

Summary
Oxytocin-expressing paraventricular hypothalamic neurons (PVNOT neurons) integrate afferent signals from the gut, including cholecystokinin (CCK), to adjust whole-body energy homeostasis. However, the molecular underpinnings by which PVNOT neurons orchestrate gut-to-brain feeding control remain unclear. Here, we show that mice undergoing selective ablation of PVNOT neurons fail to reduce food intake in response to CCK and develop hyperphagic obesity on a chow diet. Notably, exposing wild-type mice to a high-fat/high-sugar (HFHS) diet recapitulates this insensitivity toward CCK, which is linked to diet-induced transcriptional and electrophysiological aberrations specifically in PVNOT neurons. Restoring OT pathways in diet-induced obese (DIO) mice via chemogenetics or polypharmacology sufficiently re-establishes CCK’s anorexigenic effects. Last, by single-cell profiling, we identify a specialized PVNOT neuronal subpopulation with increased κ-opioid signaling under an HFHS diet, which restrains their CCK-evoked activation. In sum, we document a (patho)mechanism by which PVNOT signaling uncouples a gut-brain satiation pathway under obesogenic conditions.
Keywords: obesity, gut-brain axis, oxytocin, paraventricular hypothalamic nucleus, CCK, opioids, PVN, neuropeptide, gut hormone, NTS
Graphical abstract
Highlights
-
•
PVNOT neuron ablation triggers hyperphagic obesity and CCK insensitivity in mice
-
•
High-calorie feeding blunts electrical and transcriptional PVNOT activation upon CCK
-
•
Restoration of OT-CCK signaling rescues the suppression of feeding
-
•
Increased κ-opioid tone restrains CCK-induced PVNOT neuron activation and hypophagia
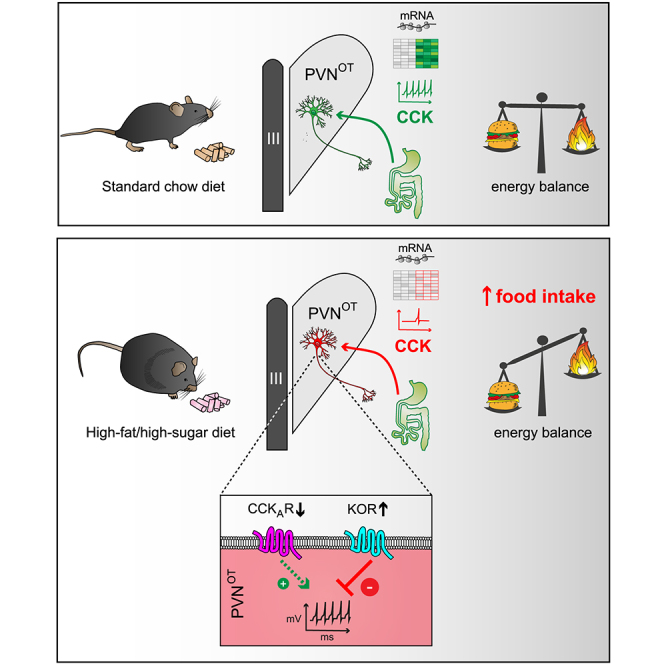
Gruber et al. reveal a mechanism that uncouples oxytocin neurons from a gut-to-brain satiation pathway upon high-calorie diet exposure. The authors show that reduced CCKAR, together with increased κ-opioid tone, blunts the activation of oxytocin neurons in response to CCK, preventing CCK-mediated food intake suppression on a high-calorie diet.
Introduction
Despite historical evidence as well as recent large-scale genetic studies1 cogently linking obesity pathogenesis to central nervous system (CNS) defects, we still do not know the exact cellular and molecular mechanism(s) involved in the initiation and progression of obesity and related metabolic disorders. Multiple studies have highlighted various sets of hypothalamic neurons that release specific neuropeptides that critically govern energy intake versus expenditure. Among these, oxytocin (OT), a nine-amino acid neuropeptide traditionally recognized for its role in reproductive physiology and social behavior, is increasingly gaining attention as an anti-obesity drug because of its favorable metabolic effects in multiple pre-clinical and clinical studies.2
In the brain, endogenous OT is exclusively synthesized by neurons of the supraoptic nucleus (SON), accessory nucleus (AN), and paraventricular nucleus (PVN) nuclei of the hypothalamus.3 Among these, the PVN has a particularly paramount role in metabolic homeostasis because electrolytic lesions as well as human genetic defects that impede PVN development result in severe obesity.4,5,6,7 In contrast to the evolutionarily more ancient, anatomically simpler SON and AN,8,9 the PVN exhibits a more complex cytoarchitecture10,11 and harbors multiple cell types, including two types of OT neurons: magnocellular and parvocellular neurons (magnOTs and parvOTs, respectively), which differ in size, morphology, electrophysiological properties, axonal projection targets, and other properties.12
Several reports document that gastric distension upon meal ingestion, and particularly the associated release of the gut peptide cholecystokinin (CCK), powerfully stimulate electrical activity of certain subsets of OT neurons.13,14,15,16,17 Chronic exposure to hypercaloric diets and the consequent obesity result in defects in this gut-brain cross-talk, and these, in turn, have been proposed to further aggravate metabolic derailment.18,19 Indeed, the food intake-suppressing effect of CCK is severely attenuated upon high-fat diet feeding, which is associated with reduced neural activation in several hypothalamic nuclei, including the PVN.20,21,22
We first found that the hypothalamic OT system undergoes maladaptive changes during chronic overnutrition in mice, including blunting the integration and propagation of the afferent CCK signal at the level of the OT system; i.e., a compromised OT response in the PVN is responsible for the loss of CCK’s food intake suppression under high-fat/high-sugar (HFHS) diet feeding. We then, by taking advantage of various gain- and loss-of-function models, systematically interrogated the physiological relevance and therapeutic potential of the OT system in obesity pathogenesis. Last, we employed single-nucleus RNA sequencing-2 (snRNA-seq2) and identified significant disruptions in the transcriptional profiles of specific OT subpopulations upon HFHS diet exposure. In sum, we show the molecular underpinnings of how OT neurons influence CCK-induced hypophagia in the orchestration of whole-body metabolic homeostasis.
Results
Selective adult-onset PVNOT neuron ablation induces rapid hyperphagic obesity
To assess whether OT neurons located in the PVN (PVNOT neurons) have a significant role in the control of energy homeostasis, we targeted these cells for selective ablation in mice using a diphtheria toxin A (DTA)-based genetic approach. Therefore, we stereotaxically injected adult male mice that carry the DTA gene downstream of a LoxP-STOP-LoxP (LSL) with adeno-associated viruses (AAVs) that drive expression of iCre or the fluorescent reporter Venus under an OT promoter23,24 (Figure 1A). As expected, iCre-mediated excision of the LSL cassette resulted in prominent induction of cleaved caspase-3 (C-CASP3; an apoptosis marker) in PVNOT neurons 5 days post AAV injection (Figures 1B and S1A–S1C; Video S1). Importantly, while PVNOT neurons were greatly reduced in number at the end of the experiment (8 weeks post AAV injection), neighboring neurons expressing arginine-vasopressin (AVP) were not (Figures S1A and S1B). At 2 weeks, DTAOT+/PVN mice had already substantially increased body weight compared with their control littermates and eventually developed pronounced obesity on a standard chow (SC) diet (Figures 1C and 1D). Despite this significantly higher body weight, however, indirect calorimetry conducted in the third and fourth week post AAV injection surprisingly did not reveal significant differences between DTAOT+/PVN mice and littermate controls with regard to uncorrected energy expenditure (Figure S1F) or in the relationship between total energy expenditure and body weight (Figure 1E). This suggests that these mice lack the normal increase in energy expenditure observed upon weight gain, which might potentially contribute to further body weight accrual. Moreover, no change was observed in the respiratory exchange ratio (RER), locomotor activity, or cumulative food intake when single housed in metabolic cages (Figures S1G–S1I). When pair housed in their habitual home cages, however, a separate cohort of DTAOT+/PVN mice displayed marked hyperphagia relative to littermate controls 4 weeks post AAV injection (Figure 1F), which led us to presume that differences in social isolation stress associated with the metabolic cages might have masked normal feeding behavior. Consistently, DTAOT+/PVN mice exhibited profoundly different body compositions at the end of the experiment (7 weeks post AAV injection), with significantly greater fat mass and lean mass relative to littermate control mice (Figure 1G). Moreover, DTAOT+/PVN mice had significantly impaired glucose tolerance (Figure 1H) and higher levels of glycated hemoglobin A1C (HbA1C), implying defective long-term glycemic control relative to control mice (Figure S1J). Presupposing that these changes in DTAOT+/PVN mice were due to diminished endogenous OT signaling, we asked whether pharmacologically resubstituting OT could normalize feeding and/or the metabolic derailments. While bidaily administration of exogenous OT (500 nmol/kg body weight [BW], subcutaneously [s.c.]) did not significantly alter BW in lean control mice, it led to a rapid BW reduction in DTAOT+/PVN mice (Figure 1I; treatment initiated 8 weeks post AAV injection). Notably, exogenous OT administration tended to reduce food intake in pair-housed mice (Figure 1J) and improved 3-h-fasted blood glucose (Figure S1K) as well as insulin sensitivity measured via Homeostatic Model Assessment of Insulin Resistance (HOMA-IR; Figure S1L). Intriguingly, the potency of exogenous OT in DTAOT+/PVN mice greatly surpassed the weight-lowering effect that has been reported previously in diet-induced obese (DIO) C57BL/6J wild-type mice of similar adiposity using comparable OT dosing.25 Next, we determined whether this heightened sensitivity of DTAOT+/PVN mice to exogenous OT is a consequence of diminished endogenous production, as suggested by a highly significant upregulation of hypothalamic Otr mRNA (encoding for the OT receptor; Figure S1M). Similarly, Ot mRNA expression tended to be reduced in whole hypothalamic lysates, which notably also includes the non-targeted SON and AN. Importantly, we did not observe any expression changes of the PVN-enriched melanocortin 4 receptor (Mc4r) mRNA (Figure S1M). In sum, the selective ablation of hypothalamic PVNOT neurons promotes hyperphagic obesity, likely via a paucity of endogenous OT signaling that can be rectified by pharmacological substitution.
Figure 1.

Virus-mediated ablation of PVNOT neurons induces hyperphagic obesity that is rectifiable by exogenous OT treatment and associated with CCK insensitivity
(A) Schematic of the experimental paradigm with an OT-specific AAV-OTp-iCre used to induce the expression of diphtheria toxin A (DTA) selectively in PVNOT neurons.
(B) Confocal micrograph depicting immunoreactivity to cleaved caspase-3 (C-CASP3; red) in PVNOT neurons (gray) 5 days post injection. Scale bar, 50 μm.
(C) BW gain of DTAOT+/PVN and control mice fed an SC diet. The arrow at 3 weeks post injection indicates single housing in metabolic cages. Data are presented as mean ± SEM. ∗∗p < 0.01, ∗∗∗∗p < 0.0001. n = 5–7 mice (two-way ANOVA).
(D) Representative images of two pairs of DTAOT+/PVN and control mice 5 weeks post injection (top and bottom, respectively) next to micrographs depicting the absence and presence of PVNOT neurons, respectively. Scale bar, 100 μm.
(E) Linear regression analysis of total energy expenditure (EE) and BW of DTAOT+/PVN and control mice 2 weeks post injection. Data are presented as individual mice. n.s., not significant. n = 5–7 mice.
(F) Daily food intake of a separate cohort of DTAOT+/PVN and control mice pair housed for mitigating isolation stress (see supplemental information). Data are presented as mean ± SEM. ∗p < 0.05. n = 4–5 pairs of mice (unpaired Student’s t test).
(G) Body composition of DTAOT+/PVN and control mice 6 weeks post injection. Data are presented as mean ± SEM. ∗∗p < 0.01, ∗∗∗∗p < 0.0001. n = 5–7 mice (unpaired Student’s t test).
(H) Blood glucose changes of DTAOT+/PVN and control mice upon a glucose tolerance test (2 g/kg BW, i.p.; left) and area under the curve (right). Data are presented as mean ± SEM. ∗∗p < 0.01, ∗∗∗∗p < 0.0001. n = 5–7 mice (two-way ANOVA).
(I) BW change in a separate cohort of DTAOT+/PVN and control mice upon treatment with exogenous OT (500 nM/kg BW, s.c., twice daily). Data are presented as mean ± SEM. ∗∗∗∗p < 0.0001. n = 8–10 mice (two-way ANOVA).
(J) Change in food intake of pair-housed DTAOT+/PVN and control mice upon treatment with exogenous OT (500 nM/kg BW, s.c., twice daily) relative to sham injections. Data are presented as mean ± SEM. ∗∗∗∗p < 0.0001. n = 4–5 pairs of mice (unpaired Student’s t test).
(K) Cumulative food intake of control mice upon vehicle versus CCK (20 μg/kg BW, i.p.). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01. n = 7 mice in a cross-over design (two-way ANOVA).
(L) Cumulative food intake of DTAPVN(OT+) mice upon vehicle versus CCK (20 μg/kg BW, i.p.). Data are presented as mean ± SEM. n = 5 mice in a cross-over design (two-way ANOVA).
3D-rendered and reconstructed PVNOT neurons as identified by OT immunoreactivity (gray) exhibit robust C-CASP3 immunosignal (red) following virus-mediated expression of Cre recombinase together with Venus, a fluorescent reporter protein (green).
Selective adult-onset PVNOT neuron ablation renders mice insensitive to systemic CCK
PVNOT neurons become strongly excited in response to meal-related gastrointestinal stimuli, including the intestinal peptide CCK.13,14,15,16,17 Given that CCK administration robustly suppresses acute feeding, we assessed the necessity of PVNOT neuron activation for CCK-elicited food intake suppression using mice devoid of PVNOT neurons fed an SC diet (4 weeks post AAV injection). After acclimating DTAOT+/PVN mice and littermate controls to single housing, SC diet removal (3–6 p.m.), and sham injections within metabolic cages for 3 days, we administered CCK (20 μg/kg BW, intraperitoneally [i.p.]) on the fourth day to all mice 10 min before dark onset and food return. Consistent with the literature,26 CCK injections at this dose produced a significant suppression of food intake in control mice (Figure 1K). In contrast, however, feeding behavior of DTAOT+/PVN mice was not significantly altered by CCK relative to sham treatments (Figure 1L). We conclude that PVNOT neurons are necessary for CCK-mediated hypophagia under physiological conditions.
Chronic exposure to an HFHS diet diminishes the activation response of PVNOT neurons to peripheral CCK
These findings suggest a prominent role of PVNOT neurons in feeding control by CCK. We then asked whether this same gut-brain communication is altered by exposure to an obesogenic diet. Indeed, it has been reported that the food intake-suppressing effect of CCK is severely attenuated upon high-fat diet feeding, and this is associated with reduced neural activation in several hypothalamic nuclei, including the PVN.20,21,22 Thus, we asked whether the hypothalamic OT system undergoes diet-induced desensitization to CCK upon HFHS diet feeding. Consistent with previous reports in rats,27,28 C57BL/6J mice fed an SC diet and receiving CCK (20 μg/kg BW, i.p.) showed robust activation of a large proportion of PVNOT neurons, as indicated by increased c-Fos immunoreactivity (Figures 2A and 2B). To discern whether peripheral CCK administration similarly affected the magnOT or parvOT subtypes, we next quantified c-Fos+ PVNOT neurons in a separate cohort of SC diet-fed reporter mice expressing tdTomato specifically in OT neurons (OT:Ai14), which received peripheral injections of Fluorogold (15 mg/kg BW, i.p.) 7 days prior to sacrifice. Fluorogold selectively marks neurons projecting beyond the blood-brain barrier and thus recognizes magnOT cells (Fluorogold [FG]+).29,30 We found that 25% of CCK-activated PVNOT neurons were FG+ magnOT neurons (Figures 2C and S2A–S2C), while CCK treatment induced c-Fos in 8% of FG− parvOT neurons. Conversely, mice chronically fed an HFHS diet failed to significantly increase c-Fos immunoreactivity in PVNOT neurons following CCK injection, suggesting a blunted activation in the course of consuming an obesogenic diet (Figures 2A and 2B).
Figure 2.

