

Significance
Ecological divergence provides compelling evidence for origin of species through natural selection that has captivated evolutionists since Darwin. In this study, we present multiple-omics analyses and lab experiments on two populations of one selfing plant growing sympatrically in contrasting environments. We unveiled distinct differentiation between two populations across phenotype to omics data. These two populations share a most recent ancestor compared with other populations and their divergence started in the early Holocene. We revealed that gene flow had continued albeit with a progressive reduction over time. The genetic divergences are dispersed across the total genome involving many unlinked coding and noncoding regions. These findings underscore the significance of natural selection in driving ecological divergence in the absence of geographic isolation.
Keywords: Brachypodium stacei, ecological divergence, functional genomics, multiomics, selfing species
Abstract
Ecological divergence without geographic isolation, as an early speciation process that may lead finally to reproductive isolation through natural selection, remains a captivating topic in evolutionary biology. However, the pattern of genetic divergence underlying this process across the genome may vary between species and mating systems. Here, we present evidence that Brachypodium stacei, an annual and highly selfing grass model species, has undergone sympatric ecological divergence without geographic isolation. Genomic, transcriptomic, and metabolomic analyses together with lab experiments mimicking the two opposite environmental conditions suggest that diploid B. stacei populations have diverged sympatrically in two slopes characterized by distinct biomes at Evolution Canyon I (ECI), Mount Carmel, Israel. Despite ongoing gene flow, primarily facilitated by seed dispersal, the level of gene flow has progressively decreased over time. This local adaptation involves the scattered divergence of many unlinked loci across the total genome that include both coding genes and noncoding regions. Additionally, we have identified significant differential expressions of genes related to the ABA signaling pathway and contrasting metabolome composition between the arid- vs. forest-adapted B. stacei populations in ECI. These results suggest that multiple small loci involved in environmental responses act additively to account for ecological adaptations by this selfing species in contrasting environments.
The adaptation of ancestral populations to contrasted environments has been assumed to trigger “origin of the species” as a major mechanism since Darwin (1). Under divergent selection, two populations accumulate multiple phenotypic and physiological traits to adapt to differentiated niches even in the presence of gene flow (2). This divergence at the same site may occur across different species which further highlights the critical role of such an ecological selection in speciation (3). Sympatric populations within the normal range of individual movement can be ecologically segregated, with ecological adaptation potentially causing pleiotropic reductions in gene flow (4). Despite many empirical studies based on genomic data investigating sympatrically diverging populations, the genetic bases of niche usage remain largely unknown for most species due to differing biogeographic patterns across entire evolutionary history (5). The ecological divergence may involve genetic differentiation across the total genome with many unlinked additive loci because of the continuous gene flow (6). However, in some cases of ecological divergence without isolation, large genomic islands containing multiple linked genes with obvious selection signals have been reported in ecological divergences of a few species in a similar manner without geographic isolation (7, 8). In inbreeding/selfing species, the reduced genetic diversity caused by strong purification, selective sweeps, and the lower effective recombination rate is expected to lead to larger and more distinct genomic islands (9), although the empirical evidence for this is still lacking.
“Evolution Canyon I” (ECI), located near Mount Carmel in Israel, provides a convenient model landscape for studying ecological divergence for diverse organisms, ranging from bacteria to plants and mammal (10). Although the overall climate and geology are essentially identical, the microclimates of the two slopes of ECI strongly differ, with the south-facing “African” slope (AS) receiving up to 300% higher solar radiation than the north-facing “European” slope (ES) (Fig. 1A) (11). Thus, the AS has a xeric biome that is tropical, hot, dry, and savannoid, whereas the ES is temperate, cool, humid, and forested. It is estimated that these distinctive microclimates have existed since the Plio-Pleistocene (5.3 Mya–11.7 Kya) (11). Evidence of ecological divergence that may lead to sympatric speciation has been found for seven species from diverse organismic groups that are distributed on both slopes of ECI (10), including the haploid soil bacterium Bacillus simplex (12), diploid fruit-fly Drosophila melanogaster (13) and Drosophila simulans (14), grain beetle Oryzaephilus surinamensis (15), spiny mouse Acomysca hirinus (16–18), wild barley Hordeum spontaneum (19), and polyploid crucifer Ricotia lunaria (20) and wild emmer wheat Triticum dicoccoides (21). The genomic divergence observed between AS and ES populations of these organisms has been found to correspond with allelic differentiation of genes that contribute to local adaptation to different environmental conditions (Dataset S10).
Fig. 1.
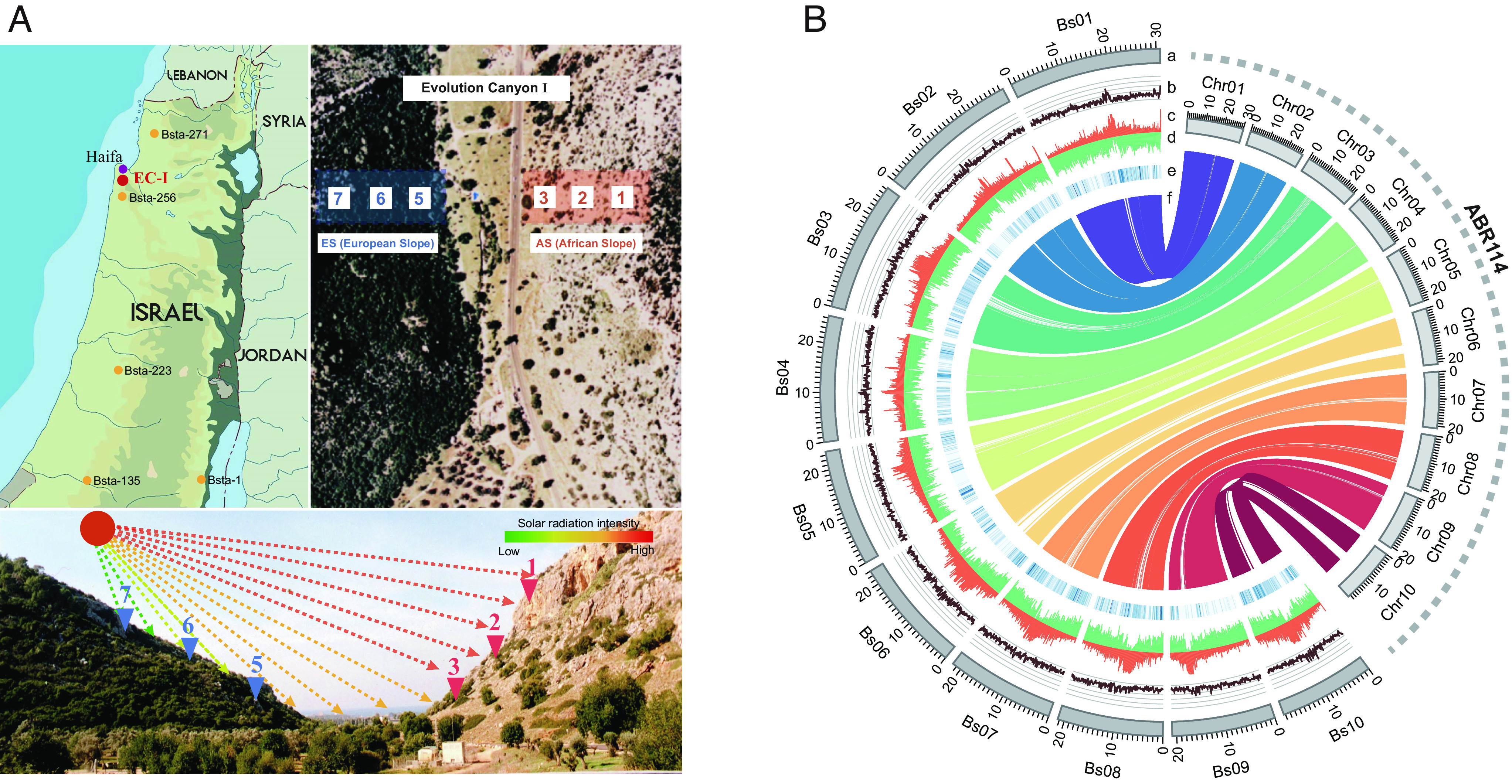
Sampling and genome organization of Brachypodium stacei (ECI). (A) Sampling locations of B. stacei in ECI and other sites in Israel. Left-most: populations of B. stacei from ECI (red) and elsewhere in Israel (yellow). Right-most: populations of B. stacei ECI from the AS (salmon) and the ES (blue). Both AS (22) and ES (16) samples were collected through spatial transects in their respective slopes (AS: 1–3, ES: 5–7), covering all potential microniches for each environment. Bottom: Despite being only 100–250 m apart in linear distance, these ES and AS slopes have strongly contrasted microclimates (e.g., solar radiation intensity differs substantially between them). (B). Overview of the newly assembled B. stacei genome from ECI (Bsta-ECI, Left) and syntenic relationships to the current B. stacei ABR114 v.1.1 reference genome (Right). The tracks indicate (a) chromosomes (Bs01–Bs10, Bsta-ECI; Chr01–Chr10, ABR114), (b) GC contents, (c) transposable element densities, (d) gene models densities, (e) single-nucleotide polymorphism (SNP) densities, and (f) collinearity of syntenic genes (Bsta-ECI vs. ABR114).
