

Abstract
Several emerging therapies kill cancer cells primarily by inducing necrosis. As necrosis activates immune cells, potentially, uncovering the molecular drivers of anticancer therapy-induced necrosis could reveal approaches for enhancing immunotherapy efficacy. To identify necrosis-associated genes, we performed a genome-wide CRISPR-Cas9 screen with negative selection against necrosis-inducing preclinical agents BHPI and conducted follow-on experiments with ErSO. The screen identified transient receptor potential melastatin member 4 (TRPM4), a calcium-activated, ATP-inhibited, sodium selective plasma membrane channel. Cancer cells selected for resistance to BHPI and ErSO exhibited robust TRPM4 downregulation, and TRPM4 re-expression restored sensitivity to ErSO. Notably, TRPM4 knockout abolished ErSO-induced regression of breast tumors in mice. Supporting a broad role for TRPM4 in necrosis, knockout of TRPM4 reversed cell death induced by four additional diverse necrosis-inducing cancer therapies. ErSO induced anticipatory unfolded protein response (a-UPR) hyperactivation, long-term necrotic cell death, and release of damage-associated molecular patterns (DAMPs) that activated macrophages and increased monocyte migration, all of which was abolished by TRPM4 knockout. Furthermore, loss of TRPM4 suppressed the ErSO-induced increase in cell volume and depletion of ATP. This data suggests that ErSO triggers initial activation of the a-UPR but that it is TRPM4-mediated sodium influx and cell swelling, resulting in osmotic stress, which sustains and propagates lethal a-UPR hyperactivation. Thus, TRPM4 plays a pivotal role in sustaining lethal a-UPR hyperactivation that mediates the anticancer activity of diverse necrosis-inducing therapies.
Keywords: Cancer Therapy, Unfolded Protein Response, Necrosis, Ion Channel, Immunogenic Cell Death
Introduction
Immunotherapy has been challenging to apply to most solid tumors that do not display neoantigens. One way to target these tumors is to combine immunotherapy agents with cytotoxic therapies whose actions activate immune cells and induce immunogenic cell death (ICD). However, most current cancer therapies are either cytostatic, or kill cancer cells by inducing apoptosis, and apoptotic cell death usually does not strongly activate immune cells. In contrast, necrotic cell death often robustly activates immune cells and induces ICD. However, unlike apoptosis, autophagy and necroptosis, specific protein mediators of anticancer therapy-induced necrosis had not been described. Despite its promise, without an established protein mediator, there has been limited development of new therapies deliberately targeting necrosis. Instead of using protein markers, unregulated necrosis was typically characterized by cell swelling (oncosis), ATP-depletion, cytoplasmic vacuole formation, release of immune cell-activating Damage Associated Molecular Patterns (DAMPs) and imaging depicting a necrotic morphology (1–3). Analysis of necrosis and development of new necrosis-inducing therapies has been challenging in part because some anticancer therapies induce a mixed death phenotype with contributions from multiple cell death pathways (1,4). In contrast, we identified promising anticancer agents, first-generation BHPI, and more potent second-generation ErSO, that kill cancer cells by inducing “pure necrosis”. Here we use BHPI and ErSO to explore necrotic cell death (5,6). BHPI and ErSO induce necrosis by strong and sustained activation of the normally tumor protective endoplasmic reticulum stress response pathway, the anticipatory unfolded protein response (a-UPR) (7); this results in ATP depletion, cell swelling and necrotic cell death. The initial steps in a-UPR hyperactivation have been extensively characterized (7–9). To initiate a-UPR activation, BHPI and ErSO engage estrogen receptor α (ERα), causing rapid phosphorylation and activation of ERα-bound c-src. Activated c-src phosphorylates and activates phospholipase C-γ (PLCγ), which enzymatically produces inositol triphosphate (IP3). The IP3 binds to and opens IP3R Ca2+ channels on the endoplasmic reticulum (EnR) membrane, causing an efflux of stored Ca2+ ions into the cytosol. This leads to robust activation of the UPR, especially its Protein Kinase R(PKR)-like ER Kinase (PERK) arm, inhibiting protein synthesis (5,6,8). Although the initial steps in lethal hyperactivation of the a-UPR pathway had been identified, specific proteins that mediate cell swelling and subsequent membrane rupture and rapid necrotic cell death remained unknown.
To probe the mechanism of necrosis, we used human breast cancer cells to perform a genome-wide CRISPR-Cas9 screen with negative selection against BHPI. In this screen, a single gene in each cell was knocked out and the cell pool was subjected to selection pressure with BHPI. Surviving cancer cells were enriched for guide RNA (gRNA) sequences related to genes whose knockout helps them survive BHPI-induced necrosis. Although we performed the screen with BHPI, most follow-on experiments use ErSO, a more potent and more therapeutically promising derivative of BHPI, with the same mechanism of action (6). A top candidate from the screen was the sodium ion channel Transient Receptor Potential Melastatin Member 4 (TRPM4), whose properties were consistent with a role in BHPI and ErSO action. TRPM4 is a unique Na+-selective, six-transmembrane ion channel activated by cytosolic Ca2+ and inhibited by intracellular ATP (10,11), both outcomes of BHPI- and ErSO-mediated a-UPR hyperactivation. Although a TRPM4 mouse knockout had no obvious phenotypic abnormalities, TRPM4 was thought to play a role in some neurodegenerative diseases and in aberrant cardiac signaling (12–14). Here, we explore the role of TRPM4 in anticancer therapy-induced necrosis and show that TRPM4 drives necrotic cell death induced by the a-UPR activators BHPI, ErSO and recently described ErSO-DFP (15). Notably, the TRPM4 pathway is also important for the action of four other necrosis-inducing anticancer therapies that are unrelated to ErSO.
Unexpectedly, our work indicates that while ErSO triggers initial a-UPR activation, it is opening of the TRPM4 channel resulting in cell swelling and osmotic stress that sustains it. Supporting this unusual mechanism of action, a brief 1 hour exposure to ErSO is nearly as effective as sustained exposure to ErSO in inducing death of breast cancer cells. This is a new mechanism for lethal activation of a stress response pathway in cancer cells. Although our target breast cancer cells contain substantial endogenous TRPM4, increasing the level of TRPM4 in cancer cells increased their sensitivity to killing by ErSO. Identification of the pivotal role of TRPM4 in necrotic cell death will allow identification of cancer patients whose TRPM4 levels make them most likely to benefit from necrosis-inducing anticancer therapies.
MATERIALS AND METHODS
Cell Culture and Reagents
T47D cells were provided by Dr. K. Horwitz, Univ. Colorado, PEO4 human ovarian cancer cells were provided by Dr. S. Kauffman, Univ. Illinois, Caov-3 cells were provided by the Mayo Clinic. Prior to use in these studies, cell lines were verified by genotyping at the University of Arizona facility and were negative for mycoplasma using a PCR assay. In general, cells were used for experiments between passages 2 and 25 after thawing. T47DERαY537S-Luciferase (TYS-Luc) were produced in our laboratory (16). MCF-7 cells used in our laboratory to produce MTKO cells were from the Michigan Cancer Foundation (MCF). MYS-Luc cells were produced in our laboratory as described (16). MCF-7 cells used by Dr. B.H. Park to generate MCF-7ERαY537S (MYS) cells were from the ATCC. All cell lines were cultured at 37°C with 5% CO2 in phenol red-free medium. Medium: T47D (MEM, 10% FBS), MCF-7 (MEM, 5% FBS), TYS-Luc (MEM, 10% charcoal:dextran-treated (CD)-FBS), MYS-Luc (DMEM, 5% FBS), Caov-3 (DMEM, 10% FBS), PEO4 (DMEM, 10% FBS, 10 μg/mL insulin, 1:250 diluted Dulbecco’s NEAA’s, 1 mM glutamine), A498 (MEM, 10% FBS). All growth media were supplemented with 1% penicillin-streptomycin. The LDH Release Assay was performed in Human Physiologically-Relevant Medium (17) supplemented with 2% FBS. Reagents used were: Doxorubicin, Staurosporine, rapamycin, Shikonin and Dimethoxycurcumin (Cayman Chemicals, MI), Fulvestrant/ICI 182,780 and Tamoxifen/z-OHT (Tocris Bioscience, MN), Ionomycin and Z-VAD-FMK (Santa Cruz Biotechnology, TX), 2-deoxyglucose, Q-VD-OPH, Nec-1 and GSK872 (MedChemExpress USA, NJ), Aprepitant and Englerin A (MilliporeSigma, MA), 35S-methionine (Perkin Elmer, MA), all other reagents were from Sigma Aldrich (MO).
Cell Proliferation Assay
Cells were plated at an appropriate density into each well of a 96-well plate. The following day, the medium was replaced with medium containing the indicated treatment. The medium was changed again and replaced with treatment medium after 2 days and proliferation was assayed after 4 days. 10 μl of Alamar Blue (ThermoFisher, MA) was added per 100 μl of medium and the cells were incubated at 37°C with 5% CO2, for 1 hour. Fluorescence intensity was measured using a PHERAstar Microplate Reader (BMG Labtech Inc., NC). MTS assays were performed as previously described (8,16).
Western Blotting
Western blotting was performed as previously described (8). The following antibodies were used: TRPM4 (Thermo Fisher Scientific, Cat # MA5-24918), Puromycin (Millipore Sigma, Cat # MABE343), phospho-src (Cell Signaling Technology (CST) # 6943), src (CST # 2109), phospho-PLCγ (CST # 14008), PLCγ (CST #5690), phospho-eIF2α (CST # 3398), eIF2α (CST #5324), β-actin (CST #3700), HMGB1 (CST #3935). ). Antibodies were probed with HRP-conjugated secondary antibodies (ThermoFisher) and imaged with the Super ECL kit (ABP Biosciences) using the iBright CL1000.
Trypan Blue Dye Exclusion Death Assay and Swelling Assay
Cell viability and cell diameter after treatment with ErSO or other compounds were measured using a Countess II cell counter (ThermoFisher) as we described (5). Briefly, 300,000 cells/well were plated in 6-well plates. The next day, vehicle or treatment was added for the indicated time. Cells were then harvested and concentrated to 2–5 million cells/ml. Trypan Blue was added immediately before counting for cell viability or measurement of cell diameter.
Lentiviral Production and Clonal Selection
Constructs for packaging vectors pMD2.G (# 12259) and psPAX2 (# 12260), Toronto KnockOut (TKO) CRISPR Library - Version 3 (#90294) and LentiCRISPRv2 puro (#98290) were from Addgene (MA). Knockout gRNA sequences were cloned into LentiCRISPRv2 puro (Sequences for TRPM4: TAGTTGACTCCACAGATCCG, CGGAGAGCTGGACTTCACGG, GTGCGGGCTGCCCAGAGCAC, GTACAGCCACACCAACATGC). TRPM4 overexpression plasmid was made by extracting mRNA from MCF-7 cells and converting it into TRPM4 cDNA using an appropriate primer (5’- GGCCCAGATGAGCCTTACTC −3’), and cloning the cDNA fragment into a lentiviral puromycin-resistant expression plasmid (pCDH-puro-cMyc #46970). Lentiviral packaging vectors were co-transfected with the required plasmid into HEK 293-T cells XtremeGene 9 (Roche). The next day, medium was changed to DMEM+1.1g/dL BSA. Virus was harvested 24 h later and used to transduce cells for 48 h before selection with cell-line specific concentrations of puromycin. For TRPM4 knockout and clones, individual colonies were picked and grown out, and validated using Western blot. The MCF-7 TRPM4 overexpression systems and the A498 TRPM4 knockout are pools of cells, not clones.
Protein Synthesis Inhibition Assay
Rates of protein synthesis were evaluated by measuring 35S-methionine incorporation into newly synthesized protein as described previously (6,8). Briefly, cells were incubated with treatment for the indicated time and 3 µCi of 35S-methionine was added for the last 30 minutes of treatment. Cells were washed with PBS and lysed with RIPA buffer. Lysates were spun and supernatants were pipetted onto Whatman 540 filter-paper discs. The filters subsequently washed 3 times each with 10% TCA, then with 5% TCA, and air dried. Protein was solubilized using NaOH, which was subsequently neutralized using HCl. A liquid scintillation cocktail was added to the solubilized protein samples and counts were measured to determine percent protein synthesis inhibition.
ATP Measurement
Assays of ATP levels were performed as previously described (6) and ATP levels were measured using the ATPlite Luminescence Assay Kit protocol (Perkin Elmer).
