

Abstract
Claudin18.2 (CLDN18.2)-specific chimeric antigen receptor (CAR-T) cells displayed limited efficacy in CLDN18.2-positive pancreatic ductal adenocarcinoma (PDAC). Strategies are needed to improve the trafficking capacity of CLDN18.2-specific CAR-T cells. PDAC has a unique microenvironment that consists of abundant cancer-associated fibroblasts (CAFs), which could secrete stromal cell-derived factor 1α (SDF-1α), the ligand of CXCR4. Then, we constructed and explored CLDN18.2-targeted CAR-T cells with CXCR4 co-expression in treating immunocompetent mouse models of PDAC. The results indicated that CXCR4 could promote the infiltration of CAR-T cells and enhance their efficacy in vivo. Mechanistically, the activation of signal transducer and activator of transcription 3 (STAT3) signaling was impaired in CXCR4 CAR-T cells, which reduced the release of inflammatory factors, such as tumor necrosis factor-α, IL-6, and IL-17A. Then, the lower release of inflammatory factors suppressed SDF-1α secretion in CAFs via the nuclear factor κB (NF-κB) pathway. Therefore, the decreased secretion of SDF-1α in feedback decreased the migration of myeloid-derived suppressor cells (MDSCs) in tumor sites. Overall, our study demonstrated that CXCR4 CAR-T cells could traffic more into tumor sites and also suppress MDSC migration via the STAT3/NF-κB/SDF-1α axis to obtain better efficacy in treating CLDN18.2-positive pancreatic cancer. Our findings provide a theoretical rationale for CXCR4 CAR-T cell therapy in PDAC.
Keywords: chimeric antigen receptor, CAR-T cells, pancreatic ductal adenocarcinoma, PDAC, myeloid-derived suppressor cells, MDSCs, cancer-associated fibroblasts, CAFs, stromal cell-derived factor 1α, SDF-1α
Graphical abstract
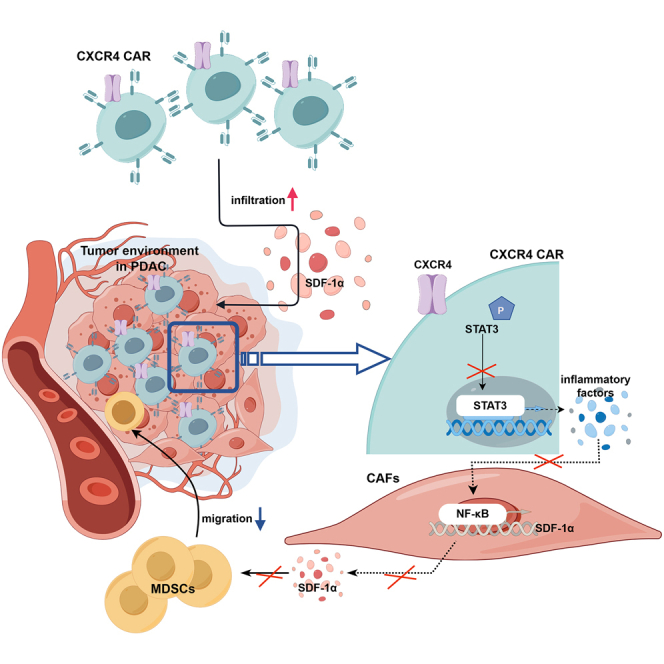
Li and colleagues found that PDAC had a unique microenvironment that consists of abundant CAFs, which secrete SDF-1α, the ligand of CXCR4. They constructed CXCR4 CAR to improve their trafficking capacity and suppress MDSCs migration via STAT3/NF-κB/SDF-1α axis, to obtained better anti-tumor efficacy against PDAC.
Introduction
Although chimeric antigen receptor (CAR-T) cells have shown significant clinical efficacy in various types of hematological malignancies, their therapeutic effect on solid tumors remains limited.1,2,3 CAR-T cells therapy in solid tumors is encumbered by numerous challenges, including fewer ideal tumor targets, immunosuppressive tumor microenvironments (TMEs), tumor heterogeneity, and particularly limited migration and persistence of CAR-T cells into tumors.4,5 Claudin18.2 (CLDN18.2), a gastric mucosa tight junction protein, has been regarded as a promising therapeutic target for gastric cancer and some other cancer types.6,7 Our previous preclinical data indicated that CT041, which contains genetically engineered autologous T cells that express the CLDN18.2-targeted CAR, could efficiently suppress the CLDN18.2-positive gastric cancer allografts tumors, which suggests it may be a promising treatment strategy for CLDN18.2-expressing solid tumors.8,9 Moreover, CT041 has promising efficacy with an acceptable safety profile in patients with CLDN18.2-positive digestive system cancers, particularly in those with gastric cancer.10
Pancreatic ductal adenocarcinoma (PDAC) continues to be one of the most lethal human malignancies with a poor prognosis because of systemic metastases and high recurrence rates. Approximately 80% of patients with PDAC present with locally advanced or metastatic disease that is not amenable to curative intent surgery. PDAC is particularly difficult to treat with cellular immunotherapy because of a pronounced desmoplastic reaction, together with poor vascularization, which limit immune cell infiltration. To date, limited success has been made in using CLDN18.2-targeted CAR cells to treat PDAC in vitro.9 The TME of pancreatic cancer in immunocompetent mice exhibits poor infiltration of CLDN18.2-specific CAR-T cells leading to their impaired anti-tumor effect, which is the main problem that needs to be solved.9 The key to lymphocyte homing is the multi-step process of rolling and adhesion. Chemokines and their receptors play a critical role in mediating cell migration to the tumor site and are important for the development and homeostasis of the immune system. Tumor tissue uses chemokine induction, integrin regulation, and enhanced tissue permeability to recruit auxiliary immune cells and migrate themselves. Similarly, tumor downregulates chemokines, which attract cytotoxic cells, such as CD8+ T and T helper type-1 cells, to escape the immune system. Chemokine gradients in tumor attracts immunosuppressive myeloid-derived suppressor cells (MDSCs) or regulatory T cells (Tregs), supporting tumor progression.11 Previous studies have reported that these characteristics of solid tumors can be used to enhance the transport of therapeutic T cells using chemokine receptors.12 However, so far, only a few chemokine receptors have been studied in this field, such as CXCR1, CXCR2, or CCR2b, that could direct the recruitment of CAR-T cells.13,14 Furthermore, the combination of radiation and CXCR1- or CXCR2-co-expressing CAR-T cells has been reported to obtain better anti-tumor efficacy in solid tumors.14 For pancreatic cancer, it has exhibited an improved anti-tumor efficiency of CCR4-co-expressing T cells.15 Recently, T cells expressing a CAR-encoding mesothelin and co-transfected with CXCR6 enhanced the efficacy of adoptive cell therapy for pancreatic tumors.16 Therefore, according to the chemokines existing in the tumor environment of pancreatic cancer, constitutive expression of chemokine receptors might promote the infiltration of CAR-T cells into tumor tissue.
PDAC has a unique microenvironment that consists of a fibrous expansion known as desmoplasia, characterized by the deposition of an abundant extracellular matrix composed of cancer-associated fibroblasts (CAFs), which represent 60% of the tumor stroma.17 Studies have demonstrated that CAFs exert immunosuppressive effects through direct exclusion of anti-tumor immune cells and recruitment of immunosuppressive immune cells to the tumor site.18,19 Pancreatic cancer cells and CAFs characteristically secrete a range of chemokines.20 Chemokines CXCL1, CXCL2, CCL2, and CCL5 secreted by CAFs are highly expressed in pancreatic cancer tissues. In addition, CAFs could secrete stromal cell-derived factor 1α (SDF-1α), also known as CXCL12.21 SDF-1α belongs to the CXC chemokine family and is the ligand of CXCR4.22 The SDF-1α/CXCR4 axis is a critical mediator of tumor-stromal interactions and has been implicated in promoting the metastatic potential of pancreatic cancer cells.23 Immune suppression by α-smooth muscle actin (α-SMA)-positive CAFs is regulated by CXCL12 binding to cancer cells and excluding T cells, which relies on the signaling of CXCR4. The SDF-1α/CXCR4 axis activates various signaling pathways that promote chemotaxis, adhesion, migration, cell proliferation, and survival.24 Besides, SDF-1α/CXCR4 axis also plays an important role in the migration of T cell precursors during development.25,26 We assumed that the SDF-1α/CXCR4 axis might be an attractive candidate axis for enhancing CAR-T cell function in PDAC because of its dual functions and the specific interactions between receptors and chemokines. Therefore, co-expression CXCR4 in CAR-T cells might promote their trafficking into the TME of PDAC, thereby enhancing their anti-tumor effects.
