

Abstract
Glioblastomas (GBM) are the most common primary brain tumors in adults and associated with poor clinical outcomes due to therapy resistances and destructive growth. Interactions of cancer cells with the extracellular matrix (ECM) play a pivotal role in therapy resistances and tumor progression. In this study, we investigate the functional dependencies between the discoidin domain receptor 1 (DDR1) and the integrin family of cell adhesion molecules for the radioresponse of human glioblastoma cells. By means of an RNA interference screen on DDR1 and all known integrin subunits, we identified co-targeting of DDR1/integrin β3 to most efficiently reduce clonogenicity, enhance cellular radiosensitivity and diminish repair of DNA double strand breaks (DSB). Simultaneous pharmacological inhibition of DDR1 with DDR1-IN-1 and of integrins αVβ3/αVβ5 with cilengitide resulted in confirmatory data in a panel of 2D grown glioblastoma cultures and 3D gliospheres. Mechanistically, we found that key DNA repair proteins ATM and DNA-PK are altered upon DDR1/integrin αVβ3/integrin αVβ5 inhibition, suggesting a link to DNA repair mechanisms. In sum, the radioresistance of human glioblastoma cells can effectively be declined by co-deactivation of DDR1, integrin αVβ3 and integrin αVβ5.
Keywords: Glioblastoma, DDR1, integrin αVβ3, integrin αVβ5, radiosensitization, DNA-PK, ATM, cilengitide, DDR1-IN-1
Introduction
IDH-wildtype glioblastomas (GBM) represent the most common type of diffuse gliomas in adults [1]. Despite an extensive multimodal standard of care regime consisting of surgical resection and radiochemotherapy, GBM remain incurable mainly due to generalized recurrences caused by the infiltration of highly therapy-resistant GBM cells into the healthy brain tissue [2,3]. In addition to heterogeneous genetic and epigenetic factors [4], networks both between GBM cells and with neighboring healthy cells, microenvironmental factors and interaction with the extracellular matrix (ECM) fuel resistance to therapeutic interventions [5-8].
Among the factors mediating cell-ECM interactions, the discoidin domain receptor 1 (DDR1) has attracted much attention as a potential therapeutic target based on its ability to promote therapy resistances in several cancer entities [9-12]. DDR1, together with DDR2, forms a receptor tyrosine kinase family with affinity to different types of fibrillar and non-fibrillar collagens [13]. They regulate a variety of cellular functions, such as proliferation and adhesion [14]. In GBM, DDR1 mRNA expression is higher than in normal brain and correlates with poorer patient survival [15-17]. Furthermore, an association between DDR1 expression and GBM cell invasion has been reported [16]. We have recently documented DDR1 to mediate pro-survival signaling via Akt serine/threonine kinase (Akt) and mechanistic target of rapamycin kinase (mTor) in established and stem-like GBM cell models [17]. Pharmacological DDR1 targeting elicited autophagy-related radiochemosensitization, linking interactions with the ECM to the GBM therapy response [17,18].
Integrins are the major family of cell adhesion receptors and act as linkers between the ECM and the cytoskeleton to transduce biochemical as well as mechanical signals that regulate many functions of healthy and malignant cells [19,20]. It is well established that integrins co-control therapy resistance in many cancer entities by sensing ECM properties such as composition or rigidity, making them suitable candidates for therapeutic exploitation [20-23]. Despite past clinical challenges with integrin-targeting agents for the treatment of cancer, new integrin inhibitors are being tested in a variety of disease settings [21]. Within the integrin family of heterodimeric cell surface receptors, many of the 18 alpha and 8 beta subunits are upregulated in GBM cells and are linked to invasive GBM growth, angiogenesis and therapy resistance [24-33]. Despite the overarching functions of integrins and DDR1, the relevance of their interplay for radiation resistance of GBM cells is unknown.
In the present study, we screen the effect of combined molecular targeting of DDR1 and all known integrin subunits together with irradiation on clonogenicity and repair of DNA double strand breaks (DSB) in human GBM cells. We demonstrate that the combined inhibition of DDR1 and integrin αVβ3 most potently decreases their survival and DSB repair capacity. Furthermore, in a panel of 2D grown GBM cultures and 3D gliospheres, pharmacological co-inhibition of DDR1 (DDR1-IN-1) and αVβ3/αVβ5 (cilengitide) elicits radiosensitization and DSB accumulation to a level superior to the single inhibitions. Mechanistically, key DNA repair proteins ATM serine/threonine kinase (ATM) and DNA-activated protein kinase (DNA-PK) are altered upon DDR1-IN-1/cilengitide treatment, suggesting a link to DNA repair mechanisms.
Materials and methods
Cell culture
The human GBM cell lines LN-229 (ATCC, Manassas, VA, USA), DD-HT7607, DD-T4 (kindly provided by A. Temme, Technische Universität Dresden, Dresden, Germany), LN-405 and U-251MG (kindly provided by L. Kunz-Schughart, Technische Universität Dresden, Dresden, Germany) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, PAN-Biotech GmbH, Aidenbach, Germany) and 1% non-essential amino acids (NEAA, Thermo Fisher Scientific) at 37°C with 8.5% CO2 and humidified atmosphere. U-343MG cells (kindly provided by A. Temme) were grown in Basal Medium Eagle (BME, Thermo Fisher Scientific), supplemented with 10% FBS (PAN-Biotech GmbH), 1% NEAA (Thermo Fisher Scientific), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 2 mM L-glutamine (both from Sigma-Aldrich) at 37°C with 5% CO2 and humidified atmosphere. For the formation of gliospheres, all cell lines were cultured in Neurobasal Medium (NBM, Thermo Fisher Scientific) supplemented with 2 mM L-glutamine, 32 U/ml heparin, 20 ng/ml human recombinant epidermal growth factor (hrEGF) (all from Sigma-Aldrich), 20 ng/ml human recombinant fibroblast growth factor (hrFGF) and 1x B27 supplement (both from Thermo Fisher Scientific) at 37°C with 5% CO2 and humidified atmosphere. All cell lines were tested as mycoplasma negative and authenticated using short-tandem repeat DNA profiling.
