

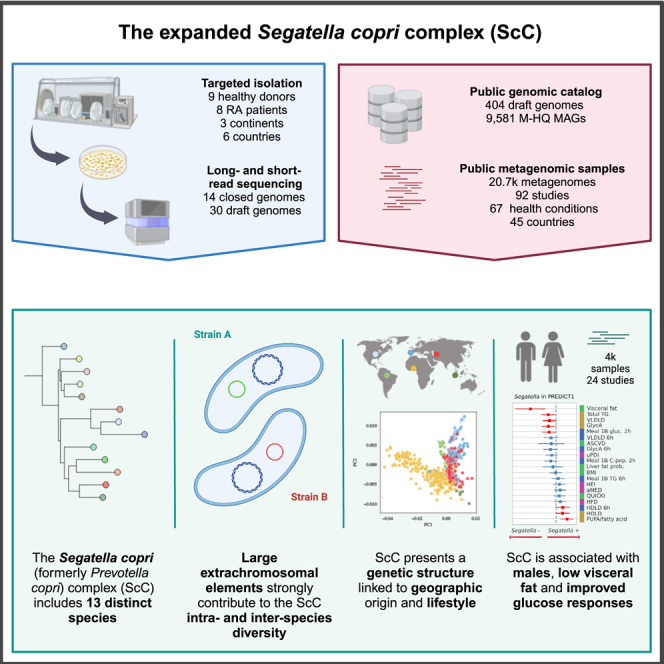
Summary
The Segatella copri (formerly Prevotella copri) complex (ScC) comprises taxa that are key members of the human gut microbiome. It was previously described to contain four distinct phylogenetic clades. Combining targeted isolation with large-scale metagenomic analysis, we defined 13 distinct Segatella copri-related species, expanding the ScC complex beyond four clades. Complete genome reconstruction of thirteen strains from seven species unveiled the presence of genetically diverse large circular extrachromosomal elements. These elements are consistently present in most ScC species, contributing to intra- and inter-species diversities. The nine species-level clades present in humans display striking differences in prevalence and intra-species genetic makeup across human populations. Based on a meta-analysis, we found reproducible associations between members of ScC and the male sex and positive correlations with lower visceral fat and favorable markers of cardiometabolic health. Our work uncovers genomic diversity within ScC, facilitating a better characterization of the human microbiome.
Keywords: Segatella copri, Prevotella copri, ScC, extrachromosomal element, bacterial isolation, human microbiome, metagenomics, cardiometabolic health
Graphical abstract
Highlights
-
•
Segatella copri (formerly Prevotella copri) complex includes 13 distinct species
-
•
Large extrachromosomal elements contribute to ScC intra- and inter-species diversities
-
•
ScC presents a genetic structure linked to geographic origin and lifestyle
-
•
ScC is associated with males, low visceral fat, and improved glucose responses
Prevalent human gut commensal bacteria feature a hidden variability hampering molecular and clinical studies. Blanco-Míguez et al. utilize cultivation-dependent and cultivation-independent approaches to describe the Segatella copri complex (ScC). They identify that ScC members contain distinct genomic elements and content as well as display variable association to host phenotypes.
Introduction
The human gut microbiome is a complex ecosystem shaped by microbial, environmental, and host-derived factors.1,2 Bacillota (formerly Firmicutes) and Bacteroidota (formerly Bacteroidetes) are the two phyla that typically dominate the bacterial fraction of these gut communities as detected by sequencing (avg. relative abundance of 51.5% ± 24% and 30% ± 23.8% in the gut of healthy adults, respectively3). Members of the families Bacteroidaceae and Prevotellaceae are the most prevalent and abundant Bacteroidota (on average 49.8% ± 32.4% and 24.6% ± 33.5% of Bacteroidota relative abundances, respectively3).4,5 Within these families, the gut microbiome of individuals living in Westernized populations are frequently dominated by species in the Bacteroides and Phocaeicola genera, whereas the genus Segatella (defined after the recent split of the genus Prevotella into seven distinct genera6) clearly dominates in individuals living in non-Westernized populations and following more traditional lifestyles.2,7,8,9,10,11 Factors such as the consumption of fiber-rich diets have been suggested to contribute to the enrichment in non-Westernized individuals and subgroups of Westernized individuals.12,13,14,15,16 Although an overabundance of Segatella spp. has been identified in various studies to be associated with specific diseases such as rheumatoid arthritis in Westernized populations,17,18,19 other studies failed to observe similar associations20,21 and even linked the presence of Segatella with an improved metabolic health after dietary interventions such as consumption of fiber-rich diets.15,22,23,24,25 Thus, whether the influence of Segatella spp., and specifically Segatella copri, on human health is positive or detrimental is yet to be clarified and may be determined by unknown genetic factors and species stratification.21
Recent advances in both metagenomic analysis and cultivation approaches are uncovering the taxonomic, genetic, and functional diversities of many prevalent gut bacteria that were not accessible with previous amplicon-based surveys. Faecalibacterium prausnitzii,26 Akkermansia muciniphila,27 and Agathobacter rectalis (formerly Eubacterium rectale)28 are some recent examples. Moreover, S. copri was shown to encompass a diversity overlooked by 16S rRNA gene analyses.20 S. copri was thus expanded into a sub-genus level taxonomic complex comprising four genetically distinct species-level clades with different functional and host profiles but highly conserved 16S rRNA gene sequences.20 The continued expansion of metagenomic and cultured bacteria collections together with novel strain-level functional assay and genetic tools29 is opening new opportunities to further characterize the S. copri-related taxa and their contribution to human health. However, the genetic diversity of the S. copri complex (ScC) is, to date, still unclear, limiting the overall understanding of its role in the gut microbiome, for instance, species- and strain-specific contributions to metabolic health and immune-mediated diseases.
To resolve the diversity within ScC, we performed large-scale studies combining (1) targeted isolation of 44 distinct strains followed by long-read sequencing of 14 strains with (2) in-depth metagenomic analyses of more than 20,000 metagenomes from humans and non-human primates (NHPs) with curated host information. This allowed us to identify 13 distinct bona fide species within ScC, expanding beyond the previously observed four clades.20 High-quality genome assembly enabled by long-read sequencing revealed the presence of a large and genetically diverse extrachromosomal element in almost all strains, with enhanced genetic diversification compared with that of the main chromosome. An analysis of metagenomic datasets corroborated the presence of these elements in S. copri-containing samples, identified striking genetic differences within species depending on their origin and lifestyle, and revealed a positive correlation of multiple but not all members of ScC with host characteristics.
Results
An integrated approach to study S. copri
We performed a targeted isolation of strains of ScC,20 obtaining 44 isolates initially identified as S. copri using S. copri-specific primers from fecal samples of human volunteers from three distinct cohorts (see STAR Methods and Table S1A). DNA from all 44 isolates was subjected to short-read sequencing, and additional PacBio long-read sequencing was obtained for thirteen representative strains. This yielded thirteen closed genomes, one genome with less than 3 contigs, and 30 high-quality draft genomes. We integrated these 44 genomes from our own isolates with (1) a collection of 404 isolate-derived genomes belonging to the Prevotella genus (prior to its recent reclassification into seven genera6) available in the NCBI GenBank database (as of June 2021)30 and (2) 9,581 medium-to-high-quality metagenome-assembled genomes (M-HQ MAGs) (completeness >50% and contamination <5%) belonging to species-level genome bins (SGBs)31 from the family-level genome bin (FGB) that contains all genomes assigned to the species previously assigned to Prevotella, including Segatella, in the MetaPhlAn 4 genomic database (version January 2021)3 (Figure 1A).
Figure 1.

The Segatella copri complex is composed of 13 different species
(A) Overall description of the data and analyses of this work.
(B) Phylogenetic tree spanning the 13 ScC species. Isolate genomes were integrated with MAGs. For each species, a maximum of 200 randomly selected genomes are shown. The tree highlights the well-defined taxonomic species based on inter-species vs. intra-species diversity. The n indicates the number of isolate genomes; the number of genomes sequenced in this work is reported in parenthesis. NHP, non-human primate.
(C) Genome characteristics for the ScC species by integrating available isolate genomes with reconstructed MAGs.
(D) Genetic distances within (intra-species) and between (inter-species) the ScC species, shown as pairwise average nucleotide identity distances (ANI distances).
(E) Jaccard distance based on pairwise gene content (UniRef90 families) within (intra-species) and between (inter-species) the ScC species. OS, other species. Boxplots in (E) show the median (center), 25th/75th percentile (lower/upper hinges), 1.5× interquartile range (whiskers), and outliers (points). See also Figure S1 and Table S1.
To investigate the species- and strain-level diversities of ScC and its association with human phenotypes and lifestyles, we leveraged 20,510 manually curated and publicly available human body metagenomes from 92 studies. These large catalogs span six human body sites (19,066 samples from the gastrointestinal tract, 743 from the oral cavity, 504 from the skin, 96 from the female urogenital tract, 93 from the airways, and 8 from breastmilk); four age categories (2,946 newborns, 2,075 children, 13,855 adults, 1,611 seniors, and 23 without reported age); sexes (8,795 males, 8,879 females, and 2,836 without reported sex); lifestyles (18,738 Westernized and 1,772 non-Westernized); and geography (45 countries).32 Additionally, we considered 24 ancient human stool metagenomes from previous studies20,33,34,35,36 (ranging from 5,300 to 200 years ago) and 203 from NHPs (22 species from 14 different countries in five continents, including primates living in the wild or held in captivity37) (Figure 1A; Table S1B).
ScC spans 13 distinct species
To characterize the diversity and structure of the Segatella genus, we computed the pairwise genetic distances between the 10,029 MAGs and isolate genomes classified as belonging to the genus Prevotella (prior to its recent reclassification into seven genera6) and clustered them into SGBs. We established a 5% genetic distance threshold for SGB definition as per previous validations31,38,39 and defined 165 SGBs (see STAR Methods). In the phylogeny built for these 165 SGBs (see STAR Methods), we identified ScC as the minimal monophyletic subtree that comprised all reference genomes labeled as S. copri in the NCBI GenBank database30 and resulted to be clearly distinct from other species (Figure S1). The expanded ScC20,21 comprised 9 additional species-level clades in addition to the four clades previously described (clades A–D),20 revealing a total of 13 distinct species (including the recently described species Segatella hominis, formerly Prevotella hominis,40 as clade E; Figure 1B).
Of the 9 additional S. copri clades, five (E–I) were mainly populated by strains reconstructed from the human gut microbiome, whereas the genomes of the other four clades (J–M) were only retrieved from NHPs. Seven clades (A–G) were supported by at least one previously sequenced isolate genome, whereas the single isolate genome of clade F has been sequenced in this work (strain RHB01T; see STAR Methods). The different clades exhibited relatively variable G + C content (GC) of genomic DNA (from 43.1% for clade E to 47.5% for clades K and I) and a number of core genes ranging from 907 for clade F to 1,567 for clade L (Figure 1C). Average nucleotide identity (ANI) between the 10,029 genomes showed limited intra-clade and high inter-clade distances in comparison with the common 5% threshold adopted to delineate species boundaries38 (Figure 1D), and this separation is also supported by distances computed on gene content (Figure 1E). Based on these results, we propose the naming of these clades as individual species: Segatella brunsvicensis (clade B), Segatella sinensis (clade C), Segatella brasiliensis (clade D), Segatella sanihominis (clade F), Segatella sinica (clade G), Candidatus Segatella caccae (clade H), Candidatus Segatella intestinihominis (clade I), Candidatus Segatella violae (clade J), Candidatus Segatella albertsiae (clade K), Candidatus Segatella mututuai (clade L), and Candidatus Segatella papionis (clade M) (protologs provided in the STAR Methods).
Complete genome reconstruction unveils LEEs and diverse extrachromosomal elements in ScC
The thirteen S. copri-related isolates, for which the combination of short- and long-read sequencing allowed us to obtain closed or nearly closed genomes, enabled us to investigate structural features of the genomes. These thirteen isolates represent seven distinct species (S. copri [clade A], S. brunsvicensis [clade B], S. sinensis [clade C], S. brasiliensis [clade D], S. hominis [clade E], S. sanihominis [clade F], and S. sinica [clade G]; Table S1C). In addition to the main replicon within the range of the expected size (from 3.32 to 4.26 Mb), we found the presence of an additional circular large extrachromosomal elements (LEEs) spanning from 72.4 to 328.8 kb (Figure 2A; Table S1C) in all species but S. brunsvicensis (clade B) and S. sanihominis (clade F), strongly contributing to the ScC intra- and inter-species diversities (Figures S2A and S2B). For S. sinensis (clade C), we noticed that one strain contained an LEE, whereas the other did not. Sequence similarity analysis of LEEs showed no significant (partial) match with previously described extrachromosomal elements or megaphages (see STAR Methods).41 Of note, various additional smaller plasmids were also detected in some of the strains (n = 0–3, ranges = 2.8–58.9 kb; Figure 2A; Table S1C).
Figure 2.

Most ScC species harbor a large extrachromosomal element
(A) Number of plasmids and variability of the main chromosome and LEE genomic characteristics between the different isolates. Main chromosome lengths are represented in terms of million (1e6) pair-bases, whereas LEE lengths are in terms of hundred thousand (1e5) pair-bases. TA, toxin-antitoxin system.
(B) Average nucleotide identity (ANI) between the main chromosome (top-right triangle) and LEE (bottom-left triangle).
(C) Pangenome presence/absence matrix of the different LEE from the 10 ScC genomes shows a highly diverse gene content between variants. Gene families are defined at 80% genomic identity. See also Table S1.