In this study, large amounts of CAFs were found to exist in the tumor tissue of PDAC, with high expression of SDF-1α. We generated a tandem construct encoding CLDN18.2-specific CAR and murine CXCR4 (CXCR4 CAR) and evaluated the anti-tumor potential of CAR-T cells in vitro and in vivo. The results demonstrated that CXCR4 could promote the recruitment of CAR-T cells into tumor sites. Compared with conventional CAR-T cells, CXCR4 CAR-T cells showed improved therapeutic effects against mouse pancreatic cancers. Furthermore, CXCR4 CAR-T cell treatment could also suppress the recruitments of MDSCs to improve the immunosuppressive microenvironment, which contributes to the survival of CAR-T cells. These findings provide a theoretical rationale for CXCR4 CAR-T cell therapy in pancreatic cancer.
Results
Large amounts of SDF-1α secreted by CAFs exist in the tumor tissue of pancreatic cancer, which is a ligand of CXCR4
Previous studies demonstrated that CLDN18.2-specific CAR-T cells (referred to as mBBZ CAR) could lead complete response in the PDX tumor model, indicating their potent tumor elimination capacity in immunodeficient mice.8,9 To better understand the anti-tumor effect of mBBZ CARs in a non-immunodeficient tumor environment, C57BL/6 mice bearing murine PANC02-A2 or KPC tumor allografts were established. After subcutaneous injection of CLDN18.2-positive tumor cells, mice were established with combined cyclophosphamide (CPA) pretreatment before the indicated T cell administration (Figures 1A and 1D). However, compared with untransduced (UTD) T cells, mBBZ CAR-T cells could not inhibit tumor growth significantly in both tumor models (Figures 1B and 1E). The CAR copy numbers also showed no differences in UTD and mBBZ CAR-T cell treatment group (Figures 1C and 1F), which indicate poor infiltration of CAR-T cells in tumor sites. The obstacle of CAR-T cell migration might suppress the anti-tumor effect of CAR-T cells. These data drove us to generate efficient CAR-T cell trafficking into TME.
Figure 1.

Large amounts of CAFs existing in tumor tissue of pancreatic cancer
In vivo experimental of (A–C) PANC02-A2 and (d–f) KPC allografts. (A, D) Experimental scheme of in vivo anti-tumor experiment under lympho depletion condition. C57BL/6 mice were inoculated subcutaneously with PANC02-A2 cells, and treated by intraperitoneal injection with CPA and then given CAR-T cells (intravenously). C57BL/6 mice: age 4–6 weeks, female, n = 5 mice per group. (B and E) The tumor volume of tumors of each treatment group. (C and F) CAR copy number in genomic DNA of residual tumors after CAR-T cells therapy was measured by RT-qPCR (TaqMan probe). (G) IHC staining of α-SMA protein expression in the representative pancreatic cancer samples. Scale bars, 400 μm or 100 μm. (H) The statistical results of the positive area and intensity of IHC staining of α-SMA protein expression in the site of cancer or para cancer. (I and J) The Cancer Genome Atlas analysis of the (I) α-SMA/ACTA2 and (J) SDF-1α/CXCL12 expression in different tumor types. (K) Western blot of SDF-1α expression in NIH3T3 cells, tumor tissues and tumor-derived CAFs. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control.
The cellular components in the TME of pancreatic cancer were complex. Immunohistochemistry (IHC) analysis of α-SMA expression in microarray containing tumor tissues from 15 PDAC patients with different grades was implemented. The results revealed that large amounts of CAFs exist in the tumor tissue of PDAC, with high expression of α-SMA (Figures 1G and 1H). Moreover, CAFs wrapped around tumor cells, forming a barrier that might limit CAR-T cell infiltration, as reported27 (Figure 1G). CAFs could secrete SDF-1α, promoting the infiltration of CXCR4-positive cells, resulting in immunosuppression in PDAC.28 In The Cancer Genome Atlas dataset, α-SMA and SDF-1α were highly expressed in pancreatic cancer (Figure 1I). Then, the expression of SDF-1α was determined in tumor tissues and tumor-derived CAFs from CLDN18.2-positive tumor-bearing mice. Compared with NIH3T3 cells (as a negative control), higher expression of SDF-1α was found in tumor tissues, especially in tumor-derived CAFs (Figure 1K). As a receptor of SDF-1α, CXCR4 was found to be associated with the features of chemokine-receptor signaling versatility and infiltration of CD8+ T cells in pancreatic cancer.29 Therefore, the co-expression of CXCR4 might improve the infiltration of CAR-T cells to the tumor sites.
Generation of mBBZ CARs co-expressing CXCR4
The murine second-generation CAR composed of CLDN18.2-specific single-chain variable fragment (scFv) fused with murine 4-1BB and CD3-ζ intracellular signaling domains (mBBZ) was constructed as previously described.8 Additionally, we developed a tandem construct encoding the mBBZ CAR and murine CXCR4 with a F2A peptide sequences between the genes (referred to as CXCR4 CAR) (Figure 2A). The transduction efficiency of mBBZ CAR-T cells was 39.5% and CXCR4 CAR-T cells was 59.5% (Figure 2B), which was determined by flow cytometry. CD3-ζ and CXCR4 protein expression were also confirmed by western blotting (Figure 2C). CXCR4 CAR-T cells showed increased CXCR4 expression, compared with UTD and mBBZ CAR-T cells (Figure 2C). Mouse pancreatic cancer cells KPC, PANC02, and CLDN18.2-overexpessing PANC02 (PANC02-A2) cells were sorted out to test the cytotoxicity of mBBZ CAR-T and CXCR4 CAR-T cells (Figure 2D). The cytotoxicity assay revealed that both CAR-T cells could specifically lyse CLDN18.2-positive target cells with equivalent efficiency, but not antigen-negative target cells in vitro (Figures 2E–2G). Higher specific lysis of target cells could be detected at a 3:1 effector/target (E/T) ratio compared with 1:1, 1:3, or 1:9 E/T ratios (Figures 2F and 2G). To evaluate the function of SDF-1α/CXCR4 axis on cytotoxicity of CAR-T cells, different concentrations of SDF-1α were added in the co-culture system. However, the cytolysis activity showed no difference between CXCR4 CAR-T cells and mBBZ CAR-T cells (Figure S1A). Cytokine release assay in vitro showed that CXCR4 CAR-T cells induced more secretion of granzyme B than mBBZ CAR-T cells (Figure 2H). However, there were no difference of IFN-γ and IL-2 release in different co-culture system (Figure 2H).
Figure 2.

Generation of mBBZ CARs co-expressing CXCR4
(A) Schematic representation of the modular composition of CLDN18.2-targeted conventional and CXCR4 CAR. This construct includes an extracellular region of antigen recognition, a transmembrane domain, an intracellular region of mouse 4-1BB costimulatory molecules, and a mouse CD3-ζ chain. (B) The transduction efficiency and CXCR4 expression of mBBZ and CXCR4 CAR-T cells on splenic T cells derived from C57BL/6 was determined by flow cytometry. Untreated T cells served as negative controls. (C) Western blot of CD3-ζ and CXCR4 expression in CAR-T cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control. (D) The expression of CLDN18.2 on PANC02, PANC02-A2. and KPC cells. Cells incubated with a mouse anti-mouse IgG antibody as negative control. (E–G) CAR-T cells were co-incubated with the CLDN18.2-negative or CLDN18.2-positive target cells at varying E:T ratios for 18 h. Cell lysis was tested using a standard nonradioactive cytotoxicity assay. (H) CAR-T cells were co-cultured with antigen-positive cells at a ratio of 1:1 for 48 h. IFN-γ, IL-2, and granzyme B in co-culture supernatants were quantified by ELISA assays (n = 3). (I) Transwell co-culture of CAR-T cells with cell culture supernatant of CAFs or SDF-1α. (J) CAR-T cells were added to the upper chamber and the cell culture supernatant of CAFs was added in the lower chamber. The cell number in the lower chamber was counted at 2 and 6 h. (K) CAR-T cells were added to the upper chamber, and 0, 50, or 100 ng/mL SDF-1α was added in the lower chamber. The cell number in the lower chamber was counted at 2 h and 6 h. (L) Transwell co-culture and LDH release tests of CAR-T cells in the upper chamber with tumor cells in the lower chamber. (M) The LDH release in the lower chamber was tested at 18 h. All data are presented as the mean ± SEM of triplicate experiments. ∗∗p < 0.01.
Next, to determine whether SDF-1α in CAFs contributed to the greater recruitment of CXCR4 CAR-T cells, transwell migration assays were performed. According to a previous study, NIH3T3 mouse fibroblasts were starved and treated with transforming growth factor β1 (TGF-β1) to induce CAFs characterized by increased expression of α-SMA30 (Figure S1B). Then, CAR-T cells were cultured in the upper chamber, with the cell culture supernatant of NIH3T3-induced CAFs or different concentrations of SDF-1α added in the lower compartment (Figure 2I). The results indicated that more CXCR4 CAR-T cells were attracted into the lower chamber compared with mBBZ CAR-T cells under the chemotaxis of cell culture supernatant or SDF-1α in a dose-dependent manner (Figures 2I and 2J). Then, CAR-T cells were added to the upper chamber and the tumor cells were added in the lower chamber (Figure 2L). However, cytotoxic assays including target cells at the bottom showed that cytotoxic activity of CXCR4 CAR-T cells was enhanced in the co-culture system (Figure 2M).