Colony and gliosphere formation assay
Clonogenicity (colony formation capacity) was analyzed by plating single cells in 24-well plates coated with 1 µg/cm2 collagen type I (BD, Franklin Lakes, NJ, USA) per well as published [28]. Gliosphere formation capacity was measured by plating single cells in 96-well ultra-low attachment plates (Merck Millipore, Billerica, MA, USA) as published [17]. In both settings, cells were irradiated with 2 Gy, 4 Gy or 6 Gy X-rays 24 hours after seeding or left unirradiated. In experiments using pharmacological inhibitors, cells were treated 1 hour prior to irradiation. Formed cell colonies were fixed with 80% EtOH and stained with Coomassie blue (Merck Millipore) after a cell line-dependent growth period of 7 days (LN-229), 10 days (DD-HT7607, DD-T4, U-251MG, U-343MG) and 11 days (LN-405). Gliosphere formation was stopped 7 days after plating with 3% formaldehyde (Carl Roth GmbH & Co. KG, Karlsruhe, Germany). Colonies containing more than 50 cells were counted using the Stemi 2000 stereo microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). Gliospheres with a diameter larger than 100 µm were counted with an Axiovert 25 microscope (Carl Zeiss Microscopy GmbH).
Radiation exposure
X-ray irradiation was delivered at room temperature using single doses of 200 kV X-rays (Yxlon Y.TU 320; Yxlon, Hamburg, Germany) filtered with 0.5 mm Cu as published [17]. The approximate dose-rate was 1.3 Gy/min at 20 mA. Dosimetry for quality assurance was performed using a Duplex dosimeter (PTW, Freiburg, Germany) prior to irradiation.
siRNA transfection
The comparative RNAi screen was performed using a Dharmacon ON-TARGETplus SMARTpool siRNA library according to the manufacturer’s instructions (Horizon Discovery, Cambridge, UK) as published [28]. Furthermore, the following siRNA were used: ITGAV siRNA (5’-GAAUAUCGGUUGGAUUAUAtt-3’), ITGB3 siRNA (5’-GAAUUGUACCUAUAAGAAUtt-3’); ITGB5 siRNA (5’-GCGUCAUGAUGUUCACCUAtt-3’) (all Thermo Fisher Scientific), DDR1 siRNA (5’-GAGCGUCUGUCUGCGGGUAtt-3’) and control siRNA (5’-GCAGCUAUAUGAAUGUUGUtt-3’) (both from Eurofins Scientific, Luxembourg, Luxembourg). All siRNA transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 24 hours, transfected cells were subjected to colony and gliosphere formation assay, foci assay and western blot analysis.
Total cell extracts and western blotting
Preparation of cell lysates and western blot analysis was performed as previously described [34]. Primary antibodies were purchased as follows: ATM (#2873), DDR1 (#5583), DNA-PKcs (4602), integrin αV (4711), integrin β3 (13166), integrin β5 (4708), phospho-ATM Ser1981 (4526) (all from Cell Signaling Technology, Danvers, MA, USA); β-actin (A5441), vinculin (V9131) (both from Sigma-Aldrich, St. Louis, MO, USA) and phospho-DNA-PKcs Ser2056 (ab18192, Abcam, Cambridge, UK). Secondary antibodies: Horseradish peroxidase (HRP)-linked sheep anti-mouse IgG (NXA931) and HRP-linked donkey anti-rabbit IgG (NA934V) (both from Cytiva, Marlborough, MA, USA). Fiji was used for densitometric analysis [35]. Total proteins were normalized to the corresponding β-actin or vinculin. Phosphorylated proteins were normalized to the corresponding total protein. Values are displayed as ratio (fold change) of the control.
Inhibitor treatment
Cells were treated 24 hours after seeding with pharmacological inhibitors of DDR1 (DDR1-IN-1, Tocris, Bristol, UK) (2D cultures: 4.99 µM, 3D gliospheres: 7.88 µM), αVβ3/αVβ5 integrin (Cilengitide, MedChemExpress, Monmouth Junction, NJ, USA) (2D cultures: 0.97 µM, 3D gliospheres: 6.05 µM); DNA-PK (Nedisertib/M3814, LKT Laboratories Inc., St. Paul, MN, USA) (2D cultures: 0.3 µM, 3D gliospheres: 1 µM) and ATM (KU55933, Biorbyt, Cambridge, UK) (2D cultures: 0.75 µM, 3D gliospheres: 3 µM) or dimethylsulfoxid (DMSO, Sigma-Aldrich) as control as published [17]. After 24 hours, inhibitor containing medium was replaced.
Foci assay
For quantification of residual DSB (foci), cells were seeded onto collagen type I coated glass coverslips for 24 hours, irradiated with 4 Gy or 6 Gy X-rays or left unirradiated as published [28]. After 24 hours, cells were fixed with 3% formaldehyde, permeabilized using 0.25% Triton X-100 (Sigma-Aldrich) and blocked with 1% bovine serum albumin fraction V (SERVA Electrophoresis GmbH, Heidelberg, Germany) in PBS. DSB were stained with anti-phospho-Histone H2A.X (Ser139) (05-636, Merck Millipore) and anti-53BP1 (NB100-304, Novus Biologicals, Centennial, CO, USA), anti-phospho-ATM Ser1981 (4526, Cell Signaling) or anti-phospho-DNA-PKcs Ser2056 (ab18192, Abcam) primary antibodies. The following secondary antibodies were used: Goat anti-mouse IgG Alexa Fluor™ 488 (A-11029); goat anti-rabbit IgG Alexa Fluor™ 488 (A-11034); goat anti-rabbit IgG Alexa Fluor™ 594 (A-11037) and goat anti-mouse IgG Alexa Fluor™ 594 (A-11032) (all from Thermo Fisher Scientific). After washing with PBS, coverslips were embedded in VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Newark, CA, USA). Representative pictures were acquired using the AxioImager M1 fluorescence microscope (Carl Zeiss Microscopy GmbH). Foci were quantified using Fiji.
Flow cytometry
After a 24-hour treatment with pharmacological inhibitors or DMSO control, cells were dissociated with Accutase (Thermo Fisher Scientific) and 2 × 105 cells were resuspended in PBS supplemented with 5% FBS. Cells were stained using FITC conjugated anti-αVβ5 (MAB1961F), FITC conjugated anti-αVβ3 (MAB1976F), FITC conjugated mouse IgG1 negative control (MABC002F) (all Merck Millipore) or a FITC conjugated RGD-containing peptide GRGDSP (AS-60619, Anaspec, Fremont, CA, USA) for 30 min on ice and protected from light. After 15 minutes of staining, propidium iodide (Sigma-Aldrich) was added for dead cell exclusion. Measurements were performed on 50,000-100,000 viable cell events using a BD Celesta flow cytometer (BD) as published [25]. Cumulative intensity was analyzed using FlowJo (Version 7.6.2, BD). Cell doublets and debris were excluded from analysis by using SSC-A versus FSC-A and SSC-H versus SSC-W.
