

Significance
Network medicine has been used to quantify disease mechanisms, comorbidities, and treatments, but most approaches have ignored interactions mediated by noncoding RNAs (ncRNAs). This study systematically combines experimentally confirmed ncRNA and protein–protein interactions to construct a comprehensive network of all physical interactions in the human cell. The inclusion of ncRNA increases the number of genes and interactions in the interactome and enhances the ability to identify disease modules and predict comorbidity patterns between diseases. Ultimately, this study shows that including noncoding interactions improves the breadth and accuracy of network medicine.
Keywords: noncoding RNA, network medicine, network science, miRNA, ncRNA
Abstract
Network medicine has improved the mechanistic understanding of disease, offering quantitative insights into disease mechanisms, comorbidities, and novel diagnostic tools and therapeutic treatments. Yet, most network-based approaches rely on a comprehensive map of protein–protein interactions (PPI), ignoring interactions mediated by noncoding RNAs (ncRNAs). Here, we systematically combine experimentally confirmed binding interactions mediated by ncRNA with PPI, constructing a comprehensive network of all physical interactions in the human cell. We find that the inclusion of ncRNA expands the number of genes in the interactome by 46% and the number of interactions by 107%, significantly enhancing our ability to identify disease modules. Indeed, we find that 132 diseases lacked a statistically significant disease module in the protein-based interactome but have a statistically significant disease module after inclusion of ncRNA-mediated interactions, making these diseases accessible to the tools of network medicine. We show that the inclusion of ncRNAs helps unveil disease–disease relationships that were not detectable before and expands our ability to predict comorbidity patterns between diseases. Taken together, we find that including noncoding interactions improves both the breath and the predictive accuracy of network medicine.
A key goal of post-genomic medicine is to translate the detailed inventory of cellular components and their disease-related mutations, offered by genomics, into mechanistic insights into disease causation and progression, ultimately resulting in novel treatments. To achieve this, we must catalog the physical interactions between all cellular components, arriving at an accurate and predictive map of the human subcellular network. Network medicine, a discipline whose goal is to exploit the predictive power of subcellular networks (1), has already improved our understanding of disease classification (2) and progression (3), disease–disease comorbidities (4), similarities (4), and treatments (5) and offered tools to identify drug repurposing opportunities (6, 7) and drug combinations (8). Some of these tools have already entered the clinical practice, resulting in network-based diagnostic tools currently used by doctors to improve treatment outcomes for rheumatoid arthritis (RA) patients (9) and the drug repurposing opportunities identified during the COVID-19 pandemic (6). These advances relied on maps of experimentally confirmed protein–protein interactions (PPI) and supported multiple foundational discoveries, like the tendency of proteins associated with the same disease to be co-localized in the same neighborhood of the PPI network (4), the finding that diseases with similar phenotypes lie in similar regions of the interactome (4) and the discovery that the network-based distance of drugs to a disease module affects drug efficacy (5, 6).
However, the current interactome maps ignore an important component of subcellular dependency, the regulatory interactions induced and mediated by noncoding RNAs (ncRNAs). In-depth transcriptome sequencing estimates that while approximately about 74% of the human genome is transcribed (10), only 2 to 3% of the human genome encodes for proteins (10–13), meaning that the remaining transcripts represent different classes of noncoding elements (13). These ncRNAs (Fig. 1A) contribute to multiple biological functions, from the maintenance and regulation of gene expression to pre- and post-processing of mRNAs, splicing, decoding mRNAs into amino acids, and the control of gene expression (14–19), ultimately contributing to multiple diseases (20).
Fig. 1.

The role of ncRNA in gene regulation and connection to the human interactome. (A) The modern central dogma of biology. A DNA strand showing the transcription process: miRNAs, lncRNAs, and mRNAs are all transcribed from the DNA; however, only processed mRNAs are translated into proteins. (B) Interaction between ncRNAs. miRNAs can bind to lncRNAs, mRNAs, and proteins. When miRNAs interact with mRNAs and lncRNAs they regulate (by activating or repressing) the gene expression process. lncRNAs can also bind to miRNAs, mRNAs, and proteins. (C) A network of protein interactions. Proteins interact with one another, forming a protein–protein interaction network. Some proteins act as TFs, which regulate gene expression. The PPI only accounts for binding interaction among protein-coding genes. (D) A Network of all interactions. ncRNAs and protein-coding RNAs interact with each other, forming a densely connected network, the PPI & NCI, which contains multiple types of physical interactions from different genomic elements. (E) PPI & NCI. Each edge on the network represents the relative frequency of all respective interactions across different element types. The PPI is a subgraph of the PPI & NCI, which only accounts for protein-coding genes and their interactions; TFs and Proteins interact with each other, responsible for 33% of the interactions in the PPI & NCI network, showing that even though protein interactions play a big role on the network, their interaction with other groups is also important. The majority of interactions occur between miRNAs and protein-coding genes and TFs. While lncRNAs interact with protein-coding genes and other TF, they interact with few other elements.
Given the important role these regulatory processes play in disease, a quantitative understanding of disease requires an accurate and comprehensive map of all physical interactions, from the interactions between the proteins, to interactions mediated by noncoding elements (Fig. 1B). The mapping and characterization of the network structure that contains both coding and noncoding elements is necessary to expand the potential of network medicine, leading to better treatments, diagnoses, and ultimately personalized therapies.
Here, we respond to this challenge by developing a comprehensive map of subcellular networks that systematically integrates information on ncRNA mediated interactions with the PPI network, resulting in a noncoding interactome (NCI). We find that the inclusion of ncRNAs increases the number of nodes by 46% and the number of links by 107% compared to the currently used PPI-based interactome. Most importantly, we find that this expansion allows us to identify disease modules for 132 diseases that lacked a statistically significant module before; hence, it could not be previously explored with the tools of network medicine. Finally, we show that the expanded interactome improves the prediction of disease–disease relationships, offering more accurate comorbidity predictions and advances that ultimately will lead to better prevention and personalized medicine.
Results
PPI Network.
The human protein–protein interactome (Fig. 1C) was derived from 21 public databases containing different types of experimentally derived PPI data (6): 1) binary PPIs, derived from high-throughput yeast-two hybrid (Y2H) experiments (HI-Union), three-dimensional (3D) protein structures (Interactome3D, Instruct, Insider) or literature curation (PINA, MINT, LitBM17, Interactome3D, Instruct, Insider, BioGrid, HINT, HIPPIE, APID, and InWeb); 2) PPIs identified by affinity purification followed by mass spectrometry present in BioPlex2, QUBIC, CoFrac, HINT, HIPPIE, APID, LitBM17, and InWeb; 3) kinase–substrate interactions from KinomeNetworkX, and PhosphoSitePlus; 4) signaling interactions from SignaLink and InnateDB; and 5) regulatory interactions derived by the ENCODE consortium. We used the curated list of PSI-MI IDs provided by Alonso-López et al. (21) to differentiate binary interactions among the several experimental methods present in the literature-curation databases. Specifically, for InWeb, interactions with curation score <0.175 (75th percentile) were not considered. All proteins were mapped to their corresponding Gene Symbol (NCBI) and proteins that could not be mapped were removed. As each database contributes with interactions between a different set of proteins (SI Appendix, Table S1), the resulting PPI network contains 536,965 interactions between 18,217 proteins (Fig. 1E). Interactions containing at least one ncRNA transcript were included in the NCI.
NCI Network.
The most studied ncRNAs are microRNAs (miRNAs), that contain ~22 nucleotides (18) and mainly act at the post-transcriptional level (19), involved in mRNA cleavage, activating or repressing translation (17, 18). The human genome accounts for approximately 2,300 miRNAs (22), each with hundreds of targets, which together regulate 10 to 30% of all genes (23). miRNA recognizes its mRNA targets by base-pairing the miRNA seed region (containing 2 to 8 nucleotides) to the complementary region on the targeted mRNA (24) (Fig. 1B). Playing a similar role as Transcription Factors (TFs), miRNAs form an intertwined network (25) that affects disease development (26) as documented in asthma (27) or schizophrenia (28), and mutated or dysregulated miRNAs are associated with the lack of function in neurogenesis (29).
