

Abstract
Herein, we report on the first implementation of charged microdroplet-based derivatization on a commercially-available cyclic ion mobility spectrometry-mass spectrometry platform. We have demonstrated the potential for our approach to improve separability of challenging isomers, but more importantly to rapidly screen derivatization reactions through droplet chemistry. Additionally, the use of cyclic ion mobility separations and tandem mass spectrometry reveals insights into product formation that would be lost with single stage mass spectrometry. Overall, we anticipate broad utility of our methodology owing to the simple design and setup for performing these droplet-based reactions and future work coupling these reactions online with liquid chromatography.
Graphical Abstract

1. Introduction
Recent work has indicated that bulk-phase reactions can be greatly accelerated (between 2 and 6 orders of magnitude) when, instead, performed in charged microdroplets generated through the electrospray ionization (ESI) process in mass spectrometry 1–13. While the mechanism for this observed acceleration in microdroplets is still unclear, it has been hypothesized that the desolvation step of ESI-based processes permits the reagents to be further concentrated and thus react in a very confined volume, resulting in lower energy barriers and faster reactions1, 2. In addition to the variables that affect bulk-phase reactions (e.g., solvent composition, stoichiometric ratios of reagents, temperature), it has been posited that the amount of product in accelerated droplet reactions also depends on droplet size and the total length/distance of the reaction region (i.e., from droplet generation via ESI to the MS inlet) 1–13. However, many of these previous literature examples of charged microdroplet-based reaction accelerations have been performed on homebuilt, or partially-modified, mass spectrometers/ionization sources3–11.
Isomers or conformers, by definition, share identical mass-to-charge (m/z) thus making them impossible to identify by mass spectrometry (MS), alone. While tandem mass spectrometry (MS/MS)-based methods have shown promise for isomer delineation, more recent efforts have focused on coupling separations to MS, instead 14–17. Ion mobility spectrometry coupled to mass spectrometry (IMS-MS) separates ions in the gas phase under the presence of an electric field based on their 3-D structures/shapes and charge (i.e., mobilities), where more compact higher mobility species will arrive first, and more elongated lower mobility species will arrive last. This relationship between size/shape (i.e., collision cross section), charge, and mobility is defined according to the Mason-Schamp equation 14–17. In recent years, IMS-MS has seen widespread use in omics-based research for the resolution of isobaric/isomeric components and provides a rapid, orthogonal, and complementary alternative to condensed-phase methods 17–20.
Recently, the development of high-resolution IMS-MS platforms (e.g., trapped; TIMS, cyclic; cIMS-MS, and structures for lossless ion manipulations; SLIM IMS-MS) has enabled advancements in the ability to resolve very structurally similar compounds, which differ in their ion-neutral rotationally averaged collision cross sections (i.e., CCS values) by <1% 21–26. Unfortunately, even with such technological advancements in resolution, the ability to resolve certain biologically-relevant isomer sets remains challenging 27–33. This presents the urgent need to develop new analytical methodologies to enable the separation of previously intractable isomeric species. Of note, derivatization (i.e., chemical modification) remains one of the most common methods for improving the separability of isomers 19, 34–35. Unfortunately, derivatization methods are often time consuming and laborious in nature. Thus, new analytical methods to enable the fast derivatization and rapid screening of reactions that could improve isomer separations are desperately needed. Herein, we assess the use droplet-based reactions performed in the charged microdroplets generated by electrospray ionization as a method for derivatizing molecules which can then be subsequently separated with high-resolution cyclic ion mobility separations and mass spectrometry. The cIMS-MS platform used in these experiments was selected as it is the highest resolution IMS platform that is currently commercially available. Our overall goal and focus of this work was to implement and assess the use of droplet-based hydrazide derivatization of bile acids on a commercial cIMS-MS platform. Figure 1 highlights the structures and hydrazide tags used in this study.
Figure 1.

Structures of the bile acids and hydrazide tags used: Girard’s Reagent T (GT), N-boc-L-histidine hydrazide, L-tyrosine hydrazide, 3-oxo deoxycholic acid (3-ODCA), 3-oxo chenodeoxycholic acid (3-OCDCA), Δ4-Dafachronic acid (4-DCA), Δ7-Dafachronic acid (7-DCA), 3-oxo allocholic acid (3-OACA), and 3-oxocholic acid (3-OCA).
