

Visual Abstract

Keywords: complement, glomerulopathy, immune complexes, MPGN (membranoproliferative GN)
Abstract
Background
C3 glomerulopathy and idiopathic immunoglobulin-mediated membranoproliferative GN (Ig-MPGN) are rare complement-mediated kidney diseases. Inherited forms of C3 glomerulopathy/Ig-MPGN are rarely described.
Methods
Three hundred ninety-eight patients with C3 glomerulopathy (n=296) or Ig-MPGN (n=102) from a national registry were screened for three complement genes: factor H (CFH), factor I (CFI), and C3. Patients with rare variant (minor allele frequency <0.1%) were included. Epidemiologic, clinical, and immunologic data at diagnosis and kidney outcomes of patients were retrospectively collected.
Results
Fifty-three different rare variants, including 30 (57%), 13 (24%), and ten (19%) in CFH, CFI, and C3 variants, were identified in 66/398 (17%) patients. Thirty-eight (72%) variants were classified as pathogenic, including 20/30 (66%) and 11/13 (84%) variants in CFH and CFI, respectively, impairing synthesis of factor H or factor I regulators. Fifteen of 53 (27%) variants were of unknown significance. At diagnosis, 69% of patients were adult (median age of 31 years). With the exception of biologic stigma of thrombotic microangiopathy, which was more frequent in patients with CFI variants (5/14 [36%] versus 1/37 [3%] and 0% in the CFH group and C3 group, respectively, P < 0.001), the clinical and histologic features were similar among the three variants groups. The kidney outcome was poor regardless of the age at onset and treatment received. Sixty-five percent (43/66) of patients with rare variant reach kidney failure after a median delay of 41 (19–104) months, compared with 28% (55/195) after a median delay of 34 (12–143) months in the nonvariant group. Among 36 patients who received a kidney transplant, 2-year recurrence was frequent, occurring in 39% (12/31), without difference between variant groups, and led to graft failure in three cases.
Conclusions
In our cohort, 17% of C3 glomerulopathy/Ig-MPGN cases were associated with rare variants in the CFH, CFI, or C3 genes. In most cases, a quantitative deficiency in factor H or factor I was identified. The presence of a rare variant was associated with poor kidney survival.
Podcast
This article contains a podcast at https://dts.podtrac.com/redirect.mp3/www.asn-online.org/media/podcast/CJASN/2023_11_08_CJN0000000000000252.mp3
Introduction
Dysfunction of the complement system, specifically the alternative pathway, has been implicated in a variety of diseases with very different clinical outcomes. Comprehensive genetic testing has demonstrated that patients with atypical hemolytic uremic syndrome (aHUS),1–3 immunoglobulin-mediated membranoproliferative GN (Ig-MPGN), C3 glomerulopathy,4–6 and age-related macular degeneration7–9 carry protein-altering variants in complement genes. Most variants are found in the complement genes CFH and CFI, which encode for two proteins implicated in the control of both fluid phase and on cell surfaces, and in the C3 gene coding for one of the proteins of the alternative pathway C3bBb convertase.
Complement genetic studies are crucial for unraveling the pathogenesis of complement in aHUS, in which rare variants in complement genes are described in 26%–62% of cases. In aHUS, genetics is a valuable tool in clinical practice to provide evidence for a causal link between complement dysregulation and the disease, to assess disease severity, and, most importantly, to predict the risk of recurrence after kidney transplantation10,11 and of disease relapse after anti-C5 treatment discontinuation.12
By contrast, in patients with C3 glomerulopathy, complement dysregulation is frequently driven by autoantibodies against a variety of complement proteins13 and complexes, while patients carrying rare variants in complement genes account for approximately 10%–20% of cases.5,14,15 When any infection, autoimmunity, or hematologic disorder is excluded in Ig-MPGN, studies have found low C3, C3 nephritic factor (C3Nef), and rare complement variants in the same proportion as found in C3 glomerulopathy,4,5,16 pleading for a role of alternative pathway in the pathogenesis.
With the exception of CFHR5 nephropathy, a unique subtype of C3 glomerulopathy, endemic to Cyprus,17 few data have been described regarding specific clinical features and kidney outcomes of C3 glomerulopathy/Ig-MPGN patients with complement gene variants.4,5,18,19 In a recent study, it has been found that the presence of variants in complement genes was an independent factor of kidney outcomes in C3 glomerulopathy.20 However, kidney survival remains heterogenous among patients with variants.21
Given the upcoming availability of new complement blockers, specifically targeting the alternative pathway, it is of paramount importance to identify the patients who will most likely benefit from these emerging therapies. Patients with genetically determined alternative pathway dysregulation seem to be a very suitable group for complement-inhibiting therapies. However, genetic testing is expensive and not broadly available. It is desirable to preselect cases for genotyping on the basis of phenotype.
The present nationwide study provides an unprecedented clinical and immunologic characterization of C3 glomerulopathy/Ig-MPGN patients in whom rare variants in CFH, CFI, and C3 genes have been identified. This will lay the groundwork for highly individualized management, targeted genetic testing, and tailored therapies.
Methods
Patients
In this study, we included patients referred to Laboratory of Immunology of European Hospital Georges Pompidou between 2002 and 2018 who met the following criteria: (1) C3 glomerulopathy or Ig-MPGN proven on kidney biopsy or on kidney graft after kidney failure of undetermined cause and (2) identification of rare variant with minor allele frequency below 0.1% in CFH, CFI, or C3 genes. All patients with positive hepatitis B or C serology, antinuclear antigen autoantibodies, anti–double-stranded DNA antibodies, hemopathy, monoclonal gammopathy, or cryoglobulinemia were excluded from the study. A national ethics committee approved the study (Nb 1921223), and all patients or patients' referent gave written informed consent for genetic analysis.
The diagnosis of C3 glomerulopathy was based on immunofluorescence following the international consensus recommendations22 with bright diffuse predominant C3 glomerular staining (≥2+), of at least two orders of magnitude greater than any other immune reactant (i.e., Ig). Patients with the MPGN pattern and C3 deposit staining below two orders of magnitude compared with Ig were defined as having Ig-MPGN.
Clinical data of patients were retrospectively collected from individual medical records, including period on native kidney and after kidney transplantation. Definitions of usual nephrologic parameters and biologic stigmata of thrombotic microangiopathy (TMA) are given in Supplemental Methods 1.
