

Abstract
Ubiquitin (Ub) and ubiquitin-like (Ubl) pathways are critical post-translational modifications that determine whether functional proteins are degraded or activated/inactivated. To date, >600 associated enzymes have been reported that comprise a hierarchical task network (e.g., E1–E2–E3 cascade enzymatic reaction and deubiquitination) to modulate substrates, including enormous oncoproteins and tumor-suppressive proteins. Several strategies, such as classical biochemical approaches, multiomics, and clinical sample analysis, were combined to elucidate the functional relations between these enzymes and tumors. In this regard, the fundamental advances and follow-on drug discoveries have been crucial in providing vital information concerning contemporary translational efforts to tailor individualized treatment by targeting Ub and Ubl pathways. Correspondingly, emphasizing the current progress of Ub-related pathways as therapeutic targets in cancer is deemed essential. In the present review, we summarize and discuss the functions, clinical significance, and regulatory mechanisms of Ub and Ubl pathways in tumorigenesis as well as the current progress of small-molecular drug discovery. In particular, multiomics analyses were integrated to delineate the complexity of Ub and Ubl modifications for cancer therapy. The present review will provide a focused and up-to-date overview for the researchers to pursue further studies regarding the Ub and Ubl pathways targeted anticancer strategies.
Key words: Ub and Ubl modifications, Ub-activating enzyme, Ub-conjugating enzyme, Ub ligase, Multiomics analyses, Drug discovery, Small molecule inhibitor, Molecular glue
Graphical abstract
This review summarizes and discusses the therapeutic targets in Ub and Ubl pathways with additional multiomics data, as well as advancements in drug discovery.

1. Introduction
Ubiquitination (or ubiquitylation) is a common and essential post-translational modification that regulates several biological processes by degrading or activating/inactivating numerous proteins. In the 1980s, Hershko et al.1,2 described ubiquitination as an ATP-dependent enzymatic reaction cascade that induced the conjugation of a highly conserved 76-residue Ub to substrates. For their contribution to the field of ubiquitination-mediated proteolysis, they were awarded the Nobel Prize in 2004. The ubiquitination reaction is sequentially catalyzed by three enzymes: Ub-activating enzyme (E1), Ub-conjugating enzyme (E2) and Ub ligase (E3)3,4. The first step of ubiquitination involves the ATP-dependent activation of Ub by an E1. Subsequently, the activated Ub is transferred to an E2 via a transthiolation reaction. Finally, a specific E3 delivers the activated Ub to the substrate via covalent attachment (Fig. 1A). To date, 2 E1s, 33 E2s, and >600 E3s have been identified in the Ub pathway. Furthermore, several Ubl proteins have been identified as post-translational modifiers, such as neural precursor cell expressed, developmentally down-regulated gene 8 (NEDD8), and small ubiquitin like modifier (SUMO)4. These proteins share a similar Ub-fold domain and the ability to conjugate to substrates via a cascade reaction driven by three evolutionarily related enzymes4.
Figure 1.

Process of ubiquitination and its associated functions. (A) Schematic diagram of the ubiquitination and deubiquitination process and their related enzymes. (B, C) Ubiquitination involves in almost all tumor associated pathways and cell death pathways.
Accumulating studies have revealed that Ub and Ubl modifications are related to the development of distinct human diseases, especially in the onset and progression of cancer. More than 90,000 references to Ub in PubMed highlight the significance of Ub and Ubl modifications for cellular homeostasis by 2022, August. Over 10,000 studies have shown that Ub and Ubl modifications play a crucial role in regulating tumor growth, proliferation, differentiation, metabolism, and the formation of tumor microenvironment3 (Fig. 1B). Furthermore, they are also involved in almost all cell death processes, such as apoptosis, autophagy, senescence, necrosis, lysosome-dependent cell death, necroptosis, and ferroptosis (Fig. 1B). These findings suggest the importance of Ub or Ubl pathways for tumorigenesis. In the present review, we have summarized the current efforts and prospects of the Ub and Ubl pathways in tumors, including oncogenic or tumor-suppressive functions, clinical significance, regulatory mechanisms, and targeted drugs. Moreover, we have summarized some important challenges that are faced by current targeting Ub or Ubl studies, and how advances in multiomics profiles can help us to deeply understand and address these challenges.
2. Advancements in the studies of Ub and Ubl-associated enzymes in cancer
Abnormality of Ub and Ubl pathways in tumors is closely related to the dysregulation of their catalytic enzymes, including E1s, E2s, E3s and deubiquitinases3,5, 6, 7, 8. These anomalies are regulated at various layers, including overexpression, depletion, or mutation. Given that the best characterized function of Ub and Ubl pathways is to maintain cellular homeostasis, these pathways have been identified as attractive targets for cancer therapy. Therefore, a comprehensive overview of the anomalies and their roles of Ub- and Ubl-associated enzymes, including E1s, E2s, E3s and deubiquitination enzymes, in tumorigenesis is essential. In the following sections, we have summarized the latest advancements in the studies of Ub and Ubl-associated enzymes in tumors. We also discussed neddylation, a representative Ubl pathway via adding NEDD8 to substrates, to provide insight into the feature of the Ubl conjugation machinery in cancer.
2.1. Targeting E1s suppresses tumor initiation or progression
Two Ub-activating enzymes UBA1 (also known as UBE1) and UBA6 have been identified so far. In 1981, UBA1 was identified as the first E1, which catalyzes Ub activation in 99% cellular ubiquitination reactions9,10. It was not until 2007 that UBA6 was identified as an alternative E1, which activated Ub with the same efficiency as that of UBA1 in approximately 1% cellular ubiquitination reactions11. UBA1 and UBA6 possess three common domains, the adenylation domain that binds with ATP, catalytic cysteine domain that forms the thioester bond with Ub, and Ub-fold domain (UFD) that interacts with E210 (Fig. 2). The difference in UFD determines the specificity of the E1–E2 pairs. UBA1 favors pairing with UBE2A, UBE2B, UBE2C, UBE2D1–4, UBE2G1–2, UBE2H, UBE2J1–2, UBE2K, UBE2L3, UBE2Q2, UBE2R1–2, UBE2S, and UBE2T, whereas UBA6 specificity activates UBE2Z (also known as Use1). Additionally, UBA6 also pairs with other UBA1 paired E2 enzymes, including UBE2G2, UBE2S, UBE2D1–4, UBE2E3, UBE2T, and UBE2L312 (Fig. 2). Recently, a genome-wide pan-cancer CRISPR/Cas9 screening was conducted to investigate the difference between UBA1 and UBA6 in suppressing tumor cell growth10. The screening results revealed that UBA1 depletion inhibited the growth of the analyzed cancer cell lines (n = 582), whereas UBA6 knockdown suppressed the growth of only 10.9% of the analyzed cancer cell lines10, thereby indicating that UBA1 is more essential than UBA6 for tumor growth.
Figure 2.

Schematic structure of E1s (UBA1 and UBA6), and the specificity of the E1–E2 pairs.
As for Ubl E1, NEDD8 activating enzyme E1 (NAE), a heterodimer of NAE1 and UBA3, is significantly up-regulated in a myriad of tumor tissues. The overexpression of NAE promotes the tumorigenesis and tumor progression, while its inhibition via pharmacological (e.g., MLN4924) or genetic approaches (e.g., siRNA) suppresses tumor growth13, 14, 15. In mechanism, targeting NAE to the inhibition of neddylation 1) induces apoptosis via inhibiting the degradation of ATF4, IκBα and NOXA13,16, 17, 18, 19, 20, 21; 2) promotes senescence via inducing the accumulation of p21 and p2722, 23, 24; 3) facilitates cell cycle arrest via suppressing the degradation of p21, p27 and Wee125, 26, 27; 4) induces the re-initiation of DNA replication (re-replication) by increasing the levels of CDT1 and ORC128,29; 5) inhibits the inflammation via inducing the accumulation of IKBα or RHBO30,31. 6) suppresses the infiltration of immune suppressive cells (e.g., MDSCs and TAMs) via NFκB–CCL2/CXCL1 axis32, 33, 34; 7) inhibits tumor angiogenesis via inducing the accumulation of RhoA35; and 8) suppresses the extravasation of cancer cells via disrupting actin cytoskeleton formation25. Furthermore, the inactivation of neddylation also induces protective autophagy in tumor cells via 1) inducing the accumulation of DEPTOR (a well-known inhibitor of mTORCs)36; 2) inhibiting the degradation of HIF1α to activate REDD1–TSC1 pathway37; and 3) blocking the degradation of IκBα to activate the ROS–ATF3 pathway38 (Fig. 3).
Figure 3.

Regulatory and pro-tumoral mechanisms of neddylation pathway. N8: NEDD8, neural precursor cell expressed, developmentally down-regulated gene 8; TSME: tumor suppressive microenvironment.
2.2. Functions and regulatory mechanisms of E2s in cancer
Up to 33 Ub-associated E2s have been identified, which share the common catalytic ubiquitin-conjugating (UBC) domain39. Through the UBC domain, a free E2 interacts with E1 to afford an activated Ub from the E1–Ub adduct. After coupling with a specific E3, E2–Ub transfers Ub to the target substrates to form Ub chains39. Notably, the type of Ub chains is determined by E2. For example, UBE2C and UBE2S are specific Lys11-linked ubiquitin-conjugating enzymes40, 41, 42, 43, whereas UBE2D and UBE2R1 catalyze the formation of Lys48-linked polyubiquitin chain44,45.
UBE2C, also known as UbcH10, delivers Ub to the substrate, which is followed by UBE2S-driven Ub extension to form the Lys11-polyubiquitination chain40, 41, 42, 43. Both UBE2C and UBE2S are overexpressed in various cancer cell lines and tumor tissues46,47 and facilitate malignant transformation of the cells or tumor growth by promoting the degradation of tumor-suppressing substrates (e.g., p27 and p53)40, 41, 42, 43,48, 49, 50, 51. UBE2C transgenic mice is susceptible to developing spontaneous tumors when exposed to certain carcinogens (e.g., anthracene derivatives)46. UBE2C and UBE2S are also activated by certain oncogenes, such as estrogen receptor 1 and androgen receptor (AR)50,52, AKT153, APC/CCDH1, and Emi142,54, whereas they are suppressed by the tumor suppressor p5350,52. Interestingly, there appears to be some divergence in the tumor-promoting functions of UBE2S and UBE2C in cancers. For instance, it has been observed that the deletion of UBE2C, but not UBE2S, regulates the degradation of DEPTOR, which in turn suppresses lung cancer cell growth55.
UBE2D (also known as UBCH5) efficiently delivers Ub to the substrate, while UBE2R1 (also known as Cdc34) promotes the extension of the Lys48-linked polyubiquitin chain44,45. According to clinical tumor sample analysis, UBE2D and UBE2R1 are overexpressed in numerous tumor tissues, such as lung cancer56,57, hepatocellular carcinoma58,59 and multiple myeloma60, and regulate the ubiquitination and degradation of several essential tumor-suppressive substrates, including IKBα, cyclin-dependent kinase inhibitor p21 and p2745,61, 62, 63. Additionally, UBE2R1 stabilizes the epidermal growth factor receptor by competing with UBE2D-matched Casitas B-lineage lymphoma (c-Cbl), thereby promoting the proliferation of lung cancer cells56. This research reveals a previously unknown function of E2s, describing that they can directly stabilize substrates instead of acting as Ub carriers to promote the ubiquitination and degradation of substrates.
In contrast to the Ub pathway, neddylation involves only two E2 enzymes, UBE2M and UBE2F. In general, UBE2M pairs with RBX1 to promote the neddylation of cullin 1, 2, 3, 4A and 4B, while UBE2F pairs with RBX2 to enhance the neddylation of cullin 5. Our studies, as well as other studies, have demonstrated that UBE2M and UBE2F are up-regulated in various tumors17,64,65. Consequently, the overexpression of UBE2M primarily leads to the accumulation of substrates on the cullin 1, 2, 3, 4A or 4B (e.g., ATF4, p21, p27, CDT1 and ORC1), whereas the overexpression of UBE2F promotes the accumulation of cullin5 substrates (e.g., NOXA) (Fig. 3). Interestingly, there is a cross-talk between these two independent E2s. The activated UBE2M serves as a dual E2 to induce the proteolysis degradation of UBE2F, thereby inducing the accumulation of pro-apoptosis protein NOXA, which promotes apoptosis in lung cancer cells66.
2.3. Functions and regulatory mechanisms of E3s in cancer
More than 600 E3 ligases have been identified in humans so far, which contribute to the functional diversity and substrate specificity of ubiquitination. According to their structures and functions, E3 ligases are grouped into three main categories: really interesting new gene (RING), homologous to E6-associated protein C-terminus (HECT), and RING-in-between-RING (RBR) E3 ligases (Fig. 4). Especially, RING E3 ligases, the largest E3 ligases with more than 600 members, were summarized in details in this section.
Figure 4.

Structures and components of RING, HECT and RBR E3 ligases. According to the active form, RING E3s are divided into 2 groups, including the single-subunit RING E3 ligases and multiple subunit E3 ligases.
2.3.1. Representative RING E3s in cancer
RING E3 ligases share a common RING domain or U-box domain for binding with E2–Ub thioester. RING E3s typically transfer Ub directly from E2 to the targeted substrates. All RING E3 ligases have an E2 binding domain, but not every RING E3 ligase possesses the substrate recognition domain. Therefore, RING E3 ligases are divided into two groups: single-subunit RING E3 ligases (e.g., c-Cbl, MDM2, and IAP) and multi-subunit RING E3 ligases (e.g., Cullin–RING ligases [CRLs])67 (Fig. 4). CRLs, also known as the largest RING E3 ligases, comprise several subunits, including RING, cullin, adaptor, and/or substrate receptor proteins68 (Fig. 4). RBX1 and RBX2, also known as the two classical RING proteins of CRLs, deliver Ub or Ubl from the E2–Ub or E2–Ubl complex to the substrate68. Cullins constitute the skeletal proteins of CRLs that assemble the RING, adaptor, and/or receptor proteins to deliver Ub to the recruited substrates68. Eight distinct cullins paired with RBX1 or RBX2 form CRL1, CRL2, CRL3, CRL4A, CRL4B, CRL5, CRL7, and CRL968, which are primarily involved in regulating certain specialized cellular functions during tumor initiation and progression.
2.3.1.1. CRL1 E3 ligases
CRL1, also known as SKP1-Cullin1-F-box protein (SCF), comprises four subunits: RBX1, cullin1, adaptor protein Skp1, and substrate receptor F-box proteins. F-box proteins have two essential functional domains, the F-box motif for Skp1-mediated cullin1 recruitment and the C-terminal domain for substrate recognition. Based on the latter, F-box proteins are divided into three categories: 10 members of FBXWs with WD40 repeats, 22 members of FBXLs with leucine-rich repeats (LRR), and 37 members of FBXOs with a different domain or no recognizable motifs69,70. Among them, S-phase kinase-associated protein 2 (Skp2), F-box and WD repeat domain-containing 7 (FBXW7), and β-transducin repeat-containing E3 ubiquitin protein ligase (β-TRCP) are well-characterized F-box proteins in cancer studies (Fig. 5).
Figure 5.