2. Experimental Section
Please refer to the Supporting Information for all experimental details and optimization of droplet-based reactions (Table S1).
3. Results and Discussion
To perform droplet-based reactions, we designed a dual syringe setup (Image S1). We utilized a dual syringe set-up instead of mixing the derivatization reagent in a single to prove the viability of our long-term goal of performing online derivatization post-column in the context of LC separations. To demonstrate and verify if our dual syringe setup was indeed capable of performing droplet-based reactions and thus reaction acceleration, we began by replicating an experiment from previous literature2. Specifically, we replicated the derivatization of cortisone on a commercial ESI source instead of a custom-built DESI source. We utilized our dual syringe approach, where one syringe contained a 10 μM cortisone solution, and the other syringe contained a 10 μM solution of Girard’s Reagent T (GT), both dissolved in an 80/20 (v/v) water/methanol with 0.5% (v/v) formic acid and at flow rates of 5 μL/min. We observed the hydrazide-tagged cortisone precursor ion at m/z 474.3 for the [M]+ species. We further verified this to be correct by performing MS/MS, which matched the MS/MS spectrum from previous literature (see Figure S1) 2.
After validating that our dual syringe setup was capable of performing droplet-based reactions, we wanted to assess if these reactions were also possible for other molecules along with other hydrazide tags to build upon the cortisone condensation reactions. Specifically, we were interested in assessing droplet-based hydrazide derivatization for bile acids because of their many possible isomeric permutations which could then be subjected to cIMS-MS and assessed for their overall separability. Initially, it was unclear whether these reactions would even occur given that bile acids are dissimilar in their structures as compared to steroids as well as the fact that we are derivatizing using hydrazide tags previously unreported in literature. Additionally, it was uncertain if cIMS-MS-based separations would enable any new information on product formation to be obtained (i.e., presence of multiple products or observation of multiple gas-phase conformers).
Figure 2A highlights our overall workflow, where bile acids and hydrazide tags are kept in separate syringes, mixed with a T-in junction and subjected to ESI, where the droplet reactions occur in the ~10 mm long microdroplet spray region. From there, we can generate the mass spectrum for each reaction and assess product formation, as well as being able to perform a cIMS-MS separation for desired product ions of interest. We assessed all combinations of bile acids with the various hydrazide tags shown in Figure 1 to determine if droplet-based reactions were possible for these systems and optimal concentration ratios, solvent compositions and impact of acid catalyst additives (see Table S1 in the Supporting Information). We would like to note that we were able to synthesize bile acid hydrazide derivatives in bulk, according to previously published hydrazide derivatization procedures 36, with keto groups in positions other than the third one, such as 7-ketolithocholic acid, 7-ketodeoxycholic acid and 12-ketolithocholic acid. However, we did not observe product formation in droplets, which we speculate can be attributed to the orientation of molecules within the droplets 13. Figure 2B shows an example of 3-OCDOCA derivatized in charged microdroplets with N-boc-L-histidine hydrazide and its corresponding MS/MS spectrum. The red trace in Figure 2B is the reaction performed in droplets with our dual syringe approach, whereas the green trace was performed in bulk. It can be observed that the inverted tandem mass spectra agree well with one another, thus demonstrating the same product was formed in both bulk and droplets. To validate that our pre-mixing step at the T-in junction prior to ESI is not responsible for any product formation, we performed a control experiment. Specifically, we derivatized 3-OCA with GT in droplets and collected its mass spectrum for the precursor ion (Figure 2C, yellow trace). Next, we subjected this same reaction to ambient bulk conditions for 1 hour (i.e., the 3-OCA and GT were left mixing in a single syringe for 1 hour at room temperature). We then subjected this ambient bulk sample to ESI-MS, and thus subjecting them to droplet reactions, and its mass spectrum (Figure 2C, purple trace) matches the absolute intensity observed for our droplet-based reactions (i.e., no bulk pre-mixing step). To determine if any product was formed during microflow, we performed an additional experiment. Specifically, we used a single syringe setup and performed an offline microflow experiment for 1 hour where we then collected the resulting material. This material was then subjected to direct infusion. Since our signal intensities were similar in magnitude between our dual syringe setup (i.e., droplets) and that of the microflow experiment plus direct infusion (i.e., microflow plus droplets), we can then conclude that our product formation was obtained in droplets with negligible contributions from microflow (see Figure S2 in the Supporting Information). Since all three experiments have the common factor of the reaction occurring in droplets and the MS signal intensities are similar, we can conclude that the contribution from the 1 hour bulk reaction and 1 hour microflow reaction are negligible. Thus, we can definitively conclude that neither the pre-mixing step nor the microflow contribute to any significant product formation and that our dual syringe setup can indeed be used to accelerate bulk-phase reactions through charged microdroplet chemistry in the ESI process. We would like to note that other reactions could potentially occur in the pre-mixing step, thus care should be taken by performing these control experiments outlined. For other droplet-based derivatization reactions performed with the other hydrazide tags and bile acids, please see the Figures S3–S5 in the Supporting Information.