These LEEs from 10 isolates have a GC% that is, on average, 3.09% ± 2.3% lower than the corresponding main chromosome (Wilcoxon signed-rank test = 0.009, Figure 2A; Table S1C). Notably, most of these elements encoded a complete copy of a ribosomal operon (5S, 16S, and 23S rRNA genes were all found in 8 of the 10 isolates), various loci of toxin-antitoxin (TA) systems, and multiple transposases (Figure 2A). However, the majority of predicted genes in LEEs have an unknown function (69.88% ± 10.22% in comparison with 56.02% ± 1.34% in the main chromosome, Wilcoxon signed-rank test = 0.002). Pangenome analysis between the complete reconstructed genomes (see STAR Methods) revealed an enrichment of unique proteins (singletons) when compared with the main chromosome (33.24% ± 9.84% vs. 12.84% ± 2.53%, Wilcoxon signed-rank test = 0.002; see Key Resources Table). LEEs are highly diverse within isolates of the same species (avg. ANI = 85.03% ± 3.48%) especially in comparison with the corresponding main chromosome divergence (96.25% ± 0.28%, Wilcoxon signed-rank test = 0.0009), and this diversity is enlarged when comparing LEEs of isolates from different species (avg. ANI = 81.19% ± 1.19%, Wilcoxon signed-rank test = 3.65e−7) (Figure 2B). These big differences between LEEs are also reflected at the functional level, where only a few gene families are shared between the LEE variants (only 19.32% are present in at least two variants and just 6.11% in more than two; Figure 2C).
We then expanded the analysis of LEEs using the dataset of 20,7000 publicly available human and NHP shotgun metagenomes (Table S1B). We mapped the LEE variants from 10 isolates against the 8,477 samples for which, at least, one of the ScC species was detected using MetaPhlAn 4,3 and then, we reconstructed the LEE variants using a consensus-based metagenomic approach (see STAR Methods). In total, 4,683 samples (55.24% of the ScC-positive samples) showed one LEE variant with a breadth of coverage above 50%. For S. copri (clade A), in which we recovered five different LEE variants, we found an enrichment of the variant HDC01 (the most predominant, reconstructed on 70.47% of the strains in the S. copri [clade A] phylogeny) in a subclade of the tree mainly populated by individuals from Westernized populations (Figure 3A). For other variants of S. copri (clade A), as well as for those of S. hominis (clade E) (with two different variants), we did not find any similar association to Westernization/non-Westernization (Figures 3A and S3). Of LEEs that were identified from genomes at a coverage of 1×, 96.6% were found at a breadth of coverage above 40% (with the exception of HDE04 variant from S. sinensis [clade C]; median = 68.48%, 25th percentile = 59.61%, 75th percentile = 76.79%; Figure 3B). This distinct pattern reflects the observation based on isolates, as an LEE was not consistently observed in isolates from S. sinensis (clade C). In addition, we found high collinearity between the depth of coverage of the main chromosome and that of LEE (when depth of coverage of the main chromosome >2× and breadth of coverage of the LEE >50%, Spearman’s r = 0.88 ± 0.084, p < 1e−6, linear regression slope = 0.81 ± 0.073; Figures 3C and S2C). Both these findings suggest that for most of the species, LEEs are always present and in the same copy number as the main chromosome.
Figure 3.

LEEs are highly prevalent and diversifies with the main chromosome
(A) Phylogenetic reconstruction of S. copri (clade A). The length of the external bar plots represents the breadth of coverage of LEE in the sample. NHP, non-human primate.
(B) Breadth of coverage of LEE when the depth of coverage of the main chromosome is above 1×.
(C) Spearman correlation between the depth of coverage of the main chromosome (x axis) and LEE (y axis) of S. copri (clade A), when the depth of coverage of the main chromosome is above 2× and the breadth of coverage of LEE is above 50% (Spearman’s r = 0.95). The color gradient represents the breadth of coverage of LEE.
(D) Spearman correlation of the single-nucleotide polymorphisms (SNPs) rates between the main chromosome (x axis) and LEE (y axis) of S. copri (clade A), when the breadth of coverage of LEE is above 80% (Spearman’s r = 0.87). SNP rates of the main chromosome were calculated using the multiple-sequence alignment (MSA) of the StrainPhlAn marker genes and those from LEE using the MSA of the full LEE alignment. Boxplots in (B) show the median (center), 25th/75th percentile (lower/upper hinges), 1.5× interquartile range (whiskers), and outliers (points). See also Figures S2–S5.
We then investigated whether there is evidence of co-diversification between the main chromosome and LEE or whether LEEs are frequently horizontally transmitted. Specifically for S. copri (clade A), we found a strong correlation between the pairwise single-nucleotide polymorphism (SNP) rates of the main chromosome and LEE (when assessing sequences sharing the same LEE variant and the breadth of coverage of LEE >80%, Spearman’s r = 0.87; Figure 3D), suggesting a potential co-diversification between them. We found similar correlations when assessing the SNP rates of the S. brasiliensis (clade D), S. hominis (clade E), and S. sinica (clade G) (Spearman’s r = 0.71, 0.58, and 0.59, respectively; Figure S2D). The lowest co-phylogenetic correlation was shown by S. sinensis (clade C) (Spearman’s r = 0.38; Figure S2D). Interestingly, we found that the mutations observed in the main chromosomes were mostly driven by neutral evolution, whereas those of LEEs produced significantly higher amino acid changes (Figure S2E). Moreover, the analysis of horizontal gene transfer (HGT) between main chromosome and LEE revealed that from 30% in S. sinica (clade G) (HDE06 strain; 22 coding sequence [CDS] of 77 total LEE-encoded CDS) to 7.73% in the HDB01 strain of S. copri (clade A) (13 CDS of the 168 total LEE-encoded CDS) were shared. The gene annotations of these shared CDS correspond mainly to hypothetical proteins, integrases, and transposases, suggesting that mobile elements actively contribute to HGT between LEE and main chromosome, albeit at low level. Thus, although their divergence even within species and their gene composition suggests that LEEs are under horizontal transfer pressure, their host co-diversification patterns and their essential need for the host suggested by their consistent presence and TA systems point at a rather stable integration in the host cell.
All ScC species are underrepresented in Westernized individuals
Westernization has been driven by industrialization and related factors over the past two centuries and is broadly characterized by urbanized dwelling, dietary changes, and access to healthcare and pharmaceuticals20,31,42 (see STAR Methods). Numerous reports associated non-Westernized populations as being rich in Prevotellaceae species compared with Westernized individuals, which are conversely rich in Bacteroidaceae species.2,7,8,9,10 Previously, it was shown that ScC was not only highly prevalent in non-Westernized populations but also characterized by multiple clades co-occurring within non-Westernized individuals. Expansion of the complex shows that at least one member of ScC is present in 49.18% Westernized compared with 92.71% non-Westernized individuals (in comparison with 48.66% and 91.85% when assessing only the original four species/clades, Figure 4A—“any clades”), and similar results are obtained when assessing each species separately (Figure 4B). Although the expanded species (clades E–I) tend to be less prevalent than the original four, S. hominis (clade E) and S. sinica (clade G) are present in more than 65% of the non-Westernized individuals (68.64% and 66.54%, respectively, Figure 4B). S. copri (clade A) and S. sinensis (clade C) are the most co-occurring species in both Westernized and non-Westernized populations (both present in 13.26% and 78.64% of the individuals, respectively; Figure S5A). Of non-Westernized individuals in which at least one member of ScC was detected, 85.31% individuals have more than one species compared with 19.59% Westernized individuals (Figure 4C), and in 15.06% of cases of non-Westernized individuals, all 9 ScC species are present (Figure 4C).
Figure 4.

Diversity and genetic stratification of the ScC species
(A) Prevalence of the ScC species across Westernized and non-Westernized populations, ancient samples, and non-human primates (NHPs). Number of samples per dataset are reported between parentheses.
(B) Percentage of Westernized and non-Westernized samples containing multiple ScC species.
(C) The prevalence of the ScC species differs between Westernized and non-Westernized populations. Only stool samples from studies with at least 30 samples were assessed.
(D) Per sample prevalence of the ScC species in ancient metagenomes. The horizontal dashed line represents the total number of samples assessed.
(E and F) Multidimensional scaling (MDS) based on the pairwise SNP rates on the StrainPhlAn species-specific marker genes for S. copri (clade A) colored by (E) lifestyle (PERMANOVA p < 0.001) and (F) continent (PERMANOVA p < 0.001).
(G) Differences in the intra-lifestyle phylogenetic distances comparison for S. copri (clade A) (Mann-Whitney U test p < 1e−10). Pairwise phylogenetic distances were calculated using the StrainPhlAn tree branch lengths normalized by the total branch length.
(H) Differences in the polymorphisms found between Westernized and non-Westernized samples for S. copri (clade A) (Mann-Whitney U test p < 1e−10). Polymorphisms were calculated using the StrainPhlAn consensus marker genes and were defined as positions in the reconstructed markers with a dominant allele frequency below 80%. Boxplots in (B), (G), and (H) show the median (center), 25th/75th percentile (lower/upper hinges), 1.5× interquartile range (whiskers), and outliers (points). See also Figures S3 and S6–S9.
In order to confirm that the decreased prevalence and diversity of ScC in Westernized populations compared with contemporary non-Westernized populations reflect the historical changes that modern Westernized populations underwent in the last few millennia, we analyzed fecal fossils (i.e., coprolites) that are currently available. We examined 24 ancient fecal samples from different ancient individuals dated from ∼3,300 BC to 1,720–1,783 AD and 5 countries and 3 continents (Table S1B). Although small in number, both prevalence and multiple species co-occurrence were akin to contemporary non-Westernized individuals (Figure 4D). In the samples in which ScC was present (95.83% of the ancient samples), we found at least three Segatella species present simultaneously and in 87.5% and 20.83% of the cases, 4 and 5 species were present (Figure S5B). Surprisingly, S. hominis (clade E), S. sanihominis (clade F), S. sinica (clade G), and Ca. S. caccae (clade H) were only found in European coprolites (Figure 4D). Ca. S. intestinihominis (clade I), instead, was never found in the ancient samples (Figure 4D). All these findings reinforce the hypothesis that the ScC species are long-standing core members of the co-evolved human microbiome but have rapidly declined in prevalence and diversity in connection with the process of Westernization.20
The ScC species present a genetic structure linked to geography and lifestyle
We further investigated the strain-level diversity and stratification of the 9 human ScC species using StrainPhlAn 43 (Figure S3; see STAR Methods). We found a phylogenetic stratification for all the species that is strongly linked with lifestyle (Westernized vs. non-Westernized; Figures 4E and S6, PERMANOVA p < 0.05) and geographic origin when comparing strains from different continents (Figures 4F and S7, PERMANOVA p < 0.01). Interestingly, strains reconstructed from Westernized individuals appear to be phylogenetically closer between them than those from non-Westernized ones (Figures 4G and S8A, Mann-Whitney U test p < 0.05). We also found a significantly lower proportion of polymorphic positions (defined as the proportion of bases in the reconstructed sequences in which the main allele dominance is below 80%) in the strains of Westernized individuals (Figures 4H, S5C, and S8B; Mann-Whitney U test p < 0.05), suggesting that individuals from non-Westernized populations might carry more strains from the same species than those from the Westernized ones. However, this increase in polymorphisms might also partially depend on the intra-population diversity of the species (Figure S9).
Strains from ancient humans and NHPs were also integrated into the phylogeny of the 9 ScC species (Figure S3). We found strains from captive macaca (housed in a Chinese research facility43) clustering close to that from Chinese individuals. Although more investigation is still needed, these potential strain-sharing events might be explained by inter-species transmission that could have been favored by the human-controlled primate’s diet in captivity and environmental conditions and contact. With the exception of Ca. S. caccae (clade H) and Ca. S. intestinihominis (clade I), ancient strains were also reconstructed and integrated into the phylogeny, and they were typically found clustering between lifestyles or forming their own distant subcluster. ScC is thus not only shaped in their prevalence and diversity by lifestyle and/or geographical origin, but these factors seem to also impact their genetic evolutionary and adaptive trajectories.
The ScC displays a divergent functional repertory between species
Next, we explored the functional diversity across the nine human ScC species using UniRef-annotated open reading frames (ORFs) of each genome44 (see STAR Methods). We observed a significant variability in the presence and absence of UniRef50 families between and even within the different species (3,230 differentially present UniRef50 families, PERMANOVA on per-genome profile, p < 0.001; Figure 5A; Table S2A). Consistent with previous observations,20 S. brunsvicensis (clade B) has the most distant functional profiles to other species, followed by Ca. S. caccae (clade H), which adjoins S. brunsvicensis (clade B) based on principal coordinate analysis (PCoA). Significantly distinguishing functional features include those involved in amino acid metabolism or transport, e.g., branched chain amino acid aminotransferase and amino acid carrier protein, which are prevalent in all species except S. hominis (clade E) (Figure 5B). Likewise, amino acid carrier proteins mediating the transfer of amino acids in and out of cells were also depleted in S. hominis (clade E). By contrast, dihydrofolate reductase (a known ubiquitous enzyme that is important for cell proliferation and growth) was found only enriched in two species, with variant U50_A0A374C8Z1 differentially abundant in S. hominis (clade E) and U50_R6XDH9 in S. sanihominis (clade F). Although dihydrofolate reductase was considered to be essential for bacterial growth, species or strains could also survive with alternative pathways or enzymes providing reduced folate derivatives.45 Interestingly, most species are found devoid of virulence proteins, but we observed multiple variants of virulence-associated protein E co-existing in S. sanihominis (clade F). In the biosynthesis of leucine, an essential amino acid, the gene 3-isopropylmalate dehydratase is common to all species but depleted in Ca. S. caccae (clade H). Compared with other species, Ca. S. intestinihominis (clade I) lacked riboflavin biosynthesis and thiamine phosphate synthase, which are responsible for Vitamin B production and thiamine metabolism, respectively (Table S2A).