Statistical analysis
Data are depicted as means ± standard deviations (SD) of at least three independent biological replicates. For statistical analysis of two groups, a two-tailed Student’s t-test was performed using Excel (Microsoft, Redmond, WA, USA). Comparisons of more than 2 groups were performed with ANOVA followed by a Dunnett post hoc test with GraphPad Prism (version 7.02 for Windows, GraphPad Software, San Diego, CA, USA). Patient survival was displayed as Kaplan-Meier curves and analyzed using a logrank test with GraphPad Prism (GraphPad Software). P values of less than 0.05 were considered statistically significant.
Results
RNAi screen identifies combined DDR1/integrin β3 depletion as the most effective approach to radiosensitize and impair DNA double strand break repair in GBM cells
Despite the fact that integrins and DDR1 family members are cell adhesion molecules [19], their functional interaction for the regulation of cellular radiosensitivity and DSB repair remains elusive. Here we performed a comparative RNAi screen in the human GBM cell lines U-251MG and LN-229 on all 18 α and 8 β integrin subunits plus DDR1 versus DDR1 alone (Figure 1A) and assessed clonogenicity (Figure 1B) and residual DSB (Figure 1C) without and in combination with X-ray irradiation. In addition to other potential DDR1/integrin combinations, the simultaneous targeting of DDR1 with integrin β3 proved to be the most promising relative to controls regarding enhancement of radiosensitivity (~1.5-fold) (Figure 2A) together with an increase in residual DSB in the absence and presence of X-ray irradiation (~2-fold) (Figure 2B). Our observations, summarized in Figure 2C and 2D, clearly demonstrate the efficacy of simultaneous integrin β3/DDR1 inactivation in terms of radiosensitization and DSB repair inhibition in both GBM cell lines. Intriguingly, among others, radiosensitizing and DSB repair impairing effects are also observable for the combinations of integrin β5/DDR1 and integrin αV/DDR1 (Figure 2A-D), integrin receptors that are inhibited by the cyclic RGD pentapeptide cilengitide.
Figure 1.

RNAi screen of DDR1 and integrins reveals regulators of clonogenicity and DNA repair in human glioblastoma cells. (A) Workflow of RNAi screen. (B) Normalized clonogenic survival and (C) residual DNA double strand breaks (DSB; γH2AX/53BP1-positive foci) of small interfering RNA (siRNA)-treated, non-irradiated or 4 Gy X-ray-irradiated U-251MG and LN-229 GBM cells. (C) Quantification of >50 cells per replicate. (B, C) Data show means ± SD (n = 3-4, one-way ANOVA, Dunnett post hoc test). n.s., not significant.
Figure 2.
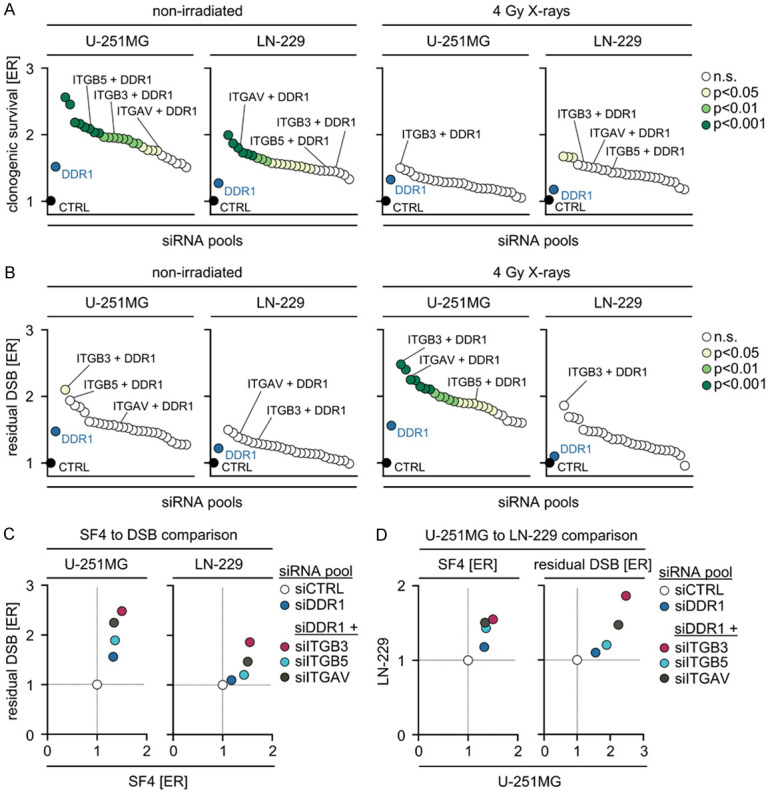
Simultaneous depletion of DDR1 and integrin β3 most effectively enhances radiosensitivity and impairs DNA repair in GBM cells relative to single DDR1 depletion. Enhancement ratios (ER) of (A) clonogenic survival and (B) residual DSB (γH2AX/53BP1-positive foci) of non-irradiated and 4 Gy X-ray-irradiated U-251MG and LN-229 GBM cells. (A, B) Data show means (n = 3-4, one-way ANOVA, Dunnett post hoc test). (C, D) Correlations of ER of surviving fraction (SF4) and residual DSB after 4 Gy X-ray irradiation in U-251MG and LN-229 cells. Data show means (n = 3-4). n.s., not significant.
Simultaneous integrin β3/DDR1 targeting mediates the strongest radiosensitization
To further untangle the therapeutic dependencies on DDR1 as well as integrins β3, β5 and αV, we investigated U-251MG cells grown under 2D adherent or 3D gliosphere conditions upon depletion of these receptors (Figure 3A, 3B). In 2D, the different knockdown approaches elicited varying degrees of basal survival reduction (ranging from 40% to 75%; Figure 3C) and radiosensitization (Figure 3D). Intriguingly, single DDR1 or ITGB5 silencing mediated strongest radiosensitization equitoxic to double DDR1/ITGB3 as well as triple DDR1/ITGB3/ITGAV and DDR1/ITGB5/ITGAV (Figure 3E). In marked contrast, gliosphere conditions per se led to a fundamental increase in cell survival and diminished the impact of targeting these receptors on basal cell survival (Figure 3C). In addition, it was found that single, double, and triple targeting generally resulted in radiosensitization, but to a lesser extent than 2D (Figure 3D). Calculation of enhancement ratios, shown in Figure 3E, summarizes these observations.