Then, we investigated the memory phenotype of T cells in response to antigen stimulation. The results indicated that more CD8+ T cells had a CD44+CD62L+ central memory phenotype in CXCR4 CAR-T cells with antigen stimulation, compared with mBBZ CAR-T cells (Figure S1C). Of note, the apoptosis and proliferation of CXCR4 CAR-T cells and CAR-T cells with antigen stimulation showed no difference (Figures S1D and S1E). Immune checkpoint molecules, such as programmed cell death protein 1 (PD-1), T cell immunoglobulin and mucin domain 3 (TIM3), and lymphocyte-activation gene 3 (LAG3) play an important role in cancer immunotherapy,31 and the immune checkpoint molecules of mouse CAR-T cells were tested. No significant differences appeared in the expression of PD-1, TIM3, and LAG3 between CXCR4 CAR-T and mBBZ CAR-T cells (Figure S1F−S1H). These results revealed that CXCR4 had no effect on the cell phenotype, exhaustion, proliferation, or cytotoxicity of CAR-T cells. However, CXCR4 could improve the infiltration of CAR-T cells toward SDF-1α.
CXCR4 enhanced anti-tumor effect of CAR-T cells in pancreatic cancer allografts
To address the anti-tumor activities of CXCR4 CAR-T cells, mice bearing CLDN18.2-positive PANC02 (PANC02-A2) tumor allografts were established (Figure S2A). The body weight of mice showed no significant difference in all groups, suggesting no that severe toxicity was caused by CAR-T cell treatment (Figure S2B). CXCR4 could significantly enhance the anti-tumor effect of CAR-T cells, as shown by the delayed tumor growth (Figures S2C and S2D). Tumor inhibition rates and tumor weights were measured and were in accordance with tumor volumes results (Figures S2E and S2F). However, in the absence of lymphocyte depletion, the tumor suppressive effect of CXCR4 CAR-T cells was limited compared with mBBZ CAR-T cells.
Previous studies have shown that combined CPA pre-conditional chemotherapy and immune T cells can inhibit tumor growth in mice, and a few clinical trials use similar chemoimmunotherapeutic strategies to treat patients with tumor.32,33 To investigate the anti-tumor effects of CAR-T cells on different mice models with lymph depleting chemotherapy in vivo, the mouse PANC02-A2 and KPC tumor models were established with CPA pretreatment. In the PANC02-A2 tumor model, after CPA lymphodepletion (Figures S3A and S3B), the tumors volume of PANC02-A2 tumor-bearing mice treated with CXCR4 CAR-T cells grew more slowly than those in the mBBZ CAR-T cell treatment group (p < 0.01) (Figure 3A), consistent with the weight of residual tumors in each treatment group (Figures 3B–3D). The tumor growth inhibition of mBBZ CAR-T cell and CXCR4 CAR-T cell treatment in PANC02-A2 tumor-bearing mice was 39.39% and 87.63%, respectively (Figure 3E). No significant difference in body weight was found in any of the groups (Figure 3F). To determine the infiltration of CAR-T cells in the tumor tissue of each treatment group, CAR copies in residual tumors were also detected. The CAR copy numbers were more significantly elevated in the CXCR4 CAR-T cell treatment group compared with those of other groups (Figure 3G).
Figure 3.

CXCR4 enhanced anti-tumor effect of CAR-T cells in pancreatic cancer
(A–G) In vivo experimental of PANC02-A2 allografts. (A) Experimental scheme of in vivo antitumor experiment under lymph depletion condition. C57BL/6 mice were inoculated subcutaneously with PANC02-A2 cells and treated with intraperitoneal injection with CPA and then given CAR-T cells intravenously. (B and C) The tumor volume of tumors of each treatment group. (d) The tumor weight at the endpoint of the animal experiment. (E) The tumor growth inhibition of each treatment group. (F) The body weight of each treatment group. (G) CAR copy number in genomic DNA of residual tumors after CAR-T cells therapy was measured by RT-qPCR (TaqMan probe). (H–N) In vivo experiment with KPC allografts. (H) Experimental scheme of in vivo antitumor experiment under lymph depletion condition. C57BL/6 mice were inoculated subcutaneously with KPC cells, and treated by intraperitoneal injection with CPA and then given CAR-T cells intravenously. (I and J) The tumor volume of tumors of each treatment group. (K) The tumor weight at the endpoint of the animal experiment. (L) The tumor growth inhibition of each treatment group. (M) The body weight of each treatment group. (N) CAR copy number in genomic DNA of residual tumors after CAR-T cells therapy was measured by RT-qPCR (TaqMan probe). C57BL/6 mice: age 4–6 weeks, female, n = 5 mice per group. All data are presented as the mean ± SEM of triplicate experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
In the KPC tumor model (Figure 3H), CXCR4 CAR-T cell treatment had a better anti-tumor effect than mBBZ CAR-T cell treatment, according to the tumor volume (p < 0.05) and residual tumor weight (Figures 3I–3K). The tumor growth inhibition in the CXCR4 CAR-T cell treatment group was 28.4%, whereas in the mBBZ CAR-T cell treatment group it was 4.29% (Figure 3L). The body weight of mice showed no significant difference in any of the groups (Figure 3M). The CAR copy numbers were more significantly elevated in the CXCR4 CAR-T cell treatment group than those in other groups (Figure 3N). These results indicate that CXCR4 promoted more CAR-T cells trafficking into tumor tissue of CLDN18.2-positive tumor-bearing mice and enhanced the anti-tumor efficiency of CAR-T cells.
CXCR4 CAR-T cell treatment reduced recruitment of MDSCs in tumor tissue of CLDN18.2-positive tumor-bearing mice
The SDF-1α/CXCR4 axis also participates in promoting the proliferation and migration of other immune cells in the TME. To evaluate the influence of CAR-T cells on immune cells, we next investigated the immune cells in tumor tissues of CLDN18.2-positive tumor-bearing mice. In PANC02-A2- and KPC-tumor-bearing mice, CXCR4 CAR-T cell treatment showed more CD8+ T cells accumulation in tumor tissue by IHC staining and flow cytometry, whereas the CD4+ T cells in each group showed no differences (Figures 4A–4C and 4F–4H). No significant differences appeared in the expression of PD-1, TIM3, and LAG3 of CD3+ T cells in tumor tissues from the CXCR4 CAR-T cell and mBBZ CAR-T cell treatment groups (Figures S3C and S3D). Then, immune cells in TME were tested. Inducible nitric oxide synthase (iNOS) and arginase 1 (Arg-1) are produced by MDSCs, which are regarded as cellular markers of MDSCs.34 The mRNA expressions of Arg-1 and iNOS were decreased in tumor tissues from the CXCR4 CAR-T cell treatment group of CLDN18.2-positive tumor-bearing mice (Figure S3E). Moreover, the ratio of MDSCs in CD45+ immune cells was significantly decreased in the CXCR4 CAR-T cell treatment group compared with the mBBZ CAR-T cell treatment group (Figures 4D, 4E, 4I, and 4J). However, there were no significant differences in the ratios of dendritic cells, natural killer cells, or other immune cells in the tumor tissues of the CXCR4 CAR-T cell and mBBZ CAR-T cell treatment groups (Figures S3F−S3G and S4A−S4H).
Figure 4.

CXCR4 CAR-T cell treatment reduced recruitment of MDSCs in tumor tissue of CLDN18.2-positive tumor-bearing mice
(A–E) Analysis the immune cells in tumor tissues of PANC02-A2 allografts. (A and B) The representative immunostaining images and quantification of CD4+ and CD8+ T cells and hematoxylin and eosin staining in tumor tissues from each treatment group. The images were obtained under original magnification ×200. Scale bars, 100 μm. (C) The quantitation of tumor-infiltrating CD8+ and CD4+ cells in CD3+ T cells of each treatment group by flow cytometry. (D and E) Representative flow cytometry plots showing the frequencies and quantitation of tumor-infiltrating CD45+ immune cells and MDSCs of each treatment group. (F–J) Analysis the immune cells in tumor tissues of KPC allografts. (F and G) Representative immunostaining images and quantification of CD4+ and CD8+ T cells and hematoxylin and eosin staining in tumor tissues from each treatment group. The images were obtained under original magnification ×200. Scale bars, 100 μm. (H) The quantitation of tumor-infiltrating CD8+ and CD4+ cells in CD3+ immune cells of each treatment group by flow cytometry. (I and J) Representative flow cytometry plots showing the frequencies and quantitation of tumor-infiltrating CD45+ immune cells and MDSCs of each treatment group. All data are presented as the mean ± SEM of triplicate experiments. ∗p < 0.05.
