

Visual Abstract

Keywords: CKD, human genetics
Abstract
Background
Sickle cell trait affects approximately 8% of Black individuals in the United States, along with many other individuals with ancestry from malaria-endemic regions worldwide. While traditionally considered a benign condition, recent evidence suggests that sickle cell trait is associated with lower eGFR and higher risk of kidney diseases, including kidney failure. The mechanisms underlying these associations remain poorly understood. We used proteomic profiling to gain insight into the pathobiology of sickle cell trait.
Methods
We measured proteomics (N=1285 proteins assayed by Olink Explore) using baseline plasma samples from 592 Black participants with sickle cell trait and 1:1 age-matched Black participants without sickle cell trait from the prospective Women's Health Initiative cohort. Age-adjusted linear regression was used to assess the association between protein levels and sickle cell trait.
Results
In age-adjusted models, 35 proteins were significantly associated with sickle cell trait after correction for multiple testing. Several of the sickle cell trait–protein associations were replicated in Black participants from two independent cohorts (Atherosclerosis Risk in Communities study and Jackson Heart Study) assayed using an orthogonal aptamer-based proteomic platform (SomaScan). Many of the validated sickle cell trait–associated proteins are known biomarkers of kidney function or injury (e.g., hepatitis A virus cellular receptor 1 [HAVCR1]/kidney injury molecule-1 [KIM-1], uromodulin [UMOD], ephrins), related to red cell physiology or hemolysis (erythropoietin [EPO], heme oxygenase 1 [HMOX1], and α-hemoglobin stabilizing protein) and/or inflammation (fractalkine, C-C motif chemokine ligand 2/monocyte chemoattractant protein-1 [MCP-1], and urokinase plasminogen activator surface receptor [PLAUR]). A protein risk score constructed from the top sickle cell trait–associated biomarkers was associated with incident kidney failure among those with sickle cell trait during Women's Health Initiative follow-up (odds ratio, 1.32; 95% confidence interval, 1.10 to 1.58).
Conclusions
We identified and replicated the association of sickle cell trait with a number of plasma proteins related to hemolysis, kidney injury, and inflammation.
Introduction
Sickle cell trait is defined as the heterozygous carrier state for the β-globin p.Glu6Val mutation that causes sickle cell anemia. Sickle cell trait affects approximately 8% of Black individuals in the United States and approximately 300 million individuals globally.1 True prevalence is currently unknown because universal newborn screening for sickle cell trait and sickle cell disease in all 50 states has only been in place since 2006.2 While previously considered a benign carrier state, sickle cell trait has been increasingly associated with various clinical sequelae, including higher risk of exertional rhabdomyolysis,3 anemia,4 CKD,5,6 diabetes,7 and venous thromboembolic disease.8 Sickle cell trait–related kidney abnormalities are likely due to red cell sickling under local conditions within the kidney inner medulla (relatively low oxygen tension, acidosis, and reduced blood flow).9,10 While there are plausible biologic pathways (e.g., hemoglobin S polymerization, red cell sickling, and consequent ischemia-reperfusion injury) that may explain the higher susceptibility of selected health outcomes among those with sickle cell trait, the underlying biologic and molecular mechanisms of these increasingly recognized sickle cell trait phenotypic associations remain poorly characterized.
Emerging proteomic technologies make it possible to simultaneously evaluate a large number of circulating proteins for disease risk prediction, to allow early detection of subclinical disease, or to provide a systems or pathway perspective to understanding disease pathogenesis. By performing proteomic profiling in a sickle cell trait case–control sample of Black participants from the Women's Health Initiative (WHI) cohort, our overall objective was to provide a more comprehensive understanding of the molecular pathobiology of sickle cell trait–related clinical and laboratory sequelae in Black individuals. We additionally evaluated whether the newly identified sickle cell trait–associated proteins, individually or in aggregate, are associated with CKD outcomes among individuals with sickle cell trait.
Methods
WHI Sickle Cell Trait Case–Control Sample and Olink Proteomic Measurement
For this proteomics study, among a total of 14,118 eligible self-reported Black participants from the prospective WHI who had undergone rs334 genotyping for the sickle cell mutation, we randomly selected 592 Black participants with sickle cell trait. We also selected 592 WHI Black participants as controls (who genotyped as negative for the rs334 sickle mutation) matched 1:1 on exact age (in years) to the sickle cell trait cases (see Supplemental Methods).
Baseline sociodemographic and lifestyle characteristics and medical history were collected using standardized questionnaires at WHI recruitment.11 Hemoglobin concentration (in grams per deciliter) and total white blood cell count (cells per microliter) at the baseline examination were measured using an automated electronic cell counter. Serum creatinine was measured at baseline using an enzymatic method that was traceable to an isotope dilution mass spectrometry reference creatinine standard. GFR was estimated (eGFR) from serum creatinine using the 2009 Chronic Kidney Disease Epidemiology Collaboration equation.12 For ascertainment of incident kidney failure during follow-up, WHI participants were linked to the United States Renal Data System13 using complete name, date of birth, and other identifiers. Kidney failure events as of 2014 are included in these analyses. Thirty-four individuals without sickle cell trait and 101 with sickle cell trait were lost to follow-up or died before developing kidney failure. The mean (SD) follow-up time was 11.3 (±3.8) years among the sickle cell trait cases and 13.7 (±1.6) years among the non–sickle cell trait controls. Among those who died before developing kidney failure, the mean follow-up time was 12.9 years among the sickle cell trait cases and 9.0 years among the non–sickle cell trait controls.
These 1184 Black WHI participants underwent proteomic profiling on stored baseline EDTA plasma samples using the Olink Explore 1536 panel.14 After removal of proteins with >20% of participants below the lower limit of detection, a total of 1285 proteins were available for analysis. To assess the association of sickle cell trait with baseline protein levels, we transformed each normalized protein expression value using rank-based inverse normal transformation. We then performed linear regression, with transformed protein level as the dependent variable and sickle cell trait as the independent variable adjusting for baseline age as a covariate. Statistical significance was assessed using a Bonferroni correction to adjust for testing 1285 individual proteins (P value < 0.05/1285=3.9×10−5). Several additional sensitivity analyses were performed, as described in Supplemental Methods.