Figure 5.

Functional characterization of the human ScC species
(A) PCoA based on the Jaccard distances of the UniRef50 families (PERMANOVA p < 0.001).
(B) Prevalence (%) of selected UniRef50 families (except carbohydrate-metabolism-related families) depleted and enriched in the ScC species. All UniRef50 families shown were significantly enriched/depleted in one species compared with all other species separately (as defined by coupled Fisher’s exact tests between each pair of species, false discovery rate [FDR] < 0.01).
(C) Prediction of total carbohydrate-active enzymes (CAZymes) in the different ScC species. (Kruskal-Wallis p = 1.7287e−68.).
(D) Prediction of total PULs in the different ScC species (Kruskal-Wallis p = 2.5194e−70).
(E) PCoA based on the Jaccard distances of the predicted CAZymes between S. brunsvicensis (clade B) MAGs reconstructed from Westernized or non-Westernized individuals (PERMANOVA p = 0.0099).
(F) PCoA based on the Jaccard distances of the predicted PULs between S. brunsvicensis (clade B) MAGs reconstructed from Westernized or non-Westernized individuals (PERMANOVA p = 0.0199). Boxplots in (C) and (D) show the median (center), 25th/75th percentile (lower/upper hinges), 1.5× interquartile range (whiskers) and outliers (points). See also Figures S10 and S11 and Table S2.
To better understand ScC’s capability of degrading dietary fiber, which was typically rich in non-Westernized diet tradition,11,46 we extended the screening for carbohydrate-active enzymes (CAZymes) analysis to all species and additionally analyzed their organization in polysaccharide utilization loci (PULs) (see STAR Methods). Nine species have considerable variability in the number of both CAZymes and PULs (Figures 5C and 5D). Specifically, genomes of S. sinica (clade G) contain the highest number of CAZymes on average and S. sanihominis (clade F) encoded more PULs compared with other species. Conversely, the lowest number of CAZymes and PULs were detected in S. brunsvicensis (clade B) and Ca. S. intestinihominis (clade I), respectively. PCoA based on the presence of CAZymes and PULs further confirmed a significant differentiation in carbohydrate and even polysaccharide degradation potential between and within species (Figures S10A and S10B). We found most CAZyme families are shared between all species, but many are present across species with a large degree of variance in prevalence (Figures S10C and S10D; Table S2B), which is consistent with previously reported observations.20 Notably, the glycoside hydrolases families GH141 (includes α-L-fucosidase and xylanase activities), GH138 (rhamnogalacturonan α-1,2-galacturonohydrolase), and GH63 (α-glucosidase and mannosidase) are exclusive to S. copri (clade A); the polysaccharide lyase family (PL) 2, important for degrading plant tissues, was found only in S. brasiliensis (clade D); the glycosyltransferase family GT10 was exclusively in Ca. S. intestinihominis (clade I). For the carriage of PULs, we obtained a similar presence pattern with Ca. S. intestinihominis (clade I) being the most distinguished, lacking nearly all PULs which were carried by at least one of the other species (Figures S10E and S10F). Intriguingly, genomes from Westernized and non-Westernized samples displayed significant differences in both CAZymes and PULs in S. hominis (clade E) and S. brunsvicensis (clade B), S. brasiliensis (clade D), and S. sinica (clade G) (Figures 5E, 5F, and S11).
ScC is strongly enriched in males and individuals with low levels of visceral fat
We investigated potential associations of the different ScC species with host characteristics such as sex, age, and body mass index (BMI). We queried the curatedMetagenomicData (cMD) repository32 for gut microbiomes of healthy, adult individuals from Westernized populations. In total, we analyzed using MetaPhlAn 4 4,095 microbiome samples for the association with sex (n = 14 studies), 3,190 (n = 11 studies) with age, and 4,783 (n = 24 studies) with BMI. For each species and study, we fitted a logistic regression model linking the presence of the ScC species with these host factors and performed meta-analysis of the resulting log-odd ratios (see STAR Methods and Tables S3A–S3C). The presence of S. copri (clade A), S. sinensis (clade C), S. sinica (clade G), S. sanihominis (clade F), and Ca. S. intestinihominis (clade I) was statistically associated with the male sex (Wald-p < 0.05; synthetic log-odd = 0.28, 95% confidence interval [CI] [0.09, 0.48], 0.4 [0.19, 0.62], 0.7 [0.34, 1.06], 0.74 [0.3, 1.18], and 1.55 [0.48, 2.61], respectively), highlighting that the ScC presence is robustly associated with the male sex in Westernized populations (Figure 6A). This association was also significant when investigating the presence of any ScC species (Figure 6A), and the total number of ScC species present simultaneously (see STAR Methods, overall standardized mean difference = 0.16, p = 1.9e−7, Figure 6E; Table S3D). We did not find any consistent statistical association with age or BMI, with the exception of age decreasing the probability of carrying Ca. S. intestinihominis (clade I) (Figures 6B and 6C).
Figure 6.

Association analysis of the ScC species with sex, age, BMI, diseases and cardiometabolic health
(A–D) Condensed forest plots for the association of S. copri (clade A), S. brunsvicensis (clade B), S. sinensis (clade C), S. sinica (clade G), Ca. S. caccae (clade H), S. sanihominis (clade F), and Ca. S. intestinihominis (clade I) or any of the species with sex, aging, BMI, and health-related conditions, using four sets of 14 studies (2,420 healthy females and 1,675 healthy males), 11 studies (3,190 healthy individuals), 24 studies (4,783 healthy individuals), and 22 studies (12 diseases, 1,635 cases, and 1,854 controls). Blue and red dots represent, respectively, non-significant and significant associations of the variable of interest in each dataset, obtained through a logistic regression model having the presence/absence of ScC species as response variable and sex, age, BMI, and depth as predictors and sex, age, BMI, depth, and health status in the disease one. Dark-blue and red diamonds represent, respectively, non-significant and significant random-effects meta-analysis coefficients used to summarize the single-dataset coefficients.
(E) Condensed forest plot showing Spearman’s partial correlation of sex, age, BMI, and health status with the number of ScC species. Each correlation is adjusted by each of the variables plus depth. Dark-blue and red diamonds represent the coefficient of a random-effect meta-analysis of the Fisher Z-transformed correlations (for aging and BMI) or standardized mean differences (for sex and the diseases).
(F) Plot showing the associations of S. copri (clade A), S. brunsvicensis (clade B), S. sinensis (clade C), or any of the species, with 19 cardiometabolic health parameters in 1,098 participants from the ZOE PREDICT 1 cohort. Each marker represents the coefficient of a logistic regression predicting ScC species presence/absence using sex, age, BMI, depth, and the corresponding cardiometabolic parameter, with its 95% confidence intervals. Wald-ps are colored according to an FDR correction using 0.2 as significance threshold. See also Table S3.
We next assessed the association between the ScC and health-related conditions. We queried cMD for microbiome studies containing samples of adult individuals from Westernized populations with, at least, 10 disease cases and 10 healthy controls (21 studies and 11 different diseases), to which we added one more study of rheumatoid arthritis,47 (1,635 cases and 1,854 controls in total; Table S3E). None of the ScC species resulted to be associated with any health condition when considering all the diseases together (Figure 6D). Two studies (QinN_201448 and FengQ_201549 focusing on cirrhosis and colorectal cancer, respectively) resulted in increased odds of observing the presence of S. copri (clade A) (log-odds = 2.56 and 3.6, Wald-p = 0.002 and 3e−8, respectively). The present analysis does not include per-disease medications, of which many have shown to have a measurable impact on commensal bacteria in vitro as well as the gut microbiome in vivo.50 The fact that these associations lean toward either the diseased and the control group and that they are relative to etiologies for which different medications are normally prescribed does not suggest treatments as common drivers of the observed increased presence of ScC in health and disease.
Finally, we investigated the association of ScC with the 19 main nutritional and cardiometabolic health markers from the ZOE PREDICT 1 cohort51 (1,098 individuals; Table S3F). Visceral fat was associated with a 60%-reduced chance of observing the presence of S. copri (clade A), S. brunsvicensis (clade B), and S. sinensis (clade C) (log-odds = −0.59, −1.22, and −1.02, Wald-Q < 0.01, Q = 0.21, Q = 0.03, respectively; Figure 6F). Total triglycerides (TGs) and very-low density lipoprotein (VLDL) were also negatively associated with the probability of having S. copri (clade A) and S. sinensis (clade C) (log-odds = −0.2, −0.35; Wald-Q = 0.07, 0.14; and log-odds = −0.15, −0.34; Wald-Q = 0.12, 0.13, respectively, avg. decrease in chance of observing the clade = 24%); another marker of inflammation, glycoprotein acetylation (GlycA), resulted also associated with a negative probability of having the S. copri (clade A) and also with the probability of carrying any ScC species (log-odds = −0.15, −0.17; Wald-Q = 0.13, 0.11). High-density lipoprotein (HDL) was instead positively associated with increased odds of having S. copri (clade A) (log-odds = 0.16, Wald-Q = 0.13), and polyunsaturated fatty acid (PUFA) was associated with the presence of S. copri (clade A) and S. sinensis (clade C) (log-odds = 0.26, 0.51, Wald-Q < 0.01 in both, avg. increase = 47%). S. sinensis (clade C) also resulted positively associated with two healthy diet scores (healthy eating index [HEI] and alternate Mediterranean diet [aMED] scores, log-odds = 0.29 and 0.41, Wald-Q = 0.12 and 0.03, avg. increase = 42%).
Discussion
Previous studies disclosed that genomes relatively close to the S. copri type strain are not part of a single lineage monophyletic species but rather form a species complex, comprising 4 genetically and functionally distinct clades.20,21 In this work, we further expanded the definition of ScC into a total of 13 different species that we characterized and named as distinct species, 4 of which are specific to NHPs. Due to the large intra-species genetic variability (Figures 1C–1E), their common co-presence within the same individual (Figure 4C), as well diverse functional and metabolic capabilities of each clade (Figure 5), the original concept of clades should now be considered deprecated and substituted with the standard definition of multiple distinct species. Notably, strains in different species within ScC have very similar 16S rRNA gene sequences, which makes them difficult to distinguish in amplicon-based microbiome analyses as it has been shown for other species.27 As previously shown20 for four ScC species, we confirmed that the human ScC species are highly prevalent in non-Westernized populations (92.71% of individuals carrying, at least, one species) with multiple species commonly co-occurring within individuals (Figures 4A–4D) closely resembling what could be recovered from ancient stool microbiome samples pre-dating the industrial era. These differences in ScC species in prevalence between lifestyles are also mirrored at the intra-species strain level, as most ScC species exhibit a genetic and phylogenetic structure associated with host lifestyle (Figure 4E). Moreover, strains carried by non-Westernized individuals tend to be more different from each other in comparison with those of individuals following a Westernized lifestyle (Figure 4G). The higher ScC microbial diversity46 together with a larger number of co-occurring ScC species in the microbiome of non-Westernized populations might reflect a more diverse availability of ScC species-promoting nutrients and fibers.52
Targeted isolation coupled with PacBio long-read sequencing revealed the presence of LEEs within the ScC genomes (Figure 2). These LEEs encode rRNA genes, have a high prevalence in most species, and, despite evidence of horizontal mobility, show clear patterns of co-diversification with the main chromosome. Although plasmids are frequent in Segatella’s genomes (including those we sequenced in this work) and secondary chromosomes have been already found in other Prevotellaeae species,53 LEEs we described in this work are difficult to be classified as either of the two. They show heterogeneous sizes between strains of the same species (clade A: 135.1–211.7 kb), contain full sets of rRNA genes, and show enrichment in transposon elements. However, they lack the dnaA genes found in the second chromosomes of other Prevotellaeae species.53 Hence, although the lengths of our LEEs ranging from 93.1 to 328.8 kb may resemble that of larger plasmids, the genomic similarities as well as the evolutionary patterns and their seemingly consistent presence might suggest that these elements are precursors to secondary chromosomes. Clear exemptions are S. brunsvicensis (clade B) and S. sanihominis (clade F), which seems to completely lack an LEE. Moreover, LEE of S. sinensis (clade C), unlike the other species, lacks an rRNA operon and LEE of its type strain displays low prevalence in metagenomic datasets, suggesting that in this species a further genomic rearrangement occurred. Further long-read assembly of diverse strains of ScC as well as functional experiments including the elucidation of the replication system will be required to clarify their distribution and their contribution to ScC species biology.
The role of S. copri in health and disease17,18,19,54,55,56,57 has been extensively explored, but so far, no consensus has been reached regarding its beneficial contribution to human health.21,58,59,60,61 Although some authors have suggested that these incongruences might be explained by its subspecies genetic variation,20,62 large-scale meta-analysis attempts have not shown any strong associations between the previously identified S. copri clades and human diseases.20 Likewise, our extension of ScC did not reveal any strong statistical association with human health conditions (Figure 6D). However, we discovered particularly strong associations between the presence of most of the ScC species and the male sex (Figure 6A). Previous studies have reported enrichment of Prevotella species in the gut microbiome of men who maintain sexual relations with other men (MSM) compared with that of women and men having sex with women.63 Although it is highly speculative, as sexual orientation is rarely recorded in microbiome studies, the consistently higher prevalence of ScC in men could be partially explained by this association as up to 6% of the Westernized male population is estimated to be MSM.64 Of note, a metagenomic study of the MSM gut microbiome revealed not only an increased relative abundance of Prevotellaceae in MSM but also an increased diversity including the co-colonization with diverse members of ScC similar to what has been observed in non-Westernized populations (K.D.H., unpublished data). Additional large-scale cohort studies will be required to further substantiate this unexpected link.