Figure 3.

siRNA-mediated depletion of DDR1 combined with ITGAV/ITGB3 elicits strongest radiosensitization. (A) Western blot analysis of indicated siRNA knockdown efficiencies from U-251MG whole cell lysates. β-actin serves as loading control. Representative images are shown (n = 3). (B) Densitometry of DDR1, integrin β3, integrin β5 and integrin αV after indicated siRNA knockdowns from western blots from (A). (C) Normalized clonogenic survival of non-irradiated and (D) surviving fractions of X-ray-irradiated U-251MG cells grown under 2D adherent and 3D gliosphere culture conditions after siRNA-mediated knockdown of indicated genes. (B-D) Data show means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05, **P<0.01, ***P<0.001). (E) ER of surviving fraction after 6 Gy X-ray irradiation (SF6) in U-251MG cells grown under 2D adherent and 3D gliosphere culture conditions after siRNA-mediated single, double or triple knockdown (KD) of indicated genes. Data show means (n = 3).
DDR1, ITGB3, ITGB5 and ITGAV are overexpressed in GBM relative to normal brain and their expression levels impact patient outcome
Although integrins αV, β3 and β5 have been intensively studied [36,37], we next analyzed their and DDR1 mRNA expression in GBM versus healthy brain and the impact of this expression on patient survival (data from The Cancer Genome Atlas (TCGA) [38]). Our evaluation revealed patients with high levels of DDR1, ITGB3 and ITGB5 to have a significant reduction in overall survival indicating a relevance of these proteins in human GBM pathology (Figure 4A-D). Furthermore, all proteins of interest are overexpressed in GBM compared with healthy brain tissue (Figure 4E) and their expression was detected in U-251MG and LN-229 cells as well as an extended panel of GBM cell lines independent of the culture growth conditions (Figure 4F, 4G).
Figure 4.

DDR1, ITGB3, ITGB5 and ITGAV are overexpressed in GBM relative to normal brain and their expression correlates with patient survival. Kaplan Meier survival analyses of GBM patients with low versus high levels of (A) DDR1, (B) ITGB3, (C) ITGB5 and (D) ITGAV. Curves were generated using the TCGA Affymetrix HT HG U133A GBM dataset [38] (https://betastasis.com) (p-values were calculated by logrank test). (E) Comparative mRNA expression analysis of DDR1, ITGB3, ITGB5 and ITGAV between GBM and normal brain using the TCGA GBM dataset (https://www.oncomine.org) [46]*. Data are shown as box and whiskers (5-95 percentile) (two-sided t-test: ***P<0.001). (F) Western blot analysis of indicated proteins in whole cell lysates from indicated GBM cell lines grown under 2D adherent and 3D gliosphere culture conditions. β-actin serves as loading control. Representative images are shown (n = 3). (G) Densitometry of DDR1, integrin β3, integrin β5 and integrin αV in indicated GBM cell lines grown under 2D adherent and 3D gliosphere conditions from western blots from (F). Data are shown as means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05, **P<0.01).
Combined DDR1-IN-1/cilengitide application elicits strongest radiosensitization in GBM cells
Next, we elucidated the radiosensitizing potential of biologicals inhibiting our identified targets, i.e. DDR1, integrin αVβ3, and integrin αVβ5. For addressing this question in a panel of 2D adherently and 3D gliosphere grown GBM cell models, we chose the biologicals DDR1-IN-1 for DDR1 and cilengitide for integrins αVβ3 and αVβ5. The data revealed similar trends regarding cytotoxicity and radiosensitization as observed upon the RNAi approach (Figure 5A-D). In brief, the biologicals presented with marginally lower cytotoxicity on clonogenicity (2D and gliosphere for U-251MG cells; Figure 5A and 5C) as well as lower radiosensitization (gliosphere for U-251MG cells; Figure 5D) relative to RNAi, while the degree of radiosensitization of 2D adherently growing U-251MG cells appeared stronger upon DDR1-IN-1/cilengitide administration compared with DMSO (Figure 5B). Across the entire GBM cell model panel, we found differential rates for cytotoxicity and enhancement of radiosensitivity under both adherent and gliosphere growth conditions relative to DMSO (Figure 5B, 5D, 5E).
Figure 5.
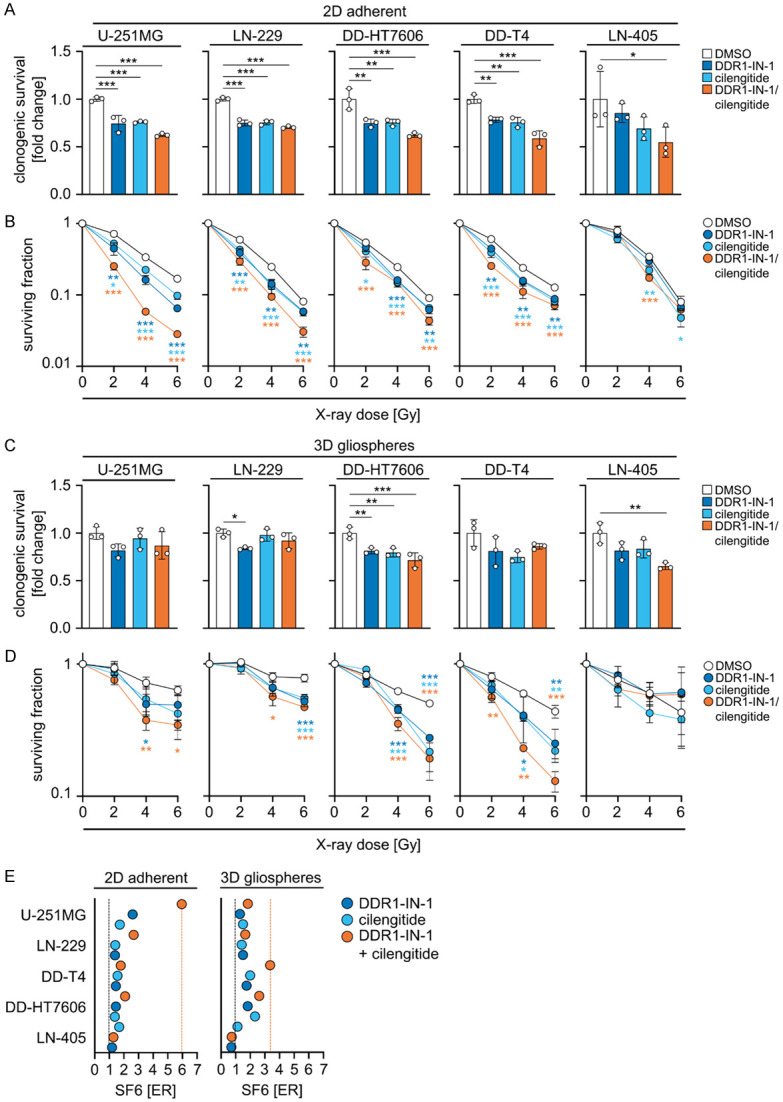
Simultaneous DDR1-IN-1/cilengitide exposure radiosensitizes glioblastoma cell lines. (A) Normalized clonogenic survival of non-irradiated and (B) surviving fractions of X-ray-irradiated GBM cell lines grown under 2D adherent culture conditions after single or combined pharmacological inhibition using DDR1-IN-1 and cilengitide compared with controls (DMSO). (C) Normalized clonogenic survival of non-irradiated and (D) surviving fractions of X-ray-irradiated GBM cell lines grown as 3D gliospheres after the indicated pharmacological treatments. (A-D) Data show means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05, **P<0.01, ***P<0.001). (E) ER of surviving fraction after 6 Gy X-ray irradiation (SF6) of the indicated GBM cell lines grown under 2D adherent and 3D gliosphere culture conditions after inhibition of DDR1 (DDR1-IN-1), αVβ3/αVβ5 (cilengitide) or a combination of both. Data show means (n = 3).