SDF-1α downregulation in CAFs by the treatment of CXCR4 CAR-T cells
MDSCs have emerged as important contributors to solid tumor immune evasion.35 Previously, we observed that MDSCs could suppress mouse T cell proliferation and inhibit the lytic activity of CAR-T cells.36,37 To explore the mechanism of reduced recruitment of MDSCs in the CXCR4 CAR-T cell treatment group, mRNA expression of chemokines in tumor tissue from CLDN18.2-positive tumor-bearing mice were tested. The mRNA and protein expression of SDF-1α in tumor tissue was significantly decreased in the CXCR4 CAR-T cell treatment group compared with the mBBZ CAR-T cell treatment group (Figures 5A and S5A). And the accumulation of CAFs was observed in tumor tissue of CLDN18.2-positive tumor-bearing mice examined by IHC (Figure S5B). As reported, SDF-1α could bind to its receptor CXCR4, which is highly expressed on the cancer cells surface.38 This binding allows the activation of numerous signaling pathways in cancer cells, such as nuclear transcription factor κB (NF-κB) and Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathways, to promote the survival, proliferation, adhesion, chemotaxis and migration of tumor cells.39,40 Here, the inactivation of the downstream signaling pathway were also found in the CXCR4 CAR-T cell treatment group compared with the mBBZ CAR-T cell treatment group from CLDN18.2-positive tumor-bearing mice (Figures 5A–5D). Our previous studies indicated that CAFs could promote the recruitment of MDSCs via the SDF-1α/CXCR4 axis in tumor tissue.37 We isolated MDSCs from the bone marrow of C57BL/6 mice and detected high expression of CXCR4 (Figure 5E). Then, MDSCs were transwell cultured in the upper chamber, with different concentrations of SDF-1α added in the lower compartment (Figure 5F). Greater migration of MDSCs was determined in the lower compartment with high concentration of SDF-1α (Figure 5G). These data suggest that reduced CAFs-derived SDF-1α might contribute to the less recruitments of MDSCs.
Figure 5.

The SDF-1α downregulation in CAFs by the treatment of CXCR4 CAR-T cells
(A–D) The protein levels of p-stat3, stat3, IkBα, p-NF-κB, NF-κB, α-SMA, and SDF-1α in the tumor tissue of (A and B) PANC02-A2-tumor bearing-mice and (C and D) KPC tumor-bearing mice from each treatment group. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control. (E) The expression and of CXCR4 on MDSCs determined by FACS. Representative flow cytometry plots showing the frequencies of MDSCs (CD11b+ Gr1+ cells) in CD45+ immune cells isolated from bone marrow (BM). (F) Transwell co-culture of MDSCs with different concentrations of SDF-1α. (G) Effects of SDF-1α on the chemotaxis of MDSCs. MDSCs were added to the upper chamber and 0, 50, or 100 ng/mL SDF-1α was added in the lower chamber. The cell number in the lower chamber was counted at 2 and 6 h. (H and I) The protein levels and of SDF-1α in the tumor tissue of (H) PANC02-A2-tumor bearing mice and (I) KPC-tumor bearing mice from each treatment group at days 0, 10, and 14. GAPDH served as a loading control. (J) The expression of CXCR4 on CD8+ T cells, CD4+ T cells, and MDSCs determined by flow cytometry. (K) The expression of CXCR4 on mBBZ, CXCR4 CAR-T cells, or MDSCs determined by flow cytometry. (M) Effects of SDF-1α on the chemotaxis of mBBZ, CXCR4 CAR-T cells, and MDSCs. mBBZ, CXCR4 CAR-T cells, or MDSCs were added to the upper chamber and 0, 50, or 100 ng/mL SDF-1α was added in the lower chamber. The cell number in the lower chamber was counted at 2 and 6 h. All data are presented as the mean ± SEM of triplicate experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Except for MDSCs, SDF-1α is also the key chemokine for CXCR4 CAR-T cell trafficking into tumor tissue. It is important to notice that SDF-1α decreased after CXCR4 CAR-T cell infusion (Figures 5H–5J). Generally, greater expression of CXCR4 was found in MDSCs than in CD4+ T and CD8+ T cells (Figure 5K). However, CXCR4 CAR-T cells showed greater expression of CXCR4 compared with MDSCs (Figure 5L). Then, CXCR4 CAR-T cells, mBBZ CAR-T cells, and MDSCs were transwell cultured in the upper chamber with different concentrations of SDF-1α added in the lower compartment. tGreater migration of CXCR4 CAR-T cells occurred in the lower compartment under the chemotaxis of SDF-1α at dose- and time-dependent contrast with MDSCs (Figure 5M). These data suggest that, in the presence of SDF-1α, CXCR4 CAR-T cells showed a stronger chemotactic ability to tumor tissue than MDSCs. Therefore, CXCR4 CAR-T cells migrated into tumor tissue at first and decreased expression of SDF-1α arose subsequently, after CXCR4 CAR-T cell infiltration.
Decreased expression of inflammatory factors contributed to SDF-1α reduction in CAFs via the NF-κB signaling pathway in the CXCR4 CAR-T cell treatment group
Next, we explored the mechanism of SDF-1α reduction in the CXCR4 CAR-T cell treatment group. The level of cytokines in the tumor tissues from CLDN18.2-positive tumor-bearing mice was determined. The secretion and mRNA expression of inflammatory factors IL-17A, IL-6, and tumor necrosis factor (TNF-α) in the tumor tissues of the CXCR4 CAR-T cell treatment group were found decreased compared with the mBBZ CAR-T cell treatment group (Figures 6A, 6B, and S5C), whereas the secretion of IL-4, IL-10, and IL-2 showed no difference between the two groups from CLDN18.2-positive tumor-bearing mice (Figures S5D and S5E).
Figure 6.

Decreased expression of inflammatory factors contributed to SDF-1α reduction in CAFs via NF-κB signaling pathway in the CXCR4 CAR-T cell treatment group
(A and B) The secretion levels of TNF-α, IL-17A, and IL-6 in tumor tissues of PANC02-A2 and KPC tumor-bearing mice from each treatment group. (C) The production of TNF-α, IL-17A, and IL-6 from different CAR-T cells and target cells co-culture system. Different CAR-T cells and target cells were cultured at a 1:1 ratio of for 24 h. (D) The activation of NF-κB signaling pathway in CAFs induced by CAR-T cells. The NIH3T3-induced CAFs were treated with the cell culture supernatant of antigen-positive tumor cells and CAR-T cells for 24, 36, or 48 h. (E and F) The expressions of p-NF-κB, NF-κB, IkBα, SDF-1α, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were determined by western blot. (E) The tumor-derived CAFs were treated with TNF-α, IL-17A, or IL-6 at the concentrations of 0, 10, 20, or 50 ng/mL for 48 h. (F) The tumor-derived CAFs were treated with 20 ng/mL TNF-α, 20 ng/mL IL-17A, or 20 ng/mL with the exposure of PDTC at the concentration for 0, 0.2, or 0.5 μM for 48 h. (G) RNA-seq was performed on CAR-T cells stimulated with antigen-positive target cells for 4 h (n = 2). Functional enrichment analysis of Kyoto Encyclopedia of Genes and Genomes pathways changed in CXCR4 CAR-T cells compared with the mBBZ CAR-T cells with or without antigen stimulation. (H) Gene set enrichment analysis of the activation of JAK/STAT3 signaling pathway between mBBZ and CXCR4 CAR-T cells with or without antigen stimulation. (I) The expression of p-stat3, stat3, caspase 3, cleaved caspase 3, and GAPDH in the mBBZ and CXCR4 CAR-T cells with antigen stimulation for 0, 5, 15, 30, or 60 min. (J) The expression of p-stat3, stat3, caspase 3, cleaved caspase 3, and GAPDH in the UTD, mBBZ, and CXCR4 CAR-T cells treated with antigen stimulation for 0, 30, or 60 min and different concentrations of SDF-1α. (K) The heatmap results shows JAK/STAT3-regulated gene expression, with red and blue representing higher and lower transcription, respectively. (L) The secretion levels of TNF-α, IL-17A, and IL-6 in mBBZ or CXCR4 CAR-T cells and antigen-positive tumor cells co-culture system. mBBZ or CXCR4 CAR-T cells were co-incubated with antigen-positive tumor cells at a 1:1 ratio with STAT3 signaling pathway inhibitor napabucasin (0, 0.01, 0.02, or 0.05 μM) for 24 h and the cytokines in supernatants were measured by CBA kits. (M) The expressions of p-NF-κB, NF-κB, IκBα, SDF-1α, and GAPDH was determined by western blot. The cell supernatants of CAR-T cells and antigen positive tumor cells with the exposure of napabucasin (0, 0.01, 0.02, or 0.05 μM) for 48 h were added into the tumor-derived CAFs and cultured for 24 h. All data are presented as the mean ± SEM of triplicate experiments. ∗p < 0.05, ∗∗∗p < 0.001.