Despite the inconclusive results in relation to human diseases, S. copri has been recurrently associated with high fiber and low fat diets,12,14,65 favorable health measures such as reduced visceral fat and improved glucose metabolism,51,66,67 and vegan dietary habits.62 Although we did not find statistical associations with any specific food group (Table S3F), re-analysis of the 1,098 deeply phenotyped individuals from the ZOE PREDICT 1 cohort51 further corroborated the beneficial associations between the presence of several ScC members and a decreased visceral fat, total triglycerides, and VLDL (Figure 6F).
In summary, this work provides the definition and comprehensive genomic characterization of ScC, extending its underestimated diversity, discovering prevalent extrachromosomal elements, and further expanding previous associations in relation to human health. Although the relation between S. copri and human diseases is still uncertainty, the strong associations with glucose homeostasis and host metabolism pave the way for future studies aiming to understand their potential anti-inflammatory role and the possibility of exploiting them as biomarkers for a healthy gut. Functional studies of many members of the ScC will be enabled by the reference strains now available in public repositories.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Segatella copri (DSM 18205) | DSMZ | DSM 18205 |
| Segatella copri (HDB01) | https://doi.org/10.15252/embj.202110828 | DSM 108419 |
| Segatella copri (HDC01) | https://doi.org/10.15252/embj.202110828 | DSM 108556 |
| Segatella copri (HDA03) | https://doi.org/10.15252/embj.202110828 | DSM 108386 |
| Segatella copri (HDD04) | https://doi.org/10.15252/embj.202110828 | DSM 108558 |
| Segatella brunsvicensis (NI025) | This study | DSM 113023 |
| Segatella sinensis (HDE04) | https://doi.org/10.15252/embj.202110828 | DSM 108151 |
| Segatella sinica (HDE06) | https://doi.org/10.15252/embj.202110828 | DSM 111807 |
| Segatella hominis (HDD15) | https://doi.org/10.15252/embj.202110828 | DSM 113020 |
| Segatella hominis (HDD12) | https://doi.org/10.15252/embj.202110828 | DSM 111806 |
| Segatella copri (HDE03) | This study | DSM 108150 |
| Segatella brasiliensis (HDD05) | https://doi.org/10.15252/embj.202110828 | DSM 112105 |
| Segatella sanihominis (RHB01) | This study | DSM 113786 |
| Deposited data | ||
| Genomes from isolates | This study | PRJEB60954 |
| Genome analysis from isolates | This study | https://doi.org/10.6084/m9.figshare.24337687 |
| Software and algorithms | ||
| MetaPhlAn (version 4) | Blanco-Míguez et al.3 | https://github.com/biobakery/MetaPhlAn/ |
| StrainPhlAn (version 4) | Blanco-Míguez et al.3 | https://github.com/biobakery/MetaPhlAn/ |
| PhyloPHlAn (version 3.0) | Asnicar et al.68 | https://github.com/biobakery/phylophlan/ |
| GraPhlAn (version 1.1.4) | Asnicar et al.69 | https://github.com/biobakery/graphlan |
| fastANI (version 1.3) | Jain et al.38 | https://github.com/ParBLiSS/FastANI |
| PyPhlAn (commit 1207314) | NA | https://github.com/SegataLab/pyphlan |
| Bowtie2 (version 2.4.2) | Longmead and Salzberg70 | https://github.com/BenLangmead/bowtie2 |
| CMSeq (version 1.0.4) | Zolfo et al.71 | https://github.com/SegataLab/cmseq |
| Prokka (version 1.14) | Seemann72 | https://github.com/tseemann/prokka |
| Diamond (version 0.9.24) | Buchfink et al.73 | https://github.com/bbuchfink/diamond |
| MMseqs2 (version cf150146df85 2b25eebf621ce0ffda2ac07d818d) |
Steinegger and Söding74 | https://github.com/soedinglab/MMseqs2 |
| dbCAN2 (version v2.0.6) | Zhang et al.75 | https://bcb.unl.edu/dbCAN2/ |
| PULpy (commit 8955cdb) | Stewart et al.76 | https://github.com/WatsonLab/PULpy |
| dbCAN-PUL (version 2020-10-30) | Ausland et al.77 | https://bcb.unl.edu/dbcan_pul/Webserver/static/DBCAN-PUL/PUL.faa |
| SPAdes (version 3.10.0) | Bankevich et al.78 | https://github.com/ablab/spades |
| BLASTn (version 2.9.0) | Altschul et al.79 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Burrows-Wheeler Aligner (version 0.7.12-r1039) | Li and Durbin80 | https://bio-bwa.sourceforge.net/ |
| Varscan (version 2.3.6) | Koboldt et al.81 | https://varscan.sourceforge.net/ |
| Roary (version 3.13.0) | Page et al.82 | https://sanger-pathogens.github.io/Roary/ |
| Biostrings (version 2.64.0) | Pages et al.83 | https://github.com/Bioconductor/Biostrings |
| FindMyFriends (version 1.17.0) | Pedersen84 | https://github.com/thomasp85/FindMyFriends |
| Barrnap (version 0.9) | Seemann and Booth85 | https://github.com/tseemann/barrnap |
| SLING (version 2.0.1) | Horesh et al.86 | https://github.com/ghoresh11/sling |
| Other | ||
| curatedMetagenomicData | Pasolli et al.32 | https://github.com/waldronlab/curatedMetagenomicData |
| UniRef | Suzek et al.44 | https://www.uniprot.org/uniref/ |
| NCBI GenBank database | Benson et al.30 | https://www.ncbi.nlm.nih.gov/genbank/ |
| CAZy-DB | Lombard et al.87 | https://github.com/linnabrown/run_dbcan |
| SMRT Portal | N/A | https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/ |
| Protologger Portal | Hitch et al.88 | http://www.protologger.de/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Till Strowig (till.strowig@helmholtz-hzi.de).
Materials availability
All strains isolated as part of this study are readily available from the DSMZ or will be provided upon reasonable request from Till Strowig (till.strowig@helmholtz-hzi.de).
Experimental model and subject details
Study design
Within this study, we included fecal samples from participants of three distinct German cohorts: MikroDivers, Nutrimmune89 and RheumaVOR.90 The Nutrimmune cohort comprises 94 healthy individuals with different dietary habits, between the age of 25 and 45 and with a BMI ranging from 20 to 28 kg/m2. Exclusion criteria for the study participation were regular consumption of tobacco, intake of antibiotics or laxatives and/or weight loss within the last six months as well as gastrointestinal or chronic cardiovascular diseases. The RheumaVOR cohort is an ongoing cohort comprising patients being newly diagnosed with different rheumatic diseases as well as their household members. Following their initial examination by a rheumatologist, patients as well as their household members were asked to donate stool samples and provided written informed consent. There were no further inclusion criteria for the RheumaVOR study participation. Until September 2023 more than 500 individuals were included. The MicroDivers cohort is an ongoing study recruiting volunteers aged 18 and above. The inclusion criteria are the willingness and ability to sign the informed consent, to provide a fecal sample, and to answer a questionnaire on geographic origin, diet preferences, age, gender, BM, weight, and height. The exclusion criteria were recent antibiotic intake (<4 weeks), intake of chemotherapeutics and immunosuppressive medications (ever), acute and chronic inflammatory bowel disease, colon cancer, recipient of fecal microbiota transplants, and being involved in direct patient care (e.g., in a hospital). Until September 2023 more than 30 individuals were included.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics commision of the Hannover Medical School (MikroDivers: 8628_BO_K_2019, RheumaVor: 8063_BO_K_2018) and of the Medical Association of Lower Saxony (Hanover, Germany; Nutriimmune). The Nutriimmune study was registered in the German Clinical Trial Register (DRKS 00019887). All patients/participants provided their written informed consent to participate in this study.
Method details
Stool sample collection and processing
Stool samples were collected freshly and were further processed inside an anaerobic chamber. A pea-sized aliquot of the sample was resuspended in 5ml BHI-S and subsequently filtered through a 70μm cell strainer. The resulting flow-through was either immediately used for the isolation of S. copri or was mixed with an equal volume of BHI/Glycerol medium, filled into sealed glass vials and kept at -80°C for long term storage.
Isolation of Segatella copri strains
Fecal samples of participants from the cohorts Mikrodivers, Nutrimmun and Rheuma-VOR were previously screened for the presence of S. copri by 16S rRNA gene sequencing. Stool from S. copri-positive donors was diluted and was streaked out in serial dilutions on BHI (brain heart infusion) blood agar plates supplemented with vancomycin. After incubation of the inoculated plates at 37°C for 48-72h inside an anaerobic chamber, individual colonies were picked into BHI medium supplemented with FBS and Vitamin K3. Following incubation at 37°C for 24-48h, resulting cultures were screened by PCR using S. copri specific primers (P_copri_69F/P_copri_853R) as well as the clade primers. S. copri-positive cultures were further passed on agar plates to obtain pure isolates, which were additionally confirmed by Sanger sequencing. To enable storage of the isolates, bacterial cultures were mixed with an equal volume of BHI + 50% glycerol, were aliquoted into sealed glass vials and were immediately cryopreserved at -80°C. Information about all the isolates was collected in Table S1A.
DNA extraction and sequencing
For short-read library preparation, total DNA was isolated from bacterial cells and stool pellets using the ZymoBIOMICS DNA MiniPrep Kit following the manufacturer’s instructions. Total DNA was quantified and diluted to 25 ng/μl.
Short read library preparation was performed using NEBNext® Ultra™ II FS DNA Library Prep Kit (New England Biolabs) for Illumina with parameters as followed: 500 ng input DNA and 5 min at 37°C for fragmentation; > 550-bp DNA fragments for size selection; primers from NEBNext Multiplex Oligos for Illumina Kit (New England Biolabs) for barcoding. The library was sequenced on the Illumina MiSeq 2 x 250 bp .
For the PacBio long-read sequencing we selected every isolate from all ScC species but S. copri (clade A). For S. copri (clade A) we selected 5 strains from different donors, as well as the type strain DSM 18205T. For long-read library preparation, high molecular weight DNA was prepared using Qiagen Genomic Tip/100 G (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
In case of sequencing on the PacBio RSII, SMRTbell™ template libraries were prepared according to the instructions from Pacific Biosciences (Menlo Park, CA, USA), following the Procedure & Checklist – Greater Than 10 kb Template Preparation. Briefly, for preparation of 15 kb libraries 8 μg genomic DNA were sheared using g-TUBEs™ from Covaris (Woburn, MA, USA) according to the instructions of the manufacturer. DNA was end-repaired and ligated overnight to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 from Pacific Biosciences. Reactions were carried out according to the instructions of the manufacturer. BluePippin™ Size-Selection to greater than 4 kb was performed according to the instructions of the manufacturer (Sage Science, Beverly, MA, USA). Annealing conditions of sequencing primers and binding of polymerase to purified SMRTbell™ template were assessed with the calculator in RS Remote. Libraries were sequenced on the PacBio RSII taking one 240-minutes movie for each SMRT cell.
In case of sequencing on the Sequel II, SMRTbell™ template library was prepared according to the instructions from Pacific Biosciences, following the Procedure & Checklist – Preparing Multiplexed Microbial Libraries Using SMRTbell™ Express Template Prep Kit 2.0. Briefly, for preparation of 10 kb libraries 1 μg genomic DNA was sheared using g-TUBEs™ from Covaris, (Woburn, MA, USA) according to the instructions of the manufacturer. DNA was end-repaired and ligated to barcoded adapters applying components from the SMRTbell™ Express Template Prep Kit 2.0 from Pacific Biosciences. Reactions were carried out according to the instructions of the manufacturer. Samples were pooled according to the calculations provided by the Microbial Multiplexing Calculator. Annealing conditions of sequencing primers and binding of polymerase to purified SMRTbell™ template were assessed with the calculator in SMRT Link . Libraries were sequenced on the Sequel II taking one 15h movie per SMRT cell.
Genome assembly
Short-read assembly was performed with SPAdes (version 3.10.0) using the “careful” mode.78 Obtained contigs were then filtered by length and coverage (contigs > 500 bp and coverage > 5x) and furthermore gene prediction and gene annotation were performed using Prokka version v1.1472 with default parameters.
Long-read genome assembly of PacBio RSII sequencing reads was carried out using the ‘RS_HGAP_Assembly.3’ protocol in SMRT Portal (version 2.3.0). Resulting contigs were trimmed and circularized by removing overlapping ends. Chromosomes were adjusted to dnaA as the starting point. Extrachromosomal elements were adjusted to their predicted replication or partitioning proteins (when possible). Long-read correction and circularization control was carried out using the ‘RS_BridgeMapper.1’ protocol implemented in SMRT Portal (version 2.3.0).
Long-read genome assembly of PacBio Sequel II sequencing reads was carried out using the SMRT Analysis Application ‘Microbial Assembly’ in SMRT Link. When circularized within the application, chromosomes and extrachromosomal elements were adjusted to dnaA or predicted replication or partitioning proteins (when possible), as in case of PacBio RSII sequenced genomes. Long-read correction was carried out using the SMRT Analysis Application ‘Resequencing’ available in SMRT Link.