Here, we explore genetic bases underlying ecological divergence of an inbreeding grass species, the annual diploid B. stacei (2n = 2x = 20), which occurs on both the ES and AS slopes of ECI (Fig. 1). This arid species is broadly distributed in the southern part of the circum-Mediterranean region (23) where it grows in shady warm places or in open habitats, usually protected by shrubs (24). By utilizing high-quality genome assemblies for B. stacei, genome-resequencing data from populations at ECI and surrounding regions in Israel, together with transcriptomic, metabolomic data, and lab experiments, we aim to test whether genomic divergence has occurred between the ES and AS populations of this selfing species in ECI. Furthermore, we investigate how the highly divergent genes are functionally related and distributed across the total genome of this species in ECI and explore how this model grass expands our understanding of sympatric speciation within an evolutionary frame.
Results
Genomic Assembly and Annotation of B. stacei in ECI.
The genome size of B. stacei ECI was estimated to be 263.59 Mb with a low heterozygosity of 0.09%, which is ~25 Mb larger than the B. stacei reference genome ABR114 (SI Appendix, Fig. S1). In order to make genomic analysis more reliable, an improved de novo assembly was generated for the B. stacei Bsta-ECI genome (SI Appendix, Tables S1 and S2). The new Bsta-ECI assembly had markedly higher contiguity than the current reference genome assembly B. stacei ABR114 v. 1.1 (https://phytozome-next.jgi.doe.gov/) (25), with ~22.57 Mb longer total contig length (256.71 Mb vs. 234.17 Mb), ~98.5% lower total contig numbers (79 vs. 3,132), and ~44-fold higher N50 length (10.19 Mb vs. 0.23 Mb) (SI Appendix, Fig S2 and Table S2). These high-quality contigs were further anchored onto 10 chromosomes assisted by 3D proximity information from Hi-C datasets (SI Appendix, Table S3). The final genome assembly captured 248.99 Mb of the genome sequences, with 96.99% anchored percentages (SI Appendix, Fig S3 and Table S2). All chromosomes show longer sizes and fewer gaps than the reference genome ABR114 v. 1.1 (SI Appendix, Table S4). The base call accuracy (QV) and assembly completeness were 39.78 and 99.18%, and Illumina pair-end reads mapping both show a >98.5% mapping rate and coverage rate in the Bsta-ECI genome assembly (SI Appendix, Table S5). Then, 98.6% of the BUSCO ortholog and homeolog genes could be completely predicted in Bsta-ECI, which were slightly higher than those in the previous assembly (SI Appendix, Fig. S4). In addition, the LAI (LTR Assembly Index) of Bsta-ECI was higher than values for the previously reported ABR114 assembly (SI Appendix, Fig. S2C), indicating that Bsta-ECI has higher long-terminal repeat (LTR) retrotransposon completeness. All assessments suggested high consistency and completeness of the Bsta-ECI genome assembly which showed great improvement in contiguity and repetitive sequence completeness compared with the reference genome assembly ABR114.
Around 103.46 Mb (40.31%) of the total Bsta-ECI genome sequence was annotated as repetitive element sequences (SI Appendix, Table S6), a percentage higher than that of the reference genome ABR114 (SI Appendix, Table S7). Most of the repetitive element sequence is transposable elements (TEs), which include SINE (~0.59%), LINE (~7.33%), LTR (~44.92%), and DNA (~17.91%) (SI Appendix, Table S7). We further predicted 32,951 high-confidence protein coding genes in the Bsta-ECI genome, slightly higher than those previously reported in ABR114 v. 1.1 (SI Appendix, Fig. S4 and Table S8). The gene structure feature (average CDS length, exon length, exon number, and intron length) are similar to those of the ABR114 v. 1.1 and B. distachyon Bd21 reference genome (SI Appendix, Fig. S4 and Table S8). More than 97% of the predicted genes of the Bsta-ECI genome had homologs in public functional databases (SI Appendix, Table S9). We also found that 99.1% BUSCOs could be completely detected (SI Appendix, Fig. S5), indicating the high completeness of the gene model annotation. Furthermore, 1,838 transcription factors encoding genes, belonging to 67 gene families representing 5.5% of the total predicted genes, were found in the Bsta-ECI genome (SI Appendix, Table S10). The high-quality chromosome-level assembly and annotation of our Bsta-ECI genome (Fig. 1B) provide robust foundations for investigating the evolutionary processes that gave rise to ecological divergence of this species in ECI.
Genome Resequencing and Population Structure.