Mouse Xenograft Studies
All studies involving animals were approved by the University of Illinois Institutional Animal Care and Use Committee (IACUC). Establishment and measurement of orthotopic mouse xenograft tumors followed the general procedures as described before (6). MCF7-ERαY537S-Luciferase (MYS-Luc) and MYS-Luc TRPM4 Knockout (MYTKO) cells were used. NSG ovariectomized mice were from Jackson Laboratory (#005557). MYS-Luc and MYTKO cells (1,000,000 cells in 1:1 Hanks’ balanced salt solution:Matrigel) were inoculated into the mammary fat pad, allowed to establish, and tumors were grown to ~300 to 400 mm3 (~21 days to establish tumors). Mice within a cell type group were then randomized and then treated with vehicle daily (n = 5) or ErSO daily (40 mg/kg, i.p.). Tumor size was measured by calipers on Day 0, 7 and 14. BLI tumor images were taken on Day 0, 3, 7 and 14. Mice were weighed on Days 0, 7 and 14. For the protein samples, mice with MYS-Luc or MYTKO breast tumors were injected with 40 mg/kg ErSO for 6 hours, following which the primary tumor tissue was excised, homogenized and lysed in RIPA buffer. The samples were run on Western blots to determine levels of proteins. For the protein synthesis inhibition assay, mice with MYS-Luc or MYTKO tumors were injected with 40 mg/kg ErSO for 6 hours, and were subsequently injected with puromycin made in sterile PBS (0.04 μmol/ per g of body weight) 45 minutes before the end of the treatment time. The primary tumor tissue was excised, homogenized and lysed in RIPA buffer. Incorporation of puromycin into proteins was determined via Western blot analysis. Protein synthesis was determined by analysis of whole lanes. Ponceau S staining was utilized as a loading control.
Quantitative RT-PCR
RNA was extracted using a QiaShredder kit (Qiagen) for cell homogenization and purified with the RNeasy Mini kit (Qiagen, CA). cDNA was prepared from 1 µg RNA using OligodT primers by reverse transcription using M-MuLV Reverse Transcription Enzyme. Quantitative PCR assays were performed using 3 biological replicates and at least 1 technical replicate. Reactions were set up to contain 10 ng of cDNA and 50 nM forward and reverse primers in 15 µl and were carried out using Power SYBR Green PCR Mastermix (Applied Biosystems). The fold change in expression of each gene was calculated using the ∆∆Ct method with the ribosomal protein 36B4 was used as the internal control.
CRISPR Screen
The TKOv3 library was amplified in Lucigen Endura electrocompetent cells to have at least 200-fold library coverage (71,090 sgRNAs). The plasmid library was then packaged into lentiviral particles forming a viral sgRNA library. 150 million T47D cells were plated evenly onto twenty-five 15 cm tissue culture dishes and were infected with the viral library at an MOI of 0.3. The cells were selected with 3.5 μg/ml puromycin for 3 days. Surviving cells were harvested and 15 million cells were frozen as the Day 0 control sample. The rest of the cells were plated evenly into four T175 flasks (four biological replicates) and treated with 100 nM BHPI for 28 days. The surviving cells were harvested and four samples of 15 million cells from each biological replicate were obtained. Genomic DNA from the four samples as well the control sample was extracted and the sgRNA region was amplified using PCR. Subsequently, i5 and i7 primers were used to amplify the sgRNA libraries which were then sequenced on Illumina HiSeq4000. The sequence data was analyzed using the MAGeCK-VISPR algorithm (18).
Sodium Influx Assay
Wild type or TRPM4 KO cells were seeded at a density of 30,000/well in 96-well black-walled clear bottomed cell culture plates in 100 μl of induction medium (DMEM+10% FBS). Cells were incubated in a 37°C incubator for 48 hours. The induction medum was replaced with 100 μl of assay buffer (140 mM NMDG‐Cl, 10 KCl mM, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES (pH 7.2) with NMDG‐OH and 290 to 300 mOsm) and the cells were incubated at RT for 45 minutes. The assay buffer was replaced with 95 μl of dye loading solution (5 μM Asante Natrium Green-II prepared in assay buffer, supplemented with 0.1% pluronic acid). The cells were incubated in the dark at RT for 45 minutes. After incubation, the dye solution was removed and replaced with 80 μl of assay buffer, and incubated again in the dark for 20 minutes. Basal fluorescence readings were recorded using a PHERAstar Microplate Reader (Ex=488 nm, Em=540 nm). To initiate Na+ influx, 20 μl of stimulus buffer (700 mM NaCl, 4 mM CaCl2, 4 mM MgCl2, 40 mM HEPES, pH 7.2 with NaOH) containing vehicle, 50 μM ErSO, or 50 μM Ionomycin was added to their respective wells. Fluorescence readings were recorded and plotted against time.
Calcium Electroporation
Calcium electroporation was performed as described by Gehl and coworkers with some modifications (19). MYS and MYTKO cells in suspension in a Bio-Rad 4 mm electroporation cuvette (Cat: 165–2088) were electroporated using a BTX T820 Square Porator. Electroporation conditions and the calcium concentration to use were determined in pilot studies. In each experiment, to identify the fraction of cancer cells killed by TRPM4-independent reversible electroporation, a no-calcium sample was also electroporated. Cells were electroporated in 5 mM calcium, and then transferred to multiwell plates in standard culture medium.
THP-1 Migration Assay
Wild type or TRPM4 knockout cells were plated in a 6-well plate at 360,000 cells/well in complete medium. The following day, the medium was replaced with serum-free medium with vehicle, 100 nM ErSO or 1 μM Raptinal. The cells were incubated with the treatments for 6 hours, after which the cells were washed once with ErSO-free medium and fresh medium containing no ErSO was supplied to the cells. The cells were then incubated for another 18 hours. After incubation, the medium was collected from all the treatment groups. THP-1 human monocytes were seeded in 5 μm transwell inserts at 200,000 cells/ well, in a 24-well plate, in no serum RPMI. 500 μl of vehicle medium, 10% FBS medium, ErSO-treated medium, or Raptinal-treated medium was pipetted into the bottom chamber as rapidly as possible. Migration was allowed to proceed for 4 hours. The bottom chamber medium was transferred to a 96-well plate and an Alamar Blue assay was performed to measure the number of live cells that passed through the membrane.
Statistical analysis
Information on statistical analysis is in the legends to individual figures. Briefly, unpaired Student’s T test was used to compare one variable between groups. Graphs of the main text figures show individual data points. All graphs are the mean plus or minus the standard error of the mean unless stated otherwise in the figure legend. Statistical significance: n.s.: not significant by Student’s T test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Data and materials availability
The data supporting the findings and conclusions of this study are available within the paper and its supplementary information. The publicly available data analyzed in this study were obtained from The Cancer Genome Atlas (TCGA) at https://www.cancer.gov/ccg/research/genome-sequencing/tcga. Resources described here and raw data are available from the corresponding authors upon reasonable request. Correspondence and requests for materials should be addressed to: David J. Shapiro: djshapir@illinois.edu or Santanu Ghosh: santanu2@illinois.edu.
Results
TRPM4 is Important for BHPI and ErSO-induced Necrotic Cell Death
To identify genes whose knockout confers resistance to a-UPR hyperactivators, we performed a genome-wide, CRISPR-Cas9 screen with negative selection against the known a-UPR hyperactivator, BHPI (Fig. 1A). Detailed screen quality information is in (Fig. S1A-C). Notably, the sodium ion channel TRPM4 was the top target to emerge from the screen (Fig. 1B). Moreover, the TRPM4 modulator KCTD5 (20) was also a top target from the screen. This suggested loss of TRPM4 in the knockout cells was associated with resistance to BHPI and similar compounds. Supporting a possible role for TRPM4 in BHPI and ErSO action, TRPM4 is activated by increased intracellular calcium and inhibited by ATP (10,11), and a-UPR hyperactivation by BHPI and ErSO increases intracellular calcium and depletes ATP (5,6,8) (model in Fig. S1D). Analysis of the TCGA database indicates that luminal breast cancers, which are typically ERα+, have a higher level of TRPM4 expression than triple negative breast cancers, (Fig. S1E). TRPM4 is also highly expressed in many other cancers (Fig. S1G); other than in prostate cancer (21,22), its role in tumors remains largely unexplored. Since TRPM4 is highly expressed in many ERα+ breast cancers and transcriptional regulation of TRPM4 was unexplored, we examined the effect of estrogen on TRPM4 mRNA levels. Interestingly in ERα+ MCF-7, human breast cancer cells, TRPM4 mRNA is down regulated by long-term (3-day) exposure to estrogen (17β-estradiol; E2) (Fig. S1F). However, exposure of breast cancer cells to E2 over the short times required for ErSO to initiate lethal hyperactivation of the a-UPR (see below) did not significantly alter levels of TRPM4 mRNA or protein (Fig. S2A-B). The selective ERα degrader (SERD), fulvestrant/ICI 182,780, reduces the level of ERα protein (Fig S2C) and therefore modestly affects the level of TRPM4 mRNA and protein (Fig S2D-E). However, pre-treatment with ICI, which will down-regulate ERα, but only modestly impact TRPM4, did not significantly alter the ability of ErSO to kill ERα+ MCF-7 breast cancer cells (Fig S2C-F). This is in agreement with our previous work that levels of ERα are usually not limiting for UPR hyperactivation (6,9). Consistent with our earlier work (6), ErSO is not a SERD and its actions are distinct from SERDs, such as fulvestrant. Unlike fulvestrant (ICI), or the selective ERα modulator (SERM), tamoxifen (OHT), ErSO is fully effective in the presence of saturating E2, indicating it does not compete with E2 for binding to ERα (Fig. S2G). Moreover, unlike the SERD fulvestrant, ErSO does not induce rapid loss of ERα, nor does it result in loss of TRPM4 expression (Fig. S2C,H-I). This indicates that ErSO activates TRPM4 through an ERα-mediated signaling pathway and not by transcriptional modulation, or by inducing rapid ERα degradation.
Figure 1.
