

Abstract
Agastache rugosa, also known as Korean mint, is a perennial plant from the Lamiaceae family that is traditionally used for various ailments and contains antioxidant and antibacterial phenolic compounds. Molecular breeding of A. rugosa can enhance secondary metabolite production and improve agricultural traits, but progress in this field has been delayed due to the lack of chromosome-scale genome information. Herein, we constructed a chromosome-level reference genome using Nanopore sequencing and Hi-C technology, resulting in a final genome assembly with a scaffold N50 of 52.15 Mbp and a total size of 410.67 Mbp. Nine pseudochromosomes accounted for 89.1% of the predicted genome. The BUSCO analysis indicated a high level of completeness in the assembly. Repeat annotation revealed 561,061 repeat elements, accounting for 61.65% of the genome, with Copia and Gypsy long terminal repeats being the most abundant. A total of 26,430 protein-coding genes were predicted, with an average length of 1,184 bp. The availability of this chromosome-scale genome will advance our understanding of A. rugosa’s genetic makeup and its potential applications in various industries.
Subject terms: Plant sciences, Genome
Background & Summary
Agastache rugosa, a perennial plant belonging to the Lamiaceae family, is widely distributed in Korea, China, Taiwan, and Japan. In Korean traditional medicine, the aerial part of A. rugosa, known as “Gwakyang”, is prescribed for various ailments, such as miasma, cholera, anorexia, and vomiting1. A. rugosa produces phenolic compounds such as rosmarinic acid, which has antioxidant and antibacterial properties2–5. In addition to its uses in traditional herbal medicine, A. rugosa leaves are used as a spice or vegetable and its flowers as a tea ingredient6. Desta et al. assessed the antioxidant activity of various parts of A. rugosa—including the flowers, leaves, stems, and roots—and found that the leaves, flowers, and roots exhibited notably strong antioxidant properties7.
Previous research on A. rugosa has primarily concentrated on its secondary metabolites3,4, phenylpropanoid-biosynthetic genes8–10, and cell culture11,12. To date, there are no whole genome sequences available for A. rugosa, and only transcriptome data have been published13. An integrated analysis of its metabolites and genome will provide insight into chemotype breeding of A. rugosa and improve its economic value in the market.
In this study, we assembled the chromosome-level genome of A. rugosa using Nanopore sequencing and Hi-C technology. The final genome assembly had a scaffold N50 of 52.15 Mbp, totaling 410.67 Mbp. With integration of Hi-C data, nine pseudochromosomes were generated, accounting for 89.1% of the entire predicted genome. The first chromosome-scale genome of A. rugosa provides a foundational genetic resource for breeding programs targeting enhanced production of secondary metabolites like rosmarinic acid and essential oils. This genome assembly bolsters the efficiency of genotyping methods such as GBS, facilitating more precise QTL analysis or GWAS, which are crucial for optimizing agricultural traits.
Methods
Sampling and sequencing
A breeding line, AG34, of A. rugosa, sourced from a specific population in the field, was chosen for reference genome sequencing and assembly. This line was derived from original natural accessions obtained from the Chungbuk National University (Korea). Young leaf samples were collected once during the vegetative stage after being grown in a greenhouse for three months. Leaf tissue samples were stored at −80 °C and used for DNA extraction, whole genome sequencing, and Hi-C library construction. DNA was extracted using the Biomedic Plant gDNA extraction kit (#BM20211222A, Korea) following the manufacturer’s instructions.
An Oxford Nanopore Technology (ONT) sequencing library was constructed using the ONT genomic ligation sequencing kit SQK-LSK110 (ONT, UK). ONT sequencing was performed using the flow cell vR9.4 (FLO-MIN106) and GridION platform operated with MinKNOW Core 4.4.3 following the manufacturer’s instructions. We obtained 55.9 Gb of raw genomic data. Guppy v5.0.17, embedded in MinKNOW14, was used to convert raw ONT sequencing data (FAST5 files) to FASTQ format using the default parameters of the high-accuracy method. All ONT sequencing procedures were conducted by Phyzen Co. (www.phyzen.com, Korea). Paired-end (PE) Illumina sequencing was also conducted with the NovaSeq6000 platform after constructing a standard Illumina paired-end library. We obtained 115.5 Gb of raw data from Illumina sequencing.