The correlation analysis results suggested that IL-17A, IL-6, and TNF-α were positively correlated with SDF-1α expression in pancreatic cancer (Figure S6A). The secretion of IL-6 and TNF-α was decreased in CXCR4 CAR-T cells co-culture system (Figure 6C). Previous studies demonstrated that stromal cells in pancreatic cancer secrete SDF-1α in an NF-κB-dependent fashion.41,42 After incubation with antigen-positive target cells (at a ratio of 1:1), CAR-T cells could release multiple cytokines. The cell culture supernatants of this co-culture system were added into NIH3T3-induced CAFs cell media for 24, 36, or 48 h, and the activity of NF-κB signaling pathway was inhibited in CXCR4 CAR-T cells co-culture system (Figure 6D). Then, the addition of IL-17A, IL-6, and TNF-α was found to stimulate the activation of NF-κB signaling pathway as reported,43 with increased expression of SDF-1α in NIH3T3-induced CAFs (Figures S6B−S6D). The tumor-derived CAFs were obtained from tumor tissue of PANC02-A2 tumor-bearing mice (Figure S6E). With the addition of IL-17A, IL-6, or TNF-α, tumor-derived CAFs presented activated NF-κB signaling pathway and increased SDF-1α expression (Figure 6E).
To confirm whether the regulatory effect of cytokines on SDF-1α expression was through the NF-κB signaling pathway, pyrrolidine dithiocarbonate (PDTC), an inhibitor of the NF-κB signaling pathway, was added to the co-culture system (Figure 6F). And the activation of the NF-κB signaling pathway induced by IL-17A, IL-6, or TNF-α could be suppressed by PDTC (Figure 6F). These data demonstrate that the decreased secretion of inflammatory factors in the CXCR4 CAR-T cell treatment group might suppress SDF-1α expression via the NF-κB signaling pathway.
Overexpression of CXCR4 in CAR-T cells exhibit suppressed activity of STAT3 signaling pathway
Next, to explore the mechanism of the decreased secretion of inflammatory factors in the CXCR4 CAR-T cell treatment group, mBBZ CAR-T cells and CXCR4 CAR-T cells were stimulated with or without antigen stimulation for 4 h. Then, the CAR-T cells were sorting for RNA-sequencing. The RNA-sequencing analysis results showed that there were 170 genes changed in both mBBZ CAR-T cells and CXCR4 CAR-T cells under antigen and non-antigen stimuli. Among the 170 genes, many of them were JAK/STAT3 signaling pathways (Figures 6G and S6F). Gene set enrichment analysis revealed that the JAK/STAT3 signaling pathway was activated by antigen stimuli (Figure S7A) and inactive in CXCR4 CAR-T cells, compared with mBBZ CAR-T cells (Figure 6H).
Then, the activity of various signaling pathways participating in regulating the function of CAR-T cells was explored (Figures S7B−S7C). JAK/STAT3 signaling is a major cytokine effector pathway critical for the development and function of different immune cell types.44 STAT3 is an important mediator of signaling for cytokines involved in the generation of T cells.45 Here, the STAT3 signaling pathway was continuously activated with prolonged antigen stimulation in mBBZ CAR-T cells (Figure 6I). However, the expression of p-STAT3 decreased in CXCR4 CAR-T cells compared with mBBZ CAR-T cells (Figure 6I and S7C). In the exposure of SDF-1α, the activation of STAT3 in CXCR4 CAR-T cells was still suppressed compared with mBBZ CAR-T cells after 30 min of antigen stimulation (Figures 6J and S7D). However, there was no difference of cleaved caspase 3 expression, a marker of cell apoptosis, between mBBZ and CXCR4 CAR-T cells (Figure 6J). Consistently, the heatmap of RNA sequencing (RNA-seq) results showed JAK/STAT3-related gene expression was downregulated in CXCR4 CAR-T cells compared with mBBZ CAR-T cells with or without antigen stimuli, including TNF-α, IL-6, and IL-17A (Figures 6K and S7E). Then, mBBZ CAR-T cells were treated with napabucasin (Napa), an inhibitor of THE STAT3 signaling pathway, and presented decreased rusecretion of inflammatory factors, such as TNF-α, IL-6, and IL-17A (Figure 6L). mBBZ and CXCR4 CAR-T cells were treated with different concentrations of napabucasin (Figure S7F). CXCR4 CAR-T cells were more resistant to napabucasin-induced cell death at low concentrations (<0.2 μM), which suggested that CXCR4 CAR-T cells were less dependent on STAT3-mediated cell survival n (Figure S7F). Moreover, the phosphorylation of STAT3 in mBBZ CAR-T cells could be significantly downregulated by A low concentration of napabucasin addition (Figure S7G). Then, co-culture system of mBBZ or CXCR4 CAR-T cells and antigen-positive tumor cells were treated with napabucasin at the concentration of 0.01, 0.02, or 0.05 μM. Of note, with exposure to napabucasin, less apoptosis of CD3+ T cells was observed in the CXCR4 CAR-T cell coculture system (Figures S7H−S7I).
To determine whether STAT3 pathway-regulated cytokine release led to altered NF-κB pathway activity in CAFs, CAR-T cells were co-cultured with antigen-positive tumor cells under the exposure of napabucasin. The secretion of TNF-α, IL-6, and IL-17A was decreased by napabucasin treatment and in a dose-dependent manner (Figure 6L). And CXCR4 CAR-T cells showed lower sensitivity to napabucasin-reduced inflammatory factors release (Figure 6L). Then, the cell culture supernatants from co-culture system were added to the tumor-derived CAFs culture media. The activation of the NF-κB signaling pathway in CAFs was blocked and the expression of SDF-1α was decreased with napabucasin pre-treatment and was dose dependent in a co-culture system (Figure 6M). Moreover, under 0.02 and 0.05 μM napabucasin pre-treatment in a CXCR4 CAR-T cell coculture system, SDF-1α expression from CAFs showed no difference compared with an mBBZ CAR-T cells coculture system (Figure 6M). These data indicated that STAT3 signaling pathway participated in the release of inflammatory factors in CAR-T cells. Further, lower activation of the STAT3 signaling pathway in CXCR4 CAR-T cells might regulate the inflammatory factor-reduced SDF-1α expression in CAFs via an NF-κB signaling pathway.
Discussion
Despite promising preclinical investigations, CAR-T cell therapy remains encumbered by significant hurdles in advanced solid cancers. One of the barriers of CAR-T cell therapy is inadequate trafficking to tumor foci.46 Previously, we demonstrated the impaired infiltration and limited efficacy of h8E5(2I)-mBBZ CAR-T cells in treating pancreatic cancer allografts.9 To improve the infiltration and survival of CAR-T cells, we engineered h8E5(2I)-mBBZ CAR-T cells to co-express CXCR4. Evidence was provided that CAR-T cells co-expressing CXCR4 triggers an anti-tumor immune response with more migration of CAR-T cells and decreased recruitment of MDSCs in mouse pancreatic cancer models. Mechanistically, low activation of the STAT3 signaling pathway in CXCR4 CAR-T cells might downregulate some inflammatory factors, leading to decreased SDF-1α expression in CAFs via the NF-κB signaling pathway, further limiting the recruitment of MDSCs into tumor tissue (Figure 7).
Figure 7.

Schema: Overexpression of CXCR4 enhances the anti-tumor ability of CAR-T cells by suppressing the infiltration of MDSCs
(Top) After co-incubating CAR-T cells with antigen-positive target cells, with activated STAT3 signaling pathway, CAR-T cells secreted more inflammatory factors such as TNF-α, IL-17A, and IL-6. These inflammatory factors activated the NF-κB signaling pathway in CAFs, thereby increasing SDF-1α expression. Excessive SDF-1α secretion induced the infiltration of immunosuppressive cells MDSCs, thereby forming a tumor immunosuppressive microenvironment and limiting the function of CAR-T cells. (Bottom) Co-expression of CXCR4 improved the migration of CAR-T cells. Moreover, CXCR4 reduced the activity of STAT3 signaling pathway in CAR-T cells. The impaired activation of STAT3 signaling pathway suppressed the secretion of TNF-α, IL-17A, and IL-6, then decreased SDF-1α expression in the CAFs via NF-κB signaling pathway, thereby limiting the infiltration of MDSCs and enhancing the anti-tumor effect of CAR-T cells.
The TME of pancreatic cancer is composed of non-malignant stromal cells, including CAFs. In this study, CAFs are determined to be the main cell types constituting the tumor stroma from 15 pancreatic cancer samples. CAFs were reported to modulate tumor development, invasion, and therapeutic resistance.47 Studies have shown that, in animal models of pancreatic cancer, complete clearance of CAFs leads to accelerated tumor progression.48 These findings suggest that it should be more inclined to use the characteristics of CAFs cells rather than completely eliminating CAFs cells when targeting therapy of CAFs in pancreatic cancer. SDF-1α secreted by CAFs could recruit a plurality of cells expressing the corresponding receptor, CXCR4, including tumor cells, neutrophils, and monocytes-macrophages.23 Therefore, we developed CLDN18.2-specfic CAR-T cells co-expressing CXCR4 aiming to promote the recruitment of effector T cells into tumor tissues. As expected, CXCR4 CAR-T cell therapy showed increased infiltration of cytotoxic cells and improved anti-tumor activity significantly compared with conventional CAR-T cell therapy.