Validation of Sickle Cell Trait–Protein Associations in the Jackson Heart Study and the Atherosclerosis Risk in Communities Study
Sickle cell trait–protein associations initially identified in the WHI case–control sample were validated using two preexisting proteomic datasets derived from two independent cohorts of Black participants: the Jackson Heart Study (JHS) and the Atherosclerosis Risk in Communities (ARIC) study (see Supplemental Methods). The validation sample included 2054 JHS participants assayed using an orthogonal aptamer-based proteomic platform (SomaScan 1.3k)15,16 and 2048 Black ARIC participants assayed using the SomaScan platform v4.17 We performed linear regression within each cohort, with inverse normal transformed protein-level as the dependent variable and sickle cell trait as the independent variable adjusting for age, sex, and batch. Because different versions of the SomaScan platform were used in the ARIC study and JHS, association results are presented separately for each study. We also provide P values for the combined ARIC study and JHS SomaScan results of each overlapping protein using fixed-effect sample size–weighted meta-analysis, which includes a total of 4102 participants.
Construction of the Sickle Cell Trait Protein Risk Score in WHI
We calculated a sickle cell trait protein risk score in the 1184 WHI Black participants with Olink data using a 70%/30% training/testing division. In the training dataset, we tested the association of sickle cell trait with each protein and selected the top 50 proteins as candidates for the protein risk score. After removing correlated proteins (Pearson r>0.7), 44 proteins were left. We evaluated the association of this 44-protein risk score in the held-out 30% of WHI samples. The protein risk score was then calculated as the sum (protein level×β from the testing set).
Assessment of Individual Sickle Cell Trait Proteins or Composite Protein Risk Score with CKD Outcomes in WHI
To assess whether identified sickle cell trait–associated proteins attenuated the association between sickle cell trait and baseline eGFR, we added each protein individually to the regression model containing sickle cell trait as the exposure variable and baseline eGFR as the outcome, adjusting for age. The weighted composite protein risk score was used to evaluate the predictive ability of sickle cell trait–related plasma proteins on risk of incident ESKD (kidney failure) (separately in those with and without sickle cell trait) using logistic regression, with several different levels of covariate adjustment.
Results
The baseline characteristics of the 1184 WHI Olink proteomics study participants, stratified by sickle cell trait status, are summarized in Table 1. Compared with those without sickle cell trait, participants with sickle cell trait had lower hemoglobin levels, lower eGFR, higher proportion of African ancestry, and higher prevalence of anemia and diabetes.
Table 1.
Baseline characteristics of Women's Health Initiative study participants stratified by sickle cell trait
| Characteristic | Sickle Cell Trait | Non–sickle Cell Trait |
|---|---|---|
| N | 592 | 592 |
| WHI observational study, n (%) | (54) | (37) |
| Age, yr | 61±7 | 61±7 |
| % African ancestry | 80% | 73% |
| US region, n (%) | ||
| Northeast | 95 (16) | 117 (20) |
| South | 278 (47) | 267 (45) |
| Midwest | 142 (24) | 146 (25) |
| West | 77 (13) | 62 (10) |
| Body mass index, kg/m2 | 31±18 | 31±6 |
| Current smoking, n (%) | 61 (11) | 50 (9) |
| Systolic BP, mm Hg | 132±18 | 131±18 |
| Treated hypertension, n (%) | 253 (45) | 257 (46) |
| Treated diabetes, n (%) | 88 (15) | 56 (9) |
| History of cardiovascular disease, n (%) | 91 (16) | 102 (18) |
| eGFR, ml/min per 1.73 m2 | 89±20 | 93±18 |
| CKD | 34% | 27% |
| Stage 1 GFR (>90 ml/min), n (%) | 215 (50) | 303 (55) |
| Stage 2 mild CKD (GFR=60–89 ml/min), n (%) | 187 (44) | 226 (41) |
| Stage 3A moderate CKD (GFR=45–59 ml/min), n (%) | 21 (5) | 22 (4) |
| Stage 3B moderate CKD (GFR=30–44 ml/min), n (%) | 4 (0.9) | 3 (0.5) |
| Stage 4 severe CKD (GFR=15–29 ml/min), n (%) | 1 (0.2) | 0 (0) |
| Stage 5 end-stage CKD (GFR <15 ml/min), n (%) | 1 (0.2) | 0 (0) |
| Hemoglobin, g/dl | 12.8±1.0 | 13.0±0.9 |
| Anemia, n (%) | 140 (24) | 89 (15) |
Data are presented as mean±SD and number (percentage). The number of participants with missing variables was as follows: body mass index, n=8; smoking status, n=21; hypertension, n=63; diabetes, n=3; cardiovascular disease, n=60; eGFR, n=201; and hemoglobin, n=16. eGFR was calculated on the basis of creatinine. African ancestry percentage was based on genetic similarity to reference panels, as described in Supplemental Material. All participants had a self-reported African American ethnicity. Anemia was defined as hemoglobin level <12.0 g/dl. WHI, Women's Health Initiative.
Protein Associations with Sickle Cell Trait in WHI
In age-adjusted models, 35 proteins (Figure 1 and Table 2) were associated with sickle cell trait (P < 3.9×10−5). Many of the 35 sickle cell trait–associated proteins are known to be related to kidney function, red cell physiology and hemolysis, and/or inflammation. The three strongest associations were for fractalkine/CX3CL1 (P = 1.1×10−11), HAVCR1/kidney injury molecule-1 or KIM-1 (P = 2.6×10−11), and heme oxygenase-1/HMOX-1 (P = 1.2×10−10). There were moderate-to-strong pairwise correlations among several of the 35 proteins. We note that such correlations would make individual protein mediation analyses (to assess which proteins may statistically mediate the relationship of sickle cell trait with kidney outcomes in WHI) difficult to interpret; such analyses are thus not presented. A heat map based on pairwise correlation shows two partially overlapping main protein clusters largely consisting of kidney injury and inflammation-related proteins and a third smaller cluster containing the red cell proteins erythropoietin (EPO), HMOX-1, and α-hemoglobin stabilizing protein (AHSP) (Figure 2). Gene set enrichment analysis using ClusterProfiler 4.018 identified cytokine–cytokine receptor interaction, death receptor activity, and inflammatory response as pathways enriched among the 35 sickle cell trait–associated proteins (Supplemental Figure 1).
Figure 1.