Total RNA was extracted from leaf tissue of the same material used for the genome sequencing of A. rugosa, and the transcriptome was sequenced on the Illumina NovaSeq6000 platform by Macrogen Co. (www.macrogen.com, Korea). The RNA reads were used for gene annotation.
Sequence trimming and genome size estimation
ONT data were trimmed using Porechop (v.0.2.3, https://github.com/rrwick/Porechop) with default parameters to remove adaptors and chimeric sequences. Raw Illumina sequencing data were trimmed using fastp (v.0.21.0, https://github.com/OpenGene/fastp) with default parameters. The amount of trimmed Illumina PE sequencing data was 97 Gb, which was used for further genome size estimation based on k-mer analysis. An optimal k-mer value of 19 was calculated by Jellyfish (v2.0)15, and the genome size was estimated using GenomeScope (v2.0)16. The estimated genome size of A. rugosa based on k-mer analysis was 460.89 Mbp, which is slightly smaller than the 520 Mb previously reported using flow cytometry17. The heterozygous rate was 0.55%, and the repeat rate was 62.21% (Fig. 1).
Fig. 1.

The result of K-mer analysis. (a) 19-mer frequency distribution in A. rugosa genome. The X-axis is the k-mer depth, and Y-axis represents the frequency of the k-mer for a given coverage. (b) Statistics of K-mer analysis.
Contig assembly
The first round of de novo assembly was performed using NextDenovo assembler (v.2.3.1, https://github.com/Nextomics/NextDenovo) with default parameters, employing only preprocessed 55,923,595,489 bp of ONT data(~121X of estimated genome size, 460Mbp). Assembled contigs were then polished using NextPolish (v1.3.1, https://github.com/Nextomics/NextPolish) with trimmed Illumina PE sequencing data. Haplotigs were removed using Purge Haplotigs18 with default parameters. The assembly statistics improved, with fewer contigs and increased minimum, average contig lengths, and N90 (see Table S1). Finally, a draft genome assembly was generated with 221 contigs totaling 410.65 Mbp, with a contig N50 of 3.85 Mbp (Table 1).
Table 1.
Assembly statistics of A. rugosa.
| De novo assembly | |
| Total contigs number | 221 |
| Total size of assembled contigs (bp) | 410,656,262 |
| Minimum length of contig (bp) | 48,164 |
| Maximum length of contig (bp) | 12,657,832 |
| Average length of contigs (bp) | 1,858,173 |
| Contig N50 (bp) | 3,851,190 |
| Contig N90 (bp) | 885,118 |
| GC contents (%) | 36.51 |
| Final statistics of Hi-C scaffolding | |
| The number of scaffolds (pseudomolecule) | 9 |
| Unscaffolded contigs | 21 |
| Total length | 410,677,362 |
| Total length of scaffolds anchored to chromosomes | 405,296,100 |
| Total length of unscaffolded contig | 5,381,262 |
| Maximum length of unscaffolded contigs | 697,320 |
| Minimum length of scaffold | 70,820 |
| Maximum length of scaffold | 73,606,202 |
| Scaffold N50 | 52,151,255 |
| Scaffold N90 | 32,072,577 |
Chromosome-level genome assembly using Hi-C data
A Hi-C library of A. rugosa was constructed for chromosome assembly using the ProximoTM Hi-C Plant Kit (Phase Genomics, United States) following the manufacturer’s instructions. A total of 30.77 Gbp of clean Hi-C data were generated and aligned to the assembled contigs using BWA-MEM (v0.7.17)19 with -5SP and -t 8 options specified. Chromosome-level scaffolding was performed with the Phase Genomics Proximo Hi-C genome scaffolding platform based on the LACHESIS method20, and sequences were anchored to nine pseudochromosomes with chromosome lengths ranging from 27.7 Mb to 73.6 Mb. Our chromosome-scale assembly coincides with that from a previous karyotype analysis, as the base chromosome number of Agastache species is reported to be nine, and A. rugosa is a diploid species21,22. Additional manual correction of the chromatin contact matrix was performed using Juicebox (https://github.com/aidenlab/Juicebox). The nine pseudochromosomes were clearly identified by distinct interaction signals in the Hi-C interaction heatmap (Fig. 2), and the final assembled genome was 410.68 Mbp, with a scaffold N50 of 52.15 Mb, accounting for 89.1% of the predicted genome size based on the k-mer analysis (Table 1 and Fig. 3). The assembled genome sizes of Lamiaceae species show a wide range of variation: A. rugosa in this study (410.68 Mbp), Perilla frutescens var. hirtella (676.94 Mbp)23, P. frutescens var. frutescens (1.2 Gbp)23, Salvia hispanica (321.47 Mbp)24, and Salvia splendens (805.9 Mbp)25.