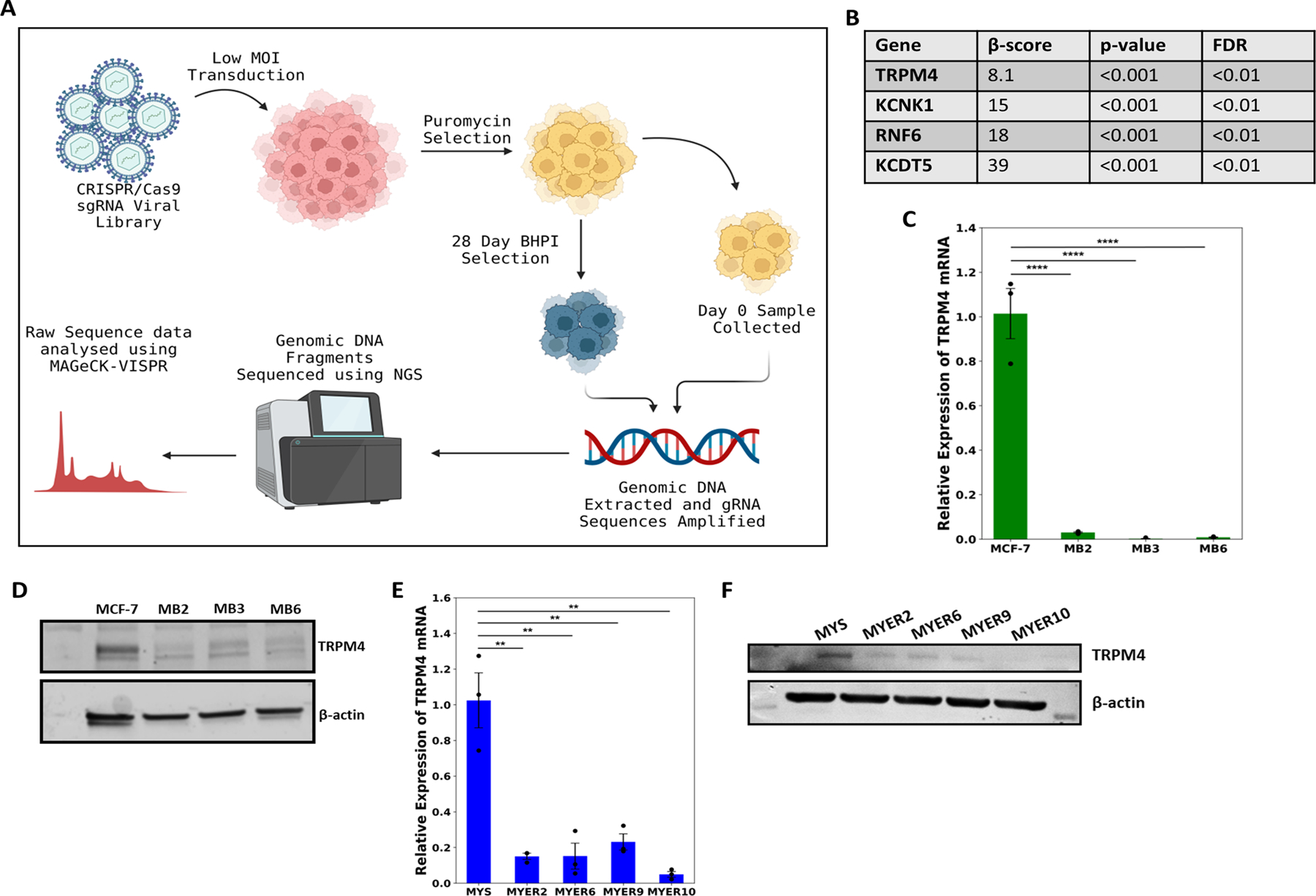
TRPM4 is important for BHPI and ErSO-induced cell death. A Schematic diagram of the CRISPR/Cas9-based genome-wide negative selection screen performed using BHPI on T47D cells. B Partial list of targets obtained from the genome-wide negative selection screen with BHPI. C Relative expression levels of TRPM4 mRNA in wild type MCF-7 and BHPI-resistant MCF-7 cells (MB) (n=3). D Western blot analysis of TRPM4 protein levels in wild type MCF-7 and MB cells. E Relative expression levels of TRPM4 mRNA in MYS (MCF7-ERαY537S) cells and ErSO-resistant MYS cells (MYER) (n=3). F Western blot analysis of TRPM4 protein levels in MYS cells and MYER cells. All data are mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
If TRPM4 is important in BHPI and ErSO-induced necrosis, a logical mechanism of resistance in BHPI- and ErSO-resistant cells would be down-regulation of TRPM4. We explored resistance to BHPI in human MCF-7 breast cancer cells and in MCF-7 cells in which one copy of wild type ERα has been replaced with the ERαY537S mutation (MCF-7ERαY537S/MYS) (6,23). This mutation, seen in patients with metastatic breast cancer, confers estrogen-independent activity on ERα and is associated with resistance to endocrine therapies and with shorter patient survival (24). In clones of MCF-7 cells selected for BHPI-resistance (MB cells), and in MYS cells selected for ErSO-resistance (MYER cells), TRPM4 mRNA and protein were robustly down-regulated (Fig. 1C-F). These data support a potential role for TRPM4 in BHPI and ErSO-induced necrotic cell death.
TRPM4 Knockout Confers Complete Resistance to ErSO-induced Necrotic Cell Death
The screen results and data from the resistant clones suggested that sensitivity of cancer cells to ErSO should increase with increasing levels of TRPM4 and decrease in the absence of TRPM4. Despite considerable endogenous TRPM4 in MCF-7 and MYS human breast cancer cells, TRPM4 overexpression (TRPM4 overexpressing MCF-7 cells: MTOV1 and MTOV2) elicited a level-dependent increase in sensitivity to killing by ErSO (Fig. 2A, S3A). We generated a series of TRPM4 knockout (TKO cells) cell lines from ERα+ T47D and MCF-7 breast cancer cells (named TTKO and MTKO respectively), and Caov-3 and PEO4 ovarian cancer cells (named CTKO and PTKO respectively). We also used T47D and MCF-7 cells expressing the ERαY537S mutation (TYS and MYS cells, TRMP4 KO: YTKO and MYTKO cells) (Fig. S3B-C). Notably, TRPM4 knockout completely abolished rapid ErSO-induced cell death in all 6 cancer cell models (Fig. 2B). Strong TRPM4-dependent cell death 24 hours after ErSO treatment was observed both with our automated Trypan Blue exclusion assay for cell death (Fig. 2B), which is based on loss of membrane integrity, and with the MTS assay, which is based on mitochondrial activity and cell energy level (Fig. S3D). To evaluate whether TRPM4 expression could restore sensitivity to ErSO, we re-expressed TRPM4 in two ErSO-resistant clones that lack detectable TRPM4 protein. Although the ErSO-resistant clones grow well in 1,000 nM ErSO, re-expression of TRPM4 re-sensitized the cells to as little as 10 nM ErSO (Fig. S3E-F). Unlike the TRPM4 knockouts, there are multiple changes in the clones selected for progressive resistance to ErSO; therefore, restoration of TRPM4 only renders them partially sensitive to ErSO. We next explored how TRPM4 plays such a pivotal role in ErSO-induced necrotic cell death.
Figure 2.
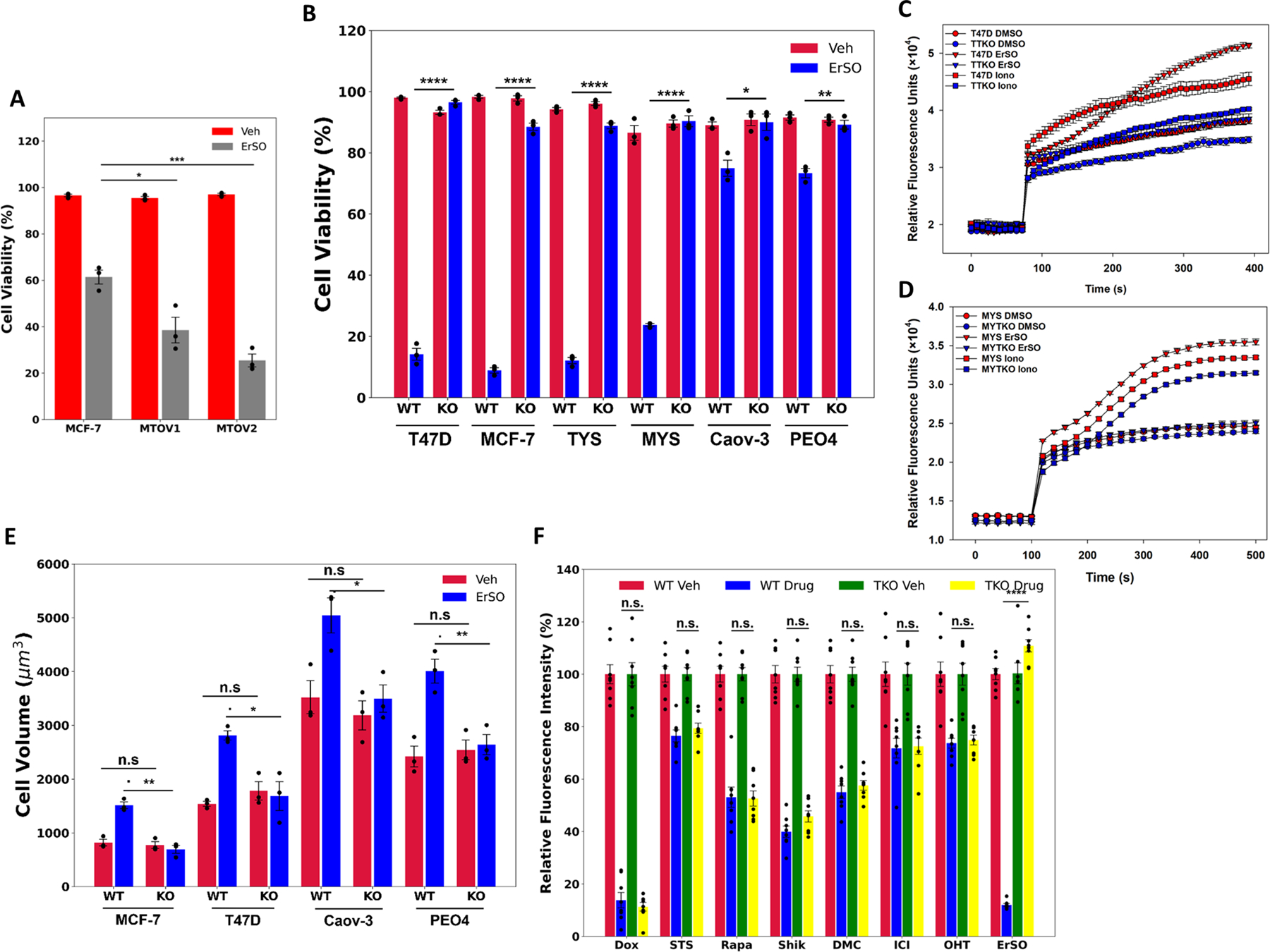
TRPM4 knockout completely blocks ErSO-induced cell death. Automated Trypan Blue dye exclusion assays comparing A wildtype MCF-7 cells and two TRPM4 overexpressing clonal pools (MTOV1 and MTOV2) treated with either vehicle or 50 nM ErSO for 24 hours (n=3), B ERα positive breast cancer cell lines, T47D, MCF-7, TYS and MYS, ovarian cancer cell lines Caov-3 and PEO4, and their TRPM4 knockout clones (TTKO, MTKO, YTKO, MYTKO, CTKO and PTKO respectively) were treated with either vehicle or ErSO (100 nM for T47D, TTKO, MCF-7, MTKO, TYS, and YTKO; 1 μM for MYS, MYTKO, Caov-3, CTKO, PEO4 and PTKO) for 24 hours (n=3). C-D Sodium influx assay comparing D wild type and TRPM4 knockout T47D cells (TTKO) cells, and E MYS and MYTKO cells treated with vehicle, 10 μM ErSO, or the ionophore ionomycin (10 μM) (n=4). E Cell volume of ERα positive breast cancer cell lines, T47D and MCF-7, and ovarian cancer cell lines, Caov-3 and PEO4 and their knockout counterparts, treated with vehicle or with 1 μM ErSO for 1 hour (n=3). F Relative fluorescence from Alamar Blue proliferation assays performed on TYS cells treated with vehicle (set to 100%), apoptosis inducers, doxorubicin (100 nM) and staurosporine (100 nM), autophoagy inducer rapamycin (1 μM), necroptosis inducer shikonin (1 μM), paraptosis inducer dimethoxycurcumin/DMC (10 μM), fulvestrant/ICI (1 μM), tamoxifen/z-OHT (1 μM) or ErSO (100 nM), for 4 days (n=8). All data are mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s. = not significant by Student’s t-test.
Since there is a ~10X higher concentration of Na+ outside cells than inside (25), we hypothesized that opening the TRPM4 channel results in an influx of Na+ ions and subsequently Cl− ions to balance the charge and water to maintain osmolality. This would swell the cells, leading to plasma membrane rupture and necrotic cell death. To test this hypothesis, we explored whether ErSO treatment opens the TRPM4 Na+ channel. In wild type T47D and MYS cells, but not in TRPM4 knockout cells, ErSO (Fig. 2C,D) and BHPI (Fig. S4A) induced a rapid increase in intracellular Na+ levels. Demonstrating that TRPM4 knockout is selective and does not result in a general loss of the cell’s ability to uptake Na+, the well-known ionophore, ionomycin (26), induced Na+ influx in both wild type and knockout cells (Fig. 2C,D). Supporting the hypothesis that the Na+ influx triggers cell swelling, rapid (1 hour) ErSO-induced cell swelling was abolished in the TRPM4 knockout cells (Fig. 2E). Thus, a-UPR activation could open the TRPM4 channel and initiate cell swelling and plasma membrane rupture, causing necrosis-like oncolytic cell death within 24 hours. This type of cellular edema was suggested in a few neuronal disease models, but was unexplored in anticancer drug-induced cell death (27,28).
