

Significance
Giant axonal neuropathy (GAN) is caused by mutations in the GAN gene encoding for Gigaxonin (GIG), an adaptor of the CUL3-RBX1-GIG (CRL3GIG) E3 ubiquitin ligase complex. GAN’s pathological hallmark is characterized by the accumulation of neurofilaments (NFs) in the axons. However, it has yet to be explored how CRL3GIG regulates the homeostasis of NFs. Here, we find that the CRL3GIG–USP15 pathway governs the destruction of NF proteins NEFL and INA. Furthermore, CRL3GIG controls actin filaments by targeting TPM1, TPM2, TAGLN, and CNN2 for proteasomal degradation. Strikingly, mutations in the Kelch domain of GIG disrupted its binding to NEFL and INA, leading to the accumulation of NF proteins in GAN-knockout cells. This accounts for the loss-of-function mutations in GAN patients.
Keywords: Gigaxonin, USP15, neurofilament, GAN
Abstract
Giant axonal neuropathy (GAN) is caused by mutations in the GAN gene encoding for gigaxonin (GIG), which functions as an adaptor of the CUL3-RBX1-GIG (CRL3GIG) E3 ubiquitin ligase complex. The pathological hallmark of GAN is characterized by the accumulation of densely packed neurofilaments (NFs) in the axons. However, there are fundamental knowledge gaps in our understanding of the molecular mechanisms by which the ubiquitin–proteasome system controls the homeostasis of NF proteins. Recently, the deubiquitylating enzyme USP15 was reported to play a crucial role in regulating ubiquitylation and proteasomal degradation of CRL4CRBN substrate proteins. Here, we report that the CRL3GIG–USP15 pathway governs the destruction of NF proteins NEFL and INA. We identified a specific degron called NEFLL12 degron for CRL3GIG. Notably, mutations in the C-terminal Kelch domain of GIG, represented by L309R, R545C, and C570Y, disrupted the binding of GIG to NEFL and INA, leading to the accumulation of these NF proteins. This accounts for the loss-of-function mutations in GAN patients. In addition to regulating NFs, CRL3GIG also controls actin filaments by directly targeting actin-filament-binding regulatory proteins TPM1, TPM2, TAGLN, and CNN2 for proteasomal degradation. Thus, our findings broadly impact the field by providing fundamental mechanistic insights into regulating extremely long-lived NF proteins NEFL and INA by the CRL3GIG–USP15 pathway and offering previously unexplored therapeutic opportunities to treat GAN patients and other neurodegenerative diseases by explicitly targeting downstream substrates of CRL3GIG.
The ubiquitin–proteasome system (UPS) plays a crucial role in almost all critical cellular pathways, and its dysregulation leads to deadly human diseases, including cancer and neurodegenerative diseases (1, 2). Ubiquitylation of protein substrates is catalyzed by the ubiquitin-specific E1, E2, and more than 600 E3 ligases, and can be reversed by ~100 deubiquitylating enzymes (DUBs) (3). The cullin-RING ubiquitin ligases (CRLs) represent a major family of multicomponent RING-class E3 complexes, composed of four subunits: a cullin scaffold, an E2-recruiting RING domain protein (RBX1 or RBX2), an adaptor, and a substrate receptor (4, 5). In mammals, there are eight human cullins (CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, CUL7, and CUL9) (6). For example, CUL3 interacts with BTB domain proteins, encoded by approximately 180 genes in the human genome (7), which function as both substrate adaptors and receptors in a single protein (8–11). The BTB domains can also facilitate self-assembly to form dimers, leading to the formation of CUL3-AdaptorBTB E3 homodimers (12–14). However, little is known about how CRL3 E3 ligases control ubiquitylation and proteasomal degradation of downstream substrates under neurodegenerative disorders.
In eukaryotes, the cytoskeleton comprises three types of filaments: microtubules, microfilaments (known as actin filaments), and intermediate filaments (IFs). IF proteins, encoded by 73 genes, are classified into six types (I–VI), based on sequence homology (15). The type I and type II groups are the acidic and basic keratins, respectively. The type III group includes vimentin (VIM), desmin (DES), glial fibrillary acidic protein (GFAP), and peripherin (PRPH), while the nuclear lamins are categorized as type V. Tanabin, transitin, and nestin are type VI (16). Neurofilaments (NFs), classified as type IV IF proteins, are composed of 4 subunits: NF heavy (200 kDa), medium (145–160 kDa), and light polypeptide (68–70 kDa) (NFH, NFM, and NFL, respectively; hereafter referred to as NEFH, NEFM, and NEFL), and a-internexin (INA, 59–65 kDa) in mature neurons in the central nervous system (CNS) (17, 18). In neurons in the peripheral nervous system (PNS), NFs include NEFH, NEFM, NEFL, and PRPH (57–58 kDa) (18). Despite recent advances in understanding neurofilament biology and pathophysiology, numerous fundamental questions remain unanswered. Specifically, how does the UPS regulate the turnover of NF subunits identified as extremely “long-lived” proteins? Which DUBs and E3 ubiquitin ligases are involved in maintaining the homeostasis of NFs? What is the significance of NF turnover in contributing to neurodegenerative diseases?
Giant axonal neuropathy (GAN), first described in 1972 by Berg and colleagues, is a rare hereditary autosomal recessive neurodegenerative disorder (19, 20). The pathological hallmark of GAN is characterized by the accumulation of densely packed NFs in the axons of PNS and CNS (21, 22). GAN patients manifest severe early-onset motor and sensory axonal neuropathy with CNS involvement and characteristic “kinky” hair around 3–5 y of age. Their death usually occurs in the second to third decade of life (23). In 2000, Bomont and colleagues identified the disease-causing gene GAN encoding Gigaxonin protein (GIG) (24). GIG comprises an amino-terminal BTB domain and a C-terminal Kelch domain (24). Based on sequence homology (7) and the structural study of CUL3-GIGBTB homodimer complex (12), GIG (also known as KLHL16) may function as an adaptor in the CUL3-RBX1-GIG (known as CRL3GIG) E3 ubiquitin ligase complex and regulate diverse substrates. However, it is not known whether CRL3GIG could ubiquitylate specific substrates in vitro and in vivo. What is a specific degradation signal on substrates (degron) that binds GIG, leading to ubiquitylation and degradation of CRL3GIG substrates? What are the mechanisms of pathogenesis in patients carrying GAN mutations?
Targeted protein degradation by the UPS is an important therapeutic modality in drug discovery to target previously undruggable disease-causing proteins for destruction (25–27). The molecular glue and PROTAC degraders hijack E3 ubiquitin ligases, leading to ubiquitylation and subsequent degradation of protein targets. A recent study by Nguyen has reported that USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase (GS) and neosubstrates, thereby preventing their degradation (28). Targeting USP15 sensitizes the immunomodulatory drug (IMiD)-sensitive and resistant multiple myeloma cells to lenalidomide by promoting the degradation of neosubstrates. USP15 diminishes the action of molecular glue Cereblon E3 ligase modulator (CELMoD) and CRBN-based PROTAC degraders by antagonizing the activity of the CRL4CRBN (28). However, whether USP15 may regulate the downstream substrates of other CRL E3 ligases is largely unknown.
In the present study, we addressed multiple “long-standing” questions. We found that the CRL3GIG-USP15 pathway is required to regulate long-lived NF proteins NEFL and INA. In addition to regulating NFs, CRL3GIG also controls another class of cytoskeleton proteins by targeting the destruction of actin filament–binding regulatory proteins such as TPM1, TPM2, TAGLN, and CNN2. We further identified a specific degron called NEFLL12 degron for CRL3GIG. This fundamental knowledge is essential because it helps us further understand the molecular basis of GAN pathogenesis and opens previously unexplored opportunities for developing novel targeted therapies to treat neurodegenerative diseases.
Results
USP15 Controls the Stability of NF Proteins.
To systemically characterize the alteration of proteome profile in wild-type (WT) versus USP15-knockout (KO) 293FT cells (28), and to search for potential USP15 substrates, we performed the proteome profiling analyses of WT and USP15-KO 293FT cells and quantified 8,757 proteins (Fig. 1A, SI Appendix, Fig. S1, and Dataset S1). A total of 22 proteins were significantly down-regulated (Fig. 1A and SI Appendix, Table S1A), while 20 proteins were up-regulated in USP15-KO 293FT cells (Fig. 1A and SI Appendix, Table S1B). GS, a well-characterized substrate of USP15, was down-regulated in USP15-KO 293FT cells (SI Appendix, Table S1A) (28). Notably, the top 10 most down-regulated proteins included two NF proteins NEFL and INA (Fig. 1A and SI Appendix, Table S1A). NF proteins are mainly expressed in neurons and present predominantly in axons, which play a crucial role in maintaining axon caliber and the transmission of electrical impulses along axons (17). Previous studies indicated that HEK293 (or derivative) cells express all 4 NF subunits (29), and they are widely used to study NFs such as translational regulation of NEFM and NEFL mRNAs by the UBR1/UBR2-mediated Arg/N-degron pathway (30) or the GIG-IF interaction network (31). Therefore, 293FT cell lines were chosen for our biochemical studies.
Fig. 1.