Figure 2.
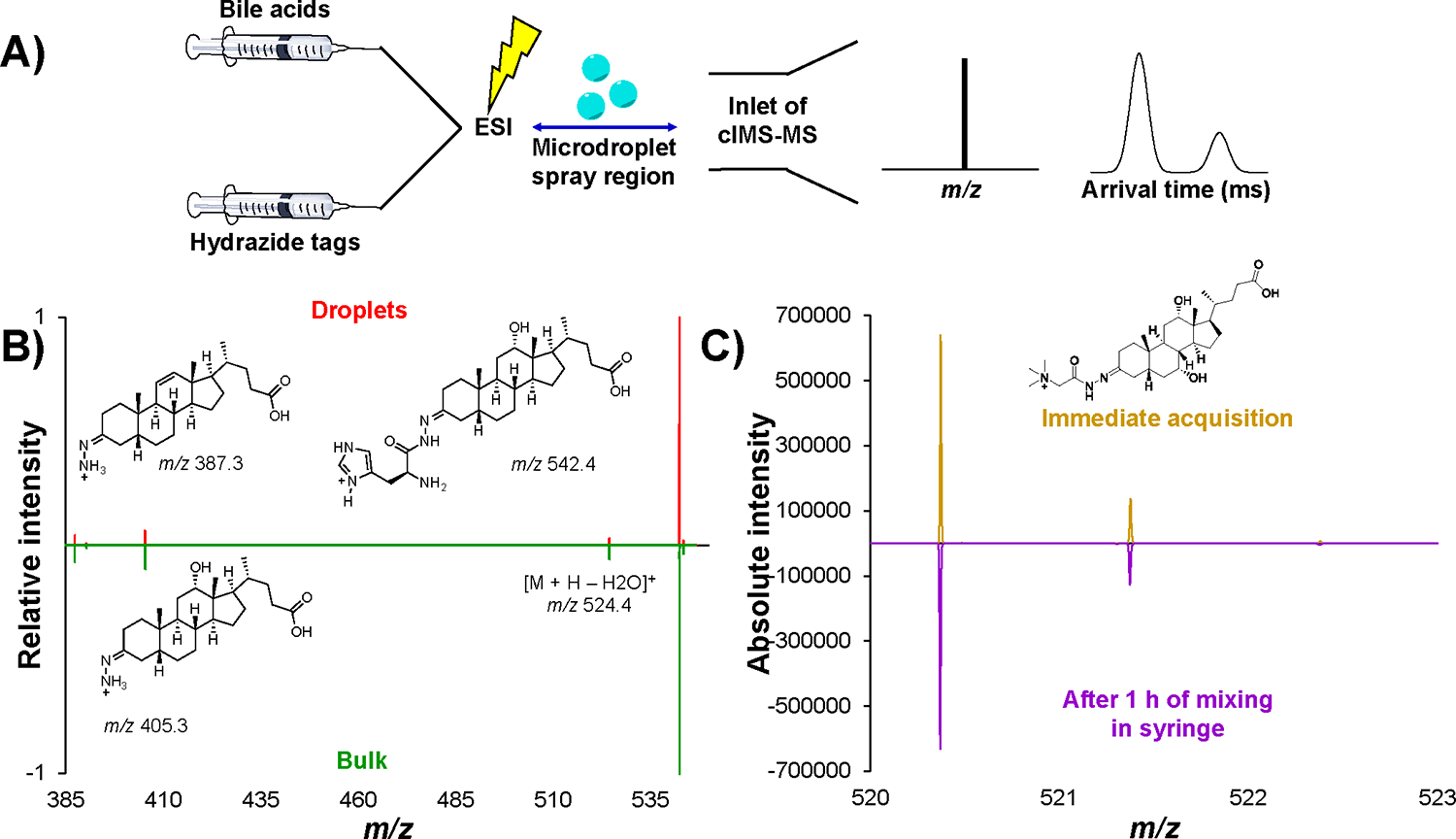
Dual syringe experimental setup (A). Inverted tandem mass spectra highlighting fragment ions for the products of the reaction of 3-OCDOCA & N-boc-L-histidine hydrazide in (red) and bulk (green) at a collision induced dissociation (CID) voltage of 40 V with signal averaging performed for two minutes for each (B). Inverted mass spectra of the product of the reaction of 3-OCA and GT with signal averaging performed for four minutes for each (C). It is noted that this data was collected in “pass-through” mode, where no cIMS-MS separation occurs.
Based on our results from Figure 2 demonstrating that droplet-based reactions can be performed on our cIMS-MS platform, we next wanted to extend our methodology to explore separations of our hydrazide-tagged bile acid isomers. Figure 3 highlights two pairs of bile acid isomers that can be separated as their N-boc-L-histidine hydrazide derivatives, which were synthesized in charged microdroplets. These demonstrations indicate that droplet-based reactions are compatible with the timescale necessary for cIMS-MS separations. From both separations shown in Figure 3, we can observe that baseline resolution is achieved for both isomeric sets as their N-boc-L-histidine hydrazide derivatives generated through droplet-based reactions. Overall, this demonstrates the additional information that can be obtained through cyclic ion mobility separations, which cannot be obtained with MS alone. In the case of the derivatization of 7-DCA with L-tyrosine hydrazide, we observed two distinct IMS peaks after a 5 m separation (Figure 4A) indicating two isomeric products were formed through our droplet-based