Immunologic characteristics, clinical features at diagnosis, and kidney survival of patients with CFH, CFI, or C3 rare variants were compared with the available data of 211 patients with C3 glomerulopathy or Ig-MPGN diagnosed and explored on native kidney, genetically explored at Laboratory of Immunology of European Hospital Georges Pompidou on the same period (2002–2018) and without rare variant in CFH, CFI, and C3 complement genes.
Assays for Complement Proteins and Complement Gene Analysis
Complement protein assays and C3b crystal structure construction are presented in Supplemental Methods 2.
Complement genetic analysis was performed as part of the usual workup in patients diagnosed with C3 glomerulopathy or idiopathic Ig-MPGN. All coding sequences of CFH, CFI, and C3 genes were analyzed by direct sequencing analysis or by next-generation sequencing. In our study, a variant was defined as rare if its minor allele frequency was below 0.1% in the European population using the GnomAD database (gnomAD—Non-Finnish European, https://gnomad.broadinstitute.org). Rare variant was defined as pathogenic if the genetic change affects the protein function (well-established in vitro functional studies supportive of a damaging effect on the gene product) and/or if the genetic change was found in a disease-related functional domain and/or affects the protein expression (well‐demonstrated lack of in vitro synthesis, large deletion, or quantitative deficiency in the patient's plasma). The other variants were classified as variants of uncertain significance (VUS). For patients (n=2) with variants identified in multiple genes, we decided to include them in the variants group considered as pathogenic or the most probably pathogenic.
Statistical Analyses
Data are expressed as median with interquartile range (IQR) for continuous variables and percentage for categorical variables. Statistical analyses were performed using Mann–Whitney and Kruskal–Wallis tests, as appropriate, for comparison of continuous variables. The Fisher exact test was used for comparison of categorical variables. Kidney survival was determined using log-rank test and censored at 240 months for native kidney. P values below 0.05 were considered significant. For each variable, the number of available data is presented in tables. The median follow-up of patients is presented in Table 1. The results were analyzed using GraphPad Prism (v8).
Table 1.
Characteristics of patients referred to the Laboratory of Immunology of European Hospital Georges Pompidou between 2002 and 2018 with C3 glomerulopathy or immunoglobulin-mediated membranoproliferative GN proven on kidney biopsy
| Characteristics of Patients | No. of Patients with Available Data | No Varianta N=211 |
CFH, CFI, or C3 Variants N=66 |
|---|---|---|---|
| Clinical data at first clinical evaluation | |||
| Male sex | 277 | 112/211 (53) | 37/66 (56) |
| Age | 275 | 21 (12–38) | 31 (15–47) |
| Children | 275 | 86/210 (41) | 20/65 (31) |
| Proteinuria, g/d | 186 | 3.9 (2.0–7.7) | 2.9 (1.5–4.3) |
| Nephrotic syndrome | 233 | 95/175 (54) | 25/58 (43) |
| eGFR, ml/min per 1.73 m2 | 241 | 80 (37–100) | 49 (28–86) |
| eGFR >60 ml/min per 1.73 m2 | 241 | 107/179 (60) | 27/62 (44) |
| eGFR 15–60 ml/min per 1.73 m2 | 241 | 55/179 (31) | 26/62 (42) |
| eGFR ≤15 ml/min per 1.73 m2 | 241 | 17/179 (9) | 9/62 (15) |
| Histologic data | |||
| C3 glomerulopathy | 277 | 179/211 (84) | 55/66 (83) |
| Ig-MPGN | 277 | 32/211 (15) | 11/66 (17) |
| Treatment | |||
| No specific treatment | 235 | 47/173 (27) | 33/62 (53) |
| Plasma exchange | 235 | 1/173 (1) | 3/62 (5) |
| Immunosuppressive treatment | 235 | 125/173 (73) | 26/62 (42) |
| Corticosteroid alone | 235 | 41/173 (24) | 16/62 (26) |
| Corticosteroid associated with other IS agent | 235 | 84/173 (49) | 10/62 (16) |
| Follow-up on native kidney | |||
| Follow-up | 222 | 65 (30–134) | 100 (60–180) |
| Kidney function at the last follow-up | |||
| eGFR >60 ml/min per 1.73 m2 | 222 | 72/156 (46) | 18/66 (27) |
| eGFR <60 ml/min per 1.73 m2 | 222 | 29/156 (19) | 5/66 (8) |
| Kidney failure (dialysis or transplantation) | 222 | 55/156 (35) | 43/66 (65) |
Qualitative variables are described as frequencies (percentages) and quantitative variables as median (interquartile range). Time is expressed in months. CFH, complement factor H; CFI, complement factor I; Ig-MPGN, immunoglobulin-mediated membranoproliferative GN; IS, immune suppressive.
Immunologic characteristics, clinical features at diagnosis, and kidney survival of patients with CFH, CFI, or C3 rare variants were compared with the available data of 211 patients with C3 glomerulopathy or immunoglobulin-mediated membranoproliferative GN diagnosed and explored on native kidney, genetically explored at Laboratory of Immunology of European Hospital Georges Pompidou on the same period (2002–2018) and without rare variant in CFH, CFI, and C3 complement genes.
Results
Complement Variants
Between 2002 and 2018, genetic screening of CFH, CFI, and C3 genes was performed in 398 patients with C3 glomerulopathy (n=296) or idiopathic Ig-MPGN (n=102) (Table 1). Rare variants in CFH, CFI, or C3 genes were identified in 66/398 (17%) patients, including 55/296 (19%) C3 glomerulopathy and 11/102 (11%) Ig-MPGN (Figure 1). A total of 53 rare variants in CFH (n=30), CFI (n=13), and C3 (n=10) were identified (Supplemental Tables 1–3).
Figure 1.

C3 glomerulopathy, Ig-MPGN, and CFH, CFI, or C3 variants: patients repartition in national French registry. *Rare variants were defined by minor allelic frequency <0.1%. **Three patients carried two variants: one carried C3 VUS (p.Ala257Thr) and homozygous pathogenic CFH variant (p.Cys431Ser) and was included in the group of patients with CFH variant. Another one carried a pathogenic variant in C3 (p.Asn1179Thr) and a CFI VUS (p.Ile306Val) and was included in the group of patients with C3 variant. The last one carried two pathogenic rare variants in CFI (p.Pro50Ala and p.Ala210Ser). C3G, C3 glomerulopathy; CFH, complement factor H; CFI, complement factor I; Ig-MPGN, immunoglobulin-mediated membranoproliferative GN; VUS, variant of uncertain significance.
