

Abstract

A product and DFT computational study on the reactions of 3-ethyl-3-(trifluoromethyl)dioxirane (ETFDO) with bicyclic and spirocyclic hydrocarbons bearing cyclopropyl groups was carried out. With bicyclo[n.1.0]alkanes (n = 3–6), diastereoselective formation of the alcohol product derived from C2–H bond hydroxylation was observed, accompanied by smaller amounts of products derived from oxygenation at other sites. With 1-methylbicyclo[4.1.0]heptane, rearranged products were also observed in addition to the unrearranged products deriving from oxygenation at the most activated C2–H and C5–H bonds. With spiro[2.5]octane and 6-tert-butylspiro[2.5]octane, reaction with ETFDO occurred predominantly or exclusively at the axial C4–H to give unrearranged oxygenation products, accompanied by smaller amounts of rearranged bicyclo[4.2.0]octan-1-ols. The good to outstanding site-selectivities and diastereoselectivities are paralleled by the calculated activation free energies for the corresponding reaction pathways. Computations show that the σ* orbitals of the bicyclo[n.1.0]alkane cis or trans C2–H bonds and spiro[2.5]octanes axial C4–H bond hyperconjugatively interact with the Walsh orbitals of the cyclopropane ring, activating these bonds toward HAT to ETFDO. The detection of rearranged oxygenation products in the oxidation of 1-methylbicyclo[4.1.0]heptane, spiro[2.5]octane, and 6-tert-butylspiro[2.5]octane provides unambiguous evidence for the involvement of cationic intermediates in these reactions, representing the first examples on the operation of ET pathways in dioxirane-mediated C(sp3)–H bond oxygenations. Computations support these findings, showing that formation of cationic intermediates is associated with specific stabilizing hyperconjugative interactions between the incipient carbon radical and the cyclopropane C–C bonding orbitals that trigger ET to the incipient dioxirane derived 1,1,1-trifluoro-2-hydroxy-2-butoxyl radical.
Introduction
The cyclopropyl group is an important and versatile motif. Because of its characteristic structural and bonding features,1 substitution of cyclopropane can modify the properties of substrates and provide access to a variety of useful synthetic transformations. Accordingly, cyclopropane-containing molecules are finding increasing application in organic synthesis,2 in drug development,3 and as functional molecules in different fields.4 The cyclopropyl group is also present in several natural products including terpenoids, steroids, and alkaloids, among which, many show biological activity and may serve as potential drug leads.5
A promising approach for structural diversification of cyclopropane containing molecules is represented by C(sp3)–H bond functionalization, a mainstream topic of modern synthetic chemistry.6 Overlap between a cyclopropane Walsh C–C bonding orbital and the σ* antibonding orbital of an α-C–H activates this bond toward functionalization (Figure 1a), providing a powerful handle to implement site-selectivity in these reactions.6a
Figure 1.
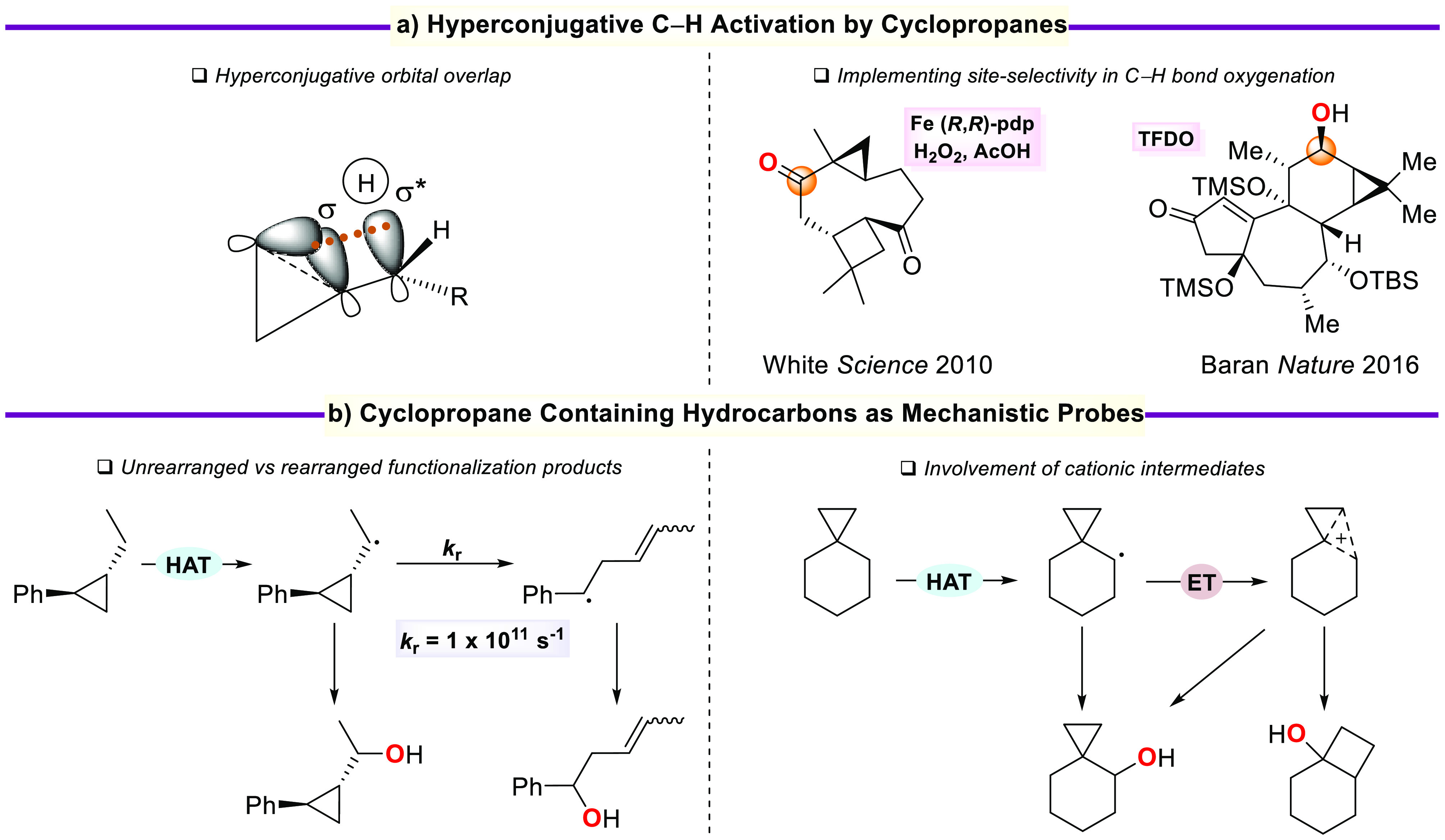
Use of cyclopropyl containing substrates (a) to induce selectivity in HAT-based C–H bond functionalization procedures and (b) as mechanistic probes.
Concerted insertion or two-step hydrogen atom transfer (HAT) strategies typically occur. In the latter case, however, because the intermediate cyclopropylcarbinyl radicals formed in the HAT step are known to undergo rapid rearrangement,7 the procedure is limited to the use of reagents that ensure very fast radical capture, preventing competitive unimolecular pathways and delivering the unrearranged functionalized product. Metal-oxo species,8 dioxiranes,9 and oxaziridines10 are examples of such reagents, able to promote stereoretentive C(sp3)–H oxygenations.
Along these lines, the C–H bond oxygenation of linear, bicyclic, and spirocyclic substrates bearing cyclopropane moieties has been studied employing a variety of oxygenation reagents.11 High selectivity for hydroxylation and ketonization at the activated α-methylenes over other sites has been generally observed. Similar selectivity patterns have been observed in dihalocarbene insertions into the C(sp3)–H bonds of hydrocarbons bearing cyclopropane moieties.12
In the framework of synthetically useful procedures, the full potential of this activation is witnessed by the results obtained by White in the site-selective C–H bond ketonization of a terpenoid derivative with H2O2 catalyzed by the Fe (R,R)-pdp complex,11f and by Baran in the site-selective and stereoselective C–H bond hydroxylation promoted by 3-methyl-3-(trifluoromethyl)dioxirane (TFDO), employed in an intermediate step of the total synthesis of (+)-phorbol (Figure 1a).13
Because of the tendency of cyclopropylcarbinyl radicals to undergo rapid rearrangement,7 cyclopropane-containing substrates are coveted mechanistic probes to study the involvement of radical intermediates in a reaction,14 to assess the concerted, radical, and/or cationic nature of enzymatic and biomimetic reaction mechanisms,8a,15 as well as to calibrate the rates of competing radical reactions (Figure 1b). For example, trans-1-ethyl-2-phenylcyclopropane has been employed as a probe to calibrate the rate constant for recombination of the radical couple formed in the first step of its reaction with dimethyldioxirane (DMDO).16 Based on a ring-opening rate constant kr = 1 × 1011 s–1, and a 40:1 unrearranged/rearranged product ratio, a rate constant k = 4 × 1012 s–1 could be estimated at room temperature corresponding to a lifetime of the radical couple of 200 fs.
With spiro[2.5]octane, the corresponding cyclopropylcarbinyl radical undergoes ring-opening with kr = 5 × 107 s–1.15a In the framework of the oxygenation of this substrate promoted by metal-oxo species,11f dioxiranes,11e ozone,11a and cytochrome P450 enzymes,15a no evidence for the formation of products deriving from radical rearrangement has been observed, in line with the relatively low value of kr that prevents competition with the radical capture or radical recombination steps.