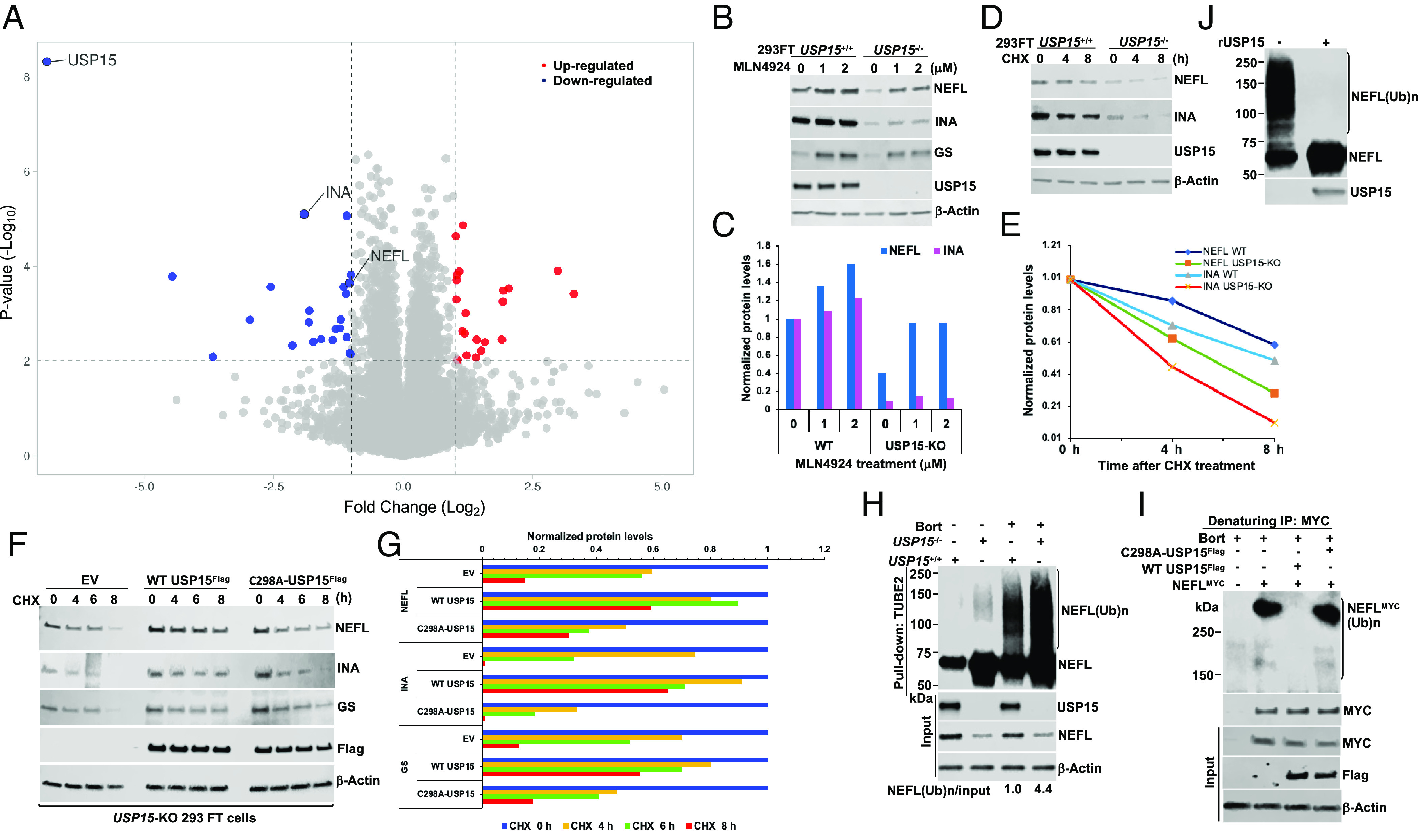
USP15 controls the stability of NF proteins. (A) Global proteome analysis of WT and USP15-KO 293FT cells. The threshold of fold changes was set to more than a twofold increase or decrease, and the –Log10 P-value was above 2; two biological and technical replicate samples were subject to mass spectrometric analysis. (B) Validation of proteomic data. WT and USP15-KO 293FT cells were treated with different concentrations of MLN4924 (0, 1, and 2 µM) for 8 h. Cell extracts were analyzed by immunoblotting (IB) with the indicated antibodies. (C) Quantification of NEFL and INA proteins from (B). (D) CHX experiment. USP15+/+ and USP15−/− 293FT cells were treated with cycloheximide (CHX, 50 µg/mL) at the indicated times. Cell extracts were analyzed by IB. (E) Quantification of NEFL and INA proteins from (D). (F) USP15-KO 293FT cells were transiently transfected with empty vector (EV, control), WT USP15Flag, or C298A-USP15Flag mutant plasmid. After 40 h, cells were treated with CHX for the indicated times. Cell extracts were analyzed by IB with the indicated antibodies. (G) Quantification of NEFL, INA, and GS proteins from (F). The relative ratios of indicated proteins:Actin, normalized to time 0, are shown. (H) USP15+/+ and USP15−/− 293FT cells were treated with bortezomib (Bort, 1 µM) for 7 h. Total ubiquitylated proteins were purified using TUBE2-agarose. Bound fractions and input were analyzed by IB. (I) USP15-KO 293FT cells were transiently transfected with NEFLMYC in the presence of EV, WT USP15Flag, or C298A-USP15Flag mutant plasmid. After 40 h, cells were treated with 2 µM bortezomib for 6 h, followed by cell lysis and MYC IP under denaturing conditions. The bound fractions and input were analyzed by IB with antibodies against ubiquitin (Top), MYC, Flag, and Actin. (J) In vitro deubiquitylation assay. USP15−/− 293FT cells were treated with 1 µM Bort for 7 h. Total ubiquitylated proteins, purified using TUBE2, were treated with recombinant (r)USP15 and then analyzed by IB with anti-NEFL and anti-USP15 antibodies. (Ub)n, polyubiquitin. The results shown are representative of three independent experiments (B, D, F, and H–J).
To validate our proteomic data, we treated WT and USP15-KO 293FT cells with the NEDD8-activating enzyme inhibitor MLN4924, which inactivates CRL activity (32) and analyzed by Western blot. Consistent with the proteomic data (Fig. 1A), the steady-state levels of NEFL and INA were significantly down-regulated in USP15-KO 293FT cells (Fig. 1 B and C). MLN4924-treated cells stabilized at least twofold the protein level of NEFL, and to a lesser extent INA protein level in USP15-KO 293FT cells, which was similar to the results observed in GS protein level (28) (Fig. 1B). Depletion of USP15 by short hairpin RNA (shRNA) knockdown (KDO) down-regulated NEFL and INA protein levels in the neuroblastoma cell line SH-SY5Y (SI Appendix, Fig. S2A). Depletion of USP15, but not other DUBs, including USP7 and USP11, caused the downregulation of NEFL, INA, and GS proteins (Fig. 1B and SI Appendix, Fig. S2 A and C). These findings suggest that USP15 might play an essential role in regulating the stability of NF proteins.
NF proteins exhibit exceptionally long half-lives (17); however, our cycloheximide (CHX) chase experiments revealed that both NEFL and INA were significantly degraded in USP15-KO 293FT cells (Fig. 1 D and E). The downregulation and the enhanced degradation rate of endogenous NEFL, INA, and GS proteins in USP15-KO 293FT cells were blunted by overexpressing WT USP15Flag, but not empty vector or a catalytically inactive C298A-USP15Flag mutant (Fig. 1 F and G and SI Appendix, Fig. S2 D and E). Our RNA-sequencing (RNA-seq) analysis showed that the mRNA levels of NEFL and INA were unchanged in USP15-KO 293FT cells and MLN4924-treated USP15-KO 293FT cells (SI Appendix, Figs. S3 and S4 and Datasets S2 and S3). These data indicated that the molecular control of USP15-mediated degradation of NEFL and INA was a posttranslational mechanism and that USP15 may control their ubiquitylation status and subsequent degradation by the proteasome. To test this idea, we performed TUBE2 pulldown experiments in cells treated with the proteasome inhibitor bortezomib (Bort) (33). We found that the ubiquitylated forms of NEFL were significantly accumulated in Bort-treated USP15-KO 293FT cells (Fig. 1H). In line with the results observed in 293FT cell lines (Fig. 1H), USP15-depleted SH-SY5Y cells increased ubiquitylated forms of NEFL and INA (SI Appendix, Fig. S2B). Overexpression of WT USP15Flag completely abrogated polyubiquitylated NEFL, while a C298A-USP15Flag mutant failed to deubiquitylate polyubiquitylated NEFL in USP15-KO 293FT cells (Fig. 1I). Strikingly, recombinant USP15 enzyme (rUSP15) purified from insect cells efficiently deubiquitylated polyubiquitylated NEFL in vitro (Fig. 1J). Collectively, our data strongly indicated that USP15 plays an important role in regulating the stability of NEFL and INA in a CRL E3 ubiquitin ligase-dependent fashion.
Depletion of GAN Resulted in the Accumulation of NF Proteins and Actin Filament–Associated Regulatory Proteins.