Chronic exposure to an HFHS diet impairs the electrical and transcriptional activation of PVNOT neurons in response to peripheral CCK
(A) Representative confocal micrographs depicting neuronal activation by means of nuclear c-Fos immunoreactivity (red) in PVNOT neurons (green) in adult male C57BL/6J mice fed either an SC or HFHS diet receiving CCK (20 μg/kg BW, i.p.). Scale bar, 10 μm.
(B) Corresponding quantification of c-Fos+ PVNOT neurons relative to total PVNOT neurons counted. Data are presented as mean ± SEM. ∗∗∗∗p < 0.0001, n = 4 mice, 4–8 hemisections per mouse (unpaired Student’s t test).
(C) Quantification of CCK-activated (c-Fos+) subpopulations of PVNOT in a separate cohort pre-treated with fluorogold (FG; 15 mg/kg BW, i.p.) to distinguish parvOT neurons (FG−) from magnOT neurons (FG+); data are represented as mean in percent relative to total parvOT and magnOT cell count, respectively. n = 3 mice, 46 hemisections, 2,486 cells.
(D) Schematic of the OT:RiboTag mouse model used to isolate actively translated mRNA specifically from OT+ neurons by IP of the HA-tagged ribosomal subunit Rpl22.
(E) Heatmap of translating mRNA enrichment of various hypothalamic neuropeptides in the immunoprecipitate (IP) relative to input. n = 4 mice.
(F) Heatmap representation of enrichment in Cckar mRNA and Cckbr mRNA in IP relative to input. n = 4 mice.
(G) Relative abundance of Cckar mRNA and Cckbr mRNA in the IP of OT:RiboTag mice fed an HFHS diet relative to an SC diet. n = 4 mice.
(H) Heatmap representation of DEGs in hypothalamic OT+ neurons of adult male OT:RiboTag mice fed either an SC (left) or HFHS (right) diet upon injection of vehicle versus CCK (20 μg/kg BW, i.p.). Rows reflect normalized (Z score) gene expression abundance. n = 4 mice.
(I) Volcano plot showing log-transformed adjusted p values plotted against fold changes for DEGs in hypothalamic OT neurons from SC diet-fed OT:RiboTag mice upon injection of vehicle versus CCK (20 μg/kg BW, i.p.).
Peripheral CCK induces a characteristic transcriptional profile specifically in hypothalamic OT neurons of lean but not obese mice
Next, we sought out to identify CCK-inducible gene expression modules in an OT neuron-specific manner. We generated OT:RiboTag mice (OT-ires-Cre mice intercrossed to floxed Rpl22HA mice), allowing us to employ translating ribosome affinity purification (TRAP) and RNA-seq of actively translated mRNA selectively in OT neurons (Figure 2D). Hemagglutinin (HA)-tagged ribosomes derived from OT neurons were immunoprecipitated from whole hypothalamic lysate (input) using an anti-HA antibody, resulting in a greater than 80-fold enrichment of Ot mRNA in the immunoprecipitation (IP) compared with the input fraction (Figure 2E). Notably, the IP fraction was significantly enriched for Cckar mRNA while being de-enriched for Cckbr mRNA compared with the input (Figure 2F). Importantly, mice chronically fed an HFHS diet tended to have lower Cckar mRNA and higher Cckbr mRNA abundance compared with lean, SC diet-fed control mice (Figure 2G). We next explored more broadly how HFHS diet exposure affects gene expression networks and pathways in OT neurons. Thus, we performed Gene Ontology (GO) enrichment analysis of a total of 3,127 differentially expressed genes (DEGs) between vehicle-treated SC diet- versus HFHS diet-fed mice (adjusted [adj.] p < 0.05). By employing the cellular component category analysis, we found that HFHS diet exposure strongly modifies pathways involved in organelle localization, Golgi apparatus function, and synaptic vesicle transport and recycling as well as exocytosis (Figure S2D). Last, we injected SC diet- or HFHS diet-fed cohorts of OT:RiboTag mice with either CCK (20 μg/kg BW, i.p.) or vehicle to conduct TRAP followed by high-throughput RNA-seq for input and IP samples. Strikingly, in mice maintained on an SC diet, administration of CCK 2 h before sacrifice elicited widespread and profound changes in the gene expression profile of OT neurons compared with vehicle (158 DEGs with adj. p < 0.05; Figures 2H and 2I). Of note, the respective input samples did not exhibit any significant gene expression signatures upon CCK delivery, further supporting the notion that OT neurons represent a major CCK-responsive population within the hypothalamus (Figures S2E and S2F). Strikingly, all CCK-related transcriptomic changes were completely abolished when mice were chronically fed an HFHS diet (Figure 2H). In sum, an acute increase in circulating CCK elicits characteristic changes in mRNA translational activity at the level of OT neurons in lean but not in obese mice.
CCK triggers the activation of PVNOT neurons via CCKAR-mediated mechanism in lean but not in obese mice
We next investigated the mechanism of action to discern whether PVNOT neurons directly sense CCK levels or whether the response is mediated via relay pathways downstream of a vagal mechanism.31,32 Selective expression of a genetically encoded Ca2+ indicator, GCaMP6f, was specifically targeted to PVNOT neurons by stereotaxically injecting AAV-DIO-EF1α-GCaMP6f into OT-ires-Cre mice. Using 2-photon excitation Ca2+ imaging of ex vivo brain slices, we found that bath application of CCK in the presence of synaptic blockers evoked robust and immediate increases in fluorescent signals in putative magnocellular PVNOT neurons (Figures 3A–3D, S3A, and S3B; Video S2). This indicates that this population is directly responding to CCK with increased cytosolic Ca2+ transients. To identify the molecular mechanisms that enable PVNOT neurons to directly sense systemic CCK, we next applied single-molecule fluorescence in situ hybridization (FISH; RNAscope) and assessed the expression of the CCKA receptor subtype in PVNOT neurons. CCKAR, which is predominantly found in vagal sensory neurons along the alimentary canal,33,34 constitutes the major subtype mediating the food intake-suppressive effect of CCK and has been implicated previously in the activation of OT neurons in rodents.31,32 Contrary to the initially proposed vagally mediated mechanism, and consistent with our ribosomal profiling results, we here detected abundant Cckar mRNA expression in PVNOT neuronal somata (Figure 3E), which was significantly lower in mice chronically exposed to an HFHS diet compared with SC diet-fed control mice (Figure 3F). To further substantiate a direct CCKAR-mediated mechanism in PVNOT neurons, we next conducted whole-cell patch-clamp recordings from electrophysiologically identified30 magnOT neurons ex vivo. Bath application of a potent and selective CCKAR agonist (A-71623, 25 nM) elicited robust increases in action potential frequency of putative magnOT neurons from SC diet-fed mice. In contrast, mice chronically fed an HFHS diet did not show any significant increases in action potential frequency when A-71623 or native CCK-8s (engaging CCKAR and CCKBR, 50 nM) were bath applied (Figures 3G and 3H). Notably, magnOT neurons from SC diet- versus HFHS diet-fed mice did not exhibit differences in electrophysiological properties, such as basal action potential frequency, FI curve (firing frequency as a function of injected current), and input resistance (Figures S3C–S3E). Based on these results, we thus conclude that mice on an SC diet can respond to systemic CCK at the level of PVNOT neurons via a direct CCKAR-mediated mechanism to suppress food intake,35,36 a pathway that becomes aberrant upon feeding mice with an HFHS diet.
Figure 3.

PVNOT neurons are activated by CCK via a direct, CCKAR-dependent mechanism in lean but not obese mice
(A) 2-photon micrograph of an acute brain slice expressing the Ca2+ indicator GCaMP6f (green, top left) in PVNOT neurons conditionally tagged by Ai14-tdTomato (red, bottom left) in a cell-type-specific manner (merge, center). Representative images were taken during the time-lapse recording before (top right) and after bath application of 50 nM CCK (bottom right) in the presence of synaptic blockers. Scale bars, 25 μm.
(B) Heatmap representation of cytosolic Ca2+ transients of individual PVNOT neurons upon bath application of CCK (50 nM) in the presence of synaptic blockers. n = 1 mouse, 49 neurons.
(C) Pie chart illustrating the percentages of PVNOT neurons that increase (yellow) or decrease their activity (purple) upon CCK application or that do not exhibit any change (gray).
(D) Quantification of Ca2+ events over time, displayed in 1-min bins. n = 1 mouse, 49 neurons.
(E) 3D-rendered, high-power confocal micrograph depicting Ot mRNA (green), Cckar mRNA (magenta), and DAPI (gray) upon FISH (fluorescence in situ hybridization; RNAscope). Individual nuclei are outlined for demarcation. Scale bar, 20 μm.
(F) Corresponding quantification of FISH using background-corrected mean fluorescence intensity (M.F.I.) of Cckar mRNA per Ot+ soma in the rostromedial and caudal PVN of adult male C57BL/6J mice fed either an SC or HFHS diet. Data are presented as mean of all somata analyzed ± SEM. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001. n = 3 mice, 4–8 hemisections per mouse (unpaired Student’s t test).
(G) Representative traces of action potential frequency of putative magnOT neurons derived from SC diet-fed versus HFHS diet-fed mice in response to increasing concentrations of bath-applied A-71623 (5, 25, and 50 nM), followed by superfusion with native CCK (50 nM).
(H) Summary of changes in action potential frequency visualized in (G). Data are presented as mean superimposed with individual data points. ∗p < 0.05, n = 2–3 mice/6 neurons per mouse (two-way ANOVA).
Time-lapse 2-photon excitation microscopy of cytosolic Ca2+ dynamics in PVNOT neurons that were virally transduced to express GcaMP6f. Despite the presence of synaptic blockers in the bath solution, bath application with native CCK (50 nM) robustly triggered increases in fluorescent signal at the somata and neurites of most PVNOT neurons within the field of view. Time-of-CCK-appearance in the bathing solution as indicated in the movie. Scale bar, 25 μm.
Targeted PVNOT neuron activation restores CCK-induced satiety in HFHS diet-fed mice
Based on our previous finding, we hypothesized that CCK’s inability to suppress high-fat diet intake at established doses20,37,38,39 is a consequence of its decoupling from the hypothalamic OT system, which we observed upon chronic HFHS diet exposure. To test the pathophysiological relevance of this gut-to-brain pathway, we circumvented this diet-associated impediment of PVNOT neuron engagement by administering systemic CCK to HFHS diet-fed mice while concomitantly activating PVNOT neurons using designer receptors exclusively activated by designer drugs (DREADDs). To accomplish this, we stereotaxically injected either AAV-DIO-hSYN1-hM3Dq-mCherry (an activating DREADD) or a control virus into the PVN of adult male OT-ires-Cre mice. After 4 weeks, mice underwent dosing acclimatization with sham i.p. injections within metabolic cages for 3 consecutive days. As expected, CCK administration on the fourth day produced a significant suppression in food intake and a prolonged feeding latency in SC diet-fed control mice relative to vehicle injection (Figure 4A); in contrast, CCK injections did not reduce food intake in obese control mice chronically fed an HFHS diet (Figure 4B), as observed previously by others.37,38,40 Next, we asked whether the chemogenetic activation of PVNOT neurons is sufficient to restore CCK-mediated hypophagia in HFHS diet-fed mice that selectively express hM3Dq (hM3DqOT+/PVN mice). To do this, we pre-injected HFHS diet-fed hM3DqOT+/PVN mice with the DREADD-activating ligand clozapine-N-oxide (CNO) 20 min prior to the systemic administration of CCK and closely assessed their feeding behavior. Strikingly, the simultaneous chemogenetic activation of PVNOT neurons readily restored the responsiveness to CCK and produced a durable suppression in HFHS diet intake over the course of 2 h (Figure 4C). Importantly, and consistent with a previous report,41 chemogenetic activation of PVNOT neurons without CCK administration did not alter SC diet or HFHS diet intake, relative to their respective control mice (Figures S4A and S4B; conducted in a separate cohort of mice). Based on these findings, we inferred that activation of PVNOT neurons is necessary to mediate CCK-induced satiation but that their activation is not sufficient to suppress HFHS diet intake on its own.
Figure 4.

Blunted suppression of food intake in response to CCK on an HFHS diet is reinstated by concomitant chemogenetic activation of PVNOT neurons
(A) Cumulative food intake of SC diet-fed control mice upon vehicle versus CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.). Data are presented as mean ± SEM. Shown is corresponding latency to feed (bottom). ∗∗p < 0.01, ∗p < 0.05. n = 5 mice in a cross-over design (two-way ANOVA and paired Student’s t test [bottom]).
(B) Cumulative food intake of HFHS diet-fed control mice upon vehicle versus CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.). Data are presented as mean ± SEM. Shown is corresponding latency to feed (bottom). n = 8 mice in a cross-over design (two-way ANOVA and paired Student’s t test [bottom]).
(C) Cumulative food intake of HFHS diet-fed hM3DqOT+/PVN mice upon vehicle versus CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.). Data are presented as mean ± SEM. Shown is corresponding latency to feed (bottom). ∗∗p < 0.01, n = 8 mice in a cross-over design (two-way ANOVA and paired Student’s t test [bottom]).
(D) Representative confocal micrographs depicting neuronal activation by means of nuclear c-Fos immunoreactivity (green) in virally transduced PVNOT neurons (mCherry+, red). Scale bar, 50 μm.
(E) Quantification of activated (c-Fos+) virally transduced PVNOT neurons (mCherry+) upon CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.) injections in HFHS diet-fed hM3DqOT+/PVN mice or control mice. ∗∗∗∗p < 0.0001, n = 4 mice/3–8 hemisections (one-way ANOVA).
(F) Quantification of activated (c-Fos+) PVN neurons overall upon CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.) injections in HFHS diet-fed hM3DqOT+/PVN mice or control mice. ∗∗p < 0.01, ##p < 0.01, n = 4 mice/3–8 hemisections (one-way ANOVA).
(G) Representative confocal micrographs depicting neuronal activation (c-Fos+, green) across the NTS of hM3DqOT+/PVN mice or control mice following either CNO or CNO+CCK; insets display co-localization with TH (gray). Scale bar, 100 μm and 20 μm (inset).
(H) Quantification of activated (c-Fos+) NTS neurons upon CCK (20 μg/kg BW, i.p.) plus CNO (1 mg/kg BW, i.p.) injections in HFHS diet-fed hM3DqOT+/PVN mice or control mice. ∗∗p < 0.01. n = 4 mice/3–8 hemisections (one-way ANOVA).
(I) Relative BW changes of male wild-type mice and tamoxifen-inducible global OTR−/− mice, all fed an HFHS diet and treated bidaily with either OT (500 nmol/kg BW, s.c.), A-71623 (30 nmol/kg BW, i.p.), or their combination. Data are presented in percent of initial BW as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n = 6–8 mice (two-way ANOVA).
(J) Cumulative food intake of the cohort shown in (H). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n = 6–8 mice (two-way ANOVA).
(K) Changes in body composition of the cohort shown in (H) and (I) at the end of study. Data are presented relative to initial body composition as mean ± SEM. ∗∗p < 0.01, ∗∗∗∗p < 0.0001. n = 5–7 mice (unpaired Student’s t test).
Concomitant PVNOT neuron stimulation with CCK administration broadly activates the PVN in HFHS diet-fed mice
To determine how PVNOT neurons are required for co-executing intake suppression with CCK, we next assessed changes in neuronal activity patterns in control mice versus hM3DqOT+/PVN mice receiving either CNO alone or CNO+CCK (Figure 4D). In control mice, HFHS diet feeding greatly blunted the induction of c-Fos immunoreactivity in virus-targeted OTmCherry+ neurons by CNO+CCK co-administration relative to CNO alone. In contrast, hM3DqOT+/PVN mice had robust and near-complete activation of virus-targeted OThM3Dq-mCherry neurons, whether given CNO only or CNO+CCK in combination (Figure 4E). Importantly, combined CNO+CCK administration in these mice also resulted in a potentiated activation of the PVN overall, including neighboring non-OT neurons (Figure 4F). Thus, this pronounced c-Fos response in the PVN upon CNO+CCK administration, which was observed in hM3DqOT+/PVN mice but not in control mice or hM3DqOT+/PVN mice given CNO only, precisely reflected the changes in feeding behavior and further suggests that PVNOT engagement is a prerequisite for broader PVN activation and suppression of food intake upon CCK.