In addition, less SDF-1α and a lower proportion of MDSCs in infiltrating leukocytes were found in the tumor tissues of the CXCR4 CAR-T group than in that of the conventional CAR-T cell treatment group. MDSCs increase in abundance under the duress of cancer and have emerged as important contributors to solid tumor immune evasion. This negative effect has been ascribed to their immunosuppressive roles and their effects on tumor cell invasion and angiogenesis.49 Tumor-associated cytokines, such as the C-C and C-X-C motif chemokines CCL2 and CXCL15, induce recruitment of MDSCs to the tumor site.50 Notably, SDF-1α produced within the tumor can attract CXCR4+ MDSCs, potentiating the tumor-promoting effect. It is worth noting that, with higher CXCR4 expression, there was greater migration of CXCR4 CAR-T cells under the chemotaxis of SDF-1α, in contrast with MDSCs. Therefore, we speculated that more CXCR4 CAR-T cells migrated into tumor tissues at first, followed by decreased expression of SDF-1α, thereby inhibiting the infiltration of MDSCs. Moreover, MDSCs could induce CD8+ T cell apoptosis and inhibited the lytic activity of CAR-T cells.36,37 Thus, in this study, greater residence of CD8 + T cells in the tumor tissue of mice treated with CXCR4 CAR-T cells might be at least partially ascribed to the lower recruitment of MDSCs. In addition, the downstream signaling of SDF-1α/CXCR4 pathway, including NF-κB and STAT3 pathways, showed limited activation in tumor tissues of the CXCR4 CAR-T cell treatment group, leading to suppressed growth of the tumor.
STAT3 plays a pivotal role in promoting cell proliferation and a plethora of tumor-infiltrating immune cells that predominantly comprise the TME.51 Previous studies had developed a novel CAR construct that activates the JAK/STAT pathway to enhance their anti-tumor efficacy.52 However, more studies emphasized that hyperactivation of STAT3 in tumor-infiltrating immune cells causes immunosuppression by inhibiting both innate and adaptive immune responses.53 Moreover, excessive STAT3 activity in innate immune cell subsets may impair the production of IFN-γ, dampen antigen presentation, and inhibit the tumor-killing activities of effector T cells,54 although with the restricted activity of STAT3 signaling pathway in CXCR4 CAR-T cells, their proliferation ability was not affected. In addition, the STAT3 signaling pathway has long been recognized as a potential therapeutic target for cancer therapy because of their roles in tumor formation, metastasis, and drug resistance. Some small-molecule STAT3 inhibitors can enhance therapeutic efficacy against tumors.55 Combination therapy is considered to be a promising direction for improving outcomes for cancer treatment. Here, CXCR4 CAR-T cells showed lower sensitivity of STAT3 inhibitors. These encouraging data triggered investigations of combination strategies, such as the combination of STAT3 inhibitors with CXCR4 CAR-T cells in treating pancreatic cancer.
Notably, the decreased activity of the STAT3 signaling pathway in CXCR4 CAR-T cells is accompanied by a reduced release of inflammatory factors such as IL-6, TNF-α, and IL-17A. Many STAT3-regulated genes encoding cytokines such as IL-6 and IL-10 could in turn activate STAT3 signaling and propagate a feedforward loop.43 Therefore, the impaired activity of the STAT3 signaling pathway in CXCR4 CAR-T cells might decrease the expression of some inflammatory factors, and these inflammatory factors might also regulate the activation of the STAT3 signaling pathway in feedback. However, the mechanisms of CXCR4-induced inactivation of the STAT3 signaling pathway in CAR-T cells still requires further investigation. In addition, in vivo, after CAR-T cell treatment, large numbers of cytokines such as IL-6 and IL-1β were released into plasma, which might lead acute toxicity and cytokine storm. However, no morphological abnormality appeared in tissue sections from heart, liver, spleen, lung, and kidney of CXCR4 CAR-T cell treatment group, even with lower inflammatory factors compared with mBBZ CAR-T cells. Therefore, CXCR4 CAR-T cells might be a safe and effective treatment for pancreatic cancer and less likely to cause cytokine storm in other normal tissues. Further, we will explore the anti-tumor effect and safety of CXCR4 CAR-T cells in pancreatic cancer models in situ.
Together, in this study, we demonstrated that co-expression CXCR4 could significantly enhance the anti-tumor efficacy of CAR-T cells in mouse pancreatic cancer model, and decrease the recruitment of MDSCs to the TME. Our findings may provide a potential therapeutic strategy for the treatment of pancreatic cancer.
Materials and methods
Cell lines and cell culture
PANC02 (obtained from the American Type Culture Collection) and KPC mice-derived PDAC cell line FC1199 (referred to as KPC, a gift from Professor Jing Xue) were cultured in DMEM medium (Gibco) with 10% FBS (Gibco) and 100 U/mL penicillin/streptomycin (Invitrogen). CLDN18.2 was cloned into the lentiviral vectors pWPT-GFP (12255; Addgen) for expression (referred to CLDN18.2 lentiviral vectors). The PANC02 cells were lentivirally transduced to stably express murine CLDN18.2 using CLDN18.2 lentiviral vectors (referred to as PANC02-A2). The 293T packaging cell lines (obtained from the Chinese Academy of Sciences) were cultured in in DMEM medium with 10% FBS. Mouse NIH3T3cell lines (obtained from the American Type Culture Collection) were cultured in DMEM with 10% NCS (Gibco) and 100 U/mL penicillin/streptomycin (Invitrogen). All cells were routinely maintained at 37°C in a 5% CO2 atmosphere incubator (Thermo Fisher Scientific).
The procedure of inducing NIH3T3 cells into CAF cells was as follows: NIH3T3 fibroblasts were incubated with free serum (starvation) for 24 h and then treated with 2.5 ng/mL TGF-β1 in 10% serum for an extra 24 h.
CAR design
The second generation of the CLDN18.2 CAR is composed of the anti-CLDN18.2 scFv (hu8E5-2I) linked in-frame to the hinge and transmembrane regions of the murine CD8α chain and intracellular murine 4-1BB and CD3ζ signaling domains, which was then cloned into the retroviral vector pMSCV-IRES-GFP (52107; Addgen) for expression. The scFv (hu8E5-2I) recognized both the human and the murine CLDN18.2 antigen. To generate CARs expressing murine CXCR4, F2A peptide sequences were intercalated among the second-generation CAR genes.
Virus production and generation of CAR-T cells
Retroviruses were obtained by transfection of 70%–80% confluent 293T cells with CAR and packaging plasmid pCL-Eco (12371, Addgen) using poly ethylenimine. Viruses were harvested 48 h later and filtered through a 0.45-μm filter unit (Millipore) to remove cell debris. Then, the recombinant murine CLDN18.2 CAR retrovirus was generated as follows. Mouse T cells were isolated from murine spleen using a mouse T cell isolation kit (Stem Cell Technologies) and subsequently simulated with anti-mouse CD3/28 magnetic beads (Thermo Fisher Scientific) at a bead-to-cell ratio of 1:1 for 24 h and then cultured in RMPI 1640 (Gibco) with 10% FBS, supplemented with 50 μM β-mercaptoethanol (Sigma-Aldrich), 100 U/mL penicillin/streptomycin, and 100 U/mL recombinant human IL-2 (Shanghai Huaxin High Biotechnology, Inc) and then infected with retrovirus in RetroNectin (Takara)-coated plates. Infected cells were incubated overnight at 37°C and 5% CO2.
Mice and animal experiments
Four- to 6-week-old female C57BL/6 mice were bought from Shanghai Sippr BK Laboratory Animal Co. Ltd. All animal experiments were performed according to protocols approved by the Shanghai Cancer Institute Experimental Animal Care Commission. We injected 2 × 106 CLDN18.2-positive tumor cells on the right flank of C57BL/6 mice. When the tumor volume reached 120 mm3, tumor-bearing mice were randomly grouped. The CPA (100 mg/kg) was administered intraperitoneally 24 h before 2.5 × 106 anti-CLDN18.2 CAR-T cells were injected. In all experiments, tumor growth was measured by caliper twice a week. Tumor volumes were calculated using the following formula: Tumor volume = Length × Width2/2. Outcomes, including death or a tumor size >2,000 mm3.
Flow cytometry
h8E5(2I) on T cells was evaluated using antibody biotin-conjugated goat-anti-human Fab (Jackson ImmunoResearch) with phycoerythrin-conjugated streptavidin (12-4317-87; Invitrogen). CLDN18.2 on tumor cells was measured by h8E5(2I) antibody, which was generated by our laboratory, and then followed by incubation with antibody fluorescein isothiocyanate-conjugated goat anti-human IgG (Jackson ImmunoResearch).