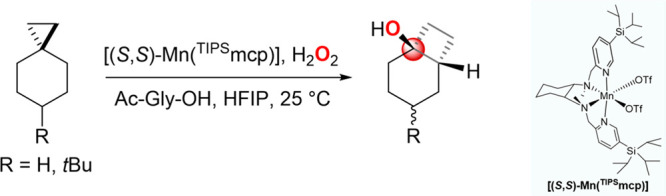
With substrates such as spiro[2.5]octane and bicyclo[4.1.0]heptane (norcarane), the product distribution can also provide information on the possible involvement of cationic intermediates, revealing the occurrence of competitive ET steps.15a In the specific case of spiro[2.5]octane, the formation of bicyclo[4.2.0]octan-1-ol can provide conclusive evidence for the involvement of a cationic intermediate. Evidence for the formation of rearranged alcohol products has been obtained in a recent study on the oxygenation of spiro[2.5]octane and 6-tert-butylspiro[2.5]octane promoted by manganese-oxo species, where leveraging on the use of fluorinated alcohol solvents and on catalyst electronics, predominant or exclusive formation of bicyclo[4.2.0]octan-1-ol and cis-4-(tert-butyl)-bicyclo[4.2.0]octan-1-ol, respectively, was observed (Scheme 1).17
Scheme 1. Results Obtained in the Oxidation of Spiro[2.5]octanes with H2O2 Catalyzed by [(S,S)-Mn(TIPSmcp)] (HFIP = 1,1,1,3,3,3-Hexafluoro-2-propanol).
Because similar mechanistic features are associated with oxygenations promoted by metal-oxo species and dioxiranes,8,18 and considering that the oxidizing ability of the intermediate α-hydroxy alkoxyl radical formed following HAT to the dioxirane (Scheme 2) can be modulated by careful choice of the precursor ketone as well as by solvent effects, we explored if these reagents in combination with fluorinated alcohol solvents could lead to the (unprecedented) involvement of cationic intermediates in dioxirane reactions.
Scheme 2. Mechanism of C(sp3)–H Bond Oxidation by Dioxiranes.

We report on the results of a detailed product and computational study of the reactions of 3-ethyl-3-(trifluoromethyl)dioxirane (ETFDO) with bicyclic (S1–S5) and spirocyclic (S7 and S8) hydrocarbons bearing cyclopropyl groups, the structures for which are displayed in Figure 2. Product studies have been also extended to 1,1-dimethylcyclohexane (S6) and to the diastereomeric alcohol couples P2a-OH, P2b-OH, P8a-OH, and P8c-OH (Figure 2).
Figure 2.

Structures of the substrates investigated in this work.
Results
Reactions with ETFDO
The reactions of substrates S1–S8 with in situ generated ETFDO were carried out at 0 °C in a 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)/H2O 3:1 solvent mixture containing the substrate (1 equiv), oxone (1 equiv), NaHCO3 (4 equiv), 1,1,1-trifluoro-2-butanone (0.2 equiv), and Bu4NHSO4 0.05 equiv, according to a previously reported procedure.19 Product yields for the oxygenation of bicyclic hydrocarbons (S1–S5), 1,1-dimethylcyclohexane (S6), and spirocyclic hydrocarbons (S7 and S8) by ETFDO are shown in Scheme 3 and Scheme 4. The schemes show the results obtained at ≥80% conversion, where the total yields of the oxygenation products approach 87%. This is accompanied by ≥90% mass balances. With S6, a 49% conversion was observed after a 48 h reaction time with a 46% total yield of oxygenation products (Scheme 4). Schemes 3 and 4 also present the product yields obtained at low conversion with substrates S1, S2, S4, and S8. The ketone products arising from overoxidation of the first formed alcohols at the C–H bonds that are α to the cyclopropyl group are not observed under low conversion conditions. Full experimental details are reported in the Supporting Information (SI) (Tables S1–S8).
Scheme 3. Oxygenation of Bicyclo[n.1.0]alkanes (n = 3–6) (S1–S5) Promoted by ETFDO.

Scheme 4. Oxygenation of 1,1-Dimethylcyclohexane (S6) and of Spiro[2.5]octanes (S7 and S8) Promoted by ETFDO.

The yield of the minor products deriving from C–H bond oxygenation at remote positions (C-3 for S1 and S2; C-3 and C-4 for S4 and S5) was calculated as the sum of the alcohol and ketone products. In the oxygenation of S3, product yields of alcohols at C-2 and C-5 are given in both cases as the sum of the cis- and trans- isomers (full details on the product distributions are displayed in the SI, Table S3). For the oxidation of S6 and S7, product yields were obtained after chromic acid oxidation of the reaction mixture (see SI, Tables S6 and S7).
The reaction with ETFDO was also extended to some of the oxygenation products of S2 and S8. The main reaction products P2a-OH and P8a-OH and the corresponding ketones P2-O and P8-O were isolated by the scale-up oxidation of S2 and S8, respectively. P2b-OH and P8c-OH (the diastereoisomer of P8a-OH, not observed in the oxidation of S8) were prepared by diastereoselective reduction of parent ketones P2-O and P8-O, respectively (see SI). Conversions and product yields observed in the oxygenation of the isomeric cis- and trans- alcohol products P2a-OH and P2b-OH by ETFDO are displayed in Scheme 5a. The results of the competitive oxygenation of a 1:1 mixture of P2a-OH and P2b-OH by ETFDO are described in Scheme 5b. Scheme 6 shows the conversions and product yields that are observed in the corresponding experiments with P8a-OH and P8c-OH.
Scheme 5. Oxygenation of cis-Bicyclo[4.1.0]heptan-2-ol (P2a-OH) and trans-Bicyclo[4.1.0]heptan-2-ol (P2b-OH).

Conversion and product yields were determined by GC and averaged over two independent experiments. (a) Reaction conditions: P2a-OH or P2b-OH 1 equiv, oxone 1 equiv, NaHCO3 4 equiv, 1,1,1-trifluoro-2-butanone 0.2 equiv, HFIP/H2O (3:1), Bu4NHSO4 0.05 equiv, T = 0 °C, 3 h. (b) P2a-OH 1 equiv, P2b-OH 1 equiv, oxone 1 equiv, NaHCO3 4 equiv, 1,1,1-trifluoro-2-butanone 0.2 equiv, HFIP/H2O (3:1), Bu4NHSO4 0.05 equiv, T = 0 °C, 6 h. rsm: recovered starting material.
Scheme 6. Oxygenation of trans-6-tert-Butylspiro[2.5]octan-2-ol (P8a-OH) and cis-6-tert-Butylspiro[2.5]octan-2-ol (P8c-OH).

Conversion and product yields were determined by GC and averaged over two independent experiments. (a) Reaction conditions: P8a-OH or P8c-OH 1 equiv, oxone 1 equiv, NaHCO3 4 equiv, 1,1,1-trifluoro-2-butanone 0.2 equiv, HFIP/H2O (3:1), Bu4NHSO4 0.05 equiv, T = 0 °C, 3 h. (b) P8a-OH 1 equiv, P8c-OH 1 equiv, oxone 1 equiv, NaHCO3 4 equiv, 1,1,1-trifluoro-2-butanone 0.2 equiv, HFIP/H2O (3.0:1.0), Bu4NHSO4 0.05 equiv, T = 0 °C, 6 h. rsm: recovered starting material.
Computational Studies
Density functional theory (DFT) computations were performed with Gaussian 16.20 The ωB97X-D functional was used to optimize molecular geometries,21 with the 6-311++G(d,p) basis set and the SMD solvation model accounting for H2O.22 Frequency calculations were conducted at the same level of theory used for the geometry optimizations to obtain thermal Gibbs free energies and characterize the stationary points on the potential energy surface. The correct unrestricted wave functions were obtained by performing a stability test with the Gaussian keyword stable = opt. Gibbs free energies were corrected using Goodvibes, which corrects the vibrational frequencies via the approximation for the quasi-harmonic correction, as proposed by Grimme.23 Intrinsic reaction coordinate (IRC) calculations were performed to verify that a transition state (TS) connects the reactant and product on the potential energy surface. CYLview was employed to visualize molecular structures.24
The computed site-selectivities for C(sp3)–H bond oxygenation of bicyclo[n.1.0]alkanes S1, S2, S4 and S5 with ETFDO are shown in Figure 3. The relative activation free energies (ΔΔG‡) for the C2–H and C3–H bonds are given in kcal mol–1. For comparison, the experimental ΔΔG‡ values, which are derived from the experiments illustrated in Scheme 3 (for which the normalized site-selectivities are displayed in Figure 8), are also shown.
Figure 3.

Difference in activation free energies (ΔΔG‡, in kcal mol–1) for HAT from the C2–H and C3–H bonds in S1, S2, S4, and S5 to ETFDO: computational and experimental studies.
Figure 8.
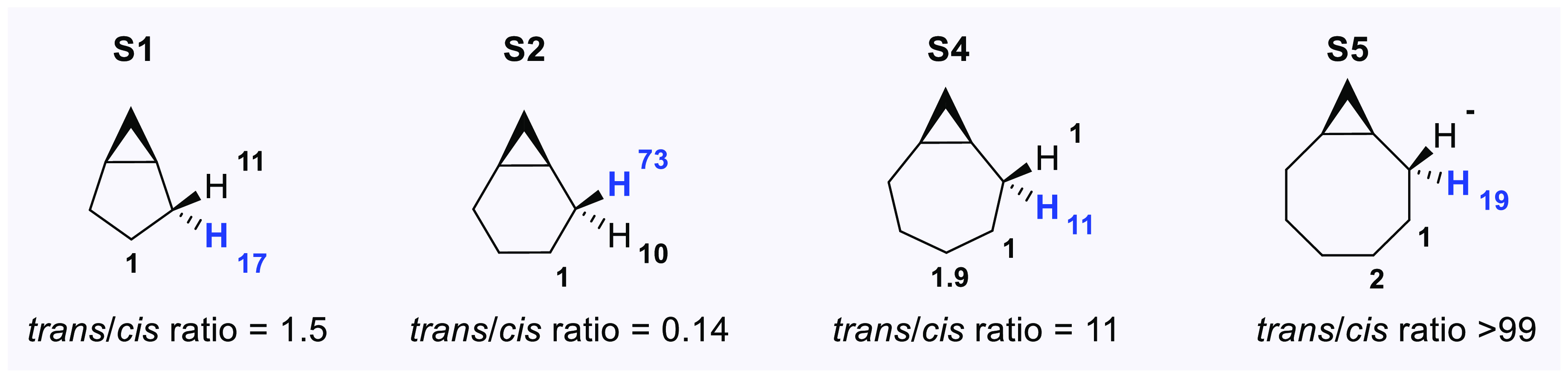
Normalized site-selectivities and diastereoselectivities observed in the hydroxylation of bicyclo[n.1.0]alkanes S1, S2, S4, and S5 by ETFDO.