It remained possible that TRPM4 might be a general pathway-independent enhancer of cell death. We therefore compared the response of TRPM4-containing (TYS) and TRPM4 knockout (YTKO) cells to cell death induced by apoptosis, autophagy, necroptosis and paraptosis inducers, as well as the breast cancer endocrine therapies, Fulvestrant/Faslodex/(ICI) and Tamoxifen (z-OHT) (29–35). At physiologically relevant concentrations, TRPM4 knockout did not reverse the effect of these drugs on cell proliferation (Fig. 2F). Additionally, in both wild type and TRPM4 knockout cells, the apoptosis-inducing UPR activator, tunicamycin, robustly induced the UPR marker, spliced XBP1 mRNA (Fig. S4B) (36), and the apoptosis inducer, staurosporine, induced cell death (Fig. S4C). This indicates that knocking out TRPM4 does not impair the UPR response to other types of activators. Moreover, the broad-spectrum caspase inhibitor, Q-VD-OPH, which reversed staurosporine-induced apoptosis (Fig. S4D) (37), had no effect on ErSO-induced cell death and inhibition of proliferation (Fig. S4E-F). Since some features of RIP kinase-dependent necroptosis, namely caspase-independent cell swelling, and immune cell activation are similar to those seen in unregulated necrosis (38,39), to evaluate a possible role for necroptosis in ErSO action, we explored the effect of the RIP kinase inhibitors, Nec-1 and GSK872 on ErSO-induced inhibition of cell proliferation and cell death (40). Neither rapid (1-day) ErSO-induced cell death nor longer-term inhibition of proliferation was blocked by the RIPK inhibitors (Fig. S4E-F). Since the RIPK inhibitors, but not TRPM4 knockout, successfully blocked shikonin-induced necroptosis, the inhibitors were functional (Fig. S4D,G). Moreover, the necroptosis inhibitor necrosulfanimide (NSA) which inhibits the Mixed Lineage Kinase Domain-like Protein (MLKL), which is downstream of RIP kinases in the necroptosis pathway, also did not block ErSO-induced cell death (Fig S4H). These data demonstrate that ErSO kills cells via pure necrosis and that TRPM4 plays a pivotal, pathway-dependent role in ErSO-induced cell death.
TRPM4 is Essential for Long-term ErSO Action in Physiologically Relevant 3D Organoids and Tumor Models
The discovery of TRPM4 and our earlier work indicated that ErSO induces a two-stage death process. Initially, there is rapid TRPM4-dependent cell swelling, leading to membrane rupture and death of many cells. Concurrently, there is a strong and sustained activation of the UPR by ErSO; this leads to ATP depletion, AMPK activation and global inhibition of protein synthesis (model in Fig. S1D). Over time, we expected this strong and sustained UPR activation to cause death of cancer cells that survive the initial wave of TRPM4-dependent necrotic cell death. Surprisingly, in all six TRPM4 knockout models, TRPM4 knockout completely blocked not only rapid cell death (Fig. 2B), but also blocked ErSO-induced inhibition of proliferation in a 4-day Alamar Blue assay (Fig. 3A). Similar results were obtained with a 4-day MTS assay (Fig. S5A). These standard 2-dimesional cell cultures do not recapitulate the 3-dimensional architecture of tumors; we therefore explored the effect of TRPM4 knockout in 3-dimensional organoids and in a mouse xenograft model. To better mimic in vivo conditions, we grew organoids from wild type and TRPM4 knockout (TKO) cells in Human Physiologically Relevant Medium (17). Using Lactate Dehydrogenase (LDH) release, an established marker of necrotic cell death, we observed that ErSO induced significant time-dependent LDH release from wild type cells, but not from TRPM4 knockout cancer cells (Fig. S5B-F).
Figure 3.
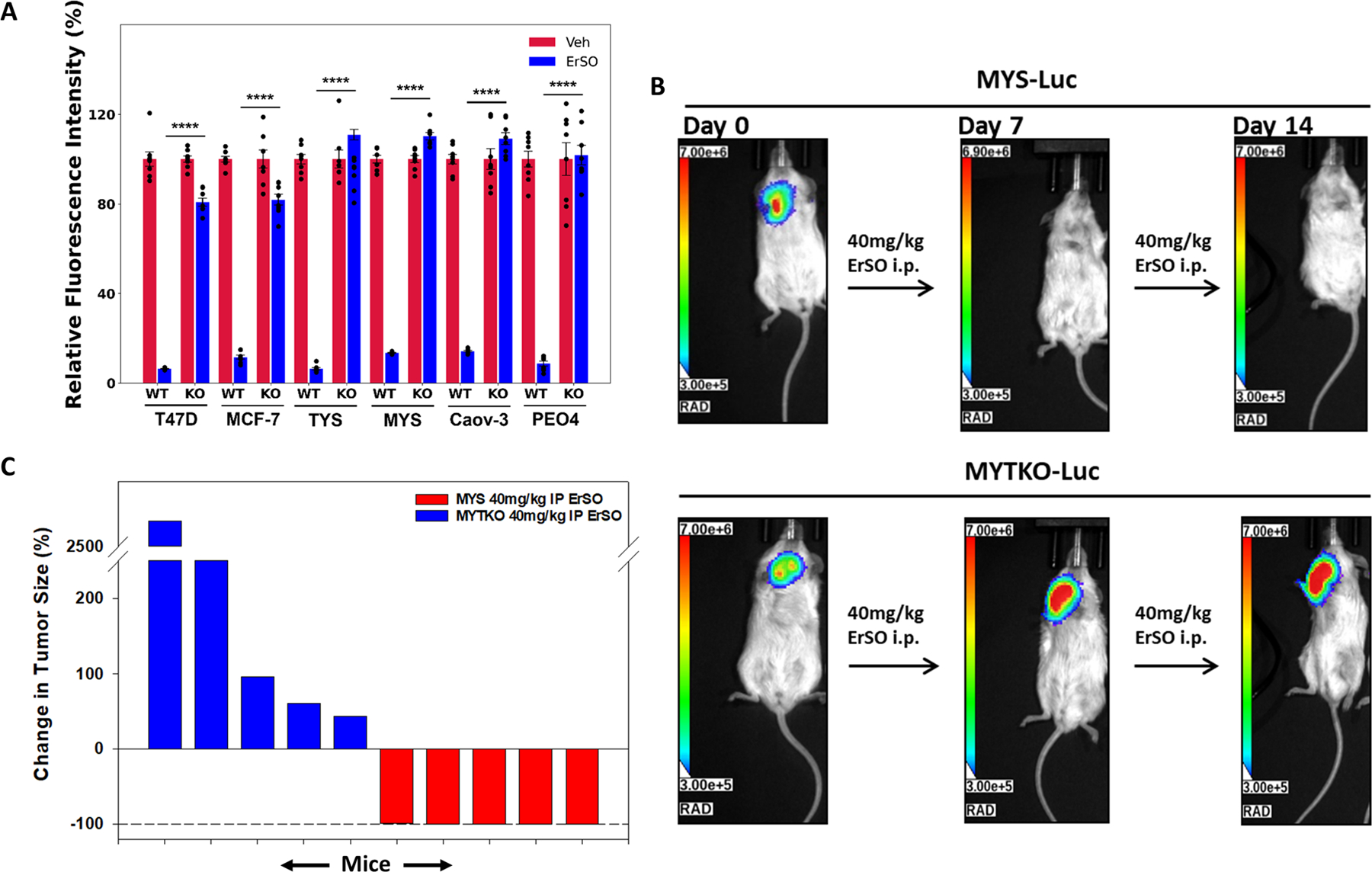
TRPM4 is essential for ErSO-induced cancer cell death in tumor models. A Alamar Blue proliferation assay in wild type and knockout breast and ovarian cancer cell lines in standard cell culture treated with either vehicle (set to 100%) or 100 nM ErSO for 4 days. B Orthotopic MCF-7ERαY537S luciferase stable (MYS-Luc) tumors and MYS-Luc TRPM4 knockout (MYTKO) tumors were established in ovariectomized Nu/J mice and grown for 21 days to ≈400mm3, randomized, and treated with ErSO (40 mg/kg) i.p. daily (n=5 mice/group). Shown are representative images of the bioluminescent tumors at the beginning of the study (day 0), on day 7 and day 14. TRPM4 KO tumors grow throughout the 14 days of treatment. C Waterfall plot depicting the change in tumor size as a percentage of the initial flux for each mouse in all mice of the treatment groups.
The absence of TRPM4 not only blocked rapid cell death but also reversed inhibition of proliferation and LDH release over 4 days. Hence, we next explored longer ErSO exposure using orthotopic mouse xenografts. We previously showed that ErSO treatment resulted in near complete regression of aggressive MCF-7ERαY537S-Luciferase (MYS-Luc) breast tumors (6). We repeated this study with TRPM4 knockout tumors (MYTKO-Luc) and found that using both luciferase-based bioluminescent imaging (BLI) and caliper measurements of tumor volume, TRPM4 knockout completely abolished the effect of ErSO; the ErSO-treated knockout tumors exhibited continued growth (Fig. 3B-C, S6A-F, S7A-G). These data indicate that TRPM4 is not only critical for rapid cell death induced by ErSO, but it must also be important for sustained UPR activation. We therefore explored the effect of TRPM4 knockout on key growth-inhibitory features of UPR activation: ATP depletion, AMPK activation, eEF2 phosphorylation and PERK activation leading to phosphorylation of eIF2α and inhibition of protein synthesis (model in Fig. S1D).
TRPM4 is Required for Sustained Lethal UPR Hyperactivation
TRPM4 knockout blocked ErSO-mediated (i) ATP depletion and therefore AMPK activation and eEF2 phosphorylation (Fig. 4A, S8A), (ii) activation of the UPR arms, as shown by formation of p-PERK and p-eIF2α, and induction of the UPR marker spliced XBP1 mRNA (Fig. S1D, 4B, S8B) and (iii) inhibition of protein synthesis (Fig. 4C). This mechanism of action through TRPM4 is shared by our recently described close ErSO relative, ErSO-DFP (Fig. S8C-E) (15).
Figure 4.
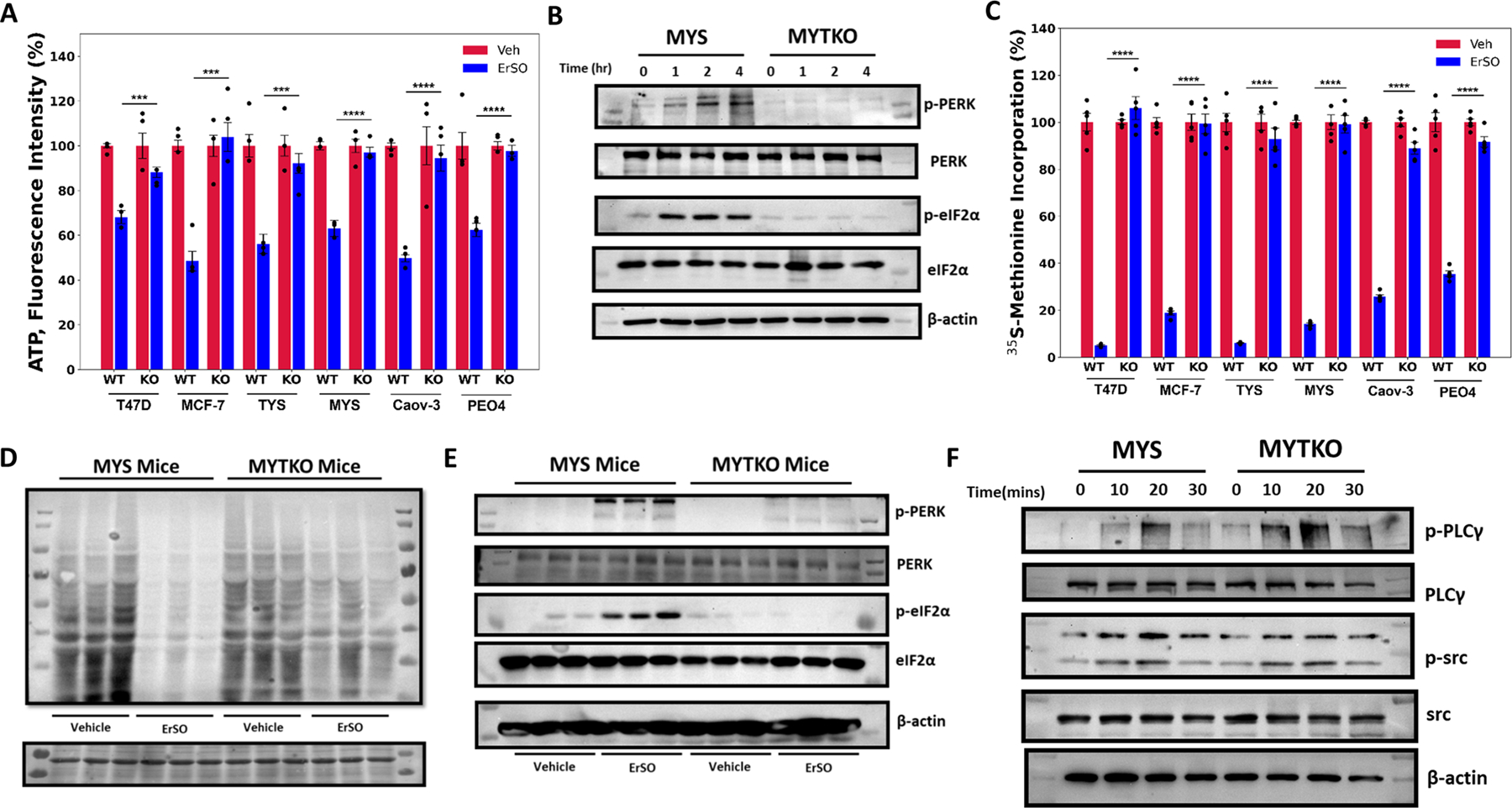
TRPM4 sustains lethal UPR hyperactivation. A Whole cell ATP levels of the indicated wild type and TRPM4 knockout breast and ovarian cancer cells treated with vehicle (set to 100%) or 1 μM ErSO for 4 hours (n=4). B Western blot analysis of p-PERK, total PERK, p-eIF2α, total eIF2α and β-actin in MYS cells and MYTKO cells at 0, 1, 2 and 4 hours after treatment with 1μM ErSO. C Incorporation of 35S-methionine into newly synthesized protein in the indicated wild type and TRPM4 knockout breast and ovarian cancer cell lines treated with vehicle (set to 100%) or 1 μM ErSO for 1 hour (n=5). D, E MYS-Luc and MYTKO-Luc cells were orthotopically grafted in NSG mice and tumors allowed to grow for 21 days. Mice were then treated with either vehicle or ErSO (40 mg/kg i.p) for 5 hours (n = 3). D Mice were then injected with puromycin in sterile PBS (0.04 μmol/ per g of body weight). After 1 hour, mice were euthanized and tumors were resected. Protein lysates from the tumors were prepared and puromycin-labeled peptides were identified using Western blot analysis. Ponceau S staining was used as a loading control (on the bottom). E Mice were euthanized and tumors were resected. Protein lysates from the tumors were prepared and Western blot analysis was performed for p-PERK, total PERK, p-eIF2α, total eIF2α and β-actin. F Western blot analysis of p-PLCγ, total PLCγ, p-src, total src and β-actin in wild type MYS cells and MYTKO cells at 0, 10, 20, and 30 minutes after addition of 1 μM ErSO. Data are mean ± s.e.m. ***p<0.001, ****p<0.0001.