Since we uncovered a pathway in the UPS, including USP15, proteasome, and a putative CRL ubiquitin ligase to control the stability of NEFL and INA, we next sought to identify the CRL E3 ligase involved in this process. Consistent with our results that MLN4924 suppressed the degradation of NEFL and INA in USP15-KO 293FT cells (Fig. 1B), a previous study by Emanuele and colleagues focused on the global identification of modular Cullin-RING ligase substrates revealed NEFL protein as one of the top candidates in the CRL3 and MLN4924 GPS screens (34). GIG was particularly interesting among all potential CUL3 adaptors because NF proteins were markedly accumulated in GAN patients (19). Notably, the crystal structure of the GIGBTB domain confirmed that GIG is a CUL3 adaptor (12). Based on these previous studies and our results (Fig. 1B), we hypothesized that CRL3GIG controls the turnover of NEFL and other potential substrates. We first generated GAN-KO 293FT cells by CRISPR/Cas9 gene editing to test this hypothesis and found that 15 of 26 clones were completely knockout (SI Appendix, Fig. S5). Interestingly, all GAN-KO 293FT cells accumulated NEFL protein levels (SI Appendix, Fig. S5).
To broadly understand how the proteome is remodeled and to search for potential GIG substrates, we performed multiplexed quantitative proteomics on total-cell extracts of WT and two GAN-KO 293FT cell lines (GAN-KO2 and GAN-KO16) and quantified 8,124 proteins (Fig. 2A, SI Appendix, Fig. S6, and Dataset S4). NF proteins NEFL and INA, along with actin filament–associated regulatory proteins TPM1, TPM2, and TAGLN, were significantly up-regulated in both GAN-KO2 and GAN-KO16 293FT cell lines (Fig. 2A). BRMS1 and CCN2 protein levels were also up-regulated (Fig. 2A). We perform RNA-seq analysis and found that the mRNA levels of NEFL, INA, TPM1, TPM2, and TAGLN remained unchanged in GAN-KO 293FT cells, suggesting that their upregulation in protein levels was a posttranslational regulation. In contrast, the mRNA level of CCN2 was significantly up-regulated (SI Appendix, Fig. S7 and Dataset S5), suggesting that its increased protein level, observed in proteomic data (Fig. 2A), was due to altered transcription.
Fig. 2.
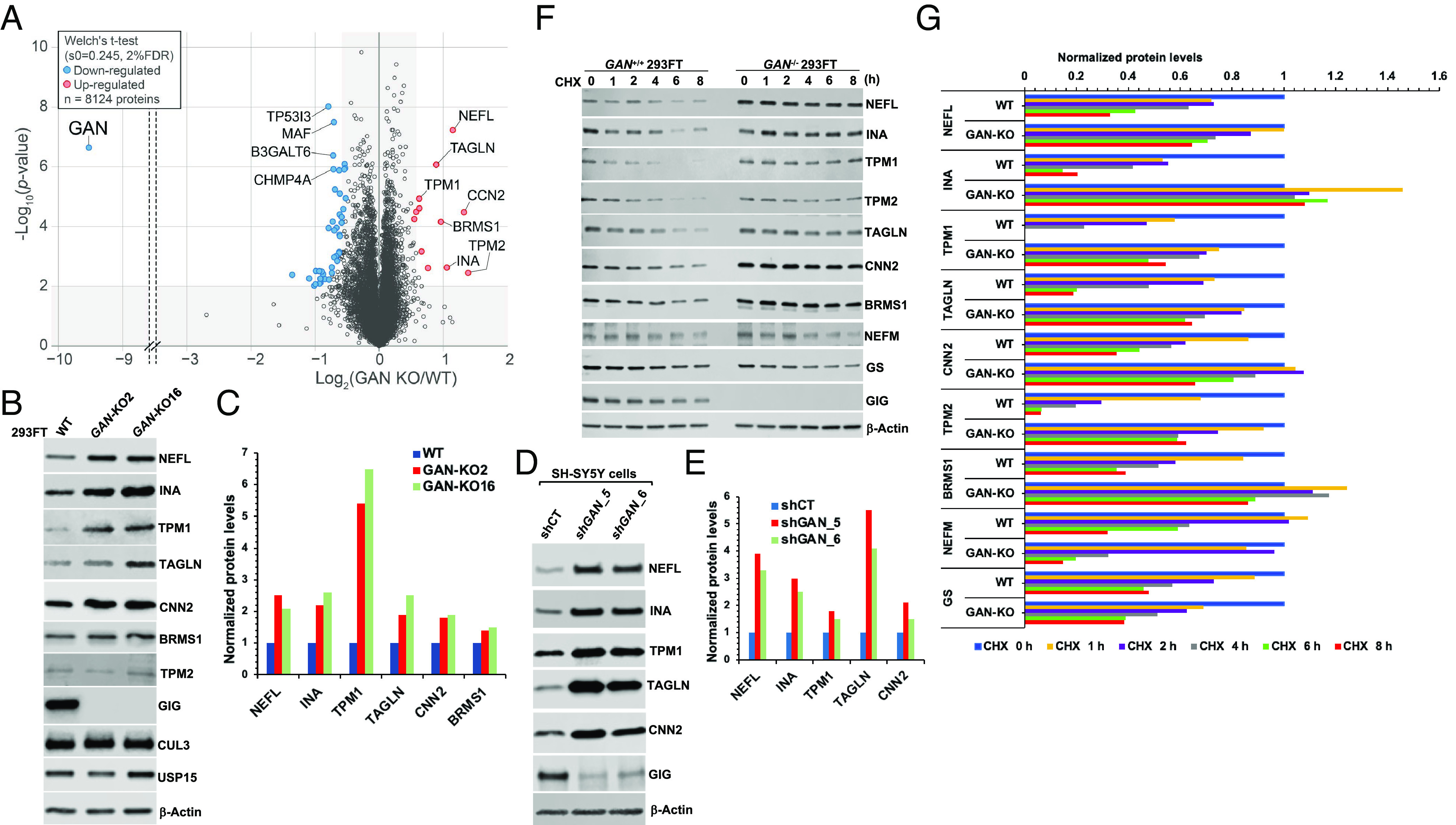
Depletion of GAN resulted in the accumulation of NF proteins and actin filament–associated regulatory proteins. (A) Comparative proteome analysis of WT (n = 4), GAN-KO2 (n = 3), and GAN-KO16 (n = 3) 293FT cell lines from 10plex TMT analysis. The data are depicted as a volcano plot where both GAN KO clones were combined (n = 6). The red and blue dots denote statistically significant enriched or reduced proteins, respectively. (B) Validation of proteomic data. Cell extracts from WT, GAN-KO2, and GAN-KO16 293FT cell lines were analyzed by IB with the indicated antibodies. (C) Quantification of NEFL, INA, TPM1, TAGLN, CNN2, and BRMS1 proteins from (B). (D) Cell extracts from SH-SY5Y cells, stably expressing shRNA control (shCT) and shRNAs targeting GAN clones 5 and 6 (shGAN_5, shGAN_6), were analyzed by IB with the indicated antibodies. (E) Quantification of NEFL, INA, TPM1, TAGLN, and CNN2 proteins from (D). (F) CHX experiment. WT and GAN-KO2 293FT cells were treated with CHX for the indicated times. Cell extracts were analyzed by IB. (G) Quantification of NEFL, INA, TPM1, TAGLN, CNN2, TPM2, BRMS1, NEFM, and GS proteins from (F). The relative ratios of indicated proteins:Actin, normalized to time 0, are shown. The results shown are representative of at least three independent experiments (B, D, and F).
To validate our proteomic data, we performed western blot using cell extracts from WT, GAN-KO2, and GAN-KO16 293FT cell lines and found that the steady-state protein levels of NEFL, INA, TPM1, and TAGLN were significantly up-regulated in two GAN-KO 293FT cell lines, whereas those of TPM2 and BRMS1 were slightly increased (Fig. 2 B and C). Although not listed as the most up-regulated protein, CNN2, another actin filament–binding regulatory protein, was elevated in two GAN-KO 293FT cell lines based on our Western blot analysis (Fig. 2 B and C). Furthermore, we adopted the SH-SY5Y cell line as a neuronal cell model to validate our findings. Consistent with the results obtained in GAN-KO 293FT cells, knock-down of GAN in SH-SY5Y cells by two independent GIPZ shRNAs up-regulated NEFL, INA, TPM1, TAGLN, and CNN2 protein levels (Fig. 2 D and E and SI Appendix, Fig. S8 A and B). Remarkably, our CHX experiments indicated that the degradation of NEFL, INA, TPM1, TPM2, TAGLN, CNN2, and BRMS1 was blunted in GAN-KO 293FT cells and GAN-KDO SH-SY5Y cells (Fig. 2 F and G and SI Appendix, Fig. S8 C and D), further confirming that the turnover of these proteins was dependent on GIG. CRL4CRBN-dependent GS degradation was unaffected (Fig. 2 F and G), indicating that the UPS was intact in GAN-KO 293FT cells. Consistent with the proteomic analysis of GAN-KO 293FT cell lines (Dataset S4), we also found in our CHX experiments that GIG was not required for the turnover of NEFM (Fig. 2 F and G), suggesting that targeting NF proteins for the CRL3GIG-mediated degradation was selective to NEFL and INA.