The pertinent transition structures obtained for these selectivity studies together with the analysis of the hyperconjugation effect on the C2–H bonds provided by the fused cyclopropane moiety are shown in Figures S7–S10 of the SI for the reactions of substrates S1, S2, S4, and S5, respectively. The computed site-selectivity for the C(sp3)–H bond oxygenation of 1-methylbicyclo[4.1.0]heptane (S3) is displayed in Figure 4.
Figure 4.
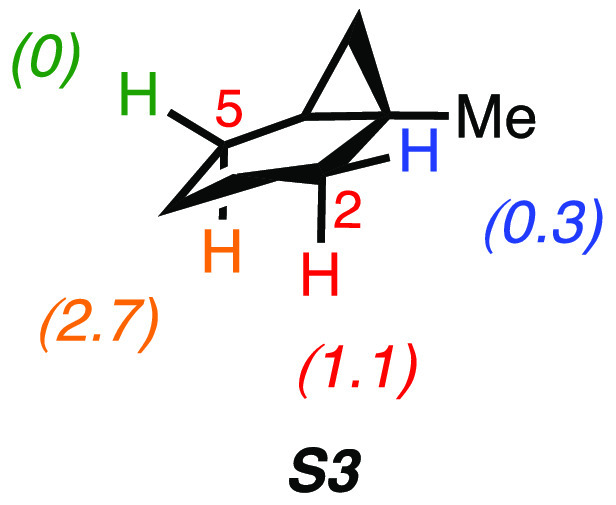
Computed difference in activation free energies (ΔΔG‡, in kcal mol–1) for HAT from the C2–H and C5–H bonds in S3 to ETFDO.
The transition structures for HAT from the C2–H and C5–H bonds of S3 to ETFDO are displayed in the SI as Figure S11. The energetics of the hydroxylation mechanisms for each of the C–H bonds at C-2 and C-5 are displayed in Figure 5.
Figure 5.
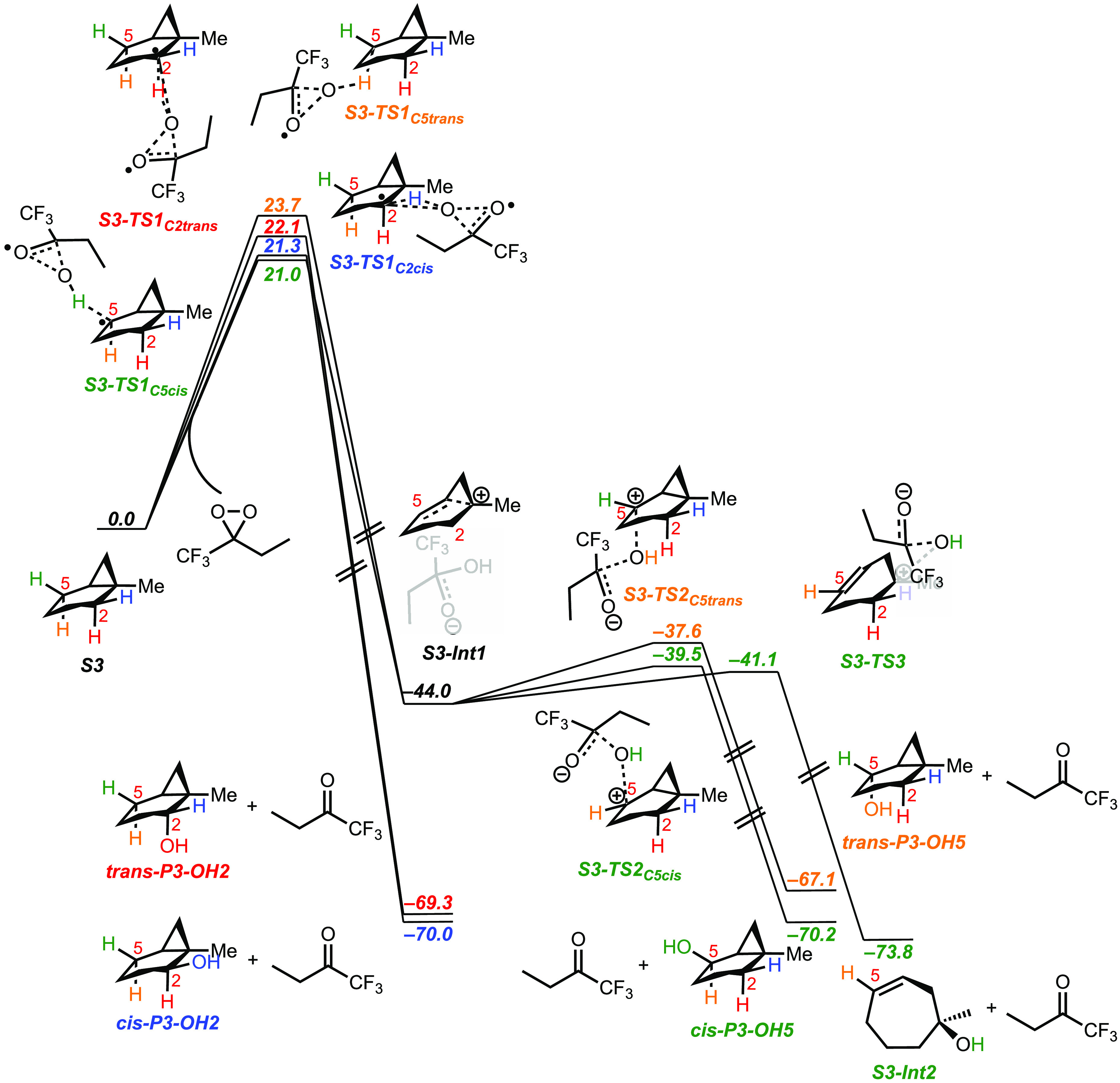
Energetics (in kcal mol–1) of C–H bond oxidation of S3 promoted by ETFDO.
The computed site-selectivities for C(sp3)–H bond oxygenation of spiro[2.5]octanes S7 and S8 by ETFDO are displayed in Figure 6 along with the experimental ΔΔG‡ values that are derived from the product distributions displayed in Scheme 4.
Figure 6.

Difference in activation free energies (ΔΔG‡, in kcal mol–1) for HAT from the C–H bonds of S7 and S8 to ETFDO: computational and experimental studies.
The transition structures for HAT from various positions of S7 and S8 to ETFDO and the analysis of the hyperconjugation effect on the C4–H bonds provided by the spiro-cyclopropane moiety are displayed in the SI as Figures S12 and S13, respectively. The energetics of the hydroxylation mechanisms for the axial and equatorial C4–H bonds of S8 are displayed in Figure 7.
Figure 7.
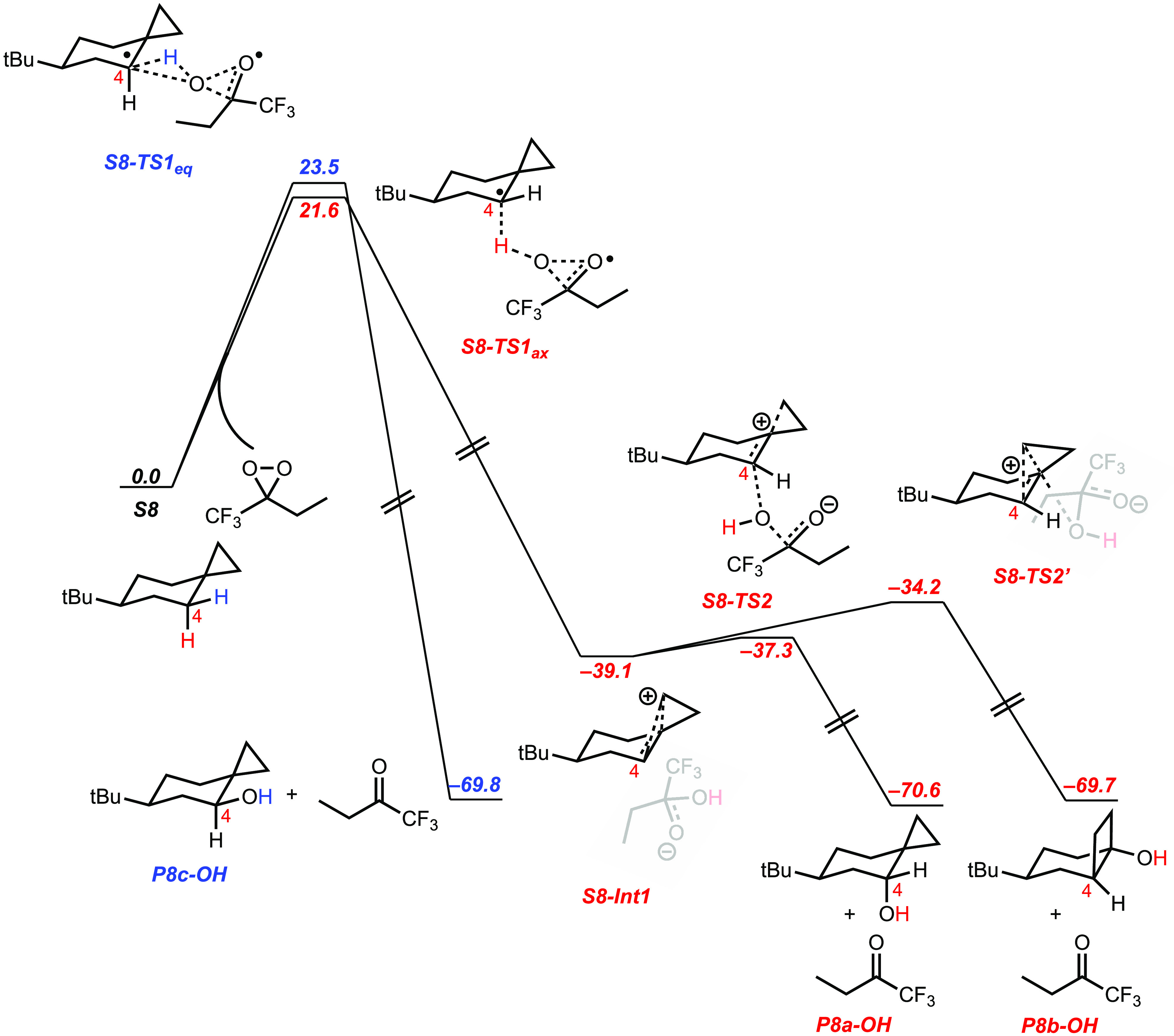
Energetics (in kcal mol–1) of C–H bond oxidation of S8 by ETFDO.
