

Abstract
Mutations in the superoxide dismutase 1 (SOD1) gene are the second most common known cause of ALS. SOD1 variants express high phenotypic variability and over 200 have been reported in people with ALS. It was previously proposed that variants can be broadly classified in two groups, ‘wild-type like’ (WTL) and ‘metal binding region’ (MBR) variants, based on their structural location and biophysical properties. MBR variants, but not WTL variants, were associated with a reduction of SOD1 enzymatic activity. In this study we used molecular dynamics and large clinical datasets to characterise the differences in the structural and dynamic behaviour of WTL and MBR variants with respect to the wild-type SOD1, and how such differences influence the ALS clinical phenotype. Our study identified marked structural differences, some of which are observed in both variant groups, while others are group specific. Moreover, collecting clinical data of approximately 500 SOD1 ALS patients carrying variants, we showed that the survival time of patients carrying an MBR variant is generally longer (∼6 years median difference, p < 0.001) with respect to patients with a WTL variant. In conclusion, our study highlighted key differences in the dynamic behaviour between WTL and MBR SOD1 variants, and between variants and wild-type SOD1 at an atomic and molecular level, that could be further investigated to explain the associated phenotypic variability. Our results support the hypothesis of a decoupling between mechanisms of onset and progression of SOD1 ALS, and an involvement of loss-of-function of SOD1 with the disease progression.
Keywords: Amyotrophic lateral sclerosis, Superoxide Dismutase type 1 (SOD1), Diseaseassociated SOD1 mutations, Molecular dynamics (MD) simulations, Performed principal component analysis, Survival analysis
Graphical Abstract
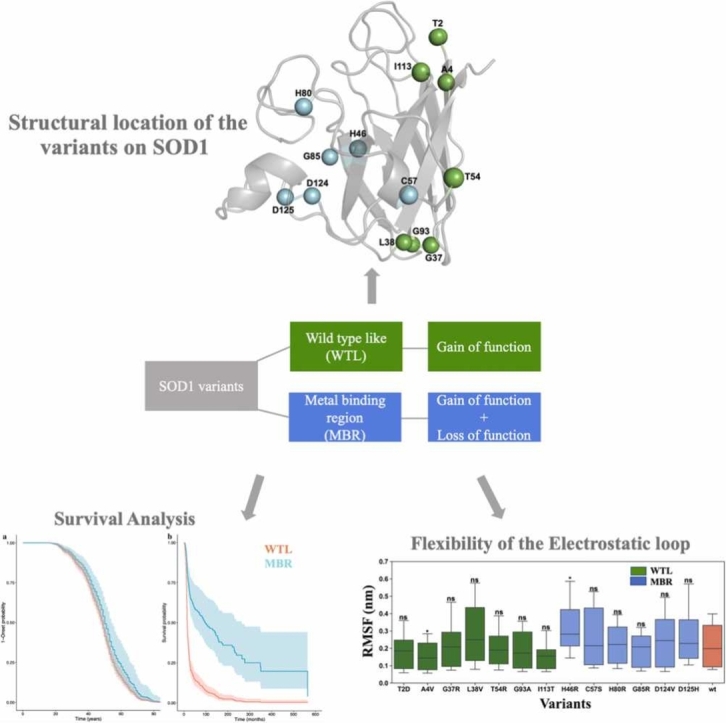
Summary
SOD1 mechanisms underlying ALS onset and progression might be decoupled and reduced SOD1 function might be associated with slower progression.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder, characterized by progressive muscle weakness and paralysis, leading to death from neuromuscular respiratory failure typically within 5 years of symptom onset [1], [2]. Mutations in the superoxide dismutase type 1 (SOD1) gene are the second most common known cause of ALS and have been found in both familial ALS and sporadic ALS [3]. SOD1 is an antioxidant metalloenzyme, which protects cells from oxidative damage. It catalyses the conversion of the superoxide O2- free radical present in the cytoplasm to H2O2 and molecular oxygen. SOD1 is a 32 kDa protein which exists as a homodimer and its structure is characterized by the presence of a β-barrel immunoglobin fold, two long loops: metal binding loop (MBL) and electrostatic loop (ESL) [4]. The fully formed SOD1 homodimer is highly stable and has a melting point of 92 °C [5], due to copper and zinc binding, an intrasubunit disulphide bond and dimerization [6].
At present, more than 220 SOD1 mutations have been reported in people with ALS [7], [8]. Although there is evidence that SOD1 mutations cause ALS, the exact underlying mechanism of disease onset and progression remains poorly understood [9]. It has been proposed that the pathogenic mutations hinder post-translational maturation, which decreases structural stability, thereby instigating the misfolding of SOD1 protein [10], [11], [12]. The ultimate outcome of SOD1 misfolding is the formation of hallmark amyloid aggregates of SOD1 in the affected tissues [13], [14], [15]. A noteworthy feature of the misfolded SOD1 is its prion-like behaviour of self-proliferation. It was proposed that a misfolded SOD1 can transverse between cells and cause misfolding of other SOD1 molecules via the protein aggregates released from dying cells [16], [17], [18].
Moreover, SOD1 variants can express high phenotypic variability. For example, A4V (A5V in the HGVS V2.0 nomenclature – traditional nomenclature will be used throughout this manuscript) is a variant which is responsible for 48% of SOD1 ALS in US familial ALS. It is associated with variable site of onset, rapid disease progression and a mean survival time of 1.1 years from clinical presentation [19], [20], [21]. In contrast, H46R is commonly observed in the Japanese, Pakistan and Norwegian populations and is invariably associated with a stereotypic phenotype with slowly ascending paresis beginning in the legs and long survival (approx. 12 years from diagnosis) [22], [23], [24], [25]. Investigating the differences between SOD1 mutations in terms of their structural effect on the protein might help to understand the mechanisms underlying such a heterogeneous clinical presentation.
Disease-associated SOD1 mutations are found in all domains of the protein and over the years attempts have been made to classify the impact of each variant based on location and subsequent effect on protein structure [26]. Based on their structural location and biophysical characteristics such as metal binding affinity and influence on SOD1 in vitro activity, it has been proposed that SOD1 variants can be classed in two broad groups [26], [27], [28]. The first group consists of the ‘wild-type like’ (WTL) variants such as: A4V, L38V, G37R, G41S, G72S, D76Y, D90A and G93A. These variants bind metal ions tightly and the catalytic activity is not affected in vitro, therefore their biophysical behaviour is expected to be similar to that of the wild-type SOD1 (wt-SOD1) [27], [29], [30]. The second group contains ‘metal binding region’ (MBR) variants, such as: H46R, H48Q, G85R, D124V, D125H, G127X and S134N. These variants are localized in the metal binding sites, metal binding loop and electrostatic loop regions, and cause reduced metal binding and diminished in vitro catalytic activity [4], [31], [32], [33].
The aim of this study was to characterize the differences in the structural and dynamic behaviour of the WTL and MBR disease-causing SOD1 variants with respect to the wt-SOD1. For this we performed all-atom molecular dynamics (MD) simulations of the wt-SOD1, 6 MBR and 7 WTL SOD1 variant monomers. We focussed on the metal-free SOD1 monomer, as the conformational changes of the metal binding loop and electrostatic loop are unravelled by the lack of metal stabilisation [33]. In order to quantify and characterise the motion of the whole protein and its loops, we studied root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration, number of hydrogen bonds and performed principal component analysis (PCA). Moreover, two additional analyses that focus on the study of individual residues were carried out. In the first we used a graph-theory based approach to study the complexity of the intermolecular connections that were captured in a simple residue-specific descriptor and in the second, we studied the covariance of the residue motions. Our results suggested that the structural and dynamic behaviour differences between WTL and MBR variants could be linked to the ALS clinical phenotype. To explore this hypothesis, we adopted a novel approach of comparing the results of simulations with the survival and age of onset analysis performed on one of the largest clinical dataset worldwide, consisting of 489 SOD1-ALS patients carrying WTL and MBR variants [34]. To the best of our knowledge this is the first MD simulations study focussing on differentiating the dynamic behaviour of the WTL and MBR variants.
