed before determining receptor concentration. The entire sample was used for nanodisc reconstitution.
Labeled receptors were reconstituted into biotinylated MSP1E3D1 nanodiscs as previously described16. Briefly, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG, Avanti) were prepared in a 3:2 POPC:POPG ratio and solubilized in ND buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 180 mM cholate). MSP1E3D1 was expressed and purified as previously described (Ritchie 2009) and biotinylated with EZ-Link NHS-polyethylene glycol 4 (PEG4)-Biotin (Thermo Fisher) per manufacturer instructions. The receptors, MSP1E3D1, and lipids were combined at a molar ratio of 0.1:1:110 for ACKR3:MSP:lipids respectively. Additional ND buffer was added to keep the final cholate concentration >20 mM. After 30 min at 4 °C, 200 mg of Biobeads (Bio-Rad) were added and incubated for 3-6 hrs. The sample was then loaded on a Superdex 200 10/300 GL column equilibrated with 25 mM HEPES pH 7.5, 150 mM NaCl and fractions containing nanodisc complexes were combined. 200 μl Talon resin was added with 20 mM imidazole (final concentration) and the samples were incubated for 16 hrs at 4 °C. The resin was then transferred to a Micro Bio-Spin Column (Bio-Rad) and washed with 25 mM HEPES pH 7.5, 150 mM NaCl, 20 mM imidazole and eluted with 25 mM HEPES pH 7.5, 150 mM NaCl, 250 mM imidazole and buffer-exchanged using 100k Da spin concentrators to remove the imidazole. Sample were concentrated to ~1 μM and stored at 4 °C until use.
CXCL12 purification from E. Coli
CXCL12 was expressed and purified as previously described15, 16. Briefly, the chemokines were expressed by IPTG induction in BL21(DE3)pLysS cells. The cells were collected by centrifugation, resuspended in 50 mM tris pH 7.5, 150 mM NaCl and lysed by sonication. Inclusion bodies were then collected by centrifugation, resuspended in equilibration buffer (50 mM tris, 6 M guanidine-HCl pH 8.0), sonicated to release the chemokines and the samples centrifuged again to pellet insoluble material. The supernatant was then passed over a Ni-nitrilotriacetic acid (NTA) column equilibrated with equilibration buffer to bind the His-tagged chemokines. The column was washed with wash buffer (50 mM MES pH 6.0, 6 M guanidine-HCl) and eluted with 50 mM acetate pH 4.0, 6 M guanidine-HCl. The chemokine-containing elutions were pooled and dithiothreitol added to a final concentration of 4 mM. After incubating 10 min, the solution was added dropwise into refolding buffer (50 mM tris pH 7.5, 500 mM arginine-HCl, 1 mM EDTA, 1 mM oxidized glutathione) and incubated at room temperature for 4 hrs before dialyzing against 20 mM tris pH 8.0, 50 mM NaCl. To remove the N-terminal purification tag, enterokinase was added and the sample incubated at 37 °C for 5 days. Uncleaved chemokine and free tags were removed by reverse Ni-NTA and eluted with wash buffer. Finally, the sample was purified on a reverse-phase C18 column equilibrated with 75% buffer A (0.1% trifluoroacetic acid (TFA)) and 25% buffer B (0.1% TFA, 90% acetonitrile) and eluted by a linear gradient of buffer B. The pure protein was lyophilized and stored at −80 °C until use.
smFRET microscopy
smFRET experiments were performed on a custom built prism-based TIRF microscope as previously described74. Briefly, a flow cell was assembled on a quartz slide passivated with PEG-biotin and coated with neutravidin75. Labeled ACKR3 or CXCR4 in biotinylated nanodiscs was diluted into trolox buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM propyl gallate, 5 mM trolox), flowed into the sample chamber and incubated for 5 min at room temperature. The samples were then washed twice with imaging buffer (trolox buffer with 2 mM protocatechuic acid, 50 nM protocatechuate-3,4-dioxygenase). Movies were collected with 100 ms integration time using a custom single-molecule data acquisition program to control the CCD camera (Andor). Single-molecule donor and acceptor emission traces were extracted from the recordings using custom IDL (Interactive Data Language) scripts. The software packages used to control the CCD camera and extract time trajectories were provided by Dr. Taekjip Ha. In all cases, five initial apo receptor movies were recorded at different locations on the slide and then ligand was flowed into the cell by two washes with chemokine or small molecule in imaging buffer at final concentrations of 500 nM for chemokines (CXCL12WT, CXCL12P2G, CXCL12LRHQ, CXCL11) and 1 μM for the small molecules (IT1t, VUF16840, VUF15485). Ten more movies were collected for each condition. The data represented are a composite of at least three individual slides and treatments.