Fig. 2.

Hi-C contact map the chromosome-level assembly of A. rugosa. The intensity of interactions was calculated using a bin size of 140 K.
Fig. 3.

Overview of genome features of the A. rugosa. Syntenic block among inter-chromosome were analyzed with MCScanX. (a) Gene distribution, (b) Repeat percentage(%), (c) Gypsy (red line) and Copia (blue line) LTR distribution (%), (d) GC content(%).
Assessment of the genome assemblies
The completeness of the assembled genome was evaluated using BWA-MEM (v0.7.17)19 and Benchmarking Universal Single-Copy Orthologs (BUSCO, v5.2.1)26 with the embryophyta_odb10 lineage dataset. Approximately, 98.04% of the Illumina short read were aligned to genome, of which 89.6% of reads were properly mapped. The BUSCO analysis showed that the assembled draft genome sequence contained 1,596 (98.9%) complete BUSCOs, including 1,533 (95.0%) single-copy BUSCOs, 63 (3.9%) duplicated BUSCOs, and 7 (0.4%) fragmented BUSCOs (Table 2).
Table 2.
Result of the BUSCO assessment of A. rugosa.
| Type | Genome | |
|---|---|---|
| Count | Ratio (%) | |
| Complete BUSCOs (C) | 1,596 | 98.9 |
| Complete and single-copy BUSCOs (S) | 1,533 | 95.0 |
| Complete and duplicated BUSCOs (D) | 63 | 3.9 |
| Fragmented BUSCOs (F) | 7 | 0.4 |
| Missing BUSCOs (M) | 11 | 0.7 |
| Total BUSCO groups searched | 1,614 | 100.0 |
Repeat annotation
The de novo repeat families were identified with RepeatModeler27, and by LTR_retriever28, then repetitive sequences were masked using RepeatMasker 4.0.9 (http://www.repeatmasker.org). A total of 561,061 repeat elements were identified, accounting for 61.65% of the A. rugosa genome. Among the various repeat elements, Copia and Gypsy, which are long terminal repeats (LTRs), were dominant in the genome, accounting for 14.98% and 13.91%, respectively (Table 3).
Table 3.
Repetitive elements annotation in A. rugosa.
| Class | Number of elements | Sequence length (bp) | Percentage of genome (%) |
|---|---|---|---|
| DNA | 37,867 | 10,748,442 | 2.62% |
| CMC-EnSpm | 3,533 | 2,472,316 | 0.68% |
| MULE-MuDR | 10,783 | 9,767,543 | 2.38% |
| PIF-Harbinger | 5,955 | 2,660,594 | 0.65% |
| TcMar-Pogo | 646 | 104,439 | 0.03% |
| TcMar-Stowaway | 578 | 510,816 | 0.12% |
| hAT-Ac | 3,793 | 2,366,869 | 0.58% |
| hAT-Tag1 | 477 | 202,820 | 0.05% |
| hAT-Tip100 | 776 | 289,420 | 0.07% |
| LINE | 1,973 | 252,866 | 0.06% |
| L1 | 3,602 | 1,630,673 | 0.40% |
| LTR | 48,566 | 12,283,572 | 2.99% |
| Caulimovirus | 5,413 | 10,430,421 | 2.54% |
| Copia | 34,703 | 61,518,387 | 14.98% |
| Gypsy | 41,357 | 57,125,275 | 13.91% |
| unkown | 27,449 | 15,268,074 | 3.72% |
| RC | — | — | — |
| Helitron | 5,338 | 2,634,800 | 0.64% |
| SINE | 5,191 | 1,120,846 | 0.27% |
| tRNA | 157 | 42,282 | 0.01% |
| Unknown | 229,870 | 57,817,026 | 14.08% |
| total interspersed | 468,027 | 249,247,481 | 60.69% |
| Low_complexity | 16,665 | 793,497 | 0.19% |
| Simple_repeat | 76,369 | 3,132,257 | 0.76% |
| Total | 561,061 | 253,173,235 | 61.65% |
Gene prediction and annotation
Gene prediction involved a combination of evidence-based annotation methods and ab initio prediction using repeat-masked assembly sequences. RNA-Seq data were assembled by Trinity and used for the transcript set. Additionally, protein data from four related Lamiaceae species were obtained from the NCBI. The first round of gene prediction was performed using MAKER (v3.01.03)29 with evidence data, the transcript set and the protein data from the four related species. The ab initio gene predictions were conducted on only the first gene models with sufficient evidence (AED of 0.25 or less) using GeneMark-ES (v4.38)30, SNAP (v2006-07-28)31, and Augustus (v3.3.2)32. Final gene predictions were confirmed again based on the first gene model and ab initio gene model using MAKER3 (v3.01.03)29 and EvidenceModeler (v1.1.1)33. In total, 26,430 protein-coding genes were predicted and annotated, with an average gene length of 1,184 bp (Table 4). The complete BUSCOs of predicted gene set were calculated as 98.9%.