reactions for an individual bile acid. To determine the structure of these two products, we performed post-cIMS-MS/MS which revealed unique fragmentation patterns for each observed peak (Figures 4B and C). This enabled us to elucidate each of their structures, which differed in the location of the hydrazide tag. We do also acknowledge the possibility that in the case of the product depicted in red, the derivatizing agent might have attached to the amine nitrogen instead of the hydrazone nitrogen. Overall, this demonstrates the power of coupling droplet-based reactions with high-resolution cIMS-MS separations to provide further insight into product formation that otherwise would not be attainable with MS-only methods.
Figure 3.

10 m cIMS-MS separation of the products of the 4-DCA and 7-DCA isomers with N-boc-L-histidine hydrazide as its [M + H]+ adducts under TW conditions of 450 m/s and 32 V with signal averaging performed for four minutes (A). 2 m cIMS-MS separation of the products of the 3-OCA and 3-OACA isomers with N-boc-L-histidine hydrazide as its [M + H]+ adducts under TW conditions of 450 m/s and 35 V with signal averaging performed for two minutes (B). The dotted black trace represents the isomeric mixture.
Figure 4.

5 m cIMS-MS separation of two observed products in the same experiment for the reaction between 7-DCA and L-tyrosine hydrazide as its [M + H]+ adduct under TW conditions of 400 m/s and 35 V with signal averaging performed for four minutes (A). The potential structures of the two products (A) were derived based on the tandem mass spectra highlighting fragment ions for the products at CID of 40 V with signal averaging performed for five minutes (B & C).