Long ncRNAs (lncRNAs), another family of nonprotein-coding RNAs, that exceed 200 nucleotides (30), present a poli-A tail and can be spliced. Even though only a small number of lncRNAs are well characterized (31), they are involved in a wide range of biological functions, from X-chromosome inactivation (32, 33), to imprinting (34, 35), and often interact with proteins (36–38)—acting as a TF, and sponges for miRNA (39), bind to chromatin (40) and enhancers (41). lncRNAs can bind to both RNAs and proteins and hence are classified into protein-focused and RNA-focused (31) (Fig. 1B). Moreover, lncRNAs are also associated with multiple diseases (42), cancers (43), autoimmune neuropathies (25), and neurodegenerative diseases (44), and the lncRNA CRNDE has been identified as a promising target for the therapeutic treatment of prostate cancer (45).
To construct the human NCI (Fig. 1D), we combine nine publicly available databases that collect and curate experimentally derived noncoding interactions: 1) miRNA-targets, derived from reporter assay, western blot, microarray, and next-generation sequencing experiments from mirTARbase; 2) miRNAs and lncRNAs interactions validated using Cross-linking and immunoprecipitation (CLIP-seq), Argonaute-crosslinking and immunoprecipitation (AGO-CLIP), Chromatin Isolation by RNA purification (ChIRP-seq), High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) and photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) from lncBook, NPinter4, DIANA Tools, RAIN, and lncRNome; 3) RNA–RNA interactions validated from transcriptome-wide sequencing-based experiments [PARIS (psoralen analysis of RNA interactions and structures), SPLASH (Sequencing of Psoralen crosslinked, Ligated, and Selected Hybrids), LIGRseq (LIGation of interacting RNA followed by high-throughput sequencing), and MARIO (Mapping RNA interactome in vivo)], and targeted studies [RIA-seq (RNA interactome analysis, followed by deep sequencing), RAP-RNA (RNA Antisense Purification to systematically map RNA-RNA interactions), and CLASH (cross-linking, ligation, and sequencing of hybrids)] from RISE; 4) Literature curated from miRNet and miRecords. Additionally, we include any PPI-derived interaction involving at least one ncRNA.
As each of the datasets we used as input relies on a different degree of evidence, we retained only experimentally validated interactions. These include data from DIANA Tools (46), that provides target prediction from algorithms and databases of experimentally verified miRNA targets on coding and ncRNAs. lncBook (47) contains experimental and predicted information on interactions of lncRNAs to proteins and miRNAs. lncRNome (48) provides predicted and experimentally validated interactions from lncRNAs and other RNAs. mirTARbase (49, 50) is a collection of experimentally validated miRNAs and their targets. miRecords (51) is a manually curated database of experimentally validated miRNA-target interactions. miRNet (52) aggregates information from miRTarBase v8.0, TarBase v8.0, and miRecords and allows for the selection of experimentally validated miRNA-targets. NPinter4 (53) contains only experimentally validated interactions from ncRNA to DNA, TF, proteins, and other RNAs. RAIN (54) contains experimentally validated and predicted interactions. RISE (55) focuses on RNA–RNA interactions, which come from transcriptome-wide sequencing-based experiments. A detailed description of the databases can be found in SI Appendix, section 2.1. Note that, during the construction of the PPI, some databases reported protein and ncRNA binds, and we included those interactions only in the NCI.
Given our focus on experimentally validated interactions between protein-coding or noncoding genes, we did not include databases limited to predicted or literature-mined interactions without experimental validation, such as mirwalk, TargetScan, PicTar, TargetMiner, TargetScanVert, miRDB, and microrna.org. Finally, several other classes of ncRNAs can help maintain the homeostasis of the cell. For example, small nuclear RNAs help pre-process mRNA, performing splicing, or intron removal; Transfer RNAs (tRNAs) help decode mRNAs into peptides or proteins; Ribosomal RNAs (rRNAs) are involved in protein translation; housekeeping RNAs, such as rRNAs, can carry modifications (e.g. methylations and pseudouridylations), guided by small nucleolar RNAs (16), and small double-stranded RNAs mediate post-transcriptional gene silencing of mRNAs, via RNA interference. Currently, we lack databases that curate their interactions with other cellular components, limiting our ability to systematically explore their role.
Network Analysis.
We begin by constructing two networks: i) the protein–protein interaction (PPI network), which contains 536,965 interactions between 18,217 protein-coding genes, and ii) the joint PPI & NCI network, which has 26,575 genes [18,358 coding, 2,134 lncRNA, 1,650 miRNAs, 3,429 pseudogenes and 1004 other ncRNAs such as piRNA, siRNA (small interfering RNA), tRNAs] connected by 1,114,777 binding interactions (Fig. 1E). The inclusion of ncRNAs increases the number of nodes by 46% and the number of links by 107%, compared to the PPI. The final interactome is fairly complete, containing 94,5% of all human proteins, 99.6% of all TFs, 86,3% of all miRNAs, and 38.5% of all lncRNAs transcripts (SI Appendix, Fig. S5). The inclusion of ncRNAs in the network increases the diameter of the PPI from 7 to 9, mainly because of the larger number of genes, also inducing an increase in the average shortest path length from 2.66 (PPI) to 2.79 (PPI & NCI; SI Appendix, Table S2). We have also analyzed the degree distribution of the PPI and the PPI & NCI networks, finding that while the inclusion of the NCI does not affect the scale-free nature of the network (56), it does alter the coefficients of the degree exponent, which is crucial for determining the properties of the network. Specifically, we find that the degree exponent for the PPI & NCI network is , less than the degree exponent for the PPI network, (SI Appendix, section 2.3), indicating that the NCI increases the role of the hubs, leading to a more degree-heterogeneous network.
While in the PPI proteins interact with 30 [12; 64] [median value (interquartile range)] other proteins, in the PPI & NCI network, the median degree increases to 54 [20; 107]. A TF in the PPI interacts with 48 proteins [20; 102] and its degree increases to 90 [41; 168] after the inclusion of the ncRNAs. miRNAs and lncRNAs, absent in the PPI, are connected to 52 [16; 224.75] and 3 [1; 6] elements (miRNAs, lncRNAs, proteins, TFs, or other ncRNAs) respectively (SI Appendix, Table S3). In the PPI & NCI network, 34.8% of the interactions are between miRNAs and protein-coding genes and 16% between miRNAs and TFs, and lncRNAs are responsible for less than 1% of the combined interactions (Fig. 1E). In summary, we find that miRNAs are responsible for the majority of the noncoding interactions with protein-coding genes, TFs, and pseudogenes, as well as 40% of the lncRNAs interactions (Fig. 1E), confirming the central role miRNAs play in the regulation of the underlying cellular network.
Disease–Disease Associations Under the Light of ncRNAs.
Relating genes and their mutations to diseases is the central question of post-genomic medicine, drug-based therapeutics, drug discovery, and drug repurposing. Traditionally, genes were associated with traits via positional cloning (57), but recently most gene–disease associations come from genome-wide association studies (GWAS) followed by functional analysis to evaluate the effect of a single nucleotide polymorphism (SNP) or gene on the trait. Genes can be also associated with diseases using differential gene expression, epigenetic, susceptibility and other association studies, and literature-based curation (followed or not by an expert review). We assembled a Gene Disease Association (GDA) database by retrieving evidence of disease associations from 15 well-curated databases [ClinGen, ClinVar, CTD, Disease Enhancer, DisGeNET, GWAS Catalog, HMDD (58), lncBook, LncRNA disease, LOVD, Monarch, OMIM, Orphanet, PheGenI, and PsyGeNet; see Material and Methods and SI Appendix, section 2.2 and Table S2].
We limit our exploration to diseases with at least 10 gene associations, arriving at a database with 861 diseases and 13,216 disease-associated genes, of which 10,764 are protein-coding and 2,452 encode ncRNAs. The diseases with most miRNAs associations are carcinoma hepatocellular (380 miRNAs), breast neoplasms (372 miRNAs), and colorectal neoplasms (347 miRNAs). Diseases with high lncRNAs involvement include astrocytoma (311 lncRNAs), breast neoplasms (114 lncRNAs), and stomach neoplasms (112 lncRNAs). We find that 250 of the 861 diseases are enriched for ncRNAs (proportions test; P-adj < 0.05, FDR corrected), i.e., they have more associated ncRNAs than we would expect by chance.