To explore the potential adaptive evolution of B. stacei in ECI, we subjected 46 B. stacei individuals [41 from ECI (25 AS, 16 ES), 5 from other regions of Israel] to whole-genome resequencing (Figs. 1A and 2A, and Dataset S1). A total of 328.87 GB of clean Illumina data were obtained and mapped to the Bsta-ECI local reference genome, with an average of ~27× coverage for each individual. More than 98.5% average mapping rate and genome coverage were obtained for both AS and ES individuals, indicating high alignment accuracy (Dataset S2). After variant calling and quality filtering, 220,321 short INDELs (2–725 bp) and 722,351 high-quality SNPs were identified for downstream analysis (SI Appendix, Table S11). The average INDEL and SNP density were 0.89/kb and 2.81/kb, which occur sparsely across the total individual genomes from both groups, thus indicating similar coverages for the called genotypes of AS and ES populations (SI Appendix, Fig. S6). We first constructed a maximum-likelihood (ML) phylogenetic tree based on the full SNP dataset (rooted with B. distachyon Bd30) to evaluate the relationships among B. stacei lineages. The phylogenetic tree separated the ECI individuals into two strongly supported clades (designated AS and ES), corresponding to their spatial distribution at ECI (Figs. 1A and 2A). The B. stacei lineages from other localities of Israel were resolved as either sister to the ECI clade or more distantly related, followed by the more divergent western and central Mediterranean lineages. Strong support was obtained for all main split nodes, and phylogenetic divergence tended to reflect overall geographic distances (Fig. 2A). The robust sister relationship between the two ECI populations indicates that the split was likely primary, rather than a result of secondary contact after allopatric divergence between geographically isolated populations (Figs. 1A and 2A). Principal component analysis (PCA) also showed that individuals collected from the ES slope clustered together and were separated from AS individuals along the first principal component axis (SI Appendix, Fig. S7A). Population structure analysis for the best K = 2 hypothetical populations was also consistent with the ML tree and PCA results (Fig. 2A and SI Appendix, Fig. S7B). Interestingly, we observed a few genomically admixed individuals in both AS and ES populations, suggesting that gene flow may still occur between these two spatially adjacent populations (Fig. 2A and SI Appendix, Fig. S7B). We further performed phylogenetic and population structure analyses using a reduced 25,471 fourfold degenerate SNP data set of exclusively neutral markers; however, the new results (SI Appendix, Fig. S8) did not show apparent differences when compared with those retrieved from the whole-genome SNP dataset (SI Appendix, Fig. S7). Using Bsta-ECI genome as reference, we identified 1,021 transposable elements’ polymorphisms (TEPs) between AS and ES populations, including 732 deletions and 289 insertions across all individuals (SI Appendix, Fig. S9 and Dataset S3). Besides, 13,811 structural variants (SVs) were also identified (SI Appendix, Table S12). A neighbor-joining tree and a PCA based, respectively, on SV data and transposable elements polymorphism (TEPs) also clearly distinguished the AS and ES populations from each other (SI Appendix, Fig. S10). These results indicate that genomic divergence has formed in multiple variation category between these two geographically close populations.
Fig. 2.
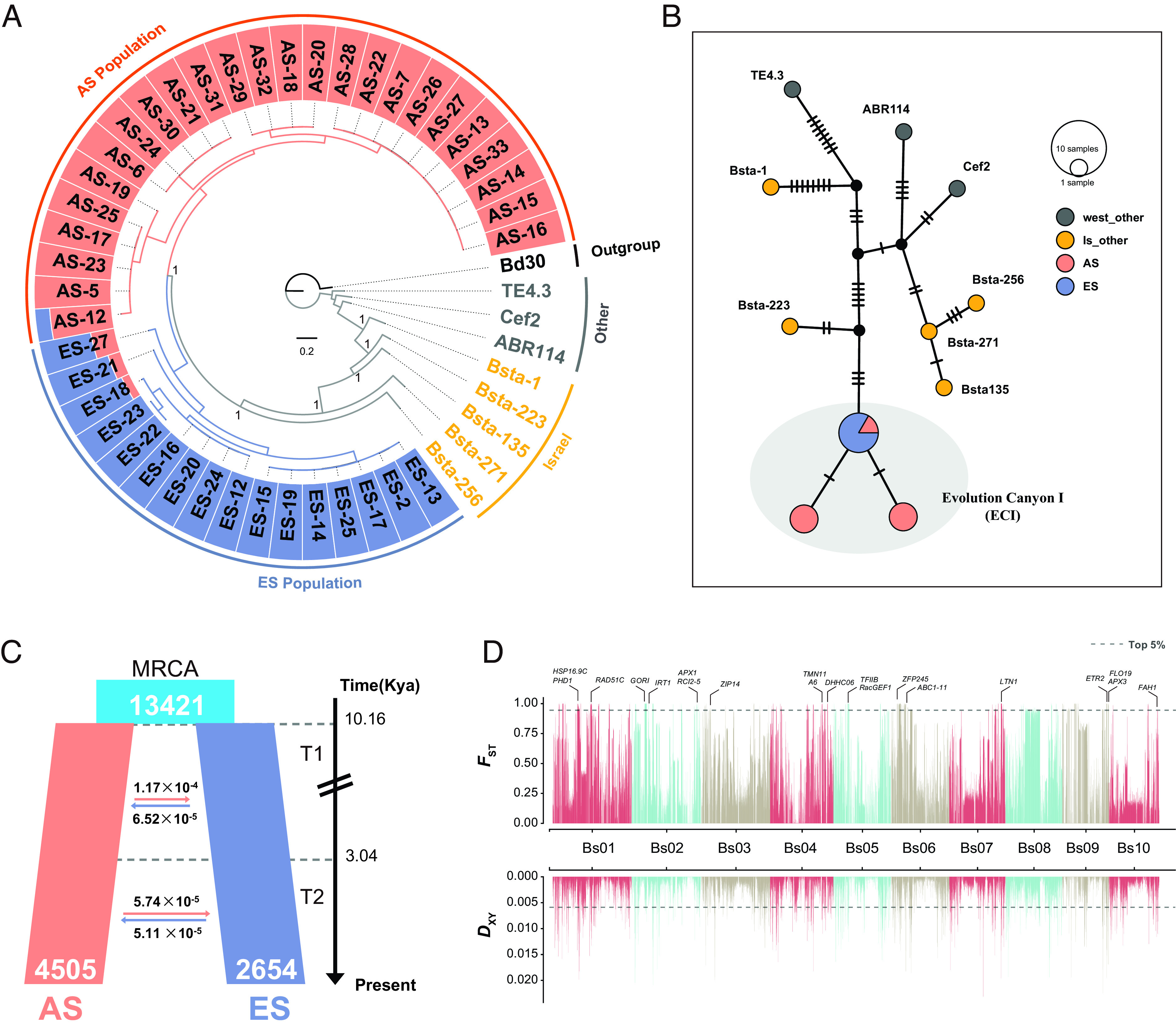
Genome divergence of B. stacei resequenced accessions from ECI. (A) Maximum-likelihood phylogenetic tree of the B. stacei accessions based on high-quality SNP data from individual genomes of B. stacei [AS (salmon), ES (blue), Israel (yellow), and other western and central Mediterranean populations (gray)], rooted with B. distachyon Bd30. The tree’s outer ring displays the population structure (with optimal K = 2) of the B. stacei AS and ES populations. (B) Statistical parsimony plastome haplotype network of B. stacei samples from Israel and other Mediterranean localities. The area of each circle in the network is proportional to the haplotype frequency, and the number of mutational steps between two nodes is indicated by short bars. (C) Demographic history of the B. stacei AS and ES populations inferred by the best fit ∂a∂i model (“asym_mig_twoepoch” model), indicating that a single split of the ancestral population (MRCA; light blue) 10.16 Ka gave rise to the modern AS and ES populations. The reciprocal average migration rates between ES and AS populations in two temporal epochs (T1 and T2) are shown by horizontal arrows and the estimated ages of T1 and T2 on the right side of the panel. (D) Genetic differentiation and divergence between the B. stacei AS and ES populations revealed by FST and DXY data. Dashed horizontal lines depict the top 5% thresholds (FST 0.92, DXY 0.0011) and rice ortholog functional genes located in highly divergent regions are marked on the respective chromosomes.