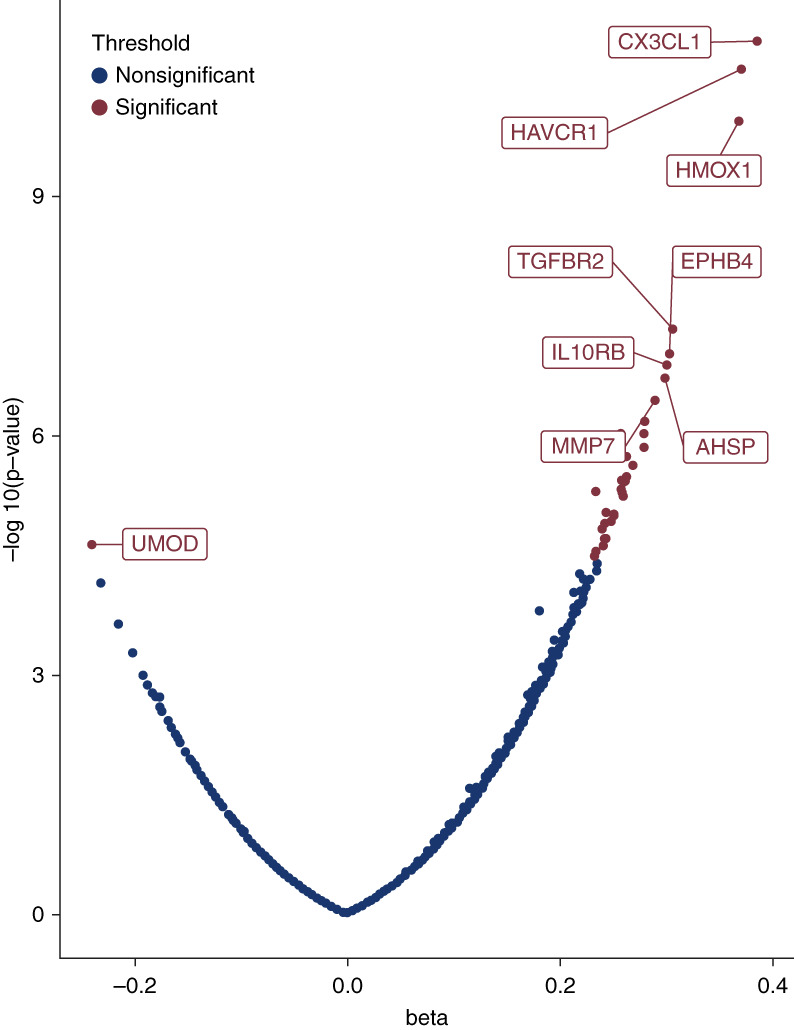
Volcano plot showing 35 significant protein associations with sickle cell trait in the WHI cohort. Effect estimates (β value) are noted on the x axis on the SD scale. All significant associations are labeled in red, but only the top eight proteins with association in the positive direction are labeled because of space constraints. AHSP, α-hemoglobin–stabilizing protein; EPHB4, ephrin type-B receptor 4; HAVCR1, hepatitis A virus cellular receptor 1; HMOX1, heme oxygenase 1; IL10RB, interleukin-10 receptor subunit β; MMP7, matrilysin; TGFBR2, TGF-β receptor type-2; UMOD, uromodulin; WHI, Women's Health Initiative.
Table 2.
Proteins associated with sickle cell trait status in Women's Health Initiative Black participants
| Name | Uniprot ID | Protein | β | SEM | P Value |
|---|---|---|---|---|---|
| CX3CL1 | P78423 | Fractalkine | 0.3875 | 0.0565 | 1.14E-11 |
| HAVCR1 | Q96D42 | Hepatitis A virus cellular receptor 1 (KIM-1) | 0.3723 | 0.0553 | 2.58E-11 |
| HMOX1 | P09601 | Heme oxygenase-1 | 0.3695 | 0.0568 | 1.16E-10 |
| TGFBR2 | P37173 | TGF-β receptor type-2 | 0.3076 | 0.0560 | 4.71E-08 |
| EPHB4 | P54760 | Ephrin type-B receptor 4 | 0.3043 | 0.0567 | 9.54E-08 |
| IL10RB | Q08334 | Interleukin-10 receptor subunit β | 0.3017 | 0.0568 | 1.31E-07 |
| AHSP | Q9NZD4 | AHSP | 0.3001 | 0.0573 | 1.96E-07 |
| MMP7 | P09237 | Matrilysin | 0.2898 | 0.0567 | 3.67E-07 |
| CCL2 | P13500 | C-C motif chemokine 2 | 0.2921 | 0.0571 | 3.73E-07 |
| PLAUR | Q03405 | Urokinase plasminogen activator surface receptor | 0.2810 | 0.0562 | 6.73E-07 |
| WFDC2 | Q14508 | WAP four-disulfide core domain protein 2 | 0.2585 | 0.0524 | 9.42E-07 |
| TNFRSF4 | P43489 | TNF receptor superfamily member 4 (OX40) | 0.2801 | 0.0569 | 9.71E-07 |
| EPO | P01588 | Erythropoietin | 0.2803 | 0.0578 | 1.40E-06 |
| SCARB2 | Q14108 | Lysosome membrane protein 2 | 0.2643 | 0.0551 | 1.84E-06 |
| B4GALT1 | P15291 | β-1,4-galactosyltransferase 1 | 0.2699 | 0.0569 | 2.38E-06 |
| DLL1 | O00548 | Delta-like protein 1 | 0.2640 | 0.0565 | 3.30E-06 |
| EFNA4 | P52798 | Ephrin-A4 | 0.2594 | 0.0558 | 3.76E-06 |
| NECTIN4 | Q96NY8 | Nectin-4 | 0.2624 | 0.0565 | 3.84E-06 |
| SPON2 | Q9BUD6 | Spondin-2 | 0.2590 | 0.0564 | 4.90E-06 |
| GDF15 | Q99988 | Growth/differentiation factor 15 | 0.2352 | 0.0514 | 5.15E-06 |
| STC1 | P52823 | Stanniocalcin-1 | 0.2606 | 0.0572 | 5.78E-06 |
| COLEC12 | Q5KU26 | Collectin-12 | 0.2448 | 0.0550 | 9.43E-06 |
| AMBP | P02760 | A1M | 0.2518 | 0.0567 | 9.72E-06 |
| SIRPB1 | O00241 | Signal-regulatory protein β-1 | 0.2520 | 0.0571 | 1.11E-05 |
| CD79B | P40259 | B-cell antigen receptor complex-associated protein β chain | 0.2502 | 0.0569 | 1.20E-05 |
| EPHA2 | P29317 | Ephrin type-A receptor 2 | 0.2434 | 0.0556 | 1.29E-05 |
| IGFBP4 | P22692 | Insulin-like growth factor-binding protein 4 | 0.2405 | 0.0554 | 1.52E-05 |
| AGRP | O00253 | Agouti-related protein | 0.2455 | 0.0573 | 1.96E-05 |
| FAS | P25445 | TNF receptor superfamily member 6 (FAS) | 0.2420 | 0.0565 | 1.99E-05 |
| TNFRSF21 | O75509 | TNF receptor superfamily member 21 (DR6) | 0.2438 | 0.0570 | 2.06E-05 |
| UMOD | P07911 | Uromodulin | −0.2394 | 0.0564 | 2.34E-05 |
| THY1 | P04216 | Thy-1 membrane glycoprotein | 0.2414 | 0.0570 | 2.44E-05 |
| FCER2 | P06734 | Low-affinity immunoglobulin epsilon Fc receptor | 0.2430 | 0.0575 | 2.52E-05 |
| CKAP4 | Q07065 | Cytoskeleton-associated protein 4 | 0.2342 | 0.0558 | 2.91E-05 |
| FOLR1 | P15328 | Folate receptor α | 0.2342 | 0.0562 | 3.25E-05 |
The β coefficient for sickle cell trait association is measured on the one-SD change scale (for rank-based inverse normal transformed protein expression values). AHSP, α-hemoglobin–stabilizing protein; A1M, α-1 microglobulin.