Chemogenetic PVNOT neuron stimulation potentiates the CCK-induced activation of hindbrain nucleus tractus solitarius (NTS) neurons in HFHS diet-fed mice
Intrigued by this broad PVN activation in CNO+CCK-injected hM3DqOT+/PVN mice, we next pondered whether this effect would extend beyond the PVN additionally re-sensitizing other critical circuit nodes of central gut hormone signaling. As described extensively elsewhere,42 peripheral CCK co-activates a dispersed neurocircuitry, including the PVN as well as the NTS in the caudal brain stem—two regions that are heavily interconnected and critical for the orchestration of the behavioral and physiological responses toward CCK.43,44 To interrogate whether HFHS diet-induced aberrations in PVNOT neurons would impact the structure and function of this hypothalamus-brain stem neurocircuit, we employed dual-color 3D whole-brain imaging (iDISCO45) of OT:Ai14 reporter mice in combination with immunostaining against tyrosine hydroxylase (TH). By this, we were able to resolve the descending axonal projections from PVNOT neurons as well as their extensive innervation of subregions throughout the NTS, including the catecholaminergic A2/C2 cell group (TH+) (Figures S4C–S4F; Video S3). Next, we analyzed the activation state of NTS neurons, including the A2/C2 cell group, upon CNO+CCK co-injections of the same cohort of hM3DqOT+/PVN mice and controls as in Figure 4. Strikingly, hM3DqOT+/PVN mice exhibited significantly more c-Fos+ neurons in the NTS upon CNO+CCK co-administration relative to control mice (Figures 4G and 4H), which was particularly prominent for the catecholaminergic A2/C2 cell group in the caudal medial portion of the NTS (Figure 4G, inset). In sum, our data suggest that defective sensing of gut-originating CCK upon chronic HFHS diet exposure is a consequence of a disturbed hypothalamus-brain stem network. Importantly, our data further highlight the fact that PVNOT neurons constitute a key population within the hierarchical structure of this network, which is evidenced by the fact that their selective activation readily re-sensitizes mice to the feeding-suppressive effect of systemic CCK. We thus conclude that PVNOT neurons function as a pivotal central hub whose targeted activation can restore the transmission of the gut-derived anorexigenic signal CCK under obesogenic conditions.
3D light-sheet fluorescence microscopy of iDISCO-cleared whole mouse brain (OT:Ai14). Descending axonal projections of OT neurons (green) can be traced to nucleus tractus solitarius (NTS) of the brainstem where they densely innervate catecholaminergic A2/C2 neurons (TH+; magenta).
Combined OTR-CCKAR co-agonism improves metabolic outcomes in DIO mice
Because the hypothalamic OT system co-executes food intake suppression together with CCK, a process that is disrupted upon HFHS diet exposure, we hypothesized that the pharmacological combination of synthetic OT and a potent and selective CCKAR agonist (A-71263)46 would confer significant metabolic improvements in DIO C57BL/6J mice. Compared with respective mono-agonism, co-treatment with OT (500 nmol/kg BW, s.c.) and A-71263 (30 nmol/kg BW, i.p.) twice daily for 10 days resulted in increased BW loss (Figure 4H) associated with a significant reduction in food intake relative to what occurred in vehicle-treated control mice (Figure 4I). Body composition analyses further revealed that the substantial weight-lowering effect of combined OT and A-71263 was primarily due to a loss in fat mass (Figure 4J). Importantly, combined OT and A-71263 treatment had no effect on BW or composition in mice that globally lack the OT receptor upon tamoxifen-induced Cre-loxP recombination in adulthood (OTRiΔ mice). Thus, we conclude that integrating OTR and CCKAR agonism promotes favorable metabolic effects in DIO mice and might emerge as a promising combination to be added to the current arsenal of anti-obesity polypharmacies.
snRNA-seq2 reveals intersectional regulation of hypothalamic OT neurons by CCKAR and κ-opioid receptors that is dependent on dietary context
We next sought to explore the transcriptional diversity across hypothalamic OT neurons at the resolution of single cells. To accomplish this, we generated mice in which the nuclei of OT neurons are tagged with super-folded (sf)GFP; specifically, we back-crossed OT-ires-Cre mice with a reporter mouse line that Cre-dependently expresses the nuclear membrane protein SUN-domain containing protein 1 (SUN1) fused to sfGFP (OT:Sun1-sfGFP mice; Figure 5A). Being therefore amenable to fluorescence-activated nucleus sorting (FANS), we then isolated hypothalamic OT neuronal nuclei from SC diet- or HFHS diet-fed mice and individually sorted them into 384-well plates (Figure 5B). We harvested approximately 500 sfGFP+ nuclei per mouse brain, which is consistent with stereological counting studies of OT neuron numbers.47 To assess the overall impact of chronic HFHS diet feeding, we performed differential gene expression analyses (123 DEGs with adjusted p < 0.05) comparing the entirety of single-nucleus transcriptomes between mice fed an SC diet versus HFHS diet (Figure 5C). We then queried our snRNA-seq2 dataset for the expression of receptors related to hormonal, neuropeptide, and low-molecular transmitter signaling implicated in energy homeostasis. We found multiple OT neurons to express varying combinations of metabolism-related receptors, reassuringly including Cckar, suggesting substantial convergence in metabolic information processing at the level of distinct OT neuronal subsets (Figure 5D). We then focused on OT neurons exhibiting transcript intersections between Cckar and Oprk1 (κ-opioid receptor 1 [KOR]), which constitutes an inhibitory Gi/Go-coupled receptor that stands out by its highly abundant expression across OT neurons. Chronic exposure to an HFHS diet increased the proportion of Cckar+ OT neurons co-expressing Oprk1 mRNA (Figures 5D and 5E).
Figure 5.

Intersectional regulation of hypothalamic OT neurons by CCKAR and κ-opioid receptors is dependent on dietary context
(A) Confocal micrographs of coronal brain section from an adult male OT:Sun1-sfGFP mouse, depicting the nuclear localization of sfGFP (green) in hypothalamic OT neurons (gray) of the PVN and SON. III, third ventricle; ot, optic tract. Scale bars, 200 μm and 25 μm (inset).
(B) Schematic of the workflow used to sort individual sfGFP+ nuclei into 384-well plates using FANS (top left) with a representative FANS plot displayed (top right); isolation was followed by low-volume pipetting robot-assisted single nucleus lysis, cDNA synthesis, and library preparation for snRNA-seq2 (bottom).
(C) Volcano plot highlighting differential gene expression changes across the sum of individual OT nuclei from adult male OT:Sun1-sfGFP mice chronically fed an HFHS diet relative to SC diet-fed littermate controls. n = 2 mice.
(D) Upset plot visualization of intersectional expression of select receptors in individual OT nuclei from SC diet-fed (top) and HFHS diet-fed (bottom) OT:Sun1-sfGFP mice. Transcripts for Cckar and Oprk1 are highlighted (red dash boxes), as well as their intersections (blue bars). Percentages of Cckar+ OT nuclei also expressing Oprk1 are visualized as pie charts for each panel.
(E) UMAP plot visualization of individual OT nuclei from SC diet-fed (top) and HFHS diet-fed (bottom) OT:Sun1-sfGFP mice, colored according to their expression of either Cckar (magenta), Oprk1 (cyan), their combination (blue), or none (gray). N = 2; 614 cells (SC diet) and 588 cells (HFHS diet).
(F) Representative traces of action potential frequency of magnOT neurons derived from adult male OT:Ai14-tdTomato reporter mice fed either an SC diet or HFHS diet in response to bath-applied A-71623 (25 nM) with or without pre-treatment with nor-BNI (200 nM). Scale bar, 25 μm.
(G) Summary of changes in action potential frequency visualized in (F). Data are presented before and after application of A-71623 as mean ± SEM. ∗p < 0.05. n = 2–3 mice/5–8 neurons per mouse (paired Student’s t test).
(H) Cumulative food intake of HFHS diet-fed male C57BL/6J wild-type mice upon injection of a CCK + nor-BNI combination (20 μg/kg BW and 10 mg/kg BW, respectively; i.p.), nor-BNI alone (10 mg/kg BW, i.p.), or vehicle. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01. n = 6–7 mice (two-way ANOVA).
κ-opioid tone restrains PVNOT neuronal activation by CCK under HFHS diet feeding
Given that signaling through KOR is associated with inhibitory neuromodulation, we hypothesized that HFHS diet-induced κ-opioid tone in Cckar+ OT neurons might indirectly and tonically blunt their responsivity to systemic CCK. To test this hypothesis, we carried out patch-clamp recordings of PVNOT neurons in acute brain slices derived from mice that selectively express tdTomato in OT neurons (OT:Ai14-tdTomato). In agreement with our previous findings, we observed that bath application of the selective CCKAR agonist A-71623 (25 nM) dramatically increased action potential frequency in magnocellular PVNOT neurons from SC diet-fed mice independent of presynaptic inputs (Figures 5F, 5G, S5A, and S5B), whereas this response was significantly blunted when mice were chronically fed an HFHS diet (Figure 5F). However, when brain sections from HFHS diet-fed mice were pre-incubated with the potent and highly selective KOR antagonist nor-binaltorphimine (nor-BNI; 200 nM in artificial cerebrospinal fluid [aCSF] for 20 min), the CCKAR agonist A-71623 was readily able to trigger significant action potentials in PVNOT neurons (Figures 5F and 5G). Intrigued by these results, we next wondered whether systemic blockade of KOR signaling could re-sensitize DIO mice toward CCK-mediated food intake suppression. Therefore, we pretreated male mice fed an HFHS diet with nor-BNI (10 mg/kg BW, i.p.) 20 min before receiving CCK injections at the dose shown previously to be ineffective in DIO mice (20 μg/kg BW, i.p.). Strikingly, we observed that their combined administration resulted in significant feeding suppression (Figure 5H), an effect that was not observed with nor-BNI injections alone or with CCK injections at various doses (Figure S5C).
Thus, we provide electrophysiological evidence underscoring the notion that increased κ-opioid signaling within CCK-sensitive OT subsets exerts tonic restraint on their excitability under an obesogenic diet. Last, we highlight systemic κ-opioid antagonism as a promising approach to reinstate CCK-mediated suppression of food intake.
Discussion
Gut-to-brain communication is pivotal for the control of whole-body energy homeostasis,18 and pharmacological targeting of select pathways within this communication network has been found recently to effectively lower BW in clinically obese populations.48 Spurred by this translational success story, we sought out to explore alternative brain-targeted gut peptide therapeutics that, when rationally combined, might better mimic the pluri-hormonal physiology of eating.49 Here, we elucidated how the anorexigenic gut peptide CCK fails to suppress food intake under obesogenic diets in mice. Specifically, we observed that feeding an HFHS diet induces aberrations in hypothalamic OT neurons on the transcriptional and electrophysiological levels by which this pivotal cell population ultimately decouples from CCK signaling. Moreover, we found that mice undergoing adult-onset ablation of PVNOT neurons fail to suppress food intake upon CCK administration, similar to what has been reported previously.50 Perhaps most strikingly, these mice rapidly developed obesity on an SC diet, suggesting an indispensable role of PVNOT neurons in energy homeostasis. While some alternative approaches employed to impair PVNOT neuron function corroborate the indispensability of PVNOT neurons in BW regulation,51,52 other investigations have reported less dramatic effects.50,53 These discrepant finding might be due to significant differences in animal models. Interestingly, both studies reporting minimal effects on BW regulation employed the OT-ires-Cre mouse to conditionally target DTA receptor expression to all Cre+ cells. Therefore, we consider it a possibility that this approach resulted in the unintended, and unnoticed, ablation of peripheral OT+ cells, such as those described recently in the gastrointestinal tract,54,55 which might have opposed the obesity-inducing effects of central ablation. Similarly, significant disagreements remain as to whether, or to what extent, the OT system interacts with the melanocortin circuit, a central pillar of energy homeostasis control within the hypothalamus.56 While several reports provide evidence for57,58 as well as against59,60 this notion, a recent study by Fenselau et al.61 might have provided a crucial clue by identifying a missing component within the melanocortin system in the form of a fast-acting glutamatergic satiation pathway; intriguingly, this pathway is formed by neurons specifically sensitive to OT,61 suggesting (1) that this pathway is organized in a more complex manner than initially claimed and (2) that OT might indirectly, but significantly, modulate the melanocortin pathway.
CCK induces a gene expression signature at the level of PVNOT neurons in lean but not obese mice
By employing affinity purification of tagged ribosomes followed by in-depth translatome profiling, we identified a highly coordinated translational program that is induced in hypothalamic OT neurons upon CCK injection. In agreement with what occurs in another population of hypothalamic neurons,62 our data add to the evidence suggesting that hormonal cues can readily induce robust and characteristic gene expression signatures in specialized cell types to adjust neuronal physiology to the nutritional state. Besides a pronounced enrichment of mRNA species involved in neuroplasticity, including retinoic acid signaling (Rdh9),63 two of the transcripts most robustly induced by CCK were Exoc3l2 and Trappc9/Nbp, genes that encode for proteins crucially involved in vesicular trafficking and neuropeptide signaling. Intriguingly, mutations in Trappc9/Nbp have been linked previously to a Prader-Willi-like obesity syndrome (PWS) in humans.64,65 We further revealed that chronic HFHS diet feeding abolishes the induction of this gene expression signature, suggesting that long-term caloric excess might uncouple episodic hormonal information from molecular processes promoting OT neuronal plasticity and cell-cell communication. However, future investigations are still required to reveal further intricacies as to how hormone-inducible gene expression signatures modulate hypothalamic function and energy homeostasis and whether they might bear therapeutic potential for dietary and/or genetic forms of obesity.
Targeted co-activation of OT and CCK signaling elicits enhanced food intake suppression and weight loss despite obesogenic diet exposure
Physiological energy homeostasis crucially depends on the gastrointestinal system informing the brain about the nutritional status quo via gut-derived humoral and neural signals such as CCK.18 CCK, as the first-described and paradigmatic anorexigenic gut hormone, suppresses feeding in a variety of species ranging from rodents66,67 to primates,68 including humans.69 Over the course of several decades, intense research efforts gave rise to a detailed anatomical blueprint highlighting a major role of the vagus nerve in CCK’s effects on gastric emptying70,71 and caloric intake regulation.72,73,74 However, and despite these extensive insights, CCK’s hoped-for therapeutic potential as an anti-obesity drug was soon questioned based on observations reporting that chronic intake of fat-rich diets greatly blunts its anorexigenic effect.20,37,38,39,75,76 While earlier work has primarily considered desensitization events at the level of the gut, such as at jejunal afferents,77 we now provide additional evidence suggesting that diet-induced perturbations of CCK signaling also operate directly in the hypothalamus, particularly on higher-order levels such as neuropeptide circuits. We then hypothesized that gaining insights into the cellular and molecular mechanisms underlying this failed signal integration might help to identify corrective countermeasures with the potential to reinstate CCK’s biological effects. Our finding that the hypothalamic OT system undergoes significant uncoupling from its CCK inputs further prompted us to explore approaches to restore the normal output from this specific neurocircuitry using chemogenetic and polypharmacological means. Consistent with previous observations in rats showing that intracerebroventricular OT delivery enhances food intake suppression upon low-dose CCK,78 we could demonstrate that artificially evoking OT signaling in mice readily restored food intake suppression in response to CCK despite chronic HFHS diet feeding. Therefore, we added another component to an already extensive body of literature, highlighting the so far underexplored role of dietary contexts in the coupling of anorexigenic CCK-OT signaling pathways in the hypothalamus.