Twenty of the 30 (66%) rare variants in CFH were absent in gnomAD (Supplemental Table 1). We used the guidelines of the American College of Medical Genetics and Genomics to classify variants.23 Missence variants of CFH implicating a cysteine residue (n=8) or CFH variants resulting in low factor H level in plasma (n=4) were classified as pathogenic as well as nonsense variant (n=4) or variant with in vitro demonstration of damaging effect (n=2)24,25 and a nonsense variant (n=4), a small deletion (n=5), or a splice site variant (n=1) were classified as pathogenic with strong or very strong evidence of pathogenicity, respectively. In the absence of pathogenic criteria, six variants were classified of undetermined significance. A total of 24 of the 30 CFH rare variants (80%) were classified as pathogenic.
Thirteen rare variants in CFI gene were identified in 17 patients, including 11 (84%) pathogenic variants with decreased factor I production, concordant with in vitro reduced recombinant expression26 and two VUS (15%) (Supplemental Table 2). Ten variants were already reported in gnomAD (10/13 [77%]).
Ten rare variants in C3 gene were found in 13 patients, including four patients from two familial forms of glomerulopathy (Supplemental Table 3). Three variants were classified with strong (n=2 with in vitro study27,28) or supporting evidence of pathogenicity (located on the surface of C3 and close to the binding site of factor H). Seven variants, located on the surface of C3b without interaction with any complement regulatory proteins, were classified as VUS in the absence of functional data (7/10 [70%]) (Supplemental Figure 1). Three patients carried two variants: one carried a C3 VUS (p.Ala257Thr) and a homozygous pathogenic CFH variant (p.Cys431Ser) and was included in the group of patients with CFH variants. Another one carried a pathogenic variant in C3 (p.Asn1179Thr) and a CFI VUS (p.Ile306Val) and was included in the group of patients with C3 variants. The last one carried two pathogenic rare variants in CFI (p.Pro50Ala and p.Ala210Ser).
Clinical Features, Laboratory Evaluation, and Pathologic Findings of Patients at the Time of Disease Onset
We identified 38, 16, and 12 patients with a rare variant in CFH, CFI, and C3 (Supplemental Table 4), respectively. Except three, all 66 patients carried one rare variant.
Except for lower proteinuria (median [IQR] 2.9 [1.5–4.3] g/d in variants associated with C3 glomerulopathy/Ig-MPGN versus 3.9 [2.0–7.7] g/d in the nonvariant group, P = 0.009) and worse kidney function (median [IQR] eGFR 49 [28–86] ml/min per 1.73 m2 in the variant group versus 80 [37–100] ml/min per 1.73 m2 in nonvariant group, P = 0.04), clinical features at the first clinical evaluation were not significantly different between patients carrying rare variants or not (Table 1). Thirty‐seven of 66 (56%) patients were of male sex (Table 2). At diagnosis, 45 (69%) patients were adults. Rare variant prevalence was 14%, 17%, and 27% in children, adults younger than 50 years, and older than 50 years, respectively (P = 0.14) (Supplemental Figure 2). The frequency of the pathogenic variants among rare variants identified was significantly higher in patients older than 50 years at diagnosis (13/13 [100%]) compared with adults younger than 50 years (23/32 [72%]) and children (11/20 [55%]) (P = 0.02) (Figure 2, Supplemental Tables 5–8). The median (IQR) eGFR was 49 (28–86) ml/min per 1.73 m2, without difference according to variant groups. The median (IQR) proteinuria was 2.9 (1.5–4.3) g/d. Biologic stigmata of TMA were more frequent in patients with CFI variants (5/14 [36%] versus 1/37 [3%] and 0% in the CFH group and C3 group, respectively, P < 0.001) (Table 2).
Table 2.
Specific characteristics of C3 glomerulopathy or immunoglobulin-mediated membranoproliferative GN patients carrying rare variants in CFH, CFI, or C3 genes explored at the Laboratory of Immunology of European Hospital Georges Pompidou between 2002 and 2018
| Characteristics of Patients | No. of Patients with Available Data |
CFH Variants N=38 |
CFI Variants N=16a |
C3 Variants N=12b |
|---|---|---|---|---|
| Clinical data at first clinical evaluation | ||||
| Male sex | 66 | 22/38 (58) | 7/16 (44) | 8/12 (67) |
| Age | 65 | 32 (15–53) | 32 (18–46) | 22 (8–41) |
| Children | 65 | 11/37 (30) | 4/16 (25) | 5/12 (42) |
| Proteinuria, g/d | 58 | 2.5 (1.1–4) | 3.0 (1.3–5.0) | 3.0 (1.9–5.4) |
| Nephrotic syndrome | 58 | 11/34 (32) | 6/12 (50) | 8/12 (67) |
| eGFR, ml/min per 1.73 m2 | 62 | 49 (28–85) | 40 (12–78) | 57 (36–98) |
| eGFR >60 ml/min per 1.73 m2 | 62 | 15/35 (43) | 6/15 (40) | 6/12 (50) |
| eGFR 15–60 ml/min per 1.73 m2 | 62 | 16/35 (46) | 5/15 (33) | 5/12 (42) |
| eGFR ≤15 ml/min per 1.73 m2 | 62 | 4/35 (11) | 4/15 (27) | 1/12 (8) |
| Biologic stigmata of TMA | 63 | 1/37 (3) | 5/14 (36) | 0/12 (0) |
| Infectious trigger | 49 | 11/29 (38) | 2/10 (20) | 2/10 (20) |
| Histologic data | ||||
| C3 glomerulopathy | 66 | 32/38 (84) | 12/16 (75) | 11/12 (92) |
| Ig-MPGN | 66 | 6/38 (16) | 4/16 (25) | 1/12 (8) |
| Treatment | ||||
| No specific treatment | 62 | 17/34 (50) | 8/16 (50) | 8/12 (67) |
| Plasma exchange | 62 | 3/34 (9) | 0 | 0 |
| Immunosuppressive treatment | 62 | 14/34 (41) | 8/16 (50) | 4/12 (33) |
| Corticosteroid alone | 62 | 8/34 (24) | 5/16 (31) | 3/12 (25) |
| Corticosteroid associated with other IS agent | 62 | 6/34 (18) | 3/16 (19) | 1/12 (8) |
| Follow-up on native kidney | ||||
| Follow-up | 105 (65–184) | 95 (36–188) | 106 (54–178) | |
| Kidney function at the last follow-up | ||||
| eGFR >60 ml/min per 1.73 m2 | 66 | 12/38 (32) | 3/16 (19) | 3/12 (25) |
| eGFR <60 ml/min per 1.73 m2 | 66 | 5/38 (13) | 0 | 0 |
| Kidney failure (dialysis or transplantation) | 66 | 21/38 (55) | 13/16 (81) | 9 (75) |
| Duration of evolution until kidney failure | 66 | 64 (20–108) | 43 (1–101) | 23 (13–84) |
Qualitative variables are described as frequencies (percentages) and quantitative variables as median (interquartile range). P value was calculated by comparing the three different groups: CFH, CFI, and C3 variants. Time is expressed in months. CFH, complement factor H; CFI, complement factor I; TMA, thrombotic microangiopathy; Ig-MPGN, immunoglobulin-mediated membranoproliferative GN; IS, immune suppressive.