To further investigate the origins of the AS and ES population at ECI, we phylogenetically analyzed the maternally inherited plastomes of this species including samples from ECI, other regions of Israel, and other native Mediterranean locations (SI Appendix, Fig. S11A). A maximum likelihood plastome tree rooted with B. distachyon Bd30 supported the strong monophyly of the ECI group (concatenated plastome genes tree, SI Appendix, Fig. S11B; whole plastome tree, SI Appendix, Fig. S11C), which was nested within a strongly supported ECI-Israel p. p. (pro partim) clade. Within the ECI lineage, all ES samples were shown to be derived from one of the AS lineages (SI Appendix, Fig. S11B) and a parsimony network of plastome haplotypes from the two slopes provided further support for this scenario (Fig. 2B).
Demographic History Inference.
To reconstruct the most plausible evolutionary scenario of divergence between AS and ES populations of B. stacei in ECI, we simulated alternative demographic history models of the two populations using forward simulation and residuals analysis in ∂a∂i. We tested seven models by fitting a site frequency spectrum (SFS) of the AS and ES populations (Fig. 2C and SI Appendix, Fig. S12A). The best-fitting model based on likelihood and AIC values suggested a single population divergence event with reciprocal and asymmetric migration rates occurring in two different time periods (T1 and T2; referred to as the asym_mig_twoepoch model) (Fig. 2C and SI Appendix, Fig. S12B and Tables S13 and S14). According to this model, the most recent common ancestor of the AS and ES populations diverged ~10.16 kya with an estimated population size of 13,421 individuals. The first epoch (T1) lasted until ~3.04 kya, when the second epoch (T2) began and continued until the present. The AS and ES populations were estimated to have populations sizes of 4,505 and 2,654 individuals, respectively (Fig. 2C and SI Appendix, Table S13). The model suggests that there was continuous and reciprocally asymmetric migration between the two populations in both time periods. However, as the divergence time increased, the gene flow and its asymmetry decreased. In epoch 1, gene flow was nearly one order of magnitude greater from the more ancestral AS to the more recently derived ES (1.17E-4) than from ES to AS (6.5E-5), compared to epoch 2 (5.7E-5 vs. 5.1E-5) (Fig. 2C). This asymmetry of gene flow in epoch 1 aligns well with the phylogenetic scenario of a founder event for ES from AS (Fig. 2B), thus supporting a greater gene flow from AS to ES than the reverse. In contrast, the reciprocal gene flow between the two populations became balanced and decreased in epoch 2, likely due to the stabilization and expansion of the ES population. These results suggest that there has been ongoing gene flow between the two populations, although the rate of gene flow has decreased over divergence time (Fig. 2C and SI Appendix, Table S14).
Genetic Parameter and Genomic Divergence of AS and ES Populations.
We observed a similar distribution pattern of population nucleotide diversity (π) in the AS population (mean π = 1.7893 × 10−3) and ES population (mean π = 1.7810 × 10−3) (t test P-value = 0.782) (SI Appendix, Fig. S13), indicating that both populations have maintained comparable levels of genetic diversity in their respective habitats. In addition, we detected similar levels of genome-wide linkage disequilibrium (LD, indicated by r2 values) between the AS and ES populations (SI Appendix, Fig. S14A). The average physical distance between unlinked SNPs within each population was ~200 kb, with half of the maximum r2 value (SI Appendix, Fig. S14A). This LD decay distance is longer compared to distances reported in other plant species (22). Additionally, we corroborated the high inbreeding rate of this species in both AS and ES ECI populations (SI Appendix, Fig. S14B), as reported for other circum-Mediterranean populations (26), which is consistent with their low recombination rate (SI Appendix, Fig. S14C).
The estimated average genome-wide genetic divergences between the AS and ES populations, expressed in terms of fixation index (FST) and mean pairwise nucleotide differentiation (DXY), were ~0.336 and 0.001, respectively (Fig. 2D and SI Appendix, Fig. S15). We identified 807 “genomic islands” (merged into 284 non-overlapping windows) exhibiting high genetic divergence (Dataset S4) as measured by FST using a permute method with 10-kb windows (22). We employed a 100-kb threshold to classify these islands, considering regions above it as “large” and below it as “small.” This threshold value is conservative, considering the similarity in sizes between the genomic islands and the LD decay distances with those observed in theoretical simulation (8). Out of the 284 genomic islands, 13 had a size of >100 kb, encompassing 28.62% of the total length of all genomic islands, and were scattered across all chromosomes (SI Appendix, Table S15).
Comparing genomic islands with the background genome, we found a significant increase in DXY but a reduction in π within these regions (SI Appendix, Fig. S16 A–C, Wilcoxon P-value < 2.2 × 10−16). We then explored genomic regions that may be resistant to gene flow or indicative of ancient polymorphisms based on the top 5% DXY value (27). Within this range, we identified a total of 2.05 Mb of genomic regions encompassing 440 genes (Fig. 2D and Dataset S5). Some of these genes, along with their rice orthologs, were found to be functionally associated with reproduction and development (OsRAD51C, GORI, OsSPL15, FLO19, and OsETR2), plant–pathogen interactions (SWAP70A, OsRacGEF1, and OsRbohB), responses to abiotic stress (OsHSP16.9C, ZFP245, OsRCI2-5, and OsAPX1), ion transport (OsTMN11, OsIRT1, and OsA6), enzyme activity (LTN1 and OsAPx3) and cell energy metabolism (PHD1 and OsABC1-11) (Fig. 2D).
As a complement to the FST and DXY approaches, we utilized Hudson–Kreitman–Aguadé (HKA) tests to detect genes under recent selection. A total of 795 genes (Dataset S6) were identified in the two populations, and out of these, 392 genes showed significant overlap with the genomic islands (SI Appendix, Fig. S17, P-value < 2.2 × 10−16, chi-square test). Notably, these genes were found to be enriched in functions related to plant responses to abiotic stress (SI Appendix, Fig. S18 and Dataset S7). For example, we found three fixed SNPs in the coding regions of PIN3A between the AS and ES populations, two of which were nonsynonymous coding mutations. A homolog of this gene encodes a putative auxin efflux carrier in rice, and its over-expression can improve the drought tolerance of rice (28). In addition, rice orthologs of the genes Sta2 (29) and HsfB2b (30) reportedly contribute to salt or drought responses. The AS and ES polymorphisms in these abiotic stress response genes may reflect selective adaptation to their respective microhabitats. These analyses suggest that the genomic divergence between the two B. stacei populations from opposite slopes of ECI resulted from ongoing local adaptation to contrasting microhabitats, which may lead to incipient sympatric speciation in these populations.
Transcriptomic and Metabolomic Divergence between AS and ES Populations.
To further elucidate potential mechanisms of local adaptation in the AS and ES populations of ECI, we applied multiple-level comparisons in laboratory experiments that mimicked the two contrasting environmental conditions. We first conducted drought tolerance experiments and root growth assay. The above-ground plant phenotypes of AS and ES individuals were similar under well-watered condition but differed slightly under the drought treatment being more affected the ES individuals (Fig. 3A). Five measured physiological parameters in leaf tissue differed between plants grown in drought and well-watered conditions; transpiration rate (E), assimilation rate (A), intracellular carbon dioxide concentration (Ci), and stomatal conductance (Gsw) values were lower, while water use efficiency (WUE) values were higher, under drought than under watered conditions (SI Appendix, Fig. S19). Both AS and ES individuals showed similar changing trends although AS plants generally exhibited higher values in most cases. Notably, we observed that root growth in AS individuals was significantly faster than in ES individuals in control conditions. However, in the presence of abscisic acid (ABA), which correlates with the existence of drought stress, root growth in AS individuals was more inhibited. This indicates that the AS population has developed significantly greater tolerance to drought than the ES population (SI Appendix, Fig. S20).