2. Results
To characterize the impact of one mutation at a time, we focussed only on SOD1 structures with one single amino acid variant. We could identify 13 WTL and MBR mutations in the Protein Data Bank (PDB) for which high quality structures were available. We divided them in the two categories based on whether they were previously reported to belong to one or whether they were in proximity of another mutation of known category. For example, we classified T2D as WTL as it is in proximity of A4V which was reported to exhibited normal SOD activity in vitro [27]. The WTL group included T2D, A4V, G37R, L38V, T54R, G93R, I113T and the MBR variants were H46R, C57S, H80R, G85R, D124V and D125H. The structural location of the variants is shown in the Fig. 1. The detailed structural information, clinical interpretation, and resolution of the PDB structures used in the study are shown in Table 1.
Fig. 1.

SOD1 monomer structure coloured in grey, the residues which form WTL variants are coloured in blue and the residues which form the MBR variants are coloured in green.
Table 1.
List of PDB structures of the wt-SOD1 and its variants used in the study.
| SOD1 variant | HGVS V2.0 nomenclature | Pathogenicity | PDB ID | Missing residues | Res (Å) | Protein chain | Run time (ns) | Replicates | |
|---|---|---|---|---|---|---|---|---|---|
| 1. | T2D | T3D | Likely pathogenic | 5K02 | - | 1.99 | A | 100 | 3 |
| 2. | A4V | A5V | Pathogenic | 6SPH | - | 2.25 | A | 100 | 3 |
| 3. | G37R | G38R | Pathogenic | 1AZV | - | 1.9 | A | 100 | 3 |
| 4. | L38V | L39V | Pathogenic | 2WYT | - | 1.0 | A | 100 | 3 |
| 5. | H46R | H47R | Pathogenic | 1OEZ | 66–77, 126–141 |
2.15 | A | 100 | 3 |
| 6. | T54R | T55R | Likely pathogenic | 3ECW | - | 2.15 | A | 100 | 3 |
| 7. | C57S | C58S | Likely pathogenic | 4MCM | - | 2.20 | A | 100 | 3 |
| 8. | H80R | H81R | Pathogenic | 3H2Q | 67–78, 134–138 |
1.85 | A | 100 | 3 |
| 9. | G85R | G86R | Pathogenic | 2VR6 | - | 1.30 | A | 100 | 3 |
| 10. | G93A | G94A | Pathogenic | 3GZO | - | 2.10 | A | 100 | 3 |
| 11. | I113T | I114T | Pathogenic | 1UXL | - | 1.60 | A | 100 | 3 |
| 12. | D124V | D125V | Pathogenic | 3H2P | 67–78, 125–139 |
1.55 | A | 100 | 3 |
| 13. | D125H | D126H | Pathogenic | 1P1V | 129–135 | 1.40 | A | 100 | 3 |
| 14. | wt-SOD1 | - | - | 2C9V | - | 1.07 | A | 100 | 3 |
We have used the monomeric apo SOD1 instead of the fully formed SOD1 homodimer, as it is the least stable form of the protein and an ideal model to study the impact of the pathogenic variants [32], [33]. We performed all-atom MD simulations of the wt-SOD1, 6 MBR and 7 WTL variants in TIP3P water for 100 ns each and each simulation was performed in triplicates (see Methods for additional detail).
2.1. RMSF comparison
The RMSF is a measure of individual protein residue flexibility. A high RMSF indicates higher flexibility and a low RMSF indicates lower flexibility. We analysed the RMSF of Cα atoms of the wt-SOD1 and 13 SOD1 variants. Our analysis illustrates that the metal binding and electrostatic loops are the most flexible regions (Fig. 2a, b), while the remaining part of the protein was relatively less flexible (Sup Fig. 1). We observed that the electrostatic loop was the most dynamic part of the SOD1 protein in both WTL, MBR variant groups and the wt-SOD1. Since the metal binding and electrostatic loops were the most flexible regions of the SOD1 protein in the two variant groups, we further assessed these regions to compare the dynamic behaviour of the two variant groups and the wt-SOD1.
Fig. 2.

Boxplots depicting the RMSF distribution of the WTL (green), MBR (blue) variants and wt-SOD1 (coral) in the a) Metal binding loop b) Electrostatic loop. RMSF is calculated for the Cα atoms. The whisker bars represent the range of minimum and maximum RMSF, the median is represented by a line subdividing the box. Wilcoxon rank-sum test, p-value annotation legend: ns: p < = 1.00e+ 00, * : 1.00e-02 < p < = 5.00e-02, * *: 1.00e-03 < p < = 1.00e-02, * ** : 1.00e-04 < p < = 1.00e-03, * ** *: p < = 1.00e-04.
For the metal binding loop (Fig. 2a), overall, the RMSF was higher for the MBR variants in comparison to the WTL variants and the highest flexibility was observed in the H46R variant. Next, we performed the Wilcoxon Rank-Sum test for statistical comparison of the RMSF of each SOD1 variant with wt-SOD1. We noted that the RMSF difference between a variant and the wt-SOD1 was significant for A4V, H46R and D125H. The H46R variant had greater flexibility in the metal binding loop than the wt-SOD1 and A4V variant. For the remaining variants the RMSF difference between the variants and the wt-SOD1 were non-significant.
Focussing on the electrostatic loop, the MBR variants such as C57S, D125H, H46R H80R (Fig. 2b), generally showed higher RMSF with respect to WTL variants, illustrating their increased flexibility. In contrast, WTL variants, such as A4V, G37R, I113T and G93A, showed RMSF values lower or very similar to that of wt-SOD1 (Fig. 2b). The only exception was L38V which had very high RMSF values in the electrostatic loop. The statistical analysis showed that the RMSF difference between A4V, H46R and wt-SOD1 were significant, while for the remaining variants the differences were not significant. The electrostatic loop is very flexible in H46R in comparison to the A4V variant, with a median RMSF difference of 0.3 nm and 0.15 nm respectively. Overall, lower flexibility of the loops in A4V in comparison to the H46R highlights the difference in the dynamic behaviour of the two variants.
Furthermore, the variation in the flexibility of these variants suggests that the increased flexibility of the H46R variant has potentially higher impact on the SOD1 activity as seen in previous studies [31], [32], [33]. In comparison low flexibility observed in A4V variant in comparison to the wt-SOD1 and H46R suggests less disruption of the SOD1 catalytic activity. This strongly supports our hypothesis of gain of function in case of WTL variants and loss of function in the MBR variants. It is further important to note that A4V causes one of the most aggressive forms of SOD1 ALS, whilst H46R results in very slow progression. Their RMSFs corresponded to the lowest and highest values among the investigated variants, suggesting that such dynamics features could be explored to explain the clinical manifestation of variants.
Finally, we tested the difference between the RMSF distributions of the entire protein for all the variants in each class, i.e., WTL and MBR, and the wt-SOD1 (Sup. Fig. 2). The RMSF of the WTL variants was not different from the wt-SOD1 (p = 0.54) while the RMSF of the MBR variants was higher than both the wt-SOD1 (p = 5.2 ×10^−07) and the WTL variants (p = 6.4 ×10^−08).
2.2. Conformational changes of the SOD1 protein
The RMSD is a quantitative measure of the similarity between two superimposed sets of atomic coordinates and is widely used in the MD analysis to highlight the conformational changes observed in the protein structures over the length of simulations. We observed that there are clear differences in the Cα RMSD distribution of the WTL and MBR variants (Fig. 3a and Table 2). The RMSD distributions of the WTL variants were similar to that of the wt-SOD1. The mean and median RMSDs observed in all WTL variants were < = than the mean and median RMSD of the wt-SOD1 (both 0.22 nm), while the mean and median RMSDs observed for all MBR variants but G85R, for which the mean was 0.20 nm and median 0.21 nm, were higher than the wt-SOD1 and WTL variants. The same trend was observed when only the RMSDs between 20 ns and 100 ns were considered to account for the equilibration phase (Sup. Fig. 2).