Representative receptors or adaptors of CRL E3 ligases and their role in tumor initiation and progression. Green: tumor suppressive role; Orange: tumor promoting role; Blue: dual roles in a context-dependent manner.
In 1995, Skp2 (also known as FBXL1, FBL1, and p45) was identified as an essential cytokinetic regulator of the cyclin A–CDK2 complex for S-phase entry71. In the past 26 years, Skp2 has been established as an oncogene that promotes the progression of various tumors, such as those of lung cancer72, nasopharyngeal carcinoma73, hepatocellular carcinoma74 and gastric carcinoma75. Mechanistically, Skp2 recognizes substrates via its LRR domain in a phosphorylation kinase-dependent manner, subsequently promoting the formation of a Lys48-linked polyubiquitin chain for substrate degradation via the 26S proteasome or the formation of a Lys63-linked polyubiquitin chain for protein–protein interaction (PPI) and activation7. Therefore, Skp2 substrates are divided into two categories: proteolytic substrates, including p2176, p2777, p5778, CDT179, ORC180 and FOXO181 and nonproteolytic substrates, including AKT73,82, YAP83, MTH184, LKB185, NBS186 and Twist87. Notably, proteolytic Skp2 substrates primarily exhibit tumor-suppressive activity, inhibiting tumor growth or promoting cell death pathways (e.g., apoptosis or senescence), whereas the nonproteolytic Skp2 substrates primarily promote tumor growth or metastasis7. Thus, Skp2 constitutes a promising pharmacological target in cancer therapy owing to its essential role in facilitating tumor initiation and progression.
FBXW7, also known as FBW7, CDC4, and SEL-10, is widely regarded as a tumor suppressor. Numerous FBXW7 substrates have been identified as oncoproteins that promote tumor growth, including Aurora A88,89, BRAF90, c-Jun91,92, c-Myc93,94, GFI195, HIF1α96,97, SHOC298, KLF599,100, MCL1101,102, NOTCH1103, 104, 105 and ZNF322A106, further highlighting the tumor-suppressive property of FBXW7. FBXW7 encodes three isoforms, namely FBXW7α, FBXW7β, and FBXW7γ, which display distinct subcellular distributions. FBXW7α is a major isoform in human tissues and cells, located in the nucleoplasm; FBXW7β resides in the cytoplasm, whereas FBXW7γ is primarily found in the nucleolus107,108. Therefore, different FBXW7 isoforms may cooperatively promote the ubiquitination of oncogenic proteins. Loss-of-function FBXW7 mutations have been frequently observed in tumors where they intensify tumor progression, chemoresistance, and radiation tolerance90,95,101,107,109,110. Additionally, FBXW7 is activated by p53111 and suppressed by some microRNA, such as miR-24112, miR-27113, miR-32114, miR-223115 and miR-367116. Thus, targeting the negative regulators or substrates of FBXW7 can serve as an effective anticancer strategy.
β-TRCP1 (also known as FBXW1, FBXW1A, and FWD1) and β-TRCP2 (also known as FBXW11, FBXW1B, and HOS) are subordinate members of the β-TRCP family that exert context-dependent oncogenic or tumor-suppressive effects. For example, β-TRCP targets numerous tumor suppressors, including IκBα117,118, Wee1119, p53120, DEPTOR121, FBXW2122, BimEL123, Mxi1124, PDCD4125, and pro-caspase3126, for degradation to facilitate tumor growth, whereas β-TRCP ubiquitinates certain important oncoproteins, including HER2127, YAP128, MDM2129, PD-L1130 and MCL1131, to suppress tumor initiation or progression (Fig. 5). Clinical tissue analysis revealed that β-TRCP is upregulated in various tumors (e.g., colorectal cancer and hepatoblastomas) and is associated with decreased overall survival in patients with tumor132,133, whereas somatic mutations of β-TRCP promote the progression of certain tumors (e.g., gastric and prostate cancers)134,135.
2.3.1.2. CRL2 E3 ligases
CRL2 comprises RBX1, cullin2, adaptor protein Elongin B/C, and substrate recognition subunit136. The most characteristic substrate recognition subunit of CRL2 is the Von Hippel-Lindau tumor suppressor (VHL). VHL was initially cloned in 1993137, and subsequently its mutations were identified in most ccRCC cases in 1994138. The classical substrates of VHL include hypoxia-inducible factors (HIFs)136. Loss-of-function VHL mutations induce HIF accumulation, which further facilitates tumor angiogenesis by promoting HIF-mediated transcription of essential oncogenes, including VEGF and EPO139. Loss-of-function VHL mutations and elevated HIF and VEGF levels have been frequently observed in tumors, especially in those of ccRCC140, 141, 142. Hence, targeting the VHL–HIF–VEGF axis constitutes a major therapeutic intervention strategy for ccRCC142,143. In addition to HIFs, VHL substrates include Zinc fingers and homeoboxes 2 (ZHX2)144, protein kinase C145, β2-adrenergic receptor146, and the RNA polymerase II subunit hsRPB7147. ZHX2, an NF-κB transcriptional activator, offers an additional therapeutic option for patients with HIF-insensitive or -resistant ccRCC144.
2.3.1.3. CRL3 E3 ligases
Unlike other CRLs, CRL3 comprises RBX1, cullin3, and broad-complex-tramtrack-bric-α-brac (BTB) adaptor protein. BTB proteins not only directly bind to cullin3 but also recognize substrates via their BTB domain (also known as the POZ domain). Functionally, BTB proteins facilitate the 26S proteasome-mediated substrate degradation by promoting the formation of Lys48-linked polyubiquitin chains. Conversely, BTB proteins catalyze Lys63-and Lys33-linked ubiquitination, affecting PPI and subcellular localization148. To date, 188 putative BTB proteins have been identified in the human genome, which are grouped into five subfamilies: the BTB-kelch (n = 55), BTB–Zn finger (n = 49), K+ voltage-gated channel (n = 27), and KCTD (n = 25) subfamilies and others (n = 32)148. Kelch-like ECH-associated protein 1 (Keap1) and speckle-type BTB/POZ protein (SPOP) constitute two classical substrate adaptor proteins of CRL3 (Fig. 5).
Keap1 was identified as a nuclear factor erythroid 2-related factor 2 (Nrf2) inhibitor. Nrf2 is a key regulator of cellular antioxidant response that stimulates the transcription of >200 antioxidant genes, including HO1 and NQO1149. Previous studies have reported that Nrf2 is ubiquitinated by cytoplasmic Keap1 under normal conditions. Conversely, the oxidation of Keap1 by reactive oxygen species or electrophiles failed to ubiquitinate Nrf2, inducing its accumulation. The accumulated Nrf2 translocated into the nucleus, thereby increasing the transcription of antioxidant genes and preventing oxidative damage to normal cells and tissues. Thus, Nrf2-knockout mice are susceptible to spontaneous tumors150,151. However, numerous studies have shown that Nrf2 hyperactivation protects cancer cells from chemotherapeutic agents or radiotherapy-induced oxidative stress149.
SPOP regulates substrate ubiquitination and affects the progression of several human cancers, including lung cancer152, prostate cancer153, 154, 155, 156, RCC157,158, endometrial cancer155,159 and breast cancer160. Generally, SPOP acts as a tumor suppressor in multiple cancers (e.g., lung and prostate cancers) by mediating the degradation of several oncoproteins, including ERG156, PD-L1161, cyclin E1162, ATF2163, c-Myc164, AR165, steroid receptor coactivator 3166, BRD2, BRD3 and BRD4153, 154, 155. However, SPOP exhibits an oncogenic function in RCC by mediating the degradation of phosphatase and tensin homologue (PTEN), proapoptotic molecule Daxx, Gli2, DUSP6, and DUSP7157. The subcellular localization of SPOP primarily affects its dual functions. SPOP accumulates in the nucleolus of cells to mediate oncoprotein degradation154,167,168. Conversely, SPOP demonstrates predominantly cytoplasmic localization in ccRCC due to the VHL mutations and hypoxic conditions in ccRCC, consequently promoting the degradation of cytoplasmic tumor suppressors (e.g., PTEN, Daxx, Gli2, DUSP6, and DUSP7)157.
2.3.1.4. CRL4 E3 ligases
CRL4 is subdivided into CRL4A and CRL4B based on the skeleton proteins of cullin 4A and cullin 4B, respectively. Damage-specific DNA-binding protein 1 (DDB1) is an essential adaptor protein of CRL4 that detects and repairs damaged DNA. Meanwhile, DDB1-cullin4 associated factors (DCAFs) serve as substrate receptors169,170. Three representative DCAFs in anticancer therapy are damage-specific DNA-binding protein 2 (DDB2), denticleless E3 ubiquitin protein ligase homolog (DTL), and cereblon (CRBN) (Fig. 5).
DDB2 is a WD40 repeat-containing protein that heterodimerizes with DDB1 to repair DNA damage or inhibit the transcription of certain oncogenes. When DDB2 is directly linked to damaged DNA, it assembles the CRL4 complex to ubiquitylate histones H2A171, H3, or H4172, thereby facilitating DNA repair by weakening the histone–DNA interaction. Furthermore, DDB2 was ubiquitinated by CRL4A at the lesion site173,174, thus inducing the accumulation of damage recognition factor XPC to remove damaged pyrimidine dimers174,175. Therefore, DDB2 knockout mice are susceptible to skin carcinogenesis, and the upregulation of DDB2 suppresses the onset and progression of UV-induced squamous cell carcinoma176, 177, 178. In addition, DDB2 inhibits the transcription of HIF1α179, ALDHA1180, Snail and ZEB1181, thus inhibiting tumor growth or epithelial-to-mesenchymal transition.
As a potential oncoprotein, DTL (also known as CDT2, DCAF2, RAMP, and L2DTL) recognizes and ubiquitinates its substrates to master genome stability182,183. Inducing the accumulation of CDT1184, p21185 and PR-set7/set8186 in the S phase in a proliferating cell nuclear antigen (PCNA)-dependent manner, CDT2 knockdown inhibits DNA replication. Knockdown of CDT2 triggers G2 cell cycle arrest by preventing the degradation of CHK1 in a PCNA-independent manner187. In contrast, overexpression of CDT2 accelerates the degradation of the substrate, CDT1, subsequently promoting cancer cell re-replication and impairing senescence or apoptosis188.
CRBN, an important substrate recognition receptor of the CRL4, is the primary target of immunomodulatory drugs (IMiDs), such as thalidomide and its derivatives (e.g., pomalidomide, lenalidomide, CC-885) for hematological malignancy treatment. CRBN facilitates the ubiquitination and degradation of the Ikaros family (IKZF1 and IKZF3) in a lenalidomide-dependent manner189, 190, 191 and that of ARID domain-containing protein 2 (ARID2) in a pomalidomide-dependent manner to suppress the survival and proliferation of multiple myeloma cells192. In acute myeloid leukemia cells, CRBN promotes the degradation of G1 to S-phase transition protein 1 to exert a broad-spectrum anticancer effect in a CC-885-dependent manner189. Furthermore, CRBN stabilizes CD147 and monocarboxylate transporter 1 (MCT1) to promote angiogenesis, tumor growth, or lactate export, whereas IMiDs interact with CRBN to disrupt the CD147–MCT1 complex and exert antitumor and teratogenic effects193. Interestingly, CRBN also functions as a transmembrane protein-specific co-chaperone molecule of HSP90 to modulate HSP90–AHA1 activity, whereas IMiDs inhibit their interaction to suppress multiple myeloma cell growth194.
2.3.1.5. CRL5 E3 ligases
CRL5 comprises the scaffold protein cullin 5, RING protein RBX2, adaptor protein Elongin B/C, and substrate recognition receptor protein SOCS. The primary substrate receptors of CRL5 include 37 members, categorized into 5 types: SOCS, ASB, SPSB, and WSB subfamilies and others (such as Rab40 and MUF1)195.
Suppressor of cytokine signaling 1 (SOCS1, also known as SSI1) and SOCS3 (also known as SSI3) are two extensively studied SOCS proteins of CRL5 that suppress inflammatory reaction and exert context-dependent effects in tumorigenesis195. SOCS1 depletion in macrophages196 or dendritic cells197 in conditional knockout mice enhances antitumor inflammation or antigen-specific antitumor immunity. Conversely, SOCS3 or cullin 5 depletion inhibits integrin β1 degradation, subsequently activating the focal adhesion kinase/SRC (FAK/SRC) signaling pathway, thereby promoting small-cell lung cancer metastasis198. Similarly, SOCS1 inhibits STAT3 phosphorylation and limits granulocyte–macrophage colony-stimulating factor and interleukin-6 production, thus inhibiting myeloid-derived suppressor cell differentiation and increasing antitumor immunity199. Additionally, elevated SOCS3 levels improve the susceptibility of castration-resistant prostate cancer cells toward natural killer cells200. Consistent with these findings, low SOCS1 and SOCS3 expression levels have been frequently observed and positively associated with poor prognosis in patients with small-cell lung198 and breast cancer201.
SplA/ryanodine receptor domain and SOCS box-containing 1 (SPSB1, also known as SSB1) ubiquitinates a key inflammatory effector, inducible NO synthase (iNOS)202,203. iNOS has multiple cellular origins and harbors both tumor prosurvival and suppressive functions204. In addition to iNOS, SPSB1 promotes p21 proteasomal degradation to increase the viability and migration of ovarian cancer cells205. Furthermore, SPSB1 promotes breast cancer recurrence by preventing chemotherapy- or HER2/neu inhibitor-induced apoptosis in tumor cells206.
2.3.2. HECT E3s in tumorigenesis
In contrast to RING E3s, HECT ligases ubiquitinate their substrates in two steps. They first load the activated Ub onto themselves to produce the E3–Ub intermediate via the HECT domain, and then they transfer Ub onto the substrates5 (Fig. 4). HECT E3s are divided into three subfamilies based on the N-terminal structure of the HECT domain: 1) Nedd4 subfamily, also known as C2–WW–HECT, whose structures contain two to four tryptophan–tryptophan (WW) domains; 2) HERC (HECT and RCC1-like domain) subfamily, which contains one or more RCC1-like domains; and 3) other HECT E3s, such as E6AP, HECTD2, G2E3, TRIP12, EDD, HACE1, and HUWE15 (Fig. 4). NEDD4, one of the brain development regulators, is the first Nedd4 subfamily member to be identified207. In tumors, NEDD4 ubiquitylates and promotes degradation of PTEN, an essential tumor suppressor, to promote tumor growth208, whereas CK1α competitively antagonizes NEDD4-mediated PTEN ubiquitination to inhibit lung tumor growth209. In addition, NEDD4 promotes the formation of K63-linked polyubiquitination chains of MDM2 to stabilize MDM2, thus promoting the degradation of p53 and inhibiting the DNA damage response210. Furthermore, NEDD4 promotes the proteasomal degradation of VDAC2/3, the voltage-dependent anion channel, to suppress erastin-induced ferroptosis211. On the contrary, the most prominent feature of NEDD4-null mice is delayed embryonic development and growth as a result of a decrease in insulin and insulin-like growth factor 1 (IGF-1)212. Meanwhile, knockout of NEDD4 upregulates PDL1 and inhibits CD8+ T cell infiltration in bladder cancer cells to avoid immune surveillance213. Besides, NEDD4 inhibits RBX2 to sensitize etoposide-induced apoptosis214. This dual function of NEDD4 in tumors justifies further research into NEDD4 for future anticancer therapy.
2.3.3. RBR E3s in tumorigenesis
RBR E3s, possessing the characteristics of both RING and HECT E3s, comprise a RING1 domain, an in-between-RING (IBR) domain, and a RING2 domain6,215. RBR E3s catalyze the ubiquitination reaction in three steps. They first recognize the E2–Ub complex with the RING1 domain, then transfer Ub onto RING2 to form the E3–Ub intermediate, and finally transfer Ub onto the substrates6,215 (Fig. 4). Herein, 14 RBR E3s, including Parkinson protein 2 (also known as Parkin), RNF19A, RNF19B, RNF144A, RNF144B, RNF216, RNF217, RBCK1, RNF31 (also known as HOIP), RNF14, ARIH1, ARIH2, PARC, and Ankib1, identified in humans (Fig. 4)6.
Parkin is a well-known RBR E3 ligase with neuroprotective function216. Parkin is often downregulated in various tumors and demonstrates tumor-suppressive activity217, 218, 219. Parkin downregulation is typically associated with poor overall survival of patients with tumors217,218. Parkin null mice are susceptible to developing carcinoma220. In mechanism, Parkin facilitates apoptosis via promoting the degradation of the antiapoptosis protein MCL1 during mitochondrial depolarization221, whereas Parkin depletion promotes the activation of PI3K–AKT pathway in a PTEN-dependent manner to facilitate tumorigenesis218. In addition, Parkin suppresses Kras-driven pancreatic tumorigenesis and HIF1α-mediated Warburg effect by promoting the degradation of SLC25A37 and SLC25A28222. Furthermore, Parkin promotes the degradation of phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme of serine synthesis, to inhibit serine synthesis and tumor progression223. Additionally, Parkin promotes the formation of the K33-linked polyubiquitination chain of RIPK3 to inhibit necrosome assembly, hence preventing necroptosis and tumorigenesis224.
2.4. Functions of representative deubiquitinases (DUBs) in cancer
Protein deubiquitination is a reversal of ubiquitination that uses deubiquitinases (DUBs) to remove Ub from substrates. Based on their sequence and domain conservation, ∼100 identified DUBs have been classified into six families8. Five families of cysteine proteases include USPs, OTUs, UCHs, MJDs, and MINDYs8,225. In addition, the JAB1/MPN/MOV34-domain-containing metalloproteases (JAMMs) comprise 16 zinc metallopeptidases8,225. DUBs primarily remove Ub from substrates to modulate protein stability, enzymatic activity, and subcellular localization8,225. Therefore, DUB disruption contributes to tumor initiation and progression by interfering with the dynamic equilibrium of ubiquitination.
Based on their roles in tumorigenesis, DUBs are classified into three types. The first type is prosurvival DUBs, such as USP2 and USP7 in the USP family and CSN5 in the JAMM family. USP2 is overexpressed in several human tumor tissues and is crucial for tumor growth as it inhibits the ubiquitination and degradation of oncoproteins, including Skp2226, Twist227, MDM2228,229, TGFBR1230, fatty acid synthase (FAS)231, PD-L1232, and cyclin D1233. USP7 activation is associated with poor overall patient survival in multiple tumors. USP7 overexpression stabilizes numerous oncoproteins, such as EZH2234, PHF8235, MDM2236, LSD1237, β-Catenin238 and DNMT1239,240, thereby stimulating tumor growth or drug resistance. CSN5 facilitates tumor growth by inhibiting the degradation of Snail241, ZEB1242, surviving243 and FOXM1244, whereas curcumin-induced CSN5 depletion decreases PD-L1 expression and enhances the anticancer efficacy of anti-CTLA4 drugs245.
Tumor-suppressive DUB families, CYLD lysine 63 deubiquitinase (CYLD) in the USP family and OTU deubiquitinase 1 (OTUD1) in the OTU family, constitute the second type of DUBs. CYLD inhibits the Wnt/β-catenin and TGF-β signaling pathways by regulating the Lys63-linked ubiquitination of ALK5, thereby suppressing tumor growth and cell invasion246,247. OTUD1 deubiquitinates the TGF-β inhibitor SMAD7 and inhibits p53, thereby increasing the cleavage of caspase-3 and PARP-dependent apoptosis and enhancing tumor suppression248,249.
Context-dependent DUBs, such as OTUD3 and USP10, constitute the third type of DUBs that exhibit contradictory functions in various tumors. OTUD3 regulates PTEN stability to inhibit PI3K/AKT signaling transduction and tumorigenesis in breast cancer250. However, OTU3 stabilizes GRP78 and promotes lung cancer cell growth and migration251, suggesting that the tumor-suppressive or -proliferative activity of OTU3 depends on the tissue specificity of the tumor251. USP10 typically functions as a tumor suppressor in lung cancer, RCC, and colon cancer, and as a tumor promoter in acute and chronic myeloid leukemia; however, in hepatocellular carcinoma, USP10 exerts multiple activities: a) it reverses the MDM2-induced nuclear export and degradation of p53 and reduces the progression of RCC252; b) it blocks the degradation of the canonical tumor suppressor PTEN and KLF4 and inhibits lung cancer growth253,254; c) it antagonizes the transcriptional activity of the oncogene c-Myc by stabilizing SIRT6 and p53, thereby promoting cell cycle arrest and inhibiting colon cancer cell proliferation255; d) it stabilizes the oncoproteins FLt3 and Skp2 to facilitate the growth of myeloid leukemia256,257; and e) in hepatocellular carcinoma, it removes the Ub chains on YAP/TAZ to promote the growth of the HepG2, SNU387, and Li7 cell lines258, whereas it stabilizes PTEN and AMPKα to suppress the growth of the Huh7, HCCLM3, MHCC97L, and Bel7402 cell lines259.
3. Multiomics analyses empower anticancer target identification in Ub and Ubl pathways
As mentioned above, Ub and Ubl pathways are attractive anticancer targets. However, the complexity of these pathways, comprising hundreds of Ub-associated enzymes that form a hierarchical task network to modulate substrates, complicates our understanding of their significance in cancer. Thus, there remains a need for a comprehensive approach to identify potential anticancer targets within the Ub and Ubl pathways. With the advancements of high-throughput sequencing and computational tools, multiomics analyses have emerged as valuable resources for unraveling the complexity of cancer. In the following sections, we will delineate the role of Ub and Ubl-associated enzymes in tumorigenesis by multiomics analyses. Furthermore, we will outline their advance in the identification of potential anticancer targets and application for precise treatment. The narrative order and associated subtitle are expanded in the order of E1, E2 and E3.
3.1. Multiomics analyses of E1s reveals that overexpression of UBA1 and UBA6 represents poor overall survival of tumor patients
First, we investigated the association between E1s in tumors and in normal tissues alongside their prognostic value using the Cancer Genome Atlas (TCGA) and Clinical Proteomic Tumor Analysis Consortium (CPTAC) databases from the UALCAN website260,261. Compared with normal tissues, >13 tumor types expressed high UBA1 and UBA6 mRNA levels, whereas only 3 tumor types exhibited low UBA1 and UBA6 mRNA levels (Fig. 6A and B). At the protein level, UBA1 and UBA6 were overexpressed in colon cancer and head and neck squamous cancer (Fig. 6C and D). These findings align with the protumoral functions of UBA1 and UBA6, as evidenced by the following observations: 1) targeting UBA1 via pharmacological and genetic approaches significantly inhibits tumor cell growth, including lung cancer, colon cancer, liver cancer, acute myeloid leukemia, non-Hodgkin lymphoma, melanoma11,262, 263, 264, 265; 2) inhibition of UBA1 overcomes drug resistance by inducing ER stress or apoptosis in myeloma266; 3) overexpression of UBA6 promotes tumor formation267. Kaplan–Meier analysis revealed that higher UBA1 in patients was associated with poorer overall survival in prostate adenocarcinoma, hepatocellular carcinoma, and lung adenocarcinoma (LUAD) (Fig. 7A). Similarly, patients with LUAD, kidney chromophobe, and ccRCC exhibited worse prognoses in case of high tumoral UBA6 levels (Fig. 7B).
Figure 6.

Multiomics analyses of E1s. (A–D) Gene expression levels of UBA1 and UBA6 in 24 tumors and normal tissues using TCGA database and CPTAC database in UALCAN website. Statistical significance was determined by the Mann–Whitney test (two-tailed): ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, n.s. indicates no significant difference.
Figure 7.

Kaplan–Meier analyses of UBA1 and UBA6 using TCGA database in UALCAN website.
3.2. Multiomics analyses of E2s reveals the potential of UBE2C as a broad-spectrum anticancer target in tumors
The expression levels and clinical importance of ubiquitination-related E2s were determined using transcriptomic, proteomic, and clinical data from patients with tumors260,261. The mRNA levels of 27 E2s (81.81%) were significantly upregulated in the tumor tissues of patients with LUAD than that in normal tissues, whereas the mRNA levels of 23 E2s (69.70%) were significantly upregulated in the tumor tissues of patients with lung squamous cell carcinoma (P ≤ 0.05) (Fig. 8A and B). Conversely, less than six E2s were significantly downregulated in the tumor tissues of patients with LUAD and lung squamous cell carcinoma (P ≤ 0.05, respectively) (Fig. 8A and B). Kaplan–Meier analysis revealed that higher UBE2C, UBE2O, UBE2S, UBE2T, UBE2V2 and UBE2Z in patients was associated with poorer overall survival in LUAD (Fig. 8A and B).
Figure 8.

Multiomics analyses of E2s. (A, B) mRNA levels and Kaplan–Meier analyses of ubiquitination-associated E2s in lung adenocarcinoma and lung squamous cell carcinoma.
The UBE2C mRNA levels were 18- and 30-fold higher in the tumor tissue of patients with LUAD than that in normal tissues and those with lung squamous cell carcinoma tissue, respectively. An overall survival analysis revealed that UBE2C (HR = 2.50) was risk factors for LUAD (P ≤ 0.05), but not in lung squamous cell carcinoma through the analysis of the TCGA database (Fig. 9A and B). UBE2C has been identified as an essential factor in KrasG12D-induced lung cancer55. In addition, in our multiomics analyses, UBE2C was considerably and consistently upregulated in the tumor tissue of patients within the enrolled 10 tumors, including lung cancer, than in the normal tissues at protein levels (Fig. 9A). UBE2C, in conjunction with the APC/C E3 ligase, promotes ubiquitylation and degradation of tumor suppressive proteins (e.g., p27, DEPTOR and p53)40, 41, 42, 43,48, 49, 50, 51,55. The mRNA levels of UBE2S, which cooperate with UBE2C for Lys11-polyubiquitination chain extension (as discussed in detail in the previous section), were significantly upregulated in the tumor tissue of patients with LUAD and lung squamous cell carcinoma tissue than that in normal tissues (Fig. 8A and B). An overall survival analysis showed that the hazard ratio of UBE2S was up to 2.71 in LUAD (Fig. 8A). Consistently, Zhang et al.55 reported that overexpression of UBE2C or UBE2S is related with the poor overall survival over 400 LUAD cases. Further protein level analysis showed that UBE2S was significantly upregulated in the tumor tissue of patients than in the normal tissues in ovarian cancer, head and neck cancer and glioblastoma, but not in lung cancer (Fig. 9B). Knockdown of UBE2C, but not UBE2S, inhibits the growth of lung cancer cells and KrasG12D-induced lung carcinoma55. This intriguing pattern suggests that UBE2C, but not UBE2S might be an attractive target for lung cancer.
Figure 9.