Earlier studies on GAN pathology revealed the accumulation of NFs in neurons and did not distinguish which NF proteins were affected (19, 20). In 1983, Ionasescu and colleagues studying NF proteins from neurons of GAN patients reported that the amount of 68,000-Da neurofilament protein (corresponding to NEFL) was two times higher in the GAN nerve than in the control nerve, while the amounts of other NF subunits with 160,000 and 210,000 Da (corresponding to NEFM and NEFH, respectively) were normal (35). This work suggested that GIG mutants in GAN patients, which caused a loss of function, could not ubiquitylate NEFL, leading to the accumulation of NEFL in neurons, whereas GIG did not regulate NEFM and NEFH. Subsequent studies by Johnson-Kerner and colleagues reported that NEFL, and to a lesser extent PRPH, but not NEFM, NEFH, and VIM, were up-regulated in induced pluripotent stem cells (iPSCs) from GAN patients (36). Consistent with these previous studies and our results (Fig. 2 A–G), the NEFL protein levels in the brains of GAN-KO mice were markedly increased (37, 38).
We also noted that a small fraction of endogenous NEFL protein levels was degraded in GAN-KO 293FT cells (Fig. 2 F and G), suggesting that other E3s may be involved in the proteasomal degradation of NEFL. In agreement with this possibility, a previous study showed that the E3-ubiquitin ligase TRIM2 regulates NEFL ubiquitylation (39). In GAN patients, aggregates of IFs in multiple tissues suggest that CRL3GIG may control a vast repertoire of substrates. In line with this argument, the GAN mRNA expression profile in diverse human tissues revealed that GAN mRNA is highly expressed in multiple tissues, including the heart, liver, kidney, skeletal muscle, and brain (SI Appendix, Fig. S9). Taken together, our results demonstrated that CRL3GIG controls the stability of at least two types of substrates, including NF proteins NEFL and INA, and actin filament–associated regulatory proteins TPM1, TPM2, TAGLN, and CNN2.
CRL3GIG Directly Ubiquitylates NF Proteins and Actin Filament–Associated Regulatory Proteins.
To explore the molecular mechanisms by which CRL3GIG regulates the stability of NF proteins, we focused on identifying the GIG-NEFL interaction. In line with the previous studies (31, 40), we also found that GIG interacted with NF proteins NEFL and INA in cells overexpressing GIGFlag (Fig. 3 A and B). Moreover, we performed endogenous IP and confirmed that endogenous NF proteins NEFL and INA interacted with endogenous GIG (Fig. 3C). The NEFL/INA-GIG interaction was enhanced in MLN4924-treated 293FT cells (Fig. 3C). Notably, the GIG-NEFL interaction was direct since it was confirmed by the in vitro pulldown experiments using recombinant proteins (Fig. 3D). We also found that GIG interacted with actin filament–associated regulatory proteins TPM1 and CNN2 in vivo and in vitro (Fig. 3 B and E), but we did not pursue further in the current work.
Fig. 3.
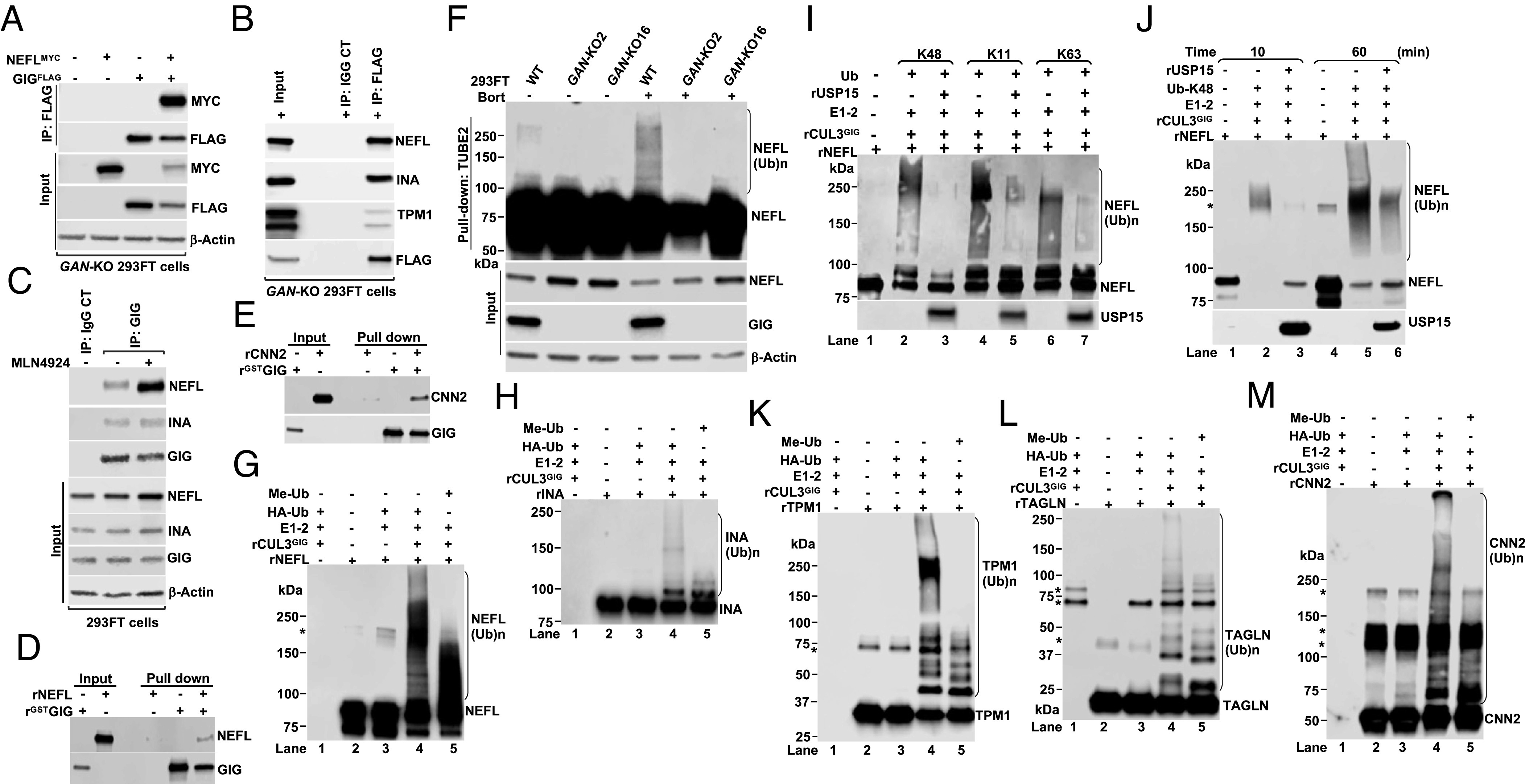
CRL3GIG directly ubiquitylates NF proteins and actin filament–associated regulatory proteins. (A) GAN-KO 293FT cells were transfected with the indicated plasmids expressing GIGFLAG and MYC-tagged NEFL (NEFLMYC) for 40 h. Cellular extracts were immunoprecipitated with anti-Flag antibody, followed by IB with antibodies against MYC and FLAG. (B) GAN-KO 293FT cells were transfected with wild-type GIGFLAG. After 48 h, cell extracts were immunoprecipitated with anti-FLAG or IgG control antibody, followed by IB with the indicated antibodies. (C) Endogenous NEFL and INA proteins interact with endogenous GIG. 293FT cells were treated with or without 2 µM MLN4924 for 4 h. Protein extracts were IP with rabbit IgG control or GIG antibody. IP and input samples were analyzed by IB with the indicated antibodies. (D and E) The direct interactions between GST-GIG and NEFL (D) or CNN2 (E) in GST pull-down assays. At least two experimental replicates were repeated to optimize conditions (A–E). (F) Cells were treated with or without bortezomib (Bort, 1 µM) for 6 h. Total ubiquitylated proteins were purified using TUBE2-agarose. Bound fractions were analyzed by IB. (G and H) In vitro ubiquitylation reactions of rNEFL (G) and rINA (H) were carried out in the presence or absence of E1, E2, HA-tagged ubiquitin (HAUb) or methylated ubiquitin (Me-Ub), and rCUL3GIG complex. *Indicates a nonspecific band. (I) In vitro competitive ubiquitylation and deubiquitylation assay of NEFL was carried out in the presence of rNEFL, E1, E2, rCUL3GIG complex, ubiquitin mutants with K48 only, K11 only or K63 only and rUSP15. (J) Same as (I), except that reactions were collected at 10 min and 60 min. (K–M) In vitro ubiquitylation reactions of rTPM1 (K), rTAGLN (L), and rCNN2 (M). The results shown are representative of at least three independent experiments (F–M).