Previous results show that proteins associated with a specific trait, phenotype, disease, or biological process tend to cluster in the same topological neighborhood of the PPI network (59), forming a sub-network known as the disease module. The phenomena that proteins linked to the same phenotype have a strong tendency to interact with each other (and to cluster in the same network neighborhood) has been documented multiple times (1, 60–63) and the occurrence of such phenomena is the basis of disease module, representing a connected sub-network of the disease-associated genes with a potential mechanistic link to a particular phenotype. The size of the largest connected component (LCC) of this sub-network measures the magnitude of the disease module, and its statistical significance is obtained by deriving the LCC distribution based on the random selection of the disease-associated genes. It is important to note that the size of the LCC must be accompanied by its statistical significance, as even a large LCC could emerge by chance. As a second measure, here we introduce the relative LCC (rLCC), defined as the ratio between the size of LCC and the number of genes associated with the disease. The rLCC captures the completeness of the disease module, allowing comparison across diseases with different numbers of genes. For example, schizophrenia has 1,458 associated genes (1,292 coding, 148 ncRNAs) of which 1,364 form an LCC in the PPI & NCI (P-adj < 0.05, FDR corrected), resulting in an rLCC of 93,55%. In contrast, even with the inclusion of ncRNAs, obsessive-compulsive disorder, with 92 genes (81 protein-coding and 11 noncoding) has an LCC of 8 in the PPI & NCI, resulting in an rLCCPPI & NCI of only 8.69%, indicating that the corresponding disease module is highly incomplete.
In the following, we focus on three diseases, RA, Chron’s disease (CD), and pre-eclampsia (PE), to illustrate the value of adding the NCI to the interactome. RA is a multisystemic, chronic inflammatory disease characterized by destructive synovitis, erosive arthritis, progressive articular damage, and systemic organ involvement (64–66), which can lead to decreased functional capacity and quality of life, increased morbidity and mortality. RA is associated with 391 genes, (302 protein-coding, 66 miRNAs, 14 lncRNAs, and 9 other ncRNAs). While there is a significant disease module (P-adj < 0.05, FDR corrected) in both the PPI and the PPI & NCI, the PPI’s disease module accounts for only 189 disease-associated genes (rLCCPPI: 62%), whereas the PPI & NCI includes 345 disease-associated genes (Fig. 2A), increasing the rLCCPPI & NCI to 88%.
Fig. 2.
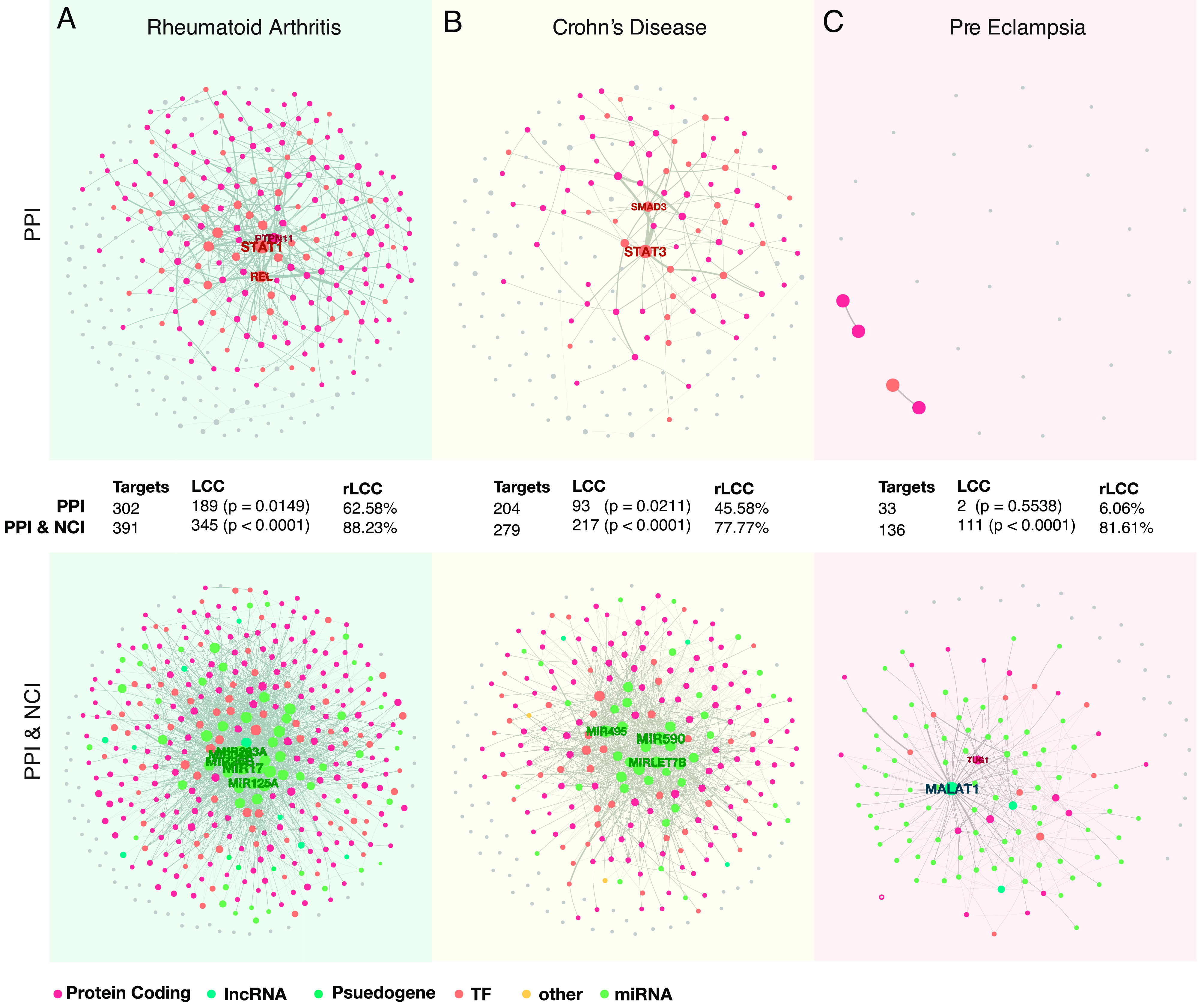
Disease modules for RA, CD, and PE. (A) RA disease module. In RA, both PPI and the PPI & NCI networks do form a significant LCC; however, the inclusion of ncRNAs into the network allows a much better retrieval of disease genes, increases the rLCC from rLCCPPI: 62% to rLCCPPI & NCI: 88%. Gray nodes are not in the LCC of the complete interactome, while the colored nodes are present. (B) CD disease module. Both PPI and the PPI & NCI identify significant disease modules. However, by including ncRNAs into the disease module of CD, the rLCC increases from rLCCPPI: 45% to rLCCPPI & NCI: 77%, increasing the disease gene retrieval. (C) PE disease module. PE shows that the inclusion of ncRNAs can change our ability to identify significant disease modules. In the PPI, we are unable to define and identify a disease module; however, when in the PPI & NCI, a disease module emerges (rLCCPPI: 6%, and rLCCPPI & NCI: 81%).
CD is an autoimmune disease, that causes inflammation of the digestive tract, which can lead to abdominal pain, severe diarrhea, fatigue, weight loss, and malnutrition. CD is associated with 279 genes (204 protein-coding, 47 miRNAs, and 14 lncRNAs). It has a significant disease module in both networks; however, the size of the LCC more than doubles with the inclusion of the ncRNA (LCCPPI: 93; LCCPPI & NCI: 217), and the proportion of disease-associated genes also increases greatly (from rLCCPPI: 45% to rLCCPPI & NCI: 77%) (Fig. 2B), collecting many more known disease genes.
Finally, PE is characterized by persistent high blood pressure during pregnancy or postpartum, and in rare cases, it can progress to severe PE very quickly, which can lead to the death of the mother and the baby. PE can also lead to premature birth, malnutrition, and lack of oxygen in the womb; adults whose mothers had PE have higher chances of developing diabetes, congestive heart failure, and hypertension (67). PE has no statistically significant disease module in the PPI network, hence, previously we could not apply network medicine tools to explore this disease. Indeed, PE is associated with 136 genes; the majority of which (95) encode miRNAs. It does, however, have a significant module in the combined PPI & NCI network (Fig. 2C). The PPI network accounts for only 33 of the 136 genes associated with the disease, of which only 2 are part of the LCC, while the PPI & NCI contains 136 genes of which 111 are in the LCC. Similarly, the ratio of disease genes found in the disease module jumps from 6% (rLCCPPI) to 81% (rLCCPPI & NCI), confirming the key role miRNAs play in regulation of PE. In other words, the PPI, historically the starting point of network medicine studies, provides a highly incomplete map of the PE disease module. However, the inclusion of ncRNAs allows us to now detect the disease module, opening up the possibility to explore the disease using the tools of network medicine.