Re-coupling of OT and CCK signaling engages neural substrates in the hypothalamus and brain stem
Besides the hypothalamus, prior studies have demonstrated that the NTS in the brain stem constitutes another pivotal brain region that mediates CCK-induced hypophagia.27,79,80,81 Within the NTS, the catecholaminergic A2/C2 cell group in particular is robustly activated by systemic CCK injections.43 Notably, this cell group is reciprocally interconnected with the hypothalamic OT system and receives dense innervation from parvOT neurons residing in the PVN;82 consistent with this neuroanatomical evidence, OT signaling in the brain stem synergizes with gut-derived signals upon a nutrient preload to promote satiety in lean, SC diet-fed animals.83 While receiving dense innervation by parvOT neurons, the NTS conversely targets the PVN via ascending catecholaminergic projections. In fact, norepinephrine release within the hypothalamus is highly correlated with systemic OT secretion following peripheral CCK injections,79 strongly implying that PVNOT neurons and the A2/C2 cell group constitute an interrelated tandem for processing CCK-mediated information. We now report that the blunted co-activation of this PVN-NTS neurocircuit in HFHS diet-fed mice can be overcome solely by chemogenetic stimulation of PVNOT neurons, thereby restoring neural activation in the NTS, particularly its catecholaminergic TH+ neurons. Besides the A2/C2 cell group, the NTS additionally harbors neurons that produce glucagon-like peptide 1 (GLP-1), another potent anorexigen, whose central signaling profoundly intersects with the OT81,84 and CCK pathways.85 Future research should discern whether GLP1-producing neurons in the NTS are involved in diet-induced OT-CCK signaling perturbation. Last, chemogenetic activation of PVNOT neurons not only induced robust c-Fos expression in the targeted population itself but, surprisingly, also broadly across the PVN when applied concomitantly with CCK administration. Given that the PVN contains multiple neuronal subtypes other than OT neurons with pivotal roles in energy homeostasis,86 future research should address which exact micro-neurocircuits in the PVN are contributing to OT-CCK-mediated food intake suppression and how they interact in the context of specific disease states such as obesity. In sum, this emergent complexity and intricate cross-talk among various gut-to-brain-pathways on peripheral and central levels make it ever clearer that we are still far from a comprehensive understanding of the polyhormonal control of eating.
Obesity is associated with increased Oprk1 mRNA expression and inhibitory κ-opioid signaling in PVNOT neurons that restrain their excitability in response to CCK
By devising single-nucleus isolation in combination with snRNA-seq2, we mapped the molecular heterogeneity across the entire hypothalamic OT system. Importantly, the hypothalamic OT system consists of a large proportion of neurons outside the PVN. These are not only found in the SON but also in lesser defined, so-called ANs of the hypothalamus as well as in adjacent areas, which exhibit profoundly species-specific expression patterns.87,88,89,90 Thus future research should explore the role and molecular profiles of these other OTergic cell groups; e.g., by spatial transcriptomics, while also considering species and sex differences. For this report, however, we focused on different dietary contexts in male mice and were able to demonstrate that multiple individual OT neurons express a series of different metabolism-related receptors. Intriguingly, these findings are indicative of distinct populations with partly overlapping sensitivities toward metabolic cues. Oprk1, which encodes for the inhibitory KOR, was significantly increased in Cckar+ OT neurons by chronic HFHS diet exposure. Oprk1 mRNA expression additionally overlapped pronouncedly with that of Fam19a1 (family with sequence similarity 19, member 1a), a brain-enriched and metabolically responsive neurokine91 that has been associated with human obesity in a recent genome-wide association study.92 Prompted by the striking Oprk1 expression levels as well as by the suggested cross-talk with CCKAR signaling, we went on to functionally probe for tonic inhibition of CCK-evoked PVNOT neuron activation under HFHS feeding using ex vivo electrophysiology. Indeed, we were able to restore excitability toward CCKAR agonism by selectively blocking KOR signaling in brain slices of HFHS diet-fed mice, while CCK’s hypophagic competence could be readily restored in DIO mice by systemic KOR antagonism. Consistent with previous reports,93,94 we now provide additional evidence of the functional significance of increased κ-opioid tone onto hypothalamic OT neurons and propose that it is a major mechanism that contextually restrains the CCK-evoked activation under obesogenic diets. The elucidation of this hitherto elusive mechanism might shed light on prior observations that pharmacological blockade of opioid signaling specifically in the PVN reduces homeostatic and hedonic feeding in rats95 and on the fact that mice globally lacking KOR (Oprk1−/− mice) are protected from developing dietary obesity.96 Further in-vivo studies will be required to disentangle the functional intersections between the OT system, CCK, endogenous opioids, and perhaps other factors, such as FAM19A1, in the context of metabolic disorders. Last, we are also intrigued by our finding suggesting that social context, such as single versus group housing, modulates the interaction among these different brain systems. Building on some of our recent publications,29 we seek to address this intersection between energy homeostasis and sociability in future work. In sum, we have shed light on the molecular heterogeneity within the OT system as well as on its (mal)adaptive plasticity that occurs under (patho)physiological contexts. Future studies will be required to ascertain whether these insights might aid to harness OT as such or in combination with synergistic compounds like CCK and/or KOR blockers for the development of next-generation precision medicine to correct metabolic physiology.
Conclusion
In conclusion, we have identified the mechanisms as to why systemic CCK fails to activate hypothalamic OT neurons under obesogenic diets and delineated approaches such as endogenous opioid modulation to recouple CCK signaling with hypothalamic OT pathways for restoration of gut-brain satiation signaling. These experiments ultimately reinforce the concept that PVNOT neurons constitute a molecularly and functionally diverse assembly of neurons that are tightly enmeshed within a widespread metabolic control network in which they occupy a key position, completing a feedback loop between afferent peripheral signals, central circuits, and behavior.
Limitations of the study
One limitation of our study is that we only provide indirect proof of a causal role of CCKAR expression in PVNOT neurons for CCK-mediated hypophagia. This open question might be addressed by future experiments using conditional gene deletion in CCKAR.flox mice. Moreover, we decided against combining CCK administration with acute silencing of PVNOT neurons by using, e.g., inhibitory DREADD (hM4Di). Although such an acute loss-of-function approach theoretically bears value to further ascertain the results obtained by PVNOT neuron ablation using DTA, we expected considerable technical challenges and confounders, such as the uncertainty whether Gi/o signaling equally suppresses OT release from the somata and dendrites (within PVN) or from axon terminals (NTS).97 Last, future work is needed to decipher the underlying neuronal networks in more detail, including the bidirectional PVN-NTS neurocircuit, as well as to identify the source of κ-opioid modulation of PVNOT neurons; e.g., by putative prodynorphin-expressing neurons.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Abcam | Cat#ab13970; RRID: AB_300798 |
| Guinea pig anti-(Arg8)-vasopressin | Peninsula Laboratories | Cat#T5048; RRID: AB_518680 |
| Mouse anti-OT-neurophysin | Dr. Harold Gainer | PS38 |
| Rabbit anti-C-CASP3 (Asp175) | Cell Signaling | Cat#9661; RRID: AB_2341188 |
| Rabbit anti-c-fos | Synaptic Systems | Cat#226003; RRID: AB_2891278 |
| Rabbit anti-dsRed (Living Colors®) | Takara Bio | Cat#632496; RRID: AB_10013483 |
| Alexa Fluor 405 goat anti-mouse IgG | Invitrogen | Cat#A31553 |
| Alexa Fluor 488 goat anti-chicken IgG | Invitrogen | Cat#A11039 |
| Alexa Fluor 568 donkey anti-rabbit IgG | Invitrogen | Cat#A10042 |
| Alexa Fluor 647 donkey anti-goat IgG | Invitrogen | Cat#A21447 |
| Alexa Fluor 647 donkey anti-rabbit IgG | Invitrogen | Cat#A31573 |
| Alexa Fluor 647 donkey anti-sheep IgG | Invitrogen | Cat#A21448 |
| Chemicals, peptides, and recombinant proteins | ||
| 4′, 6-Diamidino-2-phenylindol (DAPI) | Thermo Fisher | Cat#1306; CAS No. 28718-90-3 |
| 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) | Sigma-Aldrich | Cat#C239; CAS No. 115066-14-3 |
| A-71623 (CCKAR agonist) | Tocris | Cat#2411/1; Cas No. 130408-77-4 |
| Bovine serum albumin | Sigma-Aldrich | Cat#A8806; CAS No. 9048-46-8 |
| Calbiochem® Phosphatase and Protease Inhibitors | Merck Millipore | Cat#134544 |
| Cholecystokinin octapeptide, sulfated | Tocris | Cat#1166; CAS No. 25126-32-3 |
| Clozapine-N-oxide | Enzo | Cat#BML-NS105-0025; CAS No. 34233-69-7 |
| D-(−)-2-Amino-5-phosphonopentanoic acid (D-AP5) | Tocris | Cat#0106; CAS No. 79055-68-8 |
| Deoxycholate | Thermo Fisher | Cat#89005; CAS No. 302-95-4 |
| Dibenzyl ether (DBE) | Sigma-Aldrich | Cat#33630; CAS No. 103-50-4 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#276855; CAS No. 67-68-5 |
| DL-Dithiothreitol (DTT) | Sigma-Aldrich | Cat#D8255; CAS No. 578517 |
| Fluorogold | Santa Cruz | Cat#sc-358883; CAS No. 223769-64-0 |
| Gelatin from porcine skin (SUMI) | VWR International | Cat#SAFSG1890; CAS No. 9000-70-8 |
| Glycine | Sigma-Aldrich | Cat#G7126; CAS No. 56-40-6 |
| Heparin sodium | Sigma-Aldrich | Cat#H0200000; CAS No. 904108-1 |
| IPEGAL CR-630 | Sigma-Aldrich | Cat#18896; CAS No. 9002-93-1 |
| Methanol | Sigma-Aldrich | Cat#322415; CAS No. 67-56-1 |
| Mowiol® 4-88 | Merck Milllipore | Cat#81381-250G; CAS No. 81381 |
| NeuroTrace™ 435/455 | Thermo Fisher | Cat#N21479 |
| Nonidet™ P40 substitute | Sigma-Aldrich | Cat#74385; CAS No. 9016-45-9 |
| Paraformaldehyde | Carl Roth | Cat#0335.2; CAS No. 30525-89-4 |
| Picrotoxin | Sigma-Aldrich | Cat#P1675; CAS No. 124-87-8 |
| Pierce™ Horsereadish peroxidase | Thermo Fisher | Cat#31490; CAS No. 9003-99-3 |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat#P3911-25G; CAS No. 7447-40-7 |
| Primers | Thermo Fisher | N/A |
| RNasin Plus RNase inhibitor | Takara Clontech | Cat#2313A |
| Spermidine | Sigma-Aldrich | Cat#85553; CAS No. 124-20-9 |
| Spermine | Sigma-Aldrich | Cat#S3256; CAS No. 71-44-3 |
| SuperAsein RNase Inhibitor | Thermo Fisher | Cat#AM2696 |
| Syntocinon (Oxytocin) | Selleckchem | Cat#P1902; CAS No. 50-56-6 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648; CAS No. 10540-29-1 |
| TaqMan Gene Expression Assay | Thermo Fisher | N/A |
| TaqMan Universal Master Mix II, no UNG | Thermo Fisher | Cat#4440040 |
| Triton X-100 | Roche Diagnostics | Cat#11858620; CAS No.9036-19-5 |
| Tween 20 | Sigma-Aldrich | Cat#P1379; CAS No. 9005-64-5 |
| Critical commercial assays | ||
| Agilent RNA 6000 Pico Kit | Agilent Technologies | Cat#5067-1513 |
| Quant-IT RiboGreen Kit | Thermo Fisher | Cat#Q33210 |
| RNeasy Micro Kit | QUIAGEN | Cat#74004 |
| RNeasy Mini Kit | QUIAGEN | Cat#74104 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit | TaKaRa | Cat#6344893 |
| SMARTer® PCR cDNA synthesis kit | TaKaRa | Cat#634455 |
| Tetramethyl benzidine substrate (TMB) | BioLegend | Cat#421101 |
| Deposited data | ||
| snRNA-seq2 (raw data; GEO - NCBI) | This paper | GSE243448 |
| RiboTag-mRNA-seq (raw data; GEO – NCBI) | This paper | GSE242245 |
| RiboTag-mRNA-seq and snRNA-seq2 - code (Zenodo) | Zenodo | https://doi.org/10.5281/zenodo.7673205; https://zenodo.org/record/7673205#.Y_h_bHbMI2x |
| RiboTag-mRNA-seq and snRNA-seq2 - code (Github) | Github | https://github.com/viktormiok/snRNAseq_RiboTagRNAseq-oxytocin |
| Experimental models: Organisms/strains | ||
| Ai14 (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J) | Jackson Laboratory | RRID: IMSR_JAX:007914 |
| CAG-Sun1sfGFP (B6;129-Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat/J) | Jackson Laboratory | RRID: IMSR_JAX:021039 |
| Oxtrflox(B6.129(SJL)-Oxtrtm1.1Wsy/J) | Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Oxytocin-ires-Cre (B6;129S-Oxttm1.1(cre)Dolsn/J) | Jackson Laboratory | RRID: IMSR_JAX:024234 |
| RiboTag (B6N.129-Rpl22tm1.1Psam/J) | Jackson Laboratory | RRID: IMSR_JAX:011029 |
| ROSA-DTA (B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J) | Jackson Laboratory | RRID: IMSR_JAX:009669 |
| ROSA.CreERT2(B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J) | Jackson Laboratory | RRID: IMSR_JAX:008463 |
| Oligonucleotides | ||
| Cckar (TaqMan probe) | Thermo Fisher | Mm00438060 |
| Cckbr (TaqMan probe) | Thermo Fisher | Mm00432329 |
| Hprt (TaqMan probe) | Thermo Fisher | Mm01545399 |
| Mc4R (TaqMan probe) | Thermo Fisher | Mm00457486 |
| Ot (TaqMan probe) | Thermo Fisher | Mm01329577 |
| Otr (TaqMan probe) | Thermo Fisher | Mm01182684 |
| Sim1 (TaqMan probe) | Thermo Fisher | Mm00441390 |
| Software and algorithms | ||
| FIJI | NIH | https://ImageJ.net/Fiji/Downloads |
| FlowJo | BD | N/A |
| GraphPad Prism 9.4.1 | GraphPad Software | N/A |
| IMARIS ×64 9.1.2 and ×64 9.2 | Bitplane, Oxford Instruments | N/A |
| MacLab Chart Pro 7.0 | GE Healthcare | N/A |
| RStudio 4.21 | Posit, RStudio PBC | https://www.rstudio.com/products/rstudio/download/ |
| Other | ||
| AAV2/1-DIO-CAG-GCaMP6f | (Chen et al.)98 | Addgene viral prep #100839 |
| AAV2/1-DIO-hSYN1-hM3Dq-mCherry | (Krashes et al.)99 | Addgene viral prep #44361 |
| AAV2/1-DIO-hSYN1-mCherry | Bryan Roth lab (unpublished) | Addgene viral prep #50459 |
| AAV2/1-OTp-iCre | Custom made23 | N/A |
| AAV2/1-OTp-Venus | Custom made23 | N/A |
| Ketamine (Ketabel) | HFW/Bremer Pharma GmBH | CAS No. 6740-88-1 |
| Meloxicam | WDT/Pharma-Partner-Vetriebs GmBH | CAS No. 71125-38-7 |
| Metamizol | WDT/Pharma-Partner-Vetriebs GmBH | CAS No. 68-89-3 |
Resource availability
Lead contact
Requests for further information, reagents and resources should be directed and will be fulfilled by the lead contact, Cristina García-Cáceres (garcia-caceres@helmholtz-muenchen.de).
Materials availability
Mouse lines (ROSA26.LSL.DTA; OT-ires-Cre; and OTR.flox) and AAV vectors (e.g., AAV2/1-OTp-iCre, AAV2/1-OTp-Venus, AAV2/1-DIO-CAG-GCaMP6f, AAV2/1-hSYN1-(hM3Dq-)mCherry etc.) used in this study are available from the lead contact upon request.
Experimental model and subject details
Mouse models
Animal studies were approved by the Animal Ethics Committee of the government of Upper Bavaria (Germany) and by the Institutional Animal Care and Use Committee at the Van Andel Institute (USA). Wildtype mice (C57BL/6J, Janvier, Le Genest-Saint-Isle, France) or genetically modified mice at adult age (>12 weeks) were provided ad libitum access to either a pelleted standard chow (SC) diet (5.6% fat; LM-485, Harlan Teklad) or a high-fat, high-sucrose (HFHS) diet (D12331; 58% of calories from lipids; Research Diets, New Brunswick, NJ). Animals had continuous free access to water and were maintained at 23°C with constant humidity on a 12-h light–dark cycle. OT-ires-Cre (also known as OXT-IRES-Cre) were originally provided from Jackson Laboratory (strain name: B6; 129S-Oxttm1.1(cre)Dolsn/J; # 024234); homozygous OT-ires-Cre males were interbred with non-Cre-carrying female mice to obtain experimental cohorts of male mice containing the knock-in allele in heterozygosity. RiboTag mice (strain name: B6N.129-Rpl22tm1.1Psam/J; # 011029), Ai14 (strain name: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J; # 007914), ROSA-DTA (strain name: B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J; # 009669), and CAG-Sun1sfGFP (strain name: B6; 129-Gt(ROSA)26Sortm5(CAG−Sun1/sfGFP)Nat/J; # 021039) were all provided from Jackson Laboratory and used in heterozygosity in final cohorts. Global, inducible OTR knockout mice (OTRiΔ mice) were generated by crossing Oxtrflox (strain name: B6.129(SJL)-Oxtrtm1.1Wsy/J; # 008471) and R26-CreERT2 (strain name: B6; 129-Gt(ROSA)26Sortm5(Cre/ERT2)Tyj/J; # 008463), both provided by Jackson Laboratory, for two successive generations to obtain heterozygous R26-CreERT2 mice homozygous for Oxtrflox allele. At 6 weeks of age, male OTRiΔ mice were injected with tamoxifen (10 mg/kg BW; i.p.) for 5 consecutive days. Tamoxifen was dissolved in sunflower oil at a final concentration of 10 mg/mL at 37°C and filter sterilized.