Gene expression levels of UBE2C and UBE2S in 10 tumors and normal tissues by exploring the CPTAC database in UALCAN website. Statistical significance was determined by the Mann–Whitney test (two-tailed): ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, n.s. indicates no significant difference.
3.3. Multiomics analyses of representative E3s reveals their clinical significance and regulatory mechanisms
F-box proteins, serving as substrate-recognition subunits of CRL1 E3 ligases, play important roles in tumor initiation and progression69,70. To comprehensively delineate the role of F-box proteins in tumorigenesis, multiomics analyses were performed, encompassing all F-box proteins (n = 69), with the aim of providing insight into the characteristics of RING E3 ligases in cancer. Transcriptomic analysis showed that thirty-three F-box proteins were upregulated and twelve F-box proteins were downregulated (P < 0.05) at mRNA levels in LUAD tissues compared to normal tissues (Fig. 10A). At the mRNA level, the extensively studied F-box protein Skp2 showed a 2.72-fold increase in LUAD. This finding aligns with previous studies that have confirmed the upregulation of Skp2 in LUAD72,268. These consistent results between our omics analyses and published data further support the reliability of our findings. The expression of FBXO45, a potential oncogene that inhibits tumor cell death by degrading tumor suppressor FBXW7269, was increased by 2.10-fold in LUAD (Fig. 10A). Similarly, the expression of FBXW5, an underlying oncogene that targets kinesin-13 and LATS1/2 for proteasomal degradation270,271, was increased by 1.36-fold (Fig. 10A). At the protein level, FBXW5 was significantly increased in ccRCC and uterine corpus endometrial carcinoma compared to normal tissues, whereas FBXO45 was predominantly upregulated in breast cancer, ccRCC, head and neck squamous cell carcinoma, and glioblastoma (Fig. 10B and C). Consistently, FBXW5 and FBXO45 are overexpressed in numerous tumors and exhibit oncogenic properties270,271. There were lower mRNA and protein levels of FBXW2 (Fig. 10D). The downregulation of FBXW2 leads to the inhibition of ubiquitylation and degradation of β-catenin and Skp2, thereby promoting lung cancer cell migration and invasion122,272. In addition, the mRNA level of FBXL21 was 14.59-fold higher in LUAD tissues than in normal tissues, followed by FBXO43 (6.05-fold), FBXO32 (5.70-fold), CCNF (also known as FBXO1, 3.78-fold), FBXO41 (2.99-fold), FBXL13 (2.86-fold), and FBXL6 (2.08-fold) (Fig. 10A). These analyses offered a comprehensive perspective on the mRNA levels of F-box proteins in LUAD tissues compared to normal tissues.
Figure 10.

Multiomics analyses of F-box proteins (n = 69). (A) mRNA levels and prognostic values of F-box proteins in lung adenocarcinoma. (B–D) Protein expression levels of FBXW5, FBXO45 and FBXW2 in tumors and normal tissues by exploring the CPTAC database in UALCAN website.
High protein levels are regulated at multiple layers, including genetic amplification, transcriptional, translational, and post-translational levels. Consequently, we examined genetic alterations of Skp2, FBXW7, β-TRCP1 (gene name: BTRC), β-TRCP2 (gene name: FBXW11), FBXW5, FBXO45 and FBXW2 in thirty-three tumor types using the cBioPortal website (Fig. 11). Among these six F-box proteins, SKP2 and FBXO45 showed a higher amplification rate, whereas FBXW7 was the most frequently mutated gene (Fig. 11A). FBXW7, a well-known tumor suppressor that has been discussed in detail in the previous section, was exhibited the highest frequency of endometrial cancer (mutation rate of 19.97%), colorectal cancer (14.81%) and cervical cancer (11.78%) (Fig. 10B). These mutation patterns of FBXW7 were consistent with previous studies273. The mutations primarily occurred in the F-box domain and the substrate recognition WD40-repeat domain, particularly at three arginine residues (R465 C/H/G/L/P, R479Q/∗/G/P/L, and R505 G/C/H/L) in the WD40-repeat domain (Fig. 11C), which are essential for recognizing and binding substrates107. Additionally, Skp2, a well-established oncogene, was found to be amplified in non-small cell lung cancer (amplification rate of 9.12%), bladder cancer (6.81%), and esophagogastric cancer (6.27%) (Fig. 11D). FBXO45 surpassed Skp2 with an amplification rate of 15.75% in ovarian epithelial tumor, followed by non-small cell lung cancer (14.72%) and cervical cancer (14.14%) (Fig. 10A and E). These results suggest that the overexpression of Skp2 and FBXO45 may be attributed to genetic amplification.
Figure 11.

Multiomics analyses of F-box proteins. (A) Gene alterations of 7 F-box proteins were determined in 33 tumors via the exploration of cBioPortal website. (B) Cancer type summary about altered SKP2, FBXO45 and FBXW7. (C) Mutation site analysis of FBXW7 by exploring the cBioPortal website. (D, E) Cancer type summary about altered SKP2 and FBXO45.
4. Overview of Ub and Ubl targeted drug discovery
The Ub and Ubl pathways are integral components of the ubiquitin–proteasome system (UPS) that is responsible for the degradation of over 80% of cellular proteins. In the past two decades, accumulated evidence confirmed that targeting 26S proteasome (the downstream of UPS) had been viewed as a desirable outcome of anti-cancer therapeutics. This endeavor resulted in U.S. Food and Drug Administration (FDA) approval of the first proteasome inhibitor (PI) bortezomib (also called velcade) in 2003, for treatment of multiple myeloma and relapsed mantle cell lymphoma. Subsequently, other PIs, such as ixazomib, delanzomib, and carfilzomib, were approved, further validating anticancer reliability of UPS suppression in clinic. However, the application of PIs is hindered by limitations, including targeting specificity, oral stabilization, and low penetration into solid tumors, consistently constrain the application of PIs. For instance, PIs have shown limited effectiveness in solid tumors, even when combined with other antitumor agents. Recent rapid advances in the functions and mechanisms of Ub and Ubl pathway, offer potential solutions to address these challenges.
In this section, we categorized the current reported small molecule inhibitors targeting essential components of the Ub and Ubl pathways, including E1s, E2s, E3s, DUBs, as well as their structural characteristics, clinical research progress and limitations. We also discuss the application of E3s ligands in the studies of target protein degradation (TPD). Especially, molecular glue degraders (MGD), as a classical TPD type, are considered as a prospective therapeutic strategy in cancer therapies, to be summarized the current progress in this section. Recognizing the growing importance of bioinformatics approaches in the pharmaceutical industry, we also discussed the utilization of bioinformatics resources and methods to facilitate the TPD drug discovery based on the multiomic data.
4.1. Representative E1 and E2 inhibitors
As mentioned above, ubiquitination has two essential E1s, UAE1 (also known as UAE) and UBA6. The first successful attempt at UAE inhibition dates back to the 1990s. A nonhydrolyzable ATP analog, adenosyl-phospho-ubiquitinol, was designed and synthesized to evaluate the inhibitory activity of the UAE–Ub adduct in an ATP-competitive manner (Ki = 50 nmol/L)274. This study first demonstrated the feasibility of selectively targeting the upstream of UPS, and developing small-molecular inhibitors of UAE. For a considerable amount of time, however, the majority of reported UAE inhibitors lacked targeting specificity and potent inhibitory effects against UAE (half maximal inhibitory concentration IC50 > 1 μmol/L). Similar circumstances occurred during the development of Ubl inhibitors275. Not until 2009 did Soucy T.A. et al. develop the first NAE specific inhibitor, MLN4924 (also known as pevonedistat), with a new adenosine sulfamate (AdoS) skeleton (Fig. 12). MLN4924 is a potent and selective NAE inhibitor (IC50 = 4 nmol/L) that is unrelated to other homologous enzymes, such as UAE, UBA6, and SUMO-activating enzyme (SAE) of SUMO (IC50 = 1.50, 8.20, and 1.80 μmol/L, respectively)276. Furthermore, MLN4924 suppresses the growth of various cancer cell lines by inducing apoptosis, senescence, or sensitizing cancer cells to chemoradiation33. The crystal structure of the MLN4924–NAE–NEDD8 ternary complex showed that MLN4924 not only binds with high affinity to the ATP-binding site of NAE but also forms a covalent cysteine thioester with NEDD8 via its terminal sulfamate moiety277.
Figure 12.

Chemical structures of AdoS-derived covalent inhibitors targeting E1 and their inhibitory mechanism.
The structure of MLN4924 inspired the researchers of Millennium Pharmaceuticals to successfully identify an AdoS analog, Compound 1, with potent UAE inhibiting activity278. Consistent with the mechanism of interaction between MLN4924 and the NAE complex, Compound 1 forms a covalent bond with Ub via its sulfamate group and then suppresses the formation of UAE–Ub thioester278. However, Compound 1 cannot be developed as a UAE inhibitor due to its nonselective effect on other E1 enzymes, including NAE and SAE. To develop highly selective UAE inhibitors, Hyer et al.11 screened seven hundred small-molecular compounds and identified a potent, mechanism-based UAE inhibitor, TAK-243. Similar to Compound 1, the sulfamate moiety of TAK-243 blocks the covalent linkage between UAE and Ub. In contrast to Compound 1, TAK-243 not only inhibits UAE (IC50 = 1 nmol/L), but also UBA6 (IC50 = 7 nmol/L), NAE (IC50 = 28 nmol/L), and SAE (IC50 = 850 nmol/L)11 at a lesser extent (Fig. 12). Accumulated evidence has shown that TAK-243 completely suppresses the cellular ubiquitylation and induces ER stress-mediated apoptosis11. According to the data from the ClinicalTrials website, TAK-243 has now entered the clinical phase for treating patients with acute/chronic myeloid leukemia or myelodysplastic syndrome. Considering the potent SAE inhibition of Compound 1 and the high similarity between UAE and SAE, Langston et al.279 hypothesized that the sulfamate group of Compound 1 may also help covalently bind the SUMO–AdoS adduct. Then, several Compound 1 analogs were synthesized, and their SAE inhibitory effects were investigated using the homogenous time-resolved fluorescence assay. TAK-981 was identified to specifically bind the enzyme SAE (IC50 = 1 nmol/L) more effectively than other E1 (IC50, NAE = 0.96 μmol/L, UAE> 1 μmol/L). To date, TAK-981 has entered phase I clinical trials for patients with metastatic solid tumors in combination with pembrolizumab and non-Hodgkin's lymphoma in combination with rituximab (NCT03648372, NCT04074330, and NCT04381650)279.
Although E2s may constitute a promising target for anticancer therapeutics, comparatively fewer E2 small-molecular inhibitors have been reported than E1 inhibitors. Ceccarelli et al.280 screened and identified a small molecule, CC0651 that selectively inhibits the E2 Ub-conjugating enzyme Cdc34. The crystal structure of the CC0651–Cdc34A–Ub complex revealed that CC0651 stabilizes a low-affinity interaction with a composite binding pocket generated by Cdc34A and Ub281. Similar to TAK-243, CC0651 induces the accumulation of p27 by inhibiting its degradation (IC50 = 1.7 μmol/L) and suppresses the growth and proliferation of various cancer cell lines. Moreover, Tsukamoto et al.282 identified leucettamol A (IC50 = 105 μmol/L) from the Leucetta sponge as the first Ubc13 inhibitor, and subsequently isolated two more Ubc13 inhibitors, Manadosterols A and B, which are derived from the marine sponge Lissodendryx fibrosa (IC50 = 90 nmol/L and 130 nmol/L, respectively). In contrast to several Ub- and NEDD8-related E2s, Ubc9 is the only E2 in the SUMOylation pathway. The submicromolar Ubc9 inhibitory activity of flavonoid analog 2D08 is utilized as a positive probe to identify other new Ubc9 inhibitors283.
4.2. Small-molecule ligands targeting RING E3 ligases
The Ub and Ubl pathways regulated the degradation of various substrates by establishing a PPI network involving human E3 ligase proteins as mentioned above. Over the past two decades, the development of PPI inhibitors has emerged as a viable and important technique for drug discovery targeting E3 ubiquitin ligases8,284. However, these PPI inhibitors are still primarily based on the concept of “occupancy-driven” small-molecule modalities, also known as the inhibitor-centric approach. Unfortunately, only approximately 20% of the human proteome is druggable for an inhibitor-centric development approach. To address the critical limitations of the inhibitor-centric approach, the development of E3-associated modulators has introduced the concept of target protein degradation (TPD) as a novel pharmacologic modality285. Notably, proteolysis-targeting chimeras (PROTACs) have expanded the application of PPIs and provided insights into the degradation pathways for drug discovery286, 287, 288, 289. PROTACs are typically composed of an E3 small-molecule ligand, a connecting linker, and a target protein ligand287. The successful development of high-quality E3 small-molecule ligands is essential for PROTAC development. However, the lack of suitable E3 ligase ligands has been a major obstacle in the development of PROTAC degraders290. Fortunately, drug-like small-molecule ligands have been continuously to bind to the substrate receptor subunits of CRLs, such as CRL1Skp2, CRL2VHL, CRL3Keap1, CRL3SPOP, and CRL4CRBN. Beyond that, ligands targeting other E3 ligases, such as MDM2 and cIAP, have been developed as E3 inhibitors or as part of PROTAC molecules290,291. Among them, CRL4CRBN and CRL2VHL ligands have achieved notable success and are widely used in TPD studies and PROTAC design292. In the following, we will highlight the recent progress in small-molecule ligands targeting RING E3 ligases.
4.2.1. Small-molecule ligands of cullin RING E3 ligases
Numerous studies have confirmed that CRL1Skp2 inhibits apoptosis and cell cycle arrest by regulating the degradation of cell cycle inhibitors p21 and p27, suggesting that Skp2 is a promising anticancer target. Chan et al. conducted a high-throughput virtual screening to identify several potential Skp2 inhibitors293 and found that Compound 25 displayed better Skp2 inhibitory activity than the other hit compounds and suppressed the survival of cancer cells and cancer stem cells. This study was the first to demonstrate that the pharmacological inactivation of Skp2 constitutes a promising approach for cancer treatment293. Although Wu et al.294 had previously identified a series of Skp2 probes in silico, including thiazolidinedione derivatives C1 and C2, there was insufficient evidence to support their anticancer effect.
As a CRL2 substrate receptor, VHL regulates the degradation of HIF-1α and is involved in hypoxia adaptation. Previous studies have reported that residue hydroxy proline 564 (Hyp564) of HIF-1α is essential to bind VHL, thereby targeting Hyp of HIF-1α regulating its ubiquitin-mediated proteasomal degradation295,296. Buckley et al.297,298 modified the structure of Hyp in silico to rationally design Hyp analogs, discovering the first small-molecule inhibitor that suppressed the PPI of VHL and HIF-1α at submicromolar concentrations. Soares et al.299 further modified these Hyp analogs according to structure-guided and fragment-based drug design. Probe VH298 was identified as the first inhibitor with a double-digit nanomolar affinity for CRL2VHL binding in vitro and in vivo299. Recently, a series of bifunctional small molecules containing two homologous VHL ligases, also known as Homo-PROTACs, were designed to dimerize the enzyme VHL and induce its self-degradation. The most active Homo-PROTAC, CM11, was designed as a selective and powerful VHL degrader at a concentration of 10 nmol/L300.
CRL3 possesses two primary E3 ubiquitin ligases, Keap1 and SPOP. CRL3keap1 destabilizes its primary substrate Nrf2 to regulate the expression of antioxidant proteins and inflammatory regulators. Recently, CRL3Keap1 has steadily emerged as an attractive target for cancer therapy alongside various other diseases, such as diabetes, Alzheimer's disease, and Parkinson's disease, which involve oxidative stress and inflammation301. According to prior research, in a phase 3 trial, the clinical candidate CDDO-Me inhibited the interaction between cullin3 and Keap1. Additional research demonstrated that CDDO-Me covalently binds to the BTB domain of Keap1302. Biogene conducted a high-throughput screening to identify small molecules that directly inhibited the PPI of Keap1 and Nrf2302. In a homogeneous fluorescence polarization assay, more than 300 thousand compounds were screened and evaluated. As the most active compound, ML334 exhibited a high affinity for Keap1 (Kd = 1.00 μmol/L)303. Additionally, Jiang et al.304 modified a potential Keap1 inhibitor by screening the commercial Evotec Lead Discovery library and identified Compound 25 as an effective inhibitor of the interaction between Nrf2 and Keap1 with a half maximal effective concentration of 28.6 nmol/L. Recently, Astex Pharmaceuticals developed a series of novel oxathiazepin derivatives with potent affinity via fragment-based molecular modification and optimization. The analog KI-696 was inhibited the Keap1–Nrf2 interaction at a single nanomolar concentration (Kd = 1.30 nmol/L)305. SPOP with the BTB domain plays vital roles in the growth and progression of tumors by mediating the ubiquitination of several important substrates, such as PTEN and DUSP7157,306,307 Jiang et al.308,309 identified a series of small-molecule SPOP inhibitors by integrating virtual screening, pharmacophore modeling, and molecular docking. They further found optimal Compound 6b to inhibit SPOP-mediated PPI in ccRCC cells at a concentration of 10 μmol/L. Following optimization and SAR analyses, Compound 6lc was identified as having superior SPOP inhibition and ccRCC cytotoxicity to those of 6b308,309.
4.2.2. Small-molecule ligands of single-subunit RING E3s ligases IAPs and MDM2 ligands
Unlike CRLs, IAPs and MDM2 are two classical single-subunit protein RING E3 ubiquitin ligases. The IAP family, which consists of cIAP1/2 (cellular inhibitor of apoptosis) and XIAP (X-linked inhibitor of apoptosis), induces apoptosis by various mechanisms, including receptor-mediated, mitochondria-mediated, and TNF factor receptor 1 (TNFR1)-mediated apotosis310,311. The IAP family is typically composed of a ubiquitin-associated (UBA) domain, a RING domain, and multiple BIR domains (BIR1/2/3). Du et al.312 revealed that the second mitochondria-derived activator of caspase [SMACs, also known as direct IAP-binding protein with low pI (DIABLO)] has a promising endogenous antagonistic effect against IAPs via interacting with BIR2 and BIR3 domains. Extensive studies have shown that an N-terminal four peptide molecule (Ala1–Val2–Pro3–Ile4, AVPI) forms a similar binding model with SMAC (SMAC Kd = 420 nmol/L, AVPI Kd = 580 nmol/L)313. The binding model showed that the amino group (–NH2) and carbonyl group (–C O) of AVPI form multiple hydrogen bonds with the corresponding residues Glu314, Gln319, Trp310, Trp323, and Thr308. Based on the SARs of AVPI–BIR3, several SMAC mimetics were designed to assess the IAP inhibitory activity (Fig. 13). Genentech had advanced GDC-0152 and GDC-0917, the first-generation IAP inhibitors, through phase 1 clinical trials. Compared to pan-IAP inhibition of GDC-0152, GDC-0917 displayed a 250-fold more selectivity for cIAP over XIAP314,315. In addition to GDC-0152 and GDC-0917, Novartis has also developed the SMAC analog LCL161, a 1st generation peptidomimetic that recently passed phase 2 clinical trials for patients with relapsed or refractory multiple myeloma316. Subsequently, a series of peptidomimetic BIR3 IAP antagonists with bicyclic lactam skeletons were published by Sun et al.317, which advanced the research of second-generation IAP antagonists. Cyclization of valine and proline formed [8,5]-bicyclic Debio-1143 (also known as Xevinapant), a 2nd generation IAP antagonist. Subsequent research showed that Debio-1143 possessed superior inhibitory selectivity (IC50 XIAP and cIAP1/2 BIR3 = 66.4, 1.9, and 5.1 nmol/L, respectively) and pharmacokinetic properties than the majority of 1st generation inhibitors. Currently, a phase 3 clinical trial is recruiting patients with locally advanced squamous cell carcinoma of the head and neck treated with Debio-1143 in conjunction with platinum-based chemotherapy (NCT04459715).
Figure 13.