To provide direct evidence supporting our central hypothesis that CRL3GIG may ubiquitylate NF proteins NEFL and INA in vitro, we first performed the in vivo ubiquitylation assays using TUBE2 pulldown. As shown in Fig. 3F, the polyubiquitylated forms of NEFL, (NEFL(Ub)n) were significantly enriched in WT 293FT cells, which were treated with the proteasome inhibitor bortezomib. In contrast, NEFL(Ub)n were completely absent in two independent GAN-KO 293FT cell lines (Fig. 3F), suggesting that GIG is required for the ubiquitylation of NEFL in cells. Consistent with these findings, a similar result was observed in GAN-KDO SH-SY5Y cells (SI Appendix, Fig. S10A). USP15-mediated downregulation of NEFL and INA proteins was rescued by MLN4924 treatment in USP15-KO 293FT cells (Fig. 1 B and C) or by GAN KDO in USP15-KO 293FT cells using three independent GIPZ shRNAs targeting GAN (SI Appendix, Fig. S10 B and C), suggesting that USP15 regulates the degradation of NEFL and INA in downstream of CRL3GIG. Remarkably, our in vitro ubiquitylation assays using all recombinant proteins (rUb, rE1, rE2, rCRL3GIG, and rNEFL) strongly demonstrated that rCRL3GIG efficiently ubiquitylated rNEFL (Fig. 3G, lane 4, and SI Appendix, Fig. S11A), and the reaction was inhibited by adding methylated ubiquitin (Me-Ub) (Fig. 3G, lane 5 and SI Appendix, Fig. S11A). In addition, rINA was ubiquitylated by rCRL3GIG in vitro (Fig. 3H and SI Appendix, Fig. S11B). We next asked whether USP15 could deconjugate different polyubiquitin chains of NEFL in vitro. We performed the in vitro competitive ubiquitylation/deubiquitylation assays using all critical components of the UPS: (rUb, rE1, rE2, rCRL3GIG, rNEFL, and rUSP15). As illustrated in Fig. 3 I and J and SI Appendix, Fig. S11 C and D, USP15 substantially antagonized CRL3GIG-mediated K48-, K11-, or K63-linked polyubiquitylation of NEFL. These results indicated that the CRL3GIG-USP15 pathway directly controls the turnover of NF proteins NEFL and INA.
Although this study was mainly focused on the molecular control of NF degradation by the UPS, we also sought to determine whether CRL3GIG may ubiquitylate actin filament–associated regulatory proteins in vitro. Polyubiquitylation of rTPM1, rTAGLN, and rCNN2 was readily detected by the addition of rCRL3GIG, while it was blocked by the addition of methylated ubiquitin (Fig. 3K and SI Appendix, Fig. S11E; Fig. 3L and SI Appendix, Fig. S11F; and Fig. 3M and SI Appendix, Fig. S11G, respectively). All these actin filament–associated regulatory proteins were also natural substrates of CRL3GIG. Together, CRL3GIG controls both NFs and actin filaments, possibly in neurons and other cell types, respectively.
Mutations in GAN Patients at Amino Acid Residues L309, R545, and C570 in the Kelch Domain of GIG Fail to Interact with NF Proteins and Exhibit a Loss-of-Function Mechanism.
To further study the functional significance of GIG–NEFL interaction based on GAN mutations in human patients (24), we first analyzed the sequence alignment of Gigaxonin orthologs across species. We found that mutations in GAN patients at amino acid residues (L309, R545, and C570) in the Kelch domain of GIG were highly conserved across species (Fig. 4A and SI Appendix, Fig. S12). It was likely that these point mutations may cause detrimental effects on the turnover of NEFL, leading to its accumulation in GAN patients (19, 35). Moreover, GAN is an autosomal recessive genetic disorder, suggesting that the molecular disease mechanism is due to loss-of-function mutations. To investigate this hypothesis, we first performed IP experiments. We found that the binding of L309R, R545C, and C570Y mutants and, to a lesser extent, E486K mutant to endogenous NEFL and INA was significantly disrupted (Fig. 4 B and C). Next, we sought to test whether the GIG-dependent upregulation of endogenous NEFL protein levels may be rescued by introducing empty vector (EV, control), WT GIGFLAG, L309R, and C570Y mutants in GAN-KO 293FT cells and found that only WT GIGFLAG but not EV, L309R, or C570Y mutant greatly down-regulated the endogenous protein levels of NEFL and INA in GAN-KO 293FT cells (Fig. 4 D and E). The molecular phenotype of the C570Y mutant exerted a more robust effect since it completely lost its function in destabilizing endogenous protein levels of NEFL and INA (Fig. 4 D and E). These findings account for a loss-of-function mechanism in GAN patients.
Fig. 4.
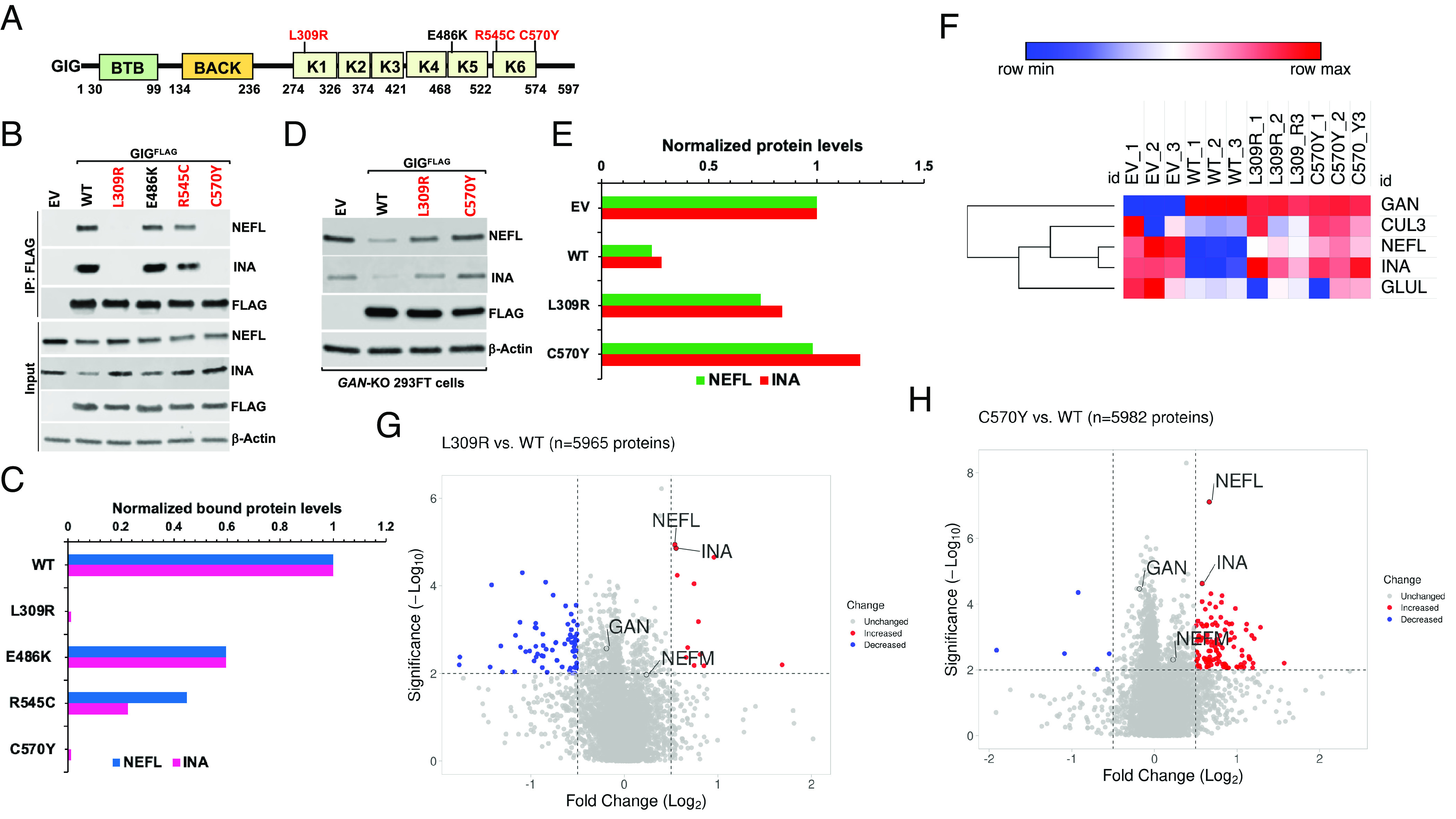
Mutations in GAN patients at amino acid residues L309, R545, and C570 in the Kelch domain of GIG exhibit a loss-of-function mechanism. (A) Schematic diagram of human GIG. GIG contains BTB, BACK, and Kelch (K1-6) domains. Highly conserved residues in the Kelch domain of GIG across species, which are mutated in GAN patients, including (L309, R545, and C570), are highlighted in red. (B) WT 293FT cells were transfected with empty vector (EV) or wild-type (WT) GIGFLAG and its mutants. After 24 h, cells were treated with 2 µM MLN4924 for 4 h. Cell extracts were immunoprecipitated with anti-FLAG antibody, followed by IB with the indicated antibodies. (C) Quantification of NEFL and INA proteins from (B). The relative ratios of endogenous NEFL and INA interacted with GIGFLAG to input NEFL and INA, normalized to that of WT in lane 2, are shown. (D) Destabilizing endogenous NEFL and INA protein levels in GAN-KO cells by WT GIG, but not its mutants. GAN-KO16 293FT cells were transfected with EV or WT GIGFLAG and its mutants. After 48 h, cell extracts were analyzed by IB with the indicated antibodies. (E) Quantification of NEFL and INA proteins from (D). At least three experimental replicates were repeated to optimize conditions (B and D). (F–H) Global proteome analyses (n = 3) of EV-, WT GIGFLAG-, L309R mutant-, and C570Y mutant-transfected GAN-KO 293FT cells. The threshold of fold changes was set to more than 1.41-fold increase or decrease and the –Log10 P-value above 2. The data were depicted as a heatmap (F) and volcano plots for L309R vs. WT (G), and C570Y vs. WT (H). The red and blue dots denote significantly enriched or reduced proteins, respectively.