Fig. 3.
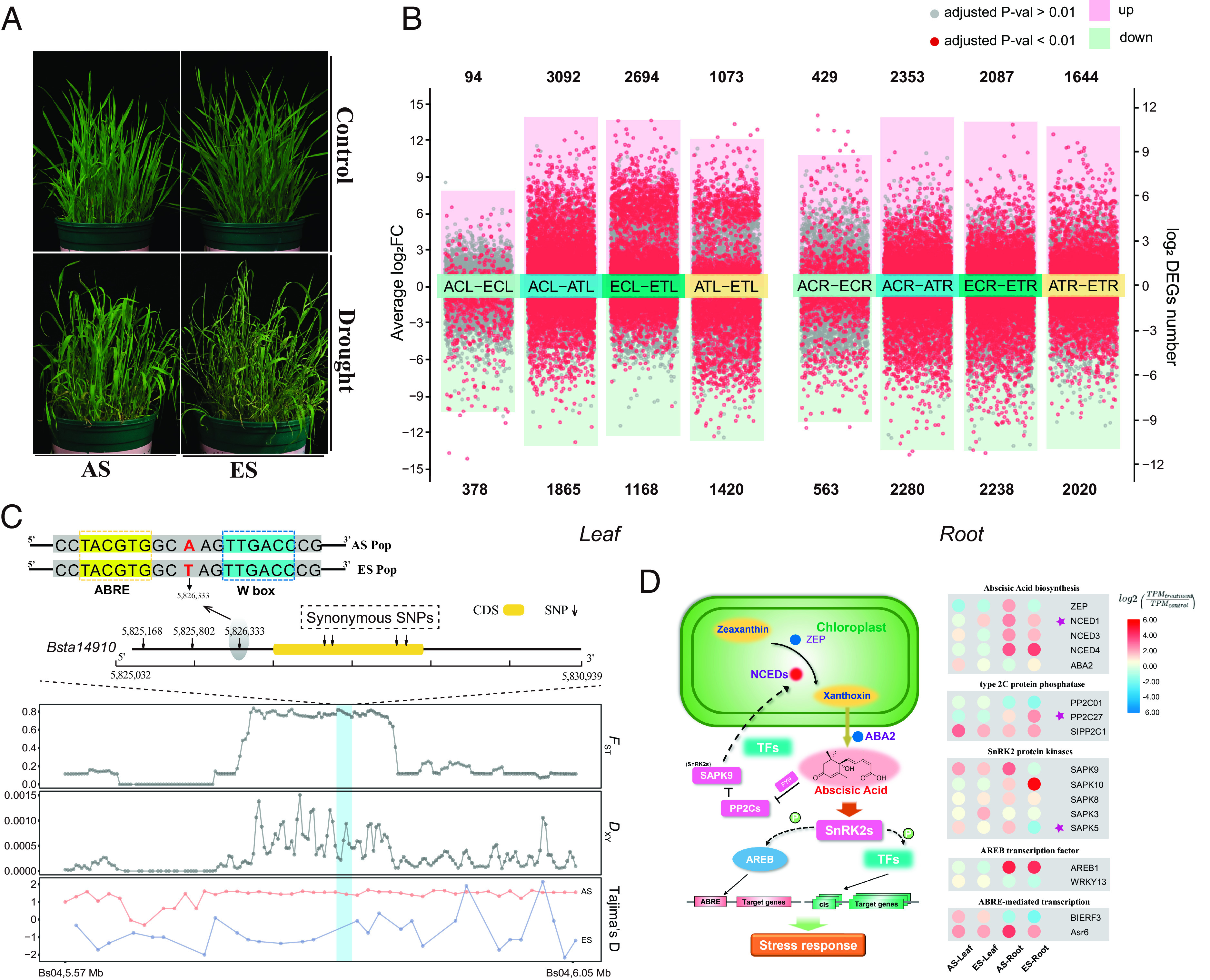
Phenotypic, transcriptomic, and genetic responses related to drought stress conditions of B. stacei plants from AS and ES populations. (A) Above-ground phenotypes of plants from AS and ES populations grown under drought and well-watered (control) conditions. (B) Up-regulated and down-regulated differentially expressed genes (DEGs) in leaf and root tissues from intra- and inter-AS and ES population comparisons; adjusted P-values < 0.01 and >0.01 are indicated by red and gray dots, respectively. (C) Genetic structure and SNP variants located between ±2 kbp (upstream and downstream) of gene Bsta14910 (ortholog of rice OsNCED1) in chromosome Bs04. Population parameters FST, DXY, and Tajima’s D of AS and ES populations. The bright blue bar in the plots represents the windows containing Bsta14910 and its polymorphisms. (D) Different expression patterns of ABA signaling pathway genes in B. stacei leaf and root tissues of samples from the AS and ES populations at ECI under drought stress treatment. Heatmaps of differential expression patterns of genes of AS and ES leaf and root tissue samples for relevant genes of the ABA signaling pathway routes. Each cell represents the Log2 fold change of DEGs between drought (treatment) and well-watered (control) conditions (TPMTreatment/TPMControl); the color codes for these values are indicated in the corresponding chart. Genes located in selective regions are marked with a star.
To identify transcriptomic dynamics in drought responses, RNA-seq analysis was applied and Pearson correlation coefficients showed good repeatability of gene expression profiles among replicates from each population (SI Appendix, Fig. S21). We compared gene expression levels in leaf and root tissues from AS and ES plants grown under control (well-watered) and drought conditions, in both inter- and intra-population comparisons (Fig. 3B). In agreement with the observed phenotypic and physiological differences, numerous differentially expressed genes (DEGs) were detected between plants subjected to contrasting conditions in the intrapopulation comparisons (Fig. 3B). In total, we detected 3,862 and 4,325 DEGs in leaves and roots of ES individuals, and more (4,957 and 4,633 DEGs) in AS plants (Fig. 3B). The sets of these DEGs in both AS and ES plants exhibited enrichment of diverse Gene Ontology (GO) terms and KEGG pathways associated with drought responses (SI Appendix, Fig. S22 and Dataset S8). In inter-population comparisons between AS and ES plants, we identified 1,630 common DEGs in leaves and 1,423 common DEGs in roots (SI Appendix, Fig. S23). The Log2 fold change (Lfc) values of these differentially expressed genes were similar for both AS and ES plants (Pearson correlation, R ≥ 0.75, P < 2.2e−16) (SI Appendix, Fig. S24), suggesting that drought stress induced comparable qualitative effects on gene expression in both leaf and root tissues of AS and ES individuals, despite significant differences in their respective expression levels.
Only 462 leaf DEGs and 992 root DEGs were identified from AS–ES interpopulation comparisons under well-watered conditions (Fig. 3B), suggesting that gene expression is quite similar in both populations under well-watered conditions. In contrast, 2,493 leaf and 3,664 root DEGs were identified from AS–ES interpopulation comparisons under drought conditions, 5.3- and 3.6-fold higher than the numbers observed in the corresponding comparisons of well-watered plants (Fig. 3B), suggesting potential differences in drought response mechanisms between individuals of the AS and ES populations.