Table 4.
Summary of gene annotation.
| Type | Number | Percent | |
|---|---|---|---|
| BLASTP (DIAMOND) | NCBI nr | 24,583 | 93.01 |
| Araport11 | 21,770 | 82.37 | |
| Protein domains (InterProScan) | 20,523 | 77.65 | |
| Gene Ontology (BLAST2GO) | 14,946 | 56.55 | |
| KEGG pathway (KAAS webtools) | 10,047 | 38.01 | |
| Annotated genes | 24,624 | 93.17 | |
| Total length of genes (bp) | 31,296,426 | ||
| Smallest gene length (bp) | 102 | ||
| Largest gene length (bp) | 15,765 | ||
| Average gene length (bp) | 1,184 | ||
| GC content (%) | 46.61 | ||
| Unannotated | 1,847 | 6.99 | |
| Total number of genes | 26,430 | ||
The predicted genes of A. rugosa were functionally annotated by comparing their similarities against those in the NCBI nonredundant (nr) protein database and the reference genome Araport11 of Arabidopsis thaliana using DIAMOND (v0.9.30.131)34 with an E-value cutoff of 1E-5. Conserved protein domains were predicted by InterProScan (v5.34-73.0)35. Gene Ontology analysis was conducted using the Blast2GO command line (v.1.4.4), and genes were assigned to metabolic pathways by comparing them to those in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database36 using the KEGG Automatic Annotation Server (KAAS) webtools (v2.1)37. A total of 24,624 genes were successfully annotated for A. rugosa, accounting for 93.2% of all predicted genes (Table 4 and Fig. 4). Predicted gene models were comparable to four other Lamiaceae species in aspects such as gene count, average CDS length, average exons per gene, and average exon and intron length (Table 5).
Fig. 4.
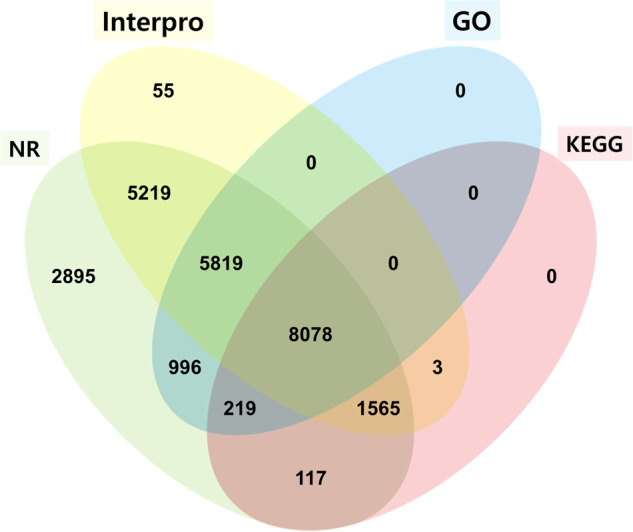
Venn diagram of the number of genes from A. rugosa with homology or functional classification using multiple public databases.
Table 5.