To generalize beyond RA, CD, and PE, we calculated the LCC, rLCC, and the significance of the disease module for all 861 diseases in the PPI and in the PPI & NCI. Of the 861 diseases, 505 have a statistically significant LCC in the PPI network and 602 have a statistically significant LCC in the PPI & NCI (permutation test, N = 1,000, P-adj < 0.05, FDR corrected; Fig. 3A). Taken together, we find that for 132 diseases the identification of a statistically significant disease module cannot be achieved without the inclusion of ncRNA-based interactions. We also find that the inclusion of ncRNAs decreases the LCC P-value (FDR corrected) for 522 diseases, hence increasing our confidence over the observed disease module (Fig. 3 B, C, and E) and also increases the median size of the significant LCCs (Wilcoxon test, P < 0.0001; Fig. 3D). Finally, we find that the rLCC in the PPI & NCI increases for 430 diseases by 26.6% on average, indicating that the NCI considerably reduces the number of unconnected disease genes by linking many previously isolated components to the LCC.
Fig. 3.
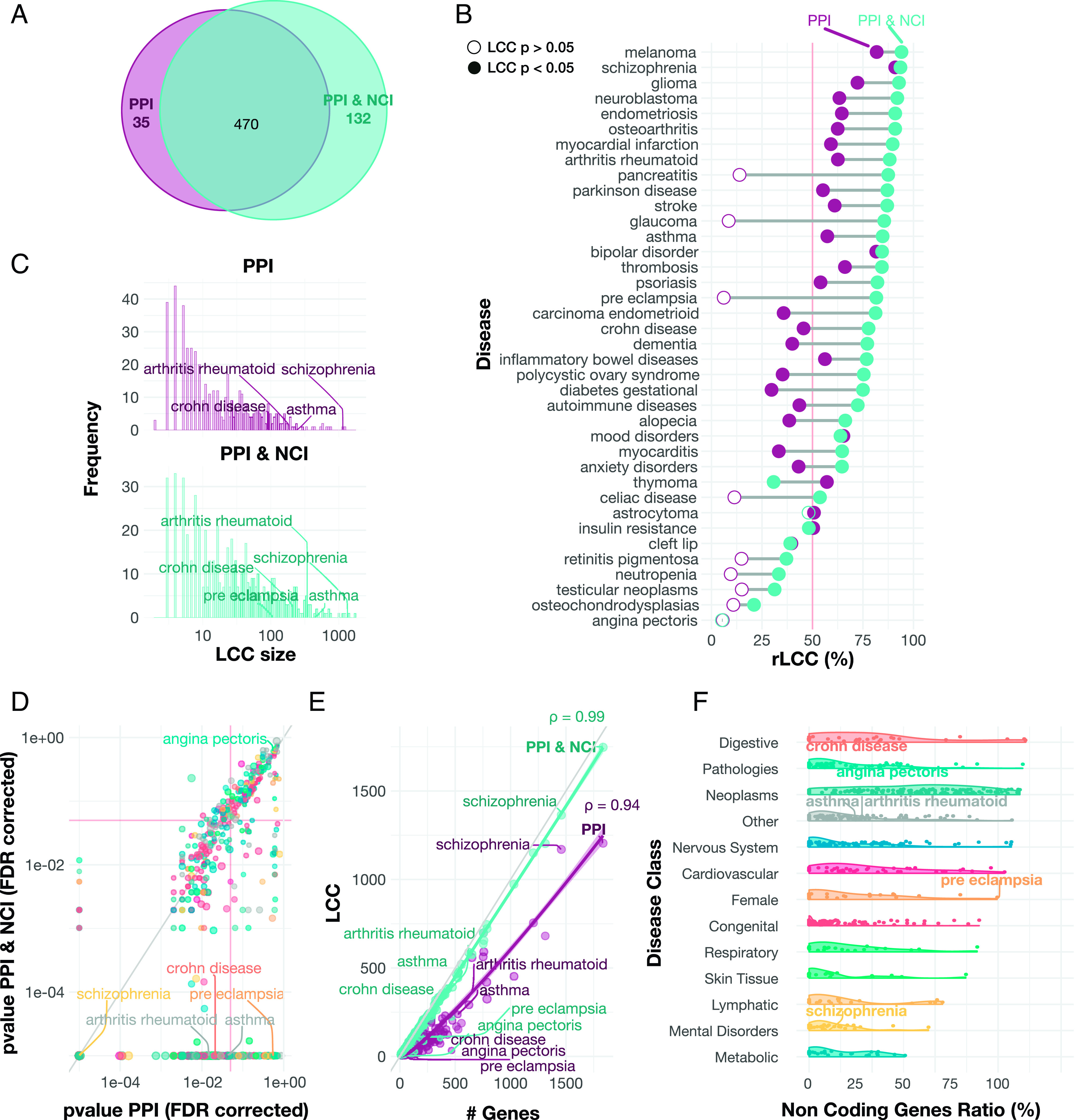
Uncovering gene–disease associations. (A) Overlap of disease modules identification. Venn diagram representing the number of significant diseases in each network. In the PPI and the PPI & NCI, we can identify 470 disease modules, while solely the PPI identifies 35 disease modules (P < 0.05, FDR corrected). However, the PPI & NCI enables us to identify disease modules for 132 diseases that could not be identified previously. (B) rLCC change in the PPI and the PPI & NCI. Each line represents the rLCC size for a disease in the PPI and the PPI & NCI, along with disease-module significance. Empty circles represent diseases that do not have a significant LCC. The rLCC using the PPI & NCI is larger for most of the diseases than the rLCC for the PPI alone. In some cases, i.e, PE and glaucoma, we can identify disease modules that did not exist in the PPI. (C) LCC Size Increases in the PPI & NCI. The histogram depicts LCC distribution for diseases in both PPI and PPI & NCI, the PPI (in purple) shows a distribution heavily shifted to the left, and the PPI & NCI (in turquoise) indicates a distribution that its values are shifting to the right, indicating that the average distribution of the LCCs in the combined network increases. (D) The relationship between the P-value on the PPI and the PPI & NCI. The scatterplot shows the p-values in the PPI and the PPI & NCI for all diseases. On average, P-values of the disease module are smaller on the combined network, suggesting that the inclusion of NC elements improves the identification of the disease modules. The red lines indicate P-value = 0.05. (E) Genes associated with disease and the LCC size. The scatterplot depicts the number of genes associated with a disease, and the disease-module size for both networks. The greater the number of known associated genes, the greater their LCC ( ; , Pearson correlation). We also find that PPI & NCI network has the LCCs closer to the total amount of genes associated with the disease, while the PPI has a smaller LCC compared to the number of genes per disease, suggesting an incompleteness of the disease module. (F) Proportion of noncoding genes. The violin plot shows the percentage of ncRNAs associated with diseases classified in each disease category. We find that Digestive, Pathologies, and Neoplasms are the top three disease categories with the highest ratio of the noncoding genes, suggesting that ncRNAs play a role in the manifestation of those disease categories.
Taken together, we find that including ncRNAs in the PPI increases the size, significance, and disease-gene retrieval of the disease module, expanding the reach of network medicine to a large number of diseases that previously could not be explored using network-based tools. Digestive problems, pathologies, and neoplasm have the highest ratio of ncRNAs (Fig. 3F), indicating that those disease classes benefit most from the inclusion of ncRNAs.
Direct and Indirect Bindings Are Supported by Co-Expression Networks.
Gene co-expression networks are often used to shed light on the molecular mechanisms that underlie biological processes (68). As gene co-expression is driven by the regulatory network, it leads to the hypothesis, supported by previous evidence (69), that interacting proteins are products of genes with higher co-expression, compared to proteins that do not physically interact. Here we extend this hypothesis to noncoding elements, asking if genes connected by noncoding interactions show higher co-expression than expected by chance. In other words, we use gene co-expression to probe the relative strength of the interactions induced by coding and noncoding elements. As bulk RNA-seq is not appropriate for measuring miRNA, due to the lack of poli-A tail in miRNAs, here we derive an indirect physical network that includes co-regulatory interactions (Fig. 4A), driven by the hypothesis that two proteins are co-expressed if they are co-regulated by the same ncRNAs. We distinguish three interactions that could modulate co-expression networks: i) Direct PPI (Fig. 4B), constructed using direct physical interaction between two proteins; ii) Indirect NCI (iNCI; Fig. 4C), connecting two proteins if proteins “A” and “B” are co-regulated by the same ncRNA; iii) Direct & iNCI (Fig. 4D), representing the combination of the PPI and the iNCI network; or iv) For control, we measure co-expression between nodes that have no known physical interaction between them.
Fig. 4.