The observed dissimilarities in genome and transcriptome between AS and ES populations were mirrored by their respective metabolomes. We analyzed metabolite profiles of leaf and root tissues from AS and ES plants under drought conditions using liquid chromatography–mass spectrometry (LC–MS). In interpopulation metabolome comparisons, we identified 158 and 120 differential metabolites (DM) in the leaf and root tissues, respectively. Orthogonal partial least-squares discriminant analysis (OPLS-DA) separated the AS and ES metabolomic profiles into two distinct clusters, indicating notable differences in the metabolite profiles of AS and ES plants grown under drought conditions (SI Appendix, Fig. S25). The DM data were consistent with the enrichment of KEGG biological pathways associated with drought stress responses in the DEGs of the AS and ES populations (SI Appendix, Fig. S26 and Datasets S8 and S9). We further investigated the expression of genes directly involved in drought and oxidative responses, such as DRO1, APX8, and ECK1, several of which exhibited different expression patterns in the AS and ES populations (SI Appendix, Fig. S27). These contrasting patterns of gene expression and metabolite data suggest that the AS and ES populations respond discordantly to drought stress, likely as a consequence of distinct local adaptations to their respective microclimates.
Key Factors Regulating Microclimate Adaptation in B. stacei ECI Populations.
The ABA phytohormone and its related downstream signaling pathway are crucial regulatory systems for plants, exerting broad effects, particularly on their responses to abiotic stress. Our root growth assay revealed a higher sensitivity to ABA in the AS population compared to ES population (SI Appendix, Fig. S20). Therefore, we investigated genes involved in the abscisic acid (ABA) signaling pathway that exhibited differential expression patterns between the AS and ES populations. The conversion of 9-cis-epoxycarotenoids to xanthoxin, by 9-cis-epoxycarotenoid dioxygenases (NCEDs), represents a key regulatory step in ABA biosynthesis (31). We identified a putative ortholog of rice OsNCED1 (Bsta14910) (SI Appendix, Table S16) that displayed substantial genetic divergence and expression changes between the AS and ES populations (Fig. 3C and SI Appendix, Fig. S28A). Notably, we identified four SNPs within the coding region of this gene, resulting in reciprocal synonymous amino acid mutations between the two populations (Fig. 3C). We also detected 16 SNPs in the 5′-upstream 2000 bp regulatory region of Bsta14910, all of which exhibited high divergence between the AS and ES populations (SI Appendix, Table S17). Among these SNPs, three were fixed in the AS and ES populations, including one located 699 bp upstream of the start codon (Fig. 3C). Further analysis showed that this SNP was located between the TACGTG (ABRE) and TTGACC (W box) motifs (Fig. 3C), suggesting that these mutations may affect the transcription of Bsta14910. Indeed, we observed significant overexpression of this gene in the leaf and root tissues of the AS samples compared to the ES samples (SI Appendix, Fig. S28A). Other NCED1 orthologs are involved in heat responses in rice (32) and Lactuca sativa (33), underscoring this gene’s potential importance for adaptation to warm conditions.
We also found that exposure to drought-induced stronger changes in the AS population compared to the ES population samples in the expression of a gene encoding a serine/threonine protein kinase (SAPK5) (Fig. 3D), which plays a key role in ABA signaling pathways activated by hyperosmotic stress (34). In addition, we identified a Glossy1 B. stacei ortholog (GL1-2) that exhibited significantly higher expression patterns in the AS population samples than in the ES populations samples (SI Appendix, Figs. S28B and S29). This gene is induced by ABA and reportedly involved in wax biosynthesis and thus may participate in important adjustments of the composition of leaf waxes that enhance resistance to abiotic stressors, such as drought, ultraviolet light, and extreme temperatures (35). Together, these results suggest that differential expression of genes encoding key proteins involved in ABA signaling and wax synthesis may play important roles in different adaptations to local environments observed in the AS and ES populations. Additionally, changes in TEs may affect the expression of nearby genes through altering or creating regulatory element during ecological divergence (36). We identified 15 TE insertion polymorphisms (TIPs) that exhibited different frequencies of absent or present between the two populations (SI Appendix, Fig. S9 and Dataset S9), and nine genes were associated with these TIPs. Among them, two genes (Bsta12548 and Bsta19483) consistently displayed differential expressions between the two populations, with higher expression levels observed in the AS samples compared to the ES samples in both tissues and conditions (SI Appendix, Fig. S30). Bsta12548 is involved in posttranslational modification and protein turnover process while Bsta19483 encodes a wall-associated receptor kinase. Therefore, mutations in regulatory motifs and coding regions of these genes and their target genes, may also contribute to disconnected local adaptations and genetic divergence observed between the arid AS and mesic ES populations at ECI.
Discussion
Ecological divergence has been frequently reported for organisms living in ECI (Dataset S10) because of the striking differences in microclimate and biomes within the canyon (10). A wide range of organisms, including bacteria, mammals, and plants, have undergone ecological divergence in ECI. The contrasting ecological conditions have facilitated prezygotic and/or postzygotic reproductive isolation, potentially leading to the emergence of distinct species. Populations of certain species, for example, wild emmer wheat and spiny mouse, on the two slopes of ECI, have been reproductively isolated through prezygotic (17, 19) (differences in flowering time for the former and mate discrimination for the latter) and postzygotic (due to chromosome rearrangements) isolation mechanisms (21), although not completely. In our study, we found that the contrasting microclimates of ECI have similarly fostered genomic, transcriptomic, and metabolomic divergence between two diploid inbreeding populations of B. stacei (Figs. 2 and 3). These findings suggest the ongoing divergence of the two populations and the potential future separation into two species with complete reproductive isolation.
Our population analysis of maternally inherited plastomes showed that both AS and ES individuals shared a common ancestor compared to other B. stacei accessions out of ECI (Fig. 2B and SI Appendix, Fig S11), supporting that the two populations diverged in situ. Furthermore, we observed that ES individuals formed a subclade nested within the AS clade in a strongly supported ML plastome tree (SI Appendix, Fig. S11), indicating that B. stacei was established first in the AS of ECI and subsequently colonized the ES. The lower divergence observed in recently expanded ES individuals, which exhibited a single plastome haplotype, compared to the greater divergence observed in more ancestral AS individuals (with three plastome haplotypes), was also supported by several nuclear genomic components (SNPs, SVs, TEPs; SI Appendix, Figs. S7A and S10). Previous evolutionary studies have reported both colonization scenarios from AS to ES and from ES to AS (10). However, our plastome data strongly support an AS to ES colonization scenario for B. stacei in ECI (Fig. 2B and SI Appendix, Fig. S11). Despite the non-reciprocally monophyletic clustering of individuals from each slope in the plastome tree, which likely reflects their colonization history, the nuclear genome data clearly differentiate the two populations (Fig. 2A and SI Appendix, Figs. S7, S8, and S10), suggesting that individuals from each population share their own genome variants. These analyses collectively demonstrate that both populations have evolved as distinct and independent lineages.
Our demographic analyses further suggest that the AS and ES populations of B. stacei diverged approximately 10,000 years ago, and gene flow gradually decreased as divergence time increased (Fig. 2C and SI Appendix, Fig. S12 and Tables S13 and S14). Although the variations in historical and recent reciprocal gene flow between the AS and ES populations (Fig. 2C) could have influenced the genomic makeup of individuals, as evidenced by the greater genomic admixture in ES individuals compared to AS individuals (Fig. 2A), the fundamental mechanisms of speciation driven by ecological adaptation may not have been significantly affected by limited gene flow, given that the estimated levels of gene flow were generally low in all instances.