Three patients carried two variants: one carried C3 variants of undetermined significance (p.Ala257Thr) and an homozygous pathogenic CFH variant (p.Cys431Ser) and was included in the group of patients with CFH variant.
Another one carried a pathogenic variant in C3 (p.Asn1179Thr) and a CFI variant of uncertain significance (p.Ile306Val) and was included in the group of patients with C3 variant. The last one carried two pathogenic rare variants in CFI (p.Pro50Ala and p.Ala210Ser).
Figure 2.
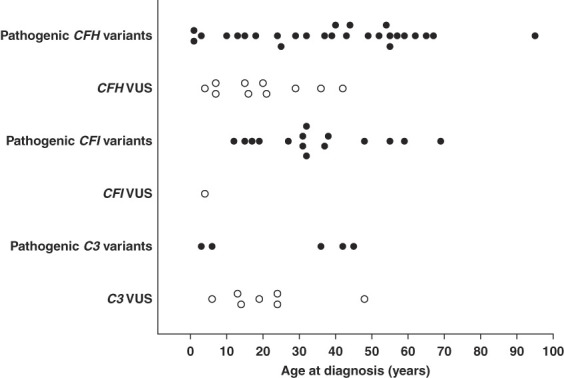
Distribution of rare variants in C3, CFI, or CFH genes according to age at diagnosis. Pathogenic variants are represented in black and VUS in white.
Fifty-five (83%) and 11 (17%) of 66 patients had criteria of C3 glomerulopathy and Ig-MPGN, respectively. Histologic features were not different between variant groups, except for greater mesangial hypercellularity in the CFH group (35/37 [97%] versus 10/16 [63%] and 7/11 [64%] in the CFI and C3 groups, respectively, P = 0.006) and great glomerulosclerosis in the C3 group (median [IQR] of % sclerotic glomeruli=18 [4–58] versus 12 [0–35] and 0 [0–14] in the CFH and CFI groups, respectively, P = 0.03) (Supplemental Table 9). Except from a younger age and lower rate of eGFR >60 ml/min per 1.73 m2 at diagnosis and a nonexclusive MPGN pattern in C3 glomerulopathy, clinical and histologic presentations were not different between C3 glomerulopathy and Ig-MPGN associated with rare variants (Supplemental Table 10). Rare variants associated with Ig-MPGN were more frequently treated with steroids (7/11 [64%] versus 9/51 [18%] in C3 glomerulopathy, P = 0.004), and the rate of kidney failure was not different between the two histologic groups.
Rare Variant Distribution in C3 Glomerulopathy/Ig-MPGN and Immunologic Characteristics of Patients at Disease Onset
A total of 48 patients carried a pathogenic variant (73%) with a lower frequency in the C3 variants (5/12 [42%]) than in the CFH (28/38 [74%]) and CFI (15/16 [94%]) (P = 0.009) (Supplemental Table 11). In comparison with controls from the 1000 genome project,29 the C3 glomerulopathy patient group demonstrated a higher frequency of pathogenic variants for the three tested genes, whereas the Ig-MPGN group had a higher frequency of CFH and CFI pathogenic variants yet not of C3 variants (Supplemental Table 12).
The frequency of low factor I and factor H was higher in patients carrying rare variants in CFI and CFH, respectively, compared with patients without rare variant (low FI found in 11/16 [69%] patients carrying rare variants in CFI versus 1/181 [1%] in patients without rare variants and low FH found in 18/38 [47%] patients carrying rare variants in CFH versus 2/184 [1%] in patients without rare variant, both P < 0.001), whereas C3, C4, and sC5b-9 levels were not different between patients with or without rare variants (Figure 3, Table 3). Eighteen (18/38 [53%]), four (4/16 [25%]), and nine (9/12 [75%]) patients with CFH, CFI, and C3 variants had a low C3 level, respectively (P = 0.04) (Supplemental Table 11). The screening of C3NeF was positive in 9/66 (13%) patients with rare variants, whereas none of the patients were positive for antifactor H antibody, compared with 49% (98/201) and 14% (24/179), respectively, in patients without rare variant (P < 0.001 and P = 0.002).
Figure 3.

Plasma level of C3, factor H, and factor I according to rare variants in CFH, CFI, or C3 complement genes. (A) Plasma C3, (B) factor H, and (C) factor I levels at diagnosis differed according to the complement variant. Dashed lines represent lower limit of normal value for each complement component protein measured. Mean±SD is represented by dark and gray lines. CFH, complement factor H; CFI, complement factor I; FH, factor H; FI, factor I; P, pathogenic.
Table 3.
Complement assays in C3 glomerulopathy/immunoglobulin-mediated membranoproliferative GN patients carrying rare variants in CFH, CFI, and C3 genes or not
| Characteristics of Patients | No. of Patients with Available Data | No Variant N=211 |
CFH, CFI, or C3 Variants N=66 |
|---|---|---|---|
| Complement activation biomarkers | |||
| C3 level,a mg/L | 274 | 675 (310–954) | 677 (493–927) |
| Low C3 level | 274 | 102/208 (49) | 31/66 (47) |
| C4 level,b mg/L | 266 | 238 (178–296) | 252 (187–309) |
| Low C4 level | 266 | 4/200 (2) | 2/66 (3) |
| Soluble C5b-9,c ng/ml | 219 | 446 (282–798) | 437 (289–758) |
| High sC5b-9 | 219 | 123/174 (72) | 33/45 (73) |
| Regulatory proteins | |||
| Factor H level (% of normal value) | 250 | 110 (90–126) | 101 (62–119) |
| Low factor Hd | 250 | 2/184 (1) | 19/66 (29) |
| Factor I level (% of normal value) | 247 | 108 (97–124) | 107 (90–122) |
| Low factor Id | 247 | 1/181 (1) | 12/66 (18) |
| Associated acquired abnormalities | |||
| Positive C3NeF | 267 | 98/201 (49) | 9/66 (13) |
| Anti-FH Ab | 245 | 24/179 (14) | 0/66 |
Qualitative variables are described as frequencies (percentages) and quantitative variables as median (interquartile range). P value was calculated by comparing the three different groups: CFH, CFI, and C3 variants. CFH, complement factor H; CFI, complement factor I; sC5b-9, soluble C5b-9; C3Nef, C3 nephritic factor; FH, factor H; FI, factor I.