To analyze the in vitro proliferation of CAR-T cells, a CellTrace Violet cell proliferation kit (Thermo Fisher Scientific) was used for labeling of cells to trace multiple generation via dye dilution. For in vivo detection of immune cells in the tumor, tumor tissues isolated from tumor-bearing mice were cut into small pieces and resuspended in digestion medium containing collagenase type IV (0.5 mg/mL; Sigma Aldrich), collagenase type I (0.5 mg/mL; Sigma Aldrich), hyaluronidase (0.5 mg/mL; Sigma Aldrich), and Dnase I (0.02 mg/mL; Stem Cell Technologies) for 30 min at 37°C. The suspensions were filtered through a 70-μM Falcon cell strainer, centrifuged, and stained with antibodies according to the manufacturer’s instructions. Next, cells were washed with FACS buffer. Then, extracellular markers were labeled by adding the antibody and incubated at room temperature for 30 min. After fixation and permeabilization (Transcription Factor Staining Buffer Set, 130-122-981, Miltenyi Biotec; Fixation Buffer, 420801, BioLegend), the cell suspensions were labeled with the intracellular markers FOXP3 for the detection of Treg cells. The antibodies used for the flow cytometry experiments are listed in Table S1. The analysis was performed by using BD FACs Celesta flow cytometer.
Cytotoxicity in vitro
Cytotoxicity was assessed by the lactate dehydrogenase (LDH) release assay using a CytoTox96 Non-Radioactive Cytotoxicity Kit (Promega) according to the manufacturer’s instruction. Target cells (1 × 104) were co-cultured with CAR-T cells at E/T ratios of 1:9, 1:3, 1:1, and 3:1 in 96-well plates at 37°C for 18 h. The supernatants were collected, and then LDH release in the supernatants was evaluated by a colorimetric reaction (absorbance at 490 nm). The spontaneous LDH release of effector and target cells and the maximum LDH release of target cells were also measured.
Cytokine measurements
Target cells were co-cultured with CAR-T cells at E/T ratios of 1:1 at 37°C for 24 h. Tumor mass was homogenized in T-PER Tissue Protein Extraction Reagent with protease inhibitor cocktail and the supernatants were collected after centrifugation. Cytokines in coculture supernatants or plasma or tumor mass were measured using cytometric bead arrays (CBA, BD Biosciences) for IFN-γ, TNF-α, IL-4, IL-10, and IL-6 according to the manufacturer’s instructions.
IHC staining
Formalin-fixed and paraffin-embedded tumor tissues were examined by IHC staining using anti-CD4 (ab183685; Abcam), anti-CD8α (98941; Cell Signaling Technologies), and anti-α-SMA antibody (19245; Cell Signaling Technologies). Briefly, the sections were exposed to 3% H2O2 in methanol after deparaffinization and rehydration and then blocked with 1% BSA for 30 min at room temperature. After blocking, the sections were incubated with primary antibody overnight at 4°C, followed by incubation with peroxidase-conjugated secondary antibodies (ChemMate DAKO EnVision Detection Kit, Peroxidase/DAB, Rabbit/Mouse, BD Biosciences) and detection reagents. CD4+ T cells and CD8+ T cells were quantified by measuring the number of stained cells in sections from three mice in each group. The mean count of the three areas per section was obtained and expressed as the absolute.
Chemotaxis assay in vitro
For migration, CAR-T or UTD cells were placed in the upper chamber of a transwell unit with an 5 μm polycarbonate filter (Corning) and the culture supernatants of CAFs or different concentration of SDF-1α were added in the lower chamber. At 2 and 6 h of migration, the cells in the lower chamber were counted.
Western blot analysis
Cells were washed three times with PBS and lysed in RIPA buffer with a protease inhibitor cocktail and PMSF (200 μg/mL). Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 5% non-fat dry milk in PBS containing 0.1% Tween 20 at room temperature for 1 h. Subsequently, the membranes were exposed to indicated primary antibodies in 5% non-fat dry milk in TBS overnight at 4°C. After washing and incubating with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h at room temperature, the membranes were incubated with Pierce ECL western Blotting Substrate (Thermo Fisher Scientific) as the substrate of HRP and membranes were scanned on film. The antibodies used were showed in Table S1.
MDSCs isolation
MDSCs were isolated via Ly6G magnetic selection from bone marrow of healthy mice by using the MDSC isolation kit (Miltrnyi Biotec). Cells from bone marrow were incubated with antibodies to mouse CD11b (Biolegend) and Ly6G (BD Bioscience) followed by red blood cell removal and then was analyzed by flow cytometry. All antibodies were used according to manufacturer’s recommendations. Live cells were gated by forward scatter/side scatter. Analysis was performed using a BD FACs Celesta flow cytometer and FlowJo software (RRID: SCR_008520).
RNA-seq analysis
RNA-seq analysis was performed as described previously.56 CAR-T cells were stimulated with antigen-positive tumor cells for 4 h and then CAR-T cells were sorted. Total RNA was extracted using the RNeasy Mini Kit (Qiagen). Sequencing libraries were generated using VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2 (Vazyme). The sequencing was performed on an Illumina Nova seq platform and analyzed at Shanghai Biochip Corporation. RNA-seq count data were quantified as transcripts per million, which were then normalized using limma and visualized with pheatmap package in R between h8E5(2I)-BBZ and h8E5(2I)-BBZ-CXCR4 CAR-T cells.
Tissue microarray
The microarray comprising 15 pancreatic tumor tissues and the paired adjacent normal tissues were purchased from Shanghai Outdo Biotech Inc. The clinical pathological information was provided by the company as well.
Statistical analysis
All data are presented as the mean ± SEM. The Student t-test was used for two samples comparisons. One-way ANOVA was used for multi-sample comparisons. Tumor growth data were analyzed with two-way ANOVA. All statistical analyses were done using GraphPad Prism software (RRID: SCR_002798). Data were presented as the mean values ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 were considered statistically significant.
Acknowledgments
We thank Prof. Jing Xue for the gift of KPC mice-derived PDAC cell line FC1199. The graphical abstract was drawn by Figdraw. This study was supported by the National Natural Science Foundation of China (ID 82073358 to H.J.; ID 82073359 to Z.L.; ID 82272920 to M.Z.) and Shanghai Municipal Health and Health Commission (ID 20234Y0193 to R.S.).
Author contributions
R.S., H.J., and Z.L. designed the study; R.S., Y.S., C.W., Y.L., Y.D., G.D., and H.L. performed experiments and analyzed data; R.S. and Y.S. performed the animal experiments; R.S. wrote the manuscript; H.J. and Z.L. reviewed and edited the manuscript; R.S., H.J., M.Z., and Z.L. supervised the project.
Declaration of interests
H.J. and Z.L. have ownership interests of CAR-T cells relating to this work and are stockholders in CARsgen Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.09.010.
Contributor Information
Hua Jiang, Email: jianghuapy@163.com.
Zonghai Li, Email: zonghaili@shsmu.edu.cn.
Supplemental information
Data and code availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Gene expression data from RNA-seq analysis were deposited in the NCBI Gene Expression Omnibus (GEO) database (GSE211750).