Physically Interacting genes are co-expressed. (A) Schematics of the complete PPI and NCI network. A noncoding RNA “X” interacts with three proteins “A”, “B” and “C”, and protein “B” binds to protein “D”. (B) The PPI network contain only interactions among proteins, and all interactions containing NC interactions are absent. (C) The induced NCI (iNCI) contains indirect interactions. ncRNA “X” interacts with genes “A” and “B”; therefore, they are co-regulated by the same ncRNA, leading to an indirect interaction. In the same fashion, proteins “A” and “C” and “B” and “C” are also co-regulated by the same ncRNA “X”, inducing a triangle among them. (D) PPI & iNCI includes direct and indirect interactions. Combining the interactions identified in (B and C). (E) Genes with direct or indirect physical binding (PPI, PPI & NCI, or co-regulated by an ncRNA) have higher co-expression values than genes that do not physically interact. The boxplot indicates that the absolute Pearson correlation is higher when there is physical interaction, compared to then nonexisting links, indicating an association between physical binding and strength of co-expression. (F) Co-expression networks can predict physical interactions. We use the correlation values between two transcripts to predict a direct or indirect binding, finding that the inclusion of ncRNAs increases the AUC in the iNCI and the PPI & iNCI.
We compare the measured co-expression for the three different interaction types (PPI, iNCI, and PPI & iNCI), relying on gene co-expression derived from whole blood samples by GTEx (70). We use both Pearson correlation (ρ) and wTO (71, 72) (ω) to ensure that the results are not biased by the methodology. We then compare the absolute co-expression weights (ρ and ω) for the three binding interaction types (PPI, iNCI, and PPI & iNCI, and no interaction). We find that the absolute co-expression weights (ρ and ω) are, on median, higher for all three types of physical interactions (Fig. 4E and SI Appendix, Fig. S7A) compared to the control (Kruskal–Wallis; Dunn’s post hoc test, P-adj < 0.05 Holm method; SI Appendix, Table S5), confirming that two protein-coding genes that interact (directly or indirectly) have higher co-expression compared to genes that do not interact. Most importantly, we find the strength of the co-expression induced by the PPI or by the iNCI to be statistically indistinguishable, indicating the comparable impact of noncoding interactions on co-expression.
The observed higher co-expression values on the physical networks (PPI, iNCI, and PPI & iNCI) prompt us to ask whether the co-expression weights are predictive of physical binding. We, therefore, calculated the area under the ROC (AUROC), which measures the ability to discriminate between binding or no binding, where an AUROC of 0.5 indicates a random choice (i.e., lack of predictive power), while an AUROC of 1 indicates accurate predictions. We find that the AUCs increased from 0.59 in the PPI to 0.63 in the iNCI and PPI & iNCI networks for ρ (Fig. 4F) and from 0.58 PPI to 0.63 in the iNCI and PPI & iNCI networks for ω (SI Appendix, Fig. S7B). In other words, the inclusion of ncRNA-induced indirect interactions improves the accuracy of correlation-based networks to predict physical interactions, demonstrating the important role NCI plays in the interpretability of co-expression patterns.
Disease Comorbidity.
Diseases with similar phenotypes tend to have common genetic roots, as captured by the Jaccard index of genes associated with different phenotypes. In addition, diseases with similar symptoms tend to share their disease network neighborhood (69), a feature captured by the network-based separation of two diseases, and , defined as (4)
where is the average shortest distance from disease to . A negative indicates that two diseases are in overlapping network neighborhoods, while implies that components associated with diseases and are in distinct network neighborhoods.
To illustrate how NCI can help improve our understanding of disease relationships, we focus first on RA and its comorbidities. If we limit the network to the PPI, RA has only one statistically overlapping disease, CD (Fig. 5 A and B—PPI). In contrast, in the extended PPI & NCI network, uncovers statistically significant network-based overlap with CD, ulcerative colitis (UC), inflammatory bowel disease, inflammation, systemic lupus erythematosus (SLE), multiple sclerosis (MS), diabetes mellitus types 1 and 2, asthma, heart failure, stroke, atherosclerosis, and multiple neoplasms (Fig. 5 A and B—PPI & NCI). Most of these diseases predicted to be in the same network neighborhood as RA have known comorbidities to RA, confirmed by clinical evidence. Indeed, RA patients present chronic inflammation (73), often develop SLE, a combination known as rhupus (74, 75); MS is a well-known comorbidity of RA (76), similarly to inflammatory bowel diseases (77) [e.g., UC and CD (78)]. Also, patients with RA have a higher risk of developing diabetes type I (79) due to insulin resistance, and patients with RA often develop diabetes mellitus (80); additionally, patients with asthma have higher risk of developing RA (81, 82). Moreover, patients with RA have reported higher rates of heart failure (83, 84), such as myocardial infarction (83) and stroke (84) in addition to atherosclerosis (85). Higher risk of multiple neoplasms has also been reported in patients with RA treated with anti-TNF drugs (86, 87), interestingly, even though multiple myeloma and colonic neoplasms (88) share the same network neighborhood, they have reported decreased risk in patients with RA (89), suggesting that two diseases in the same network vicinity might also grant protection from each other. Taken together, we find that while the PPI can identify only one comorbidity for RA, most of the clinically documented comorbidities can be detected in the joint PPI & NCI network, indicating that the inclusion of ncRNA interactions is necessary to reveal disease comorbidities.
Fig. 5.

Disease similarity on the two networks. (A) RA disease similarities and possible comorbidities. Two diseases are connected if they have a negative separation (meaning that they co-exist in the same network neighborhood) and if their overlap is significant. RA Disease–Disease network (LCC P < 0.05, < 0, P < 0.05, Jaccard index > 0.05, hypergeometric test P < 0.05; all P values are FDR corrected) is shown for both the PPI and the PPI & NCI. Diseases are colored according to literature references for comorbidity. We find in the PPI that only CD is possible comorbidity, while the PPI & NCI predicts several other comorbid diseases, and most of them have a reported RR greater than 1, indicating comorbidity, few have RR smaller than 1, indicating a protective effect from those diseases. (B) RA comorbidities are also connected. Diseases that have high comorbidity with RA are also connected. Similar to A, we find an expansion of disease associations in the PPI & NCI. The PPI alone identifies two distinct clusters: A neoplasm and an inflammation module. The inclusion of ncRNA into the PPI helps us identify how those two clusters (neoplasm and inflammation) are also interconnected with each other. (C) Complete map of disease–disease relationships. We show the complete disease–disease network, unveiling the comorbidity map between 466 diseases. Each disease with a significant module (full dots) has its node size representing the size of the disease module, and the link width is relative to the normalized absolute value. Note that the PPI & NCI network forms a connected component with all the 213 diseases that have a significant LCC; moreover, we see that neoplasm (represented in blue) are close and form a module. The PPI, in its turn, forms a connected component with only 100 diseases, and the combination of both gives us a component that includes 249 diseases.
As a second case study, we focus on PE, which lacks a disease module in the PPI; hence, we could not predict comorbidities based on the PPI alone. By using the PPI & NCI combination, the top 10 closest diseases with significant topological overlap with PE are atherosclerosis, uterine cervical neoplasms, osteosarcoma, pancreatic neoplasms, glioma, cholangiocarcinoma, multiple myeloma, triple-negative breast neoplasms, heart failure, and carcinoma pancreatic ductal. Indeed, there is clinical evidence that women with PE are at increased risk for atherosclerosis (90) and other cardiovascular diseases such as heart failure (91), women with HPV infection, the main cause for uterine cervical neoplasms, also have increased risks for PE (92), and PE has been associated with an increased risk for several types of cancer (93). In other words, the joint PPI & NCI can accurately predict the known comorbidities of PE.
Finally, we expand our investigation by mapping out the disease–disease relationships that capture the network proximity of all disease pairs. The PPI predicts 543 comorbidity links between 350 diseases, revealing distinct clusters for neoplasm, cardiovascular, and gastrointestinal disease (Fig. 5C—PPI). In the combined PPI & NCI, we find 2,659 pairwise disease links between 466 diseases (Fig. 5C—PPI & NCI). We find that by including ncRNAs into the PPI, we retrieve more complete and biologically more meaningful list of disease–disease interactions, offering a better quantitative understanding of disease similarity and comorbidity, ultimately helping us understand disease progression in patients (3).
The NCI Predicts Relative Risk (RR).