Integrin αVβ3 and αVβ5 cell surface expression and binding capacity remain unaltered under treatment with DDR1-IN-1 or cilengitide
To investigate putative interdependencies between DDR1 and integrins αVβ3 and αVβ5 contributing to cell survival and cellular radiosensitivity, we quantified the cell surface expression of these receptors as well as the integrin binding capacity in 2D adherent and 3D gliosphere cultures of LN-229 and U-251MG cells by flow cytometry (Figure 6A-D). No systematic alterations in cell surface expression or integrin binding capacity were detected after treatment with DDR1-IN-1 or cilengitide.
Figure 6.
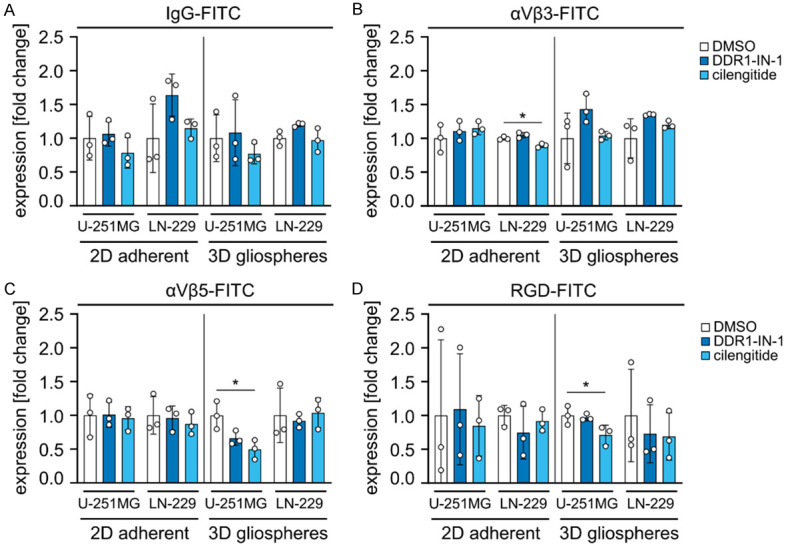
Surface expression of αVβ3 and αVβ5 integrins as well as RGD peptide binding capacity remain unaltered under DDR1-IN-1 or Cilengitide administration. (A-D) Flow cytometry analysis of U-251MG and LN-229 GBM cells treated with DMSO control, DDR1-IN-1 or cilengitide under 2D adherent or 3D gliosphere conditions. Cells were surface stained with (A) FITC-conjugated non-specific IgG antibodies (control), FITC-conjugated antibodies specific for (B) integrin αVβ3 and (C) integrin αVβ5 or (D) RGD binding. Data show means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05).
Inhibition of DDR1 and integrins αVβ3 and αVβ5 impairs DSB repair
To address the mechanisms mediated by the most efficient radiosensitizing combination composed of DDR1-IN-1 and cilengitide, residual 53BP1/γH2AX-positive foci (Figure 7A) were determined in our GBM cell model panel. DDR1-IN-1/cilengitide application alone induced a significant raise in foci numbers in four out five GBM cell models compared with DMSO (Figure 7B). Interestingly, including the non-responding LN-405 cell model, DDR1-IN-1/cilengitide treatment combined with irradiation elicited significant elevation of 53BP1/γH2AX-positive foci numbers in a cell model-dependent manner relative to DMSO (Figure 7B).
Figure 7.

Combined DDR1-IN-1/cilengitide treatment impairs DSB repair including altered ATM and DNA-PK phosphorylation. (A) Representative immunofluorescence images of γH2AX (green), 53BP1 (red) and DNA (blue) of LN-229 cells treated with DDR1-IN-1/cilengitide. Representative images are shown (n = 3). Scale bar 10 µm. (B) Number of γH2AX/53BP1-positive residual DSB (foci) per cell in indicated GBM cell lines after combined DDR1-IN-1/cilengitide treatment without and with 6 Gy X-rays. Quantification of >50 cells per replicate. Data are shown as box and whiskers (5-95 percentile) (n = 3, one-way ANOVA, Dunnett post hoc test: **P<0.01, ***P<0.001). (C) Western blot analysis of indicated proteins in whole cell lysates of 2D adherently grown U-251MG cells. Vinculin serves as loading control. Representative images are shown (n = 3). (D) Densitometry of phospho-ATM Ser1981 (pATM), ATM, phospho-DNA-PK Ser2056 (pDNA-PK) and DNA-PK in 2D adherently grown U-251MG of western blots from (C). (E) Western blot analysis of indicated proteins in whole cell lysates of 3D gliosphere grown U-251MG cells. Vinculin serves as loading control. Representative images are shown (n = 3). (F) Densitometry of pATM, ATM, pDNA-PK and DNA-PK in 3D gliosphere grown U-251MG of western blots from (E). (D, F) Data are shown as means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05, **P<0.01). Number of (G) pATM or (H) pDNA-PK foci per cell in U-251MG cells after treatment with DMSO control, DDR1-IN-1, cilengitide or a combination of both inhibitors without and with 6 Gy X-rays. (G, H) Quantification of >50 cells per replicate. Data are shown as box and whiskers (5-95 percentile) (n = 3, one-way ANOVA, Dunnett post hoc test: *P<0.05, ***P<0.001). (I) Normalized clonogenic survival of non-irradiated and (J) surviving fractions after 6 Gy X-ray-irradiation (SF6) of U-251MG cells grown under 2D adherent and 3D gliosphere culture after single pharmacolocial inhibition of ATM (KU55933) or ATM inhibition combined with either DDR1-IN-1, cilengitide or both compared to control treatment (DMSO). (I, J) Data show means ± SD (n = 3, one-way ANOVA, Dunnett post hoc test: ***P<0.001).