Various generations of IAP antagonists, 1st generation Smac peptidomimetic antagonist family, 2nd generation Smac peptidomimetic antagonist with bicyclic structure, 3rd generation Smac peptidomimetic antagonist family with bivalent structure, non-peptide IAP antagonists.
As described above, the BIR3 domain of IAPs binds to caspase-9 whereas the BIR2 domain binds to caspase-3/7318. Previously reported monovalent antagonists always bind to the BIR3 domains of XIAP and c-IAP1, and the majority exhibit higher selectivity than BIR2 selectivity. Li et al.319 proposed a 3rd generation SMAC mimetic with a distinct dimer skeleton that more effectively induced apoptosis in human cancer cells (Fig. 13). In addition, the results demonstrated that the bivalent antagonists had superior inhibitory efficacy against caspase-3/-7 and −9 by engaging both BIR2 and BIR3 domains of the IAP family. Inspired by the aforementioned discoveries, two representative bivalent IAP antagonists APG-1387320 and birinapant321, were further developed by Ascentage and TetraLogic, respectively. APG-1387 had been applied to patients recruited in phase 1 clinical trials to treat advanced solid tumors or hematologic malignancies (NCT03386526). However, various druggability problems, including physicochemical features (high MW, high TPSA) and poor oral bioavailability, limited the use of either APG-1387 or birinapant in the treatment of solid tumors. Using fragment-based drug discovery, nonalanine small molecules, as a new class of IAP antagonists have recently been developed322. Tolinapant (also known as ASTX660), a nonalanine and dual antagonist of XIAP and c-IAP1 (IC50 = 2.80 and 0.22 nmol/L, respectively), was synthesized from a simple acetylamine fragment322. Compared to previously reported antagonists, linapant exhibited reduced hERG inhibition (30% @ 30 μmol/L) and adequate oral efficacy.
MDM2 is another representative single protein RING E3 ligase323. MDM2 promotes p53 degradation to adversely regulate the tumor suppressor p53, thereby restraining p53-mediated cell cycle arrest and cell apoptosis (Fig. 14). In 2004, Roche reported the first selective MDM2 inhibitor, Nutlin-3a, that was identified from a series of cis-imidazoline-derived molecules324. Nutlin-3a inserts its two 4-chlorophenyl groups into the Trp23 and Leu26 pockets by mimicking the interaction of the p53 peptide, which directs its isopropoxy group toward the Phe19 pocket. Meanwhile, its cis-imidazoline scaffold replaces the helical backbone of the p53 peptide without polar hydrogen bonding interactions. According to the SARs, Roche further optimized the structure of Nutlin-3a to increase its potency and metabolic stability. As the first clinical MDM2 inhibitor, the analog molecule RG7112 had completed phase 1 clinical trials for patients with advanced solid tumors and hematologic neoplasms (NCT00623870, NCT00559533)325. According to a subsequent mechanistic investigation of the MDM2–p53 interaction, the indole ring of Trp23 residue in p53 is deeply inserted into a hydrophobic cavity on the surface of MDM2, and its NH group forms a hydrogen bond with residues in MDM2 (p53-Trp23/MDM2-Leu54)326 Inspired by this observational result, Ding et al.326 mimicked the structural interaction of Trp23 residue in p53 with MDM2 by deriving a unique spirooxindole alkaloids and subsequently identified a series of potent non-peptide MDM2 inhibitors327. Additionally, the optimal variant SAR405838 had completed phase 1 safety testing. In recent years, several analogs of SAR405838, such as RG7388103 from Roche328, APG-115 from Ascentage327, and DS-3032b from Daiichi Sankyo329, have entered clinical trials.
Figure 14.