The corresponding energy profiles of the hydroxylation mechanisms for the C4–H, C5–H, and C6–H bonds of S7 are displayed in the SI as Figure S14.
Discussion
Oxygenation of Bicyclic Substrates (S1–S5, P2a-OH, and P2b-OH)
The products of the reaction of ETFDO with S1–S5 are displayed in Scheme 3. With S1, S2, and S4, reactions carried out at low substrate conversion (3–6 h reaction time, 15–28% conversion) showed, in all cases, the predominant formation of the diastereomeric alcohol products deriving from C2–H bond hydroxylation, accompanied by smaller amounts of products deriving from oxygenation at the other methylenic sites. With all three substrates, no evidence for the formation of the ketone product deriving from overoxidation of the alcohols at C-2, and of products deriving from oxidation of the cyclopropane C–H bonds, was observed. The former observation can be accounted for on the basis of the strong hydrogen bond donor (HBD) ability of HFIP that, by engaging in hydrogen bonding with the hydroxyl group of the alcohol products, inverts the polarity of the adjacent C–H bond, deactivating this site toward HAT to the electrophilic ETFDO.25 The latter observation reflects the very high BDE of the cyclopropane C–H bonds,26 that are typically resistant to HAT-based functionalization. By increasing the reaction time (48 h for S1, 9 h for S2 and S4), significantly higher conversions were obtained (80–87%), forming substantial amounts of the C-2 ketone. Products are oxygenated at the C-2 position of S1, S2, and S4 with selectivities of 96%, 98%, and 72% respectively. The reaction of S5 was carried out for a 9 h reaction time (86% conversion, 76% overall product yield), with the predominant formation of trans-bicyclo[6.1.0]nonan-2-ol (P5a-OH). These selectivities result from hyperconjugative stabilization, determined by the overlap of a cyclopropane Walsh C–C bonding orbital with the σ* orbital of the adjacent C2–H (Figure 1a).6a
The analysis of the product distributions obtained for S1, S2, S4, and S5, under conditions where overoxidation is not observed, provides information about the hydroxylation diastereoselectivity. The normalized hydroxylation site-selectivities are displayed in Figure 8. The trans/cis ratios for C2–H hydroxylation are highlighted.
Preferential trans C2–H hydroxylation was observed for S1, S4, and S5, with the trans/cis ratio that increases with increasing ring size, reaching an upper limit with S5 for which the product deriving from cis C2–H hydroxylation was not detected. Preferential cis C2–H hydroxylation was instead observed with S2 (trans/cis = 0.14). Interestingly, similar diastereoselectivity patterns were observed in dihalocarbene insertions into the C2–H bonds of S1 and S2 (trans/cis = 2.8–4 and 0.23–0.25, respectively),12 because the same effects operate in dioxirane hydroxylation and carbene insertion reactions.
It is worth noting that cyclopropylcarbinyl stabilization leading to selectivity with S2 also accounts for the diastereoselectivity observed in the oxidation employed in an intermediate step of the total synthesis of (+)-phorbol.13 Within the bicyclo[4.1.0]heptane structural motif (Figure 1a), selective hydroxylation at the α-C–H bond that is cis to the cyclopropane moiety was observed.
The diastereoselectivities were also explored by computational studies on the oxygenation of S1, S2, S4, and S5 promoted by ETFDO. The activation free energy differences (ΔΔG‡) for HAT from the C2–H bonds of these substrates to ETFDO are shown in Figure 3. The corresponding transition structures are presented in the SI (Figures S7–S10). Computational results show a strong preference for the oxygenation of C2–H over C3–H bonds, supporting the effect of hyperconjugation in C–H bond activation. Moreover, the studies of the oxidation selectivity align with experimental results. Figures S7–S10 highlight the hyperconjugative interaction by the cyclopropyl group when activating the cis and/or trans C2–H bond of S1, S2, S4, and S5 toward HAT to ETFDO.
In the reaction of S1, σ* orbitals of both cis and trans C2–H bonds can interact with the Walsh orbitals activating these bonds toward HAT. As a result, the energy difference between cis and trans C2–H bond oxidation is only 0.5 kcal mol–1. The effect of hyperconjugation on trans C2–H bond activation is highlighted in Figure S7. Experiments did not differentiate the selectivity between cis and trans C3–H bonds. However, computations predict a preference for oxygenation of the cis over the trans C3–H bond (ΔΔG‡ = 1.0 and 3.1 kcal mol–1, respectively).
With S2, the experimental and computational observation of a stronger activation of the cis C2–H bond over the trans one is also corroborated by the results obtained, under the same experimental conditions, in the oxidation of cis- and trans-bicyclo[4.1.0]heptan-2-ol (P2a-OH and P2b-OH, respectively) by ETFDO (Scheme 5a). With both substrates, exclusive formation of the corresponding ketone product (P2-O) in 9.2% and 33% yield, respectively, was observed, indicating that the latter alcohol is 3.6 times more reactive than the former one. P2b-OH displays a cis C2–H bond that benefits from hyperconjugative activation, whereas with P2a-OH the trans C2–H bond cannot benefit from a similar activation. Additional support is provided by the results obtained in the competitive oxidation of a 1:1 trans–cis mixture of bicyclo[4.1.0]heptan-2-ols (P2a-OH and P2b-OH) by ETFDO (Scheme 5b). 91% of P2a-OH and 66% of P2b-OH, together with an overall 40% yield of P2-O, were obtained, indicating that the latter alcohol is 3.8 times more reactive than the former one, showing excellent agreement between the two experiments.
With S4 and S5, the trans C2–H bond (ΔΔG‡ = 0 kcal mol–1) is the most activated toward HAT to ETFDO.
Among the bicyclo[n.1.0]alkane series, the oxygenation of 1-methylbicyclo[4.1.0]heptane (S3) by ETFDO is particularly noteworthy. With this substrate, in addition to the alcohol and ketone products deriving from oxygenation at the most activated C–H bonds at C-2 (P3-OH2 + P3-O2) and C-5 (P3-OH5 + P3-O5) in 33.4% and 32.6% combined yield, respectively, cis- and trans-3-methyl-8-oxabicyclo[5.1.0]octan-3-ol (P3-1) were also observed among the reaction products in 13% combined yield (Scheme 3). Full details about the product distribution of this reaction can be found in the SI. The formation of products P3-1 can be rationalized on the basis of the mechanism proposed by Groves and co-workers in the oxygenation of bicyclo[4.1.0]heptane (S2) promoted by cytochrome P450 enzymes.15a The carbon radical formed following HAT from C-2 can undergo, in addition to the canonical OH rebound and radical rearrangement pathways, one-electron oxidation to give a cationic intermediate that, after rearrangement, is converted into the hydroxylated product by OH-transfer or nucleophilic capture by water (Scheme 7).
Scheme 7. Groves Mechanism for the Oxygenation of S2 Promoted by Cytochrome P450 Enzymes15a.

An analogous mechanism is proposed for the oxidation of S3, where the formation of 1-methylcyclohept-3-en-1-ol is initiated by HAT from the C5–H bond. The intermediate alcohol product is then rapidly converted into P3-1 as a diastereomeric mixture via epoxidation by ETFDO.27 This mechanistic hypothesis is supported well by the computational results. The oxidation site-selectivity (Figure 4) follows the order: cis C5–H (ΔΔG‡ = 0 kcal mol–1), cis C2–H (ΔΔG‡ = 0.3 kcal mol–1), trans C2–H (ΔΔG‡ = 1.1 kcal mol–1), and trans C5–H (ΔΔG‡ = 2.7 kcal mol–1), confirming the stronger activation of the cis α-C–H bonds over the corresponding trans ones. The free energy profiles (Figure 5) show concerted oxidation through asynchronous HAT from the cis and trans C2–H bonds via S3-TS1C2cis and S3-TS1C2trans (for which ΔG‡ = 21.3 and 22.1 kcal mol–1, respectively), coupled to OH-rebound to give products P3-OH2. A homoallylic tertiary carbocation intermediate (S3-Int1, – 44.0 kcal mol–1) is formed through asynchronous HAT from cis and trans C5–H bonds (S3-TS1C5cis and S3-TS1C5trans: ΔG‡ = 21.0 and 23.7 kcal mol–1, respectively) coupled to electron transfer (ET). S3-Int1 then undergoes hydroxylation at C-5 through S3-TS2C5cis (−39.5 kcal mol–1) and S3-TS2C5trans (−37.6 kcal mol–1), resulting in the formation of P3-OH5. Figure 5 shows that S3-Int1 undergoes competitive hydroxylation at C-1 through S3-TS3 (−41.1 kcal mol–1) to form 1-methylcyclohept-3-en-1-ol, S3-Int2 (−73.8 kcal mol–1). S3-Int2 is then converted into 3-methyl-8-oxabicyclo[5.1.0]octan-3-ols P3-1 by oxygen atom transfer from ETFDO. The proposed mechanistic pathways for oxidation of S3 by EFTDO are summarized in Scheme 8, which shows 3D figures of the intermediate and transition state structures.
Scheme 8. Proposed Mechanistic Pathways for the Oxygenation of S3 Promoted by ETFDO.