Subsequently, we examined the pivotal DSB repair enzymes ATM and DNA-PK at 1 h and 24 h upon irradiation followed by immunofluorescence staining for residual phospho-ATM (Ser1981) (pATM) and phospho-DNA-PK (Ser2056) (pDNA-PK) foci. While pATM and pDNA-PK levels remained stable in unirradiated adherent or gliosphere GBM cell cultures upon exposure to the DDR1-IN-1/cilengitide combination (Figure 7C-F), irradiated adherent and gliosphere cell cultures demonstrated an opposing course of phosphorylation of ATM and DNA-PK after DDR1/cilengitide (Figure 7C-F). While pATM levels increased at 1 h and 24 h, respectively, in adherent and gliosphere cell cultures compared to DMSO controls, pDNA-PK showed a steady decline over the 24 h observation period (Figure 7D and 7F). Total levels of ATM and DNA-PK were not affected by the different culture and treatment conditions (Figure 7C-F).
To provide further data for an impairment of DSB repair by DDR1 and cilengitide, foci for pATM (Figure 7G) and pDNA-PK (Figure 7H) were quantified using immunofluorescence staining. Generally, in line with changes observed on the protein level, pATM foci numbers significantly increased in unirradiated and irradiated cells treated either alone or in combination with DDR1-IN-1 and cilengitide (Figure 7G). Opposingly, these treatments induced a decline of pDNA-PK foci both in unirradiated and irradiated U-251MG cells (Figure 7H).
To explore the functional impact of elevated ATM phosphorylation, we added the pharmacological ATM inhibitor KU55933 to DDR1-IN-1/cilengitide. Despite the fact that DDR1-IN-1 and cilengitide modified phosphorylation of both enzymes, ATM inhibition dominated over single or combined DDR1-IN-1 and cilengitide (Figure 7I, 7J). Taken together, these data suggest that the deactivation of DDR1 and integrins αVβ3 and αVβ5 by DDR1-IN-1 and cilengitide, respectively, critically modulate DSB repair. The data further indicates the key DSB repair enzymes ATM and DNA-PK to be differentially influenced by this deactivation.
Discussion
GBM remains a therapeutically challenging tumor due to its intrinsic and acquired therapy resistances as well as its invasive growth. Extracellular matrix receptors such as DDR1 and the integrin family play a critical role in therapy resistances and tumor progression. In this study, we investigated the interplay between DDR1 and integrins in the regulation of the radioresponse of GBM cells. We here show that (i) combined DDR1/integrin β3 depletion has effective radiosensitizing and DSB repair-impairing potential, (ii) depletion of DDR1 in combination with integrin αVβ3 mediates strongest radiosensitization under different culture conditions, (iii) high expressions of DDR1, ITGB3 and ITGB5 in GBM associate with worse patient survival, (iv) pharmacological DDR1/αVβ3/αVβ5 inhibition with simultaneous DDR1-IN-1/cilengitide application elicits strongest radiosensitization, and (v) co-inhibition of DDR1 and integrins αVβ3/αVβ5 impairs DSB repair by differentially altering ATM and DNA-PK phosphorylations.
Previous studies have shown that DDR1 essentially contributes to therapy resistances in several cancer entities [9-12]. DDR1 inhibition in combination with standard clinical therapy enabled GBM cell radiosensitization and survival of mice bearing orthotopic GBM [17]. While most studies apply mono-targeting/mono-inhibition of DDR1 to explore cytotoxic, radiosensitizing or anti-invasive potential in GBM [16,17], putative compensatory pro-survival signaling via other transmembrane receptors, such as integrins, might be activated. Therefore, our study elucidated the potential of DDR1/integrin interactions by a genetic and a pharmacological approach. Using RNAi, we show combined depletion of DDR1 and certain integrin subunits to be superior to single DDR1 depletion in terms of radiosensitization and impairment of DSB repair. Interestingly, integrin β3/DDR1 co-depletion turned out as most effective. As the quantity of radiation-dependent residual DNA damage is a major determinant of cellular radiosensitivity, our overlap between an increased number of residual DSB and increased radiosensitivity upon DDR1/β3 inhibition further underscores our findings.
In addition, our data show a cell line-dependent enhancement of radiosensitization and residual DSB upon depletion of ITGAV, the integrin β3 partnering αV subunit, and ITGB5, as part of the αVβ5 heterodimer. These integrin pairs confer specificity for the highly conserved tripeptide Arg-Gly-Asp (RGD) recognition motif found in a variety of ECM proteins [39,40]. Analyzes of their and DDR1 mRNA expression in GBM compared to healthy brain confirm an association between high expression and patient survival, as previous studies have shown [17,41]. This indicates and corroborates once again the therapeutic potential of the targets examined here for GBM therapy. Integrins αVβ3 and αVβ5 are selectively inhibited by the RDG-blocking pentapepdide cilengitide, the first representative of integrin targeting drugs advancing to phase III clinical testing in GBM. Cilengitide combined with radiochemotherapy failed to improve overall survival of newly diagnosed GBM patients [42], often attributed to the heterogeneous expression of integrin αvβ3, unfavorable drug pharmacokinetics and its dose-dependency [43]. Hence, we focused our further investigations on the potential interplay between the αVβ3 and αVβ5 receptors and DDR1. We discovered that the cell surface expression and RGD binding capacity of integrins αVβ3 and αVβ5 remains stable by DDR1 inhibition and vice versa. Based on these observations, further translational studies are warranted that investigate the potential of pharmaceutical multi-targeting of DDR1 and αVβ3/αVβ5 in combination with radiochemotherapy in GBM as alternative to single cilengitide therapy or single DDR1 inhibition.