Chemical structures of clinical MDM2 inhibitors.
Chiral drug design is important for the development of MDM2 inhibitors. In 2012, researchers at Amgen developed a series of novel piperidinone-derived MDM2 inhibitors via de novo design and chiral synthesis330. These chiral inhibitors maintain the binding model of previous molecules that occupy with the Trp23, Leu26, and Phe19 pockets of MDM2, as well as generating an electrostatic interaction between the carbonyl linker and the imidazole side chain of His96 in MDM2330. AMG232, the preferred compound, exhibits significant MDM2 inhibitory activity (surface plasmon resonance [SPR] KD = 0.045 nmol/L, SJSA-1 EdU IC50 = 9.10 nmol/L) as well as promising pharmacokinetics characteristics and antitumor activity in the SJSA-1 osteosarcoma mice-bearing xenograft model (ED50 = 9.10 mg/kg)331. Besides, Novartis developed a series of dihydroisoquinolinone p53-MDM2 inhibitors from the virtual screening of 50,000 compounds developed in house332. Based on the binding pocket of p53-MDM2 by X-ray crystallography and molecular modeling, they identified a clinical candidate, NVP-CGM097333.
4.3. Progress and trend in the development of CRL E3 ligases-associated molecule glue degraders
Protein of interesting (POI) involves artificially inducing target protein degradation by the UPS, lysosome system, or even autophagy, which represents a R&D breakthrough of PPI to attract huge attention and funding from both academia and the pharmaceutical industry334. UPS-associated TPD involves two major approaches, PROTAC and MGD. In contrast to PROTACs, MGDs induce the proximity of ligase and POI by matching the protein surface topologies and modulating the binding attitude with ligase and POI. As a result, MGDs display comparable TPD ability while sharing the similar molecular weight and druggable structures with small-molecule agents. Although a rational approach had been reported previously to discover the MGD, largescale discovery of MGDs will require more innovations in screening assays and a deeper understanding of ligase associated PPI readout.
Over the past decades, the bioinformatics revolution has been the beneficiary of a multiomics data explosion, which contributes to improve cancer therapy strategies by extracted enormous useful information. Particularly, the multiomics data, such as genomics, transcriptomics, proteomics and metabolomics data, were generated by high-throughput technologies integrated with drug-response data, provides opportunities for identifying anticancer biomarkers and predicting drug responses. Moreover, multiple biological networks, including PPI network, drug-target network and disease-gene network, are extensively combined with multiomics data to analyze the potency of anticancer targets and facilitate PPI-based drug discovery. In the following, we want to focus on the progress in hijacking CRLs with MGDs, along with the identification of TPD via bioinformatics profiling.
4.3.1. Outline of CRL E3 ligases-associated molecule glue degraders
CRLs ubiquitinate diverse substrate proteins when brought into direct proximity to the ligase335, including CRL4CRBN and several other substrates of the CRL4 family, such as CRL4DCAF15 and CRL4DDB1, attracting considerable attention from pharmaceutical companies owing to their efficacy in the discovery of MGDs (Table 1). In early 2010, CRBN was identified as an immune-regulated target of thalidomide and pomalidomide336,337. However, the mechanism by which these thalidomide-like IMiDs regulate CRL4CRBN, thereby inducing immunomodulatory and anti-inflammatory effects, remains unknown336,337. Lenalidomide was not identified to selectively exploit the ubiquitination and degradation of two lymphoid transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) till 2014191,338. Celgene identified a potent CRL4CRBN molecular glue, thalidomide-derived CC885, that selectively degraded more GSPT1 than IKZF1/3189. Conversely, neither pomalidomide nor lenalidomide degraded GSPT1, indicating that specific neosubstrate recruitment characteristics are associated with structural differences across IMiDs. The aryl sulfonamide derivative indisulam is a clinical candidate exhibiting selective anticancer activity339. Han et al.339 identified the mechanism underlying the anticancer selectivity of indisulam, which recruited the premRNA splicing factor RBM39 (RNA-binding motif protein 39) to CRL4DCAF15 for proteasomal degradation.
Table 1.
List of representative anticancer small molecules by targeting E2s, CRLs or DUBs.
| Name | Structure | Target | In vitro | In cells | Cell viability, IC50 | Cancer cell lines | Ref. |
|---|---|---|---|---|---|---|---|
| CC0651 |  |
Cdc34 | IC50 (FP) = 2.5 μmol/L | p27 deubiquitination @ 30 μmol/L | HCT116 | 280,281 | |
| Leucettamol A |  |
Ubc13 | 105 μmol/L | 282 | |||
| 2D-08 |  |
Ubc9 | IC50 = 6 μmol/L | 100 μmol/L | BT-474 | 283 | |
| Compound 25 |  |
CRL1Skp2 | >80% inhibition@5 μmol/L | p21, p27 deubiquitination @ 5 μmol/L | <10 μmol/L | PC-3, LNCaP | 293 |
| C1, C2, C16, C20 |  |
>80% inhibition@10 μmol/L | p21, p27 deubiquitination @ 10 μmol/L | MCF-7, TD47, LNCaP | 294 | ||
| VH298 |  |
CRL2VHL | IC50 (FP) = 80 nmol/L; Kd (ITC) = 90 nmol/L |
HIF-1α destabilization @ 50 μmol/L | No cytotoxic | Hela | 299 |
| CM11 | homo-PROTAC | Kd (ITC) = 25 nmol/L | HIF-1α destabilization @ 1 μmol/L | Hela | 300 | ||
| CDDO-Me |  |
CRL3Keap1 | 58.9%@0.1 μmol/L | 50–160 nmol/L | MCF-7 | 302 | |
| ML334 |  |
Kd = ∼ 1 μmol/L | EC50 = 18 μmol/L | U2OS | 303 | ||
| Compound 2 | 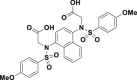 |
EC50 = 28.6 nmol/L | Nrf2–ARE induction@10 μmol/L | HepG2-ARE-C8 | 304 | ||
| KI696 | 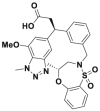 |
Kd (ITC) = 1.3 nmol/L | 305 | ||||
| 6lc | 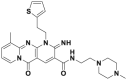 |
CRL3SPOP | KD = 30 μmol/L | PTEN deubiquitination @ 15 μmol/L | 2.1–3.5 μmol/L | A498, OS-RC-2 | 308,309 |
| CC885 | 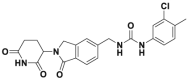 |
CRL4CRBN | GSPT1 degradation @ 1 nmol/L | NB4 | 189 | ||
| Thalidomid | 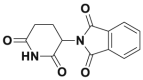 |
336 | |||||
| Pomalidomide | 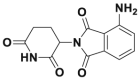 |
337 | |||||
| Lenalidomid | 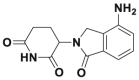 |
IKZF1 and IKZF3 degradation @ 1 μmol/L | MM1S | 191,338 | |||
| Indisulam |  |
CRL4DCAF15 | RBM39 degradation @ 2 μmol/L | HCT116 | 339 | ||
| CR8 | 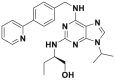 |
Cyclin K degradation @ 1 μmol/L | HCT116 | 340 | |||
| HQ461 | 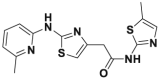 |
DC50 = 0.13 μmol/L | 1.5 μmol/L | A549 | 341 | ||
| dCeMM2/3/4 | 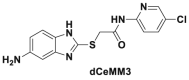 |
Cyclin K degradation @ 2.5 μmol/L | KBM7 | 342 | |||
| SJB2-043 | 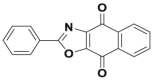 |
USP1 | 0.54 μmol/L | >80% inhibition @ 1 μmol/L | 1.1 μmol/L | K562 | 347 |
| ML323 | 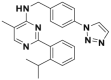 |
76 nmol/L | PCNA deubiquitination @ 20 μmol/L | 3 μmol/L | H1299 | 348 | |
| Q29 |  |
USP2 | 82% inhibition @ 0.5 μmol/L | NA | 4.7 μmol/L | DU145 | 350 |
| ML364 |  |
1.1–1.7 μmol/L | cyclin D1 destabilization @ 0.97 μmol/L | 3 μmol/L | HCT116 | 351 | |
| LCAHA |  |
9.7 μmol/L | cyclin D1 destabilization @ 20 μmol/L | 0.9 μmol/L | HCT116 | 352 | |
| XL188 |  |
USP7 | 90 nmol/L | p53 accumulation @5 μmol/L | MM.1S, MCF7 | 357 | |
| FT671 |  |
52 nmol/L | p53 accumulation @10 μmol/L | 33 nmol/L | HCT116, MM.1S, MCF7 | 358 | |
| Compound 4 |  |
6 nmol/L | p53 accumulation @1 μmol/L | 2–29 nmol/L | HCT116 | 359 | |
| Compound 41 |  |
0.44 nmol/L | p53 accumulation @25 nmol/L | 0.09–0.45 μmol/L | MM.1S, H526 | 360 | |
| GNE6640 |  |
0.75 μmol/L | p53 accumulation @10 μmol/L | >50% inhibition@5 μmol/L | HCT116, MCF7, EOL-1 | 361 | |
| Capzimin | 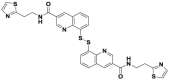 |
Rpn11 | 0.39 μmol/L | Deubiquitination @ 2–10 μmol/L | 2.1–3.8 μmol/L | 293T, A549, HCT116 | 364 |
| SOP11 |  |
1.3/0.6 μmol/L | Deubiquitination @ 10 μmol/L | GI50 = 4.7 μmol/L | HCT116 | 365 | |
| VLX1570 |  |
USP14/UCH37 | 8.1/14 μmol/L | >80% inhibition @ 25 mmol/L | 43–126 nmol/L | RPMI8226, KMS11, OPM-2 | 366 |
Previously reported molecular glues induce the degradation of specific substrates by binding to the CRL4 substrate receptors than the CRL4 adaptor proteins. Słabicki et al.340 analyzed the sensitivity of 4,518 clinical and preclinical drugs in 578 cancer cell lines via high-content screening alongside their relative E3 ligase component mRNA levels. A CDK12 inhibitor, (R)-CR8, revealed a correlation between cytotoxicity and CRL4DDB1 mRNA levels. Further studies showed that (R)-CR8 functions as a molecular glue to regulate protein cyclin K degradation340. X-ray crystallography studies of the DDB1–(R)-CR8–CDK12–cyclin K complex demonstrated that (R)-CR8 binds to the ATP-binding pocket of CDK12, forming a high affinity with the DDB1 domain. Previously, the thiazolyl derivative HQ461 was identified as a potential anticancer small molecule capable of inhibiting Nrf2 activity. To reveal the functional mechanism of HQ461, Lv et al.341 performed a pooled genome-wide CRISPR-Cas9 knockout screening in lung cancer cells A549 by targeting 19,114 genes with 4 individual sgRNAs per gene. According to the findings, HQ461 converts CDK12 into CRL4DDB1 to initiate polyubiquitination and subsequent degradation of the partner protein cyclin K of CDK12. Mayor-Ruiz et al.342 screened a library of >2000 small molecules in either wildtype or UBE2Mmut KBM7 cells and found that dCeMM1/2/3/4 displayed a correlation between cytotoxicity and UBE2M-associated cullin4 levels via scalable chemical profiling. Further studies revealed that dCeMM2/3/4 analogs regulated the ubiquitination-mediated degradation of cyclin K by prompting the interaction of CDK12 and cyclin K.
4.3.2. Multi-omics approaches empower the identification of CRL E3 ligases-associated molecule glue degraders
Interestingly, the majority of reported CRL-associated molecular glues are subordinate to CRL4 substrate receptors or adaptors, including CRBN, DCAF15, and DDB1. Notably, cell-based phenotypic assays have provided the richest source for MGD identification. It is hypothesized that numerous screening models were initially established using cell viability-based phenotypes, leading to the identification of hit compounds that interacted with CRL4 substrate receptors and adaptors. However, the traditional MDG discovery approaches face challenges in evaluating the relationship of CRL4 and cell-viability-based assay. These studies still heavily rely on the cell-viability-associated readouts to promote their MDG design, which results in the limitation that the targeted regulators are limited to those with an essential function in cell viability, such as CRL4 in possible. To overcome these limitations, it is crucial to advance phenotypic screens that can be measured, quantified, interpreted, and predicted, enabling a better understanding of MDG discovery. Modern high-throughput screening technologies, such as microarray and next-generation sequencing, have generated vast amounts of biological data that can be integrated in multiomics studies to explore the functional and mechanistic complexity of ubiquitin (Ub)-associated TPD.
Numerous existing data resources, including cancer-gene databases like Online Mendelian Inheritance in Man (OMIM) and Genetic Association Database (GAD), patient omics data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), and cancer-associated Kyoto Encyclopedia of Genes and Genomes (KEGG), along with drug-gene databases such as DrugBank, Therapeutic Target, and Clinical Trials, serve as foundational resources for bioinformatics analyses of PPI networks and biological pathways. Based on the databases, mass spectrometry (MS)-based proteomics data analysis has emerged as a key approach for identifying the E3 ligands. Quantitative expression proteomics combined with correlative transcriptomics were employed to examine the mechanisms of action (MoA) of specific MGDs, such as CR8 and dCeMM2-4. In particular, drug-affinity enrichment and MS-based chemoproteomics, such as activity-based protein profiling (ABPP) and drug affinity-responsive target stability (DARTS), have successfully been conducted to discover potential RNF114 small-molecule ligand, natural product nimbolide343, These approaches have also facilitated the identification of specific fragment–protein interactions, such as E3 covalent engaging ligands344,345. Moreover, advancement in imaging technology and precise visual analysis, such as cellular high-content imaging and small-molecule fluorescence probe, have provided additional insights into the MoA of ligases and ligands, when combined with the bioinformation profiling.
4.4. Deubiquitinases (DUBs) inhibitors
Most of the reported DUB inhibitors primarily target the proteins USP1, USP2, USP7, Rpn11, and USP14. Herein, we thus focused on novel DUB inhibitors developed between 2013 and 2021 and their potential for cancer therapy (Table 1). Among the human USP family of DUBs, USP1 has been implicated in the DNA damage response, which is responsible for deubiquitinating and stabilizing two crucial DNA repair proteins, PCNA and FANCD2 (fanconi anemia group D2)346. Chen et al.346 employed a self-established ubiquitin–rhodamine-based high-throughput screening method to identify a series of FDA-approved drugs and clinical candidates as potential USP1 inhibitors, including pimozide, GW7647, and flupenthixol. Utilizing a similar ubiquitin–rhodamine-based screening approach, Mistry et al.347 identified several USP1 inhibitors and determined their therapeutic potential for leukemia. Representative SJB2-043, derived from the hit compound C527, promotes the degradation of ID1 (a leukemic cell growth activator) and FANCD2 by inhibiting the catalytic activity of USP1 (IC50 = 0.54 μmol/L), and is currently being used as a positive control to evaluate other potential USP1 inhibitors. Furthermore, Liang et al.348 developed the first selective and potent USP1 inhibitor, ML323, based on the discoveries of pimozide and GW7647. Phenylpyrimidin-derived ML323 inhibits USP1 with a nanomolar affinity (IC50 = 76 nmol/L) and increases the monoubiquitinated PCNA (Ub-PCNA) levels (PCNA deubiquitination at 20 μmol/L in H1299)349. USP2 helps stabilize various tumor-associated substrates, including MDM2, fatty acid synthase, and cyclin D1228,231,233. Hence, numerous human cancer cell lines undergo apoptosis when USP2 is silenced. Ohayon et al.350 screened and identified a series of UCH-L3 inhibitors and found that these compounds inhibited USP2 more effectively than UCH-L3, especially several ortho-quinones analogs. The natural product β-lapachone Q29, a clinical candidate for treating pancreatic cancer, exhibits simultaneous USP1/2 inhibition (82%–92% inhibition at 20 μmol/L)350. ML364, another representative small-molecule USP2 inhibitor, accelerates cyclin D1 degradation and induces cell cycle arrest in both Mino and HCT116 cancer cell lines351. In addition, a lithocholic acid (LCA) derivative, LCAHA, destabilizes cyclin D1 in an AKT/GSK3β-independent manner, by suppressing USP2 but maintaining the expression of p27352.
Recently, USP7 has emerged as a star protein of the DUB family for cancer therapy owing to its importance in regulating p53 and MDM2 levels353,354. Several pharmaceutical companies, such as Hybrigenics, Progenra, FORMA Therapeutics, and Genentech, have contributed to the development of USP7 inhibitors355. Considering that a large number of small-molecule USP7 inhibitors have been developed during the past two decades, this review summarizes the progress of representative USP7 inhibitors from 2012 to the present. Early in 2012, Progenra developed several trisubstituted thiophene derivatives, such as P5091 and P22077348, as first-generation USP7 inhibitors356. These inhibitors covalently conjugate with the catalytic domain cysteine223 of USP7 and are applicable in cancer and immunological oncology therapies356; however, pan-deubiquitination and off-target defects limit their efficacy. By optimizing the structure of the lead compound pyrazolo-[3,4-d]-pyrimidin-4-one-piperidine (PyrzPPip), Lamberto et al.357 developed the next generation of USP7, subsequently discovering that XL188 is a highly potent and selective USP7 inhibitor (IC50 = 90 nmol/L). X-ray crystallography of the USP7–XL188 complex revealed that XL188 is a noncovalent USP7 inhibitor. Additionally, the researchers of Hybrigenics have presented a series of PyrzPPip-derived USP7 inhibitors. They first identified a noncovalent USP7 inhibitor, FT671, in the series. FT671 is capable of specifically suppressing enzyme USP7 with high affinity. Unlike other reported inhibitors, FT671 is an allosteric inhibitor that targets a dynamic pocket close to the catalytic position of USP7358. Furthermore, Almac Discovery used the surface plasmon resonance (SPR) to identify new USP7 inhibitors through a fragment-based screen. Hydroxypiperidine-derived Compound 4, the most potent inhibitor of the hit compounds, selectively inhibits USP7 in vitro (IC50 = 6 nmol/L) and in cells (IC50 = 49 nmol/L) with nanomolar inhibitory IC50 values359. Leger et al.360 identified benzofuran-amide as a potential scaffold for USP7 inhibition, revealing that the optimized scaffold has a large succinimide group that serves as a major potency-driving motif. The succinimide group forms two crucial hydrogen bonds in the allosteric position of USP7. The optimal Compound 41 inhibits USP7 with more potency and selectivity than other previously reported deubiquitinases360. In the same year, GeneTech introduced GNE6640, an additional potent allosteric USP7 inhibitor. Structural analyses revealed that GNE6640 noncovalently binds USP7 at a distance of 12 Å from catalytic cysteine and interacts with acidic residues that mediate the hydrogen bond interaction with the Lys48 side chain of ubiquitin361.
As outlined above, the proteasome comprises a 19S regulatory particle (RP) and a 20S core particle. Although approved “omibs” drugs always target the β5 active site of the 20S core particle, resistance occurs. Rpn11 is a Zn2+-dependent metalloisopeptidase derived from the JAMM zinc metalloprotease family of DUBs that hydrolyzes ubiquitin from tagged proteins and is a crucial member of the proteasome 19S regulatory particle362. Inactivating the enzymatic active site of Rpn11 by altering zinc-coordinating histidine residues inhibits substrate degradation in cells363. Recent research has shown that targeting Rpn11 as a potential cancer treatment target not only inhibits the proteasome but also increases the therapeutic efficacy of “omibs” drugs364. Li et al.364 screened 330,000 compounds and selected 8TQ as a promising lead (a thioester derivative). They subsequently optimized the structure of 8TQ using the FBDD approach to summarize SARs and identified the first potent and specific Rpn11 chemical probe, capzimin (IC50 = ∼300 nmol/L), which inhibited the proliferation of bortezomib-resistant cancer cells by stabilizing the proteasome substrates and inducing an unfolded protein response. Utilizing the positive capzimin and their inhouse screening system, they discovered an epipolythiodioxopiperazines-derived SOP11 that inhibits proteasomal deubiquitinases, such as Rpn11 and AMSH. In addition to Rpn11, the 19S RP has two more DUBs, the cysteine proteases USP14 and UCHL5 (UCH37)365. VLX1570, the first DUB inhibitor containing a reactive α and β-unsaturated carbonyl group and covalently interacting with the nucleophilic residues of USP14 and UCHL5, recently entered clinical trials366.
5. Conclusions and perspective
Recent and ongoing research demonstrates that Ub and Ubl pathways play crucial roles in tumorigenesis by degrading or activating/deactivating key regulators of tumor growth and death. The increased knowledge supports the idea that targeting aberrant Ub or Ubl pathways is a novel therapeutic strategy for multiple types of tumors. In this regard, the fundamental advances and follow-on target-based drug discoveries have been crucial in providing vital information concerning contemporary translational efforts to develop precise treatment by targeting Ub and Ubl pathways.
However, several challenges of the Ub and Ubl field are required with solution, such as 1) numerous studies have focused on identifying the roles of E1–E2–E3 ligases in tumorigenesis; however, the oncogenic or tumor-suppressive function of these enzymes is influenced by various factors, such as the cellular context (e.g., β-TRCP) and subcellular localization (e.g., SPOP). Therefore, a comprehensive understanding of the regulatory mechanisms of Ub and Ubl enzymes may provide therapeutic alternatives to inactivate/reactivate or upregulate/downregulate these enzymes in various tumors. 2) Although our multiomics analyses decipher the potential roles of particular enzymes, there is a lack of supporting in vitro or in vivo studies for many Ub- and Ubl-associated enzymes used in anticancer therapy. 3) Targeting E1, E2, or E3 may affect multiple biological processes, resulting in different phenotypes; therefore, a deeper understanding of each enzyme is required to delineate the potential tumor-promoting effects of putative inhibitors of E1–E2–E3 cascades and develop individualized treatments. 4) Some PPI interface may lack such well-defined pocket, which makes it difficult to design selective and potent small-molecule inhibitors. Overall, the present review highlighted the tumor-suppressive or tumor-promoting roles of many Ub- and Ubl-associated enzymes. An ever-growing list of substrates and upstream regulators will help us better understand the roles of these enzymes in tumors and whether they are promising anticancer targets.
Acknowledgments
This work was supported by the following funds: National Natural Science Foundation of China (Grants 81820108022, 82003297 and 22177076), Innovation Program of Shanghai Municipal Education Commission (2019-01-07-00-10-E00056, China), Shanghai Frontiers Science Center of Disease and Syndrome Biology of Inflammatory Cancer Transformation (2021KJ03-12, China), The Scientific and Technological Innovation Action Plan of Science and Technology Commission of Shanghai (20JC1411300, China), ChenGuang project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (19CG49, China).
Author contributions
Yanyu Jiang and Shuaishuai Ni collected the related papers, drafted and revised the manuscript. Biying Xiao collected the related papers. Lijun Jia revised and finalized the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
References
- 1.Hershko A., Ciechanover A., Heller H., Haas A.L., Rose I.A. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A. 1980;77:1783–1786. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciechanover A., Heller H., Elias S., Haas A.L., Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A. 1980;77:1365–1368. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng L., Meng T., Chen L., Wei W., Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Targeted Ther. 2020;5:11. doi: 10.1038/s41392-020-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cappadocia L., Lima C.D. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem Rev. 2018;118:889–918. doi: 10.1021/acs.chemrev.6b00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernassola F., Chillemi G., Melino G. HECT-type E3 ubiquitin ligases in cancer. Trends Biochem Sci. 2019;44:1057–1075. doi: 10.1016/j.tibs.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Wang P., Dai X., Jiang W., Li Y., Wei W. RBR E3 ubiquitin ligases in tumorigenesis. Semin Cancer Biol. 2020;67:131–144. doi: 10.1016/j.semcancer.2020.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Cai Z., Moten A., Peng D., Hsu C.C., Pan B.S., Manne R., et al. The Skp2 pathway: a critical target for cancer therapy. Semin Cancer Biol. 2020;67:16–33. doi: 10.1016/j.semcancer.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrigan J.A., Jacq X., Martin N.M., Jackson S.P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17:57–78. doi: 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciechanover A., Heller H., Katz-Etzion R., Hershko A. Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system. Proc Natl Acad Sci U S A. 1981;78:761–765. doi: 10.1073/pnas.78.2.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barghout S.H., Schimmer A.D. E1 enzymes as therapeutic targets in cancer. Pharmacol Rev. 2021;73:1–58. doi: 10.1124/pharmrev.120.000053. [DOI] [PubMed] [Google Scholar]
- 11.Hyer M.L., Milhollen M.A., Ciavarri J., Fleming P., Traore T., Sappal D., et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med. 2018;24:186–193. doi: 10.1038/nm.4474. [DOI] [PubMed] [Google Scholar]