Since TRPM4 knockout breast tumors exhibited continued growth, we investigated whether ErSO had also lost the ability to activate the PERK arm of the UPR and inhibit intratumoral protein synthesis. In tumors from MYS-Luc cells, but not TRPM4 knockout tumors, ErSO induced a decline in protein synthesis, as shown by a reduced level of puromycin-peptide (Fig. 4D). Moreover, wild type tumor-derived protein samples show increased p-PERK and p-eIF2α and p-src, while the TKO tumors do not (Fig. 4E,F). Taken together, these data indicate that TRPM4 is not only the executioner protein responsible for ErSO-mediated rapid cell death, but is also responsible for sustained UPR activation (model in Fig. S8F). Knocking out TRPM4 thus confers resistance not only to ErSO-induced rapid cell death, but also enables long-term cancer cell survival in normally lethal concentrations of ErSO.
Since TRPM4 is essential for strong and sustained lethal activation of the a-UPR by ErSO, we investigated where in the a-UPR pathway TRPM4 impacts activation. One possibility was that TRPM4 is required for the initial stage of a-UPR activation that results in release of calcium stored in the lumen of the endoplasmic reticulum into the cytosol (model in Fig. S1D). To test this possibility, we transfected a genetically encoded Ca2+ indicator (GECI) (41), into WT and TKO cells and used calcium-free medium to exclude possible influence of extracellular calcium. In both ErSO-treated wild type and TRPM4 knockout cells, ErSO induced a very rapid increase in intracellular calcium (Movies S1-4). Moreover, Western blot analysis showed that ErSO induced increased levels of phosphorylated c-src and PLCγ in WT and TKO cells (Fig. S1D, 4F). Thus, in TRPM4 knockout cells ErSO still triggers the initial stages of a-UPR activation, but sustained lethal hyperactivation of the a-UPR is blocked. Therefore, TRPM4 is not only executing the end step of necrosis by enabling cell swelling-induced plasma membrane rupture, but is also critical for sustained activation of the a-UPR pathway.
These data indicated that ErSO triggers initial a-UPR activation and cell swelling and osmotic stress sustains a-UPR activation. This suggested that a brief transient exposure to ErSO might be sufficient to induce cell death. We therefore explored the impact of exposing breast cancer cells to a moderate concentration of ErSO (100 nM) for different periods of time. Supporting our model, a 1 hour exposure to ErSO was almost as effective as 24 hour exposure in inducing cell death (Fig. S8G).
ATP Depletion and Osmotic Stress are Sufficient for Activation of Rapid Necrotic Cell Death
Unlike wild type cells, TRPM4 knockout cells do not exhibit rapid cell swelling (Fig. 2E). Mostly studied in plant and yeast cells, cell swelling or oncosis is linked to osmotic stress-induced UPR activation (42–44). We therefore tested whether other agents that swell cells, and deplete ATP independent of TRPM4, recapitulate aspects of ErSO-induced, TRPM4-dependent, cell death. The ionophore ionomycin is a UPR activator (45) that causes both breast cancer cells to swell continuously over 6 hours, activating UPR markers p-PERK, p-eIF2α and sp-XBP1 (Fig. S9A-D). Ionomycin killed both the wild type and TRPM4 knockout cells equally well in 24 hours (Fig. 5A-D). 2-deoxyglucose interferes with glycolysis, resulting in a rapid decline in intracellular ATP (Fig. S9E-F) (35,46). We used 2-deoxyglucose under conditions in which the decline in ATP levels it causes is in the range of ATP decline produced by ErSO. While 2-deoxyglucose alone did not induce cell death, 2-deoxyglucose used in combination with ionomycin produced a synergistic effect and nearly all MCF-7 and MCF-7 TKO (MTKO) cells died, and an increased number of T47D and T47D TKO (TTKO) cells died (Fig. 5A-D). Thus, an alternative way to swell cells and deplete ATP reproduces aspects of rapid ErSO-induced, TRPM4-dependent cell death.
Figure 5.
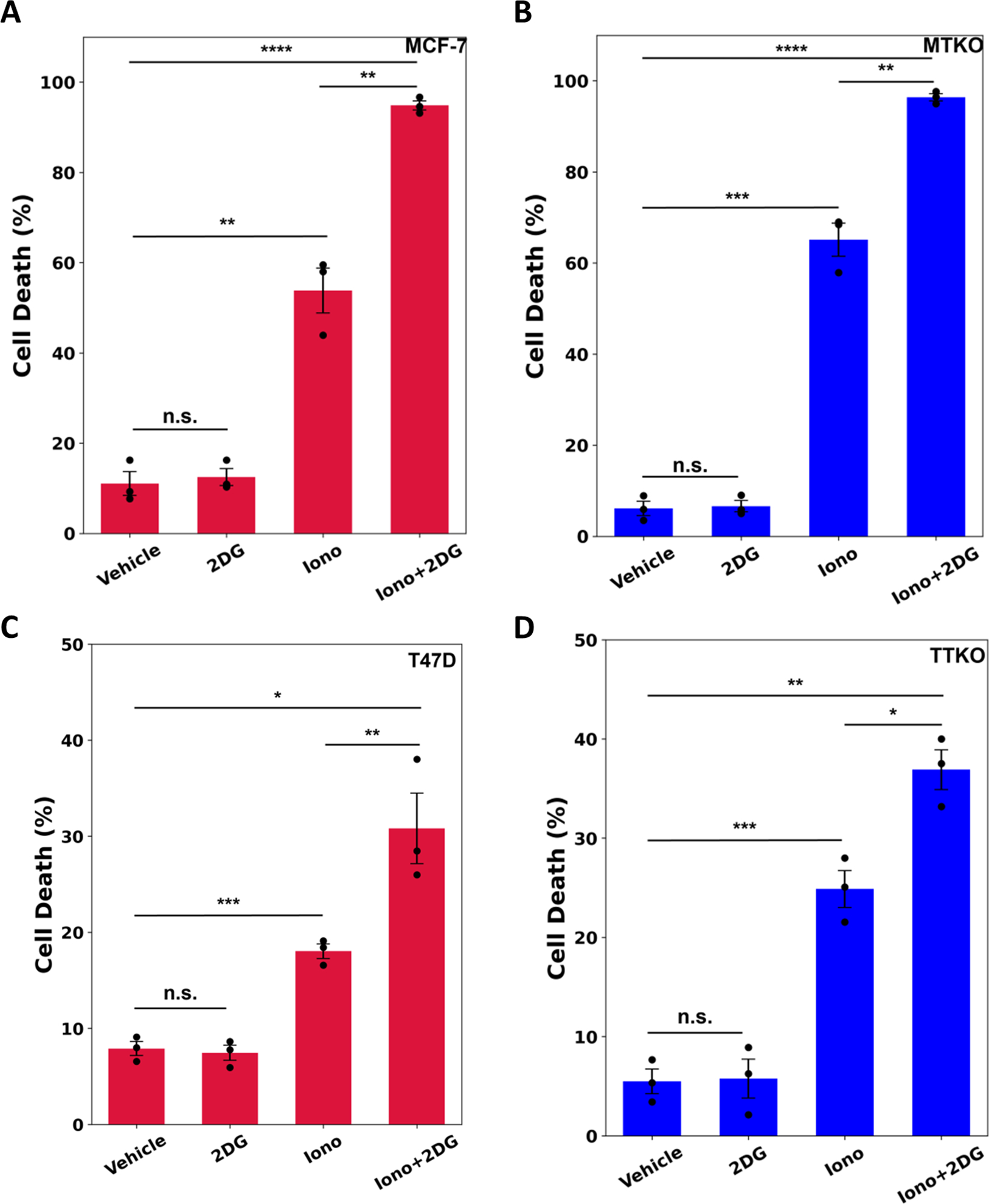
ErSO-induced rapid cell death is mimicked by ATP depletion and osmotic stress. A-D Automated Trypan Blue dye exclusion assay measuring percentage of cell death in A MCF-7, B MTKO, C T47D, and D TTKO cells pretreated for 1 hour with or without 10 mM 2-dexoyglucose (2DG), and then subsequently treated with vehicle or 10 μM ionomycin for 24 hours (n=3). Mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s. = not significant by Student’s t-test.
Diverse Necrosis-inducing Cancer Therapies use TRPM4
To explore the biological relevance of the TRPM4 necrosis pathway, we next investigated the requirement for TRPM4 in the actions of other necrosis-inducing cancer therapies. FDA-approved Apreptitant/Emend inhibits the neurokinin receptor, releasing stored calcium into the cell body (47). Widely used as an anti-nausea agent, early clinical work indicates aprepitant has strong activity against several types of cancer (48–50). In Calcium Electroporation (CaEP), electroporation transiently permeabilizes the plasma membrane allowing external calcium, provided by a calcium-containing buffer, to enter the cancer cells (51). Although mechanistically obscure, CaEP has shown great promise in European clinical trials against melanoma, lung and pancreatic cancer (19,52,53). LTX-315 is an oncolytic peptide, in clinical trials, that preferentially attacks mitochondrial membranes interfering with respiration, activating the UPR, and inducing immunologic cell death (ICD). Consistent with its ability to induce ICD, LTX-315 shows impressive effectiveness as a cancer vaccine (54,55). Preclinical Englerin A opens the plasma membrane TRPC4/C5 Ca2+ channel increasing intracellular calcium in renal cancer cells (50,56). We therefore used a renal cancer cell line, A498 and its TKO counterpart (ATKO) in Englerin experiments (Fig. S10A). Notably, TRPM4 knockout also strongly reduced the ability of each of these unrelated necrosis-inducing anticancer therapies to rapidly kill cancer cells (Fig. 6A-D). Moreover, TRPM4 knockout largely reversed the ability of most of these cancer therapies to induce cell swelling, deplete intracellular ATP, release LDH into the medium, and in the case of CaEP, blocked inhibition of cellular proliferation 24 hours post-treatment (Fig. 6E-G, S10B-G). TRPM4 knockout also significantly reduced release of the prototypical DAMP, High Mobility Group Box1 (HMGB1) that is normally released into the medium following use of these necrosis-inducing therapies (Fig. 6H-K). HMGB1 is a nuclear protein released from dying necrotic cells. Several types of immune cells contain HMGB1 receptors and the net effect of HMGB1 release on immune cells is usually activation of ICD.
Figure 6.