To shed some light on the underlying molecular mechanisms, we analyzed expression and phosphorylation of ATM and DNA-PK, two key proteins involved in DSB repair. The expression levels of both proteins were not altered upon DDR1-IN-1/cilengitide treatment. While ATM phosphorylation increased up to 24 h after treatment, a decline of DNA-PK phosphorylation was observed in the same time period. Analysis of pATM and pDNA-PK foci verified our observation. With regards to these opposing effects, an inhibitory function of DNA-PK towards ATM has been reported [44]. Thereby, DNA-PK phosphorylates ATM at multiple sites leading to the inhibition of ATM activity and concomitantly a negative regulation of ATM signaling as well as repair of DNA damages [44]. These points suggest that our observed enhancement of residual DSB by the DDR1-IN-1/cilengitide treatment may be attributable to an ATM hyperactivation upon DNA-PK deactivation, which has also been observed in other studies [44,45]. Therefore, we next investigated the effect of inhibition of ATM in combination with either DDR1-IN-1 alone, cilengitide alone or their combination. ATM deactivation demonstrated to be dominant over DDR1-IN-1 and cilengitide treatment, suggesting DDR1 and αVβ3/αVβ5 to modulate DNA repair.
In summary, our study suggests an interplay between DDR1 and integrins αVβ3/αVβ5 that influences cellular radiosensitivity and DSB repair in GBM cells. Taken together, these data are reminiscent, on the one hand, of the potential of integrin inhibition and, on the other hand, of the potential of multi-targeting approaches; in this case DDR1 and αVβ3/αVβ5. Further studies are warranted to evaluate the relevance of such approaches as an adjuvant to radiochemotherapy in GBM with regard to efficiency and safety compared to the current standard therapy.
Disclosure of conflict of interest
None.
References
- 1.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.DeWeerdt S. The genomics of brain cancer. Nature. 2018;561:S54–S55. doi: 10.1038/d41586-018-06711-8. [DOI] [PubMed] [Google Scholar]
- 5.Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, Huang L, Ratliff M, Karimian Jazi K, Kurz FT, Schmenger T, Lemke D, Gommel M, Pauli M, Liao Y, Haring P, Pusch S, Herl V, Steinhauser C, Krunic D, Jarahian M, Miletic H, Berghoff AS, Griesbeck O, Kalamakis G, Garaschuk O, Preusser M, Weiss S, Liu H, Heiland S, Platten M, Huber PE, Kuner T, von Deimling A, Wick W, Winkler F. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93–98. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- 6.Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wissmann N, Botz M, Soyka SJ, Beretta CA, Pramatarov RL, Fankhauser L, Garofano L, Freudenberg A, Wagner J, Tanev DI, Ratliff M, Xie R, Kessler T, Hoffmann DC, Hai L, Dorflinger Y, Hoppe S, Yabo YA, Golebiewska A, Niclou SP, Sahm F, Lasorella A, Slowik M, Doring L, Iavarone A, Wick W, Kuner T, Winkler F. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell. 2022;185:2899–2917. e31. doi: 10.1016/j.cell.2022.06.054. [DOI] [PubMed] [Google Scholar]
- 7.Ferrer VP, Moura Neto V, Mentlein R. Glioma infiltration and extracellular matrix: key players and modulators. Glia. 2018;66:1542–1565. doi: 10.1002/glia.23309. [DOI] [PubMed] [Google Scholar]
- 8.Faisal SM, Comba A, Varela ML, Argento AE, Brumley E, Abel C 2nd, Castro MG, Lowenstein PR. The complex interactions between the cellular and non-cellular components of the brain tumor microenvironmental landscape and their therapeutic implications. Front Oncol. 2022;12:1005069. doi: 10.3389/fonc.2022.1005069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian Y, Bai F, Zhang D. New target DDR1: a “double-edged sword” in solid tumors. Biochim Biophys Acta Rev Cancer. 2023;1878:188829. doi: 10.1016/j.bbcan.2022.188829. [DOI] [PubMed] [Google Scholar]
- 10.Sun X, Wu B, Chiang HC, Deng H, Zhang X, Xiong W, Liu J, Rozeboom AM, Harris BT, Blommaert E, Gomez A, Garcia RE, Zhou Y, Mitra P, Prevost M, Zhang D, Banik D, Isaacs C, Berry D, Lai C, Chaldekas K, Latham PS, Brantner CA, Popratiloff A, Jin VX, Zhang N, Hu Y, Pujana MA, Curiel TJ, An Z, Li R. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature. 2021;599:673–678. doi: 10.1038/s41586-021-04057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berestjuk I, Lecacheur M, Carminati A, Diazzi S, Rovera C, Prod’homme V, Ohanna M, Popovic A, Mallavialle A, Larbret F, Pisano S, Audebert S, Passeron T, Gaggioli C, Girard CA, Deckert M, Tartare-Deckert S. Targeting discoidin domain receptors DDR1 and DDR2 overcomes matrix-mediated tumor cell adaptation and tolerance to BRAF-targeted therapy in melanoma. EMBO Mol Med. 2022;14:e11814. doi: 10.15252/emmm.201911814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nokin MJ, Darbo E, Travert C, Drogat B, Lacouture A, San Jose S, Cabrera N, Turcq B, Prouzet-Mauleon V, Falcone M, Villanueva A, Wang H, Herfs M, Mosteiro M, Janne PA, Pujol JL, Maraver A, Barbacid M, Nadal E, Santamaria D, Ambrogio C. Inhibition of DDR1 enhances in vivo chemosensitivity in KRAS-mutant lung adenocarcinoma. JCI Insight. 2020;5:e137869. doi: 10.1172/jci.insight.137869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 14.Henriet E, Sala M, Abou Hammoud A, Tuariihionoa A, Di Martino J, Ros M, Saltel F. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adh Migr. 2018;12:363–377. doi: 10.1080/19336918.2018.1465156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiner HL, Huang H, Zagzag D, Boyce H, Lichtenbaum R, Ziff EB. Consistent and selective expression of the discoidin domain receptor-1 tyrosine kinase in human brain tumors. Neurosurgery. 2000;47:1400–1409. [PubMed] [Google Scholar]
- 16.Ram R, Lorente G, Nikolich K, Urfer R, Foehr E, Nagavarapu U. Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J Neurooncol. 2006;76:239–248. doi: 10.1007/s11060-005-6874-1. [DOI] [PubMed] [Google Scholar]
- 17.Vehlow A, Klapproth E, Jin S, Hannen R, Hauswald M, Bartsch JW, Nimsky C, Temme A, Leitinger B, Cordes N. Interaction of discoidin domain receptor 1 with a 14-3-3-Beclin-1-Akt1 complex modulates glioblastoma therapy sensitivity. Cell Rep. 2019;26:3672–3683. e7. doi: 10.1016/j.celrep.2019.02.096. [DOI] [PubMed] [Google Scholar]