The comparison of the gene models annotated from A. rugosa genome and other Lamiaceae.
| Species (Accession number in GenBank) | Gene Number | Average CDS length | Average exons per gene | Average exon length | Average intron length |
|---|---|---|---|---|---|
| Agastache rugosa (GCA_031470985.1) | 26,867 | 1,177 | 5.21 | 226.01 | 405.90 |
| Perilla frutescens var. frutescens (GCA_019511825.2) | 38,941 | 1,259 | 5.19 | 242.42 | 395.75 |
| Perilla frutescens var. hirtella (GCA_019512045.2) | 23,675 | 1,252 | 5.08 | 246.41 | 398.23 |
| Salvia hispanica (GCF_023119035.1) | 36,995 | 1,379 | 9.20 | 277.52 | 42.18 |
| Salvia splendens (GCF_004379255.1) | 64,211 | 1,391 | 10.54 | 276.74 | 27.35 |
Ortholog and phylogenetic analysis
Orthologs between A. rugosa and eight other plants (seven from the order Lamiales: S. hispanica24, Salvia miltiorrhiza38, P. frutescens var. hirtella23, Paulownia fortune39, Erythranthe guttata40, Andrographis paniculata41, and Genlisea aurea42, along with one outgroup, Vitis vinifera43) were identified using OrthoFinder (v2.5.4)44. The sequences for these plants were sourced from the NCBI database (http://www.ncbi.nlm.nih.gov/). From these, 371 single-copy orthologous genes were extracted, concatenated, and aligned using the Multiple Alignment program for amino acid or nucleotide sequences (MAFFT)45. We then constructed a maximum likelihood phylogenetic tree of these orthologous genes using RAxML (v8.2.12)46 under the JTT model, Gamma Distributed With Invariant Sites (G + I), with a bootstrap value of 1000. Four species, namely A. rugosa, S. hispanica, S. miltiorrhiza, and P. frutescens var. hirtella, all of which belong to the Lamiaceae family, clustered in the same clade. Notably, A. rugosa exhibited a closer relation to the two Salvia species (Fig. 5). These findings are consistent with previous phylogenetic studies based on the chloroplast genome47.
Fig. 5.

Phylogenetic relationship of Lamiales species.
Data Records
The genomic Illumina sequencing data were deposited in the Sequence Read Archive at the NCBI (SRR24282004)48.
The genomic Nanopore sequencing data were deposited in the Sequence Read Archive at the NCBI (SRR24282001)49.
The transcriptome Illumina sequencing data were deposited in the Sequence Read Archive at the NCBI (SRR24282003)50.
The Hi-C sequencing data were deposited in the Sequence Read Archive at the NCBI (SRR24282002)51.
The final chromosome assembly was deposited in GenBank at the NCBI (GCA_031470985.1)52.
The annotation result of gene structure, functional prediction, and final chromosome assembly were deposited in the Figshare database (10.6084/m9.figshare.22730084)53.
Technical Validation
The integrity and concentration of the extracted DNA and RNA were assessed with a TapeStation 2200 and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA), respectively. In a comparative context, the complete BUSCO value for A. rugosa (98.9%) exceeds those of P. frutescens var. frutescens (92.7%)23, P. frutescens var. hirtella (92.5%)23, S. splendens (92.0%)25, and S. hispanica (97.8%)24, underscoring its relative completeness and quality within the Lamiaceae family.
Supplementary information
Acknowledgements
This work was carried out with the support of “Cooperative Research Program for Agriculture Science and Technology Development (Project No. RS-2022-RD010267)” Rural Development Administration, Republic of Korea.
Author contributions
H.-S.P., Y.-S.S. and J.-W.C. conceived and designed the study. S.R. was responsible for sample collection and extraction of both genomic DNA and RNA. H.-S.P. and N.-H.K. conducted the data analysis. Interpretation and discussion of the results were carried out by H.-S.P., I.H.J., N.-H.K., Y.-S.S. and J.-W.K. The initial draft of the manuscript was written by H.-S.P. and J.-W.C. Further manuscript revisions and editing were performed by I.H.J., J.G., D.S., C.K., J.-K.Y. and Y.-S.S. All authors have reviewed, contributed to, and approved of the final version of this manuscript.