The selfing species differ in sequence evolution from outcrossing ones in the rapid fixation of variants by high levels of homozygosity, including recessive alleles and ancestral polymorphisms (9). The fixation of beneficial alleles in selfing species could contribute to environmental adaptation, but the impact of natural selection depends on the effective population size and selection coefficient (9). This is further affected by the “Hill-Robertson effect” that may affect selection efficiency on a specific mutation in a low-recombining genome due to simultaneous selection occurring on linked loci (37). Considering these multiple tangling factors, the ecological adaptation in selfing species remains ambiguous. In addition to genetic divergence, our transcriptomic, metabolomic, and physiological data provided evidence that the two populations of B. stacei have diverged in response to disruptive selection pressures on the two slopes. Individuals from the AS population have significantly more drought tolerance than those from the ES population (Fig. 3 and SI Appendix, Figs. S19–S30). Functional genomic studies (38) and metabolomics studies (39) conducted on the closely related species, B. distachyon, have also revealed a greater induction of genes and metabolites crucial for drought stress responses in individuals adapted to arid conditions as opposed to those adapted to mesic conditions. These differences at multiple dimensions between AS and ES populations suggest disruptive ecological selection could contribute to the maintenance of population separation by facilitating divergent local adaptation to contrasting habitats.
We found evidence of genetic divergence (mean FST = 0.33) between the two populations, surpassing the genetic divergence observed in other outcrossing plant and animals that have undergone sympatric speciation or divergence at this site (14, 40). This is consistent with the expectation that the elevated rates of inbreeding in B. stacei (SI Appendix, Fig. S14B) may have facilitated substantial genomic divergence. Additionally, our results suggest that gene flow between the AS and ES populations is primarily mediated by seed dispersal. However, we also identified clear indications of introgression in a small number of individuals between the AS and ES slopes (Fig. 2B and SI Appendix, Fig. S7B), indicating that interpopulation pollen exchange may still occur, albeit rarely. Both hitchhiking and background selection can lead to locally reduced polymorphism at linked sites, which are expected to affect larger genomic regions because of low recombination rates in selfing species (41). It should be noted that our findings indicate that the highly diverged and fixed allelic frequencies in both coding genes and noncoding regions (e.g., TEs) between the two populations are distributed across all chromosomes of the B. stacei genome (SI Appendix, Table S15 and Dataset S3), forming numerous small genomic islands (<30 kb) with high divergence through the process of selective sweeps. The significantly higher DXY and lower π in these genomic islands¸ compared to the background of the entire genome, also indicated a decrease in intrapopulations genetic diversity and an increase in interpopulation divergence in these regions (SI Appendix, Fig. S16), which aligns with experimental and theoretical examinations of genomic divergence under ecological differentiation with continuous gene flow and the sorting of ancient polymorphisms (26, 42). This contrasts with the ecological divergence of a few species in the sympatric site, where large-scale linked genomic divergences have led to the formation of numerous islands (8). The latter may be the result of microparapatric speciation with partly geographic isolation or secondary contacts following initial allopatric divergence in such studies. Our results reject the genomic consequence of these potential evolutionary scenarios in the microgeographic landscape and tend to support the model of sympatric speciation with gene flow for B. stacei. Prezygotic reproductive isolation resulting from ecological selection of seedling establishment in contrasting habitats should have reduced homogenization between the two populations and thereby promotes sympatric divergence of this highly selfing species. In addition, our estimation of the decreasing gene flow also supports the prediction that gradual divergence of two populations may lead to sympatric speciation (43).
In conclusion, our study provides robust sources from genomic, transcriptomic, and metabolomic analyses supporting the ecological divergence of two B. stacei populations with a most recent ancestor residing on the opposite slopes of ECI. Furthermore, it is likely that multiple unlinked loci act additively to contribute to this ecological divergence. These cumulative findings support the hypothesis of initial sympatric speciation in selfing B. stacei within ECI as indicated by previous studies with other organisms (5, 44, 45) and provide a new case for ecological speciation in selfing plants (46).
Materials and Methods
The genome was sequenced using a PacBio Sequel2 platform and assembled using NextDenovo. The whole-genome DNA resequencing data were generated by an Illumina HiSeq X Ten machine. Multiple alignment files were generated with BWA-MEM2 (v2.2.1). Population structure was analyzed using ADMIXTURE (v1.3.0). Phylogeny trees were constructed with IQ-TREE (v 1.6.12). FST and DXY were calculated by Pixy Package (v 1.2.6). Transcriptome analysis was conducted using the “HISAT2-Stringtie-DESeq” pipeline. Detailed experiments and analyses are available in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Acknowledgments
We thank three reviewers for their valuable comments on an early version of the manuscript. All the computation works were supported by the Big Data Computing Platform for Western Ecological Environment and Regional Development, and the Supercomputing Center of Lanzhou University. This study was supported by the Second Tibetan Plateau Scientific Expedition and Research program (No. 2019QZKK0502), Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB31010300), Fundamental Research Funds for the Central Universities, and International Collaboration 111 Program (BP0719040) to J.L. and E.N. P.C. was funded by the Spanish Ministry of Science and Innovation (Grant No. PID2019-108195GB-I00), the Spanish Aragon Government (Grant No. LMP82_21), and the Spanish Aragon Government-European Social Fund Bioflora (Grant No. A01-23R).
Author contributions
K.L., J.L., E.N., and P.C. designed research; W.M., Y.Y., A.B., and P.C. performed research; W.M., Y.Y., S.L., J.Y., Y.W., S.W., and M.Z. analyzed data; K.L. sampling; and W.M., J.L., E.N., and P.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: C.P., Universite de Fribourg Departement de biologie; L.L., Fudan University; and A.R.T., Washington University in St. Louis.
Contributor Information
Jianquan Liu, Email: liujq@nwipb.cas.cn.
Eviatar Nevo, Email: nevo@research.haifa.ac.il.
Pilar Catalan, Email: pcatalan@unizar.es.
Data, Materials, and Software Availability
The sequencing data are deposited at NCBI, the project number is PRJNA791186 (Brachypodium stacei Genome sequencing and assembly) (47). The genome assembly and main script used in the analyses have been uploaded at Github (https://github.com/Axolotl233/Brachypodium_stacei-EC-genome) (48).