To systemically characterize the proteome remodeling induced by WT GIGFLAG, L309R, and C570Y mutants in GAN-KO 293FT cells, we performed the proteome profiling analysis of GAN-KO 293FT cell lines transiently transfected with EV, WT GIGFLAG, L309R, and C570Y mutants. Consistent with western blot analysis (Fig. 4 D and E), our proteomic data revealed that NEFL and INA protein levels were markedly down-regulated in GAN-KO 293FT cells transfected with WT GIGFLAG, while L309R and C570Y mutants failed to destabilize NEFL and INA proteins (Fig. 4 F–H, SI Appendix, Fig. S13 A–C, and Dataset S6). We also found that the protein levels of CUL3 and glutamine synthetase (GLUL or GS), a well-known substrate of CRL4CRBN, were unaffected, suggesting that overproduction of GIG WT and mutants did not alter the UPS in GAN-KO cells (Fig. 4F). Furthermore, NEFM protein levels remained unchanged in all cell lines (Fig. 4 F–H and SI Appendix, Fig. S13 A–C), further confirming that NEFM was not an endogenous substrate of CRL3GIG. Taken together, our results indicated that CRL3GIG selectively and specifically targets NF proteins NEFL and INA for degradation by the proteasome and that mutations of GIG at amino acid residues L309 and C570 fail to interact with and degrade NEFL and INA. These findings may also translate to clinical relevance and account for the mechanisms of pathogenesis underlying GIG mutations in GAN patients, at least in part, at amino acid residues in the Kelch domain.
Mapping the Binding Site of GIG on NEFL.
To map the binding site of GIG on NEFL, we generated a series of deletion mutants with the C-terminal MYC tag (NEFLMYC). We found that deleting amino acids 237-279 (D4 mutant), including linker L12, coil 2A subdomain, and linker L2, abolished the binding to GIG (Fig. 5 A and B). We reconfirmed the D4 mutant using untagged NEFL WT and mutants with antibodies against the N- or C-terminal epitopes on NEFL (SI Appendix, Fig. S14 A and B). To identify the minimal sequence on NEFL required for binding to GIG, we designed biotinylated synthetic L12, 2A, and L2 peptides (Fig. 5C) and performed the peptide pulldown assays. As shown in Fig. 5D, the NEFLL12 peptide, but not NEFL2A and NEFLL2 peptides interacted with the GSTGIG protein in vitro. The dot blot analysis also confirmed that GSTGIG bound to NEFLL12 peptides but not NEFL2A and NEFLL2 peptides (SI Appendix, Fig. S15A). Intriguingly, the sequence alignment analysis uncovered that the L12 region was highly conserved from zebrafish to humans in both NEFL and INA orthologs (SI Appendix, Fig. S16), but it was significantly diversified from the L12 regions of NEFM and NEFH (Fig. 5A). This likely stems from evolution and diverse natural selection pressures that allows individual NF subunit to have multiple layers of regulation and functions in neurons. We hypothesized that the NEFLL12 segment contains a specific degradation signal for GIG, which does not exist in the L12 regions of NEFM and NEFH proteins. To test this idea, we generated a biotinylated synthetic NEFML12 peptide (M-L12) (Fig. 5C) and compared its binding to GIG with the NEFLL2 peptide. The peptide pulldown and dot blot analyses revealed that the NEFML12 peptide was not recognized by GIG (Fig. 5E and SI Appendix, Fig. S15B). Collectively, these findings suggested that the L12 segment of NEFL is required for the GIG-NEFL interaction.
Fig. 5.

Mapping the binding site of GIG on NEFL. (A) Schematic diagram of human NEFL protein. NEFL includes head, rod, and tail domains. The rod domain contains coil 1A, 1B, 2A, and 2B subdomains, separated by linkers L1, L12, and L2. Deletion of amino acids 237–279 (D4) highlighted in red abolished the binding to GIG, indicating that this sequence contains the NEFL degron recognized by GIG. The sequence alignment of L12, 2A, and L2 segments from human NEFL, INA, NEFM, and NEFH was shown. (B) 293FT cells were transfected with the indicated plasmids expressing GIGFLAG and full-length (FL) or deletions of NEFLMYC for 40 h. Cellular extracts were immunoprecipitated with anti-Flag antibody, followed by IB with antibodies against MYC and FLAG. A band ~ 25 kDa (top panel) represents IgG light chains (IgG-LC). (C) Design of NEFL-L12, -2A, -L2 and NEFM-L12 (M-L12) peptides. (D) Peptide pull-down assays were performed using GSTGIG protein and NEFL-L12, -2A, and –L2 peptides (lane 2 without peptide used as negative control), followed by analyzed by IB with anti-GST antibody. (E) Same as (D), except that GIGFLAG protein and NEFM-L12 (M-L12) were used. Data are representative of three independent experiments (B, D, and E).
Identification of Molecular Basis for the Specific Recognition of NEFL by GIG.
We further analyzed the sequence alignment to identify the molecular basis for the GIG-NEFL interaction. We found that the ExD/E motif on NEFL and INA is evolutionarily conserved (Fig. 6A). Interestingly, NEFL-D248 is replaced by K and R on NEFM and NEFH, respectively. We reasoned that the core NEFLL12 degron was (QISVEMDV) (Fig. 6A), which was required for its binding, ubiquitylation, and degradation. To test this hypothesis, we first performed modeling and in silico study of the NEFLL12 degron binding to GIG. The amino acid sequence of GIG (Uniprot ID: Q9H2C0) was used to build a model of the protein’s 3D structure using Alphafold2 (41). The NEFLL12 degron was observed to bind to the 6-bladed β-propeller KELCH domain of GIG, and its three residues EMD are in direct contact with C570 (Fig. 6 B and C and SI Appendix, Table S2). The binding site of the NEFLL12 degron is also located closely to L309 and R545 (Fig. 6B). Van der Waals interactions play an essential role in stabilizing the NEFLL12 degron in the binding pocket in comparison to hydrogen bonds and hydrophobic interactions. When docked to WT GIG, the NEFLL12 degron had a good docking score (SI Appendix, Table S2B). Consistent with the notion that mutations of the GAN gene in GAN patients exhibit the loss-of-function mechanism (Fig. 4 B–E), a docking score of the C570Y mutant was much higher (SI Appendix, Table S2B), indicating that this mutation affected the binding of GIG to the NEFLL12 degron. The change in binding energy was mainly observed in Van der Waals interactions (SI Appendix, Table S2B). To validate our computational data, we mutated the motif EMD into AMA called 2A mutant and found that the binding of 2A mutant to GIG was reduced (Fig. 6 D and E). Remarkably, the 2A mutant was not ubiquitylated, which substantially suppressed its degradation in cells (Fig. 6 F–H).
Fig. 6.