Further quality improvement was achieved with the Burrows-Wheeler Aligner80 (version 0.7.12-r1039) mapping the Illumina reads onto the genomes obtained by PacBio sequencing (hybrid assembly) followed by subsequent automatic detection of sequencing errors by Varscan81 (version 2.3.6) yielding a genome quality of QV60. Gene prediction and gene annotation were performed using Prokka version v1.1472 with default parameters. The genomes are deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB60954 .
Publicly available metagenomic datasets
We collected 20,737 publicly available shotgun metagenomic samples from 92 studies from modern and ancient humans as well as from non-human primates. For modern humans, we retrieved 93 samples from human airways, 19,066 from the gastrointestinal tract, 743 from oral, 504 from skin, 8 from breastmilk and 96 from the female urogenital tract. These samples span different age categories (2,946 newborns, 2,075 children, 13,855 adults, 1,611 seniors and 23 without reported age), sex (8,795 males, 8,879 females and 2,836 without reported sex), lifestyles (18,738 Westernized and 1,772 non-Westernized), 67 different health conditions and 45 countries. Stool human samples from ancient origin (N=24) were retrieved from 5 different studies, and stool samples from non-human primates (N=203) from 7 different ones (Table S1B). For all the modern human samples, metadata was retrieved from the curatedMetagenomicData 3 R package.32
Catalog of isolated and metagenomic-assembled Prevotella genomes
Starting from the original catalog of 560,084 medium-to-high quality (completeness > 50% and contamination < 5%) metagenomic-assembled genomes (MAGs) by Blanco-Miguez et al.,3 we retrieved 9,581 MAGs belonging to the sole family-level genome bin (FGB579)31 that contained all genomes assigned to the species previously assigned to Prevotella in the NCBI taxonomy database, previous to its reclassification in 7 different genera.6 We integrated this set with 458 genomes resconstructed from isolate sources, i.e. the 54 genomes generated in this work in addition to the 404 genomes assigned to the Prevotella genus (previous to its reclassification in 7 different genera6) in the NCBI RefSeq database as of June 2021. This resulted in a catalog of 10,039 genomes that were kept for downstream analyses. We computed pairwise genetic distances among these genomes using Mash (version 2.0; “-s 10000” parameters for sketching), and genomes were clustered through hierarchical clustering (python package fastcluster, single-linkage mode). Species-level genome bins (SGBs)31 were obtained by cutting the dendrogram with a 5% distance.
Westernization definition
Over the last centuries, industrialization and urbanization have had a significant effect on most human populations. This process, known as Westernization, includes changes derived from the access to pharmaceuticals products and healthcare, improved hygiene and sanitation, increased urban density, decreased exposure to livestock, and the adoption of diets enriched in fat and animal proteins, high salt and simple carbohydrates. In this study we define Westernized or non-Westernized individuals and/or populations based on either the definition given in the original publication or on the criteria described above.
Pangenome annotation of the Prevotella SGBs
Open reading frames were annotated on all 10,039 genomes assigned to Prevotella (previous to its reclassification6) using Prokka (version 1.14).72 Coding sequences (CDS) were then assigned to a UniRef50 cluster44 by aligning all the CDSs against the UniRef50 database (version 201906) using Diamond (version 0.9.24)73 and assigning a Uniref50 cluster ID in the cases the sequence identity to the cluster centroid sequence was above 50% and it covered more than 80% of the centroid sequence. Protein sequences that could not be assigned to any UniRef50 cluster were de novo clustered using MMseqs274 (version cf150146df852b25eebf621ce0ffda2ac07d818d) following the Uniclust50 criteria.91 Based on the UniRef50 and UniClust50 annotations, a pangenome was generated by collecting all the UniRef/UniClust50 clusters present in at least one genome.
Definition of the ScC and phylogenetic analysis
We considered one representative genome per Prevotella SGB to generate the tree of genomes of strains of Prevotella species (previous to its reclassification,6 see Figure S1). The representative was chosen randomly among the set of isolate genomes, when available, otherwise among the set of high quality genomes (i.e., with completeness > 90% and contamination < 5% according to CheckM estimates92). We restricted the analysis to the SGBs containing at least one isolate genome or three reconstructed genomes. The phylogenetic tree was built using PhyloPhlAn 3.068 of 400 universal markers available in PhyloPhlAn 3.0. We identified a monophyletic group composed of thirteen species and comprising exclusively all available S. copri genomes. We defined this as ScC that expanded the set of four clades identified by Tett et al.20 We named them from clade A to clade M, with clades A, B, C, D that overlapped with the ones described in Tett et al.20 In addition, we built the ScC-specific phylogeny (see Figure 1B) by considering only the genomes belonging to these thirteen species. For the PhyloPhlAn database, we retrieved ScC core genes by selecting Uniref50 clusters present in, at least, 50% of the genomes of each ScC species. A maximum of 200 randomly selected genomes were considered per species.
Genome characteristics were extracted and summary of them in terms of average values per species were considered for Figure 1C. Genetic distance between and within the ScC species were computed in terms of ANI from the whole-genomes using Mash (Figure 1D) and from the UniRef/Uniclust50 profiles (Jaccard distance; Figure 1E).
Similarity analysis between the LEE and the Lak megaphages
Similarity analysis between the LEEs and the Lak megaphages41 was performed using BLASTn version 2.9.0.79 Genomic sequences of 34 Lak megaphages were retrieved from the Crisce et al. work93 and used to build a BLAST nucleotide sequence database.
Genetic and functional analysis of the ScC closed genomes
Genome statistics were calculated for the 9 ScC complete genomes using the Biostrings83 package in R (version 2.64.0). Specifically, the function letterFrequencyInSlidingView was used with a sliding-window size of 1,000 bp to determine the percentage of GC content. For comparative genomics, a pangenome was inferred in R using the FindMyFriends84 package (version 1.17.0) with the cdhitGrouping option (kmer = 5, similarity threshold 0.6). This resulted in a pangenome consisting of a total of 30,001 genes, which were categorized into 8,324 gene groups. Of these gene groups, 1,458 were classified as core groups, 2,644 were accessory groups, and 4,222 were singletons.
The ribosomal RNA genes in genomes were predicted using Barrnap85 (version 0.9). The identification and annotation of toxin-antitoxin gene systems were performed using the SLING tool86 (version 2.0.1) with default parameters.
Average nucleotide identity between chromosomes and LEEs was calculated using fastANI (version 1.3).38 Pangenome analysis of the LEEs was performed using Roary82 (version 3.13.0) with parameter “-i 80”.
Reconstruction of the LEEs from metagenomic samples
The public 20,737 human and non-human primates metagenomes were mapped against the 9 LEE variants using Bowtie2 (version 2.4.2)70 with parameters “-a—sensitive”. Alignments against small reads (<70bp) were discarded from the mapping results. LEE consensus sequences were reconstructed from the mapping results using CMSeq’s (version 1.0.4)71 consensus.py script with parameters “--minqual 30 --mincov 1”. Breadth and depth of coverage were assessed using CMSeq’s breadth_depth.py script with parameters “--minqual 30 --mincov 1”. Breath of coverage of the main chromosome was assessed using the breadth of coverage of the MetaPhlAn 43 marker sequences of their respective ScC species. Single-nucleotide polymorphisms (SNPs) between the reconstructed LEE sequences were assessed using PhyloPhlAn 368 (--mutation rates parameter). SNPs between the main chromosomes were calculated using the multiple-sequence alignment (MSA) produced by StrainPhlAn 43 (--mutation rates parameter). Spearman correlations between main chromosome and LEE’s SNPs and depth of coverage were calculated using the “stats.spearmanr” function of the scipy python library (version 1.5.3). For depth of coverage-related comparative analysis we employed a minimum breadth of coverage threshold of 50% (i.e. to capture medium-to-high quality LEE variants). For the analysis of the correlation between SNP rates between the main chromosome and the LEEs, strongly affected by the completeness of the LEEs, we defined a minimum breadth of coverage threshold of 80%.
Prevalence and abundance of the ScC
Taxonomic profiling of the public 20,737 human and non-human primates metagenomes was performed using MetaPhlAn 4 against the Jan21 database3 with default parameters. Prevalence and abundance of the 13 ScC species (Table S1D) was assessed using datasets with more than 30 samples. For the modern human metagenomes, only stool samples from healthy adults reporting no antibiotics usage were included. For the prevalence analysis, a minimum relative abundance of 0.001% was defined.
Strain-level diversity and stratification of the ScC
Strain-level profiling with StrainPhlAn 43 was performed for all 13 ScC species on the 20,737 human and non-human primates metagenomes using parameters “--marker_in_n_samples 66 --sample_with_n_markers 66 --mutation_rates”. Isolate genomes sequences in this work were added to the phylogenetic trees using parameter “--references”. The phylogenetic trees generated by StrainPhlAn were plotted with GraPhlAn version 1.1.4.69 Phylogenetic distances were extracted based on the distance between samples in the tree and normalized by the total branch length of the tree using PyPhlAn (https://github.com/SegataLab/pyphlan). Ordination plot for each ScC species was performed with the “stats.ordination.pcoa” function of the skbio python package (version 0.5.6) using the pairwise normalized phylogenetic distances. Statistical differences between Westernized and non-Westernized phylogenetic distances and polymorphic rates were assessed using the “stats.mannwhitneyu” function of the scipy python package (version 1.5.3).
Functional characterization of the ScC
For each ScC species, a species-level pangenome was generated by collecting all the UniRef/UniClust50 clusters present in at least one genome of the species. UniRef/UniClust50 clusters were tested for the significant enrichment or depletion in at least one species relative to the other species separately, using pairwise Fisher’s exact test and an enrichment/depletion was considered significant for FDR < 0.01.
Identification of Carbohydrate-Active Enzymes (CAZymes) was performed using dbCAN275version v2.0.6(CAZy-DB version 07312019, https://github.com/linnabrown/run_dbcan). The results were post-processed with stringency cut-offs as suggested in Yin et al.94 and the family-domain assignments were defined based on the HMM database. CAZyme activities were predicted using an online database Carbohydrate-Active enZYmes Database (http://www.cazy.org). Polysaccharide utilization loci (PUL) and susC/D gene annotations were performed using PULpy76 (commit 8955cdb, https://github.com/WatsonLab/PULpy) using a sliding window of five genes and an intergenic distance 500 bp. The tool was implemented with the updated CAZy-DB (version 07312019) from dbCAN2. Putative substrates of these PULs were predicted by dbCAN-PUL (version 2020-10-30) as previously described.77 All CAZymes and PUL substrates were tested for the significant enrichment or depletion in at least one species relative to the other species separately, using Bonferroni corrected Fisher’s exact test in a manner of pairwise comparison (FDR < 0.05).
Quantification and statistical analysis
Spearman correlations between main chromosome and LEE’s SNPs and depth of coverage were calculated using the “stats.spearmanr” function of the scipy python library (version 1.5.3). Statistical differences between Westernized and non-Westernized phylogenetic distances and polymorphic rates were assessed using the “stats.mannwhitneyu” function of the scipy python package (version 1.5.3). UniRef/UniClust50 clusters were tested for the significant enrichment or depletion in at least one ScC species relative to the other species separately, using pairwise Fisher’s exact test and an enrichment/depletion was considered significant for FDR < 0.01. All CAZymes and PUL substrates were tested for the significant enrichment or depletion in at least one species relative to the other species separately, using Bonferroni corrected Fisher’s exact test in a manner of pairwise comparison (FDR < 0.05).
Association analysis
Association analyses of different human ScC species with host’s phenotypic traits (sex, age, BMI) and health condition (12 in total: colorectal cancer (CRC), ulcerative colitis (UC), Crohn’s disease (CD), type-2-diabetes (T2D), Behcet disease (BD), atherosclerotic cardiovascular disease (ACVD), asthma, migraine, schizophrenia, cirrhosis, and myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS), rheumatoid arthritis (RA)) were carried out using set of cohorts from curatedMetagenomicData 332 and designed to study these problems. In brief, we queried cMD 3 for gut microbiomes from healthy, adult, Westernized individuals present with the first time point at the most to study the sex, age, and BMI, and gut microbiomes from adult individuals that are part of case-control settings to study multiple diseases. In the sex-analysis we considered microbiomes from datasets with a balance of at least 25% of both sexes and at least 40 individuals per class for a total of 4,095 samples (1,675 males and 2,420 females, 14 studies). In the age-analysis, we considered 3,190 microbiomes (11 studies), from datasets with an age IQR of at least 15 years. For the BMI analysis, we considered 4,783 microbiomes from datasets with a BMI minimum IQR of 3 (24 studies). For the disease analysis, we considered all case-control settings having at least 10 cross-sectional microbiomes in both groups and not belonging to a population classified as non-westernized. In total, we considered 11 diseases and 21 cohorts of adult, Westernized, gut microbiomes (1,856 cases and 1,958 controls). Antibiotics usage information was available for 25 of the 32 studies analyzed. From those 25 studies, 23 excluded individuals having undergone antibiotics treatment in 3-6 months prior to sampling.