Figure 2.
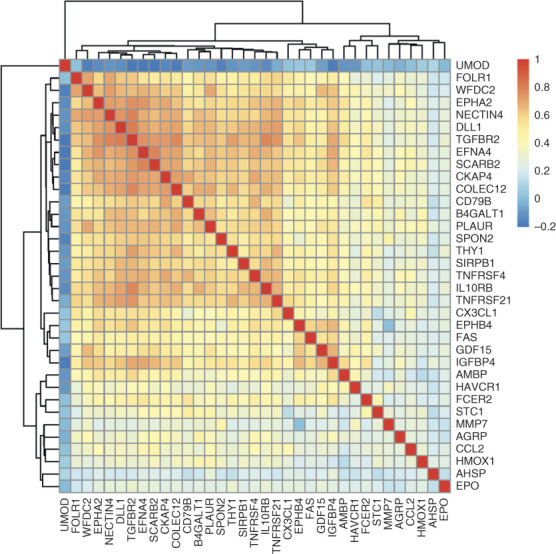
Heat map and dendrogram of pairwise correlations between 35 sickle cell trait–associated proteins.
In sensitivity analyses, the sickle cell trait–protein association results were robust to additional adjustment for proportion of African genetic ancestry (Supplemental Figure 2 and Supplemental Table 1). Many of the sickle cell trait–associated proteins were moderately inversely correlated with baseline eGFR. Most of the proteins were positively correlated with baseline white blood cell count, and several proteins showed weaker correlation with baseline hemoglobin (Supplemental Figure 3). Most protein associations were attenuated somewhat upon adjustment for baseline eGFR, including >10% reduction in effect size for uromodulin (UMOD), urokinase plasminogen activator surface receptor (PLAUR), ephrin type-A receptor 2 (EPHA2), ephrin-A4 (EFNA4), ephrin type-B receptor 4 (EPHB4), and growth/differentiation factor 15 (GDF15) (Supplemental Table 1). Six proteins CX3CL1, HAVCR1, HMOX1, TGF-β receptor type-2 (TGFBR2), matrilysin (MMP7), and WAP four-disulfide core domain protein 2 (WFDC2) remained significant after eGFR adjustment. However, we note that adjustment for common comorbidities of kidney disease (diabetes, hypertension, and smoking) or for white blood cell count did not significantly attenuate most protein associations (Supplemental Table 1).
Replication of Sickle Cell Trait–Protein Associations in the JHS and ARIC Study
We attempted replication of the 35 significant sickle cell trait–protein associations using aptamer-based (SomaScan) proteomic datasets derived from two cohorts of Black participants, from the JHS (N=2054) and the ARIC study (N=2048) (Supplemental Table 2). Together, these two datasets contain 308 individuals with sickle cell trait. Of the 35 proteins significantly associated with sickle cell trait in WHI, 21 were assayed using the aptamer-based SomaScan platform in a larger replication sample derived from the ARIC study or JHS. Of the 21 proteins available for analysis, eight (CX3CL1/fractalkine, Agouti-related protein (AGRP), EPHA2, EPHB4, EPO, GDF15, interleukin-10 receptor subunit β (IL10RB), and MMP7) or 38% were replicated in either the ARIC study or JHS (P < 0.0024) and one additional protein tumor necrosis factor receptor superfamily member 21 (DR6) in the combined ARIC study+JHS meta-analysis (Supplemental Table 3). Using more liberal criteria for replication (P < 0.05 and same direction of effect), 15 of 21 or 71% of the proteins showed consistency of results between WHI Olink discovery and ARIC study+JHS SomaScan replication panels. We note that some of the remaining proteins (e.g., TGF-β receptor type-2, lysosome membrane protein 2) have very low (<0.3) correlations between SomaScan and Olink platforms in prior reports16,19 (Supplemental Table 3) and thus would be unlikely to replicate using the SomaScan platform.
In addition to the 580 proteins that overlap between Olink Explore and SomaScan platforms, there are 742 proteins that are only present on SomaScan 1.3k, but not on Olink. Of these, three proteins were significantly associated with sickle cell trait in JHS after multiple testing correction (P < 6×10−5): hemoglobin, β2-microglobulin, and SPOCK2/testican-2 (Supplemental Table 4). The associations of sickle cell trait with higher hemoglobin, higher β2-microglobulin, and lower SPOCK2/testican-2 levels were replicated in the ARIC SomaScan dataset (Supplemental Table 4).
Relationship of Sickle Cell Trait–Associated Proteins with Kidney Function and CKD Outcomes
During WHI follow-up, kidney failure developed in 19 of those with sickle cell trait (3.2%) and eight of those without sickle cell trait (1.4%) (age-adjusted odds ratio for kidney failure=2.43; 95% confidence interval, 1.05 to 5.59; P = 0.037). Because of the relatively small number of kidney failure events and to reduce the burden of testing multiple individual proteins, we calculated an overall weighted protein risk score on the basis of the top sickle cell trait–associated proteins (see Supplemental Table 5 for protein risk score weights) and evaluated the association of the composite protein risk score with incident kidney failure among those with and without sickle cell trait. The protein risk score measured at baseline was associated with incident kidney failure among those with sickle cell trait when minimally adjusted for age only and age+baseline eGFR or when more fully adjusted for age, baseline eGFR, smoking, diabetes, and hypertension (Table 3). Among participants with sickle cell trait, the area under the curve (AUC) determined by receiver operating characteristic (ROC) statistical analysis for the protein risk score for association with incident kidney failure was 0.82 (95% confidence interval, 0.72 to 0.93). By comparison, the AUC of the age and baseline eGFR model was 0.84; the age, baseline eGFR, smoking, diabetes, and hypertension model was 0.92; and the age, baseline eGFR, smoking, diabetes, hypertension, and protein risk score model was 0.96. By contrast, the baseline protein risk score was not associated with kidney failure among those without sickle cell trait (Table 3) (interaction P value = 0.01), although we note that the overall number of kidney failure cases was very small.