Fig. 3.

a) Box plots depicting RMSD analysis of the MD simulations performed on the wt-SOD1 (coral), WTL (green) and MBR (blue) variants. The RMSD is calculated for the Cα atoms of the wt-SOD1 and the variants. The whisker bars represent the range of minimum and maximum RMSD, the median is represented by a line subdividing the box. b) Box plots depicting radius of gyration (Rg) comparison of the wt-SOD1 (coral), WTL (green) and MBR (blue) variants simulations. The whisker bars represent the range of minimum and maximum Rg, the median is represented by a line subdividing the box.
Table 2.
The mean and median of the RMSD calculated for the WTL, MBR variants and wt-SOD1.
| SOD1 variant | Mean (nm) | Median (nm) | |
|---|---|---|---|
| 1. | T2D | 0.18 | 0.17 |
| 2. | A4V | 0.20 | 0.20 |
| 3. | G37R | 0.20 | 0.22 |
| 4. | L38V | 0.22 | 0.22 |
| 5. | H46R | 0.63 | 0.64 |
| 6. | T54R | 0.17 | 0.18 |
| 7. | C57S | 0.23 | 0.24 |
| 8. | H80R | 0.30 | 0.30 |
| 9. | G85R | 0.20 | 0.21 |
| 10. | G93A | 0.22 | 0.22 |
| 11. | I113T | 0.16 | 0.17 |
| 12 | D124V | 0.36 | 0.37 |
| 13. | D125H | 0.33 | 0.34 |
| 14. | wt | 0.22 | 0.22 |
Subsequently, we investigated the conformational changes adopted by the variants and the wt-SOD1 during the simulations. We observed that the wt-SOD1 adopted one stable conformation during the simulations while some of the variants adopted multiple conformations (Sup. Fig. 3). However, no clear conformational pattern was observed between the two classes of variants or the wt-SOD1. Among the WTL variants, L38V and T54R also assumed a single conformation. A4V, G93A and I113T acquired two major conformations, whilst T2D and G37R adopted three major conformations. The MBR variants D124V and D125H presented one major conformation for the entire length of the simulation, H46R and H80R adopted two major conformations and, interestingly, C57S and G85R variants showed four major conformations during the trajectory of the simulations (Sup. Fig. 3).
Although there was no definite pattern in the conformational changes of the two variant groups, the range of fluctuations observed in the MBR variant group highlights the instability in the protein structure observed during the simulations. Moreover, the range of RMSD occupied in the different conformations attained in the WTL variants suggests that the protein structure in this group is more stable.
2.3. Radius of gyration analysis
The radius of gyration (Rg) is a measure of the compactness of a protein and is widely utilised to compare the protein’s dynamic behaviour. A high Rg is an indication that the protein structure is more extended, whilst a smaller radius of gyration is indicative of a more compact protein structure. We calculated the Rg of the WTL and MBR variants, and wt-SOD1 over the entire length of the simulations. Our analysis showed that the two variant groups have different Rg patterns. Overall MBR variants have larger Rg then the WTL variants (Fig. 3b). The largest difference with respect to the wt-SOD1 was observed in the H46R and H80R variants. This suggests that the WTL variants adopts a more compact conformation during the simulations in contrast to the more extended and open conformation acquired by the MBR variants (Fig. 3b). This indicates that the MBR variants in general are more dynamic than the WTL variants and is in sync with the behaviour shown through the RMSF analysis.
2.4. Global domain motions of SOD1
Principal Component Analysis (PCA) is a multivariate statistical technique that is frequently used to reduce the number of dimensions that describe the dominant protein domain motion [35], [36], [37]. PCA was performed on the WTL and MBR variants, and wt-SOD1. The first four eigenvectors explained > 50% of all the domain motions. The motion variance explained by PC1–4 for each variant and the wt-SOD1 is shown in Table 3. The structures corresponding to PC1 and PC2 of the T2D, A4V and I113T variants show little to no global domain motion in the entire protein. In G37R, L38V, T54R and G93A variants domain motion is primarily observed in the electrostatic loop of the SOD1 (Fig. 4a, b) and the remaining part of the SOD1 protein showed almost no domain motions. Overall, in the WTL variants no large domain motions were observed which caused huge conformational changes, but only some small fluctuations within the electrostatic loop. The only exception was the L38V variant, in which prominent domain motions were observed both in electrostatic and metal binding loops and an open (PC1) and closed (PC2) conformation was observed (porcupine plots in Fig. 4a,b).
Table 3.
Percentage of variance represented by PC1, PC2, PC3, PC4.
| SOD1 variant | PC1 (%) | PC2 (%) | PC3 (%) | PC4 (%) | |
|---|---|---|---|---|---|
| 1. | T2D | 32.74 | 9.58 | 6.5 | 5.22 |
| 2. | A4V | 28.93 | 19.89 | 9.31 | 6.25 |
| 3. | G37R | 51.56 | 12.36 | 5.74 | 3.24 |
| 4. | L38V | 40.32 | 18.38 | 7.23 | 5.29 |
| 5. | H46R | 36.99 | 25.66 | 6.52 | 4.24 |
| 6. | T54R | 27.44 | 13.15 | 9.17 | 5.71 |
| 7. | C57S | 35.31 | 24.03 | 5.63 | 3.44 |
| 8. | H80R | 29.2 | 13.27 | 9.06 | 5.21 |
| 9. | G85R | 37.39 | 10.61 | 5.84 | 4.49 |
| 10. | G93A | 22.21 | 13.09 | 11.35 | 7.04 |
| 11. | I113T | 33.31 | 20.78 | 8.04 | 5.14 |
| 12. | D124V | 30.17 | 20.63 | 5.46 | 4.97 |
| 13. | D125H | 30.88 | 15.22 | 9.25 | 5.71 |
| 14. | wt-SOD1 | 27.45 | 21.57 | 10.59 | 4.05 |
Fig. 4.

PCA analysis of the concatenated trajectories. Porcupine plots corresponding to the a) PC1 b) PC2. The wt-SOD1 and variant protein backbone is coloured in grey, the metal binding loop in blue and electrostatic loop in red. The vectors coloured in green represent the direction and magnitude of the domain motion. WTL variants are highlighted in green and MBR variants are highlighted in blue.
In the MBR SOD1 variants, considerable domain motions were observed in the metal binding loop in addition to the electrostatic loop. Moreover, the magnitude of the domain motion in the metal binding loop and electrostatic loop regions was substantially larger for the MBR variants than the WTL variants. The most pronounced domain motions were observed in H46R, H80R, D124V variants. In the H46R variant the PC1 (36.99% variance) shows the metal binding loops are extended forming an open conformation, whilst in the PC2 (25.66% variance) the two loops are present in a closed conformation. Similar open and closed conformations were observed in the H80R (PC1:29.2%, PC2:13.27%) and D124V (PC1:30.17%, PC2:20.63%) variants. In the D125H variant PC1 (30.88%) reflected a closed conformation and PC2 (15.22%) is an open conformation.
The global domain motion behaviour of WTL variants was very similar to the wt-SOD1 in which the motions were observed primarily in the electrostatic loop. The domain motions of the MBR variants were disparate from the wt-SOD1, with a distinct open and closed conformational formation in most of the MBR variants.