References
- 1.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi D., Shi Y., Kaseb A.O., Qi X., Zhang Y., Chi J., Lu Q., Gao H., Jiang H., Wang H., et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials.(2020) Clin. Cancer Res. 2020;26:3979–3989. doi: 10.1158/1078-0432.CCR-19-3259. [DOI] [PubMed] [Google Scholar]
- 3.Neelapu S.S., Locke F.L., Go W.Y. CAR T-Cell Therapy in Large B-Cell Lymphoma. N. Engl. J. Med. 2018;378:1065. doi: 10.1056/NEJMc1800913. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016;380:205–215. doi: 10.1016/j.canlet.2015.07.044. [DOI] [PubMed] [Google Scholar]
- 5.Kershaw M.H., Westwood J.A., Parker L.L., Wang G., Eshhar Z., Mavroukakis S.A., White D.E., Wunderlich J.R., Canevari S., Rogers-Freezer L., et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12(20 Pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niimi T., Nagashima K., Ward J.M., Minoo P., Zimonjic D.B., Popescu N.C., Kimura S. claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol. Cell. Biol. 2001;21:7380–7390. doi: 10.1128/MCB.21.21.7380-7390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wöll S., Schlitter A.M., Dhaene K., Roller M., Esposito I., Sahin U., Türeci Ö. Claudin 18.2 is a target for IMAB362 antibody in pancreatic neoplasms. Int. J. Cancer. 2014;134:731–739. doi: 10.1002/ijc.28400. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H., Shi Z., Wang P., Wang C., Yang L., Du G., Zhang H., Shi B., Jia J., Li Q., et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019;111:409–418. doi: 10.1093/jnci/djy134. [DOI] [PubMed] [Google Scholar]
- 9.Luo H., Su J., Sun R., Sun Y., Wang Y., Dong Y., Shi B., Jiang H., Li Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020;26:5494–5505. doi: 10.1158/1078-0432.CCR-20-0777. [DOI] [PubMed] [Google Scholar]
- 10.Qi C., Gong J., Li J., Liu D., Qin Y., Ge S., Zhang M., Peng Z., Zhou J., Cao Y., et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med. 2022;28:1189–1198. doi: 10.1038/s41591-022-01800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M., et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 12.Lim W.A., June C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y., Wang J., Yang X., Yang J., Lu P., Zhao L., Li B., Pan H., Jiang Z., Shen X., et al. Chemokine Receptor CCR2b Enhanced Anti-tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-small-cell Lung Carcinoma Model. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.628906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin L., Tao H., Karachi A., Long Y., Hou A.Y., Na M., Dyson K.A., Grippin A.J., Deleyrolle L.P., Zhang W., et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019;10:4016. doi: 10.1038/s41467-019-11869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rapp M., Grassmann S., Chaloupka M., Layritz P., Kruger S., Ormanns S., Rataj F., Janssen K.P., Endres S., Anz D., Kobold S. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology. 2016;5 doi: 10.1080/2162402X.2015.1105428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesch S., Blumenberg V., Stoiber S., Gottschlich A., Ogonek J., Cadilha B.L., Dantes Z., Rataj F., Dorman K., Lutz J., et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours.(2021) Nat. Biomed. Eng. 2021;5:1246–1260. doi: 10.1038/s41551-021-00737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn J.L., Bondre C., Gladstone H.B., Brown P.O., Chang H.Y. Anatomic demarcation by positional variation in fibroblast gene expression programs. Plos Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tjomsland V., Niklasson L., Sandström P., Borch K., Druid H., Bratthäll C., Messmer D., Larsson M., Spångeus A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin. Dev. Immunol. 2011;2011 doi: 10.1155/2011/212810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collier J.J., Sparer T.E., Karlstad M.D., Burke S.J. Pancreatic islet inflammation: an emerging role for chemokines. J. Mol. Endocrinol. 2017;59:R33–R46. doi: 10.1530/JME-17-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernard V., Semaan A., Huang J., San Lucas F.A., Mulu F.C., Stephens B.M., Guerrero P.A., Huang Y., Zhao J., Kamyabi N., et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019;25:2194–2205. doi: 10.1158/1078-0432.CCR-18-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng F., Tian W.Y., Wang Y.M., Zhang Y.F., Guo F., Zhao J., Gao C., Xue F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016;9:8. doi: 10.1186/s13045-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biasci D., Smoragiewicz M., Connell C.M., Wang Z., Gao Y., Thaventhiran J.E.D., Basu B., Magiera L., Johnson T.I., Bax L., et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA. 2020;117:28960–28970. doi: 10.1073/pnas.2013644117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dewan M.Z., Ahmed S., Iwasaki Y., Ohba K., Toi M., Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed. Pharmacother. 2006;60:273–276. doi: 10.1016/j.biopha.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Kim C.H., Pelus L.M., White J.R., Broxmeyer H.E. Differential chemotactic behavior of developing T cells in response to thymic chemokines. Blood. 1998;91:4434–4443. [PubMed] [Google Scholar]
- 26.Aiuti A., Tavian M., Cipponi A., Ficara F., Zappone E., Hoxie J., Peault B., Bordignon C. Expression of CXCR4, the receptor for stromal cell-derived factor-1 on fetal and adult human lympho-hematopoietic progenitors. Eur. J. Immunol. 1999;29:1823–1831. doi: 10.1002/(SICI)1521-4141(199906)29:06<1823::AID-IMMU1823>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 27.Lee J.J., Perera R.M., Wang H., Wu D.C., Liu X.S., Han S., Fitamant J., Jones P.D., Ghanta K.S., Kawano S., et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA. 2014;111:E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feig C., Jones J.O., Kraman M., Wells R.J.B., Deonarine A., Chan D.S., Connell C.M., Roberts E.W., Zhao Q., Caballero O.L., et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer.(2013) Proc. Natl. Acad. Sci. USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kocher F., Puccini A., Untergasser G., Martowicz A., Zimmer K., Pircher A., Baca Y., Xiu J., Haybaeck J., Tymoszuk P., et al. Multi-omic characterization of pancreatic ductal adenocarcinoma relates CXCR4 mRNA expression levels to potential clinical targets. Clin. Cancer Res. 2022;28:4957–4967. doi: 10.1158/1078-0432.CCR-22-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakravarthy A., Khan L., Bensler N.P., Bose P., De Carvalho D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018;9:4692. doi: 10.1038/s41467-018-06654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adachi K., Tamada K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015;106:945–950. doi: 10.1111/cas.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 33.Delahousse J., Skarbek C., Desbois M., Perfettini J.L., Chaput N., Paci A. Oxazaphosphorines combined with immune checkpoint blockers: dose-dependent tuning between immune and cytotoxic effects. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cimen Bozkus C., Elzey B.D., Crist S.A., Ellies L.G., Ratliff T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015;195:5237–5250. doi: 10.4049/jimmunol.1500959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Long A.H., Highfill S.L., Cui Y., Smith J.P., Walker A.J., Ramakrishna S., El-Etriby R., Galli S., Tsokos M.G., Orentas R.J., Mackall C.L. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016;4:869–880. doi: 10.1158/2326-6066.CIR-15-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di S., Zhou M., Pan Z., Sun R., Chen M., Jiang H., Shi B., Luo H., Li Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells.(2019) Front. Oncol. 2019;9:241. doi: 10.3389/fonc.2019.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun R., Luo H., Su J., Di S., Zhou M., Shi B., Sun Y., Du G., Zhang H., Jiang H., Li Z. Olaparib Suppresses MDSC Recruitment via SDF1alpha/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol. Ther. 2021;29:60–74. doi: 10.1016/j.ymthe.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katsumoto K., Kume S. The role of CXCL12-CXCR4 signaling pathway in pancreatic development. Theranostics. 2013;3:11–17. doi: 10.7150/thno.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian D., Lu Z., Xu Q., Wu P., Tian L., Zhao L., Cai B., Yin J., Wu Y., Staveley-O'Carroll K.F., et al. Galectin-1-driven upregulation of SDF-1 in pancreatic stellate cells promotes pancreatic cancer metastasis. Cancer Lett. 2017;397:43–51. doi: 10.1016/j.canlet.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Ahr B., Denizot M., Robert-Hebmann V., Brelot A., Biard-Piechaczyk M. Identification of the cytoplasmic domains of CXCR4 involved in Jak2 and STAT3 phosphorylation. J. Biol. Chem. 2005;280:6692–6700. doi: 10.1074/jbc.M408481200. [DOI] [PubMed] [Google Scholar]
- 41.Garg B., Giri B., Modi S., Sethi V., Castro I., Umland O., Ban Y., Lavania S., Dawra R., Banerjee S., et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology. 2018;155:880–891.e8. doi: 10.1053/j.gastro.2018.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu M., Fei Y., He Q., Fu J., Zhu J., Tao J., Ni C., Xu C., Zhou Q., Yao M., Ni H. Electroacupuncture Attenuates Cancer-Induced Bone Pain via NF-kappaB/CXCL12 Signaling in Midbrain Periaqueductal Gray.(2021) ACS Chem. Neurosci. 2021;12:3323–3334. doi: 10.1021/acschemneuro.1c00224. [DOI] [PubMed] [Google Scholar]
- 43.De Simone V., Franzè E., Ronchetti G., Colantoni A., Fantini M.C., Di Fusco D., Sica G.S., Sileri P., MacDonald T.T., Pallone F., et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34:3493–3503. doi: 10.1038/onc.2014.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kallal L.E., Biron C.A. Changing partners at the dance: Variations in STAT concentrations for shaping cytokine function and immune responses to viral infections. JAKSTAT. 2013;2 doi: 10.4161/jkst.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tangye S.G., Ma C.S., Brink R., Deenick E.K. The good, the bad and the ugly - TFH cells in human health and disease. Nat. Rev. Immunol. 2013;13:412–426. doi: 10.1038/nri3447. [DOI] [PubMed] [Google Scholar]
- 46.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 48.Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawanobori Y., Ueha S., Kurachi M., Shimaoka T., Talmadge J.E., Abe J., Shono Y., Kitabatake M., Kakimi K., Mukaida N., Matsushima K. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–5466. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann A., Kortylewski M., Kujawski M., Zhang C., Reckamp K., Armstrong B., Wang L., Kowolik C., Deng J., Figlin R., Yu H. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–7464. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kagoya Y., Tanaka S., Guo T., Anczurowski M., Wang C.H., Saso K., Butler M.O., Minden M.D., Hirano N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018;24:352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson D.E., O'Keefe R.A., Grandis J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018;15:234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang T., Niu G., Kortylewski M., Burdelya L., Shain K., Zhang S., Bhattacharya R., Gabrilovich D., Heller R., Coppola D., et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 55.Zou S., Tong Q., Liu B., Huang W., Tian Y., Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer. 2020;19:145. doi: 10.1186/s12943-020-01258-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Y., Dong Y., Sun R., Liu Y., Wang Y., Luo H., Shi B., Jiang H., Li Z. Chimeric anti-GPC3 sFv-CD3epsilon receptor-modified T cells with IL7 co-expression for the treatment of solid tumors. Mol. Ther. Oncolytics. 2022;25:160–173. doi: 10.1016/j.omto.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Gene expression data from RNA-seq analysis were deposited in the NCBI Gene Expression Omnibus (GEO) database (GSE211750).