Normal values for C3 plasmatic level: 660–1250 mg/L.
Normal values for C4 plasmatic level: 93–380 mg/L.
Normal values for C4 plasmatic level: <300 ng/ml.
FH and FI measurement are categorized low when <70% of normal value.
Treatment, Kidney Outcomes, and Disease Course after Transplantation
Twenty-nine patients received specific treatment without difference among the three variants groups (Table 2). Six patients were treated using eculizumab, among whom three patients had preserved kidney function at the last follow-up. Three patients with eGFR <15 ml/min at diagnosis progressed to kidney failure despite eculizumab (in 0, 2, and 40 months after diagnosis). The Kaplan–Meier estimate of the 5-year survival without kidney failure was 46%, 51%, and 75% in C3, CFI, and CFH, respectively, compared with 82% in C3 glomerulopathy/Ig-MPGN without rare variant (P < 0.001) (Figure 4A). Poorer kidney prognosis in C3 glomerulopathy/Ig-MPGN patients with rare variants compared with those without rare variant was irrespective of age at diagnosis and treatment received (Supplemental Figures 3, A and B, and 4, A and B). The Kaplan–Meier estimate of the 5-year survival without kidney failure was 89% in children compared with 53% in adults (P = 0.02) (Figure 4B).
Figure 4.

Kidney survival of patients with inherited C3 glomerulopathy/Ig-MPGN. (A) Kaplan–Meier kidney survival curve according to the complement component variant, compared with kidney survival of C3 glomerulopathy and GNMP-Ig patients without rare variant in CFH, CFI, or C3 genes. The median kidney survival was 28, 79, and 108 months, respectively, in C3, CFI, and CFH variants and undefined in C3 glomerulopathy patients without variant. (B) Kaplan–Meier kidney survival curve in rare variant associated with C3 glomerulopathy/Ig-MPGN according to age at diagnosis. The median kidney survival was 116 months in children compared with 79 months in adults. (C) Kaplan–Meier kidney survival curve in rare variant associated with C3 glomerulopathy/Ig-MPGN according to the main histologic features. The median kidney survival was 80 months in C3 glomerulopathy and 94 months in Ig-MPGN. (D) Kaplan–Meier kidney survival curve in rare variant associated with C3 glomerulopathy/Ig-MPGN according to treatment received. The median kidney survival was 79 months in patients who received conservative treatment compared with 88 months in those who received specific treatment.
Kidney survival did neither correlate with main histologic feature nor with the treatment received (P = 0.65, P = 0.58) (Figure 4, C and D).
Seventeen, ten, and nine patients with CFH, CFI, and C3 variants, respectively, underwent kidney transplantation (Supplemental Table 13). Histologic recurrence occurred in 69% of transplanted patients without difference among the three variant groups.
Discussion
Here, we reported epidemiology, exhaustive clinical, histologic, and immunologic features of a large retrospective series of patients with C3 glomerulopathy and Ig-MPGN associated with CFH, CFI, and C3 rare variants. Our results highlight the severity of inherited C3 glomerulopathy/Ig-MPGN, particularly those associated with CFI and C3 variants with poorer kidney survival compared with C3 glomerulopathy/Ig-MPGN without rare variant, and early and high frequency of recurrence after transplantation.
In this study, 17% of the 398 patients with C3 glomerulopathy/Ig-MPGN were found to carry rare variants with minor allele frequency <0.1% in CFH, CFI, and C3 genes. Our results confirm the lower frequency of inherited forms of C3 glomerulopathy/Ig-MPGN compared with aHUS,2,30 correlating with those reported in the Italian cohort of C3 glomerulopathy4 (14%, 135 patients) but not with those reported in the US cohort30 (35%, 37 patients). Recently, a large cohort of patients with the MPGN pattern was screened in the United Kingdom (C3 glomerulopathy and Ig-MPGN) by large-scale whole-genome sequencing, and the frequency of rare variants in complement genes was comparable with controls (6.8% versus 5.9%).31 In this large series, 45% of patients with Ig-MPGN had a low C4 level, suggesting that secondary forms with the activation of the classical pathway may have been included in the study. The CFH genetic abnormalities were reported at 3.7% and 16% in the Italian4 and US30 cohorts, respectively, compared with 9.5% in our cohort. Frequency of C3 rare variants was reported at 8.5% and 13% in the Italian4 and US30 cohorts, respectively, compared with 3% in our cohort. The variation of rare variant frequency between cohorts might likely be due to sampling bias inherent to cohort studies in a rare disease.
A recent study of the functional characterization of CFH variants confirms that both allele frequency and pathogenicity prediction with in silico algorithms should be used circumspectly.24 Therefore, the strongest evidence supporting a causal link between a rare variant and disease occurrence still relies on functional studies. Using the criteria of pathogenicity on the basis of known functional data or precise measurements of plasma factor H/factor I levels, two thirds of rare variants identified in this study were classified as pathogenic. The enrichment of rare and pathogenic variants in a given complement gene in a population of patients is an argument pleading for a role of complement dysregulation in the pathogenesis of the disease. Rare complement gene variants (minor allele frequency <0.1%) are present in up to 4% of healthy individuals.1 We showed that CFH, CFI, and C3 rare pathogenic variants were significantly more frequent in C3 glomerulopathy compared with healthy controls, confirming their contribution to the disease. To the contrary, in Ig-MPGN, only pathogenic variants in CFH and CFI genes but not C3 gene were significantly more frequent as compared with the general population. Interestingly, more than 90% of pathogenic CFI variants identified in the patients have previously been reported in gnomAD, while near 80% of the CFH variants are absent. These results suggest that genetic susceptibility induced by CFI variants may be lower than those of CFH variants. At the time of diagnosis, more than 50% of patients with sporadic variants in CFH and CFI had a normal C3 level, but most of them had low factor H or factor I levels in plasma. This observation suggests that the normal C3 level does not rule out the diagnosis of heterozygous factor H/factor I deficiency. However, in clinical practice, simple measurement of plasma factor H and factor I levels may be a useful tool to predict the presence of variants in CFH and CFI genes. Atypical HUS and C3 glomerulopathy are the two major kidney diseases caused by pathogenic variants in genes that encode effectors or regulators of the complement system. In patients with aHUS, CFH pathogenic variants are found throughout the gene, but most (40%–60%) are found in the C-terminal portion of the gene with normal CFH levels in plasma. By contrast, CFH pathogenic variants identified in C3 glomerulopathy are responsible for factor H quantitative deficiency in two thirds of cases. Few of the CFH variants identified in C3 glomerulopathy/Ig-MPGN patients are already described in aHUS. Therefore, the reduced interaction between factor H and cell surface is remarkable in aHUS but has a limited effect in C3 glomerulopathy. By contrast, a defect in the physiologic inactivation of fluid phase C3b or in glomerular environment by factor H is fundamental to the occurrence of C3 glomerulopathy. In both diseases, the most frequent scenario is a missense CFI variant associated with quantitative factor I deficiency. The phenotype expressed by a specific patient with CFI variant is likely influenced by additional genetic, autoimmune, or environmental factors. The frequency of C3 rare variants in our series was lower to that observed in the Italian cohort.4 However, as in the study by Iatropoulos et al., most of C3 variants identified here were classified as VUS. Indeed, only seven of ten variants of C3 gene identified here are located far from the binding site of negative regulators, highlighting the need for functional characterization to confirm their role in the occurrence of the disease.