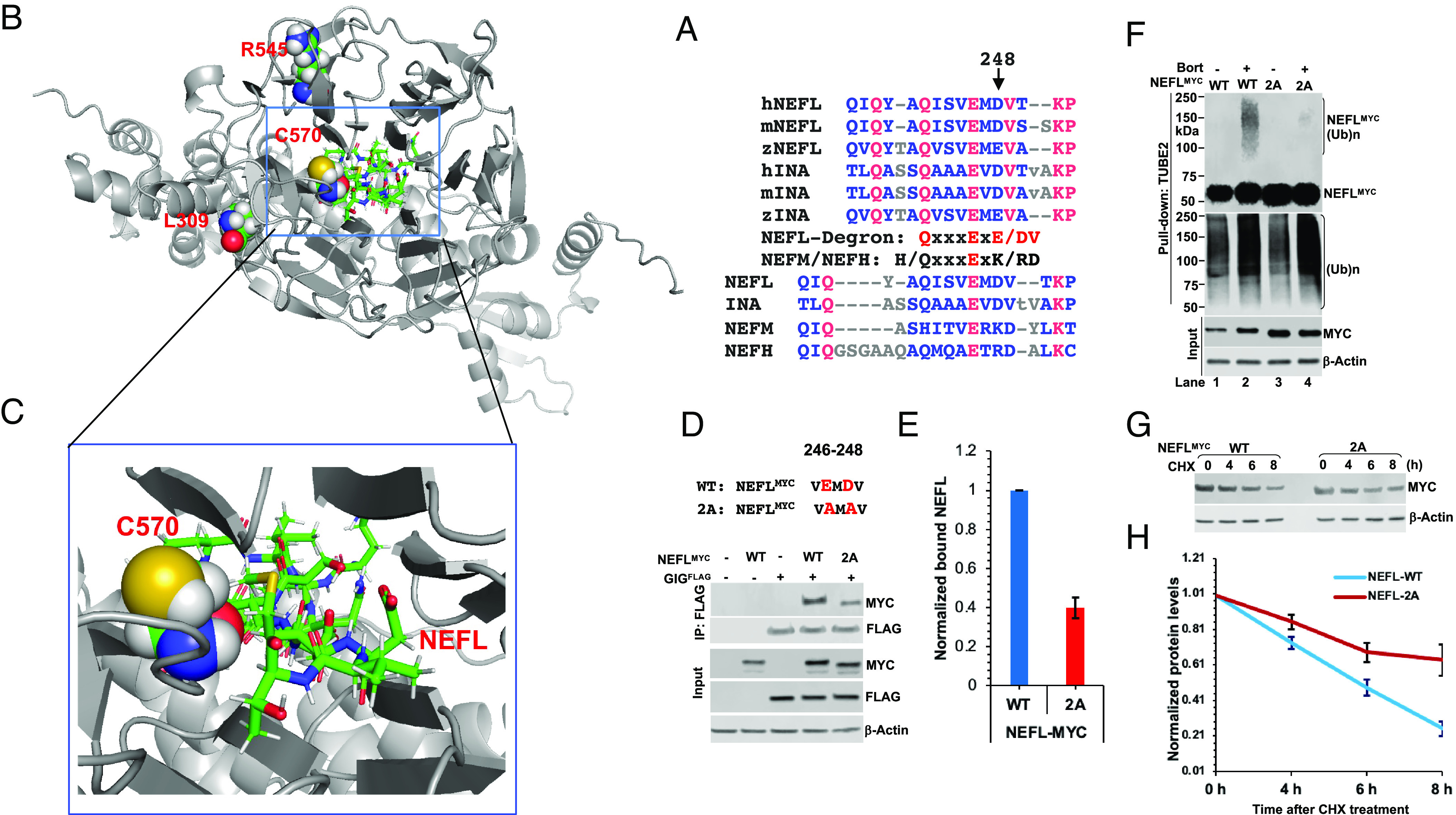
Identification of molecular basis for the specific recognition of NEFL by GIG. (A) L12 segments from human (h), mouse (m), and zebrafish (z) NEFL and INA orthologs and from human NEFL, INA, NEFM, and NEFH. (B) Docking pose of the NEFLL12 degron to GIG (n = 10). The NEFLL12 degron binds to the Kelch domain of GIG and directly interacts with residue C570 located close to residues L309 and R545. (C) A close view of electrostatic interactions between residue C570 and the motif ExD. (D) WT 293FT cells, transfected with the indicated plasmids expressing GIGFLAG and NEFLMYC WT or 2A mutant for 24 h, were treated with 2 µM MLN4924 for 4 h. Cell extracts were immunoprecipitated with anti-Flag antibody, followed by IB with antibodies against MYC and FLAG. (E) Quantification of NEFLMYC protein interacted with GIGFLAG from (D). Error bars represent ±SD; n = 2. (F) 293FT cells, transfected with NEFLMYC WT or 2A mutant for 40 h, were treated with bortezomib (Bort, 2 µM) for 7 h. Total ubiquitylated proteins were purified using TUBE2-agarose, followed by IB with anti-MYC and anti-ubiquitin antibodies. (Ub)n, polyubiquitin. (G) Same as (F), except that cells were treated with CHX at the indicated times. Cell extracts were analyzed by IB with anti-MYC and Actin. (H) Quantification of NEFL-MYC protein from (G). Error bars represent ±SD; n = 2.
Discussion
In this study, we find that the CRL3GIG–USP15 pathway governs the destruction of NF proteins NEFL and INA, as illustrated in Fig. 7. Surprisingly, CRL3GIG also regulates the stability of actin filament–associated regulatory proteins, including TPM1, TPM2, TAGLN, and CNN2 via direct ubiquitylation. This may account for mutations in GAN patients, characterized by abnormal accumulation of intermediate filaments in neuronal and non-neuronal cells. CRL3GIG may regulate a vast repertoire of substrates and plays an essential role in regulating both NF filaments and actin filaments (via their regulatory protein). Intriguingly, the present work and a recent study (28) demonstrate the biological significance of USP15 in regulating CRL activity. USP15 antagonizes the CRL3GIG-dependent ubiquitylation of NEFL and INA and the CRL4CRBN-mediated ubiquitylation of GS and neosubstrates (28). However, many fundamental questions in future research need to be addressed. For example, NFs are intermediate filaments with a diameter of 10 nm heteropolymers and micrometers in length, which extensively interact with a cytoskeletal protein network of exceptional stability (half-life of many months) and provide structural support to maintain the large calibers of myelinated axons crucial for nerve conduction velocity (18). In mature neurons, NF proteins exist mainly in stable polymers, while the pool of soluble NF proteins is small (42). How does the CRL3GIG-USP15 pathway maintain proper NF protein homeostasis under physiological and stress conditions? Does ubiquitylation of NEFL and INA proteins occur as monomer or heteropolymers and require other signals or cofactors? If NEFL and INA subunits are ubiquitylated by CRL3GIG in heteropolymers, whether the extraction of ubiquitylated NF subunits from heteropolymers requires VCP/p97 (43) or new factors so that they can be degraded by the proteasome (44, 45). Moreover, it remains to be elucidated whether USP15 or other DUB may control the turnover of actin filament–associated regulatory proteins.
Fig. 7.

Proposed model: the CRL3GIG–USP15 pathway governs the destruction of neurofilament proteins NEFL and INA. CRL3GIG also regulates the ubiquitylation of actin filament–associated regulatory proteins (TPM1, TPM2, TAGLN, and CNN2), which DUBs may deubiquitylate. Mutations in the GAN gene encoding Gigaxonin cause giant axonal neuropathy (GAN), characterized by abnormal accumulation of intermediate filaments in both neuronal and non-neuronal cells. Thus, CRL3GIG may regulate a vast repertoire of substrates. For the sake of simplicity, the homodimerization of Gigaxonin is omitted.
Actin filament–associated regulatory proteins TPM1, TPM2, TAGLN, and CNN2 are essential in many cellular pathways. Our findings that CRL3GIG directly ubiquitylates TPM1, TAGLN, and CNN2 in vitro and promotes their turnover in cells suggest that CRL3GIG regulates actin filaments. TPM1 mutations in humans cause hypertrophic cardiomyopathy (46, 47). The tension-dependent degradation of CNN2 plays a critical role in regulating the stability of lung alveolar cytoskeleton with physiological and pathological significances via an unknown mechanism (48). In addition, CNN2 was found in a lysophagy proteome landscape study (49), suggesting that it may regulate the lysophagy pathway. Consistent with this idea, a recent study by Kravic et al. demonstrated that the unknown E3-dependent ubiquitylation of CNN2 and the Arp2/3 complex translocate to damaged lysosomes and regulate actin filaments to drive phagophore formation for efficient lysophagy in a p97-dependent manner (50). Future work is needed to dissect the role of CRL3GIG-mediated ubiquitylation of actin filament–associated regulatory proteins in various biological processes under physiological and pathological conditions.
Abnormal accumulations of cytoskeleton proteins are neuropathological hallmarks of many neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, characterized by filamentous aggregates of NF proteins or intracellular inclusions of aggregated tau proteins (18, 51). Moreover, genetic mutations in genes encoding for IF proteins account for more than 80 diseases (15). For example, mutations in the NEFL gene affect the NF network, thus leading to the Charcot–Marie–Tooth disease (52, 53). Another question of interest is whether the dysfunction of the newly discovered CRL3GIG–USP15 pathway may contribute to the pathogenesis of these neurodegenerative diseases.
An interesting question is whether the degrons found in another subset of CRL3GIG substrates (TPM1, TAGLN, and CNN2) are similar to the NEFLL12 degron. We could not exclude the possibility that these degrons may not have the same motifs because E3 ligases usually regulate multiple substrates and have numerous biological functions under various physiological conditions. For example, the degrons identified among endogenous substrates of CRL4CRBN, such as MEIS2, GS, and AMPK gamma are very distinct (54–56). Future work focused on identifying the molecular basis for the NEFLL12 degron-GIG interaction requires a crystal structure of the degron bound to a C-terminal Kelch domain of GIG or a cryo-EM structure of the CUL3GIG-NEFL complex. This information is critical for developing molecular glue and PROTAC degraders based on CRL3GIG E3 ligase to treat neurodegenerative disorders.
Materials and Methods
Generation of GAN-knockout 293FT Cells by CRISPR/Cas9 Genome Editing.
USP15-KO 293FT cells were described in our recent protocol (28). This protocol was used for generating GAN-KO 293FT cells (28). Briefly, 293FT cells, cultured in a 24-well plate, were transiently transfected with 0.5 µg of human GAN CRISPR/Cas9 plasmids using Fugene HD. Two days after transfection, a single cell was seeded in a 96-well plate via serial dilutions. After 10–12 d, single clones were obtained, cultured in 24-well plates, and expanded for 5–7 d to validate the editing of GAN by western blot.