Code availability
No in-house code or scripts were used in this study. Commands and pipelines used for data processing were executed using their corresponding default parameters.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yoon-Sup So, Email: yoonsupso@chungbuk.ac.kr.
Jong-Wook Chung, Email: jwchung73@chungbuk.ac.kr.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-023-02714-x.
References
- 1.Lee B-Y, Hwang J-B. Physicochemical characteristics of Agastache rugosa O. Kuntze extracts by extraction conditions. Korean Journal of Food Science and Technology. 2000;32:1–8. [Google Scholar]
- 2.Oh Y, et al. Attenuating properties of Agastache rugosa leaf extract against ultraviolet-B-induced photoaging via up-regulating glutathione and superoxide dismutase in a human keratinocyte cell line. Journal of Photochemistry and Photobiology B: Biology. 2016;163:170–176. doi: 10.1016/j.jphotobiol.2016.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Lee J-J, et al. Agastache rugosa Kuntze extract, containing the active component rosmarinic acid, prevents atherosclerosis through up-regulation of the cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27KIP1. Journal of Functional Foods. 2017;30:30–38. doi: 10.1016/j.jff.2016.12.025. [DOI] [Google Scholar]
- 4.Yeo HJ, et al. Effects of Carbohydrates on Rosmarinic Acid Production and In Vitro Antimicrobial Activities in Hairy Root Cultures of Agastache rugosa. Plants. 2023;12:797. doi: 10.3390/plants12040797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao H, et al. DFT study on the antioxidant activity of rosmarinic acid. Journal of Molecular Structure: THEOCHEM. 2005;719:177–183. doi: 10.1016/j.theochem.2005.01.029. [DOI] [Google Scholar]
- 6.Anand S, Pang E, Livanos G, Mantri N. Characterization of physico-chemical properties and antioxidant capacities of bioactive honey produced from Australian grown Agastache rugosa and its correlation with colour and poly-phenol content. Molecules. 2018;23:108. doi: 10.3390/molecules23010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desta KT, et al. The polyphenolic profiles and antioxidant effects of Agastache rugosa Kuntze (Banga) flower, leaf, stem and root. Biomedical chromatography. 2016;30:225–231. doi: 10.1002/bmc.3539. [DOI] [PubMed] [Google Scholar]
- 8.Park WT, et al. Influence of light-emitting diodes on phenylpropanoid biosynthetic gene expression and phenylpropanoid accumulation in Agastache rugosa. Applied Biological Chemistry. 2020;63:1–9. doi: 10.1186/s13765-020-00510-4. [DOI] [Google Scholar]
- 9.Bielecka M, et al. Age-related variation of polyphenol content and expression of phenylpropanoid biosynthetic genes in Agastache rugosa. Industrial Crops and Products. 2019;141:111743. doi: 10.1016/j.indcrop.2019.111743. [DOI] [Google Scholar]
- 10.Lam VP, Kim SJ, Bok GJ, Lee JW, Park JS. The effects of root temperature on growth, physiology, and accumulation of bioactive compounds of Agastache rugosa. Agriculture. 2020;10:162. doi: 10.3390/agriculture10050162. [DOI] [Google Scholar]
- 11.Lee SY, Xu H, Kim YK, Park SU. Rosmarinic acid production in hairy root cultures of Agastache rugosa Kuntze. World Journal of Microbiology and Biotechnology. 2008;24:969–972. doi: 10.1007/s11274-007-9560-y. [DOI] [Google Scholar]
- 12.Park WT, et al. Yeast extract and silver nitrate induce the expression of phenylpropanoid biosynthetic genes and induce the accumulation of rosmarinic acid in Agastache rugosa cell culture. Molecules. 2016;21:426. doi: 10.3390/molecules21040426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang, J. et al. Comparison of Pulegone and Estragole chemotypes provides new insight into volatile oil biosynthesis of Agastache rugosa. Frontiers in Plant Science, 771 (2022). [DOI] [PMC free article] [PubMed]
- 14.Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome biology. 2019;20:1–10. doi: 10.1186/s13059-019-1727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature communications. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y, Kim S. Genome size of 15 Lamiaceae taxa in Korea. Korean Journal of Plant Taxonomy. 2017;47:161–169. doi: 10.11110/kjpt.2017.47.2.161. [DOI] [Google Scholar]
- 18.Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC bioinformatics. 2018;19:1–10. doi: 10.1186/s12859-018-2485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zielińska S, Matkowski A. Phytochemistry and bioactivity of aromatic and medicinal plants from the genus Agastache (Lamiaceae) Phytochemistry Reviews. 2014;13:391–416. doi: 10.1007/s11101-014-9349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuentes-Granados RG, Widrlechner MP, Wilson LA. An overview of Agastache research. Journal of Herbs, Spices & Medicinal Plants. 1998;6:69–97. doi: 10.1300/J044v06n01_09. [DOI] [Google Scholar]