2.5. Hydrogen bonds analysis
To further investigate the dynamics of the wt-SOD1 and its variants we analysed the total number of hydrogen bonds formed during the simulations. The average numbers of hydrogen bonds formed in the entire protein for the wt-SOD1, WTL and MBR variants are shown in Fig. 5 (full distributions in Sup. Fig. 4). Overall, there was no drastic difference between the average number of hydrogen bonds in the two variant groups. The D125H variant had the lowest number of hydrogen bonds formation. Interestingly, A4V and H46R variants, which commonly represented the two extreme opposites in the other analyses, had a similar number of hydrogen bonds as the wt-SOD1, while all other variants consistently presented a lower mean number with respect to the wt-SOD1.
Fig. 5.

Mean number of hydrogen bonds formed in the wt-SOD1 (coral), WTL (green) and MBR (blue) variants during the entire length of the simulation.
Next, we investigated the hydrogen bonds formation within the metal binding and electrostatic loops. We observed that there was not a drastic difference in the number of hydrogen bonds formed in the two variant groups, however, A4V had the largest number of hydrogen bonds formed in the metal binding loop (Sup. Fig. 5). In contrast H46R had the lowest number of hydrogen bonds in the metal binding loop. In the electrostatic loop there was more variation in the two variant groups. Overall, the MBR variant group had lower hydrogen bond formation in comparison to the WTL group. Interestingly, A4V had the highest number of hydrogen bonds formed, whilst H46R, H48R and D125H were on the lower end of the spectrum.
Although the hydrogen bonds formed in the entire protein are not too dissimilar in the two variant groups, the difference in the electrostatic loop sheds more light into the difference in the dynamic behaviour of the two variant groups. The loss in the hydrogen bond interactions in the electrostatic loop of the MBR variants explains the increased flexibility caused by the loss of stabilising interactions and explains the more dynamic nature of the MBR variants and previously seen in the RMSF and Rg analysis.
2.6. Graph-Theory based analysis
To investigate the complex nature of the intramolecular interactions of the simulated systems, we used graph-theory based descriptors to evaluate the role of each residue within their interaction network. To this end, we used a weighted network representation, where each residue of the protein was a node of the network. Two nodes were connected by a link if the distance between their side chain centroids was lower than a threshold. In addition, each link was weighted by the contact frequency as calculated from the MD simulations trajectory.
Based on this representation of the protein, we used the closeness centrality descriptor to capture the connection between residues even when they are spatially distant. The closeness centrality was defined as the reciprocal of the sum of the length of the shortest paths between a given node and all other nodes in the graph. The higher the closeness of a residue, the higher its centrality is in the protein network.
First, we compared the distributions of residue closeness centrality values analysing wt-SOD1 and its 13 variant systems. We studied separately the residues belonging to the metal binding loop and electrostatic loop. In Fig. 6a, we reported the boxplots for each system, where the boxplots represent the values obtained considering all three simulation replicas.
Fig. 6.

a) Box plots depicting Closeness centrality comparison of the wt-SOD1 and the variants. For each variant the boxplot on the left (respectively on the right) is obtained considering only the residues belonging to the metal binding loop (respectively to the electrostatic loop). The dotted line represents the mean Closeness centrality of the wt-SOD1 residues. b) Hierarchical clustering of the wt-SOD1 and its variants obtained considering the Closeness centrality indexes of the residues belonging to the MBL. c) Same as in c) but for the ESL residues. d) Mean difference (in percentage) between the closeness centrality indexes of the residues of the variants with respect to the wild-type ones for the metal binding loop. e) Same as in d) but for the electrostatic loop.
Overall, the residue closeness centrality values of MBR variants were > = than the wt-SOD1 while WTL variants were < = than the wt-SOD1. H46R and D125H showed a large difference with respect to the wt-SOD1 system considering both metal binding loop and electrostatic loop regions (Fig. 6a).
To further compare the wt-SOD1 with the variant systems, we performed a hierarchical clustering analysis (Fig. 6b-c), which allowed us to compare each system with all the others. Fig. 6b-c shows the clustering of the centrality values of their metal binding (b) and electrostatic (c) loop residues. The loops of H46R and D125H displayed the most singular behaviours since they formed a group separated from all the others. In general, the clusters of all other proteins, including the wt-SOD1, were very similar to each other, meaning that their global structure was overall maintained and highlighting the subtlety of the underlying pathogenic mechanisms.
Lastly, we investigated the contribution of each residue to the observed differences in the centrality values between variants and wt-SOD1. We calculated the mean percentage difference between the closeness centrality indexes of the residues, as calculated from the wt-SOD1 and the variants. The higher this value, the larger is the mismatch between the residues in the two classes of systems. We reported the results of this analysis in Fig. 6d-e for metal binding loop and electrostatic loop regions respectively. The red bar represents residues that underwent a mutation in one of the simulated systems. Remarkably, the most marked differences did not regard mutated residues, testifying the complexity of the interactions occurring in the protein as a result of the pathogenic mechanisms.
2.7. Analysis of the motion of the electrostatic and metal binding loops
To study the difference between the dynamics of wt-SOD1 and its variants, we analysed the correlation between the covariance of the residue motion registered in such systems. For each MD simulation we built a covariance matrix calculating the covariance of the motion between each couple of residues. Hence, for each variant we computed the Pearson correlation coefficients between the covariance values regarding the wt and the three replicates of the variant systems. We reported in Fig. 7a the mean and the standard error of the means of such correlations, where a high value means that the correlated motion between the wt and the variant systems was similar. WTL variants presented high and consistent correlations with the wt-SOD1 behaviour, while the means of the MBR variants greatly varied.
Fig. 7.

a) Mean correlation between the covariance matrices of the atomic coordinates of the wt-SOD1 and the variants. Bars represent the Standard Error of the Mean. Hierarchical clustering of the wt-SOD1 and variants obtained considering the covariances of the atomic coordinates of all the residues belonging to the b) MBL. c) ESL. d) covariances between the residues of the MBL and ESL.
In Fig. 7b-d we showed the results of the hierarchical clustering using the covariance values as descriptors. The clustering based on MBL and ESL residues (Fig. 7b-c), but not the one based on all residues (Fig. 7d) tended to cluster MBR variants away from the wt-SOD1 and WTL variants.
2.8. Phenotype analysis
In order to investigate whether WTL and MBR variants associate with different clinical outcomes, we collected clinical data of patients carrying the variants investigated in this study. We were able to retrieve clinical data of 489 ALS patients with A4V, G37R, L38V, G93R, I113T, H46R, H80R, G85R and D125H (see details of individual variants in Table 4) [34]. Cox proportional hazard analysis (Fig. 8) showed that patients with MBR variants presented a longer survival time (approximately 6 years median difference, p < 0.001) than patients with WTL variants, while no significant difference (p = 0.19) was observed for the age of onset (details in Table 4). Because A4V and I113T represented the great majority of the SOD1 ALS patients in our sample (312 and 120 respectively), we repeated the survival analysis excluding these two variants from the WTL group. This analysis confirmed a significant difference between WTL variants (without A4V and I113T) and MBR variants (approx. 2.5 years median difference, p = 0.006).
Table 4.
Characteristics of the clinical dataset.
| SOD1 variant | HGVS V2.0 nomenclature | Total sample size | Patients with disease duration | Number censored | Mean disease duration (months) | Median disease duration (months) | Patients with age of onset | Mean age of onset (years) | Median age of onset (years) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | A4V | A5V | 312 | 260 | 7 | 15.75 | 13 | 298 | 48.61 | 49.46 |
| 2. | D125H | D126H | 3 | 1 | 0 | 8.08 | 8.08 | 3 | 50 | 52 |
| 3. | G37R | G38R | 10 | 6 | 0 | 182.33 | 186 | 10 | 35.55 | 33.98 |
| 4. | G85R | G86R | 18 | 17 | 1 | 42.24 | 30 | 17 | 60 | 60 |
| 5. | G93R | G94R | 1 | 1 | 0 | 55 | 55 | 1 | 34 | 34 |
| 6. | H46R | H47R | 19 | 18 | 12 | 318.65 | 277 | 19 | 45.16 | 43 |
| 7. | H80R | H81R | 1 | 1 | 0 | 18 | 18 | 1 | 24 | 24 |
| 8. | I113T | I114T | 120 | 86 | 15 | 94.36 | 62 | 108 | 53.81 | 53 |
| 9. | L38R | L39R | 5 | 4 | 0 | 19.66 | 23.5 | 5 | 44.09 | 43 |
| 10. | All WTL | All WTL | 448 | 357 | 22 | 37.32 | 14 | 422 | 49.54 | 50 |
| 11. | All WTL (no A4V and I113T) | All WTL (no A5V and I114T) | 16 | 11 | 0 | 111.6 | 55 | 16 | 38.12 | 38.33 |
| 12. | All MBR | All MBR | 41 | 37 | 13 | 176.65 | 84.17 | 40 | 51.3 | 50 |
Fig. 8.