RNA-Sequencing Analysis.
RNA-sequencing (seg) was done using RNAs isolated from wild-type (WT), USP15-KO, MLN4924-treated USP15-KO, and GAN-KO 293FT cells as described in SI Appendix.
Proteomic Analysis.
The detailed protocols for proteomic analyses of WT, USP15-KO, and GAN-KO 293FT cell lines were described in SI Appendix.
In Vitro Ubiquitylation, Deubiquitylation, and Competitive Ubiquitylation/ Deubiquitylation Assays.
The in vitro ubiquitylation assays were performed as described previously (28, 56). Briefly, the assays were carried out for 1 h at 30 °C in a final volume of 30 µL containing 200 ng of substrates (rNEFL, rINA, rTPM1, rCNN2, or rTAGLN), E1, E2 (UbcH5a and UbcH3/Cdc34), CLR3GIG complex (200 ng NEDDylated RBX1-CUL3, mixed with 200 ng GSTGIG), and HAUb (HA-tagged wild-type ubiquitin). The in vitro deubiquitylation assays using rUSP15 (1 µg) were performed as described previously (28, 43). The in vitro competitive ubiquitylation/deubiquitylation assays were described in a recent study (28).
Peptide Pull-Down and Dot Blot Assays.
The detailed peptide pull-down and dot blot assays were described in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Acknowledgments
We would like to thank LC Sciences (Houston, TX) for assisting with RNA-sequencing experiments. This research was in part supported by the Memorial Sloan Kettering Cancer Center Support Grant P30CA008748 (A.O.), the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2021R1A6A3A14038416) to K.H.N., the Nano Material Technology Development Program (No. 2016M3A7B6908929) of NRF funded by the Ministry of Science and ICT (J.E.L.), institutional fund by the University of Missouri-Columbia (T.V.N.), and the National Institute of General Medical Sciences of the NIH Grant R01GM148470 (T.V.N.).
Author contributions
H.-M.P., L.L., A.O., J.E.L., and T.V.N. designed research; H.-M.P., L.L., T.T.N., K.H.N., and T.V.N. performed research; T.V.N. contributed new reagents/analytic tools; H.-M.P., L.L., T.T.N., K.H.N., A.O., J.E.L., and T.V.N. analyzed data; T.V.N. conceived the study and supervised research; and T.V.N. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. V.D. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Varshavsky A., The ubiquitin system, an immense realm. Annu. Rev. Biochem. 81, 167–176 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Hershko A., Ciechanover A., The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Huang X., Dixit V. M., Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 26, 484–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petroski M. D., Deshaies R. J., Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Harper J. W., Schulman B. A., Cullin-RING ubiquitin ligase regulatory circuits: A quarter century beyond the F-Box hypothesis. Annu. Rev. Biochem. 90, 403–429 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan S., Pagano M., Ubiquitin ligases in cancer: Functions and clinical potentials. Cell Chem. Biol. 28, 918–933 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stogios P. J., Downs G. S., Jauhal J. J., Nandra S. K., Prive G. G., Sequence and structural analysis of BTB domain proteins. Genome Biol. 6, R82 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furukawa M., He Y. J., Borchers C., Xiong Y., Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat. Cell Biol. 5, 1001–1007 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Pintard L., et al. , The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature 425, 311–316 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Xu L., et al. , BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 425, 316–321 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Zhang D. D., Lo S. C., Cross J. V., Templeton D. J., Hannink M., Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell Biol. 24, 10941–10953 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuang M., et al. , Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 36, 39–50 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canning P., et al. , Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 288, 7803–7814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji A. X., Prive G. G., Crystal structure of KLHL3 in complex with Cullin3. PLoS ONE 8, e60445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omary M. B., “IF-pathies”: A broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. 119, 1756–1762 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerette D., Khan P. A., Savard P. E., Vincent M., Molecular evolution of type VI intermediate filament proteins. BMC Evol. Biol. 7, 164 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan A., Rao M. V., Veeranna, Nixon R. A., Neurofilaments at a glance. J. Cell Sci. 125, 3257–3263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan A., Nixon R. A., Posttranscriptional regulation of neurofilament proteins and tau in health and disease. Brain Res. Bull. 192, 115–127 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asbury A. K., Gale M. K., Cox S. C., Baringer J. R., Berg B. O., Giant axonal neuropathy—A unique case with segmental neurofilamentous masses. Acta Neuropathol. 20, 237–247 (1972). [DOI] [PubMed] [Google Scholar]
- 20.Berg B. O., Rosenberg S. H., Asbury A. K., Giant axonal neuropathy. Pediatrics 49, 894–899 (1972). [PubMed] [Google Scholar]
- 21.Tandan R., et al. , Childhood giant axonal neuropathy. Case report and review of the literature. J. Neurol. Sci. 82, 205–228 (1987). [DOI] [PubMed] [Google Scholar]
- 22.Bruno C., et al. , Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology 62, 13–16 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Opal P., “GAN-related neurodegeneration” in GeneReviews®, Adam M. P., et al., Eds. (GeneReviews®, Seattle, WA, 1993). [Google Scholar]
- 24.Bomont P., et al. , The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 26, 370–374 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Chirnomas D., Hornberger K. R., Crews C. M., Protein degraders enter the clinic—A new approach to cancer therapy. Nat. Rev. Clin. Oncol. 20, 265–278 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Cowan A. D., Ciulli A., Driving E3 ligase substrate specificity for targeted protein degradation: Lessons from nature and the laboratory. Annu. Rev. Biochem. 91, 295–319 (2022). [DOI] [PubMed] [Google Scholar]
- 27.Sakamoto K. M., et al. , Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen T. V., USP15 antagonizes CRL4(CRBN)-mediated ubiquitylation of glutamine synthetase and neosubstrates. Proc. Natl. Acad. Sci. U.S.A. 118, e2111391118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw G., Morse S., Ararat M., Graham F. L., Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16, 869–871 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Vu T. T. M., Mitchell D. C., Gygi S. P., Varshavsky A., The Arg/N-degron pathway targets transcription factors and regulates specific genes. Proc. Natl. Acad. Sci. U.S.A. 117, 31094–31104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson-Kerner B. L., Garcia Diaz A., Ekins S., Wichterle H., Kelch domain of gigaxonin interacts with intermediate filament proteins affected in giant axonal neuropathy. PLoS ONE 10, e0140157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soucy T. A., et al. , An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Goldberg A. L., Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199, 583–588 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emanuele M. J., et al. , Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ionasescu V., et al. , Giant axonal neuropathy: Normal protein composition of neurofilaments. J. Neurol. Neurosurg. Psychiatry 46, 551–554 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson-Kerner B. L., et al. , Intermediate filament protein accumulation in motor neurons derived from giant axonal neuropathy iPSCs rescued by restoration of gigaxonin. Hum. Mol. Genet. 24, 1420–1431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dequen F., Bomont P., Gowing G., Cleveland D. W., Julien J. P., Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J. Neurochem. 107, 253–264 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganay T., Boizot A., Burrer R., Chauvin J. P., Bomont P., Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice. Mol. Neurodegener. 6, 25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balastik M., et al. , Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 105, 12016–12021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahammad S., et al. , Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J. Clin. Invest. 123, 1964–1975 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan A., Nixon R. A., Neurofilament proteins as biomarkers to monitor neurological diseases and the efficacy of therapies. Front. Neurosci. 15, 689938 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen T. V., et al. , p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc. Natl. Acad. Sci. U.S.A. 114, 3565–3571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubin D. M., Finley D., Proteolysis., The proteasome: A protein-degrading organelle? Curr. Biol. 5, 854–858 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Lowe J., et al. , Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 268, 533–539 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Karibe A., et al. , Hypertrophic cardiomyopathy caused by a novel alpha-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation 103, 65–71 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Carlus S. J., et al. , A novel homozygous TPM1 mutation in familial pediatric hypertrophic cardiomyopathy and in silico screening of potential targeting drugs. Eur. Rev. Med. Pharmacol. Sci. 24, 7732–7744 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Wang N., Ingber D. E., Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 66, 2181–2189 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eapen V. V., Swarup S., Hoyer M. J., Paulo J. A., Harper J. W., Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. eLife 10, e72328 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kravic B., et al. , Ubiquitin profiling of lysophagy identifies actin stabilizer CNN2 as a target of VCP/p97 and uncovers a link to HSPB1. Mol. Cell 82, 2633–2649.e2637 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Cairns N. J., Lee V. M., Trojanowski J. Q., The cytoskeleton in neurodegenerative diseases. J. Pathol. 204, 438–449 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossor A. M., Polke J. M., Houlden H., Reilly M. M., Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 9, 562–571 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Kim H. J., et al. , Phenotypic heterogeneity in patients with NEFL-related Charcot-Marie-Tooth disease. Mol. Genet. Genomic Med. 10, e1870 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang S. J., et al. , Ubiquitin-dependent proteasomal degradation of AMPK gamma subunit by Cereblon inhibits AMPK activity. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118729 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Fischer E. S., et al. , Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen T. V., et al. , Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 61, 809–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Data Availability Statement
All study data are included in the article and/or supporting information.