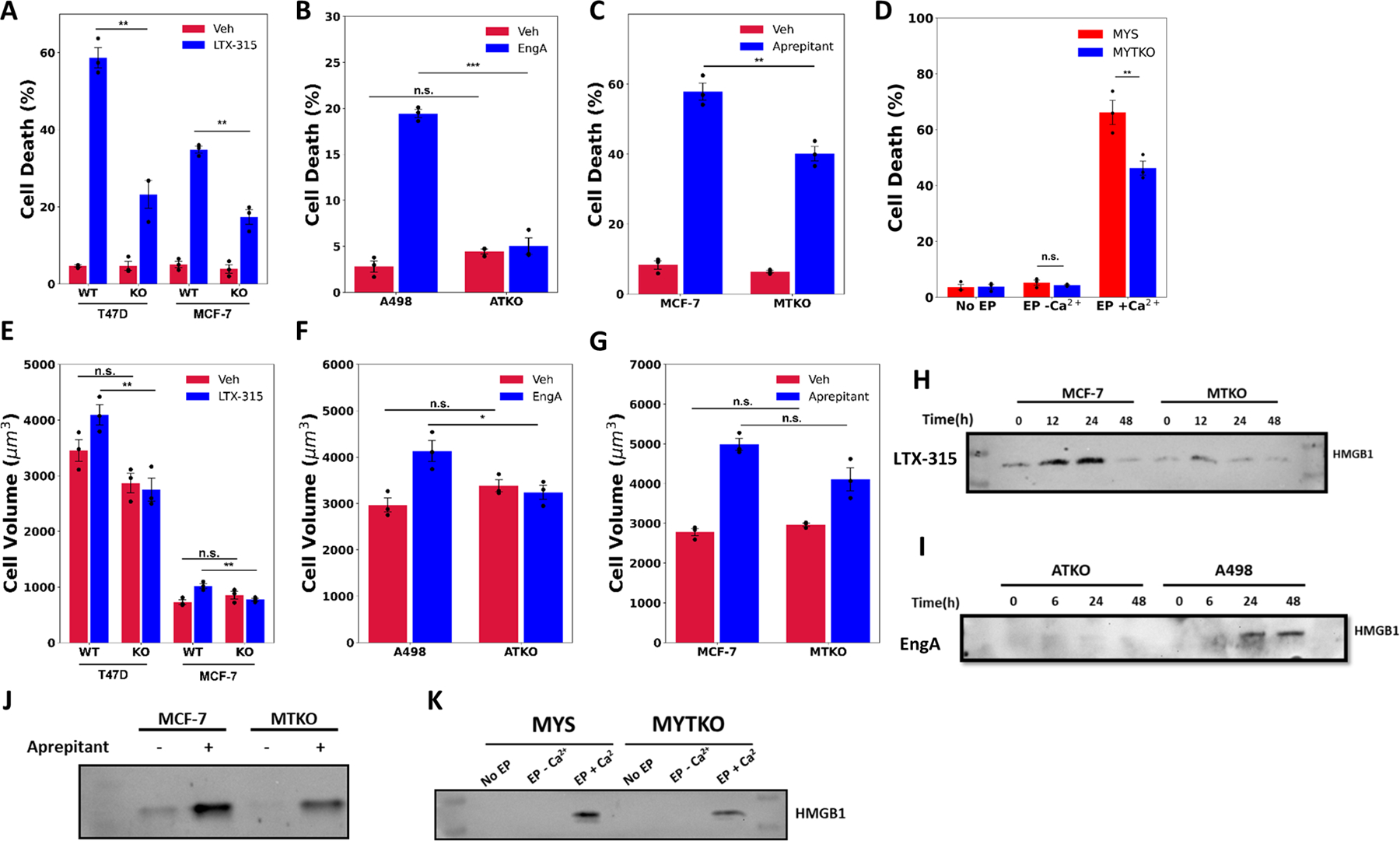
Diverse necrosis-inducing anticancer therapies work through TRPM4. A-D Automated Trypan Blue viability assay in; A T47D, MCF-7 and their TRPM4 KO clones (TTKO and MTKO) treated with either vehicle or 25 μg/ml LTX-315; B A498 and ATKO cells treated with either vehicle or 1 μM Englerin A (EngA) (note change in Y axis); C MCF-7 and MTKO cells, treated with either vehicle or 30 μM Aprepitant, all for 24 hours; and D MYS and MYTKO cells subjected to no electroporation (No EP, set to 100%), reversible electroporation in the absence of calcium (EP −Ca2+), or in the presence of calcium (EP +Ca2+). E-G Cell volume measurements in E T47D, MCF-7 and their TRPM4 KO clones treated with either vehicle or 50 μg/ml LTX-315 for 2 hours, F A498 and ATKO cells treated with either vehicle or 1 μM EngA for 2 hours, and G MCF-7 and MTKO cells, treated with either vehicle or 30 μM Aprepitant for 3 hours. H-K Western blot analysis of HMGB1 protein released into the medium from H MCF-7 and MTKO cells treated with either vehicle or 25 μg/ml LTX-315 for 0, 12, 24 and 48 hours, I A498 and ATKO cells treated with either vehicle or 1 μM EngA for 0, 6, 24 and 48 hours, and J MCF-7 and MTKO cells treated with either vehicle or 30 μM Aprepitant for 24 hours, and K MYS and MYTKO cells reversibly electroporated (EP) with (EP +Ca2+) or without (EP −Ca2+) calcium buffer, and compared to no electroporation control (No EP). N=3 biological replicates, mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, n.s. = not significant by Student’s t-test.
For these anticancer therapies, pathways other than necrosis likely contribute to cell death (47,57), making their dependence on TRPM4 less complete than with ErSO. These data indicate that several unrelated cancer therapies, with diverse ways of initiating necrosis, converge at a common TRPM4 pathway for necrotic cell death.
ErSO Induces TRPM4-dependent Release of Immune Cell Activating Components
Many solid tumors do not respond to immunotherapy, in part because they lack certain immune cells. Necrotic corpse cells, exposed calreticulin and extracellular DAMPs, such as HMGB1 and ATP all contribute to immune cell activation (58). We tested ErSO on MCF-7 breast cancer cells to see whether release of DAMPs and resulting immune cell activation are TRPM4-dependent. TRPM4 knockout largely blocked calreticulin translocation to the plasma membrane, and release of HMGB1 and ATP into the medium from ErSO-treated breast and ovarian cancer cells (Fig. 7A-B, S11A-C).
Figure 7.
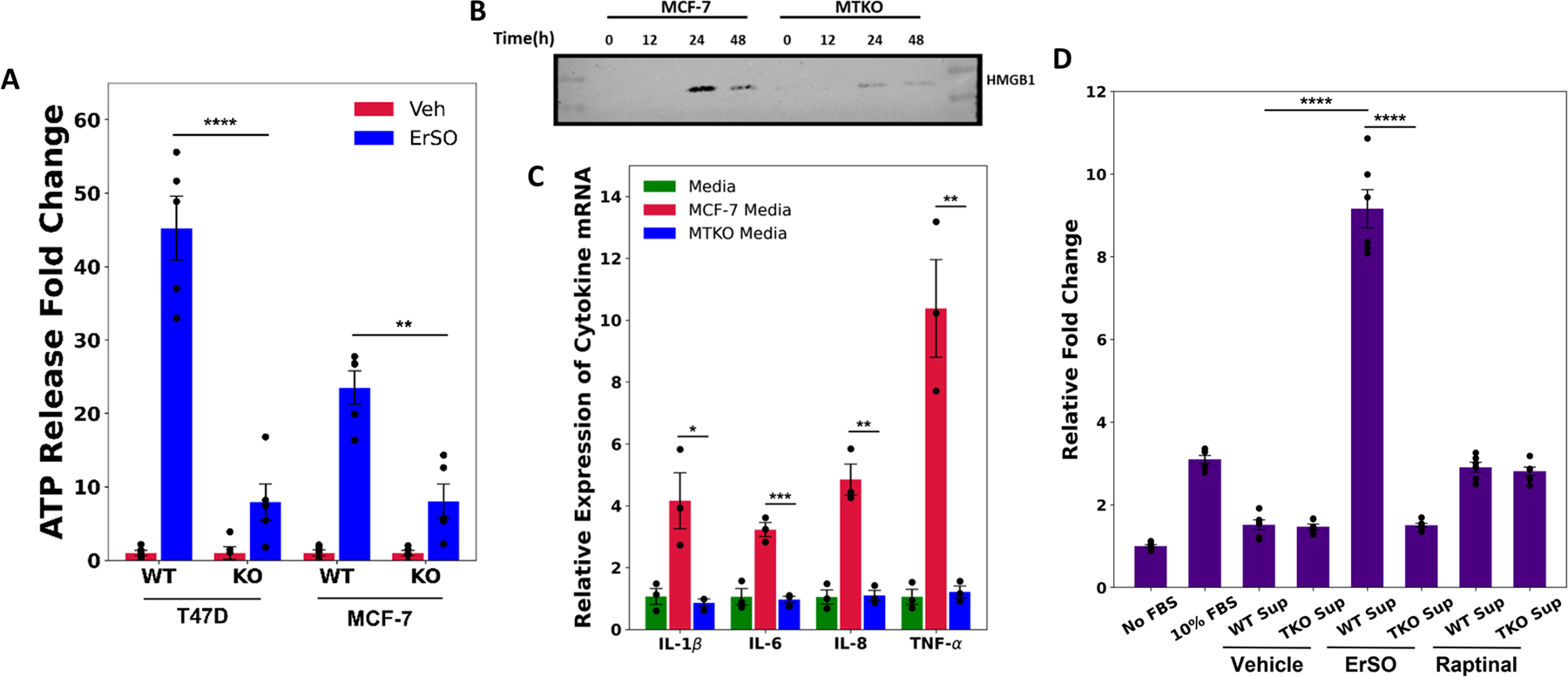
ErSO induces TRPM4-dependent immune cell activation. A Relative fold change of ATP (a DAMP) released into the medium from T47D, TTKO, MCF-7 and MTKO cells treated with either vehicle or 100 nM ErSO for 6 hours. B Western blot analysis of HMGB1 protein released into the medium from MCF-7 and MTKO cells treated with 100 nM ErSO for 0, 12, 24 and 48 hours. C Relative expression levels of IL-1β, IL-6, IL-8 and TNF-α mRNA levels from PMA-differentiated THP-1 cells treated with control fresh medium, or with medium from MCF-7 or MTKO cells pretreated with 1 μM ErSO. To ensure that the mRNA level changes observed were not due to potential effects of ErSO on differentiated THP-1 cells, ErSO was removed from the treatment medium after 4 hours and the MCF-7/MTKO were allowed to incubate in the ErSO-free medium for another 20 hours. The MCF-7 cells continue to die while the MTKO cells do not. D Undifferentiated THP-1 monocytes were analyzed by a migration assay using uncoated 5 μm membrane filters. The bottom chamber was filled with: medium with no FBS, medium containing 10% FBS, medium from MCF-7 or MTKO cells treated with vehicle or with 1 μM ErSO (as described above), or medium from MCF-7 or MTKO cells treated with 1 μM of the potent apoptosis inducer Raptinal. Migration was allowed to proceed for 4 hours. The medium from the bottom chambers were transferred to 96-well plates and an Alamar Blue assay was performed to measure the relative number of live cells that have successfully migrated across the membrane. Shown are relative fold changes in fluorescence with No FBS set to 1. A,E,F: Mean ± s.e.m; n=3. *p<0.05, **p<0.01, ***p<0.001, ****p<0.00.
To test for TRPM4-dependent release of DAMPs and subsequent immune cell activation, wild type or TRPM4 knockout breast cancer cells were treated with ErSO; ErSO was subsequently washed out and the medium was used to treat PMA-differentiated human THP-1 macrophage. Medium from the wild type cells, but not from the TRPM4 knockout cells induced a rapid and robust increase in expression of mRNAs encoding several pro-inflammatory cytokines, suggestive of an M1-polarized macrophage phenotype (Fig. 7C, S11D) (59).
We next used a variant of our Invasion-Dissociation-Rebinding assay (schematic in Fig. S11E) (34) to evaluate the ability of medium from ErSO-treated cells, to promote migration of undifferentiated THP-1 monocytes across a filter (Fig. 7D). Notably, we observed a ~9-fold increase in migration with medium from ErSO-treated wild type cells. TRPM4 knockout abolished this increase in migration. In contrast, medium from wild type and TRPM4 knockout cells treated with the apoptosis inducer, raptinal (60), only increased migration ~3-fold, about as well as fetal bovine serum. Taken together, these results highlight the TRPM4-dependent immunotherapeutic potential of ErSO and suggest a direction to addressing barriers to immunotherapy.