The PPI-based disease–disease separation is a known predictor for disease comorbidity (4), prompting us to ask whether the inclusion of ncRNAs improves not only our ability to detect clinically documented comorbidity in patients but can also help quantify comorbidity by predicting the RR between diseases. We measured the pairwise RR between all disease-pairs, relying on the disease history extracted from 13,039,018 elderly Americans enrolled in Medicare (94). RR estimates the strength of the association between two diseases, so that RR > 1 represents a risk factor, while RR < 1 indicates a protective factor. For example, UC affects 26,432 patients and Crohn’s disease (CD), 24,936. If the two diseases were independent of each other, we would expect only 45 individuals with both diseases. In contrast, we find 1,462 individuals with both UC and CD in our database. In other words, the chance of a patient developing CD is 30.55 times higher (29.0, 32.16; Wald interval, 95% confidence) in a patient with UC compared to a patient who does not have a history of UC, meaning that UC is a risk factor for CD.
Next, we investigate if diseases with a network overlap ( ) have a higher RR, meaning that diseases that are located in the same network neighborhood have a higher risk of being comorbid. We find that negative disease pairs have a statistically higher RR than the ones with positive in both the PPI and the PPI & NCI (Material and Methods and SI Appendix, Fig. S9A). The RR in the PPI is on average 8.6 (se 2.98) for diseases with an < 0 (P < 0.05), and 6.74 (se 0.41) for diseases with an > 0 (P < 0.05). For the PPI & NCI, we observe an increase in the RR average to 9.5 for negative (se 2.93), and a decrease to 6.5 (se 0.41) for positive (SI Appendix, Fig. S9A). The increase of the RR for closer diseases in the PPI & NCI networks indicates, once again, that the inclusion of ncRNAs enhances our ability to quantify comorbidity by predicting the RR for patients, offering a better understanding of the network-based roots of disease progression.
Discussion
Network medicine, with its promise to offer a better mechanistic understanding of diseases (4), their progression (3), comorbidities (4), similarities (4), and treatments, such as drug repurposing (6, 7) and drug combinations (8), traditionally relied on PPI, capturing binding interactions between proteins. Two decades after the Human Genome Project, there is overwhelming evidence that noncoding genes and ncRNAs regulate multiple biological processes and functions, playing important roles in multiple diseases, and hence must be incorporated into the network medicine framework.
Here, we show that the inclusion of ncRNAs into the PPI significantly improves the breath and the predictive power of network medicine. Protein-coding and noncoding genes are intertwined into a densely connected network, hence the inclusion of ncRNAs improves disease module identification and our ability to uncover disease–disease relationships, more accurately predicting the RR for patients. We also show that the rLCC increases when we incorporate the ncRNAs, helping us retrieve more complete disease modules, and offering biologically more interpretable identification of the molecular components contributing to a disease. We find that several neoplasms share the same noncoding neighborhoods, confirming the role ncRNAs play in neoplasm regulation. We also find improved comorbidities for many other diseases after the inclusion of ncRNAs, suggesting that noncoding elements contribute to most disease mechanisms. Disease modules are tissue-specific and are only expressed if the respective disease-genes are expressed as a connected component (95). In our study, we do not explore the tissue specificity of the specific diseases, but rather, consider the completeness of the disease modules in a tissue-independent network. Disease-focused studies should filter the PPI and the PPI & NCI to contain only genes expressed in a particular tissue, cell line, or even disease.
The more accurate disease module detection enabled by the inclusion of ncRNAs can lead to the development and identification of novel drug–targets, that hit closer to the disease module. They also raise the possibility that for some diseases targeting ncRNAs in the disease module may have a better therapeutic potential than targeting proteins. The clinical relevance of such intervention is illustrated by Bevasiranib (96), a siRNA that targets the VEGF-A gene, currently in clinical trial for treating macular degeneration and diabetic retinopathy, or Inclisiran, an LDL cholesterol lowering siRNA that targets PCSK9, the first approved ncRNA-based drug (97, 98). We also found an improved comorbidity prediction when considering ncRNAs, suggesting that the systematic inclusion of noncoding elements can offer a better understanding of disease progression, potentially opening a path toward precision medicine.
Materials and Methods
GDAs.
We surveyed around 130 databases with GDA and selected those that i) were not compiled from other data sources and ii) provided at least one kind of evidence type classified as: Strong (functional evidence using an experimental essay); Weak (GWAS evidence but no experimental validation); Inferred (relying on bioinformatics or SNPs from imputation in GWAS); not compatible [(l)ncRNA, miRNA and other transcripts with or without experimental validation]. For each database we kept the disease name, gene converted to HGNC names (HUGO Gene Nomenclature Committee), and evidence level. At the end, we combined the following data sources: GWAS from ClinGen, ClinVar, CTD, Disease Enhancer, DisGeNET, GWAS Catalog, HMDD (58), lncBook, LncRNA disease, LOVD, Monarch, OMIM, Orphanet, PheGenI, and PsyGeNet (SI Appendix, section 2.2).
We searched for datasets that provide noncoding interactions derived from experimental evidence (ncRNA vs. proteins or ncRNA vs. ncRNA). We kept only databases that provided experimental evidence for their binding interactions and removed all interactions without strong evidence (SI Appendix, section 2). To validate the collected interactions, we assessed each database’s interactions and gene’s overlap.
Combining Data.
All disease names were converted into MeSH terms after a word2vec embedding and all gene IDs into Gene Symbols. Gene names were normalized to HGNC symbol using biomaRt (99), Gene Cards (100), and gene2ensembl from NCBI. Genes were classified into coding, and noncoding (miRNA, ncRNA, etc) based on their classification from Gene Cards. Coding genes were classified as TF according to Perdomo-Sabogal 2019 (101).
To normalize disease names, we first converted all strings to low-case and kept only alphanumeric characters. We next removed diseases with keywords that represented measures (such as “body mass”, “volume”, “count”, “susceptibility”, etc). The renamed diseases were combined into clusters based on their similarity to MeSH C or F terms and synonyms based on a word2vec embedding trained on PUBMED (102). Disease names with a cosine distance lower than 0.8 to a MeSH term or a MeSH synonym were removed, and the term with higher similarity was selected. Diseases are classified into different classes based on the MeSH’s first level of classification. As some diseases may have multiple first-level classifications, we define their first level as a level with more single diseases in it. Let us take RA as an example, which is defined as “Immune System Diseases”, “Musculoskeletal Diseases” and “Skin and Connective Tissue Diseases”, to avoid counting RA three times, we classify it as one of the three classes. We do so by selecting the class with more diseases, meaning that, 69 diseases are classified as Immune System Diseases, 84 are Musculoskeletal Diseases and 78 are “Skin and Connective Tissue Diseases”, in the RA case, the class is therefore classified as Musculoskeletal Diseases. After disease and gene names normalization, we filtered for diseases that have at least five associated genes, with at least one strong, weak, or incompatible experimental evidence.
Network Medicine Tools.
Disease modules were inferred from the gene–disease association curated database, by calculating the LCC of each disease and deriving its P-value from the density of 1,000 simulations of a permutation test. The P-values are computed based on the empirical distribution derived from selecting random genes in the network and calculating the LCC size. The gene set is of the same size as the original set of genes, following a uniform distribution, i.e., each gene has the same probability of being associated with a disease. We can assume a uniform distribution here because genes are associated with diseases a high throughput setup, such as GWAS, differential gene expression, epigenetic, etc.; in other words, genes are not pre-filtered for the association study. Results using both degree-preserving and nondegree-preserving randomization are presented in SI Appendix.
Diseases separation was calculated using the measure proposed by Menche et al. (4). Separation significance is calculated by resampling 1,000 times the genes in each disease, and calculating the , one-sided P-values are obtained as approximations of the area under the empirical density curve from to the found .
To assess the disease similarity network, we selected overlapping elements given their disease separation (4) ( ) < 0, significance of the (permutation test, N = 1,000; P < 0.05), significance of the gene overlap (hypergeometric test, P-adj < 0.05; FDR corrected) and Jaccard index > 0 for each disease pair that forms a significant LCC using the two pre-defined networks (PPI, and PPI & NCI).
LCCs and were estimated using the NetSci R package.
Drug–Targets and Gene Co-Expression.
Drug–target interactions were retrieved from DrugBank (103) (version 5.1.9), keeping all target types (polypeptides, enzymes, carriers, and transporters). We filtered for drugs that have at least one drug–target described.
We analyzed gene expression in whole blood samples from GTEx (70) by selecting genes that are present in PPI & NCI database and have expression levels between the 10th and 90th quantiles. This approach enabled us to remove any outliers. We further filtered the GTEx data to focus on blood samples and retained only those genes that were expressed in at least 80% of the selected samples and had a SD greater than 0.01. Gene co-expression networks were inferred using the association between pairwise gene expression, measured using RNAseq or microarray, and the association is often derived from a correlation, such as Pearson correlation, or a transformation, such as the Weighted Topological Overlap (wTO) (71, 72). Gene expression was accessed using whole blood samples from GTEx (70), and the co-expression was constructed using Pearson Correlation and the wTO, from the wTO R Package (71), which calculates the co-expression between two genes based on their normalized correlation and further removes false positives.