We assessed the associations of each ScC species with sex, age, BMI, and diseases in the corresponding set of cohorts as follows: for each set, the ScC species presence/absence was predicted in each dataset independently by a logistic regression (statsmodels python library, ver. 0.11.1) having sex, age, BMI, depth (and disease-state in the disease analysis) and the species as response variable. The coefficients for the variable of interest (sex, age, BMI, or disease-state) were then extracted and pooled in random-effects meta-analyses with Paule-Mandel heterogeneity estimator. P-values of the single datasets are computed as Wald-test for the variable of interest in each dataset Meta-analysis P-value was computed as a Z-score over the hypothesis of null average association. The significance threshold adopted was P-value < 0.05 in the single-dataset analyses and in the meta-analysis.We investigated the correlation between the number of ScC species with sex, age, BMI, and disease in the corresponding set of cohorts similarly: Spearman’s partial correlation (pingouin python library v0.3.7) between the number of ScC species in the cases of aging and BMI, and a Standardized Mean Difference meta-analysis for sex and the twelve diseases. In all cases the variables of interest were adjusted by the other variables plus depth. In aging and BMI, ingle datasets correlations coefficients were Fisher-Z transformed, synthesized in a random-effects meta-analysis with Paule-Mandel heterogeneity, and then reverted back. In the case of sex and the twelve diseases, meta-analysis by SMD was conducted. The significance threshold adopted was P-value < 0.05 in the single-dataset analyses and in the meta-analysis. The code to run this and the previous meta-analysis is available at https://github.com/waldronlab/curatedMetagenomicDataAnalyses.
To study the association between the ScC species and 19 pre-selected cardiometabolic health parameters from the ZOE PREDICT 1 study51 we run logistic regression models predicting the presence of clades A, B, C, and any species (statsmodels python library, ver. 0.11.1) using sex, age, BMI, depth, and the standardized parameter of interest (BMI was also standardized and analyzed among the parameters). P-values for each of the cardiometabolic health parameters were obtained by Wald-test and FDR corrected, and the significance threshold adopted was FDR < 0.2 and 0.05 for assessing clinical relevance (statsmodels python library, ver. 0.11.1).
Protologues of novel Segatella species
The novel species represented by the clades for which isolates could be obtained are described below. These descriptions were generated based on functional insights from Protologger88 using gut metabolic models,95 and ANI comparisons using fastANI38 (version 1.3).
Description of Segatella brunsvicensis
Segatella brunsvicensis (bruns.vic.en’sis. N.L. fem. adj. brunsvicensis, pertaining to Brunswick). The description of this species is based on the features of the type strain, NI025T, as the only cultured representative of this species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 82.64 ± 0.48 %; clade C, 83.21 ± 0.22 %; clade D, 83.01 ± 0.0 %; Segatella hominis (clade E), 83.89 ± 0.03 %; clade F, 81.54 ± 0.0 %; clade G, 83.02 ± 0.39 %. Species separation was confirmed by GTDB-Tk assignment of the strains as “Prevotella sp900313215”. Genomic analysis identified the functional potential for the degradation of arabinoxylan (MF0001), fructan (MF0002), starch (MF0005), lactose (MF0006), melibiose (MF0009), mannose (MF0018), galacturonate (MF0022), aspartate (MF0028, MF0029), cysteine (MF0044), glutamine (MF0047), serine (MF0048), threonine (MF0049), arginine (MF0051), and the production of acetate (MF0086). The type strain, NI025T (=DSM 113023T = JCM 35353T) (Genome: PRJEB60954), was isolated from human feces. Its genome size is 3.69 Mbp with a G+C content of DNA of 45.60 %.
Description of Segatella sinensis
Segatella sinensis (si.nen’sis. N.L. fem. adj. sinensis, pertaining to China). The description of this species is based on the features of seven isolates (strains: HDE04T; HDD14; HDD11; HDD10; HDD16; HDD13; HDD03). These isolates were observed to share an average ANI of 98.81 ± 1.82 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 86.94 ± 0.29 %; clade B, 83.30 ± 0.24 %; clade D, 89.17 ± 0.22 %; Segatella hominis (clade E), 85.08 ± 0.26 %; clade F, 82.82 ± 0.09 %; clade G, 89.06 ± 0.32 %. Species separation was confirmed by GTDB-Tk assignment of the strains as “Prevotella copri_A”. Genomic analysis identified that all strains contained the functional potential for degradation of arabinoxylan (MF0001), fructan (MF0002), starch (MF0005), lactose (MF0006), melibiose (MF0009), mannose (MF0018), galacturonate (MF0022), aspartate (MF0028, MF0029), alanine (MF0034), cysteine (MF0044), glutamine (MF0047), serine (MF0048), threonine (MF0049), arginine (MF0051), and the production of acetate (MF0086). Uniquely all strains belonging to this clade, and no strains belonging to other clades, were observed to encode lactaldehyde degradation (MF0078). The type strain, HDE04T (= DSMZ 108151T =JCM 35349T) (Genome: PRJEB60954), was isolated from human feces. Its genome size is 4.24 Mbp with a G+C content of DNA of 45.37 %. It also contains a large extrachromosomal element of 0.18 Mbp.
Description of Segatella brasiliensis
Segatella brasiliensis (bra.si.li.en’sis. N.L. fem. adj. brasiliensis, from Brazil). The description of this species is based on the features of two isolates (strains: HDD05T, HDD06). These isolates share an average ANI of 99.95 ± 0.00 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 87.42 ± 0.24 %; clade B, 83.36 ± 0.11 %; clade C, 89.03 ± 0.24 %; Segatella hominis (clade E), 85.41 ± 0.42 %; clade F, 82.38 ± 0.04 %; clade G, 88.15 ± 0.18 %. Species separation was confirmed by GTDB-Tk assignment of the strains as “Prevotella sp900546535”. Genomic analysis identified that all strains contained the functional potential for degradation of arabinoxylan (MF0001), fructan (MF0002), starch (MF0005), lactose (MF0006), melibiose (MF0009), mannose (MF0018), rhamnose (MF0019), galacturonate (MF0022), aspartate (MF0028, MF0029), alanine (MF0034), cysteine (MF0044), glutamine (MF0047), serine (MF0048), threonine (MF0049), arginine (MF0051), and the production of acetate (MF0086). The type strain, HDD05T (=DSM 112105T, =JCM 35352T) (Genome: PRJEB60954), was isolated from human feces. Its genome size is 3.99 Mbp with a G+C molecular content of DNA of 44.88 %.
Description of Segatella sanihominis
Segatella sanihominis (sa.ni.ho'mi.nis. L. masc. adj. sanus, healthy; L. masc./fem. n. homo, a human being, a man; N.L. gen. n. sanihominis, of a healthy person). The description of this species is based on the features of the type strain, RHB01T, as the only cultured representative of this species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 82.46 ± 0.17 %; clade B, 81.67 ± 0.0 %; clade C, 82.69 ± 0.1 %; clade D, 82.45 ± 0.01 %; Segatella hominis (clade E), 83.43 ± 0.04 %; clade G, 82.58 ± 0.01 %. Species separation was confirmed by GTDB-Tk assignment of the strains as “Prevotella sp000436035”. Genomic analysis identified the functional potential for the degradation of arabinoxylan (MF0001), fructan (MF0002), starch (MF0005), lactose (MF0006), melibiose (MF0009), mannose (MF0018), rhamnose (MF0019), galacturonate (MF0022), aspartate (MF0028, MF0029), cysteine (MF0044), glutamine (MF0047), serine (MF0048), threonine (MF0049), arginine (MF0051), and the production of acetate (MF0086). The type strain, RHB01T (=DSM 113786T = JCM 35979T) (Genome: PRJEB60954), was isolated from human feces. Its genome size is 3.77 Mbp with a G+C content of 45.35 %.
Description of Segatella sinica
Segatella sinica (si’ni.ca. N.L. fem. adj. sinica, pertaining to China). The description of this species is based on the features of two isolates (strains: HDE06T, HDD09). These isolates share an average ANI of 95.66 ± 0.06 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 87.02 ± 0.29 %; clade B, 83.04 ± 0.33 %; clade C, 89.13 ± 0.28 %; clade D, 88.31 ± 0.12 %; Segatella hominis (clade E), 84.92 ± 0.56 %; clade F, 82.49 ± 0.17 %. Species separation was confirmed by GTDB-Tk assignment of the strains as “Prevotella sp900767615”. Genomic analysis identified that all strains contained the functional potential for degradation of arabinoxylan (MF0001), fructan (MF0002), starch (MF0005), lactose (MF0006), melibiose (MF0009), mannose (MF0018), rhamnose (MF0019), galacturonate (MF0022), aspartate (MF0028, MF0029), cysteine (MF0044), glutamine (MF0047), serine (MF0048), threonine (MF0049), arginine (MF0051), and the production of acetate (MF0086). The type strain, HDE06T (=DSM 111807T, =JCM 35351T) (Genome: PRJEB60954), was isolated from human feces. Its genome size is 4.26 Mbp with a G+C content of 46.17 %, including also contains a large extrachromosomal element of 0.09 Mbp.
Description of Candidatus “Segatella caccae”
Candidatus ‘Segatella caccae’ (cac’cae. Gr. fem. n. kakke, human ordure, feces; N.L. gen. n. caccae, of feces). The description of this species is based on the features of 36 genomes. These genomes share an average ANI of 97.54 ± 0.45 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 80.30 ± 0.89 %; clade C, 80.40 ± 0.99 %; clade D, 80.21 ± 0.84 %; Segatella hominis (clade E), 80.56 ± 0.96 %; clade F, 79.99 ± 0.58 %; clade G, 80.32 ± 0.86 %; clade I, 79.15 ± 0.53 %; clade J, 78.98 ± 0.57 %; clade K, 78.95 ± 0.48 %; clade L, 81.20 ± 0.31 %; clade M, 78.82 ± 0.55 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp900556395”. Genomes assigned to this species have been reconstructed from human faecal samples belonging to individuals from Argentina, Guinea, Tanzania, Madagascar, Denmark, Mongolian and Cameroon. The type genome, ‘RubelMA_2020__CM.378_WGS__bin.6’, was reconstructed from a fecal sample taken from a healthy human female from Cameroon (Sample ID: SRR9293009). Its size is 2.90 Mbp, with a G+C molecular content of DNA of 47.21 %, completeness of 95.12% and contamination of 2.10%.
Description of Candidatus “Segatella intestinihominis”
Candidatus ‘Segatella intestinihominis’ (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, the intestine; L. masc. n. homo, a man; N.L. gen. n. intestinihominis, of the human intestine). The description of this species is based on the features of 28 genomes. These genomes share an average ANI of 97.84 ± 0.56 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 82.86 ± 0.77 %; clade C, 83.01 ± 0.67 %; clade D, 83.24 ± 0.70 %; Segatella hominis (clade E), 81.34 ± 0.76 %; clade F, 82.78 ± 0.55 %; clade G, 83.34 ± 0.60 %; clade H, 79.14 ± 0.58 %; clade J, 82.43 ± 0.42 %; clade K, 80.74 ± 0.43 %; clade L, 79.64 ± 0.43 %; clade M, 81.85 ± 0.47 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp900548535”. Genomes assigned to this species have been reconstructed from human faecal samples belonging to individuals from China, Ethiopia, Ghana, Madagascar, Tanzania, and Cameroon. The type genome, ‘Obregon-TitoAJ_2015__SM18__bin.46’, was reconstructed from a fecal sample taken from a healthy human female from Peru (Sample ID: SRR1761761;SRR1761703[TH1] ). Its size is 2.90 Mbp, with a G+C molecular content of DNA of 44.66 %, completeness of 92.48% and contamination of 2.05%.
Description of Candidatus “Segatella violae”
Candidatus ‘Segatella violae’ (vi.o’lae. L. gen. n. violae, of Viola, the name of a social group of baboons from which the type genome was reconstructed). The description of this species is based on the features of ten genomes. These genomes share an average ANI of 98.80 ± 0.46 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 81.82 ± 0.26 %; clade C, 81.84 ± 0.29 %; clade D, 81.91 ± 0.28 %; Segatella hominis (clade E), 80.76 ± 0.39 %; clade F, 81.84 ± 0.34 %; clade G, 82.20 ± 0.25 %; clade H, 78.87 ± 0.58 %; clade I, 82.42 ± 0.41 %; clade K, 83.83 ± 1.08 %; clade L, 81.67 ± 0.76 %; clade M, 87.87 ± 1.19 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp002440225”. Genomes assigned to this species have only been reconstructed from metagenomic samples originating from faecal samples of Baboons. The type genome, ‘TungJ_2015__F03__bin.23’, was reconstructed from a faecal sample from a female Baboon (Sample ID: SRS812635). Its size is 3.37 Mbp, with a G+C molecular content of DNA of 47.32 %, completeness of 97.87% and contamination of 1.58%.
Description of Candidatus “Segatella albertsiae”
Candidatus ‘Segatella albertsiae’ (al.bert’si.ae. N.L. gen. n. albertsiae, of Alberts, named after Susan Alberts for her work on the social structure and biology of Baboons). The description of this species is based on the features of seven genomes. These genomes share an average ANI of 99.31 ± 0.32 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 80.59 ± 0.36 %; clade C, 80.62 ± 0.30 %; clade D, 80.67 ± 0.29 %; Segatella hominis (clade E), 81.19 ± 0.20 %; clade F, 82.08 ± 0.32 %; clade G, 80.81 ± 0.30 %; clade H, 78.92 ± 0.50 %; clade I, 80.75 ± 0.41 %; clade J, 83.81 ± 1.08 %; clade L, 81.55 ± 0.55 %; clade M, 83.47 ± 1.07 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp002451555”. Genomes assigned to this species have only been reconstructed from metagenomic samples originating from faecal samples of Baboons. The type genome, ‘ParksDH_2017__UBA789’, was reconstructed from a faecal sample from an adult female Baboon (Sample ID: SRX834651). Its size is 3.36 Mbp, with a G+C molecular content of DNA of 44.99 %, completeness of 85.92% and contamination of 1.23%.