Discussion
Although most current cancer therapies are either cytostatic or trigger apoptotic cell death, several diverse emerging therapies trigger necrotic cell death. While cell responses to these necrosis-inducing therapies share some common features including cell swelling and ATP depletion, and usually elevated intracellular calcium, whether these therapies converged on a common protein-based pathway for necrotic cell death was unknown. We demonstrate that the calcium-activated, ATP-inhibited sodium channel, TRPM4, plays a major role in necrotic cell death induced by 6 anticancer therapies (BHPI, ErSO, LTX-315, Englerin A, calcium electroporation and Aprepitant). What these diverse therapies share mechanistically is that they typically elevate intracellular calcium (6,8,47,52,61,62). This elevated calcium acts via the TRPM4-bound calcium sensor calmodulin to open the TRPM4 Na+ channel, initiating a common pathway for necrotic cell death (model in Fig. S8F) (10,63). This pathway exploits the ~10 fold higher concentration of Na+ outside the cell. Na+ enters the cell through the open TRPM4 channel; to balance the charge, Cl− enters through passive chloride channels and water enters to maintain osmolality. This swells the cells. Unexpectedly, our data indicates that it is this cell swelling and the resultant osmotic stress that sustains the lethal hyperactivation of the anticipatory UPR induced by BHPI and ErSO.
These studies highlight an important difference between ErSO and widely used inhibitor therapies for ERα positive breast cancer, such as aromatase inhibitors, tamoxifen and fulvestrant, in that ErSO activates a specific ERα linked pathway. Most inhibitory anticancer drugs must be continuously present at a therapeutically relevant concentration to block proliferation of cancer cells. In striking contrast, since ErSO acts to trigger a-UPR activation, a brief 1-hour exposure to ErSO was nearly as effective as continuous exposure in inducing death of breast cancer cells (Fig. S8G). This is consistent with our earlier work that two or three doses of ErSO a week apart induced strong regression of breast tumors in mice (6). These data support a path to clinical transition for ErSO that might minimize toxic effects by employing a dosing regimen based on brief intermittent exposure to transiently reach the desired blood level of ErSO, rather than a more typical dosing regimen that aims at sustaining a persistent serum concentration in the therapeutic range.
ErSO is fully effective in ERα positive breast cancer cells in the presence and absence of estrogen – indicating it is not competing with estrogens for binding to ERα and unlike tamoxifen and fulvestrant, ErSO is not a selective estrogen receptor modulator or degrader (SERM or SERD). Since a brief transient exposure to ErSO triggers death of breast cancer cells, the ability of estrogen-ERα to modestly down-regulate TRPM4 mRNA and protein over several days is unlikely to impact the response to ErSO. It is likely that once a-UPR activation is triggered above a threshold level, TRPM4-mediated sodium influx, cell swelling and osmotic stress sustain the necrosis pathway.
Interestingly, estrogen moderately down-regulated TRPM4 mRNA and protein. We previously showed that estrogen binding to ERα activates a protective pro-proliferation anticipatory UPR pathway (7) and does not induce cell death. It is possible that the modest down-regulation of TRPM4 by estrogen-ERα helps orient the activation of the anticipatory UPR pathway toward a pro-proliferation survival mode and away from activation of the necrosis inducing opening of the TRPM4 channel.
Although the anticancer therapies we investigated are all reported to induce necrotic cell death, cell death is often a continuum with both necrotic and apoptotic characteristics (1,4,64). BHPI and ErSO exhibit a nearly pure necrosis phenotype, in part because they robustly inhibit protein synthesis (Fig. S1D). By blocking protein synthesis at both initiation and elongation, ErSO and BHPI block production of CHOP protein (C/EBP Homologous Protein), a normally UPR-induced apoptosis activator (5). For ErSO and BHPI, since sustained lethal UPR hyperactivation is abolished by TRPM4 knockout, their action exhibits complete dependence on TRPM4. For therapies, such as Aprepitant, where cell death has both necrotic and apoptotic characteristics (49,57), the dependence on TRPM4 is partial. Although cell death induced by the oncolytic peptide LTX-315 is by necrosis (55,61), since it only preferentially targets mitochondrial membranes and also non-specifically permeabilizes other cell membranes, the dependence on TRPM4 is also only partial. Less is known about the detailed mechanisms of action of Englerin A and CaEP, which exhibit strong, but not complete, TRPM4 dependence. While diverse therapies use the TRPM4-mediated necrosis pathway to target cancer cells, necrosis is currently broadly defined and a few therapies, such as irreversible electroporation without added calcium, which non-specifically opens pores in membranes, can induce TRPM4-independent necrosis (19,52).
There are several limitations to our study that merit discussion. While the level of TRPM4 is related to response to ErSO, since all the necrosis-inducing therapies act indirectly to open the TRPM4 channel, levels of other pathway components will also impact responses. Therefore, while the level of intratumoral TRPM4 will help identify patients in current and future clinical trials most likely to benefit from these necrosis-inducing immunogenic therapies, it will only be one factor in predicting patient response. Although our data shows that TRPM4 plays a pivotal role in anticancer therapy-induced necrosis, given the complexity of other cell death pathways, there are almost certainly additional currently unidentified proteins that impact the necrosis pathway. These studies were carried out in human cells in culture and in human tumors in mice; responses in cancer patients may differ.
ATP depletion is common in necrotic cell death and contributes to BHPI-, ErSO- and ionomycin-induced membrane rupture and cell death (1,65). ATP depletion likely contributes to necrotic cell death in several ways. It helps maintain TRPM4 in the open state facilitating sodium entry and sustaining cell swelling. Since ATP is required for apoptosome formation (66), ATP depletion may orient cell death away from apoptosis and toward necrosis. Moreover, ATP depletion activates AMPK, inhibiting protein synthesis at elongation by phosphorylation of eEF2, blocking synthesis of pro-apoptotic CHOP (8). We speculate that ATP depletion may also impair the cell’s ability to carry out ATP-dependent reorganization of the cytoskeleton needed to adapt to the increase in cell volume and maintain plasma membrane integrity (67,68).
TRPM4 knockout completely abolished ErSO-induced regression of human breast cancer in mice and ErSO-treated TRPM4 knockout tumors exhibited continued robust growth. This might suggest loss of TRPM4 expression as a potential mechanism for development of resistance to ErSO. Notably, in our recent study demonstrating that ErSO induces near complete regression in several orthotopic models of primary and metastatic human breast cancer in mice, in more than 100 mice, we did not observe any ErSO-resistant tumors that would suggest possible loss of TRPM4 expression (6). In our mouse xenografts ErSO induced rapid near-complete tumor regression (Figs. S6 and S7G, See Day 3); thus the pool of surviving cells in which loss of TRPM4 expression might occur is small. To produce ErSO-resistant cells exhibiting TRPM4 down-regulation in culture was challenging and required use of stepwise selection with progressively increasing concentrations of BHPI or ErSO that did not induce near-complete cell death. With the identification of the pivotal role of TRPM4 in several necrosis-inducing anticancer therapies that are either approved or in clinical trials, it will be interesting to see if loss of TRPM4 ultimately emerges as a resistance mechanism.
Unlike apoptotic cell death, which usually does not strongly activate immune cells, necrotic cell death often robustly activates immune cells and induces immunogenic cell death (ICD) (69,70). Consistent with this, ErSO-treated cancer cells exhibited TRPM4-dependent DAMP release which robustly activated immune cells and enhanced their migration (Fig. 7C-D). Coupled with the rapid death induced by ErSO, which usually kills >98% of breast cancer cells in 3 days (Fig. S7G), the potential for acquired resistance by silencing TRPM4 is significantly reduced. Necrosis-inducing therapies that use DAMPs released from dying cancer cells and corpse cells to activate the patient’s immune system have an important therapeutic advantage (58,70); they likely only need to kill a fraction of the patient’s cancer cells to stimulate ICD of residual cancer cells. In part because necrotic cell death was characterized largely by changes in morphology and lacked a protein mediator, there has been limited interest in developing new cancer therapies targeting necrosis. Identifying TRPM4 as the key executioner protein in anticancer therapy-induced necrosis will catalyze development of new necrosis-inducing therapies and the successful application of necrosis-inducing cancer therapies in the clinic.
Supplementary Material
Significance:
A genome-wide CRISPR screen reveals a pivotal role for TRPM4 in cell death and immune activation following treatment with diverse necrosis-inducing anticancer therapies, which could facilitate development of necrosis-based cancer immunotherapies.
ACKNOWLEDGEMENTS
Fluorescence microscopy was performed in the Dept. of Cell and Developmental Biology Microscopy Facility; we are grateful to C.V. Neha for training in fluorescence microscopy and to Dr. Sandra McMasters of the Cell/Media Facility for preparation of human physiologically relevant medium.
Funding:
This research was supported by:
National Institutes of Health grant RO1DKO71909 (DJS)
National Institutes of Health grant RO1CA265333 (DJS)
National Institutes of Health grant R01CA258746 (PJH)
National Institutes of Health grant R01CA234025 (ERN)
Department of Defense Breast Cancer Research Program grant (ERN)
University of Illinois (PJH)
Cancer Center at Illinois Seed Grant (DJS)
Susan G. Komen Foundation and the Breast Cancer Research Foundation (BHP)
Michael Recny Predoctoral Fellowship (SG)
Herbert Carter Predoctoral Fellowship (DD)
US-Pak Knowledge Corridor Scholarship (MJ)
NSF Graduate Research Fellowship DGE 1144245 (MRL)
NIH Chemistry-Biology Interface Traineeship T32-GM136629 (MWB)
ACS Medicinal Chemistry Predoctoral Fellowship (MWB)
NIH-NCI F99 Predoctoral Fellowship F99-CA253731 (MWB)
Amend Family Foundation (DJS)
Footnotes
Conflict of Interest Disclosure Statement
The Univ. of Illinois has filed patent applications on some compounds described here on which DJS, MWB and PJH are co-inventors (US 20200190029; U.S. Patent Application No. 63/055,583). Some compounds described herein have been licensed to Systems Oncology. PJH is a consultant for Systems Oncology and is on the Systems Oncology SAB.