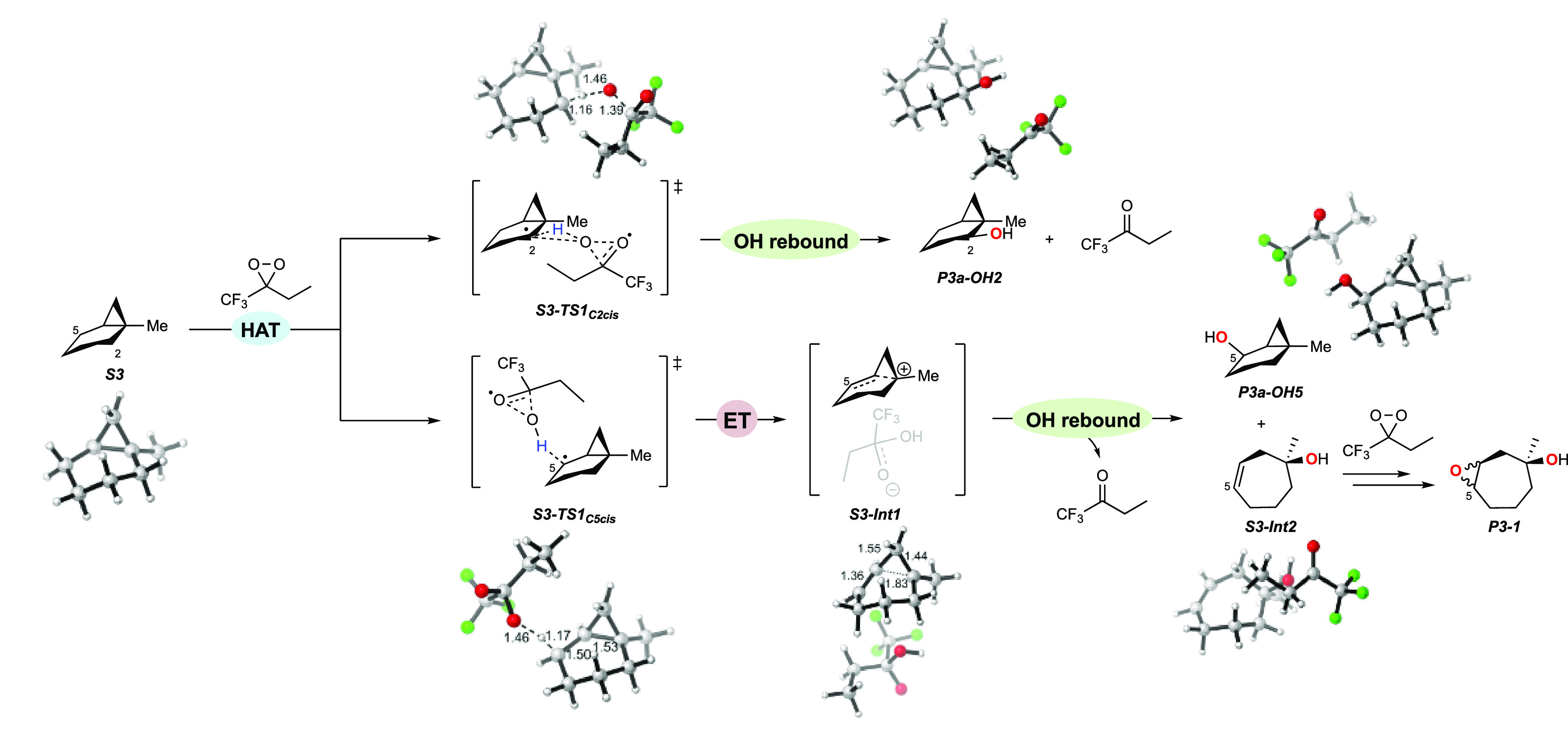
For the sake of simplicity, only the pathways initiated by HAT from the cis C2–H and C5–H bonds are displayed.
Interestingly, rearranged products (P3-1) are formed via initial HAT at the C5–H bond, and analogous isomeric products are not produced by HAT at the C2–H bond. This difference is caused by the distinct stabilization between the tertiary and secondary homoallylic cations. Hyperconjugation from the C-1 methyl group supports the emerging cationic intermediate at C-1 after HAT at the C5–H bond.28,29 Supportive evidence in favor of an ET pathway was also gained by investigating solvent effects on the oxygenation of S3 by ETFDO. By analyzing the products deriving from initial HAT at C-5, a decrease in the ratio between rearranged (P3-1) and unrearranged (P3-OH5 and P3-O5) products with decreasing solvent HBD ability was observed, i.e., going from HFIP to 2,2,2-trifluoroethanol (TFE) and MeCN (P3-1/(P3-OH5 + P3-O5) = 0.40, 0.16, and <0.01, respectively) (see SI, Table S9). This behavior can be associated with the strong HBD ability of fluorinated alcohols that, compared to non-HBD or weaker HBD solvents, can promote ET reactions via an increase in the oxidizing power of ET reagents and the ability to stabilize cationic intermediates.30
Oxygenation of Spirocyclic Substrates (S7, S8, P8a-OH, and P8c-OH)
The results obtained in the oxidation of spiro[2.5]octane (S7) and 6-tert-butylspiro[2.5]octane (S8) promoted by ETFDO were compared with those obtained for the corresponding reaction of 1,1-dimethylcyclohexane (S6) taken as a reference substrate, and are displayed in Scheme 4. With S6, the reaction carried out for 48 h, followed by treatment with chromic acid, afforded the ketone products deriving from oxidation at C-2 (P6-O), C-3 (P6-O3), and C-4 (P6-O4), in 14%, 21%, and 11% yield, respectively. Under the same conditions, the reaction of S7 led to the ketone products deriving from oxidation at C-4 (P7-O), C-5 (P7-O5), and C-6 (P7-O6), in 66%, 9.2%, and 7% yield, respectively, accompanied by the rearranged product bicyclo[4.2.0]octan-1-ol (P7b-OH) in 4.8% yield. With S8, the reaction mixture was not subjected to follow-up treatment with chromic acid, and the reaction carried out for 3 h showed the exclusive formation of the axial alcohol at C-4 (P8a-OH) in an 8.8% yield. By increasing the reaction time to 48 h, P8a-OH was formed in 54% yield, accompanied by the corresponding ketone (P8-O) and the rearranged alcohol cis-4-(tert-butyl)-bicyclo[4.2.0]octan-1-ol (P8b-OH) in 20% and 5.5% yield, respectively. With this substrate, oxygenation products at C-5 and C-6 as well as the equatorial alcohol at C-4, P8c-OH, were never observed. The formation of the rearranged alcohols P7b-OH and P8b-OH in the oxygenation of S7 and S8 by ETFDO (Scheme 4) provides conclusive evidence for the involvement of a cationic intermediate, uncovering the contribution of ET pathways to the overall reactivity.15a,17 This hypothesis is further supported by computational studies.
The energetics of the oxidation of S8 by ETFDO are shown in Figure 7. The axial C4–H bond undergoes asynchronous HAT to ETFDO through S8-TS1ax (21.6 kcal mol–1), which is coupled to ET to directly form the ion-pair S8-Int1 (−39.1 kcal mol–1). S8-Int1 undergoes either OH rebound via (S8-TS2, −37.3 kcal mol–1) or hydroxylation at C–3 via S8-TS2′ (−34.2 kcal mol–1). This observation accounts for the formation of P8b-OH through charged species S8-Int1 (Scheme 4).31 The activation energy of S8-TS2′ is slightly higher in comparison with S8-TS2 (ΔΔG‡ = 3.1 kcal mol–1). This energy difference qualitatively matches the experiment, explaining the low yield of P8b-OH. Hydroxylation of the equatorial C2–H bond (S8-TS1eq, 23.5 kcal mol–1) occurs concertedly without generating charged intermediates.
DFT calculations play a pivotal role by providing a qualitative approximation of the reaction outcomes. In a previous study,18b molecular dynamics revealed a 90% barrierless oxygen-rebound mechanism and 10% radical pair formation, while DFT predicted only a barrierless oxygen-rebound mechanism. This highlights the value of DFT and IRC in capturing the essence of the reaction mechanism, albeit with a degree of approximation. In order to confirm that S8-Int1 is the ion-pair intermediate, the CM5 calculation is employed to check the distribution of charges (Figure 9). The charge is evenly distributed in the 6-tert-butylspiro[2.5]octanylium cation (+0.94) and trifluoro-2-hydroxybutan-2-olate anion (−0.90). Moreover, a hypothetical triplet radical pair S8-Int1a is noticeably unstable compared to ion-pair S8-Int1 by 48.9 kcal mol–1. An open-shell singlet radical pair is not obtained in the computations with ωB97X-D/6-311++G(d,p)/SMD(H2O). Open-shell initial guesses led to the closed-shell result. Consequently, a hypothetical triplet radical pair S8-Int1a was employed to compare its energies with the S8-Int1 ion-pair. It is also worth mentioning that formation of a delocalized cation following ET within the hypothetical radical pair S8-Int1a strongly contributes to the reaction exergonicity. Isodesmic reaction calculations show a 10.7 kcal mol–1 thermodynamic advantage for delocalized S8-Int1 over the corresponding localized secondary carbocation (see Table S13 and Scheme S2 in the SI).
Figure 9.

(a) Charge distribution of S8-Int1 by CM5 and the electrostatic potential on a constant electron density surface. The regions of positive and negative potential are indicated in blue and red. (b) Spin density of the hypothetical triplet radical pair S8-Int1a. (c) In silico redox potentials of 1,1,1-trifluoro-2-hydroxybutoxy and 6-(tert-butyl)spiro[2.5]octan-4-yl radicals.
We also determined in silico redox potentials for the formation of 1,1,1-trifluoro-2-hydroxybutan-2-olate and 6-(tert-butyl)spiro[2.5]octan-4-ylium from the hypothetical radical pair S8-Int1a (Figure 9c). We found that the reduction potential of the 1,1,1-trifluoro-2-hydroxybutoxy radical is +1.33 V vs SCE (MeCN), and the oxidation potential from 6-(tert-butyl)spiro[2.5]octan-4-yl radical to 6-(tert-butyl)spiro[2.5]octan-4-ylium cation is −0.13 V vs SCE (MeCN). The redox potentials suggest the formation of the charged species via an exergonic redox process.