4. Conclusions
Herein, we have showcased the first implementation of droplet-based reactions on a commercial IMS-MS platform. It is notable to mention that unpublished work from the Chouinard group has also demonstrated similar reactions on an IMS-MS platform 37. Our dual syringe setup requires no instrument modifications and is simple in nature. Our analyses focused on studying the droplet-based hydrazide derivatization of various bile acid isomers. We successfully confirmed that our reactions were indeed occurring in droplets through a control experiment as well as validating that the same product was formed in bulk. By coupling these droplet-based reactions with cIMS-MS separations, we demonstrated that challenging bile acid isomers could indeed be resolved as their hydrazide derivatives. In one instance, we observed two distinct peaks for one individual bile acid that was derivatized. By performing post-cIMS-MS/MS, we were able to determine potential structures for each of the two products. Overall, our presented dual syringe approach for performing droplet-based reactions on a cIMS-MS platform has several advantages. Specifically, we envision that our setup can quickly be used to screen synthetic reactions and enable easily swapping out reagents between both syringes. Additionally, we envision that post-column derivatization can be performed with online chromatographic separations using a T-in junction with the derivatizing reagent 38–40. This could potentially enable higher resolution IMS-MS separations to be achieved for key isomers that otherwise would be unresolvable as their unlabelled versions. We also note that a setup where starting material and derivatization reagents are mixed in a single syringe can be useful when only performing direction infusion.
Overall, we envision that our proof-of-concept demonstration highlights a new analytical methodology and workflow for enabling the ability to rapidly screen derivatization reactions and use cIMS separations and MS/MS to understand product formation. We also envision future applications from our described method to develop derivatization methods to resolve previously intractable isomeric compounds in the context of more realistic biological samples. Additionally, we anticipate that our developed analytical methodology can help enable the rapid screening of other synthetic reactions and thus help guide which reaction conditions can effectively be scaled up in the bulk phase.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (1R35GM146671-01).
Footnotes
Conflicts of Interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Yan X, Bain RM and Cooks RG, Angew Chem Int Ed Engl, 2016, 55, 12960–12972. [DOI] [PubMed] [Google Scholar]
- 2.Girod M, Moyano E, Campbell DI and Cooks RG, Chem. Sci, 2011, 2, 501–510. [Google Scholar]
- 3.Heiss DR and Badu-Tawiah AK, Anal Chem, 2022, 94, 14071–14078. [DOI] [PubMed] [Google Scholar]
- 4.Heiss DR and Badu-Tawiah AK, Anal Chem, 2021, 93, 16779–16786. [DOI] [PubMed] [Google Scholar]
- 5.Holden DT, Morato NM and Cooks RG, Proc Natl Acad Sci U S A, 2022, 119, e2212642119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobs MI, Davis RD, Rapf RJ and Wilson KR, J Am Soc Mass Spectrom, 2019, 30, 339–343. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz M, Ovchinnikova OS and Van Berkel GJ, Rapid Commun Mass Spectrom, 2014, 28, 1312–1320. [DOI] [PubMed] [Google Scholar]
- 8.Sahota N, AbuSalim DI, Wang ML, Brown CJ, Zhang Z, El-Baba TJ, Cook SP and Clemmer DE, Chem Sci, 2019, 10, 4822–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobreira TJP, Avramova L, Szilagyi B, Logsdon DL, Loren BP, Jaman Z, Hilger RT, Hosler RS, Ferreira CR, Koswara A, Thompson DH, Cooks RG and Nagy ZK, Anal Methods, 2020, 12, 3654–3669. [DOI] [PubMed] [Google Scholar]