- 23.Zhang Y, et al. Incipient diploidization of the medicinal plant Perilla within 10,000 years. Nature Communications. 2021;12:5508. doi: 10.1038/s41467-021-25681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alejo-Jacuinde G, et al. Multi-omic analyses reveal the unique properties of chia (Salvia hispanica) seed metabolism. Communications Biology. 2023;6:820. doi: 10.1038/s42003-023-05192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia, K.-H. et al. Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Horticulture Research8 (2021). [DOI] [PMC free article] [PubMed]
- 26.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 27.Chen N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics. 2004;5:4.10. 11–14.10. 14. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- 28.Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiology. 2018;176:1410–1422. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC bioinformatics. 2011;12:1–14. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic acids research. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaharia, M. et al. Faster and more accurate sequence alignment with SNAP. arXiv preprint arXiv:1111.5572, (2011).
- 32.Stanke M, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology. 2008;9:1–22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 35.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du J, et al. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Molecular BioSystems. 2014;10:2441–2447. doi: 10.1039/C4MB00287C. [DOI] [PubMed] [Google Scholar]
- 37.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic acids research. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan X, et al. Chromosome-level genome assembly of Salvia miltiorrhiza with orange roots uncovers the role of Sm2OGD3 in catalyzing 15, 16-dehydrogenation of tanshinones. Horticulture Research. 2023;10:uhad069. doi: 10.1093/hr/uhad069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Y, et al. Genomic insights into the fast growth of paulownias and the formation of Paulownia witches’ broom. Molecular Plant. 2021;14:1668–1682. doi: 10.1016/j.molp.2021.06.021. [DOI] [PubMed] [Google Scholar]
- 40.2014. NCBI GenBank. GCA_000504015.1
- 41.Liang Y, et al. Chromosome level genome assembly of Andrographis paniculata. Frontiers in Genetics. 2020;11:701. doi: 10.3389/fgene.2020.00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leushkin EV, et al. The miniature genome of a carnivorous plant Genlisea aurea contains a low number of genes and short non-coding sequences. BMC genomics. 2013;14:1–11. doi: 10.1186/1471-2164-14-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.2009. NCBI GenBank. https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_030704535.1/
- 44.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome biology. 2019;20:1–14. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in bioinformatics. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Wang H, Zhou B, Yue Z. The complete chloroplast genomes of Lycopus lucidus and Agastache rugosa, two herbal species in tribe Mentheae of Lamiaceae family. Mitochondrial DNA Part B. 2021;6:89–90. doi: 10.1080/23802359.2020.1847617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.2023. NCBI Sequence Read Archive. SRR24282004
- 49.2023. NCBI Sequence Read Archive. SRR24282001
- 50.2023. NCBI Sequence Read Archive. SRR24282003
- 51.2023. NCBI Sequence Read Archive. SRR24282002
- 52.2023. NCBI GenBank. GCA_031470985.1
- 53.Park H-S, Chung J-W. 2023. Agastache rugosa genome. figshare. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2014. NCBI GenBank. GCA_000504015.1
- 2009. NCBI GenBank. https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_030704535.1/
- 2023. NCBI Sequence Read Archive. SRR24282004
- 2023. NCBI Sequence Read Archive. SRR24282001
- 2023. NCBI Sequence Read Archive. SRR24282003
- 2023. NCBI Sequence Read Archive. SRR24282002
- 2023. NCBI GenBank. GCA_031470985.1
- Park H-S, Chung J-W. 2023. Agastache rugosa genome. figshare. [DOI]
Supplementary Materials
Data Availability Statement
No in-house code or scripts were used in this study. Commands and pipelines used for data processing were executed using their corresponding default parameters.