Genotyping of mouse lines
Ear tags were obtained from mice at the age of 3 weeks and DNA was isolated by boiling the ear tags for 30 min in 200 μL 50 mM NaOH at 95°C (ThermoMixer C, Eppendorf). Afterward, 20 μL 1 M Tris was added to normalize the pH. 2 μL of isolated genomic DNA was used for the genotyping PCR (Promega) using respective protocols.
Method details
Physiological measures and metabolic phenotyping
For assessing acute feeding behavior, mice were individually housed in metabolic cages (TSE PhenoMaster; TSE systems). After 48 h acclimatization, food was removed daily for 3 h (3p.m.–6p.m.) and mice received sham injections with Vehicle (0.9% NaCl) for two consecutive days. On Day three, all mice received first CNO (1 mg/kg BW; i.p.; 25 min before dark onset) and then CCK-8s (20 μg/kg BW; i.p.; 10 min before dark onset). With dark onset, food hoppers were returned and food intake was automatically measured every 5 min. Normal baseline food intake (“Vehicle”) is represented as the calculated mean intake upon the two sham injections. Mice that displayed food spilling were excluded from analysis. Cumulative long-term food intake was measured by co-housing mice (two mice of the same group and of similar body weight per cage); daily food intake was measured per cage, divided by two, and represented as single data point. Body composition (fat and lean mass) was assessed by using a magnetic resonance whole-body composition analyzer (Echo-MRI, Houston, TX). Energy expenditure and respiratory exchange ratio (RER) of individual mice were analyzed within metabolic cages of a combined indirect calorimetry system (TSE PhenoMaster; TSE systems). Food and water intake, O2 consumption, CO2 production, and locomotor activity (i.e., horizontal and vertical beam breaks) were measured in 5-min intervals. EE (kcal/h) was calculated based on the Weir Equation 3.815∗10−3∗VO2+1.232∗10−3∗VCO2) and total EE (kcal) was correlated to the mean of body weight measured before and after the measurement.100 Latency to feed was measured by counting the elapsed time from the moment of food presentation until the first meal was consumed by the mouse (≥10 mg as measured via the food sensor). Glucose tolerance was assessed by the intraperitoneal administration of a glucose bolus (2 g/kg BW; 20% w/v in 1xPBS pH 7.4). Before the glucose tolerance test, mice were fasted for 4 h and glycemia was measured by sampling blood from the tail vein before (0 min) and at 15, 30, 60 and 120 min post injection via a handheld glucometer (Abbott, Wiesbaden, Germany). To assess insulin sensitivity, additional blood was collected at 0 min using EDTA-coated microvette tubes (Sarstedt, Nürnbrecht, Germany) to obtain plasma (5,000 x g for 10 min at 4°C). Insulin concentration was determined using a commercial insulin ELISA following the manufacturer’s instructions (Ultra-sensitive Mouse Insulin ELISA Kit, #90080 Crystalchem, Netherlands). HOMA-IR was calculated using the formula: HOMA-IR = [fasting insulin (mU/l) ∗ fasting glucose (mg/dL)]/405 (Matthews et al., 1985). Glycated HbA1C was analyzed using DCA Vantage Analyzer (Siemens, City, Germany).
Ex-vivo brain slices preparation
Adult male mice were sacrificed by cervical dislocation and the brain quickly ablated and placed in ice-cold artificial cerebrospinal fluid (aCSF) modified for slicing, containing (in mM): 87 NaCl, 2.69 KCl, 1.25 NaH2PO4, 26 NaHCO3, 7 MgCl2, 0.2 CaCl2, 25 D-glucose, and 75 sucrose (330 mOsm/Kg H2O, pH 7.4 bubbled with a carbogen mixture of 95% O2 and 5% CO2). The specimen was glued to a sectioning stage and submerged in ice-cold slicing aCSF in a vibratome (VT1200; Leica Biosystems) chamber. Coronal brain slices (250 μm) containing the paraventricular nucleus of the hypothalamus were sectioned and incubated at 32°-33° for 30 min in aCSF, containing (in mM): 124 NaCl, 2.69 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1.2 MgCl2, 2 CaCl2, 2.5 D-glucose, and 7.5 sucrose (298 mOsm/Kg H2O, pH 7.35 constantly bubbled with a carbogen mixture of 95% O2 and 5% CO2). After this period, the slices were kept under the same conditions but at room temperature for at least 40 min until electrophysiological recordings.
2-Photon excitation calcium imaging
Cytosolic calcium levels from PVNOT neurons conditionally tagged by Ai14-tdTomato within acute coronal brain slices (250 μm) of mice were monitored by 2-photon excitation microscopy using the genetically encoded calcium indicator GCaMP6f. Single brain slices were transferred to a chamber mounted on a stage of an upright multiphoton laser scanner microscope (FVMPE-RS system, Olympus) and continuously perfused with bubbled-aCSF using a gravity-driven perfusion system at a rate of ∼3 mL/min in the presence of synaptic blockers (20 μM CNQX, 50 μM D-AP5, and 100 μM picrotoxin). Neurons were visualized with a 25× water immersion objective. Excitation illumination was generated by an InSight X3 DUAL tunable laser system (Spectra-Physics). The FluoView image acquisition software (FV31S-SW, Olympus) was used to tune laser emission wavelength to 930 and 1045 nm in order to obtain 2-photon absorption signals from GCaMP6f and tdTomato fluorophores, respectively, at an acquisition rate of 0.5 Hz. Calcium imaging from PVNOT neurons consisted of a 3-min baseline recording followed by bath application of CCK (50 nM) and drug washout. At the end of each experiment, 20 mM KCl was bath-applied to check neuronal viability and calcium signal integrity. Only neurons that responded to KCl were used for analysis. Calcium transients were estimated as changes in GCaMP6f-based fluorescence intensity over the baseline (ΔF/F0), considering a calcium event when ΔF/F0 > 3 standard deviations greater than the baseline fluorescence signal. The number of calcium events were then plotted over time grouped into 1-min bins in order to quantify changes in the frequency of calcium events in PVNOT neurons in response to CCK application.
Electrophysiological recordings
Single brain slices were transferred to a chamber mounted on the stage of an upright microscope (BX51WI; Olympus) coupled with a video camera (Prime 95B; Teledyne Photometrics) and continuously perfused with carbogen-bubbled aCSF at a rate of ∼2 mL/min by a gravity-driven perfusion system. Neurons were visualized under infrared differential interference contrast (IR-DIC) optics with a 20× immersion objective (2× post-magnification) using the μManager 1.4 software.101 Accordingly, Ai14-tdTomato+ neurons were identified by epifluorescence-based signals excited at 555 nm wavelength (LedHUB; Omicron-Laserage Laserprodukte GmBH). Patching pipettes were made with thick-walled borosilicate glass (GC150TF-10; Harvard Apparatus) pulled using a horizontal puller (P-97; Sutter Instruments) and filled with an internal solution, containing (in mM): 128 K-gluconate, 8 KCl, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 10 Na-phosphocreatine (295 mOsm/Kg H2O, pH 7.3), resulting in a pipette tip resistance between 4 and 10 MΩ. Whole-cell recordings were performed with a MultiClamp 700B amplifier (Molecular Devices) in current-clamp mode. Data were acquired at 10–20 kHz and low-pass filtered at 5 kHz (Bessel) with a Digidata 1550B (Molecular Devices) using the Clampex 11.2 acquisition software (pClamp; Molecular Devices). Parvocellular neurons were identified by the presence of a transient outward rectifying current triggered by the application of a hyperpolarizing pulse (ranging from −60 to −40 pA for 1 s) followed by positive current injections (from 20 to 100 pA in five steps of 1 s).29 Membrane potential was monitored in response to bath-applied drugs at I = 0. In some experiments, 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μΜ), D-(−)-2-Amino-5-phosphonopentanoic acid (D-AP5; 50 μM), and picrotoxin (100 μM) were applied to block fast neurotransmission. Membrane input resistance was measured by the slope of the curve obtained by the response of the membrane potential to injected negative currents (from −40 to −60 pA in three steps of 1 s). Data were visualized and analyzed using custom-written codes in MATLAB (MathWorks).
Adeno-associated viruses (AAV) and stereotaxic surgery
In order to assess PVNOT neuronal activity, we conducted stereotaxic surgeries to target the fluorescent Ca2+ indicator GCaMP6f to PVNOT neurons in OT-ires-Cre mice. AAV2/1-DIO-CAG-GCaMP6f was purchased from Addgene (#100839,98 titer: 1.4 x 1013 gc/ml). To chemogenetically activate PVNOT neurons by means of DREADD we employed AAV2/1-DIO-hSYN1-hM3Dq-mCherry (Addgene # 4436199; titer: 1.6 x 1013 gc/ml) versus a respective control virus AAV2/1-DIO-hSYN1-mCherry (Addgene # 50459; titer: 9 x 1012 gc/ml). To ablate PVNOT neurons, we produced AAV2/1-OTp-iCre (titer: 2.3 x 1012 gc/ml) and AAV2/1-OTp-Venus (titer: 1.2 x 1013 gc/ml) according to our previously published protocol,23 which contain a synthetic 2.6 kb OT promoter element (OTp) faithfully restricting viral transgene expression to OT neurons. Respective AAVs were injected bilaterally (300 nL per hemisphere) using a stereotaxic system combined with a binocular 3.5x-90x stereomicroscope (AMScope, USA). Mouse skull was exposed via a small incision and a craniotomy above the viral injection sites was performed using a micro-precision drill. A pulled glass pipette with a 20–40 μm tip diameter was carefully lowered to reach the PVN on each side of the brain, respectively, and AAVs were applied at low speed using manually-applied air pressure. Undesired diffusion of viral particles was further prevented by slow retraction of the glass pipette 5 min post injection. The following stereotaxic coordinates were obtained from ‘The Mouse Brain in Stereotaxic Coordinates’ (Franklin and Paxinos, 2019) and used to target the PVN of the mouse brain: −0.7 mm posterior and ±0.2 mm lateral to the bregma and −4.75 mm ventral from the dura mater. Anesthesia was performed by a mixture of ketamine and xylazine (100 mg/kg body weight and 7 mg/kg bodyweight, respectively) while acute Metamizol (200 mg/kg, subcutaneous) followed by Meloxicam (1 mg/kg, on three consecutive days, subcutaneous) was administered for postoperative analgesia.
Transcardial perfusion, brain sectioning and immunohistochemistry
Animals were sacrificed with CO2 and transcardially perfused with 20 mL phosphate-buffered saline (PBS) (GibcoTM, pH 7.4) by using a peristaltic pump at 120 mmHG (Instech, High Flow P720 equipped with 21G canula). Perfusions were finalized with 20 mL of 4% paraformaldehyde (PFA) in PBS, pH 7.4, brains were removed and post-fixed in 4% PFA at 4°C overnight. The following day, brains were then equilibrated with 30% sucrose in Tris-buffered saline (TBS, pH 7.2) for 48 h before being sectioned into 40 μm coronal slices using a cryostat (CM3050S; Leica, Germany). Three to four brain sections per mouse were selected containing the middle portion of the PVN or NTS and further subjected to additional immunohistochemistry. Brain sections were first washed with TBS and incubated overnight at 4°C with primary antibodies in a solution containing 0.25% porcine gelatin and 0.5% Triton X-100 in TBS, pH 7.2. The next morning, sections were serially rinsed in TBS, pH 7.2, and incubated with respective secondary antibodies diluted in TBS, pH 7.2 containing 0.25% porcine gelatin and 0.5% Triton X-100 for 2h. Sections were serially washed in TBS with the last washing additionally containing fluorescent Nissl stain (1:100 NeuroTrace 435/455; ThermoFisher, Germany) to identify neuronal cells and anatomical demarcations. Lastly, brain sections were mounted on gelatin-coated glass slides, dried and cover-slipped with a polyvinyl alcohol (mowiol, Merck, Germany) mounting medium supplemented with DABCO (Merck, Germany).
IMARIS-assisted reconstruction of cellular surface
Three-dimensional reconstruction of cellular surfaces was performed using IMARIS.29,102 In brief, 5 male ROSA-DTA mice (8 weeks of age) were stereotaxically injected with a mix of AAV2/1-OTp-iCre and AAV2/1-OTp-Venus, perfused 5 days post-injection, and brains were subjected to immunohistochemical staining. After high-resolution confocal microscopy image acquisition (60× objective), raw immunosignal of fluorescent channels (i.e., OT, Venus and C-CASP3) was used as a template for the subsequent surface reconstruction using the surface reconstruction mode in IMARIS 9.2 (Bitplane, Oxford Instruments). The following settings were used: Smooth surface, Surface detail: 0.6 μm, Background Subtraction: 1.6 μm and automatic threshold selection via Imaris. Reconstructed particles/debris with a cell volume <20 μm3 were considered unspecific background signal and filtered out in a subsequent step.
Analysis of co-localization C-CASP3-ir and OT-ir
For co-localization of C-CASP3 and OT immunoreactivity, we first assessed the average background intensity of each individual image by randomly drawing three ROIs in spots where no C-CASP3 signal was observed. The average intensity (AU) of these ROIs was used as a reference point for the subsequent co-localization analysis. Next, we drew individual ROIs for each OT neuron and measured the respective C-CASP3 immunoreactivity. An OT neuron was considered apoptotic (i.e., C-CASP3-positive) when the respective C-CASP3 immunoreactivity (AU) was at least 4-times higher than the previously established background signal.
Fluorogold injections
To distinguish magnOT and parvOT neurons, fluorogold (15 mg/kg BW; i.p.) was administered to adult male OT:Ai14 reporter mice maintained on SC diet. Mice were perfused 7 days post injection of fluorogold following 3 h of food removal plus two additional hours after CCK-8s injection (20 μg/kg BW; i.p.).
CCK-8s injections
For all in-vivo experiments, food was removed for 3 h before dark onset (3–6 p.m.) in order to normalize feeding behavior. Cholecystokinin-octapeptide, sulfated (CCK-8s; Tocris, St. Louis, USA) was reconstituted in 0.9% NaCl and administered 10 min before dark onset (20 μg/kg BW; i.p.).
iDISCO-based whole-brain clearing
Clearing protocol was adopted from45 with slight adjustments. In brief, OT:Ai14 mice were transcardially perfused with 1xPBS (pH 7.4) followed by 4% PFA and their brains were carefully removed. Following post-fixation in 4% PFA overnight at 4°C, brains were washed three times in 1 x PBS (0.2% Triton X-100; v/v) for 1 h at room temperature. Brains were pre-treated by incubation in 1 x PBS (0.2% Triton X-100 and 20% DMSO; v/v) for 48 h shaking at 37°C followed by 1 x PBS (0.1% Tween 20, 0.1% Triton X-100, 0.1% Deoxycholate, 0.1% NP40 and 20% DMSO; v/v and w/v, respectively) for 48 h shaking at 37°C. After washing brains three times in 1 x PBS (0.2% Triton X-100; v/v) for 1 h at 37°C, they were incubated in permeablization solution (1 x PBS with 0.2% Triton X-100, 0.3 mM glycine, and 20% DMSO; v/v and w/v, respectively) for 48 h shaking at 37°C. Thereafter, brains were incubated in blocking solution (1 x PBS with 0.2% Triton X-100, 3% donkey serum, 3% rabbit serum, and 10% DMSO; v/v) for 48 h shaking at 37°C. After being washed four to five times in 1 x PBS (0.2% Triton X-100, and 10 μg/ml heparin; v/v), brains were incubated with the secondary antibody (donkey-anti-rabbit Alexa Fluor 647) in 1 x PBS with 0.2% Triton X-100, 10 μg/mL heparin, 3% donkey serum, and 5% DMSO (v/v) for four days shaking at 37°C. After washing brains four to five times in 1 x PBS (0.2% Triton X-100, and 10 μg/ml heparin; v/v) they were incubated with the secondary antibody (donkey-anti-rabbit Alexa Fluor 647) in 1 x PBS with 0.2% Triton X-100, 10 μg/mL heparin, 3% donkey serum, and 5% DMSO (v/v) for four days shaking at 37°C. After immunolabeling, brains were washed again for four to five times in 1 x PBS (0.2% Triton X-100, and 10 μg/mL heparin; v/v). Brains were cleared by an ascending dilution of methanol/H2O (20%, 40%, 60%, 80%, 100%) for 1 h each at room temperature and were left in fresh 100% methanol overnight. Delipidation was achieved by incubation in 66% dichloromethane (DCM) and 33% methanol for 6 h at room temperature. After short incubations of brains in 100% DCM, they were finally placed in the refractive index matching solution consisting of 100% dibenzyl ether (DBE) until imaging.