- 12.Jin J., Li X., Gygi S.P., Harper J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- 13.Li L., Wang M., Yu G., Chen P., Li H., Wei D., et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106:dju083. doi: 10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Wang C., Xu T., Pan P., Yu Q., Xu L., et al. Discovery of a small molecule inhibitor of cullin neddylation that triggers ER stress to induce autophagy. Acta Pharm Sin B. 2021;11:3567–3584. doi: 10.1016/j.apsb.2021.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou L., Zhang W., Sun Y., Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102. doi: 10.1016/j.cellsig.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen P., Hu T., Liang Y., Li P., Chen X., Zhang J., et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4–CHOP–DR5 axis in human esophageal cancer cells. Clin Cancer Res. 2016;22:4145–4157. doi: 10.1158/1078-0432.CCR-15-2254. [DOI] [PubMed] [Google Scholar]
- 17.Zhou W., Xu J., Li H., Xu M., Chen Z.J., Wei W., et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–1116. doi: 10.1158/1078-0432.CCR-16-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Jiang Y., Wu J., Zhang W., Liang Y., Jia L., et al. NEDD8-activating enzyme inhibitor, MLN4924 (Pevonedistat) induces NOXA-dependent apoptosis through up-regulation of ATF-4. Biochem Biophys Res Commun. 2017;488:1–5. doi: 10.1016/j.bbrc.2017.04.122. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Wang S., Zhang W., Wang X., Liu X., Liu L., et al. Targeting neddylation pathway with MLN4924 (Pevonedistat) induces NOXA-dependent apoptosis in renal cell carcinoma. Biochem Biophys Res Commun. 2017;490:1183–1188. doi: 10.1016/j.bbrc.2017.06.179. [DOI] [PubMed] [Google Scholar]
- 20.Milhollen M.A., Traore T., Adams-Duffy J., Thomas M.P., Berger A.J., Dang L., et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 21.Godbersen J.C., Humphries L.A., Danilova O.V., Kebbekus P.E., Brown J.R., Eastman A., et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576–1589. doi: 10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan Y., Xu H., Liu R., Jia L. Induction of cell senescence by targeting to Cullin-RING ligases (CRLs) for effective cancer therapy. Int J Biochem Mol Biol. 2012;3:273–281. [PMC free article] [PubMed] [Google Scholar]
- 23.Jia L., Li H., Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13:561–569. doi: 10.1593/neo.11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y., Luo Z., Pan Y., Wang W., Zhou X., Jeong L.S., et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. 2015;16:420–429. doi: 10.1080/15384047.2014.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y., Liang Y., Li L., Zhou L., Cheng W., Yang X., et al. Targeting neddylation inhibits intravascular survival and extravasation of cancer cells to prevent lung-cancer metastasis. Cell Biol Toxicol. 2019;35:233–245. doi: 10.1007/s10565-019-09472-w. [DOI] [PubMed] [Google Scholar]
- 26.Mackintosh C., Garcia-Dominguez D.J., Ordonez J.L., Ginel-Picardo A., Smith P.G., Sacristan M.P., et al. WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene. 2013;32:1441–1451. doi: 10.1038/onc.2012.153. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y., Cheng W., Li L., Zhou L., Liang Y., Zhang W., et al. Effective targeting of the ubiquitin-like modifier NEDD8 for lung adenocarcinoma treatment. Cell Biol Toxicol. 2020;36:349–364. doi: 10.1007/s10565-019-09503-6. [DOI] [PubMed] [Google Scholar]
- 28.Milhollen M.A., Narayanan U., Soucy T.A., Veiby P.O., Smith P.G., Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042–3051. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- 29.Pan W.W., Zhou J.J., Yu C., Xu Y., Guo L.J., Zhang H.Y., et al. Ubiquitin E3 ligase CRL4CDT2/DCAF2 as a potential chemotherapeutic target for ovarian surface epithelial cancer. J Biol Chem. 2013;288:29680–29691. doi: 10.1074/jbc.M113.495069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L., Liu B., Dong T., Lee H.W., Yu J., Zheng Y., et al. Neddylation pathway regulates the proliferation and survival of macrophages. Biochem Biophys Res Commun. 2013;432:494–498. doi: 10.1016/j.bbrc.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y., Zhang W., Wang S., Cai L., Jiang Y., Pan Y., et al. Cullin3-TNFAIP1 E3 ligase controls inflammatory response in hepatocellular carcinoma cells via ubiquitination of RhoB. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.617134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou L., Jiang Y., Liu X., Li L., Yang X., Dong C., et al. Promotion of tumor-associated macrophages infiltration by elevated neddylation pathway via NF-kappaB–CCL2 signaling in lung cancer. Oncogene. 2019;38:5792–5804. doi: 10.1038/s41388-019-0840-4. [DOI] [PubMed] [Google Scholar]
- 33.Zhou L., Jiang Y., Luo Q., Li L., Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer. 2019;18:77. doi: 10.1186/s12943-019-0979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y., Li L., Li Y., Liu G., Hoffman R.M., Jia L. Neddylation regulates macrophages and implications for cancer therapy. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.681186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao W.T., Wu J.F., Yu G.Y., Wang R., Wang K., Li L.H., et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014;5:e1059. doi: 10.1038/cddis.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Z., Yu G., Lee H.W., Li L., Wang L., Yang D., et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72:3360–3371. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y., Xiong X., Jia L., Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1–REDD1–TSC1–mTORC1–DEPTOR axis. Cell Death Dis. 2012;3:e386. doi: 10.1038/cddis.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang Y., Jiang Y., Jin X., Chen P., Heng Y., Cai L., et al. Neddylation inhibition activates the protective autophagy through NF-kappaB–catalase–ATF3 axis in human esophageal cancer cells. Cell Commun Signal. 2020;18:72. doi: 10.1186/s12964-020-00576-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart M.D., Ritterhoff T., Klevit R.E., Brzovic P.S. E2 enzymes: more than just middle men. Cell Res. 2016;26:423–440. doi: 10.1038/cr.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Chacin R.C., Bodrug T., Bolhuis D.L., Kedziora K.M., Bonacci T., Ordureau A., et al. Ubiquitin chain-elongating enzyme UBE2S activates the RING E3 ligase APC/C for substrate priming. Nat Struct Mol Biol. 2020;27:550–560. doi: 10.1038/s41594-020-0424-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garnett M.J., Mansfeld J., Godwin C., Matsusaka T., Wu J., Russell P., et al. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11:1363–1369. doi: 10.1038/ncb1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson A., Wickliffe K.E., Mellone B.G., Song L., Karpen G.H., Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A. 2009;106:18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T., Merbl Y., Huo Y., Gallop J.L., Tzur A., Kirschner M.W. UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci U S A. 2010;107:1355–1360. doi: 10.1073/pnas.0912802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chong R.A., Wu K., Spratt D.E., Yang Y., Lee C., Nayak J., et al. Pivotal role for the ubiquitin Y59-E51 loop in lysine 48 polyubiquitination. Proc Natl Acad Sci U S A. 2014;111:8434–8439. doi: 10.1073/pnas.1407849111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu K., Kovacev J., Pan Z.Q. Priming and extending: a UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol Cell. 2010;37:784–796. doi: 10.1016/j.molcel.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Ree J.H., Jeganathan K.B., Malureanu L., van Deursen J.M. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188:83–100. doi: 10.1083/jcb.200906147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dastsooz H., Cereda M., Donna D., Oliviero S. A comprehensive bioinformatics analysis of UBE2C in cancers. Int J Mol Sci. 2019;20:2228. doi: 10.3390/ijms20092228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamoto Y., Ozaki T., Miyazaki K., Aoyama M., Miyazaki M., Nakagawara A. UbcH10 is the cancer-related E2 ubiquitin-conjugating enzyme. Cancer Res. 2003;63:4167–4173. [PubMed] [Google Scholar]
- 49.Presta I., Novellino F., Donato A., La Torre D., Palleria C., Russo E., et al. UbcH10 a major actor in cancerogenesis and a potential tool for diagnosis and therapy. Int J Mol Sci. 2020;21:2041. doi: 10.3390/ijms21062041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y., Zhao R., Chi S., Zhang W., Xiao C., Zhou X., et al. UBE2C is upregulated by estrogen and promotes epithelial-mesenchymal transition via p53 in endometrial cancer. Mol Cancer Res. 2020;18:204–215. doi: 10.1158/1541-7786.MCR-19-0561. [DOI] [PubMed] [Google Scholar]
- 51.Zhang R.Y., Liu Z.K., Wei D., Yong Y.L., Lin P., Li H., et al. UBE2S interacting with TRIM28 in the nucleus accelerates cell cycle by ubiquitination of p27 to promote hepatocellular carcinoma development. Signal Transduct Targeted Ther. 2021;6:64. doi: 10.1038/s41392-020-00432-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bajaj S., Alam S.K., Roy K.S., Datta A., Nath S., Roychoudhury S. E2 Ubiquitin-conjugating enzyme, UBE2C gene, is reciprocally regulated by wild-type and gain-of-function mutant p53. J Biol Chem. 2016;291:14231–14247. doi: 10.1074/jbc.M116.731398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu L., Li X., Liu Q., Xu J., Ge H., Wang Z., et al. UBE2S, a novel substrate of Akt1, associates with Ku70 and regulates DNA repair and glioblastoma multiforme resistance to chemotherapy. Oncogene. 2017;36:1145–1156. doi: 10.1038/onc.2016.281. [DOI] [PubMed] [Google Scholar]
- 54.Wang W., Kirschner M.W. Emi1 preferentially inhibits ubiquitin chain elongation by the anaphase-promoting complex. Nat Cell Biol. 2013;15:797–806. doi: 10.1038/ncb2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang S., You X., Zheng Y., Shen Y., Xiong X., Sun Y. The UBE2C/CDH1/DEPTOR axis is an oncogene and tumor suppressor cascade in lung cancer cells. J Clin Invest. 2023;133 doi: 10.1172/JCI162434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao X.C., Wang G.Z., Wen Z.S., Zhou Y.C., Hu Q., Zhang B., et al. Systematic identification of CDC34 that functions to stabilize EGFR and promote lung carcinogenesis. EBioMedicine. 2020;53 doi: 10.1016/j.ebiom.2020.102689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou L., Li Y., Wang Y., Xu D., Cui H., Xu X., et al. UBE2D1 RNA expression was an independent unfavorable prognostic indicator in lung adenocarcinoma, but not in lung squamous cell carcinoma. Dis Markers. 2018;2018 doi: 10.1155/2018/4108919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka K., Kondoh N., Shuda M., Matsubara O., Imazeki N., Ryo A., et al. Enhanced expression of mRNAs of antisecretory factor-1, gp96, DAD1 and CDC34 in human hepatocellular carcinomas. Biochim Biophys Acta. 2001;1536:1–12. doi: 10.1016/s0925-4439(01)00026-6. [DOI] [PubMed] [Google Scholar]
- 59.Zhou C., Bi F., Yuan J., Yang F., Sun S. Gain of UBE2D1 facilitates hepatocellular carcinoma progression and is associated with DNA damage caused by continuous IL-6. J Exp Clin Cancer Res. 2018;37:290. doi: 10.1186/s13046-018-0951-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chauhan D., Li G., Hideshima T., Podar K., Shringarpure R., Mitsiades C., et al. Blockade of ubiquitin-conjugating enzyme CDC34 enhances anti-myeloma activity of bortezomib/proteasome inhibitor PS-341. Oncogene. 2004;23:3597–3602. doi: 10.1038/sj.onc.1207458. [DOI] [PubMed] [Google Scholar]
- 61.Zhang S., Sun Y. Targeting CDC34 E2 ubiquitin conjugating enzyme for lung cancer therapy. EBioMedicine. 2020;54 doi: 10.1016/j.ebiom.2020.102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sandoval D., Hill S., Ziemba A., Lewis S., Kuhlman B., Kleiger G. Ubiquitin-conjugating enzyme Cdc34 and ubiquitin ligase Skp1–cullin–F-box ligase (SCF) interact through multiple conformations. J Biol Chem. 2015;290:1106–1118. doi: 10.1074/jbc.M114.615559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu H.T., Liu S., Liu L., Ma R.R., Gao P. EGR1-mediated transcription of lncRNA–HNF1A–AS1 promotes cell-cycle progression in gastric cancer. Cancer Res. 2018;78:5877–5890. doi: 10.1158/0008-5472.CAN-18-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang S., Xian J., Li L., Jiang Y., Liu Y., Cai L., et al. NEDD8-conjugating enzyme UBC12 as a novel therapeutic target in esophageal squamous cell carcinoma. Signal Transduct Targeted Ther. 2020;5:123. doi: 10.1038/s41392-020-00226-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li L., Kang J., Zhang W., Cai L., Wang S., Liang Y., et al. Validation of NEDD8-conjugating enzyme UBC12 as a new therapeutic target in lung cancer. EBioMedicine. 2019;45:81–91. doi: 10.1016/j.ebiom.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou W., Xu J., Tan M., Li H., Li H., Wei W., et al. UBE2M is a stress-inducible dual E2 for neddylation and ubiquitylation that promotes targeted degradation of UBE2F. Mol Cell. 2018;70:1008–10024. doi: 10.1016/j.molcel.2018.06.002. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Ther. 2003;2:623–629. [PubMed] [Google Scholar]
- 68.Zhao Y., Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharmaceut Des. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Z., Liu P., Inuzuka H., Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin J., Cardozo T., Lovering R.C., Elledge S.J., Pagano M., Harper J.W. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang H., Kobayashi R., Galaktionov K., Beach D. p19Skp1 and p45Skp2 are essential elements of the cyclin A–CDK2 S phase kinase. Cell. 1995;82:915–925. doi: 10.1016/0092-8674(95)90271-6. [DOI] [PubMed] [Google Scholar]
- 72.Zhu C.Q., Blackhall F.H., Pintilie M., Iyengar P., Liu N., Ho J., et al. Skp2 gene copy number aberrations are common in non-small cell lung carcinoma, and its overexpression in tumors with ras mutation is a poor prognostic marker. Clin Cancer Res. 2004;10:1984–1991. doi: 10.1158/1078-0432.ccr-03-0470. [DOI] [PubMed] [Google Scholar]
- 73.Yu X., Wang R., Zhang Y., Zhou L., Wang W., Liu H., et al. Skp2-mediated ubiquitination and mitochondrial localization of Akt drive tumor growth and chemoresistance to cisplatin. Oncogene. 2019;38:7457–7472. doi: 10.1038/s41388-019-0955-7. [DOI] [PubMed] [Google Scholar]
- 74.Lu M., Ma J., Xue W., Cheng C., Wang Y., Zhao Y., et al. The expression and prognosis of FOXO3a and Skp2 in human hepatocellular carcinoma. Pathol Oncol Res. 2009;15:679–687. doi: 10.1007/s12253-009-9171-z. [DOI] [PubMed] [Google Scholar]
- 75.Masuda T.A., Inoue H., Sonoda H., Mine S., Yoshikawa Y., Nakayama K., et al. Clinical and biological significance of S-phase kinase-associated protein 2 (Skp2) gene expression in gastric carcinoma: modulation of malignant phenotype by Skp2 overexpression, possibly via p27 proteolysis. Cancer Res. 2002;62:3819–3825. [PubMed] [Google Scholar]
- 76.Yu Z.K., Gervais J.L., Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21CIP1/WAF1 and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carrano A.C., Eytan E., Hershko A., Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 78.Kamura T., Hara T., Kotoshiba S., Yada M., Ishida N., Imaki H., et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A. 2003;100:10231–10236. doi: 10.1073/pnas.1831009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishitani H., Sugimoto N., Roukos V., Nakanishi Y., Saijo M., Obuse C., et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25:1126–1136. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mendez J., Zou-Yang X.H., Kim S.Y., Hidaka M., Tansey W.P., Stillman B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol Cell. 2002;9:481–491. doi: 10.1016/s1097-2765(02)00467-7. [DOI] [PubMed] [Google Scholar]
- 81.Huang H., Regan K.M., Wang F., Wang D., Smith D.I., van Deursen J.M., et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan C.H., Li C.F., Yang W.L., Gao Y., Lee S.W., Feng Z., et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–1111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yao F., Zhou Z., Kim J., Hang Q., Xiao Z., Ton B.N., et al. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat Commun. 2018;9:2269. doi: 10.1038/s41467-018-04620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J.Y., Liu G.Z., Wilmott J.S., La T., Feng Y.C., Yari H., et al. Skp2-mediated stabilization of MTH1 promotes survival of melanoma cells upon oxidative stress. Cancer Res. 2017;77:6226–6239. doi: 10.1158/0008-5472.CAN-17-1965. [DOI] [PubMed] [Google Scholar]
- 85.Lee S.W., Li C.F., Jin G., Cai Z., Han F., Chan C.H., et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell. 2015;57:1022–1033. doi: 10.1016/j.molcel.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu J., Zhang X., Zhang L., Wu C.Y., Rezaeian A.H., Chan C.H., et al. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol Cell. 2012;46:351–361. doi: 10.1016/j.molcel.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruan D., He J., Li C.F., Lee H.J., Liu J., Lin H.K., et al. Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene. 2017;36:4299–4310. doi: 10.1038/onc.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Finkin S., Aylon Y., Anzi S., Oren M., Shaulian E. Fbw7 regulates the activity of endoreduplication mediators and the p53 pathway to prevent drug-induced polyploidy. Oncogene. 2008;27:4411–4421. doi: 10.1038/onc.2008.77. [DOI] [PubMed] [Google Scholar]
- 89.Kwon Y.W., Kim I.J., Wu D., Lu J., Stock W.A., Jr., Liu Y., et al. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol Cancer Res. 2012;10:834–844. doi: 10.1158/1541-7786.MCR-12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yeh C.H., Bellon M., Wang F., Zhang H., Fu L., Nicot C. Loss of FBXW7-mediated degradation of BRAF elicits resistance to BET inhibitors in adult T cell leukemia cells. Mol Cancer. 2020;19:139. doi: 10.1186/s12943-020-01254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nateri A.S., Riera-Sans L., Da Costa C., Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- 92.Wei W., Jin J., Schlisio S., Harper J.W., Kaelin W.G., Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 93.Yada M., Hatakeyama S., Kamura T., Nishiyama M., Tsunematsu R., Imaki H., et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Welcker M., Orian A., Jin J., Grim J.E., Harper J.W., Eisenman R.N., et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kuai X., Li L., Chen R., Wang K., Chen M., Cui B., et al. SCF(FBXW7)/GSK3beta-mediated GFI1 degradation suppresses proliferation of gastric cancer cells. Cancer Res. 2019;79:4387–4398. doi: 10.1158/0008-5472.CAN-18-4032. [DOI] [PubMed] [Google Scholar]
- 96.Cassavaugh J.M., Hale S.A., Wellman T.L., Howe A.K., Wong C., Lounsbury K.M. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011;112:3882–3890. doi: 10.1002/jcb.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Flugel D., Gorlach A., Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119:1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xie C.M., Tan M., Lin X.T., Wu D., Jiang Y., Tan Y., et al. The FBXW7–SHOC2–Raptor axis controls the cross-talks between the RAS–ERK and mTORC1 signaling pathways. Cell Rep. 2019;26:3037–3050.e4. doi: 10.1016/j.celrep.2019.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao D., Zheng H.Q., Zhou Z., Chen C. The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer Res. 2010;70:4728–4738. doi: 10.1158/0008-5472.CAN-10-0040. [DOI] [PubMed] [Google Scholar]
- 100.Liu N., Li H., Li S., Shen M., Xiao N., Chen Y., et al. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J Biol Chem. 2010;285:18858–18867. doi: 10.1074/jbc.M109.099440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tong J., Tan S., Zou F., Yu J., Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017;36:787–796. doi: 10.1038/onc.2016.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Inuzuka H., Shaik S., Onoyama I., Gao D., Tseng A., Maser R.S., et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu G., Lyapina S., Das I., Li J., Gurney M., Pauley A., et al. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gupta-Rossi N., Le Bail O., Gonen H., Brou C., Logeat F., Six E., et al. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem. 2001;276:34371–34378. doi: 10.1074/jbc.M101343200. [DOI] [PubMed] [Google Scholar]
- 105.Fryer C.J., White J.B., Jones K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509–520. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 106.Liao S.Y., Chiang C.W., Hsu C.H., Chen Y.T., Jen J., Juan H.F., et al. CK1delta/GSK3beta/FBXW7alpha axis promotes degradation of the ZNF322A oncoprotein to suppress lung cancer progression. Oncogene. 2017;36:5722–5733. doi: 10.1038/onc.2017.168. [DOI] [PubMed] [Google Scholar]
- 107.Yumimoto K., Nakayama K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol. 2020;67:1–15. doi: 10.1016/j.semcancer.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 108.Matsumoto A., Onoyama I., Nakayama K.I. Expression of mouse Fbxw7 isoforms is regulated in a cell cycle- or p53-dependent manner. Biochem Biophys Res Commun. 2006;350:114–119. doi: 10.1016/j.bbrc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Cui D., Xiong X., Shu J., Dai X., Sun Y., Zhao Y. FBXW7 confers radiation survival by targeting p53 for degradation. Cell Rep. 2020;30:497–509.e4. doi: 10.1016/j.celrep.2019.12.032. [DOI] [PubMed] [Google Scholar]
- 110.Ye M., Zhang Y., Zhang X., Zhang J., Jing P., Cao L., et al. Targeting FBW7 as a strategy to overcome resistance to targeted therapy in non-small cell lung cancer. Cancer Res. 2017;77:3527–3539. doi: 10.1158/0008-5472.CAN-16-3470. [DOI] [PubMed] [Google Scholar]
- 111.Mao J.H., Perez-Losada J., Wu D., Delrosario R., Tsunematsu R., Nakayama K.I., et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 112.Zhao J., Hu C., Chi J., Li J., Peng C., Yun X., et al. miR-24 promotes the proliferation, migration and invasion in human tongue squamous cell carcinoma by targeting FBXW7. Oncol Rep. 2016;36:1143–1149. doi: 10.3892/or.2016.4891. [DOI] [PubMed] [Google Scholar]
- 113.Spruck C. miR-27a regulation of SCFFbw7 in cell division control and cancer. Cell Cycle. 2011;10:3232–3233. doi: 10.4161/cc.10.19.17125. [DOI] [PubMed] [Google Scholar]
- 114.Xia W., Zhou J., Luo H., Liu Y., Peng C., Zheng W., et al. MicroRNA-32 promotes cell proliferation, migration and suppresses apoptosis in breast cancer cells by targeting FBXW7. Cancer Cell Int. 2017;17:14. doi: 10.1186/s12935-017-0383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu Y., Sengupta T., Kukreja L., Minella A.C. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. J Biol Chem. 2010;285:34439–34446. doi: 10.1074/jbc.M110.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu J., Wu W., Wang J., Huang C., Wen W., Zhao F., et al. miR-367 promotes the proliferation and invasion of non-small cell lung cancer via targeting FBXW7. Oncol Rep. 2017;37:1052–1058. doi: 10.3892/or.2016.5314. [DOI] [PubMed] [Google Scholar]
- 117.Yaron A., Hatzubai A., Davis M., Lavon I., Amit S., Manning A.M., et al. Identification of the receptor component of the IκBα-ubiquitin ligase. Nature. 1998;396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 118.Zhang B., Zhang Z., Li L., Qin Y.R., Liu H., Jiang C., et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-κB signaling. Nat Commun. 2018;9:1423. doi: 10.1038/s41467-018-03716-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Watanabe N., Arai H., Nishihara Y., Taniguchi M., Watanabe N., Hunter T., et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xia Y., Padre R.C., De Mendoza T.H., Bottero V., Tergaonkar V.B., Verma I.M. Phosphorylation of p53 by IκB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A. 2009;106:2629–2634. doi: 10.1073/pnas.0812256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao Y., Xiong X., Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu J., Zhou W., Yang F., Chen G., Li H., Zhao Y., et al. The β-TrCP–FBXW2–SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nat Commun. 2017;8 doi: 10.1038/ncomms14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dehan E., Bassermann F., Guardavaccaro D., Vasiliver-Shamis G., Cohen M., Lowes K.N., et al. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33:109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang Y., Hu K., Zhang S., Dong X., Yin Z., Meng R., et al. S6K1 phosphorylation-dependent degradation of Mxi1 by β-Trcp ubiquitin ligase promotes Myc activation and radioresistance in lung cancer. Theranostics. 2018;8:1286–1300. doi: 10.7150/thno.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dorrello N.V., Peschiaroli A., Guardavaccaro D., Colburn N.H., Sherman N.E., Pagano M. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 126.Tan M., Gallegos J.R., Gu Q., Huang Y., Li J., Jin Y., et al. SAG/ROC-SCF β-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8:1042–1054. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang H.M., Xu Y.F., Ning S.L., Yang D.X., Li Y., Du Y.J., et al. The catalytic region and PEST domain of PTPN18 distinctly regulate the HER2 phosphorylation and ubiquitination barcodes. Cell Res. 2014;24:1067–1090. doi: 10.1038/cr.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao B., Li L., Tumaneng K., Wang C.Y., Guan K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Inuzuka H., Tseng A., Gao D., Zhai B., Zhang Q., Shaik S., et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCFβ-TRCP ubiquitin ligase. Cancer Cell. 2010;18:147–159. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li C.W., Lim S.O., Xia W., Lee H.H., Chan L.C., Kuo C.W., et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7 doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ding Q., He X., Hsu J.M., Xia W., Chen C.T., Li L.Y., et al. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ougolkov A., Zhang B., Yamashita K., Bilim V., Mai M., Fuchs S.Y., et al. Associations among β-TrCP, an E3 ubiquitin ligase receptor, β-catenin, and NF-κB in colorectal cancer. J Natl Cancer Inst. 2004;96:1161–1170. doi: 10.1093/jnci/djh219. [DOI] [PubMed] [Google Scholar]