Table 3.
Association of sickle cell trait protein risk score with incident kidney failure in those with and without sickle cell trait in Women's Health Initiative
| Covariate Adjustment | With Sickle Cell Trait | Without Sickle Cell Trait | P for Interaction | ||||
|---|---|---|---|---|---|---|---|
| Cases/Total | OR (95% CI) | P Value | Cases/Total | OR (95% CI) | P Value | ||
| Age | 19/592 | 1.42 (1.27 to 1.59) | <0.001 | 8/592 | 1.10 (0.94 to 1.27) | 0.24 | 0.007 |
| Age and baseline eGFR | 13/429 | 1.38 (1.16 to 1.63) | <0.001 | 8/554 | 1.06 (0.90 to 1.25) | 0.50 | 0.01 |
| Age, baseline eGFR, smoking, diabetes, and hypertension | 13/414 | 1.32 (1.10 to 1.58) | 0.003 | 7/541 | 0.94 (0.77 to 1.14) | 0.51 | 0.003 |
Note that the sample size of the analyses declined with each subsequent model because of missing covariate data. There were 201 individuals (38 with sickle cell trait and 157 without sickle cell trait) who were dropped from the second model because of missing baseline eGFR. An additional 28 individuals (15 with sickle cell trait and 13 without sickle cell trait) were dropped from the third model because of missing data for smoking, diabetes, or hypertension. OR, odds ratio; CI, confidence interval.
Discussion
In a case–control study of sickle cell trait among Black WHI participants, we identified 35 sickle cell trait–associated plasma proteins, many of which are related to red cell hemolysis, kidney function, tubulointerstitial injury/fibrosis, and/or inflammation and immune function. Several of these sickle cell trait–protein associations were replicated in an independent Black sample assayed using an orthogonal aptamer-based proteomic profiling platform. Moreover, a sickle cell trait composite protein risk score measured at baseline was associated with incident CKD-related outcomes in individuals with sickle cell trait. Together, these results provide further support for the relationship between sickle cell trait, subclinical hemolysis, immune function, and risk of sickle cell trait nephropathy.
In sickle cell disease, hemoglobin S polymerization after deoxygenation is the primary pathophysiologic event that ultimately leads to hemolysis, vaso-occlusion, and end-organ damage. The association of sickle cell trait with lower red cell hemoglobin (ref. 4 and Table 1); higher free plasma hemoglobin (see Supplemental Table 4); and higher levels of α-hemoglobin stabilizing protein, α-1 microglobulin (A1M), and HMOX1 (see Table 2) lends further evidence that subclinical chronic intravascular hemolysis occurs in individuals with sickle cell trait,4,20 which may contribute to sickle cell trait nephropathy. The release of free hemoglobin and its metabolite heme into the circulation results in vasoconstriction, oxidative stress, and a proinflammatory state,21 all of which may contribute to end-organ damage. Extracellular heme also has the ability to trigger a type 1 interferon inflammatory response22 and inflammasome activation.23 Toxicity of free hemoglobin and heme is ordinarily limited by binding of haptoglobin to free hemoglobin, clearance of free heme and reactive oxygen species by A1M, and enzymatic degradation of free heme by HMOX-1.24 HMOX-1 and A1M both have nephroprotective effects in heme-related models of kidney injury.25 However, catabolism of free heme by HMOX-1 also results in the production of free iron, which has been associated with ferroptosis, which has been implicated in progression of fibrotic diseases, including CKD.26 Finally, analogous to what has been reported in patients with sickle cell disease,27 common genetic variants, such as the HMOX1 GT-tandem repeat polymorphism,28 may influence HMOX-1 expression or activity and may mitigate the end-organ response to subclinical hemolysis, including sickle cell trait–related CKD risk. This would be interesting to further explore in other cohorts with genotyping data for these short tandem repeats (which is not available here).
Chronic inflammation due to innate and adaptive immune cell mechanisms also plays a role in the pathogenesis of sickle nephropathy.29 More subtle alterations of immune and coagulation laboratory parameters have also been recently observed among individuals with sickle cell trait.15,30,31 Our results in WHI confirm and extend these observations to include several additional sickle cell trait–associated inflammatory or kidney tubule injury biomarkers (MCP-1, PLAUR, HAVCR1/KIM-1, UMOD, and A1M),32–35 some of which may play important roles in sickle nephropathy.9,36,37 PLAUR, MCP-1, and fractalkine are key mediators of inflammation and mediate monocyte release of innate immune cells and migration from the bone marrow to tissue sites of injury, such as the kidney. It is also interesting to note the association of sickle cell trait with several markers of lymphocyte development, differentiation, and function (e.g., tumor necrosis factor receptor superfamily member 4 [OX40], DR6, tumor necrosis factor receptor superfamily member 6 [FAS], low affinity immunoglobulin epsilon Fc receptor [FCER2], given the reported association of sickle cell trait with lower lymphocyte count.30 Soluble TNF receptor levels have been previously associated with risk of CKD progression and kidney failure.38,39 KIM-1 and UMOD are expressed in the kidney proximal tubule and loop of Henle epithelial tubular cells, respectively, and are well-characterized markers of tubular injury35 and tubule synthetic function.40 MMP7, WFDC2, TGFBR2, and ephrins are involved in kidney fibrosis, and plasma levels of some of these fibrosis biomarkers have been prognostic for CKD progression.34,41,42 While markers of subclinical inflammation or fibrosis likely are released in response to tubular and/or glomerular injury caused by red cell sickling, it is also possible that a genetic predisposition to inflammation or fibrosis may directly contribute to ongoing organ damage and disease progression. Mendelian randomization approaches jointly assessing genetically determined protein levels in relation to CKD outcomes in patients with sickle cell trait may eventually reveal such causal relationships, but much larger sample sizes will be required.43
In addition to providing further insight into pathophysiologic mechanisms involved in sickle nephropathy, proteomic profiling may enable the identification of biomarkers for early detection of subclinical disease. In this regard, several sickle cell trait–protein associations remained significant even after eGFR adjustment, reflecting early proteomic changes driven by inflammatory and hematologic effects. Recent studies have used proteomic profiling to discover new biomarkers for early-stage disease or identification of individuals at risk of progression for various types of kidney diseases,44,45 including diabetic or IgA nephropathy, but these studies were largely conducted among individuals of European ancestry. While kidney injury is also a key potential complication of sickle cell disease, some mechanisms may be shared between sickle cell trait and sickle cell disease–associated nephropathy, and others are likely distinct. Future proteomics studies may help clarify the shared and distinct proteomic predictors of these and other forms of nephropathy. In sickle cell disease nephropathy, which should be examined in future proteomics work, early detection is particularly important because glomerular hyperperfusion and hyperfiltration occur during the initial stages, and standard kidney function tests, such as eGFR, become abnormal only when kidney damage is extensive and therapeutic interventions may no longer be able to prevent progression to kidney failure.9 In sickle cell trait, early detection may provide the opportunity for closer attention to modification or treatment of additional CKD risk factors and comorbidities, including the use of renoprotective agents, such as angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, which have been beneficial in reducing eGFR decline or urinary albumin excretion in sickle cell nephropathy. The results presented here may help guide hypotheses as to the mechanism of sickle cell trait–associated kidney injury as well as guide nephrologists in identifying patients with sickle cell trait who are at particularly high risk of CKD development.