Translating ribosome affinity purification (TRAP)
Adult male OT:RiboTag mice with both transgene alleles in heterozygosity were fed either SC diet or HFHS diet for 3 months. On the day of experiments, food was removed for 3 h (3 p.m.–6 p.m.) and mice received either vehicle or CCK-8s (20 μg/kg BW; i.p.; Tocris, St. Louis, USA) injection at dark onset (6 p.m.) and were sacrificed 2 h post injection. Hypothalami were rapidly removed, snap-frozen and stored at −80°C until further processing. Per sample, two hypothalami were combined and processed according to previously published protocol.103 Yield of input and immunoprecipitate were independently quantified using a Bioanalyzer (Agilent RNA 6000 Pico Kit; Agilent Technologies, Santa Clara, USA) and Quant-IT RiboGreen Kit (ThermoFisher Scientific Inc., Rockford, IL USA) according to the manufacturer’s instructions.
Samples were amplified and synthesized into cDNA according to the manufacturer’s protocol using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara Bio Inc., Shiga, Japan) and amplification yield was checked using a Bioanalyzer (Agilent High Sensitivity DNA Kit, Agilent Technologies, Santa Clara, USA). Single-indexed libraries were generated using ThruPLEX DNA-seq Kit (Takara Bio Inc., Shiga, Japan), pooled and checked again using a Bioanalyzer (Agilent High Sensitivity DNA Kit, Agilent Technologies, Santa Clara, USA).
TRAP transcriptomics analyses
Sequencing was performed at the Helmholtz Zentrum München (HMGU) by the NGS-Core Facility. After a final quality control, the libraries were sequenced in a paired-end mode (2x150 bases) in the Novaseq6000 sequencer (Illumina) with a depth of ≥40 million paired reads per sample. BCL files were converted to FASTQ using Bcl2fastq v.2.20. The alignment was done using STAR v.7.2a and was mapped to the mouse reference genome GRCm38 (mm10). The counts and FPKM were generated using HTSeq v0.11.2 For transcriptomics downstream analysis, sample distance matrix was generated using pheatmap package from R.104 Differential expression analysis results were illustrated in the form of volcano plots using R-package EnhancedVolcano.105 The expression profile of DEGs over the samples was shown in the heatmap using R-package pheatmap.104 Wald significance test was employed for FDR correction. GO enrichment analyses were performed, and the results were illustrated using clusterProfiler106 and ggplot2107 R-packages, respectively.
RNA isolation and qPCR analysis
RNA was isolated from tissues using a commercially available kit (MicroRNeasy Kit, Qiagen, Hilden, Germany). Identical amounts of RNA were reverse-transcribed to cDNA using Superscript III (Invitrogen, Darmstadt, Germany) and gene expression was analyzed using TaqMan probes (ThermoFisher Scientific Inc., Rockford, IL USA) using a ViiATM7 Real-Time PCR System or QuantStudio 6 FLEX Real-Time PCR System (ThermoFisher Scientific Inc., Rockford, IL USA). Expression changes were calculated using the 2−ΔΔCt method normalized by Hprt as housekeeping gene. When indicated, qPCR expression analysis was conducted on cDNA derived from immunoprecipitated RNA of OT:RiboTag mice following reverse (SMARTer PCR cDNA synthesis kit; Takara Bio Inc., Shiga, Japan).
Fluorescence-activated nuclei sorting (FANS)
CAG-Sun1-sfGFP mice were crossed with OT-ires-Cre mice to generate heterozygous mice.108 Whole hypothalami were individually processed to obtain single nuclei following a previously described protocol109 with slight modifications. Briefly, frozen hypothalami were transferred to a Dounce homogenizer containing 1 mL of freshly prepared ice-cold nuclei isolation buffer (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tris pH 8.0, 0.4% IGEPAL-630, 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, 1x phosphatase & protease inhibitors, 0.4 units RNasin Plus RNase Inhibitor, 0.2 units SuperAsin RNase inhibitor). Homogenization was achieved by carefully douncing 10 strokes with the loose pestle, incubating on ice for 5 min and further douncing 15 more strokes with the tight pestle. The homogenate was filtered through a 20 μm cell strainer, centrifuged at 1000 x g for 10 min at 4°C, the nuclei pellet resuspended in 450 μL of staining buffer (PBS, 0.15 mM spermine, 0.5 mM spermidine, 0.4 units RNasin Plus RNase Inhibitor, 0.4% IGEPAL-630, 0.5% BSA) and incubated for 15min on ice. Nuclei pellets were resuspended in 1 mL of fresh staining buffer supplemented with DAPI 1 μg/μL. Nuclei integrity was assessed in the DAPI channel under a Zeiss microscope (Axio Scope, Zeiss, Germany). Doublet discrimination and DAPI staining were used for appropriate gating of single nuclei and the signal on the 488 (FITC) channel of the IgG-isotype control determined the adequate gating of GFP− and GFP+ nuclei.
Fluorescence-activated nuclei sorting
GFP+ nuclei were sorted with a 70 μm nozzle into 384-well PCR plates (thin-walled, BioRad, HSP3901) prepared freshly with 940 nL of Lysis Buffer 1 (1 μL of 10X reaction buffer is diluted in 2.75 μL of water) (SMART-Seq v4 Ultra Low input RNA kit; Takara Bio Inc.) per well, aliquoted with the Mosquito HV (STP Labtech) liquid handling robot. The Reaction buffer was prepared following the manufacturer’s instruction adding 1 μL of RNAse Inhibitor in 19 μL of 10X Lysis Buffer.
We ensured maximum sorting accuracy into the wells of the 384-well plate using a colorimetric assay with tetramethyl benzidine substrate (TMB, BioLegend, ref. 421501) and 50 μg/mL of Horseradish Peroxidase (HRP, Life Technologies, ref. 31490).110
In the plate layout, we sorted nuclei from SC diet-fed animals in half of the 384-well plate and nuclei from HFHS diet-fed animals in the remaining half. After sorting, every plate was firmly sealed (MicroAmp Thermo Seal lid, #AB0558), shortly vortexed for 10 s, centrifuged (4°C, 2000 x g for 1 min), flash-frozen on dry ice, and stored at −80°C, until cDNA synthesis. A total of four 384-well plates were sorted for this study.
snRNA-seq2
The single-nucleus RNA-seq2 methodology was used to capture a high number of transcripts from frozen tissues, allowing for the generation of double-stranded full-length cDNA as described and detailed by Richter et al.111 In brief, the reaction volumes were miniaturized with the aid of the Mosquito HV robot. Per well, 2190 nL of Lysis Buffer 2 (LB2) were dispensed. The final volume of the mixture of Lysis Buffer (LB1 and LB2) was 3.125 μL, containing NP40 2%, Triton X-100 1%, 1/300,000 diluted ERCC RNA spike-in, 3′ SMART-seq CDS Primer II A and RNAse-free water.
Every flash frozen sorted plate was thawed directly on a −20°C chilled metallic holder while LB2 was added by the Mosquito HV robot. The plate was immediately sealed, vortexed 20 s at 2000 rpm, centrifuged at 2000 x g for 30 s at 4°C and placed in 72°C for 6 min. ERCC spike-ins (Thermo Fischer Scientific, ref. 4456740; Lot no. 00892098) were diluted 1 in 10, with RNAse-free water with 0.4 U/μL Recombinant RNase Inhibitor (Takara Clontech, ref. 2313A) and a fresh dilution of 1 in 300,000 was prepared before the first strand synthesis.
Reverse transcription and Pre-PCR amplification steps were followed as described by the manufacturer with four-times reduced volumes for all steps. The PCR program for the cDNA amplification was performed in a total of 21 cycles of: 1 min at 95°C, [20 s at 95°C, 4 min at 58°C, 6 min at 68°C] × 5, [20 s at 95°C, 30 s at 64°C, 6 min at 68°C] × 9, [30 s at 95°C, 30 s at 64°C, 7 min at 68°C] × 7, 10 min at 72°C. After cDNA synthesis, the yield was assessed in an Agilent Bioanalyzer with a High Sensitivity DNA kit.
Library preparation for snRNA-seq2 and sequencing
Sequencing libraries were prepared using the Illumina Nextera XT DNA Sample Preparation kit (Illumina, Ref. FC-131-1096) and the combination of 384 Combinatorial Dual Indexes (Illumina- Set A to D, Ref. FC-131-2001 to FC-131-2004). Using the Mosquito HV robot, the reaction volumes of the Nextera XT chemistry were miniaturized, and the steps followed minutely as described by Richter et al. 2021.111,112
In brief, 500 nL of the undiluted cDNA were transferred to a new 384 well-plate containing 1500 nL of Tagmentation Mix (TD and ATM reagents). Accordingly, all Nextera XT reagents (NT, NPM and i5/i7 indexes) were added stepwise to a final library volume of 5 μL per well. The final PCR amplification was performed through 12 cycles. Once the libraries were prepared, 500 nL from each well were pooled together into a tube (total volume of ∼192 μL) to perform a final AMPure XP bead (Beckman Coulter, Ref. A63882) clean-up step. For our single nuclei libraries, two consecutive clean-ups with a ratio of sample to bead 0,9X led to library sizes between 200 and 1000 bp. The final libraries were assessed using an HS DNA kit in the Agilent Bioanalyzer, and prior to sequencing, the libraries were quantified using a Collibri library quantification kit (Thermo Fischer Scientific, Ref. A38524100) in a QuantStudio 6 Flex (Life Technologies) for higher accuracy. Each plate, counting with a total of 384 libraries, was pooled together into one final library. A total of 4 final libraries were sequenced using an Illumina NovaSeq 6000 NGS sequencer in an SP XP flowcell, in a paired-end 150 bases length. Sequencing was performed at the Helmholtz Zentrum München (HMGU) by the NGS-Core Facility.
snRNA-seq2 analysis
The snRNA-Seq2 pipeline used in this study was generated using nextflow.113 In the first step, technical replicates of the samples were merged, and reads were then mapped to rRNA (from Ensembl, GRCm38 release 102) using Bowtie2 (version 2.3.4.3114) and subsequently, unmapped reads were mapped to the mm10 genome (from Ensembl, GRCm38 release 102) using STAR (2.7.0115). Count matrices were generated by counting reads corresponding to gene exons using featureCounts (version 1.6.3116). Counts were transformed to FPKM and TPM using the actual feature length as described by Wagner et al.117 The single-nuclei transcriptomics analysis of raw count matrix loaded into python and stored as AnnData object was performed using Scanpy version 1.7.1.118 Filtering of the cells was based on a minimum count number of 250. Filtering was applied to the matrix of 1536 single nuclei and 55579 genes. Nuclei that had >1000 and <4500 detected genes were kept. Genes detected in fewer than 25 cells and with read count below 250 were filtered out. Hence, only the nuclei having less than 4500 genes detected and a library size between 10000 and 200000 reads were kept. Applying this filtering strategy, the final matrix comprises 1202 single nuclei and 13867 genes. The remaining cell vectors were normalized using the R-package scran119 in the default setting, employing the ERCC spik-ins. Batch effect corrections were performed employing combat118 using plates as a covariate. To generate UMAP plots, we took the top 15 PCs and used the PC space to compute a k-nearest neighbor (kNN) graph (k = 50, metho = umap). Leiden clustering (resolution = 0.5, flavor = vtraag) was computed based on the kNN graph. Differential gene expression analysis compared the groups of interest using Welch’s t test while applying Benjamini-Hochberg for multiplicity correction. In order to identify transcript intersections as well as their aggregates at single-cell resolution, the novel UpSet visualization technique was applied according to the previously published protocol120 using R-package UpSet.121
Statistics
Data analysis was conducted using GraphPad Prism (Version 5). Normally distributed data were analyzed by student’s T test or one- or two-way analysis of variance (ANOVA) with Bonferroni or Tukey post-hoc analyses to determine statically significant differences. Data were screened using the maximum normal residual Grubb’s test to screen for singular, statistically significant outliers. p-values ≤0.05 were considered statistically significant. All data are presented as mean ± standard error (SEM).
Quantification and statistical analysis
Data analysis was conducted using GraphPad Prism (Version 9.4.1) and Rstudio (4.2.1) No power calculations were used to predetermine sample size. Sets of data were analyzed by linear regression, Student’s t-test, or one- or two-way analysis of variance (ANOVA) with Turkey post-hoc analyses to determine statistically significant differences. Normal distribution of populations at the 0.05 levels was tested using the D’Agostino-Pearson omnibus normality test. Data were screened using the maximum normal residual Grubb’s test to identify singular, statistically significant outliers. Exclusion criteria of samples included mistargeting of AAV injections. Mice stereotaxically injected with AAV were randomly assigned to respective groups across multiple litters. All data are represented as mean ± standard error of the mean an p values as well as n are reported in the individual figure legends.
Acknowledgments
The authors thank Cassie Holleman, Clarita Layritz, Nicole Klas, and Elisavet Lola for excellent technical assistance. We thank Dr. Sandrine Lefort for contributing to Ca2+ imaging as well Maria Richter for critical feedback regarding bioinformatic analyses. This work was supported in part by funding to R.E.C. and P.T.P. from a Marie Skłodowska-Curie grant (ChroMe 675610). I.G.-G. is a recipient of a fellowship from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Sklodowska-Curie Actions (842080 – H2020-MSCA-IF-2018). R.H.W. received funding from the European 748 Research Council ERC under the European Union’s Horizon 2020 Research and Innovation 749 Program (715933). C.P.M.-J. and E.S.-Q. were supported by the Helmholtz Pioneer Campus. T.D.M. received funding from the German Research Foundation (DFG; TRR296, TRR152, SFB1123 and GRK 2816/1), the German Center for Diabetes Research (DZD e.V.), and the European Research Council ERC-CoG Trust no. 101044445. V.G. received funding from the German Research Foundation (DFG) (GR 3619/13-1, GR 3619/15-1, GR 3619/16-1, SFB Consortium 1158-3, and Germany-Israel Excellence Program grant GR 3619/19-1) and the European Research Council (ERC) (Synergy ERC grant OxytocINspace 101071777). C.G.-C. received funding from the European Research Council (ERC) (STG grant AstroNeuroCrosstalk 757393) and the German Research Foundation (DFG) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) and Helmholtz Excellence Network. T.G. and C.G.-C. received funding from the German Center for Diabetes Research (DZD) twinning grant 2020.
Author contributions
T.G., C.G.-C., and V.G. conceptualized and designed all experiments. T.G. (surgeries, immunohistochemistry, imaging, and qPCR); T.G., F.L., I.G.-G., L.M.D., and O.L.T. (metabolic phenotyping); R.E.C. (FACS); E.S.Q. (library preparation for snRNA-seq2); V.M., K.M., and D.L. (bioinformatics); F.A. and Q.K. (IMARIS); and C.M. and E.G.-C. (electrophysiology and Ca2+ imaging) conducted the experiments and collected and analyzed the data. T.G., C.G.-C., and V.G. wrote the manuscript in discussion with M.H.T., S.C.W., C.P.M.-J., R.H.W., J.A.P., R.G.J., T.D.M., and P.T.P., who revised the article critically for important intellectual content. All authors have read and approved the final version of the manuscript.