- 10.Wleklinski M, Loren BP, Ferreira CR, Jaman Z, Avramova L, Sobreira TJP, Thompson DH and Cooks RG, Chem Sci, 2018, 9, 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao P, Gunawardena HP, Zhong X, Zare RN and Chen H, Anal Chem, 2021, 93, 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davey SG, Nat Rev Chem, 2020, 4, 111. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee S, Gnanamani E, Yan X, Zare RN, Analyst, 2017, 142, 1399. [DOI] [PubMed] [Google Scholar]
- 14.Bowers MT, Int J Mass Spectrom, 2014, 370, 75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dodds JN and Baker ES, J Am Soc Mass Spectrom, 2019, 30, 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanucara F, Holman SW, Gray CJ and Eyers CE, Nat Chem, 2014, 6, 281–294. [DOI] [PubMed] [Google Scholar]
- 17.Burnum-Johnson KE, Zheng X, Dodds JN, Ash J, Fourches D, Nicora CD, Wendler JP, Metz TO, Waters KM, Jansson JK, Smith RD and Baker ES, Trends Analyt Chem, 2019, 116, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hines KM, May JC, McLean JA and Xu L, Anal Chem, 2016, 88, 7329–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paglia G, Smith AJ and Astarita G, Mass Spectrom Rev, 2022, 41, 722–765. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Wang Z, Zhang Q, Mei Y, Li L, Liu H, Wang Z and Yang L, Anal Chem, 2023, 95, 134–151. [DOI] [PubMed] [Google Scholar]
- 21.Garimella SVB, Nagy G, Ibrahim YM and Smith RD, Trends Analyt Chem, 2019, 116, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giles K, Ujma J, Wildgoose J, Pringle S, Richardson K, Langridge D and Green M, Anal Chem, 2019, 91, 8564–8573. [DOI] [PubMed] [Google Scholar]
- 23.Delvaux A, Rathahao-Paris E, Guillon B, Cholet S, Adel-Patient K, Fenaille F, Junot C and Alves S, Anal Chim Acta, 2021, 1180, 338878. [DOI] [PubMed] [Google Scholar]
- 24.Miller SA, Jeanne Dit Fouque K, Ridgeway ME, Park MA and Fernandez-Lima F, J Am Soc Mass Spectrom, 2022, 33, 1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williamson DL, Trimble TK and Nagy G, J Am Soc Mass Spectrom, 2023, 34, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habibi SC and Nagy G, Anal Chem, 2023, 95, 8028–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy G, Attah IK, Garimella SVB, Tang K, Ibrahim YM, Baker ES and Smith RD, Chem Commun (Camb), 2018, 54, 11701–11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warnke S, Ben Faleh A, Scutelnic V and Rizzo TR, J Am Soc Mass Spectrom, 2019, 30, 2204–2211. [DOI] [PubMed] [Google Scholar]
- 29.Both P, Green AP, Gray CJ, Sardzik R, Voglmeir J, Fontana C, Austeri M, Rejzek M, Richardson D, Field RA, Widmalm G, Flitsch SL and Eyers CE, Nat Chem, 2014, 6, 65–74. [DOI] [PubMed] [Google Scholar]
- 30.Kemperman RHJ, Chouinard CD and Yost RA, J Am Soc Mass Spectrom, 2023, 34, 1477–1490. [DOI] [PubMed] [Google Scholar]
- 31.Poland JC, Leaptrot KL, Sherrod SD, Flynn CR and McLean JA, J Am Soc Mass Spectrom, 2020, 31, 1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williamson DL, Bergman AE, Heider EC and Nagy G, Anal Chem, 2022, 94, 2988–2995. [DOI] [PubMed] [Google Scholar]
- 33.Zheng X, Smith FB, Aly NA, Cai J, Smith RD, Patterson AD and Baker ES, Anal Bioanal Chem, 2019, 411, 4673–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velosa DC, Dunham AJ, Rivera ME, Neal SP, Chouinard CD, J Am Soc Mass Spectrom, 2022, 33, 1761. [DOI] [PubMed] [Google Scholar]
- 35.Neal SP, Hodges WN, Velosa DC, Aderorho R, Lucas SW, Chouinard CD, Anal Bioanal Chem, 2023, DOI: 10.1007/s00216-023-04953-8. [DOI] [PubMed] [Google Scholar]
- 36.Kalmar JG, Butler KE, Baker ES and Muddiman DC, Anal Bioanal Chem, 2020, 412, 7569–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velosa DC, Neal SP, Dunham AJ, Rivera ME, Silva JGA, Aldrich GJ, Peverati R, Chouinard CD, Proceedings of the American Society for Mass Spectrometry Conference, 2022, Abstract 308873. [Google Scholar]
- 38.Wooke Z, Nagy G, Barnes LF and Pohl NLB, J Am Soc Mass Spectrom, 2019, 30, 419–425. [DOI] [PubMed] [Google Scholar]
- 39.Feng X, Xiang P, Chen H and Shen M, J Anal Toxicol, 2017, 41, 735–743. [DOI] [PubMed] [Google Scholar]
- 40.Onishi S, Itoh S and Ishida Y, Biochem J, 1982, 204, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.