- 133.Koch A., Waha A., Hartmann W., Hrychyk A., Schuller U., Waha A., et al. Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clin Cancer Res. 2005;11:4295–4304. doi: 10.1158/1078-0432.CCR-04-1162. [DOI] [PubMed] [Google Scholar]
- 134.Kim C.J., Song J.H., Cho Y.G., Kim Y.S., Kim S.Y., Nam S.W., et al. Somatic mutations of the beta-TrCP gene in gastric cancer. APMIS. 2007;115:127–133. doi: 10.1111/j.1600-0463.2007.apm_562.x. [DOI] [PubMed] [Google Scholar]
- 135.Gerstein A.V., Almeida T.A., Zhao G., Chess E., Shih Ie M., Buhler K., et al. APC/CTNNB1 (β-catenin) pathway alterations in human prostate cancers. Genes Chromosomes Cancer. 2002;34:9–16. doi: 10.1002/gcc.10037. [DOI] [PubMed] [Google Scholar]
- 136.Liu X., Zurlo G., Zhang Q. The roles of Cullin-2 E3 ubiquitin ligase complex in cancer. Adv Exp Med Biol. 2020;1217:173–186. doi: 10.1007/978-981-15-1025-0_11. [DOI] [PubMed] [Google Scholar]
- 137.Latif F., Tory K., Gnarra J., Yao M., Duh F.M., Orcutt M.L., et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 138.Gnarra J.R., Tory K., Weng Y., Schmidt L., Wei M.H., Li H., et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 139.Semenza G.L. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 140.Kim W.Y., Kaelin W.G. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 141.Sato Y., Yoshizato T., Shiraishi Y., Maekawa S., Okuno Y., Kamura T., et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 142.Hsieh J.J., Purdue M.P., Signoretti S., Swanton C., Albiges L., Schmidinger M., et al. Renal cell carcinoma. Nat Rev Dis Prim. 2017;3 doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gossage L., Eisen T., Maher E.R. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 144.Zhang J., Wu T., Simon J., Takada M., Saito R., Fan C., et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science. 2018;361:290–295. doi: 10.1126/science.aap8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Okuda H., Saitoh K., Hirai S., Iwai K., Takaki Y., Baba M., et al. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem. 2001;276:43611–43617. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- 146.Xie L., Xiao K., Whalen E.J., Forrester M.T., Freeman R.S., Fong G., et al. Oxygen-regulated β2-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2:ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Na X., Duan H.O., Messing E.M., Schoen S.R., Ryan C.K., di Sant'Agnese P.A., et al. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. EMBO J. 2003;22:4249–4259. doi: 10.1093/emboj/cdg410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang P., Song J., Ye D. CRL3s: the BTB–CUL3–RING E3 ubiquitin ligases. Adv Exp Med Biol. 2020;1217:211–223. doi: 10.1007/978-981-15-1025-0_13. [DOI] [PubMed] [Google Scholar]
- 149.Menegon S., Columbano A., Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578–593. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 150.Romero R., Sayin V.I., Davidson S.M., Bauer M.R., Singh S.X., LeBoeuf S.E., et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362–1368. doi: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Khor T.O., Huang M.T., Prawan A., Liu Y., Hao X., Yu S., et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res. 2008;1:187–191. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Luo J., Bao Y.C., Ji X.X., Chen B., Deng Q.F., Zhou S.W. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochem Biophys Res Commun. 2017;483:880–884. doi: 10.1016/j.bbrc.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 153.Dai X., Gan W., Li X., Wang S., Zhang W., Huang L., et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063–1071. doi: 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang P., Wang D., Zhao Y., Ren S., Gao K., Ye Z., et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT–mTORC1 activation. Nat Med. 2017;23:1055–1062. doi: 10.1038/nm.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Janouskova H., El Tekle G., Bellini E., Udeshi N.D., Rinaldi A., Ulbricht A., et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23:1046–1054. doi: 10.1038/nm.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gan W., Dai X., Lunardi A., Li Z., Inuzuka H., Liu P., et al. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol Cell. 2015;59:917–930. doi: 10.1016/j.molcel.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li G., Ci W., Karmakar S., Chen K., Dhar R., Fan Z., et al. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell. 2014;25:455–468. doi: 10.1016/j.ccr.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Adelaiye-Ogala R., Damayanti N.P., Orillion A.R., Arisa S., Chintala S., Titus M.A., et al. Therapeutic targeting of sunitinib-induced AR phosphorylation in renal cell carcinoma. Cancer Res. 2018;78:2886–2896. doi: 10.1158/0008-5472.CAN-17-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang P., Gao K., Jin X., Ma J., Peng J., Wumaier R., et al. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-α protein turnover. Cell Death Dis. 2015;6:e1687. doi: 10.1038/cddis.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kim B., Nam H.J., Pyo K.E., Jang M.J., Kim I.S., Kim D., et al. Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3–SPOP E3 ubiquitin ligase complex. Biochem Biophys Res Commun. 2011;415:720–726. doi: 10.1016/j.bbrc.2011.10.154. [DOI] [PubMed] [Google Scholar]
- 161.Zhang J., Bu X., Wang H., Zhu Y., Geng Y., Nihira N.T., et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ju L.G., Zhu Y., Long Q.Y., Li X.J., Lin X., Tang S.B., et al. SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 2019;26:1156–1168. doi: 10.1038/s41418-018-0198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma J., Chang K., Peng J., Shi Q., Gan H., Gao K., et al. SPOP promotes ATF2 ubiquitination and degradation to suppress prostate cancer progression. J Exp Clin Cancer Res. 2018;37:145. doi: 10.1186/s13046-018-0809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Geng C., Kaochar S., Li M., Rajapakshe K., Fiskus W., Dong J., et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017;36:4767–4777. doi: 10.1038/onc.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Geng C., Rajapakshe K., Shah S.S., Shou J., Eedunuri V.K., Foley C., et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–5643. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Geng C., He B., Xu L., Barbieri C.E., Eedunuri V.K., Chew S.A., et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang Z., Song Y., Ye M., Dai X., Zhu X., Wei W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat Rev Urol. 2020;17:339–350. doi: 10.1038/s41585-020-0314-z. [DOI] [PubMed] [Google Scholar]
- 168.Nagai Y., Kojima T., Muro Y., Hachiya T., Nishizawa Y., Wakabayashi T., et al. Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett. 1997;418:23–26. doi: 10.1016/s0014-5793(97)01340-9. [DOI] [PubMed] [Google Scholar]
- 169.Lee J., Zhou P. DCAFs, the missing link of the CUL4–DDB1 ubiquitin ligase. Mol Cell. 2007;26:775–780. doi: 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 170.He Y.J., McCall C.M., Hu J., Zeng Y., Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4–ROC1 ubiquitin ligases. Genes Dev. 2006;20:2949–2954. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kapetanaki M.G., Guerrero-Santoro J., Bisi D.C., Hsieh C.L., Rapic-Otrin V., Levine A.S. The DDB1–CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang H., Zhai L., Xu J., Joo H.Y., Jackson S., Erdjument-Bromage H., et al. Histone H3 and H4 ubiquitylation by the CUL4–DDB–ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 173.Cazzalini O., Perucca P., Mocchi R., Sommatis S., Prosperi E., Stivala L.A. DDB2 association with PCNA is required for its degradation after UV-induced DNA damage. Cell Cycle. 2014;13:240–248. doi: 10.4161/cc.26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.El-Mahdy M.A., Zhu Q., Wang Q.E., Wani G., Praetorius-Ibba M., Wani A.A. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281:13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 175.Ribeiro-Silva C., Sabatella M., Helfricht A., Marteijn J.A., Theil A.F., Vermeulen W., et al. Ubiquitin and TFIIH-stimulated DDB2 dissociation drives DNA damage handover in nucleotide excision repair. Nat Commun. 2020;11:4868. doi: 10.1038/s41467-020-18705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yoon T., Chakrabortty A., Franks R., Valli T., Kiyokawa H., Raychaudhuri P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene. 2005;24:469–478. doi: 10.1038/sj.onc.1208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alekseev S., Kool H., Rebel H., Fousteri M., Moser J., Backendorf C., et al. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res. 2005;65:10298–10306. doi: 10.1158/0008-5472.CAN-05-2295. [DOI] [PubMed] [Google Scholar]
- 178.Huang S., Fantini D., Merrill B.J., Bagchi S., Guzman G., Raychaudhuri P. DDB2 is a novel regulator of Wnt signaling in colon cancer. Cancer Res. 2017;77:6562–6575. doi: 10.1158/0008-5472.CAN-17-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bommi P.V., Chand V., Mukhopadhyay N.K., Raychaudhuri P., Bagchi S. NER-factor DDB2 regulates HIF1α and hypoxia-response genes in HNSCC. Oncogene. 2020;39:1784–1796. doi: 10.1038/s41388-019-1105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cui T., Srivastava A.K., Han C., Wu D., Wani N., Liu L., et al. DDB2 represses ovarian cancer cell dedifferentiation by suppressing ALDH1A1. Cell Death Dis. 2018;9:561. doi: 10.1038/s41419-018-0585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bommi P.V., Ravindran S., Raychaudhuri P., Bagchi S. DDB2 regulates epithelial-to-mesenchymal transition (EMT) in oral/head and neck squamous cell carcinoma. Oncotarget. 2018;9:34708–34718. doi: 10.18632/oncotarget.26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Havens C.G., Walter J.C. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568–1582. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Panagopoulos A., Taraviras S., Nishitani H., Lygerou Z. CRL4Cdt2: coupling genome stability to ubiquitination. Trends Cell Biol. 2020;30:290–302. doi: 10.1016/j.tcb.2020.01.005. [DOI] [PubMed] [Google Scholar]
- 184.Jin J., Arias E.E., Chen J., Harper J.W., Walter J.C. A family of diverse Cul4–Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 185.Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–2506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A. CRL4Cdt2 regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huh J., Piwnica-Worms H. CRL4CDT2 targets CHK1 for PCNA-independent destruction. Mol Cell Biol. 2013;33:213–226. doi: 10.1128/MCB.00847-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liontos M., Koutsami M., Sideridou M., Evangelou K., Kletsas D., Levy B., et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007;67:10899–10909. doi: 10.1158/0008-5472.CAN-07-2837. [DOI] [PubMed] [Google Scholar]
- 189.Matyskiela M.E., Lu G., Ito T., Pagarigan B., Lu C.C., Miller K., et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 190.Sievers Q.L., Petzold G., Bunker R.D., Renneville A., Slabicki M., Liddicoat B.J., et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science. 2018;362 doi: 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lu G., Middleton R.E., Sun H., Naniong M., Ott C.J., Mitsiades C.S., et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yamamoto J., Suwa T., Murase Y., Tateno S., Mizutome H., Asatsuma-Okumura T., et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat Chem Biol. 2020;16:1208–1217. doi: 10.1038/s41589-020-0645-3. [DOI] [PubMed] [Google Scholar]
- 193.Eichner R., Heider M., Fernandez-Saiz V., van Bebber F., Garz A.K., Lemeer S., et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat Med. 2016;22:735–743. doi: 10.1038/nm.4128. [DOI] [PubMed] [Google Scholar]
- 194.Heider M., Eichner R., Stroh J., Morath V., Kuisl A., Zecha J., et al. The IMiD target CRBN determines HSP90 activity toward transmembrane proteins essential in multiple myeloma. Mol Cell. 2021;81:1170–1186. doi: 10.1016/j.molcel.2020.12.046. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhao Y., Xiong X., Sun Y. Cullin-RING ligase 5: functional characterization and its role in human cancers. Semin Cancer Biol. 2020;67:61–79. doi: 10.1016/j.semcancer.2020.04.003. [DOI] [PubMed] [Google Scholar]
- 196.Hashimoto M., Ayada T., Kinjyo I., Hiwatashi K., Yoshida H., Okada Y., et al. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 2009;100:730–736. doi: 10.1111/j.1349-7006.2009.01098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shen L., Evel-Kabler K., Strube R., Chen S.Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 198.Zhao G., Gong L., Su D., Jin Y., Guo C., Yue M., et al. Cullin5 deficiency promotes small-cell lung cancer metastasis by stabilizing integrin beta1. J Clin Invest. 2019;129:972–987. doi: 10.1172/JCI122779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang C.X., Ye S.B., Ni J.J., Cai T.T., Liu Y.N., Huang D.J., et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019;26:2314–2328. doi: 10.1038/s41418-019-0302-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yoneda T., Kunimura N., Kitagawa K., Fukui Y., Saito H., Narikiyo K., et al. Overexpression of SOCS3 mediated by adenovirus vector in mouse and human castration-resistant prostate cancer cells increases the sensitivity to NK cells in vitro and in vivo. Cancer Gene Ther. 2019;26:388–399. doi: 10.1038/s41417-018-0075-5. [DOI] [PubMed] [Google Scholar]
- 201.Sasi W., Jiang W.G., Sharma A., Mokbel K. Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer. 2010;10:178. doi: 10.1186/1471-2407-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lewis R.S., Kolesnik T.B., Kuang Z., D'Cruz A.A., Blewitt M.E., Masters S.L., et al. TLR regulation of SPSB1 controls inducible nitric oxide synthase induction. J Immunol. 2011;187:3798–3805. doi: 10.4049/jimmunol.1002993. [DOI] [PubMed] [Google Scholar]
- 203.Nishiya T., Matsumoto K., Maekawa S., Kajita E., Horinouchi T., Fujimuro M., et al. Regulation of inducible nitric-oxide synthase by the SPRY domain- and SOCS box-containing proteins. J Biol Chem. 2011;286:9009–9019. doi: 10.1074/jbc.M110.190678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vannini F., Kashfi K., Nath N. The dual role of iNOS in cancer. Redox Biol. 2015;6:334–343. doi: 10.1016/j.redox.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kim H.J., Kim H.J., Kim M.K., Bae M.K., Sung H.Y., Ahn J.H., et al. SPSB1 enhances ovarian cancer cell survival by destabilizing p21. Biochem Biophys Res Commun. 2019;510:364–369. doi: 10.1016/j.bbrc.2019.01.088. [DOI] [PubMed] [Google Scholar]
- 206.Feng Y., Pan T.C., Pant D.K., Chakrabarti K.R., Alvarez J.V., Ruth J.R., et al. SPSB1 promotes breast cancer recurrence by potentiating c-MET signaling. Cancer Discov. 2014;4:790–803. doi: 10.1158/2159-8290.CD-13-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kumar S., Tomooka Y., Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- 208.Wang X., Trotman L.C., Koppie T., Alimonti A., Chen Z., Gao Z., et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cai J., Li R., Xu X., Zhang L., Lian R., Fang L., et al. CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat Cell Biol. 2018;20:465–478. doi: 10.1038/s41556-018-0065-8. [DOI] [PubMed] [Google Scholar]
- 210.Xu C., Fan C.D., Wang X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene. 2015;34:281–289. doi: 10.1038/onc.2013.557. [DOI] [PubMed] [Google Scholar]
- 211.Yang Y., Luo M., Zhang K., Zhang J., Gao T., Connell D.O., et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020;11:433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cao X.R., Lill N.L., Boase N., Shi P.P., Croucher D.R., Shan H., et al. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci Signal. 2008;1:ra5. doi: 10.1126/scisignal.1160940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jing W., Wang G., Cui Z., Xiong G., Jiang X., Li Y., et al. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder cancer immune surveillance. Cancer Res. 2022;82:114–129. doi: 10.1158/0008-5472.CAN-21-2362. [DOI] [PubMed] [Google Scholar]
- 214.Zhou W., Xu J., Zhao Y., Sun Y. SAG/RBX2 is a novel substrate of NEDD4-1 E3 ubiquitin ligase and mediates NEDD4-1 induced chemosensitization. Oncotarget. 2014;5:6746–6755. doi: 10.18632/oncotarget.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Walden H., Rittinger K. RBR ligase-mediated ubiquitin transfer: a tale with many twists and turns. Nat Struct Mol Biol. 2018;25:440–445. doi: 10.1038/s41594-018-0063-3. [DOI] [PubMed] [Google Scholar]
- 216.Bernardini J.P., Lazarou M., Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017;36:1315–1327. doi: 10.1038/onc.2016.302. [DOI] [PubMed] [Google Scholar]
- 217.Liu J., Zhang C., Zhao Y., Yue X., Wu H., Huang S., et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8:1823. doi: 10.1038/s41467-017-01947-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gupta A., Anjomani-Virmouni S., Koundouros N., Dimitriadi M., Choo-Wing R., Valle A., et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol Cell. 2017;65:999–1013. doi: 10.1016/j.molcel.2017.02.019. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yeo C.W., Ng F.S., Chai C., Tan J.M., Koh G.R., Chong Y.K., et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72:2543–2553. doi: 10.1158/0008-5472.CAN-11-3060. [DOI] [PubMed] [Google Scholar]
- 220.Fujiwara M., Marusawa H., Wang H.Q., Iwai A., Ikeuchi K., Imai Y., et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene. 2008;27:6002–6011. doi: 10.1038/onc.2008.199. [DOI] [PubMed] [Google Scholar]
- 221.Carroll R.G., Hollville E., Martin S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014;9:1538–1553. doi: 10.1016/j.celrep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 222.Li C., Zhang Y., Cheng X., Yuan H., Zhu S., Liu J., et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46:441–455. doi: 10.1016/j.devcel.2018.07.012. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Liu J., Zhang C., Wu H., Sun X.X., Li Y., Huang S., et al. Parkin ubiquitinates phosphoglycerate dehydrogenase to suppress serine synthesis and tumor progression. J Clin Invest. 2020;130:3253–3269. doi: 10.1172/JCI132876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee S.B., Kim J.J., Han S.A., Fan Y., Guo L.S., Aziz K., et al. The AMPK–Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol. 2019;21:940–951. doi: 10.1038/s41556-019-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mevissen T.E.T., Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- 226.Zhang F., Zhao Y., Sun Y. USP2 is an SKP2 deubiquitylase that stabilizes both SKP2 and its substrates. J Biol Chem. 2021;297 doi: 10.1016/j.jbc.2021.101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.He J., Lee H.J., Saha S., Ruan D., Guo H., Chan C.H. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019;10:285. doi: 10.1038/s41419-019-1512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Stevenson L.F., Sparks A., Allende-Vega N., Xirodimas D.P., Lane D.P., Saville M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–986. doi: 10.1038/sj.emboj.7601567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Allende-Vega N., Sparks A., Lane D.P., Saville M.K. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene. 2010;29:432–441. doi: 10.1038/onc.2009.330. [DOI] [PubMed] [Google Scholar]
- 230.Zhao Y., Wang X., Wang Q., Deng Y., Li K., Zhang M., et al. USP2a supports metastasis by tuning TGF-beta signaling. Cell Rep. 2018;22:2442–2454. doi: 10.1016/j.celrep.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 231.Graner E., Tang D., Rossi S., Baron A., Migita T., Weinstein L.J., et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253–261. doi: 10.1016/s1535-6108(04)00055-8. [DOI] [PubMed] [Google Scholar]
- 232.Wang Z., Kang W., Li O., Qi F., Wang J., You Y., et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B. 2021;11:694–707. doi: 10.1016/j.apsb.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shan J., Zhao W., Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell. 2009;36:469–476. doi: 10.1016/j.molcel.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zheng N., Chu M., Lin M., He Y., Wang Z. USP7 stabilizes EZH2 and enhances cancer malignant progression. Am J Cancer Res. 2020;10:299–313. [PMC free article] [PubMed] [Google Scholar]
- 235.Wang Q., Ma S., Song N., Li X., Liu L., Yang S., et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest. 2016;126:2205–2220. doi: 10.1172/JCI85747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Li M., Brooks C.L., Kon N., Gu W. A dynamic role of HAUSP in the p53–Mdm2 pathway. Mol Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 237.Yi L., Cui Y., Xu Q., Jiang Y. Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol Rep. 2016;36:2935–2945. doi: 10.3892/or.2016.5099. [DOI] [PubMed] [Google Scholar]
- 238.Novellasdemunt L., Foglizzo V., Cuadrado L., Antas P., Kucharska A., Encheva V., et al. USP7 is a tumor-specific WNT activator for APC-mutated colorectal cancer by mediating beta-catenin deubiquitination. Cell Rep. 2017;21:612–627. doi: 10.1016/j.celrep.2017.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cheng J., Yang H., Fang J., Ma L., Gong R., Wang P., et al. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat Commun. 2015;6:7023. doi: 10.1038/ncomms8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Du Z., Song J., Wang Y., Zhao Y., Guda K., Yang S., et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal. 2010;3:ra80. doi: 10.1126/scisignal.2001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Watanabe K., Yokoyama S., Kaneto N., Hori T., Iwakami Y., Kato S., et al. COP9 signalosome subunit 5 regulates cancer metastasis by deubiquitinating SNAIL. Oncotarget. 2018;9:20670–20680. doi: 10.18632/oncotarget.25060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhang S., Hong Z., Chai Y., Liu Z., Du Y., Li Q., et al. CSN5 promotes renal cell carcinoma metastasis and EMT by inhibiting ZEB1 degradation. Biochem Biophys Res Commun. 2017;488:101–108. doi: 10.1016/j.bbrc.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 243.Li J., Li Y., Wang B., Ma Y., Chen P. CSN5/Jab1 facilitates non-small cell lung cancer cell growth through stabilizing survivin. Biochem Biophys Res Commun. 2018;500:132–138. doi: 10.1016/j.bbrc.2018.03.183. [DOI] [PubMed] [Google Scholar]
- 244.Mao L., Le S., Jin X., Liu G., Chen J., Hu J. CSN5 promotes the invasion and metastasis of pancreatic cancer by stabilization of FOXM1. Exp Cell Res. 2019;374:274–281. doi: 10.1016/j.yexcr.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 245.Lim S.O., Li C.W., Xia W., Cha J.H., Chan L.C., Wu Y., et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tauriello D.V., Haegebarth A., Kuper I., Edelmann M.J., Henraat M., Canninga-van Dijk M.R., et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell. 2010;37:607–619. doi: 10.1016/j.molcel.2010.01.035. [DOI] [PubMed] [Google Scholar]
- 247.Shinriki S., Jono H., Maeshiro M., Nakamura T., Guo J., Li J.D., et al. Loss of CYLD promotes cell invasion via ALK5 stabilization in oral squamous cell carcinoma. J Pathol. 2018;244:367–379. doi: 10.1002/path.5019. [DOI] [PubMed] [Google Scholar]
- 248.Zhang Z., Fan Y., Xie F., Zhou H., Jin K., Shao L., et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat Commun. 2017;8:2116. doi: 10.1038/s41467-017-02029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Piao S., Pei H.Z., Huang B., Baek S.H. Ovarian tumor domain-containing protein 1 deubiquitinates and stabilizes p53. Cell Signal. 2017;33:22–29. doi: 10.1016/j.cellsig.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 250.Yuan L., Lv Y., Li H., Gao H., Song S., Zhang Y., et al. Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat Cell Biol. 2015;17:1169–1181. doi: 10.1038/ncb3218. [DOI] [PubMed] [Google Scholar]
- 251.Du T., Li H., Fan Y., Yuan L., Guo X., Zhu Q., et al. The deubiquitylase OTUD3 stabilizes GRP78 and promotes lung tumorigenesis. Nat Commun. 2019;10:2914. doi: 10.1038/s41467-019-10824-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Yuan J., Luo K., Zhang L., Cheville J.C., Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–396. doi: 10.1016/j.cell.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sun J., Li T., Zhao Y., Huang L., Sun H., Wu H., et al. USP10 inhibits lung cancer cell growth and invasion by upregulating PTEN. Mol Cell Biochem. 2018;441:1–7. doi: 10.1007/s11010-017-3170-2. [DOI] [PubMed] [Google Scholar]