Declaration of interests
M.H.T. participated in a scientific advisory board meeting of ERX Pharmaceuticals, Inc. (Cambridge, MA, USA) in 2019. He was a member of the Research Cluster Advisory Panel (ReCAP) of the Novo Nordisk Foundation between 2017 and 2019. He attended a scientific advisory board meeting of the Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, in 2016. He received funding for his research projects by Novo Nordisk (2016–2020) and Sanofi-Aventis (2012–2019). He consulted for Böhringer Ingelheim Pharma GmbH & Co. KG (2020). He delivered a scientific lecture for Sanofi-Aventis Deutschland GmbH in 2020. R.G.J. is a scientific advisor to Servier, Agios Pharmaceuticals, and is a shareholder and member of the scientific advisory board of Immunomet Therapeutics.
Published: October 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113305.
Contributor Information
Tim Gruber, Email: tim.gruber@vai.org.
Valery Grinevich, Email: valery.grinevich@zi-mannheim.de.
Cristina García-Cáceres, Email: garcia-caceres@helmholtz-muenchen.de.
Supplemental information
Data and code availability
-
•
RiboTag-RNA-seq and snRNA-seq2 data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Locke A.E., Kahali B., Berndt S.I., Justice A.E., Pers T.H., Day F.R., Powell C., Vedantam S., Buchkovich M.L., Yang J., et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCormack S.E., Blevins J.E., Lawson E.A. Metabolic Effects of Oxytocin. Endocr. Rev. 2020;41:121–145. doi: 10.1210/endrev/bnz012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swanson L.W., Sawchenko P.E. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- 4.Cox J.E., Sims J.S. Ventromedial hypothalamic and paraventricular nucleus lesions damage a common system to produce hyperphagia. Behav. Brain Res. 1988;28:297–308. doi: 10.1016/0166-4328(88)90132-5. [DOI] [PubMed] [Google Scholar]
- 5.Michaud J.L., Boucher F., Melnyk A., Gauthier F., Goshu E., Lévy E., Mitchell G.A., Himms-Hagen J., Fan C.M. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 2001;10:1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 6.Faivre L., Cormier-Daire V., Lapierre J.M., Colleaux L., Jacquemont S., Geneviéve D., Saunier P., Munnich A., Turleau C., Romana S., et al. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J. Med. Genet. 2002;39:594–596. doi: 10.1136/jmg.39.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tolson K.P., Gemelli T., Gautron L., Elmquist J.K., Zinn A.R., Kublaoui B.M. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J. Neurosci. 2010;30:3803–3812. doi: 10.1523/JNEUROSCI.5444-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grinevich V., Desarménien M.G., Chini B., Tauber M., Muscatelli F. Ontogenesis of oxytocin pathways in the mammalian brain: late maturation and psychosocial disorders. Front. Neuroanat. 2015;8 doi: 10.3389/fnana.2014.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knobloch H.S., Grinevich V. Evolution of oxytocin pathways in the brain of vertebrates. Front. Behav. Neurosci. 2014;8 doi: 10.3389/fnbeh.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanson L.W., Sawchenko P.E. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31:410–417. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 11.Biag J., Huang Y., Gou L., Hintiryan H., Askarinam A., Hahn J.D., Toga A.W., Dong H.W. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J. Comp. Neurol. 2012;520:6–33. doi: 10.1002/cne.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Althammer F., Grinevich V. 2017. Diversity of oxytocin neurons: beyond magno- and parvocellular cell types? J Neuroendocrinol. [DOI] [PubMed] [Google Scholar]
- 13.Verbalis J.G., McCann M.J., McHale C.M., Stricker E.M. Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science. 1986;232:1417–1419. doi: 10.1126/science.3715453. [DOI] [PubMed] [Google Scholar]
- 14.Renaud L.P., Tang M., McCann M.J., Stricker E.M., Verbalis J.G. Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. Am. J. Physiol. 1987;253:R661–R665. doi: 10.1152/ajpregu.1987.253.4.R661. [DOI] [PubMed] [Google Scholar]
- 15.Leng G., Way S., Dyball R.E. Identification of oxytoxin cells in the rat supraoptic nucleus by their response to cholecystokinin injection. Neurosci. Lett. 1991;122:159–162. doi: 10.1016/0304-3940(91)90847-m. [DOI] [PubMed] [Google Scholar]
- 16.Kutlu S., Aydin M., Alcin E., Ozcan M., Bakos J., Jezova D., Yilmaz B. Leptin modulates noradrenaline release in the paraventricular nucleus and plasma oxytocin levels in female rats: a microdialysis study. Brain Res. 2010;1317:87–91. doi: 10.1016/j.brainres.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 17.Caquineau C., Douglas A.J., Leng G. Effects of cholecystokinin in the supraoptic nucleus and paraventricular nucleus are negatively modulated by leptin in 24-h fasted lean male rats. J. Neuroendocrinol. 2010;22:446–452. doi: 10.1111/j.1365-2826.2010.01982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clemmensen C., Müller T.D., Woods S.C., Berthoud H.R., Seeley R.J., Tschöp M.H. Gut-Brain Cross-Talk in Metabolic Control. Cell. 2017;168:758–774. doi: 10.1016/j.cell.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandsma E., Houben T., Fu J., Shiri-Sverdlov R., Hofker M.H. The immunity-diet-microbiota axis in the development of metabolic syndrome. Curr. Opin. Lipidol. 2015;26:73–81. doi: 10.1097/MOL.0000000000000154. [DOI] [PubMed] [Google Scholar]
- 20.French S.J., Murray B., Rumsey R.D., Fadzlin R., Read N.W. Adaptation to high-fat diets: effects on eating behaviour and plasma cholecystokinin. Br. J. Nutr. 1995;73:179–189. doi: 10.1079/bjn19950022. [DOI] [PubMed] [Google Scholar]
- 21.Covasa M. Deficits in gastrointestinal responses controlling food intake and body weight. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R1423–R1439. doi: 10.1152/ajpregu.00126.2010. [DOI] [PubMed] [Google Scholar]
- 22.Troy A.E., Simmonds S.S., Stocker S.D., Browning K.N. High fat diet attenuates glucose-dependent facilitation of 5-HT3 -mediated responses in rat gastric vagal afferents. J. Physiol. 2016;594:99–114. doi: 10.1113/JP271558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knobloch H.S., Charlet A., Hoffmann L.C., Eliava M., Khrulev S., Cetin A.H., Osten P., Schwarz M.K., Seeburg P.H., Stoop R., Grinevich V. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron. 2012;73:553–566. doi: 10.1016/j.neuron.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 24.Grinevich V., Knobloch-Bollmann H.S., Roth L.C., Althammer F., Domanskyi A., Vinnikov I.A., Eliava M., Stanifer M., Boulant S. In: Transgenesis (Viral Vectors). In Molecular Neuroendocrinology. Murphy D., Gainer H., editors. 2016. From Genome to Physiology. John Wiley & Sons. [Google Scholar]
- 25.Snider B., Geiser A., Yu X.P., Beebe E.C., Willency J.A., Qing K., Guo L., Lu J., Wang X., Yang Q., et al. Long-Acting and Selective Oxytocin Peptide Analogs Show Antidiabetic and Antiobesity Effects in Male Mice. J. Endocr. Soc. 2019;3:1423–1444. doi: 10.1210/js.2019-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan W., Ellacott K.L.J., Halatchev I.G., Takahashi K., Yu P., Cone R.D. Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat. Neurosci. 2004;7:335–336. doi: 10.1038/nn1214. [DOI] [PubMed] [Google Scholar]
- 27.Olson B.R., Hoffman G.E., Sved A.F., Stricker E.M., Verbalis J.G. Cholecystokinin induces c-fos expression in hypothalamic oxytocinergic neurons projecting to the dorsal vagal complex. Brain Res. 1992;569:238–248. doi: 10.1016/0006-8993(92)90635-m. [DOI] [PubMed] [Google Scholar]
- 28.Motojima Y., Kawasaki M., Matsuura T., Saito R., Yoshimura M., Hashimoto H., Ueno H., Maruyama T., Suzuki H., Ohnishi H., et al. Effects of peripherally administered cholecystokinin-8 and secretin on feeding/drinking and oxytocin-mRFP1 fluorescence in transgenic rats. Neurosci. Res. 2016;109:63–69. doi: 10.1016/j.neures.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Tang Y., Benusiglio D., Lefevre A., Hilfiger L., Althammer F., Bludau A., Hagiwara D., Baudon A., Darbon P., Schimmer J., et al. Social touch promotes interfemale communication via activation of parvocellular oxytocin neurons. Nat. Neurosci. 2020;23:1125–1137. doi: 10.1038/s41593-020-0674-y. [DOI] [PubMed] [Google Scholar]
- 30.Eliava M., Melchior M., Knobloch-Bollmann H.S., Wahis J., da Silva Gouveia M., Tang Y., Ciobanu A.C., Triana Del Rio R., Roth L.C., Althammer F., et al. A New Population of Parvocellular Oxytocin Neurons Controlling Magnocellular Neuron Activity and Inflammatory Pain Processing. Neuron. 2016;89:1291–1304. doi: 10.1016/j.neuron.2016.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller T.R., Bianchi B.R., Witte D.G., Lin C.W. Peripheral cholecystokinin type A receptors mediate oxytocin secretion in vivo. Regul. Pept. 1993;43:107–112. doi: 10.1016/0167-0115(93)90413-3. [DOI] [PubMed] [Google Scholar]
- 32.Luckman S.M., Hamamura M., Antonijevic I., Dye S., Leng G. Involvement of cholecystokinin receptor types in pathways controlling oxytocin secretion. Br. J. Pharmacol. 1993;110:378–384. doi: 10.1111/j.1476-5381.1993.tb13820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dourish C.T., Hill D.R. Classification and function of CCK receptors. Trends Pharmacol. Sci. 1987;8:207–208. [Google Scholar]
- 34.Williams E.K., Chang R.B., Strochlic D.E., Umans B.D., Lowell B.B., Liberles S.D. Sensory Neurons that Detect Stretch and Nutrients in the Digestive System. Cell. 2016;166:209–221. doi: 10.1016/j.cell.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blevins J.E., Hamel F.G., Fairbairn E., Stanley B.G., Reidelberger R.D. Effects of paraventricular nucleus injection of CCK-8 on plasma CCK-8 levels in rats. Brain Res. 2000;860:11–20. doi: 10.1016/s0006-8993(99)02478-6. [DOI] [PubMed] [Google Scholar]
- 36.Mohammad S., Ozaki T., Takeuchi K., Unno K., Yamoto K., Morioka E., Takiguchi S., Ikeda M. Functional compensation between cholecystokinin-1 and -2 receptors in murine paraventricular nucleus neurons. J. Biol. Chem. 2012;287:39391–39401. doi: 10.1074/jbc.M112.416214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Covasa M., Ritter R.C. Rats maintained on high-fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides. 1998;19:1407–1415. doi: 10.1016/s0196-9781(98)00096-5. [DOI] [PubMed] [Google Scholar]
- 38.Covasa M., Ritter R.C. Adaptation to high-fat diet reduces inhibition of gastric emptying by CCK and intestinal oleate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;278:R166–R170. doi: 10.1152/ajpregu.2000.278.1.R166. [DOI] [PubMed] [Google Scholar]
- 39.Torregrossa A.M., Smith G.P. Two effects of high-fat diets on the satiating potency of cholecystokinin-8. Physiol. Behav. 2003;78:19–25. doi: 10.1016/s0031-9384(02)00888-0. [DOI] [PubMed] [Google Scholar]
- 40.Swartz T.D., Savastano D.M., Covasa M. Reduced sensitivity to cholecystokinin in male rats fed a high-fat diet is reversible. J. Nutr. 2010;140:1698–1703. doi: 10.3945/jn.110.124149. [DOI] [PubMed] [Google Scholar]
- 41.Sutton A.K., Pei H., Burnett K.H., Myers M.G., Jr., Rhodes C.J., Olson D.P. Control of food intake and energy expenditure by Nos1 neurons of the paraventricular hypothalamus. J. Neurosci. 2014;34:15306–15318. doi: 10.1523/JNEUROSCI.0226-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blevins J.E., Baskin D.G. Hypothalamic-brainstem circuits controlling eating. Forum Nutr. 2010;63:133–140. doi: 10.1159/000264401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rinaman L., Verbalis J.G., Stricker E.M., Hoffman G.E. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J. Comp. Neurol. 1993;338:475–490. doi: 10.1002/cne.903380402. [DOI] [PubMed] [Google Scholar]
- 44.Ueta Y., Kannan H., Higuchi T., Negoro H., Yamaguchi K., Yamashita H. Activation of gastric afferents increases noradrenaline release in the paraventricular nucleus and plasma oxytocin level. J. Auton. Nerv. Syst. 2000;78:69–76. doi: 10.1016/s0165-1838(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 45.Renier N., Wu Z., Simon D.J., Yang J., Ariel P., Tessier-Lavigne M. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 2014;159:896–910. doi: 10.1016/j.cell.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Asin K.E., Bednarz L., Nikkel A.L., Gore P.A., Jr., Nadzan A.M. A-71623, a selective CCK-A receptor agonist, suppresses food intake in the mouse, dog, and monkey. Pharmacol. Biochem. Behav. 1992;42:699–704. doi: 10.1016/0091-3057(92)90017-a. [DOI] [PubMed] [Google Scholar]
- 47.Lewis E.M., Stein-O'Brien G.L., Patino A.V., Nardou R., Grossman C.D., Brown M., Bangamwabo B., Ndiaye N., Giovinazzo D., Dardani I., et al. Parallel Social Information Processing Circuits Are Differentially Impacted in Autism. Neuron. 2020;108:659–675.e6. doi: 10.1016/j.neuron.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilding J.P., Batterham R.L., Calanna S., Davies M., Van Gaal L.F., Lingvay I., McGowan B.M., Rosenstock J., Tran M.T., Wadden T.A., et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021;384:989–1002. doi: 10.1056/NEJMoa2032183. [DOI] [PubMed] [Google Scholar]
- 49.Gribble F.M., O'Rahilly S. Obesity therapeutics: The end of the beginning. Cell Metabol. 2021;33:705–706. doi: 10.1016/j.cmet.2021.03.012. [DOI] [PubMed] [Google Scholar]
- 50.Xi D., Long C., Lai M., Casella A., O'Lear L., Kublaoui B., Roizen J.D. Ablation of Oxytocin Neurons Causes a Deficit in Cold Stress Response. J. Endocr. Soc. 2017;1:1041–1055. doi: 10.1210/js.2017-00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang G., Bai H., Zhang H., Dean C., Wu Q., Li J., Guariglia S., Meng Q., Cai D. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. 2011;69:523–535. doi: 10.1016/j.neuron.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inada K., Tsujimoto K., Yoshida M., Nishimori K., Miyamichi K. Oxytocin signaling in the posterior hypothalamus prevents hyperphagic obesity in mice. Elife. 2022;11 doi: 10.7554/eLife.75718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Z., Xu Y., Zhu Y., Sutton A.K., Zhao R., Lowell B.B., Olson D.P., Tong Q. An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS One. 2012;7:e45167. doi: 10.1371/journal.pone.0045167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welch M.G., Tamir H., Gross K.J., Chen J., Anwar M., Gershon M.D. Expression and developmental regulation of oxytocin (OT) and oxytocin receptors (OTR) in the enteric nervous system (ENS) and intestinal epithelium. J. Comp. Neurol. 2009;512:256–270. doi: 10.1002/cne.21872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paiva L., Lozic M., Allchorne A., Grinevich V., Ludwig M. Identification of peripheral oxytocin-expressing cells using systemically applied cell-type specific adeno-associated viral vector. J. Neuroendocrinol. 2021;33:e12970. doi: 10.1111/jne.12970. [DOI] [PubMed] [Google Scholar]