Patients with inherited C3 glomerulopathy/Ig-MPGN were mostly male and diagnosed in adulthood, in opposition to aHUS that mainly occurred before 50 years and in females, in case of adult onset. This suggests that, independently of amino acid exchange in complement protein sequence, age-dependent or sex-dependent factors may contribute to determine the specificity of kidney lesions. Most of the rare variants were identified in all age categories, even in patients older than 50 years, highlighting the fact that genetic screening needs to be proposed to all patients. Inherited C3 glomerulopathy patients are mainly associated with severe phenotype, particularly in cases of C3 or CFI variants, with poorer kidney survival than C3 glomerulopathy/Ig-MPGN patients without rare variant. As described by Caravaca-Fontán et al., we report a limited response to immunosuppressive regimen32 with early and high frequency of recurrence after transplantation. Except for the presence of a low factor H or factor I level in case of CFH or CFI variants, respectively, clinical and biologic features of patients at diagnosis do not allow predicting the presence of variant in a specific gene. From the histologic point of view, except for concomitant TMA, which was more frequent in patients with CFI and CFH variants, most of histologic features were similar among patients' groups. Familial forms are rare, accounting for 10% of C3 glomerulopathy. A total of 12 variants were identified in more than one patient. In most of the cases, clinical features, profiles of complement biomarkers of patients with the same variant, were heterogeneous, confirming that other factors probably contribute to the phenotype of the disease.
Our study has several limitations. This is a retrospective study. Despite a wide period of inclusion, it included a limited number of patients because of the rarity of the disease. This made it difficult to conclude on the real effect of different therapeutic approaches on disease course, particularly plasma exchange and eculizumab treatment. We were not able to provide functional data to clearly demonstrate the contribution of VUS identified in one quarter of the patients.
In conclusion, this study provided new insights on inherited C3 glomerulopathy/Ig-MPGN associated with CFH, CFI, and C3 rare variants. Our study demonstrated that genetic form of C3 glomerulopathy/Ig-MPGN has a severe prognosis, particularly those associated with factor I and C3 rare variants. It reveals that screening of genetic variants in complement genes needs to be proposed to all patients with C3 glomerulopathy and Ig-MPGN whatever the age of onset. In particular, we recommend genetic screening to be promptly performed in case of factor H or factor I deficiency, the absence of C3NeF in children and rapidly progressive kidney disease in adult, to discuss therapeutic strategy. In the area of new complement inhibitors in pipeline, functional studies are needed to better define mechanisms of complement overactivation and guide the therapeutic strategy.
Supplementary Material
Acknowledgments
We thank clinicians who referred patients: Dr. Aldigier (Hopital Dupuytren, Department of Nephrology, Limoges, France), Dr. Buchler (Hopital Bretonneau, Department of Nephrology, Tours, France), Prof. Caillard-Ohlmann (Hopital Civil, Department of Nephrology, Strasbourg, France), Dr. Delmas (Hopital Pellegrin, Department of Nephrology, Bordeaux, France), Prof. Deschene (Hopital Robert-Debré, Department of Pediatric Nephrology, Paris, Fance), Prof. Durbach and Prof. Francois (Hopital Bicêtre, Department of Nephrology, Kremlin-Bicêtre, France), Prof. Fakhouri (Hopital Hotel-Dieu, Department of Nephrology, Nantes, France), Prof. Frimat (Hopital de Brabois, Department of Nephrology, Nancy, France), Dr. Garnier (Hopital des enfants, Department of Pediatric Nephrology, Toulouse, France), Dr. Greze (Hopital Montpied, Department of Nephrology, Clermont Ferrand, France), Dr. Laurent (Hopital de Bois-Guillaume, Department of Nephrology, Bois-Guillaume, France), Prof. Legendre and Dr. Sberro-Soussan (Hopital Necker, Department of Nephrology and renal transplantation, Paris, France), Dr. Lionet (Hopital Huriet, Department of Nephrology, Lille, France), Dr. Mac Namara (Hopital de Bethune, Department of Nephrology, Bethune, France), Dr. Mangenot and Dr. Stolz (Hopital Robert Schuman, Department of Nephrology, Nouilly, France), Dr. Matignon (Hopital Henry Mondor, Department of Nephrology, Créteil, France), Dr. Steiz-Polski and Favre (Hopital Pasteur, Department of Nephrology, Nice, France), and Prof. Touchard (Hopital de Poitier, Department of Nephrology, Poitier, France).