Direct and Indirect Physical Interactions.
We created an indirect-physical interaction network, which identifies the co-regulation of any two protein-coding genes by the same noncoding gene. For that, we constructed a bipartite network, based on the PPI & NCI, where one set of nodes represented noncoding genes and the other set represented the protein-coding genes. From this network, we created the projection on the protein-coding network, identifying if two protein-coding genes are targets of the same ncRNAs. We combine the original PPI with the indirect-physical interaction network.
Statistical Analysis and RR.
Statistical analyses were performed using the R environment. Tests and CIs have been reported along with their P-values. All tests, unless stated, were FDR-corrected.
To access the RR of two diseases (comorbidities), we used the data from patients enrolled in MediCare (4, 94). To analyze the network of diseases represented by 3-digit codes from the International Classification of Diseases (ICD), we converted these codes into MeSH codes using the procedure outlined in Material and Methods: Combining Data. Since each MeSH code can correspond to multiple ICD codes, we combined patients with different ICD codes that mapped to the same MeSH code into the identified MeSH category. This enabled us to examine the data at a higher level of abstraction and identify patterns and connections between different disease categories. Relative risk and CI were assessed using the RelRisk function from the DescTools R package, using the Wald test.
To access the difference between the RR and the , we select RR > 1 with statistical significance (P < 0.05, FDR corrected) and focus on diseases that affect at least 5% of the population, avoiding inflated RR. Differences between groups were assessed using the Mann–Whitney test.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Italo F. do Valle and Xiao Gan for fruitful discussions. This research was supported in part by a NIH award 1P01HL132825, a Department of Veterans Affairs award Contract No. 36C24120D0027, and by Scipher Inc. Agreement 21-C-01472. A.-L.B. is supported by thethe European Union’s Horizon 2020 research and innovation programme under grant agreement No 810115 – DYNASNET.
Author contributions
D.M.G. and A.-L.B. designed research; D.M.G. performed research; D.M.G. contributed new reagents/analytic tools; D.M.G. analyzed data; and D.M.G. and A.-L.B. wrote the paper.
Competing interests
A.-L.B. is co-scientific founder of Scipher Medicine, Inc., which applies network medicine strategies to biomarker development and personalized drug selection, and founder of Naring, Inc. which applies data science to health.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The data and code utilized in the preparation of this manuscript are publicly accessible and can be obtained from the following sources: Code and Data Repository: The code and data used for data analysis and generating the results presented in this paper is available on GitHub at the following repository: https://github.com/Barabasi-Lab/NonCoding (104). Online Tool for Disease–Disease Exploration: An interactive online tool for exploring disease–disease relationships using both interactomes is accessible at the following web address: https://deisygysi.shinyapps.io/Network_NCI/ (105).
Supporting Information
References
- 1.Barabási A. L., Gulbahce N., Loscalzo J., Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loscalzo J., Kohane I., Barabasi A. L., Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol. Syst. Biol. 3, 124 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.do Valle I. F., et al. , Network-medicine framework for studying disease trajectories in U.S. veterans. Sci. Rep. 12, 1–10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menche J., et al. , Uncovering disease-disease relationships through the incomplete interactome. Science 347, 841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guney E., Menche J., Vidal M., Barábasi A.-L. L., Network-based in silico drug efficacy screening. Nat. Commun. 7, 10331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gysi D. M., et al. , Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. U.S.A. 118, e2025581118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovács I. A., et al. , Network-based prediction of protein interactions. Nat. Commun. 10, 1240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng F., Kovács I. A., Barabási A. L., Network-based prediction of drug combinations. Nat. Commun. 10, 1197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellors T., et al. , Clinical validation of a blood-based predictive test for stratification of response to tumor necrosis factor inhibitor therapies in rheumatoid arthritis patients. Netw. Syst. Med. 3, 91–104 (2020). [Google Scholar]
- 10.Djebali S., et al. , Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattick J. S., Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2, 986–991 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Human Genome Sequencing Consortium, Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 13.St Laurent G., Laurent Y., Kapranov P., Dark matter RNA illuminates the puzzle of genome-wide association studies. BMC Med. 12, 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hombach S., Kretz M., Non-coding RNAs: Classification, biology and functioning. Adv. Exp. Med. Biol. 937, 3–17 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Lambert S. A., et al. , The human transcription factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Matera A. G., Terns R. M., Terns M. P., Non-coding RNAs: Lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 8, 209–220 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Pillai R. S., Bhattacharyya S. N., Filipowicz W., Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol. 17, 118–126 (2007). [DOI] [PubMed] [Google Scholar]
- 18.John B., et al. , Human microRNA targets. PLoS Biol. 2, 661–663 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui C., Cui Q., The relationship of human tissue microRNAs with those from body fluids. Sci. Rep. 10, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gates A. J., Gysi D. M., Kellis M., Barabási A. L., A wealth of discovery built on the Human Genome Project—By the numbers. Nature 590, 212–215 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Alonso-López D., et al. , APID database: redefining protein–protein interaction experimental evidences and binary interactomes. Database 2019, baz005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alles J., et al. , An estimate of the total number of true human miRNAs. Nucleic Acids Res. 47, 3353–3364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis B. P., Burge C. B., Bartel D. P., Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Zhuo Y., Gao G., Shi J. A., Zhou X., Wang X., MiRNAs: Biogenesis, origin and evolution, functions on virus-host interaction. Cell. Physiol. Biochem. 32, 499–510 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Martinez N. J., Walhout A. J. M., The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 31, 435–445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghafouri-Fard S., et al. , The interaction between miRNAs/lncRNAs and nuclear factor-κB (NF-κB) in human disorders. Biomed. Pharmacother. 138, 111519 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Mattes J., Collison A., Plank M., Phipps S., Foster P. S., Antagonism of microRNA-126 suppresses the effector function of T H2 cells and the development of allergic airways disease. Proc. Natl. Acad. Sci. U.S.A. 106, 18704–18709 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topol A., et al. , Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Rep. 15, 1024–1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kutsche L. K., et al. , Combined experimental and system-level analyses reveal the complex regulatory network of miR-124 durinG human neurogenesis. Cell Syst. 7, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahariya S., et al. , Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 112, 82–92 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Ferrè F., Colantoni A., Helmer-Citterich M., Revealing protein–lncRNA interaction. Brief. Bioinform. 17, 106–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian D., Sun S., Lee J. T., The long noncoding RNA, Jpx, Is a molecular switch for X chromosome inactivation. Cell 143, 390–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J. T., Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes. Dev. 23, 1831–1842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyle R., et al. , The imprinted antisense RNA at the lgf2r locus overlaps but does not imprint Mas1. Nat. Genet. 25, 19–21 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Williamson C. M., et al. , Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 7, e1001347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J. J., Fu H. J., Wu Y. G., Zheng X. F., Function of lncRNAs and approaches to lncRNA-protein interactions. Sci. China Life Sci. 56, 876–885 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Wang X., et al. , Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geisler S., Coller J., RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 14, 699–712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cesana M., et al. , A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rinn J. L., et al. , Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K. C., et al. , A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–126 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y., Li Z., Chen X., Zhang S., Long non-coding RNAs: From disease code to drug role. Acta Pharm. Sinica B 11, 340–354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C. H., Chen Y., Targeting long non-coding RNAs in cancers: Progress and prospects. Int. J. Biochem. Cell Biol. 45, 1895–1910 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Wu P., et al. , Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 97, 69–80 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Fu D., et al. , Long non-coding RNA CRNDE regulates the growth and migration of prostate cancer cells by targeting microRNA-146a-5p. Bioengineered 12, 2469–2479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karagkouni D., et al. , DIANA-LncBase v3: Indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 48, D101–D110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma L., et al. , Lncbook: A curated knowledgebase of human long non-coding rnas. Nucleic Acids Res. 47, D128–D134 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhartiya D., et al. , LncRNome: A comprehensive knowledgebase of human long noncoding RNAs. Database 2013, bat034 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu S.-D., et al. , MiRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 39, D163–D169 (2011), 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang H. D. Y., et al. , MiRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 48, D148–D154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao F., et al. , miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 37, D105–D110 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang L., Zhou G., Soufan O., Xia J., miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 48, W244–W251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teng X., et al. , NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 48, D160–D165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Junge A., et al. , RAIN: RNA-protein association and interaction networks. Database 2017, baw167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong J., et al. , RISE: A database of RNA interactome from sequencing experiments. Nucleic Acids Res. 46, D194–D201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barabási A. L., Albert R., Emergence of scaling in random networks. Science 286, 509–512 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Osborne J. D., et al. , Annotating the human genome with disease ontology. BMC Genomics 10, S6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang Z., et al. , HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 47, D1013–D1017 (2019), 10.1093/nar/gky1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahn Y. Y., Bagrow J. P., Lehmann S., Link communities reveal multiscale complexity in networks. Nature 466, 761–764 (2010), 10.1038/nature09182. [DOI] [PubMed] [Google Scholar]