Description of Candidatus “Segatella mututuai”
Candidatus ‘Segatella mututuai’ (mu.tu.tu.a’i. N.L. gen. n. mututuai, named after Raphael Mututua for his work conducting research with Baboons). The description of this species is based on the features of six genomes. These genomes share an average ANI of 99.62 ± 0.24 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 79.94 ± 0.17 %; clade C, 80.10 ± 0.09 %; clade D, 80.02 ± 0.17 %; Segatella hominis (clade E), 80.09 ± 0.17 %; clade F, 80.32 ± 0.09 %; clade G, 80.10 ± 0.14 %; clade H, 81.27 ± 0.32 %; clade I, 79.63 ± 0.44 %; clade J, 81.67 ± 0.76 %; clade K, 81.57 ± 0.55 %; clade M,81.42 ± 0.77 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp002439605”. Genomes assigned to this species have only been reconstructed from metagenomic samples originating from faecal samples of Baboons. The type genome, ‘ParksDH_2017__UBA6442’, was reconstructed from a faecal sample from an adult male Baboon (Sample ID: SRX834620). Its size is 2.68 Mbp, with a G+C molecular content of DNA of 46.88 %, completeness of 80.74% and contamination of 0.34%.
Description of Candidatus “Segatella papionis”
Candidatus ‘Segatella papionis’ (pa.pi.o’nis. N.L. gen. n. papionis, of the baboon (genus Papio)). The description of this species is based on the features of five genomes. These genomes share an average ANI of 98.99 ± 0.58 % between each other, confirming they form a single species. The assignment as a novel species was made based on detailed genomic analysis, including ANI values below 95.0% to the studied isolates assigned to other clades within the ScC; Segatella copri (clade A), 81.38 ± 0.49 %; clade C, 82.17 ± 0.39 %; clade D, 81.83 ± 0.36 %; Segatella hominis (clade E), 80.27 ± 0.38 %; clade F, 81.30 ± 0.49 %; clade G, 82.29 ± 0.36 %; clade H, 78.73 ± 0.53 %; clade I, 81.81 ± 0.56 %; clade J, 87.74 ± 1.41 %; clade K, 83.32 ± 1.09 %; clade L, 81.31 ± 0.66 %. ANI values could not be calculated for comparison against clade B. Species separation was confirmed by GTDB-Tk assignment of the genomes as “Prevotella sp002297965”. Genomes assigned to this species have only been reconstructed from metagenomic samples originating from faecal samples of Baboons. The type genome, ‘ParksDH_2017__UBA731’, was reconstructed from a faecal sample from an adult female Baboon (Sample ID: SRX834658). Its size is 2.94 Mbp, with a G+C molecular content of DNA of 46.45 %, completeness of 77.52% and contamination of 0.51%.
Acknowledgments
We thank Boyke Bunk, Cathrin Spröer, Nicole Heyer, and Marzena Wyschkon at the Leibniz Institute DSMZ for helpful advice and technical assistance. We would also like to thank Aharon Oren for his help and advice in providing correctly formed names for each of the proposed novel species and Candidatus species. The graphical abstract was created with BioRender.com.This work was co-funded by the European Research Council (ERC-STG project MetaPG-716575 and ERC-CoG microTOUCH-101045015) to N.S., by the European Union’s Horizon 2020 program (ONCOBIOME-825410 project, MASTER-818368 project, and IHMCSA-964590) to N.S., by the European Union NextGenerationEU (Interconnected Nord-Est Innovation program, INEST; Views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or The European Research Executive Agency; neither the European Union nor the granting authority can be held responsible for them) to N.S., by the National Cancer Institute of the National Institutes of Health (1U01CA230551) to N.S., and by the Premio Internazionale Lombardia e Ricerca 2019 to N.S. Further funding was provided to T.S. by the Deutsche Forschungsgemeinschaft under Germany’s Excellence Strategy – EXC 2155 “RESIST” – Project ID 390874280 and the European Research Council (ERC- COG 865466).
Author contributions
A.B.-M. and E.J.C.G. designed bioinformatic analyses. A.B.-M., E.J.C.G., E.P., F.D.F., K.D.H., P.M., T.-R.L., T.R., L.C., M.P., T.C.A.H., and A.T. conducted bioinformatic analyses. L.A. designed and analyzed experiments. L.A. and I.S. conducted experiments. A.M.T., M.V.-C., T.C., S.E.B., R.D., J.W., T.D.S., J.O., A.T., and D.E. discussed data analysis and interpretation. D.E., N.S., and T.S. designed and coordinated the study. A.B.-M., E.J.C.G., E.P., T.C.A.H., N.S., and T.S. wrote the manuscript. All authors revised and approved the latest version of the manuscript.
Declaration of interests
T.D.S. and J.W. are co-founders of ZOE Ltd. (ZOE). S.E.B. and T.D.S. receive payments as consultants to ZOE. R.D. is employed by ZOE. J.W., R.D., S.E.B., and T.D.S. receive options in ZOE.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2023.09.013.
Supplemental information
(A) Description of the ScC isolates sequenced in this work.
(B) Description of the metagenomic samples analyzed in this work.
(C) Description of the ScC PacBio sequenced genomes.
(D) Correspondence between MetaPhlAn 4 SGBs and the ScC species.
(A) Differentially prevalent UniRef50 families between the ScC species.
(B) Prevalence of all CAZy families in each of the ScC species. CAZyme activities were annotated using Carbohydrate-Active enZYmes Database.
(A) Logistic meta-analysis of sex in 4,095 individuals.
(B) Logistic meta-analysis of age in 3,190 individuals.
(C) Logistic meta-analysis of BMI in 4,783 individuals.
(D) Meta-analysis of the number of ScC species in sex, age, BMI, and diseases.
(E) Meta-analysis of 12 diseases in 3,814 individuals.
(F) Regression analysis of the ScC species on the PREDICT 1 study.
Data and code availability
-
•
The genome assemblies for the isolates produced in this study are publicly available in the European Nucleotide Archive (ENA) at EMBL-EBI: PRJEB60954. Curated metadata for the public metagenomic samples is available in Table S1B.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
References
- 1.Lozupone C.A., Stombaugh J.I., Gordon J.I., Jansson J.K., Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P., et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanco-Míguez A., Beghini F., Cumbo F., McIver L.J., Thompson K.N., Zolfo M., Manghi P., Dubois L., Huang K.D., Thomas A.M., et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023;4 doi: 10.1038/s41587-023-01688-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hitch T.C.A., Bisdorf K., Afrizal A., Riedel T., Overmann J., Strowig T., Clavel T. A taxonomic note on the genus Prevotella: description of four novel genera and emended description of the genera Hallella and Xylanibacter. Syst. Appl. Microbiol. 2022;45:126354. doi: 10.1016/j.syapm.2022.126354. [DOI] [PubMed] [Google Scholar]
- 7.Smits S.A., Leach J., Sonnenburg E.D., Gonzalez C.G., Lichtman J.S., Reid G., Knight R., Manjurano A., Changalucha J., Elias J.E., et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357:802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnorr S.L., Candela M., Rampelli S., Centanni M., Consolandi C., Basaglia G., Turroni S., Biagi E., Peano C., Severgnini M., et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014;5 doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obregon-Tito A.J., Tito R.Y., Metcalf J., Sankaranarayanan K., Clemente J.C., Ursell L.K., Zech Xu Z., Van Treuren W., Knight R., Gaffney P.M., et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015;6 doi: 10.1038/ncomms7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen M.E.B., Rubel M.A., Bailey A.G., Ranciaro A., Thompson S.R., Campbell M.C., Beggs W., Dave J.R., Mokone G.G., Mpoloka S.W., et al. Population structure of human gut bacteria in a diverse cohort from rural Tanzania and Botswana. Genome Biol. 2019;20 doi: 10.1186/s13059-018-1616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Filippo C., Cavalieri D., Di Paola M., Ramazzotti M., Poullet J.B., Massart S., Collini S., Pieraccini G., Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu G.D., Chen J., Hoffmann C., Bittinger K., Chen Y.-Y., Keilbaugh S.A., Bewtra M., Knights D., Walters W.A., Knight R., et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ou J., Carbonero F., Zoetendal E.G., DeLany J.P., Wang M., Newton K., Gaskins H.R., O’Keefe S.J.D. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013;98:111–120. doi: 10.3945/ajcn.112.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Filippis F., Pellegrini N., Vannini L., Jeffery I.B., La Storia A., Laghi L., Serrazanetti D.I., Di Cagno R., Ferrocino I., Lazzi C., et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut. 2016;65:1812–1821. doi: 10.1136/gutjnl-2015-309957. [DOI] [PubMed] [Google Scholar]
- 15.Haro C., García-Carpintero S., Rangel-Zúñiga O.A., Alcalá-Díaz J.F., Landa B.B., Clemente J.C., Pérez-Martínez P., López-Miranda J., Pérez-Jiménez F., Camargo A. Consumption of two healthy dietary patterns restored microbiota dysbiosis in obese patients with metabolic dysfunction. Mol. Nutr. Food Res. 2017;61 doi: 10.1002/mnfr.201700300. [DOI] [PubMed] [Google Scholar]
- 16.Precup G., Vodnar D.-C. Gut Prevotella as a possible biomarker of diet and its eubiotic versus dysbiotic roles: a comprehensive literature review. Br. J. Nutr. 2019;122:131–140. doi: 10.1017/S0007114519000680. [DOI] [PubMed] [Google Scholar]
- 17.Scher J.U., Sczesnak A., Longman R.S., Segata N., Ubeda C., Bielski C., Rostron T., Cerundolo V., Pamer E.G., Abramson S.B., et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2013;2 doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pianta A., Arvikar S., Strle K., Drouin E.E., Wang Q., Costello C.E., Steere A.C. Evidence of the immune relevance of Prevotella copri, a gut microbe, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017;69:964–975. doi: 10.1002/art.40003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alpizar-Rodriguez D., Lesker T.R., Gronow A., Gilbert B., Raemy E., Lamacchia C., Gabay C., Finckh A., Strowig T. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2019;78:590–593. doi: 10.1136/annrheumdis-2018-214514. [DOI] [PubMed] [Google Scholar]
- 20.Tett A., Huang K.D., Asnicar F., Fehlner-Peach H., Pasolli E., Karcher N., Armanini F., Manghi P., Bonham K., Zolfo M., et al. The Prevotella copri complex comprises four distinct clades underrepresented in westernized populations. Cell Host Microbe. 2019;26:666–679.e7. doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tett A., Pasolli E., Masetti G., Ercolini D., Segata N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 2021;19:585–599. doi: 10.1038/s41579-021-00559-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marungruang N., Tovar J., Björck I., Hållenius F.F. Improvement in cardiometabolic risk markers following a multifunctional diet is associated with gut microbial taxa in healthy overweight and obese subjects. Eur. J. Nutr. 2018;57:2927–2936. doi: 10.1007/s00394-017-1563-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benítez-Páez A., Kjølbæk L., Gómez Del Pulgar E.M.G., Brahe L.K., Astrup A., Matysik S., Schött H.-F., Krautbauer S., Liebisch G., Boberska J., et al. A multi-omics approach to unraveling the microbiome-mediated effects of arabinoxylan oligosaccharides in overweight humans. mSystems. 2019;4:e00209–e00219. doi: 10.1128/mSystems.00209-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roager H.M., Vogt J.K., Kristensen M., Hansen L.B.S., Ibrügger S., Mærkedahl R.B., Bahl M.I., Lind M.V., Nielsen R.L., Frøkiær H., et al. Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: a randomised cross-over trial. Gut. 2019;68:83–93. doi: 10.1136/gutjnl-2017-314786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh T.S., Rampelli S., Jeffery I.B., Santoro A., Neto M., Capri M., Giampieri E., Jennings A., Candela M., Turroni S., et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: the NU-AGE 1-year dietary intervention across five European countries. Gut. 2020;69:1218–1228. doi: 10.1136/gutjnl-2019-319654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Filippis F., Pasolli E., Ercolini D. Newly explored Faecalibacterium diversity is connected to age, lifestyle, geography, and disease. Curr. Biol. 2020;30:4932–4943.e4. doi: 10.1016/j.cub.2020.09.063. [DOI] [PubMed] [Google Scholar]
- 27.Karcher N., Nigro E., Punčochář M., Blanco-Míguez A., Ciciani M., Manghi P., Zolfo M., Cumbo F., Manara S., Golzato D., et al. Genomic diversity and ecology of human-associated Akkermansia species in the gut microbiome revealed by extensive metagenomic assembly. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02427-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karcher N., Pasolli E., Asnicar F., Huang K.D., Tett A., Manara S., Armanini F., Bain D., Duncan S.H., Louis P., et al. Analysis of 1321 Eubacterium rectale genomes from metagenomes uncovers complex phylogeographic population structure and subspecies functional adaptations. Genome Biol. 2020;21 doi: 10.1186/s13059-020-02042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J., Gálvez E.J.C., Amend L., Almási É., Iljazovic A., Lesker T.R., Bielecka A.A., Schorr E.-M., Strowig T. A versatile genetic toolbox for Prevotella copri enables studying polysaccharide utilization systems. EMBO J. 2021;40 doi: 10.15252/embj.2021108287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2012;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P., et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasolli E., Schiffer L., Manghi P., Renson A., Obenchain V., Truong D.T., Beghini F., Malik F., Ramos M., Dowd J.B., et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods. 2017;14:1023–1024. doi: 10.1038/nmeth.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagan R.W., Hofman C.A., Hübner A., Reinhard K., Schnorr S., Lewis C.M., Jr., Sankaranarayanan K., Warinner C.G. Comparison of extraction methods for recovering ancient microbial DNA from paleofeces. Am. J. Phys. Anthropol. 2020;171:275–284. doi: 10.1002/ajpa.23978. [DOI] [PubMed] [Google Scholar]