Disclosures
V. Audard reports consultancy for Advisory boards of Addmledica, Alnylam, AstraZeneca, Bayer, and Vifor pharma; honoraria for Advisory boards for Addmledica, Alnylam, AstraZeneca, Bayer, and Vifor pharma; and advisory or leadership roles on Advisory boards for Addmledica, Alnylam, AstraZeneca, Bayer, and Vifor pharma. F. Bridoux reports research funding from Baxter and Janssen; honoraria from AstraZeneca, Janssen, and Sanofi; advisory or leadership roles for AstraZeneca, Attralus, Janssen, Novartis, and Prothena; and speakers bureau for AstraZeneca, Janssen, and Sanofi. V. Esnault reports consultancy from Alexion, AstraZeneca, Bayer, BMS-Pfizer, Boehringer-Ingelheim, and Novartis; research funding from AstraZeneca, BMS-Pfizer, Boehringer-Ingelheim, Hemotech, and Novartis; honoraria from Alexion, Amgen, Bayer, BMS-Pfizer, Boehringer-Ingelheim, Fresenius, Lilly, and Novartis; and advisory or leadership roles for AstraZeneca, Bayer, and Boehringer-Ingelheim. V. Frémeaux-Bacchi served as consultant for Alexion Pharmaceuticals, Apellis, BioCryps, Novartis, Roche, Sobi, and UCB; reports research funding from Alexion Pharmaceuticals; honoraria from Alexion Pharmaceuticals, Apellis, BioCryps, Novartis, Roche, Sobi, and UCB; and advisory or leadership roles for Alexion Pharmaceuticals, Apellis, BioCryps, Novartis, and Sobi. N. Jourde-Chiche reports consultancy for Alexion, GSK, Otsuka, and Vifor and role as a Otsuka advisory board member (paid) and Vifor advisory board member (paid). A. Karras reports consultancy for Alnylam, GSK, Novartis, Otsuka, and Vifor; honoraria from AstraZeneca, Bohringer-Ingelheim, GSK, Novartis, Otsuka, Pfizer, and Vifor; advisory or leadership role for Novartis, Otsuka, and Vifor; and speakers bureau for AstraZeneca, Boehringer-Ingelheim, Otsuka, Pfizer, and Vifor. M. Le Quintrec reports consultancy for Alexion, GSK, Novartis, and Sanofi; research funding from Alexion and Sanofi; honoraria from Alexion, Astellas, GSK, Novartis, and Sanofi; and advisory or leadership role for Novartis French board. F. Provot reports consultancy from Alexion, Astellas, and Sanofi and advisory or leadership roles for Novartis, Sanofi, and Takeda. P. Remy reports honoraria from Biogen and BMS. A. Servais reports consultancy for Chiesi; honoraria from Alexion, Chiesi, Recordati, and Vertex; and advisory or leadership role for Novartis. E. Thervet reports consultancy for Pfizer and Vifor; research funding from Alexion and GSK; and honoraria from Pfizer and Vifor. J. Zuber reports consultancy for Alexion Pharma and honoraria from Alexion Pharmaceuticals and BMS Pharmaceuticals. All remaining authors have nothing to disclose.
Funding
S. Chauvet: EURenOmics (2012-305608), Kidneeds (research grant 2019), ANR (ANR-20-CE17), and FRM (Prix 2012 FDR). M.S. Meuleman: SFNDT (2019 research grant).
Author Contributions
Conceptualization: Sophie Chauvet, Véronique Frémeaux-Bacchi.
Data curation: Vincent Audard, Véronique Baudouin, Dominique Bertrand, Frank Bridoux, Claire Dossier, Carine El Sissy, Vincent Esnault, Noémie Jourde-Chiche, Alexandre Karras, Moglie Le Quintrec, Férielle Louillet, Paula Vieira-Martins, Marie Sophie Meuleman, Marie-Pascale Morin, François Provot, Philippe Remy, David Ribes, Caroline Rousset-Rouviere, Aude Servais, Eric Thervet, Leila Tricot, Alain Wynckel, Mohamad Zaidan, Julien Zuber.
Formal analysis: Sophie Chauvet, Marie Sophie Meuleman.
Funding acquisition: Sophie Chauvet.
Investigation: Sophie Chauvet.
Methodology: Sophie Chauvet, Véronique Frémeaux-Bacchi.
Resources: Véronique Frémeaux-Bacchi, Paula Vieira-Martins,
Supervision: Sophie Chauvet, Véronique Frémeaux-Bacchi.
Writing – original draft: Sophie Chauvet, Véronique Frémeaux-Bacchi, Marie Sophie Meuleman.
Writing – review & editing: Sophie Chauvet, Véronique Frémeaux-Bacchi, Marie Sophie Meuleman.
Data Sharing Statement
All data are included in the manuscript and/or supporting information.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B802.
Supplemental Table 1. Description of the 30 CFH rare variants identified in 38 patients with C3 glomerulopathy or Ig-MPGN.
Supplemental Table 2. Description of the 13 CFI rare variants identified in 17 patients with C3 glomerulopathy or Ig-MPGN.
Supplemental Table 3. Description of the ten C3 rare variants identified in 13 patients with C3 glomerulopathy or Ig-MPGN.
Supplemental Table 4. Exhaustive genetic, immunologic, and histologic characteristics of the 66 C3 glomerulopathy/Ig-MPGN patients carrying complement rare variants.
Supplemental Table 5. Characteristics of inherited C3 glomerulopathy/Ig-MPGN according to age at diagnosis.
Supplemental Table 6. Characteristics of C3 glomerulopathy/Ig-MPGN children patients carrying rare variants in CFH, CFI, and C3 genes or not.
Supplemental Table 7. Characteristics of C3 glomerulopathy/Ig-MPGN adults younger than 50 years patients carrying rare variants in CFH, CFI, and C3 genes or not.
Supplemental Table 8. Characteristics of C3 glomerulopathy/Ig-MPGN adults older than 50 years patients carrying rare variants in CFH, CFI, and C3 genes or not.
Supplemental Table 9. Detailed histologic characteristics in inherited C3 glomerulopathy/Ig-MPGN.
Supplemental Table 10. Characteristics of patients with inherited C3 glomerulopathy/Ig-MPGN according to histologic diagnosis.
Supplemental Table 11. Detailed immunologic characteristics of inherited C3 glomerulopathy/Ig-MPGN.
Supplemental Table 12. Cumulative frequency of the rare and pathogenic variants between C3 glomerulopathy/Ig-MPGN cohorts and 503 controls from the 1000 Genomes Project.
Supplemental Table 13. Kidney transplant outcomes in patients with inherited C3 glomerulopathy/Ig-MPGN.
Supplemental Figure 1. C3b molecular structure with the position of rare variants of C3 identified in patients with C3 glomerulopathy/Ig-MPGN.
Supplemental Figure 2. C3 glomerulopathy, Ig-MPGN, and CFH, CFI, and C3 rare variants: patients repartition in national French registry according to age at diagnosis.
Supplemental Figure 3. Kidney survival of children and adults with inherited C3 glomerulopathy/Ig-MPGN.
Supplemental Figure 4. Kidney survival of patients with inherited C3 glomerulopathy/Ig-MPGN compared with kidney survival of patients without complement rare variant receiving or not specific treatment.