Supportive experimental evidence in favor of an ET pathway was gained from the study of the solvent effects on the oxidation reaction. Oxygenation of this substrate by ETFDO was studied in HFIP, TFE, and MeCN. As the solvent HBD ability was reduced, the ratio between rearranged (P8b-OH) and unrearranged (P8a-OH + P8-O) products was diminished, leading to the following P8b-OH/(P8a-OH + P8-O) ratios: 0.065, 0.028, <0.01, for HFIP, TFE, and MeCN, respectively (see SI, Table S10), pointing again toward the ability of fluorinated alcohols to promote ET reactions via an increase in the oxidizing power of ET reagents and to stabilize cationic intermediates.30,32
Based on these mechanistic studies and on previous findings,17 the oxidation mechanism of S8 by ETFDO is proposed in Scheme 9. An analogous mechanism is found for the oxygenation pathways initiated by HAT from the C4–H bond of S7 (Figure S14).
Scheme 9. Proposed Mechanism for the Oxygenation of S8 Promoted by ETFDO.
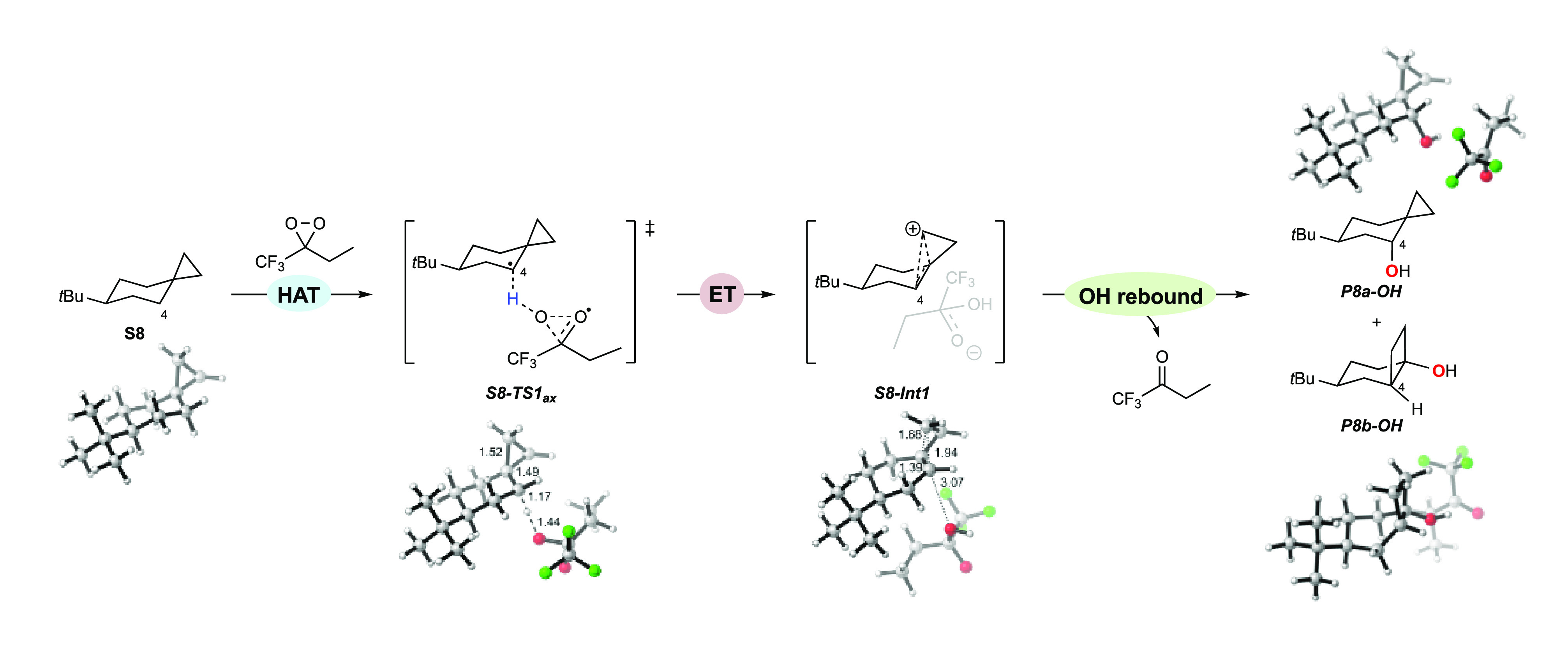
Grabovskiy et al. presented a concerted molecule-induced homolytic/rebound process of cage hydrocarbons using dioxiranes.33 Notably, our findings suggest that the generation of the cationic intermediate is associated with a specific stabilizing hyperconjugative interaction between the incipient carbon radical and the cyclopropane C–C bonding orbitals. This causes ET to the incipient 1,1,1-trifluoro-2-hydroxy-2-butoxyl radical.28
The ΔΔG‡ values for HAT from the C–H bonds of S7 and S8 to ETFDO are displayed in Figure 6. HAT from the C4–H bond of S7 presents the lowest energy barrier (ΔΔG‡ = 0 kcal mol–1), in comparison with the energy barriers for C5–H and C6–H bonds (ΔΔG‡ = 0.6 and 1.6 kcal mol–1, respectively). Furthermore, we find that the activation barriers of axial C–H bonds are lower than those of equatorial ones. The transition state structures are shown in Figure S12 in the SI. In S7-TS1C4ax, hyperconjugation leads to a slightly extended C1–C2 distance (1.52 Å) and a reduced C1–C4 distance (1.49 Å), differing from the other transition states that lack Walsh orbital interactions. Moreover, efficient hyperconjugation between the axial C4–H bond and the Walsh orbital in the transition state S7-TS1C4ax is evidenced.
With S8, oxygenation of the axial C4–H bond is favored over the equatorial one by 1.9 kcal mol–1, in good agreement with the experimental studies. Based on the analysis of the transition state structures, a hyperconjugative interaction by cyclopropane Walsh orbitals lowers the barrier of the axial C4–H bond. Compared to S8-TS1eq, S8-TS1ax exhibits a slightly longer C1–C2 distance (1.52 Å) and a shorter C1–C4 distance (1.49 Å) due to hyperconjugation.
The observation of a stronger hyperconjugative activation of the axial C4–H bond over the equatorial one is also corroborated by the results obtained, under the same experimental conditions, in the oxidation of trans- and cis-6-tert-butylspiro[2.5]octan-4-ol (P8a-OH and P8c-OH, respectively) by ETFDO (Scheme 6a). With both substrates, exclusive formation of the ketone product (P8-O) in 5% and 22% yield, respectively, was observed, indicating that the latter alcohol is 4.4 times more reactive than the former one. P8c-OH displays an axial C4–H bond that benefits from hyperconjugative activation, whereas with P8a-OH the equatorial C4–H bond C-4 cannot benefit from a similar activation. Additional support comes again from the results obtained in the competitive oxidation of a 1:1 trans–cis mixture of 6-tert-butylspiro[2.5]octan-4-ols (P8a-OH and P8c-OH) promoted by ETFDO (Scheme 6b): 94% recovery of P8a-OH and 74% recovery of P8c-OH, together with an overall 31% yield of P8-O were obtained, indicating that the latter alcohol is 4.3 times more reactive than the former one, showing an excellent agreement between the two experiments.
For the site-selectivities observed in the reactions of ETFDO with substrates S6–S8, the normalized product distributions are displayed in Figure 10. With S6, comparable selectivities were observed for the three methylenic sites (C-2:C-3:C-4 = 1.0:1.5:1.5). The slightly lower selectivity for oxygenation at C-2 over C-3 and C-4 can be reasonably explained on the basis of steric effects, where the presence of the two methyl groups limits the accessibility of the adjacent C2–H bonds to ETFDO.
Figure 10.

Normalized site-selectivities observed in the oxygenation of 1,1-dimethylcylohexane (S6), spiro[2.5]octane (S7) and 6-tert-butylspiro[2.5]octane (S8) promoted by ETFDO.
With S7, taking into account that the rearranged alcohol product P7b-OH derives from initial HAT from the C4–H bond, the normalized product distributions (C-4:C-5:C-6 = 7.6:1.0:1.5) point toward a significant activation of the C4–H bonds compared to the other methylenic sites. These results are in good agreement with those obtained previously in the oxidation of S7 promoted by the H2O2/(S,S)-Fe(pdp) and H2O2/(S,S)-Mn(TIPSpdp) systems and by TFDO.11e,11f,17 As mentioned above, this behavior reflects activation of the axial C4–H bonds via overlap with the Walsh C–C cyclopropane bonding orbitals. The site-selectivity observed in the oxygenation of S8, for which exclusive formation of products deriving from initial HAT at this site, reflects the synergistic cooperation of two effects: hyperconjugative C4–H bond activation together with C5–H and C6–H bond deactivation by torsional and steric effects determined by the presence of the bulky tert-butyl group at C-6.17,19
Conclusions
The results of product and computational studies on the C(sp3)–H bond oxygenation of bicyclic and spirocyclic hydrocarbons bearing cyclopropyl moieties promoted by ETFDO have led to a deeper understanding of the factors that govern selectivity in these processes. Activation of the C–H bonds that are α to the cyclopropyl group occurs when there is strong overlap between the cyclopropane Walsh C–C bonding orbitals and the C–H σ* orbitals. Diastereoselective hydroxylation is typically observed, reflecting preferential activation of one α-C–H bond, with the exclusive detection of a single diastereoisomer in the reactions of bicyclo[6.1.0]nonane (S5) and 6-tert-butylspiro[2.5]octane (S8). The experimental site-selectivities and diastereoselectivities are paralleled by the calculated activation free energies for the corresponding reaction pathways. The detection of rearranged oxygenation products in the oxidation of 1-methylbicyclo[4.1.0]heptane (S3), spiro[2.5]octane (S7), and 6-tert-butylspiro[2.5]octane (S8) provides unambiguous evidence for the involvement of cationic intermediates in these reactions, representing the first examples on the operation of ET pathways in dioxirane-mediated C(sp3)–H bond oxygenations.34,35 With these substrates, calculations predict the direct formation of an intermediate ion pair via HAT from a substrate C–H bond to ETFDO coupled to ET, highlighting the role of specific stabilizing interactions able to assist cation formation and divert the reaction from the canonical HAT/rebound pathway.