- 60.Oti M., Snel B., Huynen M. A., Brunner H. G., Predicting disease genes using protein-protein interactions. J. Med. Genet. 43, 691–698 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y., et al. , Variations in DNA elucidate molecular networks that cause disease. Nature 452, 429–435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghiassian S. D., Menche J., Barabási A. L., A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 11, e1004120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma A., et al. , A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma. Hum. Mol. Genet. 24, 3005–3020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graf J., Scherzer R., Grunfeld C., Imboden J., Levels of C-reactive protein associated with high and very high cardiovascular risk are prevalent in patients with rheumatoid arthritis. PLoS One 4, e6242 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W. C., et al. , Identification of differentially expressed genes in synovial tissue of rheumatoid arthritis and osteoarthritis in patients. J. Cell. Biochem. 120, 4533–4544 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Firestein G. S., Evolving concepts of rheumatoid arthritis. Nature 423, 356–361 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Al-Jameil N., Khan F. A., Khan M. F., Tabassum H., A brief overview of preeclampsia. J. Clin. Med. Res. 6, 1–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gysi D. M., Nowick K., Construction, comparison and evolution of networks in life sciences and other disciplines. J. R Soc. Interface 17, 20190610 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goh K.-I., et al. , The human disease network. Proc. Natl. Acad. Sci. U.S.A. 104, 8685–8690 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ardlie K. G., et al. , The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gysi D. M., Voigt A., de Miranda Fragoso T., Almaas E., Nowick K., wTO: An R package for computing weighted topological overlap and a consensus network with integrated visualization tool. BMC Bioinformatics 19, 392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nowick K., Gernat T., Almaas E., Stubbs L., Differences in human and chimpanzee gene expression patterns define an evolving network of transcription factors in brain. Proc. Natl. Acad. Sci. U.S.A. 106, 22358–22363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choy E. H. S., Panayi G. S., Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 344, 907–916 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Fernández A., et al. , Lupus arthropathy: A case series of patients with rhupus. Clin. Rheumatol. 25, 164–167 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Devrimsel G., Serdaroglu Beyazal M., Three case reports of rhupus syndrome: An overlap syndrome of rheumatoid arthritis and systemic lupus erythematosus. Case Rep. Rheumatol. 2018, 6194738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tseng C. C., et al. , Increased incidence of rheumatoid arthritis in multiple sclerosis. Medicine (Baltimore) 95, e3999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muñiz Pedrogo D. A., et al. , An increased abundance of clostridiaceae characterizes arthritis in inflammatory bowel disease and rheumatoid arthritis: A cross-sectional study. Inflamm. Bowel Dis. 25, 902–913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y., et al. , The risk of rheumatoid arthritis among patients with inflammatory bowel disease: A systematic review and meta-analysis. BMC Gastroenterol. 20, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verma A. K., et al. , Association of rheumatoid arthritis with diabetic comorbidity: Correlating accelerated insulin resistance to inflammatory responses in patients. J. Multidiscip. Healthc. 14, 809 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tian Z., McLaughlin J., Verma A., Chinoy H., Heald A. H., The relationship between rheumatoid arthritis and diabetes mellitus: A systematic review and meta-analysis. Cardiovasc. Endocrinol. Metab. 10, 125–131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Charoenngam N., et al. , Patients with asthma have a higher risk of rheumatoid arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 50, 968–976 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Sheen Y. H., et al. , Association of asthma with rheumatoid arthritis: A population-based case-control study. J. Allergy Clin. Immunol. Practice 6, 219–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fischer L. M., Schlienger R. G., Matter C., Jick H., Meier C. R., Effect of rheumatoid arthritis or systemic lupus erythematosus on the risk of first-time acute myocardial infarction. Am. J. Cardiol. 93, 198–200 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Subedi A., Strauss J., Gupta S., Erosive deforming inflammatory arthritis in a patient with cervical adenocarcinoma. JAMA Oncol. 5, 1628–1629 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Libby P., Role of inflammation in atherosclerosis associated with rheumatoid arthritis. Am. J. Med. 121, S21–S31 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Bongartz T., et al. , Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295, 2275–2285 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Dreyer L., et al. , Risk of second malignant neoplasm and mortality in patients with rheumatoid arthritis treated with biological DMARDs: A Danish population-based cohort study. Ann. Rheum. Dis. 77, 510–514 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Gridley G., et al. , Incidence of cancer among patients with rheumatoid arthritis. J. Natl. Cancer Inst. 85, 307–311 (1993). [DOI] [PubMed] [Google Scholar]
- 89.Eriksson M., Rheumatoid arthritis as a risk factor for multiple myeloma: A case-control study. Eur. J. Cancer 29A, 259–263 (1993). [DOI] [PubMed] [Google Scholar]
- 90.McDonald S. D., et al. , Measures of cardiovascular risk and subclinical atherosclerosis in a cohort of women with a remote history of preeclampsia. Atherosclerosis 229, 234–239 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Wu P., et al. , Preeclampsia and future cardiovascular health. Circ. Cardiovasc. Qual. Outcomes 10, e003497 (2017). [DOI] [PubMed] [Google Scholar]
- 92.McDonnold M., et al. , High risk human papillomavirus at entry to prenatal care and risk of preeclampsia. Am. J. Obstetrics Gynecol. 210, 138.e1-5 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Calderon-Margalit R., et al. , Preeclampsia and subsequent risk of cancer: Update from the Jerusalem Perinatal Study. Am. J. Obstetrics Gynecol. 200, 63.e1-5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hidalgo C. A., Blumm N., Barabási A.-L. L., Christakis N. A., A dynamic network approach for the study of human phenotypes. PLoS Comput. Biol. 5, e1000353 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kitsak M., et al. , Tissue specificity of human disease module. Sci. Rep. 6, 35241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singerman L., Combination therapy using the small interfering RNA bevasiranib. Retina 29, S49–S50 (2009). [DOI] [PubMed] [Google Scholar]
- 97.Ray K. K., et al. , Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 382, 1507–1519 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Zhang M. M., Bahal R., Rasmussen T. P., Manutou J. E., Zhong X. B., The growth of siRNA-based therapeutics: Updated clinical studies. Biochem. Pharmacol. 189, 114432 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Durinck S., Spellman P. T., Birney E., Huber W., Mapping identifiers for the integration of genomic datasets with the R/ Bioconductor package biomaRt. Nat. Protocols 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stelzer G., et al. , The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics 2016, 1.30.1–1.30.33 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Perdomo-Sabogal A., Nowick K., Genetic variation in human gene regulatory factors uncovers regulatory roles in local adaptation and disease. Genome Biol. Evol. 11, 2178–2193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mardinoglu A., Nielsen J., New paradigms for metabolic modeling of human cells. Curr. Opin. Biotechnol. 34, 91–97 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Wishart D. S., et al. , DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018), 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gysi D. M., Barabasi A. -L., Non-coding RNAs improve the predictive power of network medicine. GitHub. https://github.com/Barabasi-Lab/NonCoding/. Deposited 28 April 2023. [DOI] [PMC free article] [PubMed]
- 105.Gysi D. M., Barabasi A. -L., Non-coding RNAs improve the predictive power of network medicine. Disease-Disease Association Dashboard. https://deisygysi.shinyapps.io/Network_NCI/. Deposited 31 July 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The data and code utilized in the preparation of this manuscript are publicly accessible and can be obtained from the following sources: Code and Data Repository: The code and data used for data analysis and generating the results presented in this paper is available on GitHub at the following repository: https://github.com/Barabasi-Lab/NonCoding (104). Online Tool for Disease–Disease Exploration: An interactive online tool for exploring disease–disease relationships using both interactomes is accessible at the following web address: https://deisygysi.shinyapps.io/Network_NCI/ (105).