A unique feature of our study is the application of antibody-based proteomic profiling in a relatively large number of Black women with sickle cell trait, giving us statistical power to discover novel sickle cell trait–protein associations. The availability of complementary aptamer-based proteomic profiling in nearly 4000 independent Black participants from the ARIC study and JHS (including 308 sickle cell trait cases) allowed us to replicate several sickle cell trait–protein associations using orthogonal proteomic technologies. On the other hand, the proteins represented on each platform only partially overlap. For example, an association of sickle cell trait with lower SPOCK2/testican-2, a kidney podocyte-derived protein, was only detected using SomaScan because the protein is not present on the Olink Explore panel. Lower plasma testican-2 has been associated with higher risk of kidney failure46 and also associated with APOL1 risk variants in Black individuals.47
Another limitation of our study is the small number of kidney failure cases in WHI; the protein risk score interaction with sickle cell trait requires replication in larger studies. In addition, urinary albumin and creatinine measurements are not available in the WHI participants, so we were unable to assess whether proteomic profiling could detect kidney disease before microalbuminuria. Levels of circulating proteomes at a single time point may not accurately reflect dynamic changes or persistent elevations across the lifetime, including recurrent AKI events that ultimately contribute to the occurrence of CKD. We were also limited by the lack of serial longitudinal eGFR data in WHI. Finally, additional studies using antibody-based proteomics are required to generalize our findings to men with sickle cell trait, although we do note that the ARIC study and JHS SomaScan replication cohorts comprise women and men. In summary, we identified 35 sickle cell trait–associated plasma proteins, which are enriched in pathways related to hemolysis, kidney function, and inflammation/immunity. We provide additional evidence that the newly identified sickle cell trait–associated proteins are associated with CKD outcomes among those with sickle cell trait. Together, these results begin to provide a mechanistic understanding of the clinical and laboratory sequelae of sickle cell trait as well as potential therapeutic targets for sickle-related nephropathy.
Supplementary Material
Acknowledgments
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program were supported by the National Heart, Lung and Blood Institute (NHLBI). Proteomic data generation in WHI was supported by X01HL153408 “A Multi-omics Resource for Sickle Cell Trait in African Americans from the Women's Health Initiative,” and the data were generated by the TOPMed Broad Institute and Beth Israel Deaconess Medical Center Proteomics platform. Genome sequencing for “NHLBI TOPMed: The Jackson Heart Study” (phs000964.v1.p1) was performed at the Northwest Genomics Center (HHSN268201100037C). Core support, including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering, was provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support, including phenotype harmonization, data management, sample-identity QC, and general program coordination, was provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I).
The WHI program is funded by the National Heart, Lung, and Blood Institute; the National Institutes of Health; and the US Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, and 75N92021D00005. For a list of all the investigators who have contributed to WHI science, please visit: https://s3-us-west-2.amazonaws.com/www-whi-org/wp-content/uploads/WHI-Investigator-Long-List.pdf.
The Jackson Heart Study (JHS) was supported by and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I), and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD). The authors also wish to thank the staff and participants of the JHS.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the US Department of Health and Human Services.
All data have been submitted or are pending submission to dbGaP (phs000200 and phs001237 WHI, phs000964 and phs000286 JHS, phs001211 and phs000280 ARIC) or BioLINCC (JHS HLB00891219a, ARIC HLB00020021); data are also available through study coordinating centers (https://www.whi.org/WHI, https://www.jacksonheartstudy.org/JHS, https://sites.cscc.unc.edu/aric/ARIC) with approved manuscript proposals.
All participants in the current study provided written informed consent, and the study was approved by the institutional review board of the WHI coordinating center at the Fred Hutchinson Cancer Center. All replication cohort participants also provided written informed consent for JHS/ARIC study activities, at the University of Mississippi Medical Center and University of North Carolina at Chapel Hill, respectively.
Footnotes
L.M.R. and A.P.R. contributed equally to this work.
See related editorial, “Sickle Cell Trait and Circulating Proteome,” on pages 1391–1392.
Disclosures
J. Coresh consultancy for Healthy.io and SomaLogic, ownership interest in Healthy.io, research funding from the National Institute of Health and National Kidney Foundation, and an advisory or leadership role for SomaLogic. M.W. Foster reports research funding from Biomilq, Circumvent Pharmaceuticals, and Oerth Bio. N. Franceschini reports advisory or leadership roles for Women's Health Initiative Chair of Ancillary Committee and Women's Health Initiative Publication and Presentation Committee. M.E. Grams reports advisory or leadership roles for the American Journal of Kidney Diseases, ASN Publication Committee, CJASN, JASN Editorial Fellowship Committee, KDIGO Executive Committee (co-chair elect), NKF Scientific Advisory Board, and USRDS Scientific Advisory Board; grant funding from NKF, which receives funding from multiple pharmaceutical companies; grant funding from NIH; payment from academic institutions for grand rounds; and payment from NephSAP. S. Liu reports consultancy for Novo Nordisk and TwinHealth. J.E. Manson reports research funding from Mars Edge. L.M. Raffield reports consultancy for TOPMed Administrative Coordinating Center (through Westat). M.J. Telen reports consultancy from Novartis and Pfizer, Inc.; research funding from BioMedomics, CSL Behring, NIH, Novartis, Novo Nordisk, and Pfizer; and honoraria from Novartis and Pfizer. All remaining authors have nothing to disclose.