Acknowledgments
We are grateful to the National Science Foundation (CHE-1764328 and CHE-2153972 to K.N.H.) and the University of Rome “Tor Vergata” (Projects ORIENTATE E83C22002690005 and IseeFunND E83C22002710005) for financial support of this research. M.B. also thanks Miquel Costas for helpful discussions. The assistance of Daniel Oscar Cicero and Greta Petrella in the NMR analysis of the rearranged products observed in the oxidation of S3 is gratefully acknowledged. Calculations were performed on computational resources provided by the UCLA Institute of Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c07163.
Details on the preparation of the substrates, their oxidation reactions by ETFDO, on the isolation and characterization of reaction products, and on the computational studies. (PDF)
Author Contributions
∥ M.G. and W.L. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- De Meijere A. Bonding Properties of Cyclopropane and Their Chemical Consequences. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. 10.1002/anie.197908093. [DOI] [Google Scholar]
- a Schneider T. F.; Kaschel J.; Werz D. B. A New Golden Age for Donor–Acceptor Cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. 10.1002/anie.201309886. [DOI] [PubMed] [Google Scholar]; b Cohen Y.; Cohen A.; Marek I. Creating Stereocenters within Acyclic Systems by C–C Bond Cleavage of Cyclopropanes. Chem. Rev. 2021, 121, 140–161. 10.1021/acs.chemrev.0c00167. [DOI] [PubMed] [Google Scholar]; c Pirenne V.; Muriel B.; Waser J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. 10.1021/acs.chemrev.0c00109. [DOI] [PubMed] [Google Scholar]
- Talele T. T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- a de Meijere A.; Kozhushkov S. I.; Schill H. Three-Membered-Ring-Based Molecular Architectures. Chem. Rev. 2006, 106, 4926–4996. 10.1021/cr0505369. [DOI] [PubMed] [Google Scholar]; b Mizuno A.; Matsui K.; Shuto S. From Peptides to Peptidomimetics: A Strategy Based on the Structural Features of Cyclopropane. Chem.—Eur. J. 2017, 23, 14394–14409. 10.1002/chem.201702119. [DOI] [PubMed] [Google Scholar]
- a Wessjohann L. A.; Brandt W.; Thiemann T. Biosynthesis and Metabolism of Cyclopropane Rings in Natural Compounds. Chem. Rev. 2003, 103, 1625–1648. 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]; b Chen D. Y.-K.; Pouwer R. H.; Richard J.-A. Recent advances in the total synthesis of cyclopropane-containing natural products. Chem. Soc. Rev. 2012, 41, 4631–4642. 10.1039/c2cs35067j. [DOI] [PubMed] [Google Scholar]; c Fan Y.-Y.; Gao X.-H.; Yue J.-M. Attractive natural products with strained cyclopropane and/or cyclobutane ring systems. Sci. China Chem. 2016, 59, 1126–1141. 10.1007/s11426-016-0233-1. [DOI] [Google Scholar]; d Brill Z. G.; Condakes M. L.; Ting C. P.; Maimone T. J. Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev. 2017, 117, 11753–11795. 10.1021/acs.chemrev.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Li X.; Shimaya R.; Dairi T.; Chang W.-C.; Ogasawara Y. Identification of Cyclopropane Formation in the Biosyntheses of Hormaomycins and Belactosins: Sequential Nitration and Cyclopropanation by Metalloenzymes. Angew. Chem. Int. Ed. 2022, 61, e202113189 10.1002/anie.202113189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Newhouse T.; Baran P. S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yamaguchi J.; Yamaguchi A. D.; Itami K. C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; c Hartwig J. F. Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]; d White M. C.; Zhao J. Aliphatic C–H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Li J.; Zhang Z.; Wu L.; Zhang W.; Chen P.; Lin Z.; Liu G. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 2019, 574, 516–521. 10.1038/s41586-019-1655-8. [DOI] [PubMed] [Google Scholar]; f Davies H. M. L.; Liao K. Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization. Nature Rev. Chem. 2019, 3, 347–360. 10.1038/s41570-019-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Galeotti M.; Salamone M.; Bietti M. Electronic control over site-selectivity in hydrogen atom transfer (HAT) based C(sp3)–H functionalization promoted by electrophilic reagents. Chem. Soc. Rev. 2022, 51, 2171–2223. 10.1039/D1CS00556A. [DOI] [PubMed] [Google Scholar]; h Capaldo L.; Ravelli D.; Fagnoni M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. 10.1021/acs.chemrev.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonhebel D. C. The chemistry of cyclopropylmethyl and related radicals. Chem. Soc. Rev. 1993, 22, 347–359. 10.1039/cs9932200347. [DOI] [Google Scholar]
- a Huang X.; Groves J. T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. 10.1021/acs.chemrev.7b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vicens L.; Olivo G.; Costas M. Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. 10.1021/acscatal.0c02073. [DOI] [Google Scholar]
- a Curci R.; D’Accolti L.; Fusco C. A Novel Approach to the Efficient Oxygenation of Hydrocarbons under Mild Conditions. Superior Oxo Transfer Selectivity Using Dioxiranes. Acc. Chem. Res. 2006, 39, 1–9. 10.1021/ar050163y. [DOI] [PubMed] [Google Scholar]; b D’Accolti L.; Annese C.; Fusco C. Continued Progress towards Efficient Functionalization of Natural and Non-natural Targets under Mild Conditions: Oxygenation by C–H Bond Activation with Dioxirane. Chem.—Eur. J. 2019, 25, 12003–12017. 10.1002/chem.201901687. [DOI] [PubMed] [Google Scholar]
- a Adams A. M.; Du Bois J. Organocatalytic C–H hydroxylation with Oxone enabled by an aqueous fluoroalcohol solvent system. Chem. Sci. 2014, 5, 656–659. 10.1039/C3SC52649F. [DOI] [Google Scholar]; b Rotella M. E.; Dyer R. M. B.; Hilinski M. K.; Gutierrez O. Mechanism of Iminium Salt-Catalyzed C(sp3)–H Amination: Factors Controlling Hydride Transfer versus H-Atom Abstraction. ACS Catal. 2020, 10, 897–906. 10.1021/acscatal.9b03588. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hahn P. L.; Lowe J. M.; Xu Y.; Burns K. L.; Hilinski M. K. Amine Organocatalysis of Remote, Chemoselective C(sp3)–H Hydroxylation. ACS Catal. 2022, 12, 4302–4309. 10.1021/acscatal.2c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Proksch E.; de Meijere A. Oxidation of Cyclopropyl Hydrocarbons with Ozone. Angew. Chem., Int. Ed. Engl. 1976, 15, 761–762. 10.1002/anie.197607611. [DOI] [Google Scholar]; b Banwell M. G.; Haddad N.; Huglin J. A.; MacKay M. F.; Reum M. E.; Ryan J. H.; Turner K. A. The Chromium Trioxide-3,5-Dimethylpyrazole Complex: a Mild and Selective Reagent for the Oxidation of Cyclopropyl Hydrocarbons. J. Chem. Soc. Chem. Commun. 1993, 954–957. 10.1039/C39930000954. [DOI] [Google Scholar]; c Coudret J. L.; Zollner S.; Ravoo B. J.; Malara L.; Hanisch C.; Dorre K.; de Meijere A.; Waegell B. Role of Cyclopropanes as Activating Groups during Oxidation Reactions with RuO 4 Generated in situ. Tetrahedron Lett. 1996, 37, 2425–2428. 10.1016/0040-4039(96)00325-5. [DOI] [Google Scholar]; d Dehmlow E. V.; Heiligenstädt N. Dimethyldioxirane Oxidations of Some Cyclopropanes. Tetrahedron Lett. 1996, 37, 5363–5364. 10.1016/0040-4039(96)01107-0. [DOI] [Google Scholar]; e D’Accolti L.; Dinoi A.; Fusco C.; Russo A.; Curci R. Oxyfunctionalization of Non-Natural Targets by Dioxiranes. 5. Selective Oxidation of Hydrocarbons Bearing Cyclopropyl Moieties. J. Org. Chem. 2003, 68, 7806–7810. 10.1021/jo034768o. [DOI] [PubMed] [Google Scholar]; f Chen M. S.; White M. C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]; g Sedenkova K. N.; Andriasov K. S.; Stepanova S. A.; Gloriozov I. P.; Grishin Y. K.; Kuznetsova T. S.; Averina E. B. Direct Oxidation of Cyclopropanated Cyclooctanes as a Synthetic Approach to Polycyclic Cyclopropyl Ketones. Eur. J. Org. Chem. 2018, 2018, 879–884. 10.1002/ejoc.201701671. [DOI] [Google Scholar]
- Brinker U. H.; Lin G.; Xu L.; Smith W. B.; Mieusset J.-L. Dihalocarbene Insertion Reactions into C-H Bonds of Compounds Containing Small Rings: Mechanisms and Regio- and Stereoselectivities. J. Org. Chem. 2007, 72, 8434–8451. 10.1021/jo7013356. [DOI] [PubMed] [Google Scholar]