LIST OF SUPPLEMENTARY MATERIALS
Supplementary table 1, List of Primers
Caption for Supplementary Movies S1-S4
References
REFERENCES
- 1.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997. May 15;57(10):1835–40. [PubMed] [Google Scholar]
- 2.Westman J, Grinstein S, Marques PE. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Frontiers in Immunology [Internet] 2020. [cited 2022 Aug 11];10. Available from: 10.3389/fimmu.2019.03030 [DOI] [PMC free article] [PubMed]
- 3.Shubin AV, Demidyuk IV, Komissarov AA, Rafieva LM, Kostrov SV. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016. Aug 23;7(34):55863–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2013. Dec 1;1833(12):3448–59. [DOI] [PubMed] [Google Scholar]
- 5.Livezey M, Huang R, Hergenrother PJ, Shapiro DJ. Strong and sustained activation of the anticipatory unfolded protein response induces necrotic cell death. Cell Death Differ 2018. Oct;25(10):1796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudreau MW, Duraki D, Wang L, Mao C, Kim JE, Henn MA, et al. A small-molecule activator of the unfolded protein response eradicates human breast tumors in mice. Sci Transl Med 2021. Jul 21;13(603):eabf1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ. Anticipatory Estrogen Activation of the Unfolded Protein Response is Linked to Cell Proliferation and Poor Survival in Estrogen Receptor α Positive Breast Cancer. Oncogene 2015. Jul;34(29):3760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andruska ND, Zheng X, Yang X, Mao C, Cherian MM, Mahapatra L, et al. Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A 2015. Apr 14;112(15):4737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu L, Wang L, Kim JE, Mao C, Shapiro DJ. Src couples estrogen receptor to the anticipatory unfolded protein response and regulates cancer cell fate under stress. Biochim Biophys Acta Mol Cell Res 2020. Jun 2;118765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, Kaschitza DR, Essers M, Arullampalam P, Fujita T, Abriel H, et al. Pathological activation of CaMKII induces arrhythmogenicity through TRPM4 overactivation. Pflugers Arch 2021. Mar;473(3):507–19. [DOI] [PubMed] [Google Scholar]
- 11.Nilius B, Prenen J, Voets T, Droogmans G. Intracellular nucleotides and polyamines inhibit the Ca2+-activated cation channel TRPM4b. Pflugers Arch 2004. Apr;448(1):70–5. [DOI] [PubMed] [Google Scholar]
- 12.Feng J, Zong P, Yan J, Yue Z, Li X, Smith C, et al. Upregulation of transient receptor potential melastatin 4 (TRPM4) in ventricular fibroblasts from heart failure patients. Pflugers Arch 2021. Mar;473(3):521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J, Bengtson CP, Buchthal B, Hagenston AM, Bading H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020. Oct 9;370(6513):eaay3302. [DOI] [PubMed] [Google Scholar]
- 14.Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, et al. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med 2009. Feb;15(2):185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boudreau MW, Mulligan MP, Shapiro DJ, Fan TM, Hergenrother PJ. Activators of the Anticipatory Unfolded Protein Response with Enhanced Selectivity for Estrogen Receptor Positive Breast Cancer. J Med Chem 2022. Mar 10;65(5):3894–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao C, Livezey M, Kim JE, Shapiro DJ. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Sci Rep 2016. Oct 7;6:34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017. Apr 6;169(2):258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Köster J, Xu H, Chen CH, Xiao T, Liu JS, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biology 2015. Dec 16;16(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frandsen SK, Gissel H, Hojman P, Tramm T, Eriksen J, Gehl J. Direct therapeutic applications of calcium electroporation to effectively induce tumor necrosis. Cancer Res 2012. Mar 15;72(6):1336–41. [DOI] [PubMed] [Google Scholar]
- 20.Rivas J, Díaz N, Silva I, Morales D, Lavanderos B, Álvarez A, et al. KCTD5, a novel TRPM4-regulatory protein required for cell migration as a new predictor for breast cancer prognosis. FASEB J 2020. Jun;34(6):7847–65. [DOI] [PubMed] [Google Scholar]
- 21.Sagredo AI, Sagredo EA, Cappelli C, Báez P, Andaur RE, Blanco C, et al. TRPM4 regulates Akt/GSK3-β activity and enhances β-catenin signaling and cell proliferation in prostate cancer cells. Mol Oncol 2018. Feb;12(2):151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sagredo AI, Sagredo EA, Pola V, Echeverría C, Andaur R, Michea L, et al. TRPM4 channel is involved in regulating epithelial to mesenchymal transition, migration, and invasion of prostate cancer cell lines. J Cell Physiol 2019. Mar;234(3):2037–50. [DOI] [PubMed] [Google Scholar]
- 23.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 2015. Oct;12(10):573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun 2017. Nov 30;8:1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leslie TK, James AD, Zaccagna F, Grist JT, Deen S, Kennerley A, et al. Sodium homeostasis in the tumour microenvironment. Biochim Biophys Acta Rev Cancer 2019. Dec 1;1872(2):188304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tashiro M, Tursun P, Konishi M. Intracellular and Extracellular Concentrations of Na+ Modulate Mg2+ Transport in Rat Ventricular Myocytes. Biophysical Journal 2005. Nov 1;89(5):3235–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hazalin NAMN, Liao P, Hassan Z. TRPM4 inhibition improves spatial memory impairment and hippocampal long-term potentiation deficit in chronic cerebral hypoperfused rats. Behav Brain Res 2020. Sep 1;393:112781. [DOI] [PubMed] [Google Scholar]
- 28.Woo SK, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Gerzanich V, Simard JM. SUR1-TRPM4 channels, not KATP, mediate brain swelling following cerebral ischemia. Neurosci Lett 2020. Jan 23;718:134729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilco-Ferreto N, Calaf GM. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol 2016. Aug;49(2):753–62. [DOI] [PubMed] [Google Scholar]
- 30.Yao W, Yu X, Fang Z, Yin P, Zhao C, Li N, et al. Profilin1 facilitates staurosporine-triggered apoptosis by stabilizing the integrin β1-actin complex in breast cancer cells. J Cell Mol Med 2012. Apr;16(4):824–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dankó T, Petővári G, Sztankovics D, Moldvai D, Raffay R, Lőrincz P, et al. Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells. Int J Mol Sci 2021. Jul 27;22(15):8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shahsavari Z, Karami-Tehrani F, Salami S. Shikonin Induced Necroptosis via Reactive Oxygen Species in the T-47D Breast Cancer Cell Line. Asian Pac J Cancer Prev 2015;16(16):7261–6. [DOI] [PubMed] [Google Scholar]
- 33.Yoon MJ, Kang YJ, Lee JA, Kim IY, Kim MA, Lee YS, et al. Stronger proteasomal inhibition and higher CHOP induction are responsible for more effective induction of paraptosis by dimethoxycurcumin than curcumin. Cell Death Dis 2014. Mar 13;5:e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L, Wang L, Mao C, Duraki D, Kim JE, Huang R, et al. Estrogen-independent Myc overexpression confers endocrine therapy resistance on breast cancer cells expressing ERαY537S and ERαD538G mutations. Cancer Lett 2019. Feb 1;442:373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seyedabadi M, Ghahremani MH, Ostad SN. ATP depletion as a consequence of hypoxia enhances tamoxifen antiproliferative effects in T47D breast carcinoma cells. Oncol Res 2009;18(5–6):221–8. [DOI] [PubMed] [Google Scholar]
- 36.Luhr M, Torgersen ML, Szalai P, Hashim A, Brech A, Staerk J, et al. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J Biol Chem 2019. May 17;294(20):8197–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 2003. Aug;8(4):345–52. [DOI] [PubMed] [Google Scholar]
- 38.Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis 2017. Oct 1;22(10):1189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sprooten J, De Wijngaert P, Vanmeerbeerk I, Martin S, Vangheluwe P, Schlenner S, et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020. Aug 1;9(8):E1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pawlikowska M, Jędrzejewski T, Brożyna AA, Wrotek S. Protein-Bound Polysaccharides from Coriolus Versicolor Induce RIPK1/RIPK3/MLKL-Mediated Necroptosis in ER-Positive Breast Cancer and Amelanotic Melanoma Cells. Cell Physiol Biochem 2020. Jun 13;54(4):591–604. [DOI] [PubMed] [Google Scholar]
- 41.Ohkura M, Sasaki T, Sadakari J, Gengyo-Ando K, Kagawa-Nagamura Y, Kobayashi C, et al. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. PLoS One 2012;7(12):e51286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alves MS, Reis PAB, Dadalto SP, Faria JAQA, Fontes EPB, Fietto LG. A novel transcription factor, ERD15 (Early Responsive to Dehydration 15), connects endoplasmic reticulum stress with an osmotic stress-induced cell death signal. J Biol Chem 2011. Jun 3;286(22):20020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dragosits M, Stadlmann J, Graf A, Gasser B, Maurer M, Sauer M, et al. The response to unfolded protein is involved in osmotolerance of Pichia pastoris. BMC Genomics 2010. Mar 26;11:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dragosits M, Mattanovich D, Gasser B. Induction and measurement of UPR and osmotic stress in the yeast Pichia pastoris. Methods Enzymol 2011;489:165–88. [DOI] [PubMed] [Google Scholar]
- 45.Kerr SR, Katz SG. Activation of the Unfolded Protein Response Pathway in Cytotoxic T Cells: A Comparison Between in vitro Stimulation, Infection, and the Tumor Microenvironment. Yale J Biol Med 2019. Dec 20;92(4):675–85. [PMC free article] [PubMed] [Google Scholar]
- 46.Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer 2002. Sep;87(7):805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muñoz M, González-Ortega A, Salinas-Martín MV, Carranza A, Garcia-Recio S, Almendro V, et al. The neurokinin-1 receptor antagonist aprepitant is a promising candidate for the treatment of breast cancer. Int J Oncol 2014. Oct;45(4):1658–72. [DOI] [PubMed] [Google Scholar]
- 48.Robinson P, Rosso M, Muñoz M. Neurokinin-1 Receptor Antagonists as a Potential Novel Therapeutic Option for Osteosarcoma Patients. J Clin Med 2023. Mar 9;12(6):2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Y, Wang X, Meng Y, Ma J, Zhang Q, Shao G, et al. A Novel Mechanism of Endoplasmic Reticulum Stress- and c-Myc-Degradation-Mediated Therapeutic Benefits of Antineurokinin-1 Receptor Drugs in Colorectal Cancer. Adv Sci (Weinh) 2021. Nov;8(21):e2101936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sulzmaier FJ, Li Z, Nakashige ML, Fash DM, Chain WJ, Ramos JW. Englerin a selectively induces necrosis in human renal cancer cells. PLoS One 2012;7(10):e48032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frandsen SK, Vissing M, Gehl J. A Comprehensive Review of Calcium Electroporation -A Novel Cancer Treatment Modality. Cancers (Basel) 2020. Jan 25;12(2):E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudno-Rudzińska J, Kielan W, Guziński M, Kulbacka J. Effects of calcium electroporation, electrochemotherapy, and irreversible electroporation on quality of life and progression-free survival in patients with pancreatic cancer: IREC clinical study. Adv Clin Exp Med 2021. Jul;30(7):765–70. [DOI] [PubMed] [Google Scholar]
- 53.Kraemer MM, Tsimpaki T, Berchner-Pfannschmidt U, Bechrakis NE, Seitz B, Fiorentzis M. Calcium Electroporation Reduces Viability and Proliferation Capacity of Four Uveal Melanoma Cell Lines in 2D and 3D Cultures. Cancers (Basel) 2022. Jun 11;14(12):2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Camilio KA, Rekdal O, Sveinbjörnsson B. LTX-315 (Oncopore™): A short synthetic anticancer peptide and novel immunotherapeutic agent. Oncoimmunology 2014;3:e29181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sveinbjørnsson B, Camilio KA, Haug BE, Rekdal Ø. LTX-315: a first-in-class oncolytic peptide that reprograms the tumor microenvironment. Future Med Chem 2017. Aug;9(12):1339–44. [DOI] [PubMed] [Google Scholar]
- 56.Batova A, Altomare D, Creek KE, Naviaux RK, Wang L, Li K, et al. Englerin A induces an acute inflammatory response and reveals lipid metabolism and ER stress as targetable vulnerabilities in renal cell carcinoma. PLoS One 2017;12(3):e0172632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Javid H, Afshari AR, Zahedi Avval F, Asadi J, Hashemy SI. Aprepitant Promotes Caspase-Dependent Apoptotic Cell Death and G2/M Arrest through PI3K/Akt/NF-κB Axis in Cancer Stem-Like Esophageal Squamous Cell Carcinoma Spheres. Biomed Res Int 2021;2021:8808214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012. Dec;12(12):860–75. [DOI] [PubMed] [Google Scholar]
- 59.Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 2018. Sep;233(9):6425–40. [DOI] [PubMed] [Google Scholar]
- 60.Palchaudhuri R, Lambrecht MJ, Botham RC, Partlow KC, van Ham TJ, Putt KS, et al. A Small Molecule that Induces Intrinsic Pathway Apoptosis with Unparalleled Speed. Cell Reports 2015. Dec 1;13(9):2027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H, Forveille S, Sauvat A, Yamazaki T, Senovilla L, Ma Y, et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis 2016. Mar 10;7:e2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carson C, Raman P, Tullai J, Xu L, Henault M, Thomas E, et al. Englerin A Agonizes the TRPC4/C5 Cation Channels to Inhibit Tumor Cell Line Proliferation. PLoS One 2015;10(6):e0127498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nilius B, Prenen J, Tang J, Wang C, Owsianik G, Janssens A, et al. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J Biol Chem 2005. Feb 25;280(8):6423–33. [DOI] [PubMed] [Google Scholar]
- 64.Arakawa S, Nakanomyo I, Kudo-Sakamoto Y, Akazawa H, Komuro I, Shimizu S. Identification of a novel compound that inhibits both mitochondria-mediated necrosis and apoptosis. Biochemical and Biophysical Research Communications 2015. Nov 27;467(4):1006–11. [DOI] [PubMed] [Google Scholar]
- 65.Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 1998. Dec 28;102–103:139–42. [DOI] [PubMed] [Google Scholar]
- 66.Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 2007. Jan;14(1):56–65. [DOI] [PubMed] [Google Scholar]
- 67.Bershadsky AD, Gelfand VI. Role of ATP in the regulation of stability of cytoskeletal structures. Cell Biology International Reports 1983. Mar 1;7(3):173–87. [DOI] [PubMed] [Google Scholar]
- 68.Carlier MF. Actin polymerization and ATP hydrolysis. Adv Biophys 1990;26:51–73. [DOI] [PubMed] [Google Scholar]
- 69.Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol 2020. Dec;14(12):2994–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gamrekelashvili J, Greten TF, Korangy F. Immunogenicity of necrotic cell death. Cell Mol Life Sci 2015. Jan;72(2):273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.