Funding
N. Franceschini: NIDDK (R01-DK117445) and National Institute on Minority Health and Health Disparities (R01-MD012765). A.P. Reiner: National Human Genome Research Institute (U01-HG011720). L.M. Raffield: National Human Genome Research Institute (U01-HG011720).
Author Contributions
Conceptualization: Yanwei Cai, Laura M. Raffield, Alex P. Reiner.
Data curation: Allison E. Ashley-Koch, Josef Coresh, Melanie E. Garrett, Robert E. Gerszten, Li Hsu, Charles Kooperberg, Alex P. Reiner, Usman A. Tahir, Marilyn J. Telen.
Formal analysis: Allison E. Ashley-Koch, Paul L. Auer, Yanwei Cai, Matthew W. Foster, Nora Franceschini, Melanie E. Garrett, Morgan E. Grams, Li Hsu, Charles Kooperberg, Yun Li, Simin Liu, JoAnn E. Manson, Laura M. Raffield, Alex P. Reiner, Aditya Surapaneni, Usman A. Tahir, Hua Tang, Marilyn J. Telen, Genevieve L. Wojcik, Bing Yu.
Funding acquisition: Alex P. Reiner.
Methodology: Yanwei Cai.
Project administration: Alex P. Reiner.
Writing – original draft: Yanwei Cai, Laura M. Raffield, Alex P. Reiner.
Writing – review & editing: Allison E. Ashley-Koch, Paul L. Auer, Yanwei Cai, Josef Coresh, Matthew W. Foster, Nora Franceschini, Melanie E. Garrett, Robert E. Gerszten, Morgan E. Grams, Li Hsu, Charles Kooperberg, Yun Li, Simin Liu, JoAnn E. Manson, Laura M. Raffield, Alex P. Reiner, Aditya Surapaneni, Usman A. Tahir, Hua Tang, Marilyn J. Telen, Genevieve L. Wojcik, Bing Yu.
Data Sharing Statement
Original data created for the study are or will be available in a persistent repository upon publication.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B794 and http://links.lww.com/CJN/B795.
Supplemental Figure 1. Pathway enrichment analysis of SCT-associated proteins.
Supplemental Figure 2. SCT–protein association P-values with and without adjustment for genetic ancestry (i.e., PC1 or % African ancestry).
Supplemental Figure 3. Correlation between 35 SCT-associated proteins and baseline eGFR or hemoglobin in WHI.
Supplemental Table 1. Full summary statistics for protein associations with SCT in the discovery WHI cohort.
Supplemental Table 2. Demographics table for replication cohorts.
Supplemental Table 3. Replication results using the SomaScan platform in the ARIC study and JHS, including information on estimated correlation between Olink and SomaScan from prior publications.
Supplemental Table 4. Additional proteins that were assay-wide significant for association with SCT in JHS (with replication in the ARIC study).
Supplemental Table 5. Protein risk score (PrRS) weights.
References
- 1.Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med. 2009;122(6):507–512. doi: 10.1016/j.amjmed.2008.12.020 [DOI] [PubMed] [Google Scholar]
- 2.National Academies of Sciences. In: Engineering, and Medicine, Health and Medicine Division, Board on Population Health and Public Health Practice, Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. Addressing Sickle Cell Disease. Martinez RM, Osei-Anto HA, McCormick M, eds. National Academies Press; 2021. [PubMed] [Google Scholar]
- 3.Nelson DA, Deuster PA, Carter R, III, Hill OT, Wolcott VL, Kurina LM. Sickle cell trait, rhabdomyolysis, and mortality among U.S. Army soldiers. N Engl J Med. 2016;375(5):435–442. doi: 10.1056/nejmoa1516257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raffield LM Ulirsch JC Naik RP, et al. Common α-globin variants modify hematologic and other clinical phenotypes in sickle cell trait and disease. PLoS Genet. 2018;14(3):e1007293. doi: 10.1371/journal.pgen.1007293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naik RP Derebail VK Grams ME, et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA. 2014;312(20):2115–2125. doi: 10.1001/jama.2014.15063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naik RP Irvin MR Judd S, et al. Sickle cell trait and the risk of ESRD in blacks. J Am Soc Nephrol. 2017;28(7):2180–2187. doi: 10.1681/ASN.2016101086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulsizer J Resurreccion WK Shi Z, et al. Sickle cell trait and risk for common diseases: evidence from the UK Biobank. Am J Med. 2022;135(8):e279–e287. doi: 10.1016/j.amjmed.2022.03.024 [DOI] [PubMed] [Google Scholar]
- 8.Folsom AR Tang W Roetker NS, et al. Prospective study of sickle cell trait and venous thromboembolism incidence. J Thromb Haemost. 2015;13(1):2–9. doi: 10.1111/jth.12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol. 2022;18(6):361–377. doi: 10.1038/s41581-022-00540-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naik RP, Derebail VK. The spectrum of sickle hemoglobin-related nephropathy: from sickle cell disease to sickle trait. Expert Rev Hematol. 2017;10(12):1087–1094. doi: 10.1080/17474086.2017.1395279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Women's Health Initiative study group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials. 1998;19(1):61–109. doi: 10.1016/s0197-2456(97)00078-0 [DOI] [PubMed] [Google Scholar]
- 12.Levey AS, Stevens LA. Estimating GFR using the CKD Epidemiology Collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis. 2010;55(4):622–627. doi: 10.1053/j.ajkd.2010.02.337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.United States Renal Data System. Excerpts from the 2003 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Am J Kidney Dis. 2003;42(6 suppl 5):1–230. doi: 10.1053/j.ajkd.2003.09.004 [DOI] [PubMed] [Google Scholar]
- 14.Assarsson E Lundberg M Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9(4):e95192. doi: 10.1371/journal.pone.0095192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz DH Tahir UA Bick AG, et al. Whole genome sequence analysis of the plasma proteome in black adults provides novel insights into cardiovascular disease. Circulation. 2022;145(5):357–370. doi: 10.1161/circulationaha.121.055117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz DH Robbins JM Deng S, et al. Proteomic profiling platforms head to head: leveraging genetics and clinical traits to compare aptamer- and antibody-based methods. Sci Adv. 2022;8(33):eabm5164. doi: 10.1126/sciadv.abm5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grams ME Surapaneni A Chen J, et al. Proteins associated with risk of kidney function decline in the general population. J Am Soc Nephrol. 2021;32(9):2291–2302. doi: 10.1681/ASN.2020111607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu T Hu E Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietzner M Wheeler E Carrasco-Zanini J, et al. Synergistic insights into human health from aptamer- and antibody-based proteomic profiling. Nat Commun. 2021;12(1):6822. doi: 10.1038/s41467-021-27164-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinberg MH, Embury SH. Alpha-thalassemia in blacks: genetic and clinical aspects and interactions with the sickle hemoglobin gene. Blood. 1986;68(5):985–990. doi: 10.1182/blood.v68.5.985.985 [DOI] [PubMed] [Google Scholar]