Supporting Information
References
- 1.Darwin C., On the Origins of Species (John Murray, London, UK, 1859). [Google Scholar]
- 2.Schluter D., Ecology and the origin of species. Trends Ecol. Evol. 16, 372–380 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Richards E. J., Servedio M. R., Martin C. H., Searching for sympatric speciation in the genomic era. BioEssays 41, 1900047 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mallet J., Meyer A., Nosil P., Feder J. L., Space, sympatry and speciation. J. Evol. Biol. 22, 2332–2341 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Foote A. D., Sympatric speciation in the genomic era. Trends Ecol. Evol. 33, 85–95 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Arnegard M. E., et al. , Genetics of ecological divergence during speciation. Nature 511, 307–311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michel A. P., et al. , Widespread genomic divergence during sympatric speciation. Proc. Natl. Acad. Sci. U.S.A. 107, 9724–9729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun N., et al. , Sympatric or micro-allopatric speciation in a glacial lake? Genomic islands support neither. Natl. Sci. Rev. 9, nwac291 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charlesworth D., Meagher T. R., Effects of inbreeding on the genetic diversity of populations. Philos. Trans. R. Soc. B Biol. Sci. 358, 1051–1070 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nevo E., “Evolution Canyons model: Biodiversity, adaptation, and incipient sympatric ecological speciation across life: A revisit” in New Horizons in Evolution (Elsevier, 2021), pp. 291–348. [Google Scholar]
- 11.Nevo E., Asian-African and European biota meet at ‘Evolution Canyon’ Israel: Local tests of global biodiversity and genetic diversity patterns. Proc. R. Soc. London, Ser. B Biol. Sci. 262, 149–155 (1995). [Google Scholar]
- 12.Sikorski J., Nevo E., Adaptation and incipient sympatric speciation of Bacillus simplex under microclimatic contrast at “Evolution Canyons” I and II, Israel. Proc. Natl. Acad. Sci. U.S.A. 102, 15924–15929 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hübner S., et al. , Genome differentiation of Drosophila melanogaster from a microclimate contrast in Evolution Canyon, Israel. Proc. Natl. Acad. Sci. U.S.A. 110, 21059–21064 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang L., et al. , Genomic divergence and adaptive convergence in Drosophila simulans from Evolution Canyon, Israel. Proc. Natl. Acad. Sci. U.S.A. 116, 11839–11844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong W., et al. , Genome-wide analysis revisits incipient sympatric and allopatric speciation in a beetle. Isr. J. Ecol. Evol. 67, 69–80 (2020). [Google Scholar]
- 16.Hadid Y., et al. , Sympatric incipient speciation of spiny mice Acomys at “Evolution Canyon”, Israel. Proc. Natl. Acad. Sci. U.S.A. 111, 1043–1048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K., et al. , Sympatric speciation of spiny mice, Acomys, unfolded transcriptomically at Evolution Canyon, Israel. Proc. Natl. Acad. Sci. U.S.A. 113, 8254–8259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., et al. , Sympatric speciation of the spiny mouse from Evolution Canyon in Israel substantiated genomically and methylomically. Proc. Natl. Acad. Sci. U.S.A. 119, e2121822119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W., et al. , Genome architecture and diverged selection shaping pattern of genomic differentiation in wild barley. Plant Biotechnol. J. 21, 46–62 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian C., et al. , Transcriptomes divergence of Ricotia lunaria between the two micro-climatic divergent slopes at “Evolution Canyon” I, Israel. Front. Genet. 9, 506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., et al. , Sympatric speciation of wild emmer wheat driven by ecology and chromosomal rearrangements. Proc. Natl. Acad. Sci. U.S.A. 117, 5955–5963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma T., et al. , Ancient polymorphisms and divergence hitchhiking contribute to genomic islands of divergence within a poplar species complex. Proc. Natl. Acad. Sci. U.S.A. 115, E236–E243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholthof K. B. G., Irigoyen S., Catalan P., Mandadi K. K., Brachypodium: A monocot grass model genus for plant biology. Plant Cell 30, 1673–1694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catalán P., López-Álvarez D., Bellosta C., Villar L., Updated taxonomic descriptions, iconography, and habitat preferences of Brachypodium distachyon, B. stacei, and B. hybridum (Poaceae). An. Jard. Bot. Madrid 73, e028 (2016). [Google Scholar]
- 25.Gordon S. P., et al. , Gradual polyploid genome evolution revealed by pan-genomic analysis of Brachypodium hybridum and its diploid progenitors. Nat. Commun. 11, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiposha V., Catalán P., Olonova M., Marques I., Genetic structure and diversity of the selfing model grass Brachypodium stacei (Poaceae) in Western Mediterranean: Out of the Iberian Peninsula and into the islands. PeerJ 8, e2407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han F., et al. , Gene flow, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence among Darwin’s finches. Genome Res. 27, 1004–1015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q., et al. , The putative auxin efflux carrier OsPIN3t is involved in the drought stress response and drought tolerance. Plant J. 72, 805–816 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Kumar M., Choi J., An G., Kim S.-R., Ectopic expression of OsSta2 enhances salt stress tolerance in rice. Front. Plant Sci. 8, 316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang J., et al. , Heat shock factor OsHsfB2b negatively regulates drought and salt tolerance in rice. Plant Cell Rep. 32, 1795–1806 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Nambara E., Marion-Poll A., Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 56, 165–185 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Zhou H., et al. , Comparative analysis of heat-tolerant and heat-susceptible rice highlights the role of OsNCED1 gene in heat stress tolerance. Plants 11, 1062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huo H., Dahal P., Kunusoth K., Mccallum C. M., Bradford K. J., Expression of 9-cis-epoxycarotenoid dioxygenase4 is essential for thermoinhibition of lettuce seed germination but not for seed development or stress tolerance. Plant Cell 25, 884–900 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi Y., Yamamoto S., Minami H., Kagaya Y., Hattori T., Differential activation of the rice sucrose nonfermenting1-related protein kinase2 family by hyperosmotic stress and abscisic acid. Plant Cell 16, 1163–1177 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Islam M. A., Du H., Ning J., Ye H., Xiong L., Characterization of Glossy1-homologous genes in rice involved in leaf wax accumulation and drought resistance. Plant Mol. Biol. 70, 443–456 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Domínguez M., et al. , The impact of transposable elements on tomato diversity. Nat. Commun. 11, 4058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill W. G., Robertson A., The effect of linkage on limits to artificial selection. Genet. Res. (Camb) 8, 269–294 (1966). [PubMed] [Google Scholar]
- 38.Decena M. A., et al. , Comparative genomics, evolution, and drought-induced expression of dehydrin genes in model Brachypodium grasses. Plants 10, 2664 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher L. H. C., et al. , Linking dynamic phenotyping with metabolite analysis to study natural variation in drought responses of Brachypodium distachyon. Front. Plant Sci. 7, 1751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kossover O., Frenkel Z., Korol A., Nevo E., Genetic diversity and stress of Ricotia lunaria in “Evolution Canyon”, Israel. J. Hered. 100, 432–440 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Hartfield M., Glémin S., Limits to adaptation in partially selfing species. Genetics 203, 959–974 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cruickshank T. E., Hahn M. W., Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Bolnick D. I., Fitzpatrick B. M., Sympatric speciation: Models and empirical evidence. Annu. Rev. Ecol. Evol. Syst. 38, 459–487 (2007). [Google Scholar]
- 44.Savolainen V., et al. , Sympatric speciation in palms on an oceanic island. Nature 441, 210–213 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Barluenga M., Stölting K. N., Salzburger W., Muschick M., Meyer A., Sympatric speciation in Nicaraguan crater lake cichlid fish. Nature 439, 719–723 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Huber C. D., Nordborg M., Hermisson J., Hellmann I., Keeping it local: Evidence for positive selection in Swedish Arabidopsis thaliana. Mol. Biol. Evol. 31, 3026–3039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mu W., et al. , PRJNA791186 (Brachypodium stacei Genome sequencing and assembly). Data for “Scattered differentiation of unlinked loci across the genome underlines ecological divergence of the selfing grass Brachypodium stacei.” NCBI. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA791186/. Accessed 19 October 2023. [DOI] [PMC free article] [PubMed]
- 48.Mu W., et al. , Data for “Scattered differentiation of unlinked loci across the genome underlines ecological divergence of the selfing grass Brachypodium stacei.” Github. https://github.com/Axolotl233/Brachypodium_stacei-EC-genome. Accessed 19 October 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Data Availability Statement
The sequencing data are deposited at NCBI, the project number is PRJNA791186 (Brachypodium stacei Genome sequencing and assembly) (47). The genome assembly and main script used in the analyses have been uploaded at Github (https://github.com/Axolotl233/Brachypodium_stacei-EC-genome) (48).