FRET trajectories were generated and analyzed using custom software written in-house (https://github.com/rpauszek/smtirf). Donor and acceptor traces for each molecule were corrected for donor bleed through and background signal and FRET efficiencies were calculated as E = IA/(IA+ID), where E is the apparent FRET efficiency at each time point and ID and IA are the corresponding donor and acceptor fluorophore intensities, respectively. All traces for a particular protein/ligand combination were analyzed globally by a single Hidden Markov Model assuming two, three, four, or five states and shared variance as previously described74. The Bayesian Information Criterion (BIC) was calculated as described76. Binned histograms of FRET efficiency were compiled in each case and normalized to an integrated area of 1 to obtain probability densities. Transition density probability plots were calculated as in McKinney et al. 200677 Gaussian Mixture Model analysis to determine appropriate model complexity was performed using the Python package scikit-learn (version 1.2.2).
Data availability
All relevant data supporting this study are included in the Article or Supplementary files and are available from the authors upon request.
Supplementary Material
Acknowledgements
We would like to acknowledge the initial work on this project undertaken by Chunxia Zhao and Rajan Lamichhane. Additionally, we thank Handel lab members Cheyanne Shinn, Catherina Salanga, and Nicholas Chimileski for providing the chemokines CXCL11 and CXCL12P2G, respectively. R. Leurs (Vrije Universiteit Amsterdam) for the small molecules VUF16840 and VUF15485. N. Heveker (Université de Montréal) for the β-arrestin2_GFP10 plasmid. This work was supported by R01 GM133157 (D.P.M./T.M.H), R01 CA254402 (T.M.H.), R01 AI161880 (T.M.H.), F32 GM137505 (C.T.S.), Robertson Foundation/Cancer Research Institute Irvington Postdoctoral Fellowship (M.G.), F32 GM115017 and T32 AI007354 (R.F.P.).
Footnotes
Competing Interests
T.M.H. is a cofounder of Lassogen Inc. and serves on the Scientific Advisory Boards of Artica, Abilita Bio, and Abalone Bio. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The other authors declare that they have no competing interests.
References
- 1.Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res 124, 31–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol 14, 171–179 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Domanska UM, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer 49, 219–230 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi N, Zhang TT, Nakanishi T. Involvement of CXCR4 in Normal and Abnormal Development. Cells 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufareva I, Gustavsson M, Zheng Y, Stephens BS, Handel TM. What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annu Rev Biophys 46, 175–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32, 659–702 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Nibbs RJ, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol 13, 815–829 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Vacchini A, Locati M, Borroni EM. Overview and potential unifying themes of the atypical chemokine receptor family. J Leukoc Biol 99, 883–892 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Saaber F, et al. ACKR3 Regulation of Neuronal Migration Requires ACKR3 Phosphorylation, but Not beta-Arrestin. Cell Rep 26, 1473–1488 e1479 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Lau S, et al. A negative-feedback loop maintains optimal chemokine concentrations for directional cell migration. Nat Cell Biol 22, 266–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong M, et al. Dynamic Buffering of Extracellular Chemokine by a Dedicated Scavenger Pathway Enables Robust Adaptation during Directed Tissue Migration. Dev Cell 52, 492–508 e410 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Odemis V, et al. The presumed atypical chemokine receptor CXCR7 signals through G(i/o) proteins in primary rodent astrocytes and human glioma cells. Glia 60, 372–381 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Fumagalli A, et al. The atypical chemokine receptor 3 interacts with Connexin 43 inhibiting astrocytic gap junctional intercellular communication. Nat Commun 11, 4855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajagopal S, et al. Beta-arrestin-but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A 107, 628–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer CT, Chen Q, Tesmer JJG, Handel TM. Atypical Chemokine Receptor 3 ‘Senses’ CXC Chemokine Receptor 4 Activation Through GPCR Kinase Phosphorylation. Mol Pharmacol, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yen YC, et al. Structures of atypical chemokine receptor 3 reveal the basis for its promiscuity and signaling bias. Sci Adv 8, eabn8063 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Q, et al. ACKR3-arrestin2/3 complexes reveal molecular consequences of GRK-dependent barcoding. bioRxiv, (2023). [Google Scholar]
- 18.Hopkins BE, et al. Effects of Small Molecule Ligands on ACKR3 Receptors. Mol Pharmacol 102, 128–138 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naumann U, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One 5, e9175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanes MS, et al. Dual targeting of the chemokine receptors CXCR4 and ACKR3 with novel engineered chemokines. J Biol Chem 290, 22385–22397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaracz-Ros A, et al. Differential activity and selectivity of N-terminal modified CXCL12 chemokines at the CXCR4 and ACKR3 receptors. J Leukoc Biol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stephens BS, Ngo T, Kufareva I, Handel TM. Functional anatomy of the full-length CXCR4-CXCL12 complex systematically dissected by quantitative model-guided mutagenesis. Provisionally Accepted to Science Signaling, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngo T, et al. Crosslinking-guided geometry of a complete CXC receptor-chemokine complex and the basis of chemokine subfamily selectivity. PLoS Biol 18, e3000656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wescott MP, et al. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc Natl Acad Sci U S A 113, 9928–9933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burns JM, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203, 2201–2213 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein KR, et al. Decoy receptor CXCR7 modulates adrenomedullin-mediated cardiac and lymphatic vascular development. Dev Cell 30, 528–540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda Y, Kumagai H, Skach A, Sato M, Yanagisawa M. Modulation of circadian glucocorticoid oscillation via adrenal opioid-CXCR7 signaling alters emotional behavior. Cell 155, 1323–1336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyrath M, et al. The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat Commun 11, 3033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wingler LM, et al. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478 e411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fay JF, Farrens DL. Structural dynamics and energetics underlying allosteric inactivation of the cannabinoid receptor CB1. Proc Natl Acad Sci U S A 112, 8469–8474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao X, et al. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol 2, 417–422 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Elgeti M, Hubbell WL. DEER Analysis of GPCR Conformational Heterogeneity. Biomolecules 11, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wingler LM, et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367, 888–892 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray AP, Thakur N, Pour NG, Eddy MT. Dual mechanisms of cholesterol-GPCR interactions that depend on membrane phospholipid composition. Structure 31, 836–847 e836 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer CT, Fay JF, Janz JM, Farrens DL. Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc Natl Acad Sci U S A 113, 11961–11966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregorio GG, et al. Single-molecule analysis of ligand efficacy in beta(2)AR-G-protein activation. Nature 547, 68–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishna Kumar K, et al. Negative allosteric modulation of the glucagon receptor by RAMP2. Cell 186, 1465–1477 e1418 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandes DD, et al. Ligand modulation of the conformational dynamics of the A(2A) adenosine receptor revealed by single-molecule fluorescence. Sci Rep 11, 5910 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maslov I, et al. Sub-millisecond conformational dynamics of the A(2A) adenosine receptor revealed by single-molecule FRET. Commun Biol 6, 362 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsukamoto H, Sinha A, DeWitt M, Farrens DL. Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J Mol Biol 399, 501–511 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eberle SA, Gustavsson M. A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine-Chemokine Receptor Interactions. Cells 11, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otun O, et al. Conformational dynamics underlying Atypical Chemokine Receptor 3 activation. BioRxiv, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Kleist AB, et al. Conformational selection guides beta-arrestin recruitment at a biased G protein-coupled receptor. Science 377, 222–228 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zarca A, et al. Pharmacological characterization and radiolabeling of VUF15485, a high-affinity small-molecule agonist for the atypical chemokine receptor ACKR3. BioRxiv, (2023). [DOI] [PubMed] [Google Scholar]
- 50.Perpina-Viciano C, et al. Kinetic Analysis of the Early Signaling Steps of the Human Chemokine Receptor CXCR4. Mol Pharmacol 98, 72–87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mona CE, et al. Design, synthesis, and biological evaluation of CXCR4 ligands. Org Biomol Chem 14, 10298–10311 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg EM Jr., et al. Characterization, Dynamics, and Mechanism of CXCR4 Antagonists on a Constitutively Active Mutant. Cell Chem Biol 26, 662–673 e667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gustavsson M, Dyer DP, Zhao C, Handel TM. Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor N terminus in chemokine binding. Sci Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han M, Smith SO, Sakmar TP. Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry 37, 8253–8261 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Han X, Tachado SD, Koziel H, Boisvert WA. Leu128(3.43) (l128) and Val247(6.40) (V247) of CXCR1 are critical amino acid residues for g protein coupling and receptor activation. PLoS One 7, e42765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cui M, et al. Crystal structure of a constitutive active mutant of adenosine A(2A) receptor. IUCrJ 9, 333–341 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsukamoto H, Farrens DL. A constitutively activating mutation alters the dynamics of a key conformational change in a ligand-free GPCR. J Biol Chem, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vogel R, Mahalingam M, Ludeke S, Huber T, Siebert F, Sakmar TP. Functional role of the “ionic lock”--an interhelical hydrogen-bond network in family A heptahelical receptors. J Mol Biol 380, 648–655 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Deupi X, et al. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc Natl Acad Sci U S A 109, 119–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao Z, et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov 8, 44 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qin L, et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu K, et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Staus DP, et al. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 579, 297–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang W, et al. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pulvermuller A, Palczewski K, Hofmann KP. Interaction between photoactivated rhodopsin and its kinase: stability and kinetics of complex formation. Biochemistry 32, 14082–14088 (1993). [DOI] [PubMed] [Google Scholar]
- 67.Zhou XE, et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 170, 457–469 e413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang Y, et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Q, et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600–605 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burg JS, et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miles TF, et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Casarosa P, et al. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem 276, 1133–1137 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Gilliland CT, Salanga CL, Kawamura T, Trejo J, Handel TM. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, beta-arrestin-mediated internalization. J Biol Chem 288, 32194–32210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pauszek RF 3rd, Lamichhane R, Rajkarnikar Singh A, Millar DP. Single-molecule view of coordination in a multi-functional DNA polymerase. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lamichhane R, Solem A, Black W, Rueda D. Single-molecule FRET of protein-nucleic acid and protein-protein complexes: surface passivation and immobilization. Methods 52, 192–200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwarz G. Estimating the Dimension of a Model. Ann Statist 6, 461–464 (1978). [Google Scholar]
- 77.McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys J 91, 1941–1951 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data supporting this study are included in the Article or Supplementary files and are available from the authors upon request.