- 21.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. doi: 10.1172/jci89741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y Pal M Bao W, et al. Type I interferon is induced by hemolysis and drives antibody-mediated erythrophagocytosis in sickle cell disease. Blood. 2021;138(13):1162–1171. doi: 10.1182/blood.2021011629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roumenina LT, Rayes J, Lacroix-Desmazes S, Dimitrov JD. Heme: modulator of plasma systems in hemolytic diseases. Trends Mol Med. 2016;22(3):200–213. doi: 10.1016/j.molmed.2016.01.004 [DOI] [PubMed] [Google Scholar]
- 24.Bergwik J, Kristiansson A, Allhorn M, Gram M, Åkerström B. Structure, functions, and physiological roles of the lipocalin α1-microglobulin (A1M). Front Physiol. 2021;12:645650. doi: 10.3389/fphys.2021.645650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunenwald A, Roumenina LT, Frimat M. Heme oxygenase 1: a defensive mediator in kidney diseases. Int J Mol Sci. 2021;22(4):2009. doi: 10.3390/ijms22042009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Wang J. Ferroptosis, a rising force against renal fibrosis. Oxid Med Cell Longev. 2022;7686956:1–12. doi: 10.1155/2022/7686956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saraf SL Viner M Rischall A, et al. HMOX1 and acute kidney injury in sickle cell anemia. Blood. 2018;132(15):1621–1625. doi: 10.1182/blood-2018-05-853929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. 2004;37(8):1097–1104. doi: 10.1016/j.freeradbiomed.2004.07.008 [DOI] [PubMed] [Google Scholar]
- 29.de Azevedo JTC, Malmegrim KCR. Immune mechanisms involved in sickle cell disease pathogenesis: current knowledge and perspectives. Immunol Lett. 2020;224:1–11. doi: 10.1016/j.imlet.2020.04.012 [DOI] [PubMed] [Google Scholar]
- 30.Mikhaylova AV McHugh CP Polfus LM, et al. Whole-genome sequencing in diverse subjects identifies genetic correlates of leukocyte traits: the NHLBI TOPMed program. Am J Hum Genet. 2021;108(10):1836–1851. doi: 10.1016/j.ajhg.2021.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raffield LM Zakai NA Duan Q, et al. D-dimer in African Americans: whole genome sequence analysis and relationship to cardiovascular disease risk in the Jackson Heart Study. Arterioscler Thromb Vasc Biol. 2017;37(11):2220–2227. doi: 10.1161/atvbaha.117.310073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudhini YR, Wei C, Reiser J. suPAR: an inflammatory mediator for kidneys. Kidney Dis (Basel). 2022;8(4):265–274. doi: 10.1159/000524965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cormican S, Griffin MD. Fractalkine (CX3CL1) and its receptor CX3CR1: a promising therapeutic target in chronic kidney disease? Front Immunol. 2021;12:664202. doi: 10.3389/fimmu.2021.664202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zabetian A, Coca SG. Plasma and urine biomarkers in chronic kidney disease: closer to clinical application. Curr Opin Nephrol Hypertens. 2021;30(6):531–537. doi: 10.1097/mnh.0000000000000735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karmakova ТА, Sergeeva NS, Kanukoev КY, Alekseev BY, Kaprin AD. Kidney injury molecule 1 (KIM-1): A multifunctional glycoprotein and biological marker. Sovrem Tekhnologii Med. 2021;13(3):64–78. doi: 10.17691/stm2021.13.3.08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.dos Santos TEdJ, Gonçalves RP, Barbosa MC, da Silva GB, Jr., Daher EDF. Monocyte chemoatractant protein-1: a potential biomarker of renal lesion and its relation with oxidative status in sickle cell disease. Blood Cell Mol Dis. 2015;54(3):297–301. doi: 10.1016/j.bcmd.2014.11.019 [DOI] [PubMed] [Google Scholar]
- 37.Unal S, Ozdemir O, Ozcimen AA, Oztas Y. Increase of serum fractalkine and fractalkine gene expression levels in sickle cell disease patients. Int J Hematol. 2015;101(2):114–118. doi: 10.1007/s12185-014-1718-4 [DOI] [PubMed] [Google Scholar]
- 38.Niewczas MA Pavkov ME Skupien J, et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med. 2019;25(5):805–813. doi: 10.1038/s41591-019-0415-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen TK Estrella MM Appel LJ, et al. Biomarkers of immune activation and incident kidney failure with replacement therapy: findings from the African American Study of Kidney Disease and Hypertension. Am J Kidney Dis. 2021;78(1):75–84.e1. doi: 10.1053/j.ajkd.2020.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devuyst O, Bochud M, Olinger E. UMOD and the architecture of kidney disease. Pflugers Arch. 2022;474(8):771–781. doi: 10.1007/s00424-022-02733-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- 42.Huang Z Liu S Tang A, et al. Key role for EphB2 receptor in kidney fibrosis. Clin Sci. 2021;135(17):2127–2142. doi: 10.1042/cs20210644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J Haberland V Baird D, et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat Genet. 2020;52(10):1122–1131. doi: 10.1038/s41588-020-0682-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi H Looker HC Satake E, et al. Results of untargeted analysis using the SOMAscan proteomics platform indicates novel associations of circulating proteins with risk of progression to kidney failure in diabetes. Kidney Int. 2022;102(2):370–381. doi: 10.1016/j.kint.2022.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Govender MA, Brandenburg JT, Fabian J, Ramsay M. The use of ’omics for diagnosing and predicting progression of chronic kidney disease: a scoping review. Front Genet. 2021;12:682929. doi: 10.3389/fgene.2021.682929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen D Zhou L Zheng Z, et al. Testican-2 is associated with reduced risk of incident ESKD. J Am Soc Nephrol. 2023;34(1):122–131. doi: 10.1681/ASN.2022020216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen TK Surapaneni AL Arking DE, et al. APOL1 kidney risk variants and proteomics. Clin J Am Soc Nephrol. 2022;17(5):684–692. doi: 10.2215/CJN.14701121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data created for the study are or will be available in a persistent repository upon publication.