- Kawamura S.; Chu H.; Felding J.; Baran P. S. Nineteen-step total synthesis of (+)-phorbol. Nature 2016, 532, 90–93. 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu W.; Groves J. T. Manganese Porphyrins Catalyze Selective C–H Bond Halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]; b Aguila M. J. B.; Badiei Y. M.; Warren T. H. Mechanistic Insights into C–H Amination via Dicopper Nitrenes. J. Am. Chem. Soc. 2013, 135, 9399–9406. 10.1021/ja400879m. [DOI] [PubMed] [Google Scholar]; c Pitts C. R.; Bloom S.; Woltornist R.; Auvenshine D. J.; Ryzhkov L. R.; Siegler M. A.; Lectka T. Direct, Catalytic Monofluorination of sp3 C–H Bonds: A Radical-Based Mechanism with Ionic Selectivity. J. Am. Chem. Soc. 2014, 136, 9780–9791. 10.1021/ja505136j. [DOI] [PubMed] [Google Scholar]; d Guo S.; Zhang X.; Tang P. Silver-Mediated Oxidative Aliphatic C-H Trifluoromethylthiolation. Angew. Chem. Int. Ed. 2015, 54, 4065–4069. 10.1002/anie.201411807. [DOI] [PubMed] [Google Scholar]; e Huang X.; Bergsten T. M.; Groves J. T. Manganese-Catalyzed Late-Stage Aliphatic C–H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]; f Liu W.; Cheng M.-J.; Nielsen R. J.; Goddard W. A. III; Groves J. T. Probing the C–O Bond-Formation Step in Metalloporphyrin-Catalyzed C–H Oxygenation Reactions. ACS Catal. 2017, 7, 4182–4188. 10.1021/acscatal.7b00655. [DOI] [Google Scholar]; g Kariofillis S. K.; Jiang S.; Żurański A. M.; Gandhi S. S.; Alvarado J. I. M.; Doyle A. G. Using Data Science to Guide Aryl Bromide Substrate Scope Analysis in a Ni/Photoredox-Catalyzed Cross-Coupling with Acetals as Alcohol-Derived Radical Sources. J. Am. Chem. Soc. 2022, 144, 1045–1055. 10.1021/jacs.1c12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Auclair K.; Hu Z.; Little D. M.; Ortiz de Montellano P. R.; Groves J. T. Revisiting the Mechanism of P450 Enzymes with the Radical Clocks Norcarane and Spiro[2,5]octane. J. Am. Chem. Soc. 2002, 124, 6020–6027. 10.1021/ja025608h. [DOI] [PubMed] [Google Scholar]; b Baik M.-H.; Newcomb M.; Friesner R. A.; Lippard S. J. Mechanistic Studies on the Hydroxylation of Methane by Methane Monooxygenase. Chem. Rev. 2003, 103, 2385–2419. 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- Simakov P. A.; Choi S.-Y.; Newcomb M. Dimethyldioxirane hydroxylation of a hypersensitive radical probe: Supporting evidence for an oxene insertion pathway. Tetrahedron Lett. 1998, 39, 8187–8190. 10.1016/S0040-4039(98)01871-1. [DOI] [Google Scholar]
- Galeotti M.; Vicens L.; Salamone M.; Costas M.; Bietti M. Resolving Oxygenation Pathways in Manganese-Catalyzed C(sp3)–H Functionalization via Radical and Cationic Intermediates. J. Am. Chem. Soc. 2022, 144, 7391–7401. 10.1021/jacs.2c01466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zou L.; Paton R. S.; Eschenmoser A.; Newhouse T.; Baran P. S.; Houk K. N. Enhanced Reactivity in Dioxirane C–H Oxidations via Strain Release: A Computational and Experimental Study. J. Org. Chem. 2013, 78, 4037–4048. 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang Z.; Yu P.; Houk K. N. Molecular Dynamics of Dimethyldioxirane C–H Oxidation. J. Am. Chem. Soc. 2016, 138, 4237–4242. 10.1021/jacs.6b01028. [DOI] [PubMed] [Google Scholar]
- Martin T.; Galeotti M.; Salamone M.; Liu F.; Yu Y.; Duan M.; Houk K. N.; Bietti M. Deciphering Reactivity and Selectivity Patterns in Aliphatic C–H Bond Oxygenation of Cyclopentane and Cyclohexane Derivatives. J. Org. Chem. 2021, 86, 9925–9937. 10.1021/acs.joc.1c00902. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobfayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- a Grimme S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.—Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]; b Luchini G.; Alegre-Requena J. V.; Guan Y.; Funes-Ardoiz I.; Paton R. S. GoodVibes. GoodVibes 3.0.1 2019, 10.5281/zenodo.595246. [DOI] [Google Scholar]
- Legault C. Y.CYLview, 1.0b; Université de Sherbrooke, 2009; http://www.cylview.org (accessed on July 01 2023).
- a Dantignana V.; Milan M.; Cussó O.; Company A.; Bietti M.; Costas M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. 10.1021/acscentsci.7b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bietti M. Activation and Deactivation Strategies Promoted by Medium Effects for Selective Aliphatic C–H Bond Functionalization. Angew. Chem. Int. Ed. 2018, 57, 16618–16637. 10.1002/anie.201804929. [DOI] [PubMed] [Google Scholar]; c Borrell M.; Gil-Caballero S.; Bietti M.; Costas M. Site-Selective and Product Chemoselective Aliphatic C–H Bond Hydroxylation of Polyhydroxylated Substrates. ACS Catal. 2020, 10, 4702–4709. 10.1021/acscatal.9b05423. [DOI] [Google Scholar]
- Recommended BDE C–H value for cyclopropane: 106.3 kcal mol–1. See: Luo Y.-R.Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007, Chapter 3, page 31. [Google Scholar]
- In oxidations promoted by dioxiranes, epoxidations have been shown to be strongly favored over C(sp3)–H hydroxylations. See for example:; a Cicala G.; Curci R.; Fiorentino M.; Laricchiuta O. Stereo- and Regioselectivities in the Epoxidation of Some Allylic Alcohols by the Dioxirane Intermediate Generated in the Reaction of Potassium Caroate with Acetone. J. Org. Chem. 1982, 47, 2670–2673. 10.1021/jo00134a033. [DOI] [Google Scholar]; b Legros J.; Crousse B.; Bourdon J.; Bonnet-Delpon D.; Bégué J.-P. An efficient and robust fluoroketone catalyst epoxidation. Tetrahedron Lett. 2001, 42, 4463–4466. 10.1016/S0040-4039(01)00751-1. [DOI] [Google Scholar]
- a Soler J.; Gergel S.; Klaus C.; Hammer S. C.; Garcia-Borràs M. Enzymatic Control over Reactive Intermediates Enables Direct Oxidation of Alkenes to Carbonyls by a P450 Iron-Oxo Species. J. Am. Chem. Soc. 2022, 144, 15954–15968. 10.1021/jacs.2c02567. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gergel S.; Soler J.; Klein A.; Schülke K.; Hauer B.; Garcia-Borràs M.; Hammer S. C. Engineered cytochrome P450 for direct arylalkene-to-ketone oxidation via highly reactive carbocation intermediates. Nature Catal. 2023, 6, 606–617. 10.1038/s41929-023-00979-4. [DOI] [Google Scholar]
- As a matter of comparison, when the methyl-norcaranyl-2-yl cation was generated under superacidic conditions at −140 °C from cis-1-methylbicyclo[4.1.0]heptan-2-ol, rearrangement to the 2-methyl-norcaran-2-yl cation was observed. See:; Olah G. A.; Prakash G. K. S.; Rawdah T. N. Degenerate Cyclopropylcarbinyl Cation Rearrangement in 2-Bicyclo[n.1.0]alkyl Cations. J. Org. Chem. 1980, 45, 965–969. 10.1021/jo01294a010. [DOI] [Google Scholar]
- a Colomer I.; Batchelor-McAuley C.; Odell B.; Donohoe T. J.; Compton R. G. Hydrogen Bonding to Hexafluoroisopropanol Controls the Oxidative Strength of Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 8855–8861. 10.1021/jacs.6b04057. [DOI] [PubMed] [Google Scholar]; b Colomer I.; Chamberlain A. E. R.; Haughey M. B.; Donohoe T. J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. 10.1038/s41570-017-0088. [DOI] [Google Scholar]
- a Olah G. A.; Fung A. P.; Rawdah T. N.; Prakash G. K. S. The Spiro[2.5]oct-4-yl Cation, a Long-Lived Secondary Cyclohexyl Cation. J. Am. Chem. Soc. 1981, 103, 4646–4647. 10.1021/ja00405a090. [DOI] [Google Scholar]; b Olah G. A.; Reddy V. P.; Prakash G. K. S. Long-Lived Cyclopropylcarbinyl Cations. Chem. Rev. 1992, 92, 69–95. 10.1021/cr00009a003. [DOI] [Google Scholar]
- In the [2 + 2] cycloaddition of styrenes initiated by ET to phenyliodine(III)diacetate (PIDA), voltammetric studies showed that a change in solvent from MeCN to HFIP determined a 0.85 V increase in the oxidizing ability of PIDA, accompanied by a 0.18 V decrease in the oxidative peak potential of a reference substrate such as trans-anethole.30a
- Grabovskiy S. A.; Antipin A. V.; Ivanova E. V.; Dokichev V. A.; Tomilov Y. V.; Kabal’nova N. N. Oxidation of some cage hydrocarbons by dioxiranes. Nature of the transition structure for the reaction of C–H bonds with dimethyldioxirane: a comparison of B3PW91 density functional theory with experiment. Org. Biomol. Chem. 2007, 5, 2302–2310. 10.1039/B707753J. [DOI] [PubMed] [Google Scholar]
- Within this framework, it is worth mentioning that in a detailed mechanistic study on the oxygenation of 2-substituted adamantanes by TFDO, a remarkable electron deficiency at the reacting carbon atom in the transition state has been proposed.34 However, in contrast to S3, S7, and S8, the structure of the chosen substrates did not allow a probe into the possible involvement of cationic intermediates in these reactions.
- Gonzalez-Nunez M. E.; Royo J.; Mello R.; Baguena M.; Ferrer J. M.; Ramirez de Arellano C.; Asensio G.; Prakash G. K. S. Oxygenation of Alkane C–H Bonds with Methyl(trifluoromethyl)dioxirane: Effect of the Substituents and the Solvent on the Reaction Rate. J. Org. Chem. 2005, 70, 7919–7924. 10.1021/jo0509511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.