Age at onset (a) and survival (b) curves from the cox proportional hazard analysis (all WTL variants VS all MBR variants).
3. Discussion
In this study we investigated the differences in the structural and dynamic behaviours of the wt-SOD1 and sets of its WTL and MBR variants associated with ALS. To the best of our knowledge there are no previous computational studies focussing on the differentiation of these two variant groups. Furthermore, it is the first study illustrating the difference in the survival of ALS patients belonging to the two SOD1 variant groups. The novelty of our research is using the insights from the MD simulations and combining it with clinical data from ALS patients. Our results might support new strategies for the stratification of patients in clinical trials for SOD1-ALS based on the type of SOD1 variant participants carry. This is highly timely given the recent developments in antisense drugs targeting SOD1-ALS [38].
Our results showed the disparate nature of dynamic properties of WTL and MBR variants. We showed that the MBR variants are more flexible and dynamic in the metal binding and electrostatic loops, in contrast to the WTL variants. In addition, the electrostatic loop was found to be the most flexible region of the protein in all the SOD1 variants we studied. This is very interesting as it has been shown previously that an enervating electrostatic loop can lead to the gain of interaction, thereby increasing the formation of SOD1 amyloid fibrils [16], [39], [40]. Destabilization of the electrostatic loop due to disease causing mutations has long been linked to ALS pathogenesis [41], [42]. Akin to these previous studies, we also observed a substantial flexibility in the electrostatic loop in wt-SOD1, WTL and MBR variants. This high flexibility of the electrostatic loop leads to the formation of rather extended or open conformations in the MBR variants. This is further supported by the higher Rg observed in the MBR variants.
Disease-causing mutations in the SOD1 protein can lead to a range of misfolded states [42] which are challenging to sample computationally. The PCA performed on the trajectories of the simulations showed that the conformational changes observed in the WTL and MBR variants were led by the domain motions in the electrostatic loop and metal binding loop regions. PCA also highlighted a higher magnitude of the domain motion for MBR variants that was particularly evident in the metal binding and electrostatic loops. These larger domain motions in the electrostatic loop of the MBR variants resulted in the formation of an open and closed conformation. Whilst in the WTL variants the domain motions in the electrostatic loop were much smaller and the WTL variants mostly adopted rather closed conformations. Collectively, the motion and flexibility of the electrostatic loop was the prominent dynamic feature observed in most of the SOD1 variants we studied.
The hydrogen bond analysis highlighted differences in the behaviour of the wt-SOD1, WTL and MBR variants at an atomic level. Previous studies have pointed out that the pathogenic mutations in SOD1 might alter the protein hydrogen bonds and promote the formation of new ones which can increase the probability of misfolding and aggregation in SOD1 [43], [44]. Our results complement these by showing that apart from A4V, SOD1 variants generally form fewer hydrogen bonds which might affect the protein stability and favour misfolding. Our hydrogen bond analysis also sheds light into the increased dynamic nature of the metal binding and electrostatic loops. Overall, fewer hydrogen bonds were formed in the electrostatic loop of most of the MBR variants in comparison to the WTL variants. The loss of these stabilising interactions results in an increased flexibility in the electrostatic loop of the MBR variants. Moreover, A4V had the highest number of the hydrogen bonds in the metal binding and electrostatic loops in comparison to the other variants and wt-SOD1. These hydrogen bonds most likely increase the stability of the two loops, therefore making them less dynamic. In comparison the H46R variant had substantially less hydrogen bonds in the metal binding and electrostatic loops and the highest flexibility in these regions.
We also showed that the dynamic behaviour of the L38V variant was unexpected. Although prior studies have grouped L38V as a WTL variant [27] and L38V and wt-SOD1 have very similar crystal structures (RMSD = 0.074 Å), the difference between their dynamic behaviours was highly noticeable. In our study we observed that the increased flexibility and dominant motion of the electrostatic loop of L38V were similar to the MBR variants. This might be due to the disruption of the β-barrel plug [27]. Interestingly, L38V has been associated with early onset of ALS (mean age of onset = 38 years) [45].
Our study highlighted that H46R had the most flexible metal binding and electrostatic loops. In addition, the protein structure was the least compact in comparison to the other variants and wt-SOD1. Furthermore, we also showed that there was a significant difference in the flexibility of the metal binding loop and electrostatic loop in A4V and H46R variants with respect to the wt-SOD1. These two variants represented the two flexibility extremes across the variants that we studied. In addition, A4V remains in a somewhat closed conformation for most of the simulation, in comparison the H46R adopts an open conformation from an initial closed conformation. Interestingly, these two variants are known to represent extremes of the phenotypic spectrum of SOD1 ALS. A4V variant causes the fast progression of ALS, with a survival time of 2 years [19], [20], [21] whilst H46R causes a slow progressing form of ALS, with a mean survival of ∼12 years [22], [23], [24], [25].
Driven by the consistent differences between WTL and MBR variants highlighted in our analyses, and by the recurrent positioning of A4V and H46R at the far ends of such differences, mirroring the extreme clinical outcomes these two variants are associated with (fast and slow progressing forms of ALS), we attempted to investigate if WTL and MBR variants led to different clinical outcomes. Using a large clinical dataset (almost 500 SOD1-ALS patients) we were able to show that the survival of patients with a WTL variant was substantially shorter than the survival of patients with an MBR variant (median difference approximately 6 years, p < 0.001).
Experimental data suggests that ALS arises from a toxic gain-of-function mechanism and loss of SOD1 function alone has been linked to a severe phenotype distinct from ALS [46], [47], [48]. However, loss of function of SOD1 in ALS has been proposed as a potential modifier. A key difference between WTL and MBR variants is their effect on the SOD1 function. MBR variants, but not WTL variants, cause reduction of SOD1 enzymatic function in in vitro experiments of the isolated and engineered SOD1. On such a basis, it follows that our results support the hypothesis of decoupling between the role of gain and loss of SOD1 function in ALS affecting disease onset and duration respectively and potentially independently.
Identifying and understanding which mechanisms contribute to the development of the disease and which to its progression can improve genetic counselling, the development of new therapies and the design of more effective trials. This is particularly timely for SOD1 given the current clinical trials that are testing the efficacy of treatments based on SOD1 antisense oligonucleotides [49]. The great difference in the survival time between the patients carrying WTL and MBR variants, and the structural features that we found associated with the two classes, could be used to improve the classification into fast and slow progression of the patients that take part to trials, and to generate better estimates of their expected survival. A correct estimation of the expected survival is essential for the design of trials and for the interpretation of its results, as potential beneficial effects on slow-progressing variants would require lengthier trials to prove them.