- 56.Krashes M.J., Lowell B.B., Garfield A.S. Melanocortin-4 receptor-regulated energy homeostasis. Nat. Neurosci. 2016;19:206–219. doi: 10.1038/nn.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabatier N., Caquineau C., Dayanithi G., Bull P., Douglas A.J., Guan X.M.M., Jiang M., Van der Ploeg L., Leng G. Alpha-melanocyte-stimulating hormone stimulates oxytocin release from the dendrites of hypothalamic neurons while inhibiting oxytocin release from their terminals in the neurohypophysis. J. Neurosci. 2003;23:10351–10358. doi: 10.1523/JNEUROSCI.23-32-10351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atasoy D., Betley J.N., Su H.H., Sternson S.M. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shah B.P., Vong L., Olson D.P., Koda S., Krashes M.J., Ye C., Yang Z., Fuller P.M., Elmquist J.K., Lowell B.B. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc. Natl. Acad. Sci. USA. 2014;111:13193–13198. doi: 10.1073/pnas.1407843111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garfield A.S., Li C., Madara J.C., Shah B.P., Webber E., Steger J.S., Campbell J.N., Gavrilova O., Lee C.E., Olson D.P., et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat. Neurosci. 2015;18:863–871. doi: 10.1038/nn.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fenselau H., Campbell J.N., Verstegen A.M.J., Madara J.C., Xu J., Shah B.P., Resch J.M., Yang Z., Mandelblat-Cerf Y., Livneh Y., Lowell B.B. A rapidly acting glutamatergic ARC-->PVH satiety circuit postsynaptically regulated by alpha-MSH. Nat. Neurosci. 2017;20:42–51. doi: 10.1038/nn.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cedernaes J., Huang W., Ramsey K.M., Waldeck N., Cheng L., Marcheva B., Omura C., Kobayashi Y., Peek C.B., Levine D.C., et al. Transcriptional Basis for Rhythmic Control of Hunger and Metabolism within the AgRP Neuron. Cell Metabol. 2019;29:1078–1091.e5. doi: 10.1016/j.cmet.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shearer K.D., Stoney P.N., Morgan P.J., McCaffery P.J. A vitamin for the brain. Trends Neurosci. 2012;35:733–741. doi: 10.1016/j.tins.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Marangi G., Leuzzi V., Manti F., Lattante S., Orteschi D., Pecile V., Neri G., Zollino M. TRAPPC9-related autosomal recessive intellectual disability: report of a new mutation and clinical phenotype. Eur. J. Hum. Genet. 2013;21:229–232. doi: 10.1038/ejhg.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang Z.S., Cimino I., Yalcin B., Raghupathy N., Vancollie V.E., Ibarra-Soria X., Firth H.V., Rimmington D., Farooqi I.S., Lelliott C.J., et al. Trappc9 deficiency causes parent-of-origin dependent microcephaly and obesity. PLoS Genet. 2020;16:e1008916. doi: 10.1371/journal.pgen.1008916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibbs J., Young R.C., Smith G.P. Cholecystokinin decreases food intake in rats. J. Comp. Physiol. Psychol. 1973;84:488–495. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 67.Crawley J.N., Hays S.E., Paul S.M., Goodwin F.K. Cholecystokinin reduces exploratory behavior in mice. Physiol. Behav. 1981;27:407–411. doi: 10.1016/0031-9384(81)90324-3. [DOI] [PubMed] [Google Scholar]
- 68.Falasco J.D., Smith G.P., Gibbs J. Cholecystokinin suppresses sham feeding in the rhesus monkey. Physiol. Behav. 1979;23:887–890. doi: 10.1016/0031-9384(79)90196-3. [DOI] [PubMed] [Google Scholar]
- 69.Sturdevant R.A., Goetz H. Cholecystokinin both stimulates and inhibits human food intake. Nature. 1976;261:713–715. doi: 10.1038/261713a0. [DOI] [PubMed] [Google Scholar]
- 70.Wickbom J., Herrington M.K., Permert J., Jansson A., Arnelo U. Gastric emptying in response to IAPP and CCK in rats with subdiaphragmatic afferent vagotomy. Regul. Pept. 2008;148:21–25. doi: 10.1016/j.regpep.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 71.Moran T.H., Baldessarini A.R., Salorio C.F., Lowery T., Schwartz G.J. Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am. J. Physiol. 1997;272:R1245–R1251. doi: 10.1152/ajpregu.1997.272.4.R1245. 10.1152/ajpregu.1997.272.4.R1245. [DOI] [PubMed] [Google Scholar]
- 72.Sullivan C.N., Raboin S.J., Gulley S., Sinzobahamvya N.T., Green G.M., Reeve J.R., Jr., Sayegh A.I. Endogenous cholecystokinin reduces food intake and increases Fos-like immunoreactivity in the dorsal vagal complex but not in the myenteric plexus by CCK1 receptor in the adult rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1071–R1080. doi: 10.1152/ajpregu.00490.2006. [DOI] [PubMed] [Google Scholar]
- 73.Diepenbroek C., Quinn D., Stephens R., Zollinger B., Anderson S., Pan A., de Lartigue G. Validation and characterization of a novel method for selective vagal deafferentation of the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2017;313:G342–G352. doi: 10.1152/ajpgi.00095.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han W., Tellez L.A., Perkins M.H., Perez I.O., Qu T., Ferreira J., Ferreira T.L., Quinn D., Liu Z.W., Gao X.B., et al. A Neural Circuit for Gut-Induced Reward. Cell. 2018;175:887–888. doi: 10.1016/j.cell.2018.10.018. 10.1016/j.cell.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 75.Beutler L.R., Corpuz T.V., Ahn J.S., Kosar S., Song W., Chen Y., Knight Z.A. Obesity causes selective and long-lasting desensitization of AgRP neurons to dietary fat. Elife. 2020;9 doi: 10.7554/eLife.55909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cavanaugh A.R., Schwartz G.J., Blouet C. High-fat feeding impairs nutrient sensing and gut brain integration in the caudomedial nucleus of the solitary tract in mice. PLoS One. 2015;10 doi: 10.1371/journal.pone.0118888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daly D.M., Park S.J., Valinsky W.C., Beyak M.J. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J. Physiol. 2011;589:2857–2870. doi: 10.1113/jphysiol.2010.204594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blevins J.E., Thompson B.W., Anekonda V.T., Ho J.M., Graham J.L., Roberts Z.S., Hwang B.H., Ogimoto K., Wolden-Hanson T., Nelson J., et al. Chronic CNS oxytocin signaling preferentially induces fat loss in high-fat diet-fed rats by enhancing satiety responses and increasing lipid utilization. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;310:R640–R658. doi: 10.1152/ajpregu.00220.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown C.H., Murphy N.P., Munro G., Ludwig M., Bull P.M., Leng G., Russell J.A. Interruption of central noradrenergic pathways and morphine withdrawal excitation of oxytocin neurones in the rat. J. Physiol. 1998;507:831–842. doi: 10.1111/j.1469-7793.1998.831bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ho J.M., Anekonda V.T., Thompson B.W., Zhu M., Curry R.W., Hwang B.H., Morton G.J., Schwartz M.W., Baskin D.G., Appleyard S.M., Blevins J.E. Hindbrain oxytocin receptors contribute to the effects of circulating oxytocin on food intake in male rats. Endocrinology. 2014;155:2845–2857. doi: 10.1210/en.2014-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rinaman L., Rothe E.E. GLP-1 receptor signaling contributes to anorexigenic effect of centrally administered oxytocin in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283:R99–R106. doi: 10.1152/ajpregu.00008.2002. [DOI] [PubMed] [Google Scholar]
- 82.Blevins J.E., Eakin T.J., Murphy J.A., Schwartz M.W., Baskin D.G. Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res. 2003;993:30–41. doi: 10.1016/j.brainres.2003.08.036. [DOI] [PubMed] [Google Scholar]
- 83.Ong Z.Y., Alhadeff A.L., Grill H.J. Medial nucleus tractus solitarius oxytocin receptor signaling and food intake control: the role of gastrointestinal satiation signal processing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015;308:R800–R806. doi: 10.1152/ajpregu.00534.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brierley D.I., Holt M.K., Singh A., de Araujo A., McDougle M., Vergara M., Afaghani M.H., Lee S.J., Scott K., Maske C., et al. Central and peripheral GLP-1 systems independently suppress eating. Nat. Metab. 2021;3:258–273. doi: 10.1038/s42255-021-00344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Borgmann D., Ciglieri E., Biglari N., Brandt C., Cremer A.L., Backes H., Tittgemeyer M., Wunderlich F.T., Brüning J.C., Fenselau H. Gut-brain communication by distinct sensory neurons differently controls feeding and glucose metabolism. Cell Metabol. 2021;33:1466–1482.e7. doi: 10.1016/j.cmet.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li C., Navarrete J., Liang-Guallpa J., Lu C., Funderburk S.C., Chang R.B., Liberles S.D., Olson D.P., Krashes M.J. Defined Paraventricular Hypothalamic Populations Exhibit Differential Responses to Food Contingent on Caloric State. Cell Metabol. 2019;29:681–694.e5. doi: 10.1016/j.cmet.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rhodes C.H., Morrell J.I., Pfaff D.W. Immunohistochemical analysis of magnocellular elements in rat hypothalamus: distribution and numbers of cells containing neurophysin, oxytocin, and vasopressin. J. Comp. Neurol. 1981;198:45–64. doi: 10.1002/cne.901980106. [DOI] [PubMed] [Google Scholar]
- 88.Castel M., Morris J.F. The neurophysin-containing innervation of the forebrain of the mouse. Neuroscience. 1988;24:937–966. doi: 10.1016/0306-4522(88)90078-4. [DOI] [PubMed] [Google Scholar]
- 89.Rinaman L. Oxytocinergic inputs to the nucleus of the solitary tract and dorsal motor nucleus of the vagus in neonatal rats. J. Comp. Neurol. 1998;399:101–109. doi: 10.1002/(sici)1096-9861(19980914)399:1<101::aid-cne8>3.0.co. [DOI] [PubMed] [Google Scholar]
- 90.Duque-Wilckens N., Torres L.Y., Yokoyama S., Minie V.A., Tran A.M., Petkova S.P., Hao R., Ramos-Maciel S., Rios R.A., Jackson K., et al. Extrahypothalamic oxytocin neurons drive stress-induced social vigilance and avoidance. Proc. Natl. Acad. Sci. USA. 2020;117:26406–26413. doi: 10.1073/pnas.2011890117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lei X., Liu L., Terrillion C.E., Karuppagounder S.S., Cisternas P., Lay M., Martinelli D.C., Aja S., Dong X., Pletnikov M.V., Wong G.W. FAM19A1, a brain-enriched and metabolically responsive neurokine, regulates food intake patterns and mouse behaviors. Faseb. J. 2019;33:14734–14747. doi: 10.1096/fj.201901232RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rausch J.C., Lavine J.E., Chalasani N., Guo X., Kwon S., Schwimmer J.B., Molleston J.P., Loomba R., Brunt E.M., Chen Y.D.I., et al. Genetic Variants Associated With Obesity and Insulin Resistance in Hispanic Boys With Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2018;66:789–796. doi: 10.1097/MPG.0000000000001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Onaka T., Luckman S.M., Guevara-Guzman R., Ueta Y., Kendrick K., Leng G. Presynaptic actions of morphine: blockade of cholecystokinin-induced noradrenaline release in the rat supraoptic nucleus. J. Physiol. 1995;482:69–79. doi: 10.1113/jphysiol.1995.sp020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leng G., Dye S., Bicknell R.J. Kappa-opioid restraint of oxytocin secretion: plasticity through pregnancy. Neuroendocrinology. 1997;66:378–383. doi: 10.1159/000127263. [DOI] [PubMed] [Google Scholar]
- 95.Koch J.E., Glass M.J., Cooper M.L., Bodnar R.J. Alterations in deprivation, glucoprivic and sucrose intake following general, mu and kappa opioid antagonists in the hypothalamic paraventricular nucleus of rats. Neuroscience. 1995;66:951–957. doi: 10.1016/0306-4522(95)00001-y. [DOI] [PubMed] [Google Scholar]
- 96.Czyzyk T.A., Nogueiras R., Lockwood J.F., McKinzie J.H., Coskun T., Pintar J.E., Hammond C., Tschöp M.H., Statnick M.A. kappa-Opioid receptors control the metabolic response to a high-energy diet in mice. Faseb. J. 2010;24:1151–1159. doi: 10.1096/fj.09-143610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qian T., Wang H., Wang P., Geng L., Mei L., Osakada T., Wang L., Tang Y., Kania A., Grinevich V., et al. A genetically encoded sensor measures temporal oxytocin release from different neuronal compartments. Nat. Biotechnol. 2023 doi: 10.1038/s41587-022-01561-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen T.W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V., et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krashes M.J., Koda S., Ye C., Rogan S.C., Adams A.C., Cusher D.S., Maratos-Flier E., Roth B.L., Lowell B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tschöp M.H., Speakman J.R., Arch J.R.S., Auwerx J., Brüning J.C., Chan L., Eckel R.H., Farese R.V., Jr., Galgani J.E., Hambly C., et al. A guide to analysis of mouse energy metabolism. Nat. Methods. 2011;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14. Unit14. 2010;20 doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Althammer F., Ferreira-Neto H.C., Rubaharan M., Roy R.K., Patel A.A., Murphy A., Cox D.N., Stern J.E. Three-dimensional morphometric analysis reveals time-dependent structural changes in microglia and astrocytes in the central amygdala and hypothalamic paraventricular nucleus of heart failure rats. J. Neuroinflammation. 2020;17 doi: 10.1186/s12974-020-01892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanz E., Yang L., Su T., Morris D.R., McKnight G.S., Amieux P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kolde R. 2015. pheatmap: Pretty heatmaps. [Google Scholar]
- 105.Blighe K., Rana S., Lewis M. 2021. publication-ready volcano plots with enhanced colouring and labeling. EnhancedVolcano. [Google Scholar]
- 106.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wickham H. 2016. ggplot2-Elegant Graphics for Data Analysis (Springer-Verlag New York978-3-319-24277-4) [Google Scholar]
- 108.Deal R.B., Henikoff S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011;6:56–68. doi: 10.1038/nprot.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Krishnaswami S.R., Grindberg R.V., Novotny M., Venepally P., Lacar B., Bhutani K., Linker S.B., Pham S., Erwin J.A., Miller J.A., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc. 2016;11:499–524. doi: 10.1038/nprot.2016.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rodrigues O.R., Monard S. A rapid method to verify single-cell deposition setup for cell sorters. Cytometry A. 2016;89:594–600. doi: 10.1002/cyto.a.22865. [DOI] [PubMed] [Google Scholar]
- 111.Richter M.L., Deligiannis I.K., Yin K., Danese A., Lleshi E., Coupland P., Vallejos C.A., Matchett K.P., Henderson N.C., Colome-Tatche M., Martinez-Jimenez C.P. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-24543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mora-Castilla S., To C., Vaezeslami S., Morey R., Srinivasan S., Chousal J.N., Cook-Andersen H., Jenkins J., Laurent L.C. Miniaturization Technologies for Efficient Single-Cell Library Preparation for Next-Generation Sequencing. J. Lab. Autom. 2016;21:557–567. doi: 10.1177/2211068216630741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 114.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 117.Wagner G.P., Kin K., Lynch V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor. Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 118.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19 doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lun A.T.L., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5 doi: 10.12688/f1000research.9501.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014;20:1983–1992. doi: 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou W., Hinoue T., Barnes B., Mitchell O., Iqbal W., Lee S.M., Foy K.K., Lee K.H., Moyer E.J., VanderArk A., et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom. 2022;2 doi: 10.1016/j.xgen.2022.100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D-rendered and reconstructed PVNOT neurons as identified by OT immunoreactivity (gray) exhibit robust C-CASP3 immunosignal (red) following virus-mediated expression of Cre recombinase together with Venus, a fluorescent reporter protein (green).
Time-lapse 2-photon excitation microscopy of cytosolic Ca2+ dynamics in PVNOT neurons that were virally transduced to express GcaMP6f. Despite the presence of synaptic blockers in the bath solution, bath application with native CCK (50 nM) robustly triggered increases in fluorescent signal at the somata and neurites of most PVNOT neurons within the field of view. Time-of-CCK-appearance in the bathing solution as indicated in the movie. Scale bar, 25 μm.
3D light-sheet fluorescence microscopy of iDISCO-cleared whole mouse brain (OT:Ai14). Descending axonal projections of OT neurons (green) can be traced to nucleus tractus solitarius (NTS) of the brainstem where they densely innervate catecholaminergic A2/C2 neurons (TH+; magenta).
Data Availability Statement
-
•
RiboTag-RNA-seq and snRNA-seq2 data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.