References
- 1.Fakhouri F, Frémeaux-Bacchi V. Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics. Nat Rev Nephrol. 2021;17(8):543–553. doi: 10.1038/s41581-021-00424-4 [DOI] [PubMed] [Google Scholar]
- 2.Fremeaux-Bacchi V Fakhouri F Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554–562. doi: 10.2215/CJN.04760512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kavanagh D Richards A Noris M, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol. 2008;45(1):95–105. doi: 10.1016/j.molimm.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 4.Iatropoulos P Noris M Mele C, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol. 2016;71:131–142. doi: 10.1016/j.molimm.2016.01.010 [DOI] [PubMed] [Google Scholar]
- 5.Servais A Noël L-H Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82(4):454–464. doi: 10.1038/ki.2012.63 [DOI] [PubMed] [Google Scholar]
- 6.Osborne AJ Breno M Borsa NG, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol. 2018;200(7):2464–2478. doi: 10.4049/jimmunol.1701695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallam TM Marchbank KJ Harris CL, et al. Rare genetic variants in complement factor I lead to low FI plasma levels resulting in increased risk of age-related macular degeneration. Invest Opthalmol Vis Sci. 2020;61(6):18. doi: 10.1167/iovs.61.6.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Triebwasser MP Roberson EDO Yu Y, et al. Rare variants in the functional domains of complement factor H are associated with age-related macular degeneration. Invest Opthalmol Vis Sci. 2015;56(11):6873. doi: 10.1167/iovs.15-17432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fritsche LG Igl W Bailey JNC, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48(2):134–143. doi: 10.1038/ng.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuber J Frimat M Caillard S, et al. Use of highly individualized complement blockade has revolutionized clinical outcomes after kidney transplantation and renal epidemiology of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2019;30(12):2449–2463. doi: 10.1681/ASN.2019040331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Quintrec M Zuber J Moulin B, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome: genetic complement in renal transplantation. Am J Transplant. 2013;13(3):663–675. doi: 10.1111/ajt.12077 [DOI] [PubMed] [Google Scholar]
- 12.Fakhouri F Fila M Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438–2449. doi: 10.1182/blood.2020009280 [DOI] [PubMed] [Google Scholar]
- 13.Marinozzi M-C Chauvet S Le Quintrec M, et al. C5 nephritic factors drive the biological phenotype of C3 glomerulopathies. Kidney Int. 2017;92(5):1232–1241. doi: 10.1016/j.kint.2017.04.017 [DOI] [PubMed] [Google Scholar]
- 14.Ravindran A, Fervenza FC, Smith RJH, De Vriese AS, Sethi S. C3 glomerulopathy: ten years’ experience at Mayo clinic. Mayo Clinic Proc. 2018;93(8):991–1008. doi: 10.1016/j.mayocp.2018.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bomback AS Santoriello D Avasare RS, et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int. 2018;93(4):977–985. doi: 10.1016/j.kint.2017.10.022 [DOI] [PubMed] [Google Scholar]
- 16.Lomax-Browne HJ Medjeral-Thomas NR Barbour SJ, et al. Association of histologic parameters with outcome in C3 glomerulopathy and idiopathic immunoglobulin-associated membranoproliferative glomerulonephritis. Clin J Am Soc Nephrol. 2022;17(7):994–1007. doi: 10.2215/CJN.16801221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gale DP de Jorge EG Cook HT, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376(9743):794–801. doi: 10.1016/s0140-6736(10)60670-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ault BH Schmidt BZ Fowler NL, et al. Human factor H deficiency: mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem. 1997;272(40):25168–25175. doi: 10.1074/jbc.272.40.25168 [DOI] [PubMed] [Google Scholar]
- 19.Dragon-Durey M-A Frémeaux-Bacchi V Loirat C, et al. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15(3):787–795. doi: 10.1097/01.ASN.0000115702.28859.a7 [DOI] [PubMed] [Google Scholar]
- 20.Chauvet S Hauer JJ Petitprez F, et al. Results from a nationawide retrospective cohort measure the impact of C3 and soluble C5b-9 levels on kidney outcomes in C3 glomerulopathy. Kidney Int. 2022;102(4):904–916. doi: 10.1016/j.kint.2022.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figuères M-L Frémeaux-Bacchi V Rabant M, et al. Heterogeneous histologic and clinical evolution in 3 cases of dense deposit disease with long-term follow-up. Hum Pathol. 2014;45(11):2326–2333. doi: 10.1016/j.humpath.2014.07.021 [DOI] [PubMed] [Google Scholar]
- 22.Pickering MC D’Agati VD Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84(6):1079–1089. doi: 10.1038/ki.2013.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S Aziz N Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin Merinero H Zhang Y Arjona E, et al. Functional characterization of 105 factor H variants associated with atypical HUS: lessons for variant classification. Blood. 2021;138(22):2185–2201. doi: 10.1182/blood.2021012037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong EKS Hallam TM Brocklebank V, et al. Functional characterization of rare genetic variants in the N-terminus of complement factor H in aHUS, C3G, and AMD. Front Immunol. 2020;11:602284. doi: 10.3389/fimmu.2020.602284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Jong S Volokhina EB de Breuk A, et al. Effect of rare coding variants in the CFI gene on Factor I expression levels. Hum Mol Genet. 2020;29(14):2313–2324. doi: 10.1093/hmg/ddaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauvet S Roumenina LT Bruneau S, et al. A familial C3GN secondary to defective C3 regulation by complement receptor 1 and complement factor H. J Am Soc Nephrol. 2016;27(6):1665–1677. doi: 10.1681/ASN.2015040348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schramm EC Roumenina LT Rybkine T, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125(15):2359–2369. doi: 10.1182/blood-2014-10-609073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Auton A Abecasis GR Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bu F Borsa NG Jones MB, et al. High-throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. J Am Soc Nephrol. 2016;27(4):1245–1253. doi: 10.1681/ASN.2015040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine AP Chan MMY Sadeghi-Alavijeh O, et al. Large-scale whole-genome sequencing reveals the genetic architecture of primary membranoproliferative GN and C3 glomerulopathy. J Am Soc Nephrol. 2020;31(2):365–373. doi: 10.1681/ASN.2019040433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caravaca-Fontán F Díaz-Encarnación MM Lucientes L, et al. Mycophenolate mofetil in C3 glomerulopathy and pathogenic drivers of the disease. Clin J Am Soc Nephrol. 2020;15(9):1287–1298. doi: 10.2215/CJN.15241219 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the manuscript and/or supporting information.