A potential limitation of the design and interpretation of our study is the definition of the two classes of variants. The distinction between WTL and MBR variants was based on whether or not the variants were expected to affect the catalytic activity of SOD1. However, this effect was based on in vitro studies of isolated and engineered proteins. An analysis of SOD1 activity in blood samples from ALS patients showed that SOD1 activity was approximately halved in patients carrying a wide range of variants including some of the variants we classified as WTL [50]. Although these results are apparently in contrast with the in vitro studies we have based the classification of WTL and MBR variants on, a possible explanation could be that the loss of SOD1 function observed in the blood of patients is not an intrinsic effect of the variants, but the result of a secondary effect linked to other disease pathogenic mechanisms, e.g. SOD1 aggregation. Another possibility is that both WTL and MBR variants affect SOD1 function but with different effect sizes. Although the differences between WTL and MBR variants identified by our MD and clinical analyses are consistent across experiments, another limitation of our study is the limited number of variants studied. The 13 variants used in this work represent a small proportion of the over 200 SOD1 mutations discovered in patients. Our choice to use only high-quality structures of SOD1 mutants solved in situ, was based on the anticipated complexity of the concurrent pathogenic mechanisms underlying SOD1 ALS (gain and loss of functions), that we expected to be poorly modelled within the timeframe of our simulations if the wt-SOD1 protein were used as backbone for an in silico mutagenesis experiment.
4. Conclusions
In this study we compared the dynamic behaviour of the SOD1 variants which are associated with ALS. The variants were classified into two groups based on the structural location, and whether or not they affect metal binding affinity and catalytic activity of SOD1. The MD simulations highlighted that the electrostatic loop was the most flexible part of the SOD1 protein in both the WTL and MBR variant groups. Furthermore, the electrostatic loop was considerably more flexible in the MBR variants. This increased flexibility of the electrostatic loop can be attributed to the loss of stabilising hydrogen bond interactions in the MBR variants. The MBR variants in turn adopt an open and closed conformation due to the high flexibility of the electrostatic loop. Finally, the phenotype analysis of one of the largest clinical datasets worldwide consisting of 493 SOD1-ALS patients further illustrated that the patients with the MBR variants survive approximately 6 years longer than the patients with WTL variants. This difference in the survival of the two variant groups suggests that the effect on SOD1 catalytic activity of the variants might be linked to disease progression in ALS. In summary, our study highlights key differences in the dynamics of the WTL and MBR SOD1 variants, and wt-SOD1. It sheds light into the behaviour of SOD1 variants at an atomic and a molecular level, suggesting interesting structural features that could be investigated to explain their associated phenotypic variability, and it supports the role of loss of function of SOD1 as a modifier of disease progression in ALS.
5. Methods
5.1. Protein structures
The protein structures of wt-SOD1 and 13 variants were obtained from Protein Data Bank (PDB) [51]; the detailed structural information is shown in Table 1. SOD1 variant structures were carefully selected; only those variants which had a single point mutation were included. An in-house python script Mutatpipe.py was utilised to select and filter the variants (https://github.com/Utilon/MutaPipe_Repo). For the variants which had more than one crystal structure, the protein structure with the highest resolution and without missing residues was selected. SOD1 exists in a homodimer form in nature, however, for the scope of this study we focused only on the SOD1 monomer; the additional chains and the copper/zinc ions present in the PDB structures were manually removed. The missing residues in 1OEZ, 3H2Q, 3H2P and 1P1V monomer structures were modelled in with Modeller 10.2 program [52].
5.2. Variants nomenclature
The old nomenclature was used to name SOD1 variants as this reflects the actual position of the amino acids in the chains of the SOD1 structures we have used in our study. The new Human Genome Variation Society (HGVS) v2 nomenclature numbers the amino acids according to the mRNA reference sequence (GenBank: NM_000454.4) and it includes the start methionine which is cleaved post-translationally. Table 1 reports both nomenclatures for each variant.
5.3. MD simulations protocol
We employed MD package GROMACS (version 2020.1) [53], [54], [55] to perform all atom MD simulations of wt-SOD1 protein, WTL and MBR variants. All the MD simulations were performed on the GPUs at Rosalind (https://rosalind.kcl.ac.uk), the high-performance computing (HPC) facility at King’s College London. AMBER99SB-ILDN force field was employed for all the MD simulations performed in the study [56]. The structure was solvated in the TIP3P water model in a rhombic dodecahedron box; the protein was placed at least 1.4 nm from the edges in all directions. Na+ and Cl- ions were then added to neutralise the system and then the system was energy minimised over 50000 steps using the steepest descent method. The whole system consisted of ∼ 30237 atoms; the number slightly varied in different variants.
After energy minimising the system the solvent and ions around the protein were equilibrated in two steps. The first step was carried out under the NVT ensemble (constant number of particles, volume and temperature) for a length of 100 ps, at 300 K, using the Berendsen thermostat. The second step was carried under NPT ensemble (constant number of particles, pressure and temperature), using the Parrinello-Rahman barostat [57] for a length of 100 ps at 1 atm and 300 K.
The final production was performed in triplicates for a length of 100 ns at 300 K for the 13 variants. The covalent bonds were constrained using Linear constraint solver (LINCS) algorithm [58] and Particle Mesh Ewald (PME) algorithm was used to calculate long range interactions [59]. Leapfrog integrator was used to calculate the equation of motion with a timestep of 2 fs [59]. The detailed information about the protein structures and total number of MD simulations performed in this study are summarised in Table 1.
5.4. MD trajectory analysis
The visual analysis was performed using PyMOL [60] and Visual Molecular Dynamics (VMD) program [61]. We calculated the Cα RMSF, Cα RMSD, radius of gyration and hydrogen bonds using the GROMACS modules; gmx rmsf, gmx rms, gmx gyrate, gmx hbond.
5.5. Principal component analysis
PCA is a highly used technique to reduce the dimensionality of the data obtained from MD simulations and extract the dominant motion in the proteins [35], [36]. For PCA, the trajectories from three replicates were concatenated into one trajectory and the protein backbone was selected for analysis. gmx covar was then used to calculate and diagonalize the mass weighted covariance matrix. gmx anaeig was used to analyse the eigenvectors. The structures corresponding to PC1 and PC2 were extracted. To make the porcupine plots the structures corresponding to PC1 and PC2 were loaded to PyMOL. The vectors representing domain motion and direction were constructed after uploading the python script Modevectors.py on PyMOL (https://pymolwiki.org/index.php/Modevectors).
5.6. Statistical analysis
All the statistical analyses in the study were performed using SciPy.Stats computing library in Python [62]. Wilcoxon Rank-sum Test was employed to calculate the differences in the statistical significance of the RMSF between the variants and the wt-SOD1. Age of onset and survival time from disease onset analyses were performed using Cox proportional hazards regression. Models were adjusted for site of onset and sex. The coxph() function was utilised with tie resolution at the default setting. When modelling survival time from onset, age of onset was also included as a covariate. For all statistical analyses, p < 0.05 was considered significant.
5.7. Graph theory analysis
We modelled each protein structure as a network, schematizing each residue as a node of the network and each intramolecular interaction as a network edge [63], [64]. In particular, we defined a weighted graph for each system, weighing each link connecting two residues with the corresponding contact frequency calculated from the molecular dynamics, defining, in this case, contact between two residues if their distance is less than 8.5 °A, similar to previously described procedures [65].
5.8. Covariance analysis
Covariance matrices of the atomic positions of all the atoms of the metal binding loop and electrostatic loop were computed from molecular dynamics trajectories. Covariance between couples of residues were then obtained averaging the covariances of all the couples of atoms belonging to the considered residue couple.