- 34.Borry M., Cordova B., Perri A., Wibowo M., Prasad Honap T., Ko J., Yu J., Britton K., Girdland-Flink L., Power R.C., et al. CoproID predicts the source of coprolites and paleofeces using microbiome composition and host DNA content. PeerJ. 2020;8 doi: 10.7717/peerj.9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maixner F., Sarhan M.S., Huang K.D., Tett A., Schoenafinger A., Zingale S., Blanco-Míguez A., Manghi P., Cemper-Kiesslich J., Rosendahl W., et al. Hallstatt miners consumed blue cheese and beer during the Iron Age and retained a non-Westernized gut microbiome until the Baroque period. Curr. Biol. 2021;31:5149–5162.e6. doi: 10.1016/j.cub.2021.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wibowo M.C., Yang Z., Borry M., Hübner A., Huang K.D., Tierney B.T., Zimmerman S., Barajas-Olmos F., Contreras-Cubas C., García-Ortiz H., et al. Reconstruction of ancient microbial genomes from the human gut. Nature. 2021;594:234–239. doi: 10.1038/s41586-021-03532-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manara S., Asnicar F., Beghini F., Bazzani D., Cumbo F., Zolfo M., Nigro E., Karcher N., Manghi P., Metzger M.I., et al. Microbial genomes from non-human primate gut metagenomes expand the primate-associated bacterial tree of life with over 1000 novel species. Genome Biol. 2019;20 doi: 10.1186/s13059-019-1923-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parks D.H., Chuvochina M., Chaumeil P.-A., Rinke C., Mussig A.J., Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020;38:1079–1086. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 40.Liou J.-S., Huang C.-H., Ikeyama N., Lee A.-Y., Chen I.-C., Blom J., Chen C.-C., Chen C.-H., Lin Y.-C., Hsieh S.-Y., et al. Prevotella hominis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2020;70:4767–4773. doi: 10.1099/ijsem.0.004342. [DOI] [PubMed] [Google Scholar]
- 41.Devoto A.E., Santini J.M., Olm M.R., Anantharaman K., Munk P., Tung J., Archie E.A., Turnbaugh P.J., Seed K.D., Blekhman R., et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019;4:693–700. doi: 10.1038/s41564-018-0338-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brewster R., Tamburini F.B., Asiimwe E., Oduaran O., Hazelhurst S., Bhatt A.S. Surveying gut microbiome research in Africans: toward improved diversity and representation. Trends Microbiol. 2019;27:824–835. doi: 10.1016/j.tim.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X., Liang S., Xia Z., Qu J., Liu H., Liu C., Yang H., Wang J., Madsen L., Hou Y., et al. Establishment of a Macaca fascicularis gut microbiome gene catalog and comparison with the human, pig, and mouse gut microbiomes. GigaScience. 2018;7 doi: 10.1093/gigascience/giy100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzek B.E., Wang Y., Huang H., McGarvey P.B., Wu C.H., UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myllykallio H., Leduc D., Filee J., Liebl U. Life without dihydrofolate reductase FolA. Trends Microbiol. 2003;11:220–223. doi: 10.1016/s0966-842x(03)00101-x. [DOI] [PubMed] [Google Scholar]
- 46.Segata N. Gut microbiome: westernization and the disappearance of intestinal diversity. Curr. Biol. 2015;25:R611–R613. doi: 10.1016/j.cub.2015.05.040. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X., Zhang D., Jia H., Feng Q., Wang D., Liang D., Wu X., Li J., Tang L., Li Y., et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015;21:895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 48.Qin N., Yang F., Li A., Prifti E., Chen Y., Shao L., Guo J., Le Chatelier E., Yao J., Wu L., et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 49.Feng Q., Liang S., Jia H., Stadlmayr A., Tang L., Lan Z., Zhang D., Xia H., Xu X., Jie Z., et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015;6 doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- 50.Forslund S.K., Chakaroun R., Zimmermann-Kogadeeva M., Markó L., Aron-Wisnewsky J., Nielsen T., Moitinho-Silva L., Schmidt T.S.B., Falony G., Vieira-Silva S., et al. Combinatorial, additive and dose-dependent drug-microbiome associations. Nature. 2021;600:500–505. doi: 10.1038/s41586-021-04177-9. [DOI] [PubMed] [Google Scholar]
- 51.Asnicar F., Berry S.E., Valdes A.M., Nguyen L.H., Piccinno G., Drew D.A., Leeming E., Gibson R., Le Roy C., Khatib H.A., et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat. Med. 2021;27:321–332. doi: 10.1038/s41591-020-01183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walter J., Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- 53.Naito M., Ogura Y., Itoh T., Shoji M., Okamoto M., Hayashi T., Nakayama K. The complete genome sequencing of Prevotella intermedia strain OMA14 and a subsequent fine-scale, intra-species genomic comparison reveal an unusual amplification of conjugative and mobile transposons and identify a novel Prevotella-lineage-specific repeat. DNA Res. 2016;23:11–19. doi: 10.1093/dnares/dsv032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dillon S.M., Lee E.J., Kotter C.V., Austin G.L., Dong Z., Hecht D.K., Gianella S., Siewe B., Smith D.M., Landay A.L., et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7:983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J., Zhao F., Wang Y., Chen J., Tao J., Tian G., Wu S., Liu W., Cui Q., Geng B., et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5 doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wen C., Zheng Z., Shao T., Liu L., Xie Z., Le Chatelier E., He Z., Zhong W., Fan Y., Zhang L., et al. Correction to: quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017;18 doi: 10.1186/s13059-017-1271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iljazovic A., Amend L., Galvez E.J.C., de Oliveira R., Strowig T. Modulation of inflammatory responses by gastrointestinal Prevotella spp. - From associations to functional studies. Int. J. Med. Microbiol. 2021;311 doi: 10.1016/j.ijmm.2021.151472. [DOI] [PubMed] [Google Scholar]
- 58.Ley R.E. Gut microbiota in 2015: Prevotella in the gut: choose carefully. Nat. Rev. Gastroenterol. Hepatol. 2016;13:69–70. doi: 10.1038/nrgastro.2016.4. [DOI] [PubMed] [Google Scholar]
- 59.Cani P.D. Human gut microbiome: hopes, threats and promises. Gut. 2018;67:1716–1725. doi: 10.1136/gutjnl-2018-316723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Claus S.P. The strange case of Prevotella copri: dr. Jekyll or Mr. Hyde? Cell Host Microbe. 2019;26:577–578. doi: 10.1016/j.chom.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 61.Metwaly A., Haller D. Strain-level diversity in the gut: the P. copri Case. Cell Host Microbe. 2019;25:349–350. doi: 10.1016/j.chom.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 62.De Filippis F., Pasolli E., Tett A., Tarallo S., Naccarati A., De Angelis M., Neviani E., Cocolin L., Gobbetti M., Segata N., et al. Distinct genetic and functional traits of human intestinal Prevotella copri Strains Are associated with different habitual diets. Cell Host Microbe. 2019;25:444–453.e3. doi: 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 63.Armstrong A.J.S., Shaffer M., Nusbacher N.M., Griesmer C., Fiorillo S., Schneider J.M., Preston Neff C., Li S.X., Fontenot A.P., Campbell T., et al. An exploration of Prevotella-rich microbiomes in HIV and men who have sex with men. Microbiome. 2018;6 doi: 10.1186/s40168-018-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grey J.A., Bernstein K.T., Sullivan P.S., Purcell D.W., Chesson H.W., Gift T.L., Rosenberg E.S. Estimating the population sizes of men who have sex with men in US states and counties using data from the American community survey. JMIR Public Health Surveill. 2016;2 doi: 10.2196/publichealth.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.David L.A., Maurice C.F., Carmody R.N., Gootenberg D.B., Button J.E., Wolfe B.E., Ling A.V., Devlin A.S., Varma Y., Fischbach M.A., et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovatcheva-Datchary P., Nilsson A., Akrami R., Lee Y.S., De Vadder F., Arora T., Hallen A., Martens E., Björck I., Bäckhed F. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 2015;22:971–982. doi: 10.1016/j.cmet.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 67.De Vadder F., Kovatcheva-Datchary P., Zitoun C., Duchampt A., Bäckhed F., Mithieux G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 2016;24:151–157. doi: 10.1016/j.cmet.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 68.Asnicar F., Thomas A.M., Beghini F., Mengoni C., Manara S., Manghi P., Zhu Q., Bolzan M., Cumbo F., May U., et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asnicar F., Weingart G., Tickle T.L., Huttenhower C., Segata N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 2015;3 doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Longmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zolfo M., Pinto F., Asnicar F., Manghi P., Tett A., Bushman F.D., Segata N. Detecting contamination in viromes using ViromeQC. Nat. Biotechnol. 2019;37:1408–1412. doi: 10.1038/s41587-019-0334-5. [DOI] [PubMed] [Google Scholar]
- 72.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 73.Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 74.Steinegger M., Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 75.Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P.K., Xu Y., Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stewart R.D., Auffret M.D., Roehe R., Watson M. 2018. Open prediction of polysaccharide utilisation loci (PUL) in 5414 public Bacteroidetes genomes using PULpy. [DOI] [Google Scholar]
- 77.Ausland C., Zheng J., Yi H., Yang B., Li T., Feng X., Zheng B., Yin Y. dbCAN-PUL: a database of experimentally characterized CAZyme gene clusters and their substrates. Nucleic Acids Res. 2021;49:D523–D528. doi: 10.1093/nar/gkaa742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 80.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Page A.J., Cummins C.A., Hunt M., Wong V.K., Reuter S., Holden M.T.G., Fookes M., Falush D., Keane J.A., Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pages H., Aboyoun P., Gentleman R., DebRoy S. Biostrings: string objects representing biological sequences, and matching algorithms R Package Version. [DOI]
- 84.Pedersen T.L. 2015. FindMyFriends: microbial comparative genomics in R.http://bioconductor.org/packages/FindMyFriends/ R package version 1.0.2. [Google Scholar]
- 85.Seemann, T., and Booth, T. Barrnap: basic rapid ribosomal RNA predictor. GitHub repository. https://github.com/tseemann/barrnap
- 86.Horesh G., Harms A., Fino C., Parts L., Gerdes K., Heinz E., Thomson N.R. SLING: a tool to search for linked genes in bacterial datasets. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hitch T.C.A., Riedel T., Oren A., Overmann J., Lawley T.D., Clavel T. Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. ISME Commun. 2021;1 doi: 10.1038/s43705-021-00017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bruns A., Mueller M., Schneider I., Hahn A. Application of a modified healthy eating index (HEI-flex) to compare the diet quality of flexitarians, vegans and omnivores in Germany. Nutrients. 2022;14 doi: 10.3390/nu14153038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amend L., Gilbert B.T.P., Pelczar P., Böttcher M., Huber S., Witte T., Finckh A., Strowig T. Characterization of serum biomarkers and antibody responses against Prevotella spp. in preclinical and new-onset phase of rheumatic diseases. Front. Cell. Infect. Microbiol. 2022;12 doi: 10.3389/fcimb.2022.1096211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mirdita M., von den Driesch L., Galiez C., Martin M.J., Söding J., Steinegger M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017;45:D170–D176. doi: 10.1093/nar/gkw1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crisci M.A., Chen L.-X., Devoto A.E., Borges A.L., Bordin N., Sachdeva R., Tett A., Sharrar A.M., Segata N., Debenedetti F., et al. Closely related Lak megaphages replicate in the microbiomes of diverse animals. iScience. 2021;24 doi: 10.1016/j.isci.2021.102875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yin Y., Mao X., Yang J., Chen X., Mao F., Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vieira-Silva S., Falony G., Darzi Y., Lima-Mendez G., Garcia Yunta R., Okuda S., Vandeputte D., Valles-Colomer M., Hildebrand F., Chaffron S., et al. Species-function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol. 2016;1 doi: 10.1038/nmicrobiol.2016.88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Description of the ScC isolates sequenced in this work.
(B) Description of the metagenomic samples analyzed in this work.
(C) Description of the ScC PacBio sequenced genomes.
(D) Correspondence between MetaPhlAn 4 SGBs and the ScC species.
(A) Differentially prevalent UniRef50 families between the ScC species.
(B) Prevalence of all CAZy families in each of the ScC species. CAZyme activities were annotated using Carbohydrate-Active enZYmes Database.
(A) Logistic meta-analysis of sex in 4,095 individuals.
(B) Logistic meta-analysis of age in 3,190 individuals.
(C) Logistic meta-analysis of BMI in 4,783 individuals.
(D) Meta-analysis of the number of ScC species in sex, age, BMI, and diseases.
(E) Meta-analysis of 12 diseases in 3,814 individuals.
(F) Regression analysis of the ScC species on the PREDICT 1 study.
Data Availability Statement
-
•
The genome assemblies for the isolates produced in this study are publicly available in the European Nucleotide Archive (ENA) at EMBL-EBI: PRJEB60954. Curated metadata for the public metagenomic samples is available in Table S1B.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.