- 254.Wang X., Xia S., Li H., Wang X., Li C., Chao Y., et al. The deubiquitinase USP10 regulates KLF4 stability and suppresses lung tumorigenesis. Cell Death Differ. 2020;27:1747–1764. doi: 10.1038/s41418-019-0458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Lin Z., Yang H., Tan C., Li J., Liu Z., Quan Q., et al. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013;5:1639–1649. doi: 10.1016/j.celrep.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Weisberg E.L., Schauer N.J., Yang J., Lamberto I., Doherty L., Bhatt S., et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat Chem Biol. 2017;13:1207–1215. doi: 10.1038/nchembio.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liao Y., Liu N., Xia X., Guo Z., Li Y., Jiang L., et al. USP10 modulates the SKP2/Bcr–Abl axis via stabilizing SKP2 in chronic myeloid leukemia. Cell Discov. 2019;5:24. doi: 10.1038/s41421-019-0092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhu H., Yan F., Yuan T., Qian M., Zhou T., Dai X., et al. USP10 promotes proliferation of hepatocellular carcinoma by deubiquitinating and stabilizing YAP/TAZ. Cancer Res. 2020;80:2204–2216. doi: 10.1158/0008-5472.CAN-19-2388. [DOI] [PubMed] [Google Scholar]
- 259.Lu C., Ning Z., Wang A., Chen D., Liu X., Xia T., et al. USP10 suppresses tumor progression by inhibiting mTOR activation in hepatocellular carcinoma. Cancer Lett. 2018;436:139–148. doi: 10.1016/j.canlet.2018.07.032. [DOI] [PubMed] [Google Scholar]
- 260.Chandrashekar D.S., Karthikeyan S.K., Korla P.K., Patel H., Shovon A.R., Athar M., et al. UALCAN: an update to the integrated cancer data analysis platform. Neoplasia. 2022;25:18–27. doi: 10.1016/j.neo.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Chandrashekar D.S., Bashel B., Balasubramanya S.A.H., Creighton C.J., Ponce-Rodriguez I., Chakravarthi B., et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Majeed S., Aparnathi M.K., Nixon K.C.J., Venkatasubramanian V., Rahman F., Song L., et al. Targeting the ubiquitin-proteasome system using the UBA1 inhibitor TAK-243 is a potential therapeutic strategy for small-cell lung cancer. Clin Cancer Res. 2022;28:1966–1978. doi: 10.1158/1078-0432.CCR-21-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Best S., Hashiguchi T., Kittai A., Bruss N., Paiva C., Okada C., et al. Targeting ubiquitin-activating enzyme induces ER stress-mediated apoptosis in B-cell lymphoma cells. Blood Adv. 2019;3:51–62. doi: 10.1182/bloodadvances.2018026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Barghout S.H., Aman A., Nouri K., Blatman Z., Arevalo K., Thomas G.E., et al. A genome-wide CRISPR/Cas9 screen in acute myeloid leukemia cells identifies regulators of TAK-243 sensitivity. JCI Insight. 2021;6 doi: 10.1172/jci.insight.141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Shan Y., Yang G., Huang H., Zhou Y., Hu X., Lu Q., et al. Ubiquitin-like modifier activating enzyme 1 as a novel diagnostic and prognostic indicator that correlates with ferroptosis and the malignant phenotypes of liver cancer cells. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.592413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhuang J., Shirazi F., Singh R.K., Kuiatse I., Wang H., Lee H.C., et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood. 2019;133:1572–1584. doi: 10.1182/blood-2018-06-859686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kim S.J., Hyeong Lee T., Hee Nam S., Kim J.H., Oh S., Sook Cho Y., et al. Association of Uba6-specific-E2 (USE1) with lung tumorigenesis. J Natl Cancer Inst. 2017;109:1–11. doi: 10.1093/jnci/djw224. [DOI] [PubMed] [Google Scholar]
- 268.Osoegawa A., Yoshino I., Tanaka S., Sugio K., Kameyama T., Yamaguchi M., et al. Regulation of p27 by S-phase kinase-associated protein 2 is associated with aggressiveness in non-small-cell lung cancer. J Clin Oncol. 2004;22:4165–4173. doi: 10.1200/JCO.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 269.Richter K.T., Kschonsak Y.T., Vodicska B., Hoffmann I. FBXO45-MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 2020;27:758–772. doi: 10.1038/s41418-019-0385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Schweiggert J., Habeck G., Hess S., Mikus F., Beloshistov R., Meese K., et al. SCFFbxw5 targets kinesin-13 proteins to facilitate ciliogenesis. EMBO J. 2021;40 doi: 10.15252/embj.2021107735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yao Y., Liu Z., Huang S., Huang C., Cao Y., Li L., et al. The E3 ubiquitin ligase, FBXW5, promotes the migration and invasion of gastric cancer through the dysregulation of the Hippo pathway. Cell Death Dis. 2022;8:79. doi: 10.1038/s41420-022-00868-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Yang F., Xu J., Li H., Tan M., Xiong X., Sun Y. FBXW2 suppresses migration and invasion of lung cancer cells via promoting beta-catenin ubiquitylation and degradation. Nat Commun. 2019;10:1382. doi: 10.1038/s41467-019-09289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Shimizu K., Nihira N.T., Inuzuka H., Wei W. Physiological functions of FBW7 in cancer and metabolism. Cell Signal. 2018;46:15–22. doi: 10.1016/j.cellsig.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wilkinson K.D., Smith S.E., O'Connor L., Sternberg E., Taggart J.J., Berges D.A., et al. A specific inhibitor of the ubiquitin activating enzyme: synthesis and characterization of adenosyl-phospho-ubiquitinol, a nonhydrolyzable ubiquitin adenylate analogue. Biochemistry. 1990;29:7373–7380. doi: 10.1021/bi00484a004. [DOI] [PubMed] [Google Scholar]
- 275.Yu Q., Jiang Y., Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. 2020;10:746–765. doi: 10.1016/j.apsb.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Soucy T.A., Smith P.G., Milhollen M.A., Berger A.J., Gavin J.M., Adhikari S., et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 277.Brownell J.E., Sintchak M.D., Gavin J.M., Liao H., Bruzzese F.J., Bump N.J., et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic. in situ. Mol Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 278.Chen J.J., Tsu C.A., Gavin J.M., Milhollen M.A., Bruzzese F.J., Mallender W.D., et al. Mechanistic studies of substrate-assisted inhibition of ubiquitin-activating enzyme by adenosine sulfamate analogues. J Biol Chem. 2011;286:40867–40877. doi: 10.1074/jbc.M111.279984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Langston S.P., Grossman S., England D., Afroze R., Bence N., Bowman D., et al. Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J Med Chem. 2021;64:2501–2520. doi: 10.1021/acs.jmedchem.0c01491. [DOI] [PubMed] [Google Scholar]
- 280.Ceccarelli D.F., Tang X., Pelletier B., Orlicky S., Xie W., Plantevin V., et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–1087. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 281.Huang H., Ceccarelli D.F., Orlicky S., St-Cyr D.J., Ziemba A., Garg P., et al. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat Chem Biol. 2014;10:156–163. doi: 10.1038/nchembio.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Tsukamoto S. Search for inhibitors of the ubiquitin-proteasome system from natural sources for cancer therapy. Chem Pharm Bull (Tokyo) 2016;64:112–118. doi: 10.1248/cpb.c15-00768. [DOI] [PubMed] [Google Scholar]
- 283.Kim Y.S., Nagy K., Keyser S., Schneekloth J.S., Jr. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem Biol. 2013;20:604–613. doi: 10.1016/j.chembiol.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Blaquiere N., Villemure E., Staben S.T. Medicinal chemistry of inhibiting RING-type E3 ubiquitin ligases. J Med Chem. 2020;63:7957–7985. doi: 10.1021/acs.jmedchem.9b01451. [DOI] [PubMed] [Google Scholar]
- 285.Duran-Frigola M., Cigler M., Winter G.E. Advancing targeted protein degradation via multiomics profiling and artificial intelligence. J Am Chem Soc. 2023;145:2711–2732. doi: 10.1021/jacs.2c11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chamberlain P.P., Hamann L.G. Development of targeted protein degradation therapeutics. Nat Chem Biol. 2019;15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 287.Wang Y., Jiang X., Feng F., Liu W., Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Li Z., Lin Y., Song H., Qin X., Yu Z., Zhang Z., et al. First small-molecule PROTACs for G protein-coupled receptors: inducing α1A-adrenergic receptor degradation. Acta Pharm Sin B. 2020;10:1669–1679. doi: 10.1016/j.apsb.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Lu B., Ye J. Commentary: PROTACs make undruggable targets druggable: challenge and opportunity. Acta Pharm Sin B. 2021;11:3335–3336. doi: 10.1016/j.apsb.2021.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Diehl C.J., Ciulli A. Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem Soc Rev. 2022;51:8216–8257. doi: 10.1039/d2cs00387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.He S., Ma J., Fang Y., Liu Y., Wu S., Dong G., et al. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm Sin B. 2021;11:1617–1628. doi: 10.1016/j.apsb.2020.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Luo G., Li Z., Lin X., Li X., Chen Y., Xi K., et al. Discovery of an orally active VHL-recruiting PROTAC that achieves robust HMGCR degradation and potent hypolipidemic activity in vivo. Acta Pharm Sin B. 2021;11:1300–1314. doi: 10.1016/j.apsb.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chan C.H., Morrow J.K., Li C.F., Gao Y., Jin G., Moten A., et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wu L., Grigoryan A.V., Li Y., Hao B., Pagano M., Cardozo T.J. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19:1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Hon W.C., Wilson M.I., Harlos K., Claridge T.D., Schofield C.J., Pugh C.W., et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 296.Min J.H., Yang H., Ivan M., Gertler F., Kaelin W.G., Jr., Pavletich N.P. Structure of an HIF-1alpha–pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 297.Buckley D.L., Van Molle I., Gareiss P.C., Tae H.S., Michel J., Noblin D.J., et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Buckley D.L., Gustafson J.L., Van Molle I., Roth A.G., Tae H.S., Gareiss P.C., et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Soares P., Gadd M.S., Frost J., Galdeano C., Ellis L., Epemolu O., et al. Group-based optimization of potent and cell-active inhibitors of the von Hippel-Lindau (VHL) E3 ubiquitin ligase: structure–activity relationships leading to the chemical probe (2S,4R)-1-((S)-2-(1-cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy -N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298) J Med Chem. 2018;61:599–618. doi: 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Maniaci C., Hughes S.J., Testa A., Chen W., Lamont D.J., Rocha S., et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun. 2017;8:830. doi: 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ma B., Lucas B., Capacci A., Lin E.Y., Jones J.H., Dechantsreiter M., et al. Design, synthesis and identification of novel, orally bioavailable non-covalent Nrf2 activators. Bioorg Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2019.126852. [DOI] [PubMed] [Google Scholar]
- 302.Cleasby A., Yon J., Day P.J., Richardson C., Tickle I.J., Williams P.A., et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One. 2014;9 doi: 10.1371/journal.pone.0098896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hu L., Magesh S., Chen L., Wang L., Lewis T.A., Chen Y., et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1–Nrf2 protein–protein interaction. Bioorg Med Chem Lett. 2013;23:3039–3043. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Jiang Z.Y., Lu M.C., Xu L.L., Yang T.T., Xi M.Y., Xu X.L., et al. Discovery of potent Keap1–Nrf2 protein–protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014;57:2736–2745. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 305.Heightman T.D., Callahan J.F., Chiarparin E., Coyle J.E., Griffiths-Jones C., Lakdawala A.S., et al. Structure–activity and structure-conformation relationships of aryl propionic acid inhibitors of the Kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2 (KEAP1/NRF2) protein-protein interaction. J Med Chem. 2019;62:4683–4702. doi: 10.1021/acs.jmedchem.9b00279. [DOI] [PubMed] [Google Scholar]
- 306.Pintard L., Willis J.H., Willems A., Johnson J.L., Srayko M., Kurz T., et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- 307.Zhuang M., Calabrese M.F., Liu J., Waddell M.B., Nourse A., Hammel M., et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36:39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Dong Z., Wang Z., Guo Z.Q., Gong S., Zhang T., Liu J., et al. Correction to structure–activity relationship of SPOP inhibitors against kidney cancer. J Med Chem. 2021;64:905. doi: 10.1021/acs.jmedchem.0c02084. [DOI] [PubMed] [Google Scholar]
- 309.Guo Z.Q., Zheng T., Chen B., Luo C., Ouyang S., Gong S., et al. Small-molecule targeting of E3 ligase adaptor SPOP in kidney cancer. Cancer Cell. 2016;30:474–484. doi: 10.1016/j.ccell.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 310.Lopez J., John S.W., Tenev T., Rautureau G.J., Hinds M.G., Francalanci F., et al. CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol Cell. 2011;42:569–583. doi: 10.1016/j.molcel.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 311.Varfolomeev E., Blankenship J.W., Wayson S.M., Fedorova A.V., Kayagaki N., Garg P., et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 312.Du C., Fang M., Li Y., Li L., Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 313.Wu G., Chai J., Suber T.L., Wu J.W., Du C., Wang X., et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 314.Flygare J.A., Beresini M., Budha N., Chan H., Chan I.T., Cheeti S., et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152) J Med Chem. 2012;55:4101–4113. doi: 10.1021/jm300060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Peng Y., Sun H., Nikolovska-Coleska Z., Qiu S., Yang C.Y., Lu J., et al. Potent, orally bioavailable diazabicyclic small-molecule mimetics of second mitochondria-derived activator of caspases. J Med Chem. 2008;51:8158–8162. doi: 10.1021/jm801254r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Baggio C., Gambini L., Udompholkul P., Salem A.F., Aronson A., Dona A., et al. Design of potent pan-IAP and Lys-covalent XIAP selective inhibitors using a thermodynamics driven approach. J Med Chem. 2018;61:6350–6363. doi: 10.1021/acs.jmedchem.8b00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Sun H., Nikolovska-Coleska Z., Lu J., Meagher J.L., Yang C.Y., Qiu S., et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc. 2007;129:15279–15294. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Eckelman B.P., Salvesen G.S., Scott F.L. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Li L., Thomas R.M., Suzuki H., De Brabander J.K., Wang X., Harran P.G. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 320.Li N., Feng L., Han H.Q., Yuan J., Qi X.K., Lian Y.F., et al. A novel Smac mimetic APG-1387 demonstrates potent antitumor activity in nasopharyngeal carcinoma cells by inducing apoptosis. Cancer Lett. 2016;381:14–22. doi: 10.1016/j.canlet.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 321.Condon S.M., Mitsuuchi Y., Deng Y., LaPorte M.G., Rippin S.R., Haimowitz T., et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57:3666–3677. doi: 10.1021/jm500176w. [DOI] [PubMed] [Google Scholar]
- 322.Johnson C.N., Ahn J.S., Buck I.M., Chiarparin E., Day J.E.H., Hopkins A., et al. A fragment-derived clinical candidate for antagonism of X-linked and cellular inhibitor of apoptosis proteins: 1-(6-[(4-fluorophenyl)methyl]-5-(hydroxymethyl)-3,3-dimethyl-1H,2H,3H-pyrrolo[3,2-b]pyridin-1-yl)-2-[(2R,5R)-5-methyl-2-([(3R)-3-methylmorpholin-4-yl]methyl)piperazin-1-yl]ethan-1-one (ASTX660) J Med Chem. 2018;61:7314–7329. doi: 10.1021/acs.jmedchem.8b00900. [DOI] [PubMed] [Google Scholar]
- 323.Fang Y., Liao G., Yu B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: advances and perspectives. Acta Pharm Sin B. 2020;10:1253–1278. doi: 10.1016/j.apsb.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 325.Vu B., Wovkulich P., Pizzolato G., Lovey A., Ding Q., Jiang N., et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;4:466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ding K., Lu Y., Nikolovska-Coleska Z., Qiu S., Ding Y., Gao W., et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 327.Aguilar A., Lu J., Liu L., Du D., Bernard D., McEachern D., et al. Discovery of 4-((3′R,4′S,5′R)-6′′-chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2′′-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3′′-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic acid (AA-115/APG-115): a potent and orally active murine double minute 2 (MDM2) inhibitor in clinical development. J Med Chem. 2017;60:2819–2839. doi: 10.1021/acs.jmedchem.6b01665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Wang S., Sun W., Zhao Y., McEachern D., Meaux I., Barriere C., et al. SAR405838: an optimized inhibitor of MDM2–p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Ishizawa J., Nakamaru K., Seki T., Tazaki K., Kojima K., Chachad D., et al. Predictive gene signatures determine tumor sensitivity to MDM2 inhibition. Cancer Res. 2018;78:2721–2731. doi: 10.1158/0008-5472.CAN-17-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Chessari G., Hardcastle I.R., Ahn J.S., Anil B., Anscombe E., Bawn R.H., et al. Structure-based design of potent and orally active isoindolinone inhibitors of MDM2–p53 protein–protein interaction. J Med Chem. 2021;64:4071–4088. doi: 10.1021/acs.jmedchem.0c02188. [DOI] [PubMed] [Google Scholar]
- 331.Sun D., Li Z., Rew Y., Gribble M., Bartberger M.D., Beck H.P., et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2–p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 332.Gessier F., Kallen J., Jacoby E., Chene P., Stachyra-Valat T., Ruetz S., et al. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53–MDM2 interaction with a distinct binding mode. Bioorg Med Chem Lett. 2015;25:3621–3625. doi: 10.1016/j.bmcl.2015.06.058. [DOI] [PubMed] [Google Scholar]
- 333.Holzer P., Masuya K., Furet P., Kallen J., Valat-Stachyra T., Ferretti S., et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58:6348–6358. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- 334.Yang Z., Sun Y., Ni Z., Yang C., Tong Y., Liu Y., et al. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021;31:1315–1318. doi: 10.1038/s41422-021-00533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kozicka Z., Thoma N.H. Haven't got a glue: protein surface variation for the design of molecular glue degraders. Cell Chem Biol. 2021;28:1032–1047. doi: 10.1016/j.chembiol.2021.04.009. [DOI] [PubMed] [Google Scholar]
- 336.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 337.Zhu Y.X., Braggio E., Shi C.X., Bruins L.A., Schmidt J.E., Van Wier S., et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118:4771–4779. doi: 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kronke J., Udeshi N.D., Narla A., Grauman P., Hurst S.N., McConkey M., et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Han T., Goralski M., Gaskill N., Capota E., Kim J., Ting T.C., et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356 doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- 340.Slabicki M., Kozicka Z., Petzold G., Li Y.D., Manojkumar M., Bunker R.D., et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. 2020;585:293–297. doi: 10.1038/s41586-020-2374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lv L., Chen P., Cao L., Li Y., Zeng Z., Cui Y., et al. Discovery of a molecular glue promoting CDK12–DDB1 interaction to trigger cyclin K degradation. Elife. 2020;9 doi: 10.7554/eLife.59994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Mayor-Ruiz C., Bauer S., Brand M., Kozicka Z., Siklos M., Imrichova H., et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat Chem Biol. 2020;16:1199–1207. doi: 10.1038/s41589-020-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Spradlin J.N., Hu X., Ward C.C., Brittain S.M., Jones M.D., Ou L., et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol. 2019;15:747–755. doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Henning N.J., Manford A.G., Spradlin J.N., Brittain S.M., Zhang E., McKenna J.M., et al. Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J Am Chem Soc. 2022;144:701–708. doi: 10.1021/jacs.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Backus K.M., Correia B.E., Lum K.M., Forli S., Horning B.D., Gonzalez-Paez G.E., et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Chen J., Dexheimer T.S., Ai Y., Liang Q., Villamil M.A., Inglese J., et al. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Mistry H., Hsieh G., Buhrlage S.J., Huang M., Park E., Cuny G.D., et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Therapeut. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., et al. A selective USP1–UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Dexheimer T.S., Rosenthal A.S., Luci D.K., Liang Q., Villamil M.A., Chen J., et al. Synthesis and structure-activity relationship studies of N-benzyl-2-phenylpyrimidin-4-amine derivatives as potent USP1/UAF1 deubiquitinase inhibitors with anticancer activity against nonsmall cell lung cancer. J Med Chem. 2014;57:8099–8110. doi: 10.1021/jm5010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Ohayon S., Refua M., Hendler A., Aharoni A., Brik A. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew Chem Int Ed Engl. 2015;54:599–603. doi: 10.1002/anie.201408411. [DOI] [PubMed] [Google Scholar]
- 351.Davis M.I., Pragani R., Fox J.T., Shen M., Parmar K., Gaudiano E.F., et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chem. 2016;291:24628–24640. doi: 10.1074/jbc.M116.738567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Magiera K., Tomala M., Kubica K., De Cesare V., Trost M., Zieba B.J., et al. Lithocholic acid hydroxyamide destabilizes Cyclin D1 and induces G0/G1 arrest by inhibiting deubiquitinase USP2a. Cell Chem Biol. 2017;24:458–470. doi: 10.1016/j.chembiol.2017.03.002. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Tavana O., Gu W. Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol. 2017;9:45–52. doi: 10.1093/jmcb/mjw049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Zhang L., Ye B., Chen Z., Chen Z.S. Progress in the studies on the molecular mechanisms associated with multidrug resistance in cancers. Acta Pharm Sin B. 2023;13:982–997. doi: 10.1016/j.apsb.2022.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Wu J., Kumar S., Wang F., Wang H., Chen L., Arsenault P., et al. Chemical approaches to intervening in ubiquitin specific protease 7 (USP7) function for oncology and immune oncology therapies. J Med Chem. 2018;61:422–443. doi: 10.1021/acs.jmedchem.7b00498. [DOI] [PubMed] [Google Scholar]
- 356.Chauhan D., Tian Z., Nicholson B., Kumar K.G., Zhou B., Carrasco R., et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Lamberto I., Liu X., Seo H.S., Schauer N.J., Iacob R.E., Hu W., et al. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem Biol. 2017;24:1490–1500. doi: 10.1016/j.chembiol.2017.09.003. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Turnbull A.P., Ioannidis S., Krajewski W.W., Pinto-Fernandez A., Heride C., Martin A.C.L., et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481–486. doi: 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Gavory G., O'Dowd C.R., Helm M.D., Flasz J., Arkoudis E., Dossang A., et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14:118–125. doi: 10.1038/nchembio.2528. [DOI] [PubMed] [Google Scholar]
- 360.Leger P.R., Hu D.X., Biannic B., Bui M., Han X., Karbarz E., et al. Discovery of potent, selective, and orally bioavailable inhibitors of USP7 with in vivo antitumor activity. J Med Chem. 2020;63:5398–5420. doi: 10.1021/acs.jmedchem.0c00245. [DOI] [PubMed] [Google Scholar]
- 361.Kategaya L., Di Lello P., Rouge L., Pastor R., Clark K.R., Drummond J., et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550:534–538. doi: 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- 362.Verma R., Aravind L., Oania R., McDonald W.H., Yates J.R. 3rd, Koonin E.V., et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 363.Sherman D.J., Li J. Proteasome inhibitors: harnessing proteostasis to combat disease. Molecules. 2020;25:671. doi: 10.3390/molecules25030671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Li J., Yakushi T., Parlati F., Mackinnon A.L., Perez C., Ma Y., et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13:486–493. doi: 10.1038/nchembio.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Li J., Zhang Y., Da Silva Sil Dos Santos B., Wang F., Ma Y., Perez C., et al. Epidithiodiketopiperazines inhibit protein degradation by targeting proteasome deubiquitinase Rpn11. Cell Chem Biol. 2018;25:1350–1358. doi: 10.1016/j.chembiol.2018.07.012. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Ward J.A., Pinto-Fernandez A., Cornelissen L., Bonham S., Diaz-Saez L., Riant O., et al. Re-evaluating the mechanism of action of α,β-unsaturated carbonyl DUB inhibitors b-AP15 and VLX1570: a paradigmatic example of unspecific protein cross-linking with Michael acceptor motif-containing drugs. J Med Chem. 2020;63:3756–3762. doi: 10.1021/acs.jmedchem.0c00144. [DOI] [PMC free article] [PubMed] [Google Scholar]