- 18.Vehlow A, Cordes N. DDR1 (discoidin domain receptor tyrosine kinase 1) drives glioblastoma therapy resistance by modulating autophagy. Autophagy. 2019;15:1487–1488. doi: 10.1080/15548627.2019.1618540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 20.Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019;20:457–473. doi: 10.1038/s41580-019-0134-2. [DOI] [PubMed] [Google Scholar]
- 21.Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234–240. doi: 10.1016/j.tcb.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deville SS, Cordes N. The extracellular, cellular, and nuclear stiffness, a trinity in the cancer resistome-a review. Front Oncol. 2019;9:1376. doi: 10.3389/fonc.2019.01376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z, Gu Y, Zhao N, Xiang Q, Cui Y. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. 2023;8:1. doi: 10.1038/s41392-022-01259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vehlow A, Cordes N. Invasion as target for therapy of glioblastoma multiforme. Biochim Biophys Acta. 2013;1836:236–244. doi: 10.1016/j.bbcan.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Cordes N, Hansmeier B, Beinke C, Meineke V, van Beuningen D. Irradiation differentially affects substratum-dependent survival, adhesion, and invasion of glioblastoma cell lines. Br J Cancer. 2003;89:2122–2132. doi: 10.1038/sj.bjc.6601429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, Shen S, Hou F, Yan Y. Pathophysiological roles of integrins in gliomas from the perspective of glioma stem cells. Front Cell Dev Biol. 2022;10:962481. doi: 10.3389/fcell.2022.962481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao J, Wang L. Integrin alpha3 mediates stemness and invasion of glioblastoma by regulating POU3F2. Curr Protein Pept Sci. 2023;24:247–256. doi: 10.2174/1389203724666230224115459. [DOI] [PubMed] [Google Scholar]
- 28.Korovina I, Vehlow A, Temme A, Cordes N. Targeting integrin alpha2 as potential strategy for radiochemosensitization of glioblastoma. Neuro Oncol. 2023;25:648–661. doi: 10.1093/neuonc/noac237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghochani Y, Muthukrishnan SD, Sohrabi A, Kawaguchi R, Condro MC, Bastola S, Gao F, Qin Y, Mottahedeh J, Iruela-Arispe ML, Rao N, Laks DR, Liau LM, Mathern GW, Goldman SA, Carmichael ST, Nakano I, Coppola G, Seidlits SK, Kornblum HI. A molecular interactome of the glioblastoma perivascular niche reveals integrin binding sialoprotein as a mediator of tumor cell migration. Cell Rep. 2022;41:111511. doi: 10.1016/j.celrep.2022.111511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Xu X, Zhang Y, Mo Y, Sun X, Shu L, Ke Y. Paradoxical role of beta8 integrin on angiogenesis and vasculogenic mimicry in glioblastoma. Cell Death Dis. 2022;13:536. doi: 10.1038/s41419-022-04959-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bae E, Huang P, Muller-Greven G, Hambardzumyan D, Sloan AE, Nowacki AS, Marko N, Carlin CR, Gladson CL. Integrin alpha3beta1 promotes vessel formation of glioblastoma-associated endothelial cells through calcium-mediated macropinocytosis and lysosomal exocytosis. Nat Commun. 2022;13:4268. doi: 10.1038/s41467-022-31981-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sani S, Pallaoro N, Messe M, Bernhard C, Etienne-Selloum N, Kessler H, Marinelli L, Entz-Werle N, Foppolo S, Martin S, Reita D, Dontenwill M. Temozolomide-acquired resistance is associated with modulation of the integrin repertoire in glioblastoma, impact of alpha5beta1 integrin. Cancers (Basel) 2022;14:369. doi: 10.3390/cancers14020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanzani E, Pedrosa L, Bourmeau G, Anezo O, Noguera-Castells A, Esteve-Codina A, Passoni L, Matteoli M, de la Iglesia N, Seano G, Martinez-Soler F, Tortosa A. Dual role of integrin alpha-6 in glioblastoma: supporting stemness in proneural stem-like cells while inducing radioresistance in mesenchymal stem-like cells. Cancers (Basel) 2021;13:3055. doi: 10.3390/cancers13123055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vehlow A, Cordes N. Growth factor receptor and beta1 integrin signaling differentially regulate basal clonogenicity and radiation survival of fibroblasts via a modulation of cell cycling. In Vitro Cell Dev Biol Anim. 2022;58:169–178. doi: 10.1007/s11626-022-00656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slack RJ, Macdonald SJF, Roper JA, Jenkins RG, Hatley RJD. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discov. 2022;21:60–78. doi: 10.1038/s41573-021-00284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 40.Kapp TG, Rechenmacher F, Neubauer S, Maltsev OV, Cavalcanti-Adam EA, Zarka R, Reuning U, Notni J, Wester HJ, Mas-Moruno C, Spatz J, Geiger B, Kessler H. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci Rep. 2017;7:39805. doi: 10.1038/srep39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, Thon N, Goetz C, Kretzschmar HA, Tonn JC, Goldbrunner RH. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol. 2008;18:378–386. doi: 10.1111/j.1750-3639.2008.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Hau P, Weyerbrock A, Taphoorn MJ, Shen CC, Rao N, Thurzo L, Herrlinger U, Gupta T, Kortmann RD, Adamska K, McBain C, Brandes AA, Tonn JC, Schnell O, Wiegel T, Kim CY, Nabors LB, Reardon DA, van den Bent MJ, Hicking C, Markivskyy A, Picard M, Weller M European Organisation for Research and Treatment of Cancer (EORTC); Canadian Brain Tumor Consortium; CENTRIC study team. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100–1108. doi: 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- 43.Chinot OL. Cilengitide in glioblastoma: when did it fail? Lancet Oncol. 2014;15:1044–1045. doi: 10.1016/S1470-2045(14)70403-6. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, Lee JH, Jiang W, Crowe JL, Zha S, Paull TT. Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM. Mol Cell. 2017;65:91–104. doi: 10.1016/j.molcel.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caron P, Choudjaye J, Clouaire T, Bugler B, Daburon V, Aguirrebengoa M, Mangeat T, Iacovoni JS, Alvarez-Quilon A, Cortes-Ledesma F, Legube G. Non-redundant functions of ATM and DNA-PKcs in response to DNA double-strand breaks. Cell Rep. 2015;13:1598–1609. doi: 10.1016/j.celrep.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]