5.9. Clinical dataset
Data on people with SOD1-ALS were collected from the ALS Online Database (https://alsod.ac.uk) [7], [8], the Project MinE whole-genome sequencing dataset [66], and a number of centres. The corresponding authors of the AlSoD database entries with missing data were contacted to fill the gaps. Anonymised records of people with SOD1-ALS were obtained from the following centres: Macquarie University, ANZAC Research Institute, University of Massachusetts, University Hospitals of Montpellier, King’s College London, Washington University School of Medicine in St Louis, Peking University Third Hospital, Università degli Studi di Siena, Northwestern Medicine – Feinberg School of Medicine, Istituto Auxologico Italiano IRCCS-University of Milan, University of Belgrade and University of Calabria. Individuals were eligible to be included if their diagnosis of ALS was made by a consultant neurologist, or their data was reported in the literature with an ALS diagnosis. The clinical and demographic features utilised in this study were country of origin, sex at birth, age of onset (in years) of first motor symptoms, site of onset, survival time (in months) defined as time from diagnosis to death or latest visit. In each analysis, individuals with missing data among the subset of clinical features required were discarded. Individuals with the same SOD1 variant, country of origin, sex, age and site of onset were considered to be duplicates.
Funding
The authors are supported by South London and Maudsley NHS Foundation Trust; Wellcome Trust; MND Scotland; Motor Neurone Disease Association; National Institute for Health Research; Darby Rimmer MND Foundation; Spastic Paraplegia Foundation; Rosetrees Trust; Alzheimer’s Research UK; Italian Ministry of Health and Medical Research Council (MRC). M.M. and G.R. acknowledge support from European Research Council Synergy grant ASTRA (n. 855923). G.R acknowledges support from European Innovation Council through its Pathfinder Open Programme, project ivBM-4PAP (grant agreement No 101098989).
CRediT authorship contribution statement
Munishikha Kalia: Conceptualization, Methodology, Investigation, Writing, Visualization. Mattia Miotto: Methodology, Investigation, Writing, Review. Deborah Ness: Investigation, Writing, Review. Sarah Opie-Martin: Review, Writing, Resources. Thomas P Spargo: Writing, Methodology, Review. Lorenzo Di Rienzo: Writing, Methodology, Review. Tommaso Biagini: Writing, Methodology, Review. Francesco Petrizzelli: Methodology Ahmad Al Khleifat: Data acquisition, Review, Resources. Renata Kabiljo: Investigation, Writing, Review. Tommaso Mazza: Investigation, Review, Resources. Giancarlo Ruocco: Methodology, Review, Resources. Edoardo Milanetti: Writing, Review, Methodology. Richard JB Dobson: Writing, Review, Editing, Ammar Al-Chalabi: Data acquisition, Resources, Review, Editing. Alfredo Iacoangeli: Conceptualization, Writing, Review, Editing, Funding Acquisition. All the authors approved the final version of the manuscript and have agreed to be accountable for all aspects of the work related to the accuracy and integrity of the work.
Declaration of Competing Interest
None.
Acknowledgements
We would like to thank Simon Topp, Keith Mayl, Isabella Fogh, Puja R Mehta, Kelly L Williams, Jennifer Jockel-Balsarotti, Taha Bali, Wade Self, Lyndal Henden, Garth A Nicholson, Nicola Ticozzi, Diane McKenna-Yasek, Lu Tang, Pamela Shaw, Adriano Chio, Albert Ludolph, Jochen H Weishaupt, John E Landers, Jonathan D Glass, Jesus S Mora, Wim Robberecht, Philip Van Damme, Russell McLaughlin, Orla Hardiman, Leonard H van den Berg, Jan H Veldink, Phillippe Corcia, Zorica Stevic, Nailah Siddique, Antonia Ratti, Vincenzo Silani, Ian P Blair, Dong-sheng Fan, Florence Esselin, Elisa de la Cruz, William Camu, A Nazli Basak, Teepu Siddique, Timothy Miller, Robert H Brown, Peter M Andersen and Christopher E Shaw for providing and collecting clinical and genetic data of people living with SOD1-ALS. We acknowledge use of the research computing facility at King's College London, Rosalind (https://rosalind.kcl.ac.uk), which is delivered in partnership with the National Institute for Health Research (NIHR) Biomedical Research Centres at South London & Maudsley and Guy's & St. Thomas’ NHS Foundation Trusts and part-funded by capital equipment grants from the Maudsley Charity (award 980) and Guy's and St Thomas' Charity (TR130505). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, King's College London, or the Department of Health and Social Care. We would like to thank people with ALS and their families for their participation in this project. We thank the Project MinE Sequencing Consortium members for their collaboration and support.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.09.016.
Appendix 1
Project MinE ALS Sequencing Consortium group authors
Philip van Damme, Philippe Corcia, Philippe Couratier, Orla Hardiman, Russell McLaughlin, Marc Gotkine, Vivian Drory, Vincenzo Silani, Nicola Ticozzi, Jan H Veldink, Leonard H van den Berg, Mamede Carvalho, Susana Pinto, Jesus Mora Pardina, Monica Povedano, Peter M Andersen, Markus Weber, Nazli Başak, Ammar Al-Chalabi, Chris Shaw, Pamela Shaw, Jonathan Cooper-Knock, Alfredo Iacoangeli, Karen Morrison, John Landers, Jonathan Glass, Patrick Vourc'h.
SOD1-ALS clinical and genetic data collection group authors
Simon Topp, Keith Mayl, Isabella Fogh, Puja R Mehta, Kelly L Williams, Jennifer Jockel-Balsarotti, Taha Bali, Wade Self, Lyndal Henden, Garth A Nicholson, Nicola Ticozzi, Diane McKenna-Yasek, Lu Tang, Pamela Shaw, Adriano Chio, Albert Ludolph, Jochen H Weishaupt, John E Landers, Jonathan D Glass, Jesus S Mora, Wim Robberecht, Philip Van Damme, Russell McLaughlin, Orla Hardiman, Leonard H van den Berg, Jan H Veldink, Phillippe Corcia, Zorica Stevic, Nailah Siddique, Antonia Ratti, Vincenzo Silani, Ian P Blair, Dong-sheng Fan, Florence Esselin, Elisa de la Cruz, William Camu, A Nazli Basak, Teepu Siddique, Timothy Miller, Robert H Brown, Peter M Andersen and Christopher E Shaw.
Appendix A. Supplementary material
Supplementary material
References
- 1.Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(11):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 2.Boillée S., Velde C.V., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Perrone B., Conforti F.L. Common mutations of interest in the diagnosis of amyotrophic lateral sclerosis: how common are common mutations in ALS genes? Expert Rev. Mol. Diagn. 2020;20(7):703–714. doi: 10.1080/14737159.2020.1779060. [DOI] [PubMed] [Google Scholar]
- 4.Antonyuk S., et al. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci. 2005;14(5):1201–1213. doi: 10.1110/ps.041256705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stathopulos P.B., et al. Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J Biol Chem. 2006;281(10):6184–6193. doi: 10.1074/jbc.M509496200. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa Y., et al. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci. 2006;103(18):7148–7153. doi: 10.1073/pnas.0602048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abel O., et al. ALSoD: a user‐friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33(9):1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
- 8.Wroe R., et al. ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Lateral Scler. 2008;9(4):249–250. doi: 10.1080/17482960802146106. [DOI] [PubMed] [Google Scholar]
- 9.Wright G.S., Antonyuk S.V., Hasnain S.S. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q Rev Biophys. 2019;52 doi: 10.1017/S003358351900012X. [DOI] [PubMed] [Google Scholar]
- 10.Huai J., Zhang Z. Structural properties and interaction partners of familial ALS-associated SOD1 mutants. Front Neurol. 2019;10:527. doi: 10.3389/fneur.2019.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa Y., et al. Conformational disorder of the most immature Cu, Zn-superoxide dismutase leading to amyotrophic lateral sclerosis. J Biol Chem. 2016;291(8):4144–4155. doi: 10.1074/jbc.M115.683763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothstein J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(S1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 13.Banci L., et al. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc Natl Acad Sci. 2007;104(27):11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oztug Durer Z.A., et al. Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PloS One. 2009;4(3) doi: 10.1371/journal.pone.0005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray S.S., et al. An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43(17):4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 16.Healy E.F. A prion-like mechanism for the propagated misfolding of SOD1 from in silico modeling of solvated near-native conformers. PloS One. 2017;12(5) doi: 10.1371/journal.pone.0177284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grad L.I., et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci. 2011;108(39):16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grad L.I., et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and-independent mechanisms. Proc Natl Acad Sci. 2014;111(9):3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saeed M., et al. Age and founder effect of SOD1 A4V mutation causing ALS. Neurology. 2009;72(19):1634–1639. doi: 10.1212/01.wnl.0000343509.76828.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6(1):37–46. doi: 10.1007/s11910-996-0008-9. [DOI] [PubMed] [Google Scholar]
- 21.Hays A.P., et al. Sporadic amyotrophic lateral sclerosis and breast cancer: hyaline conglomerate inclusions lead to identification of SOD1 mutation. J Neurol Sci. 2006;242(1–2):67–69. doi: 10.1016/j.jns.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Zou Z.-Y., et al. H46R SOD1 mutation is consistently associated with a relatively benign form of amyotrophic lateral sclerosis with slow progression. Amyotroph Lateral Scler Front Degener. 2016;17(7–8):610–613. doi: 10.1080/21678421.2016.1199698. [DOI] [PubMed] [Google Scholar]
- 23.Holmøy T., Bjørgo K., Roos P.M. Slowly progressing amyotrophic lateral sclerosis caused by H46R SOD1 mutation. Eur Neurol. 2007;58(1):57. doi: 10.1159/000102170. [DOI] [PubMed] [Google Scholar]
- 24.Arisato T., et al. Clinical and pathological studies of familial amyotrophic lateral sclerosis (FALS) with SOD1 H46R mutation in large Japanese families. Acta Neuropathol. 2003;106(6):561–568. doi: 10.1007/s00401-003-0763-5. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki N., et al. Genetics of amyotrophic lateral sclerosis: seeking therapeutic targets in the era of gene therapy. J Hum Genet. 2022:1–22. doi: 10.1038/s10038-022-01055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valentine J.S., Doucette P.A., Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 27.Hayward L.J., et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis* 210. J Biol Chem. 2002;277(18):15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 28.Valentine J.S., Hart P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci. 2003;100(7):3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardo C.A., et al. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci. 1995;92(4):954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakavayev S., et al. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H2O2 via cysteine-dependent redox short-circuit. Sci Rep. 2019;9(1):1–13. doi: 10.1038/s41598-019-47326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hörnberg A., et al. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J Mol Biol. 2007;365(2):333–342. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 32.Vassall K.A., et al. Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc Natl Acad Sci. 2011;108(6):2210–2215. doi: 10.1073/pnas.0913021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sekhar A., et al. Probing the free energy landscapes of ALS disease mutants of SOD1 by NMR spectroscopy. Proc Natl Acad Sci. 2016;113(45):E6939–E6945. doi: 10.1073/pnas.1611418113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opie-Martin S., et al. The SOD1-mediated ALS phenotype shows a decoupling between age of symptom onset and disease duration. Nat Commun. 2022;13(1):1–9. doi: 10.1038/s41467-022-34620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amadei A., et al. An efficient method for sampling the essential subspace of proteins. J Biomol Struct Dyn. 1996;13(4):615–625. doi: 10.1080/07391102.1996.10508874. [DOI] [PubMed] [Google Scholar]
- 36.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Protein: Struct, Funct, Bioinforma. 1993;17(4):412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi H., et al. Essential dynamics of DNA containing a cis. syn cyclobutane thymine dimer lesion. Nucleic Acids Res. 1998;26(8):1939–1946. doi: 10.1093/nar/26.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw P., et al. BMJ Publishing Group Ltd; 2022. Tofersen in adults with SOD1-ALS: Phase 3 VALOR trial and open-label extension results. [Google Scholar]
- 39.Healy E.F. A mechanism for propagated SOD1 misfolding from frustration analysis of a G85R mutant protein assembly. Biochem Biophys Res Commun. 2016;478(4):1634–1639. doi: 10.1016/j.bbrc.2016.08.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souza P.C., et al. An allosteric pathway in copper, zinc superoxide dismutase unravels the molecular mechanism of the G93A amyotrophic lateral sclerosis-linked mutation. J Phys Chem Lett. 2019;10(24):7740–7744. doi: 10.1021/acs.jpclett.9b02868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molnar K.S., et al. A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the copper/zinc superoxide dismutase electrostatic loop. J Biol Chem. 2009;284(45):30965–30973. doi: 10.1074/jbc.M109.023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prudencio M., Borchelt D.R. Superoxide dismutase 1 encoding mutations linked to ALS adopts a spectrum of misfolded states. Mol Neurodegener. 2011;6(1):1–19. doi: 10.1186/1750-1326-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alemasov N.A., et al. Molecular mechanisms underlying the impact of mutations in SOD1 on its conformational properties associated with amyotrophic lateral sclerosis as revealed with molecular modelling. BMC Struct Biol. 2018;18(1):1–14. doi: 10.1186/s12900-018-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alemasov N.A., et al. Computer analysis of the relation between hydrogen bond stability in SOD1 mutants and the survival time of amyotrophic lateral sclerosis patients. J Mol Graph Model. 2022;110 doi: 10.1016/j.jmgm.2021.108026. [DOI] [PubMed] [Google Scholar]
- 45.Régal L., et al. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol. 2006;63(2):262–267. doi: 10.1001/archneur.63.2.262. [DOI] [PubMed] [Google Scholar]
- 46.Baskoylu S.N., et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018;14(10) doi: 10.1371/journal.pgen.1007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saccon R.A., et al. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136(8):2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park J.H., et al. SOD1 deficiency: a novel syndrome distinct from amyotrophic lateral sclerosis. Brain. 2019;142(8):2230–2237. doi: 10.1093/brain/awz182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller T.M., et al. Trial of antisense oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099–1110. doi: 10.1056/NEJMoa2204705. [DOI] [PubMed] [Google Scholar]
- 50.Keskin I., et al. Comprehensive analysis to explain reduced or increased SOD1 enzymatic activity in ALS patients and their relatives. Amyotroph Lateral Scler Front Degener. 2017;18(5–6):457–463. doi: 10.1080/21678421.2017.1301481. [DOI] [PubMed] [Google Scholar]
- 51.Berman H.M., et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eswar, N., et al., Comparative protein structure modeling using Modeller. Current protocols in bioinformatics, 2006. 15(1): p. 5.6. 1–5.6. 30. [DOI] [PMC free article] [PubMed]
- 53.Bekker H., et al. World Scientific Publishing; 1993. Gromacs-a parallel computer for molecular-dynamics simulations. in 4th International Conference on Computational Physics (PC 92) [Google Scholar]
- 54.Pronk S., et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham M.J., et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 56.Lindorff‐Larsen K., et al. Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Protein: Struct, Funct, Bioinforma. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. [Google Scholar]
- 58.Hess B., et al. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 59.Essmann U., et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 60.Janson G., et al. PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics. 2017;33(3):444–446. doi: 10.1093/bioinformatics/btw638. [DOI] [PubMed] [Google Scholar]
- 61.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 62.Virtanen P., et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17(3):261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miotto M., et al. Insights on protein thermal stability: a graph representation of molecular interactions. Bioinformatics. 2019;35(15):2569–2577. doi: 10.1093/bioinformatics/bty1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miotto M., et al. Simulated epidemics in 3d protein structures to detect functional properties. J Chem Inf Model. 2020;60(3):1884–1891. doi: 10.1021/acs.jcim.9b01027. [DOI] [PubMed] [Google Scholar]
- 65.Miotto M., et al. Inferring the stabilization effects of SARS-CoV-2 variants on the binding with ACE2 receptor. Commun Biol. 2022;5(1):1–13. doi: 10.1038/s42003-021-02946-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet. 2018;26(10):1537–1546. doi: 10.1038/s41431-018-0177-4. Project MinE ALS Sequencing Consortium. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material